
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Template-free synthesis of mesoporous and amorphous transition metal phosphate materials†
Stephanos
Karafiludis
 *ab,
Ana Guilherme
Buzanich
a,
Christian
Heinekamp
ab,
Annett
Zimathies
a,
Glen J.
Smales
a,
Vasile-Dan
Hodoroaba
a,
Johan E.
ten Elshof
c,
Franziska
Emmerling
ab and
Tomasz M.
Stawski
*a
*ab,
Ana Guilherme
Buzanich
a,
Christian
Heinekamp
ab,
Annett
Zimathies
a,
Glen J.
Smales
a,
Vasile-Dan
Hodoroaba
a,
Johan E.
ten Elshof
c,
Franziska
Emmerling
ab and
Tomasz M.
Stawski
*a
aFederal Institute for Materials Research and Testing, Unter den Eichen 87, 12205 Berlin, Germany. E-mail: tomasz.stawski@bam.de; stephanos.karafiludis@bam.de
bDepartment of Chemistry, Humboldt-Universität zu Berlin, Brook-Taylor-Straße 2, 12489 Berlin, Germany
cMESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE Enschede, the Netherlands
First published on 13th January 2023
Abstract
We present how mesoporosity can be engineered in transition metal phosphate (TMPs) materials in a template-free manner. The method involves the transformation of a precursor metal phosphate phase, called M-struvite (NH4MPO4·6H2O, M = Mg2+, Ni2+, Co2+, NixCo1−x2+). It relies on the thermal decomposition of crystalline M-struvite precursors to an amorphous and simultaneously mesoporous phase, which forms during degassing of NH3 and H2O. The temporal evolution of mesoporous frameworks and the response of the metal coordination environment were followed by in situ and ex situ scattering and diffraction, as well as X-ray spectroscopy. Despite sharing the same precursor struvite structure, different amorphous and mesoporous structures were obtained depending on the involved transition metal. We highlight the systematic differences in absolute surface area, pore shape, pore size, and phase transitions depending on the metal cation present in the analogous M-struvites. The amorphous structures of thermally decomposed Mg-, Ni- and NixCo1−x-struvites exhibit high surface areas and pore volumes (240 m2 g−1 and 0.32 cm−3 g−1 for Mg and 90 m2 g−1 and 0.13 cm−3 g−1 for Ni). We propose that the low-cost, environmentally friendly M-struvites could be obtained as recycling products from industrial and agricultural wastewaters. These waste products could be then upcycled into mesoporous TMPs through a simple thermal treatment for further application, for instance in (electro)catalysis.
Introduction
Transition metal phosphates (TMPs) have been gaining interest as materials for catalysis, electrocatalysis and electrochemistry due to their low cost, stability, and tunability.1–4 For instance, it has been reported that the performance of ammonium-bearing manganese, iron, nickel, and cobalt phosphates in the oxygen evolution reaction (OER) in neutral/alkaline solutions is comparable to IrO2/RuO2.5–8 Ammonium-free Ni and NixCo1−x phosphates demonstrate excellent supercapacitor characteristics,9–11 zirconium and vanadium phosphates are used in organic catalysis, and silver phosphate is used in photocatalysis.12–14 In some electrocatalytical applications, the ammonium ion may play a role, and it can be beneficial for ion transport, e.g. in proton conducting materials. The chemical stability of TMPs, over a wide range of temperatures, makes them ideal candidates, e.g. for fuel cell applications (the optimal T-range for many fuel cells is 150 °C ≤ T ≤ 350 °C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 15 and electrolysis.16–19
15 and electrolysis.16–19
Typically, TMPs are synthesized by precipitation from solution, hydrothermal methods, molten salt, microwave, or mechanochemical syntheses.20–25 Thereby, highly crystalline and/or amorphous bulk TMPs can be obtained. In particular, the amorphous state is often desired, because the absence of long-range ordering in TMPs enhances their reaction kinetics compared to the crystalline equivalents.26,27 Even assuming a similar chemical composition, crystalline and amorphous compounds may in general exhibit completely different physicochemical properties such as solubility, reactivity, or adsorption. For example, amorphization of pharmaceutical ingredients is a well-known strategy to improve their solubility and hence biomedical activity.28 In the case of electrocatalysis, the amorphicity of a material enhances the overall free Gibbs energy of formation which could potentially lead to a significantly improved cell potential and higher open circuit voltages in amorphous electrodes compared to the crystalline bulk counterparts.29 However, the as-synthesized TMP solids, crystalline or amorphous, typically exhibit a low surface area, which must be increased by engineering the porosity into the material.
In general, for all applications, a highly porous material with a high surface area and pore volume significantly increases the performance majorly because of the enhanced reaction kinetics and more active sites. In this context, a stable mesoporous TMP material (pore sizes between 2–50 nm, IUPAC30), which is also amorphous could combine all advantages for applications. These are for example a high number of active metal sites, significantly enhanced mass transfer through the compound and a more negative Gibbs energy of formation compared to the crystalline analogue. Typically, mesoporosity is achieved via template-assisted synthesis routes. Despite the huge potential, template-based synthesis of TMPs is highly challenging and expensive limiting the currently available metal compositions and the (up)scalability.31–33 A major issue is the incomplete removal of the template/surfactant through acid etching or high temperature calcination which could harm the target phosphate mesostructure and its purity. These hurdles have stimulated the development of new strategies for the synthesis of TMPs, with a focus on template-free methods.34,35
We present a template-free synthesis method for amorphous and mesoporous TMPs through a simple thermal treatment of a crystalline precursor called M-struvite NH4MPO4·6H2O (Fig. 1). In M-struvite M2+ is usually Mg2+, but other divalent cations such as Ni2+, or Co2+ can easily substitute Mg2+ in the struvite structure due to similar ionic radii (where other examples considered elsewhere include Zn2+, Fe2+, Mn2+).36–39 In our recent study, we showed how to precipitate and engineer Ni- and Co-struvite crystals from aqueous solutions and we explored their phase diagrams and growth mechanisms.20 Importantly, it has also been recently shown that common Mg-struvite converts directly to amorphous regular mesostructured frameworks of high surface area (∼300 m2 g−1)35 by thermal heating. Such a thermal decomposition method has not been used so far to prepare mesostructured TMPs containing Ni and Co. In this study, we show how the mesoporous metal phosphate compounds systematically differ in their absolute surface, pore size distribution and temporal evolution from each other although they share the same initial isostructural precursor struvite. A direct relationship between amorphicity and mesoporosity is demonstrated. Our findings show that the evolution of mesoporosity is not just a simple case of degassing of ammonia and water from the crystal, but it involves complex restructuring of amorphous solids. By adjusting the reaction conditions, a high degree of control over the formed porous structure is achieved.
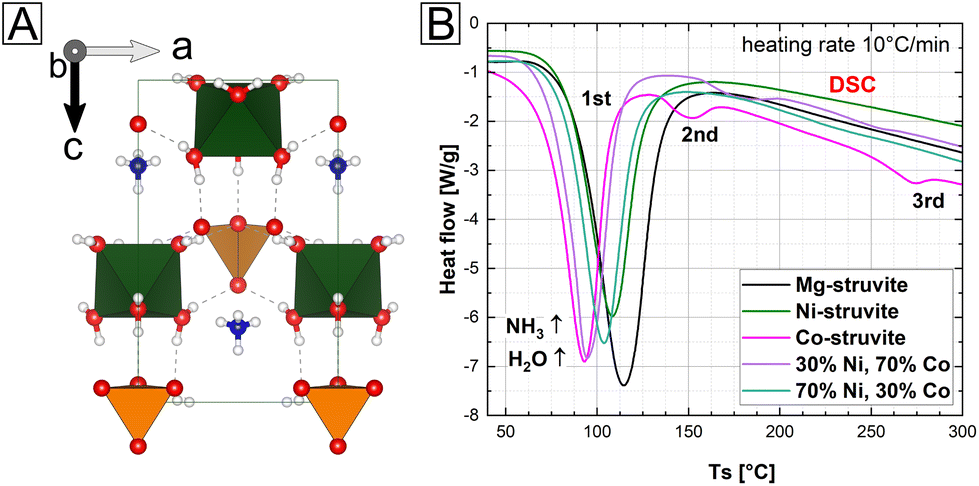 | ||
| Fig. 1 (A) Crystal structure of precursor M-struvite, color of elements: hydrogen = white, nitrogen = dark blue, oxygen = red, P = orange, metal cation = light blue (B) DSC graphs show endothermic originating from degassing of volatile components for pure M-struvite (M = Mg, Ni, and Co) and two NixCo1−x-struvite mixtures (x = 0.3 and 0.7). | ||
Methods
Synthesis
(NH4)2HPO4 (DAP) (ChemSolute, 99%), MgCl2·6H2O, NiSO4·6H2O (ChemSolute, 99%) and CoSO4·7H2O (Alfa Aesar, 98%) were used to synthesize precursor M-struvites (M = Ni, Co, NixCo1−x, and Mg).The complete synthesis procedure can be found elsewhere.20 The different salts had been dissolved in deionized water to make stock solutions, which were further diluted for the experiment. For further discussion on the (Ni,Co)-mixtures, where specifically c(Ni2++ Co2+) = 0.02 M, c(DAP) = 0.1 M, we define Ni numbers (Ni#) as they are used (see eqn (1)) from 0.1 (Co-rich) to 0.9 (Ni-rich).
 | (1) |
The precipitation reaction of M-struvite occurred according to the following mass-balance equation (eqn (2)):
| NH4+(aq) + HxPO43−x(aq) + M2+(aq) + 6H2O → NH4MPO4·6H2O(s) + xH+(aq) | (2) |
In our previous work, we already characterized the optimal conditions for the crystallization of these materials.20 Moreover, Ni-dittmarite was synthesized by heating Ni-struvite at 90 °C at 100% relative humidity (RH) in a hydrothermal reactor for t = 24 h based on the synthesis procedures in the literature.40,41
X-ray diffraction
Powder X-ray diffraction (PXRD, XRD) measurements on the precipitated powders were performed on a D8 Bruker Diffractometer equipped with a LYNXEYE XE-T detector. The diffraction data were collected with Cu-Kα-radiation (1.5406 Å, 40 kV and 40 mA) from 5–60° using a step size of 0.015° (2Θ) and a scanning time of 0.5 s per step. All samples were prepared on silicon specimen holders, to minimize the instrumental background, which was especially important for the amorphous phase.SEM
The scanning electron microscopy (SEM) analysis was performed on an FEI XL 30 tungsten cathode scanning electron microscope operating with 20 keV acceleration voltage and using a secondary electron (SE) detector. Prior to the analysis, all samples were coated with a 30 nm-thick layer of gold.The elemental-dispersive X-ray spectroscopy (EDS) analysis and SEM imaging of the NixCo1−x-mixtures were performed under a Zeiss Supra 40 (Zeiss, Oberkochen, Germany) equipped with a Schottky-field emitter. All samples were coated with carbon before analysis. A 5 kV beam voltage was applied for imaging of all samples. An energy-dispersive X-ray (EDS) system of type UltraDry SDD (Silicon Drift Detector) with a nominal crystal area of 100 mm2 from Thermo Fisher Scientific (Waltham, MA, USA) was used for the qualitative and quantitative analyses of the elemental composition. For this, highly flat sample areas (10 μm × 10 μm) were selected for an EDS quantitative analysis with the software Pathfinder (v. 1.3, 2018). The corresponding EDS spectra were collected at 15 keV and a standardless quantification was performed. Due to the rough sample surface morphology, the obtained elemental compositions are estimated to exhibit an error of <15 at. % based on multiple measurements. However, quantitative comparisons of the relative results for different samples obtained under the same experimental conditions (including sample geometries) are more accurate than the absolute values. Qualitative EDS maps were acquired over selected individual crystals, indicating the spatial distribution of the elements.
TGA/DSC
Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed in a combined mode on dry M-struvite powders using a heat flux TGA/DSC 3+ device from Mettler Toledo. All measurements were carried out under a nitrogen atmosphere (flow of 100 ml min−1). As a reference material for the heat flux DSC an α-Al2O3 corundum crucible was used. All samples were heated from room temperature to 850 °C with a heating rate of 10 °C min−1. The different endo- or exothermic peaks were fitted with Pseudo-Voigt functions to determine the peak onset and centroid temperature.N2 gas sorption measurements
Nitrogen adsorption/desorption isotherms at −196 °C were obtained with a Micromeritics ASAP 2020 V4.04 surface area and porosity analyzer. A pre-treatment of vacuum exposure at room temperature was performed for only one hour to avoid any interactions with the mesoporous sample. By applying the BET (Brunauer–Emmett–Teller) method, the specific surface area of the samples was calculated. The pore size distribution was determined by using the BJH (Barrett–Joyner–Halenda) method with the ASAP 2020 V4.04 software equipped with the analysis equipment.SAXS/WAXS
In situ and time-resolved small- and wide-angle X-ray scattering (SAXS/WAXS) measurements to follow the evolution of mesoporosity were performed at the beamline BM2642 of the European Synchrotron Radiation Facility (ESRF). Experiments were performed using a monochromatic X-ray beam at 12.4 keV (λ = 1.00 Å) aligned with a capillary (borosilicate glass, Glass Müller, Berlin ID = 1.498 mm) of the furnace LINKAM DCS 600. All the samples and backgrounds were measured with this setup.
Scattered intensities were collected at small angles with a Dectris Pilatus 1M and at wide angles with a Dectris Pilatus 300k. Sample transmission factors were measured by means of a photodiode installed in the beam-stop of the SAXS detector, and an ionization chamber was used as a primary beam monitor. The sample-to-detector distances allowed for a q-range of 0.11 < q < 7.36 nm−1 in SAXS and of 5.76 < q < 52.64 nm−1 in WAXS. The scattering q-range at small angles was calibrated against silver behenate and the intensity was calibrated to absolute units against water. The q-range in WAXS was calibrated with a silicon powder reference material (NIST SRM640f). The recorded 2D scattering data from the SAXS and WAXS detectors were reduced using the software package BUBBLE v. 4.2.43 The reduction steps involved data normalizations and instrumental corrections, correction for transmission, subtraction of a background, followed by integration to 1D scattering curves. Since the covered q-ranges of the SAXS and WAXS overlapped between 5.76 and 7.36, the WAXS data could be normalized against the SAXS data.
In a typical experiment, a capillary filled with a crystalline powder was put into a LINKAM DCS 600 furnace and heated at 20 °C min−1 to 90 °C. The scattering patterns were measured continuously at 2 fps. SAXS scattering curves were used to extract pore size-distributions as a function of time.
Ex situ SAXS/WAXS measurements were conducted using the MOUSE instrument.44 In a microfocus X-ray tube attached with parallelizing and monochromatising multilayer optics, X-rays of a distinct wavelength of Cu Kα (λ = 0.15406 nm) were generated. An in-vacuum Dectris Pilatus 1M was used to measure the scattered signal. The sample-to-detector distances allowed for a q-range of 0.015 < q < 40 nm−1 in a combined SAXS/WAXS approach. The synthesized powders were sealed on a sample holder from both sides with Scotch© tape and held in the beam path. The obtained data were corrected according to the procedure in the literature.45
Scattering model
The measured in- and ex situ SAXS data were fitted by McSAS v. 1.3 software46 under the simplified assumption that the pores could be modelled as hard-spheres.47,48 A mutual interference of the developing pores is assumed to occur in agreement with other SAXS studies of phosphate materials.35,49,50 Considering this and other factors, the hard-sphere model outputs the most meaningful fitting data. A full evaluation of the fitting model can be found in the ESI.† WAXS was used to monitor crystalline-to-amorphous transitions. The hard sphere structure factor can be described by the Percus-Yevick structure factor:48| Sc(q) = (1 + 24νG(2qRHS)/(2qRHS))−1 | (3) |
XAS
XAS (X-ray absorption spectroscopy) measurements both at the near edge structure (XANES) and extended fine structure (EXAFS) were performed at the BAMline (BESSY-II, Helmholtz Centre Berlin for Materials and Energy Berlin, Germany)51 on the solid powders to determine and compare the coordination environment of the different phases. The beam was monochromatized using a double-crystal monochromator (DCM) with a Si crystallographic orientation of [111]. The size of the beam was 3 mm (h) × 1 mm (v). The measurements were performed at Co–K edge (7709 eV) and Ni–K edge (8333 eV) in transmission geometry, with two ionization chambers as detectors. The excitation energy was varied from 7606 eV to 8678 eV for Co and 8230 eV to 9302 eV for Ni, with varying energy steps. For the pre-edge region, the energy was varied in 10 eV steps; for the region around the edge, energy was tuned first in 0.5 eV steps, then in 1 eV steps and in the EXAFS region with a constant step in the k-space of 0.04 Å−1. The associated uncertainties were experimentally determined by measuring the cobalt and nickel metal foils, 10 times each. A value of ±0.3 eV was obtained for both systems. For the measurement, the samples were mixed with boron nitride, placed in polycarbonate hole plates with a thickness of 1 mm and sealed with polyimide tape (Kapton) on both sides. Before collecting the sample spectra, a cobalt and nickel foil was used as references for the respective K edges. The energy shift is typically calibrated with such metallic foils. The relative energies of the spectra were calibrated against the first inflection point from the first derivative of the cobalt/nickel metal absorption edge. In addition, M2+ (M2+ = Ni2+, Co2+) standards such as oxides and sulfates were also measured for comparison.The XAS data were processed by using ATHENA (for normalization and background removal) and ARTEMIS (to fit models to the experimental EXAFS data). These two programs belong to the main package IFEFFIT (v. 1.2.11).52 The respective Ni-phosphate (Ni-struvite NH4NiPO4·6H2O, Ni-dittmarite NH4NiPO4·H2O) phases were used as model structures, and the scattering paths for the first and second coordination spheres were theoretically modelled and fitted to the measured spectra.
Results
Crystalline phase composition and their amorphization
In a simple one pot synthesis, the crystalline single-phase M-struvite with M = Mg, Ni Co and NixCo1−x-struvite powders were obtained as starting materials and they shared the same precursor structure (Fig. 1 and 2, ESI: Fig. S1†). M-struvite contains water and ammonia in its structure, and therefore potential phase transitions should occur at elevated temperatures as a result of dehydration and degassing processes. Based on this assumption, to evaluate such thermally-induced phase transitions, a combined TGA/DSC analysis was performed on the dry crystalline struvite powders at T ≤ 300 °C (Fig. 1B, ESI: Fig. S1†).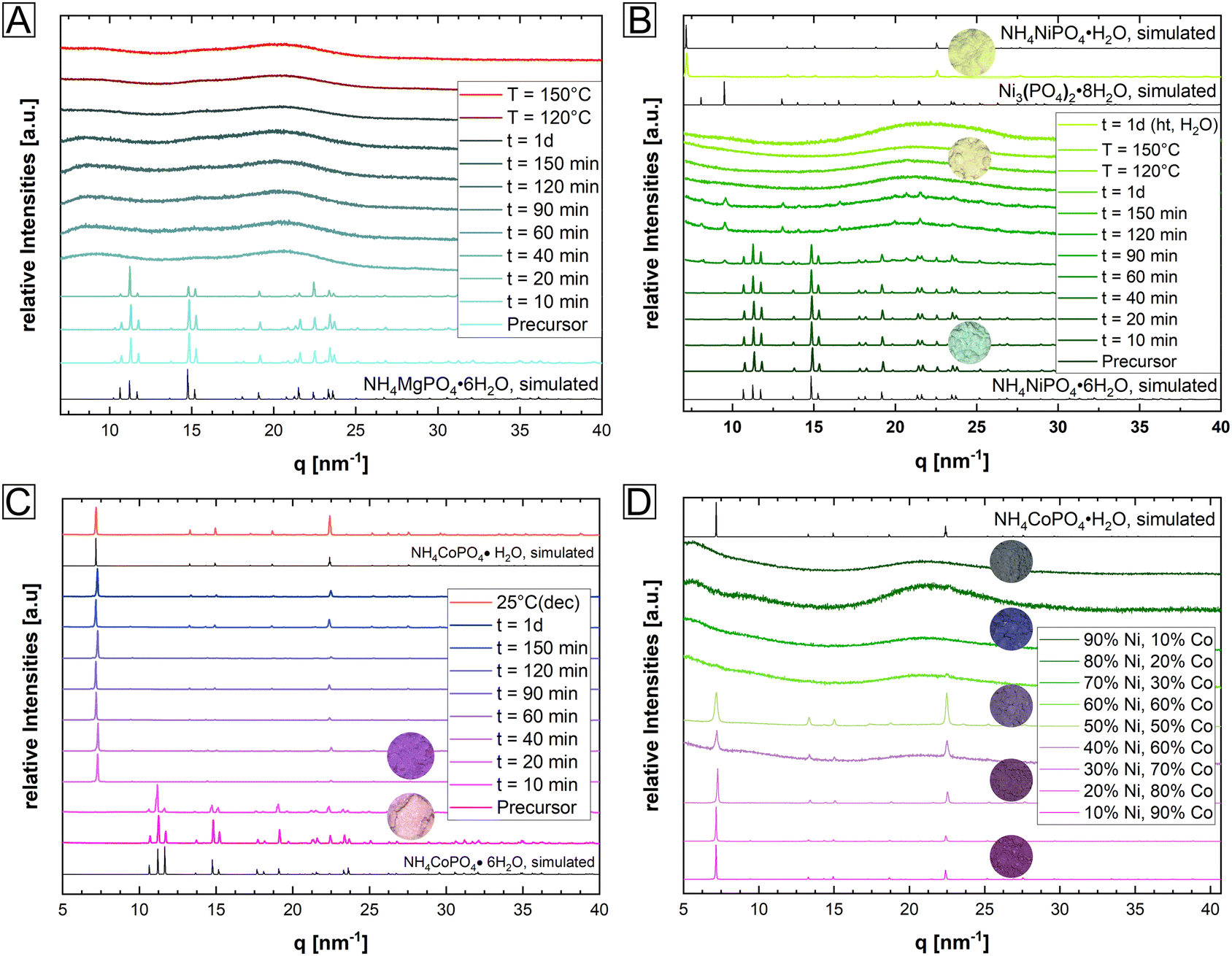 | ||
| Fig. 2 XRD patterns of an isothermally heated (A) Mg-struvite, (B) Ni-struvite, and (C) Co-struvite and (D) NixCo1-x-struvite for t ≤ 1 day at T = 90 °C. For comparison, diffractograms of reference structures were simulated. Different temperatures (T = 120 °C, 150 °C) and humidity conditions (t = 1 d, ht = hydrothermal, ~ 100% H2O) were also evaluated in the Mg- and Ni-system; photographs in circles for differently colored powder samples were added for selected Ni- and Co diffraction patterns (Mg-samples all white); Mg-struvite reference COD 9007674; Ni-struvite (NiS) reference database ICSD 403058; Co-struvite reference ICSD 170042; Co-dittmarite (COD) reference database COD 2008122; Ni-phosphate octahydrate reference database ICSD 240946. | ||
Based on the characterization, we see that the thermal decomposition behavior of the different M-struvites vary significantly. While Co-struvite transformed in the course of a quick phase transition to Co-dittmarite with an Tonset,1 = 72 °C, Ni-and Mg-struvite lost their volatile compounds at slightly higher temperatures at Tonset,1 = 83 °C (Ni) and Tonset,1 = 88 °C (Mg) (ESI: Table S1†). Interestingly, Co- and Co-rich NixCo1−x-struvite show multiple phase transitions indicated by multiple events in DSC (the 1st, 2nd, and 3rd thermal event in Fig. 1B). The resulting order of the 1st M-struvite decomposition is the following: TCo < TNi ≈ TMg (ESI: Table S2†). Considering the mass-loss analysis associated with DSC (ESI: Fig. S1†), the first thermal event corresponds in all compounds to a major stage of degassing with the release of five water molecules and minorly ammonia. Therefore, since all struvite compounds show their main degassing event at around 90 °C, the phase composition of the heated M-struvite samples was measured in a time series t = 10, 20, 40, 60, 90, 120, 150 minutes, and 1 day at this temperature. Due to the broad T-interval of the 1st thermal event, higher temperatures T = 120 °C and 150 °C were also evaluated for the different M-struvite systems to observe potential differences.
At temperatures below T < 200 °C, Mg- and Ni-struvite transformed to amorphous phases in dry air, beginning after t = 40 min and completed at t = 120 min, while Co-struvite recrystallized quickly after 20 min to Co-dittmarite (Fig. 2, ESI: Fig. S2†). In fact, simple color changes of the transition metal phosphate phases indicate phase transformations associated with different coordination environments (photographs of powders in circles, in Fig. 2B, C and D). When heated, Ni- and Co-phosphates exhibited a color change from green to yellow (Ni) and from pink to purple/violet (Co). In the Ni-system during degassing of volatile components a transitional stability of Ni3(PO4)2·8H2O had been observed which disappeared again upon further heating (Bragg reflections below 10 nm−1 in Fig. 2). At high temperatures, the amorphous Ni-phases showed a high stability up to 500 °C, while in the Co-system multiple crystalline-crystalline phase transitions were observed (ESI: Fig. S2†).
In the case of single-element struvites, all three considered compounds exhibited different thermally induced phase transitions despite sharing the same initial crystal structure. Importantly, Ni-struvite turned out to convert into a stable amorphous phase and Co-struvite turned into crystalline Co-dittmarite. The question arose how mixed NixCo1−x-struvites would evolve under similar thermal conditions; in particular the mixed transition metal struvites had different thermal profiles (Fig. 1D, ESI: Fig. S1†) from their mono-cationic analogues. In the first step, we confirmed that for a broad range of compositions we indeed synthesized phase-pure NixCo1−x-struvites with x = 0–1 (ESI: Fig. S1†). Based on EDS maps, a complete solid solution of NixCo1−x-struvite with a homogenous distribution of both metals regardless of the Ni# could be identified at least at the sub-μm-scale (ESI: Fig. S3,† Table S2†). Upon heating, the synthesized NixCo1−x struvite transformed to either amorphous or crystalline phases depending on the Ni# after the thermal treatment (eqn (1), Fig. 2D). Ni-rich NixCo1−x-struvites above Ni# >0.5 decomposed to the amorphous phase while Co-rich compositions below Ni# ≤ 0.5 transformed to M-dittmarite NH4MPO4·H2O which coexisted with the amorphous phase (Fig. 2D). Thus, the materials became phase-separated, despite originating from struvite solid solutions. Heated NixCo1−x-struvites with high Co content, Ni# ≥ 0.8, showed no amorphous phase. In a hydrothermal synthesis in a saturated H2O environment at 90 °C, Ni-struvite transformed to Ni-dittmarite NH4NiPO4·H2O (Fig. 2B). Summing up the thermal history of the compounds, Mg-, Ni- and Ni-rich (Ni# > 0.5) NixCo1−x-struvites decomposed to amorphous phases under dry conditions, whereas Co- and Co-rich (Ni# ≤ 0.5) NixCo1−x-struvite transformed inter alia to crystalline Co-dittmarite. In the thermal decomposition of the NixCo1−x-struvite series, Ni seems to be preferentially incorporated into the amorphous phase, while Co is found in the M-dittmarite (Co-dominated) based on the Bragg reflections in XRD. The occurring phase transitions, crystalline–crystalline or crystalline–amorphous are strongly dependent on the humidity, absolute temperature, heating rate and on the choice of the metal cation in the struvite precursor. We show further that the evolution of amorphicity is a key property of the material. As a stable amorphous phase prevents the structural reconfiguration of the compound and the closing of the pores, it is a necessary condition for the development of mesoporous frameworks.
Characterization of mesoporous structures
Our results show that thermal evolution of M-struvites is driven by the removal of volatile components from the crystal structure (Fig. 1, ESI: Fig. S1†). This in turn suggests that porosity should develop in the materials over time. To test this, we characterized the evolution of porosity in our samples using gas adsorption. For the evaluation of BET surface areas and pore sizes, N2 gas adsorption–desorption measurements were performed on thermally treated M-struvites (M = Mg, Ni, Co, and NixCo1−x). Based on these measurements, it was clear that the porosity would indeed develop at the mesoscale, but only in M-struvites which would become simultaneously amorphous (i.e. Mg, Ni, NixCo1−x). On the other hand, Co-struvite and Co-rich NixCo1−x-struvite showing thermally induced crystalline transformations did not develop any significant porosity.In Fig. 3, we present the BET measurement results from the Ni-struvite thermal series. Four different stages can be defined in all obtained isotherms (inset in Fig. 3A). Firstly, in stage (I) at low partial pressures (p/p0 < 0.4) a constant adsorption of N2 can be observed, associated with the formation of a mono- to multilayer on the surface. In the intermediate stage (II) at partial pressures between 0.4 < p/p0 < 0.55 progressive multilayer condensation and filling of pores smaller in size with N2 gas occur probably. The third (III) and fourth (IV) stages show the absence of significant amounts of macropores as a plateau region occurs at high relative pressures (0.55 < p/p0 < 0.92) followed by a steep increase due to bulk condensation of N2 at the highest partial pressures (p/p0 > 0.92).
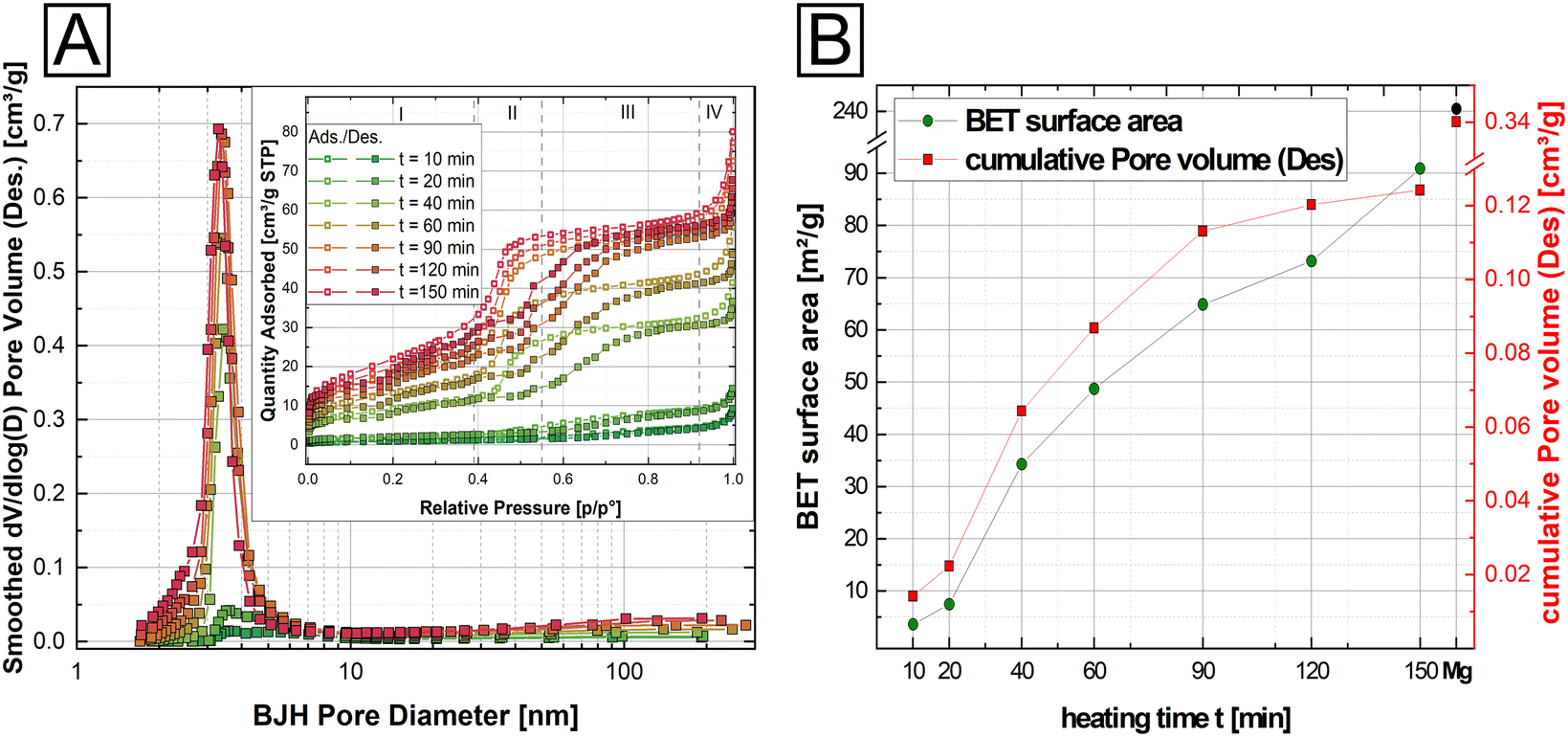 | ||
| Fig. 3 N2 adsorption/desorption BET surface measurements for a time series (10–150 min) of isothermally heated Ni-struvite at 90 °C (A) average desorption BJH pore size change (dV/dlog(D) pore volume [cm3 g−1] vs. average pore diameter [nm]), inset: evolution of type IV adsorption/desorption isotherms of the t-series (B) Temporal evolution of BET surface area [m2 g−1] (green circles) and des. cumulative pore volume [cm3 g−1] (red squares) of thermally treated Ni-struvite with heated Mg-struvite as a reference (black circle: BET surface area, red circle: cumulative pore volume). | ||
Based on the slope changes in the observed isotherms in the stage (II) and the BJH pore size distribution (Fig. 3A), heated Ni-struvite demonstrated type IV isotherms/hysteresis loops indicative of a significant amount of mesopores. The surface area and the cumulative pore volume increased from 3.61(1) m2 g−1 for t = 10 min heating time to 90.9(5) m2 g−1 for t = 150 min heating time, while the average desorption BJH pore size decreased from 12.3 nm to 5.2 nm for a longer heating time (Fig. 3B). After extended heating times t = 120–150 min the BET surface area remained nearly the same. As a reference, and for comparison of the BET data, Mg-struvite was heated at T = 90 °C for one day and measured (last data point in Fig. 3B). At a BET surface area of 240.5(9) m2 g−1, it demonstrated a significantly higher value, accompanied by an average desorption BJH pore size of 4.2 nm compared to the thermally treated Ni-struvite (Fig. 3B, ESI: Table S3†). The adsorption/desorption BJH pore size distribution of the Ni t-series clearly showed a shift to lower average pore sizes for longer heating times. Considering higher temperatures T = 120 °C, 150 °C (ESI: Fig. S4, S5 and Table S3†), the Mg and Ni system showed a systematically lower BET surface area (Mg: 195.9(2) m2 g−1 for T = 120 °C, 138.2(2) m2 g−1 for T = 150 °C; Ni: m2 g−1 for T = 120 °C and 54.5(1) m2 g−1 for T = 150 °C).
Clearly, a direct correlation between the amorphicity and mesoporosity of the material exists as only the amorphous compounds were also mesoporous. This correlation starkly demonstrated itself in heated NixCo1−x-struvite mixtures, where Ni# ≥ 50% all showed BET surface areas of around 80–90 m2 g−1 and average pore sizes of 4 nm with type IV hysteresis loops similar to Ni-struvite (ESI: Fig. S6, Table S4†). Below Ni# ≤ 50%, the BET surface area decreased to 20–30 m2 g−1 while the average pore size increased rapidly from 3–4 nm to tens of nanometers. The heated Co-rich NixCo1−x-struvite and pure Co-struvite demonstrated no significant mesoporosity and a low surface area (1.1 m2 g−1), indicated by type II isotherms (ESI: Fig. S6, Table S4†). All summarized BET data, additional adsorption/desorption pore size distributions and isotherms of M-struvite can be found in the ESI (ESI: Fig. S5 and S6, Tables S3 and S4†).
However, the presented N2 BET data were collected ex situ, while the associated BJH calculations were based on arguable assumptions, i.e. the occurrence of only disconnected parallel cylindrical pores. Therefore, to gain complementary information of the pore structure, in situ SAXS/WAXS measurements of isothermally heated M-struvite at T = 90 °C were performed allowing a view on the temporal evolution of pores during the thermal treatment (Fig. 4). Characteristic steps in the in situ SAXS data of the mesoporous Mg- and Ni-system were selected and fitted to determine the pore size distribution. By fitting the SAXS curves of selected time steps, a quantitative comparison of the calculated pore size distribution and a temporal evolution of the mesoporous framework can be obtained. All details concerning the fitting procedure can be found in Methods: SAXS/WAXS (ESI Note 1, eqn (S1) and (S2)†). All SAXS fits and pore size distributions of selected time steps of the heated Mg-, Ni- and NixCo1−x-struvites can be found in the ESI (ESI: Fig. S7–S13, Tables S5 and S6†).
 | ||
| Fig. 4 In situ SAXS/WAXS data of isothermally treated M-struvite (M = Mg, Ni and Co) at 90 °C with associated WAXS heatmap q [nm−1] vs. time [s] of the distinct experiment; WAXS heatmap: normalized WAXS intensities plotted with the simulated crystal structures and final WAXS signal; on the right side: normalized intensity of a characteristic reflection of precursor struvite (002) marked with a shadowed frame; in situ SAXS/WAXS data of thermally treated (A and B) Mg-struvite, (C and D) Ni-struvite and (E and F) Co-struvite at T = 90 °C are shown; (E) inset: in situ WAXS signal (Mg-struvite reference COD 9007674, Ni-struvite reference ICSD 403058, Co-struvite reference ICSD 170042 and Co-dittmarite reference COD 2008122). | ||
Looking at the pore size distribution of the thermally treated M-struvite, the mesoporous frameworks of heated Mg- and Ni-struvite differ significantly from each other. Considering the WAXS signal, the Mg- and Ni-system evolve pure amorphous phases during thermal treatments, while Co-struvite transforms into crystalline Co-dittmarite (Fig. 4). Again, this is the reason for the absence of a stable mesoporosity in the thermally treated Co-struvite. Therefore, only the Mg and Ni SAXS patterns were fitted with the model describing the mesoporosity (see Methods, ESI Note 1†). Upon heating, Mg-struvite exhibits initially a multimodal distribution, a mesoporous population at >38 nm pore diameter and a meso- to microporous population below 20 nm, respectively, at t = 400 s (Fig. 5A and C, ESI: Fig. S8†). Upon heating after t = 2000 s, the mesoporous population of large pore size with diameters of around 38 nm disappears, while the population of mesopores of small diameter below 20 nm shifts significantly from 13 ± 3 nm to 8 ± 1 nm in size. At the final stage at t = 5500 s, the mesoporous population becomes narrower with time, as is indicated by a decreasing width of size distribution. The final maxima (1st, 2nd and 3rd maximum in Fig. 4) point out to an average and uniform pore size (ESI: Fig. S7, S8 and S11†). Although the microporous population evolves quickly already at t = 398 s, its average pore size remains similar at around 1 nm (1.3 ± 0.3 nm at t = 400 s and 1.0 ± 0.3 nm at t = 5500 s). In the Ni-system, beginning with a broad multimodal population in the micro- and mesoporous range at t = 2200 s with a mean pore diameter of 22 ± 6 nm, a bimodal population of micro- and mesopores establishes itself after t = 5500 s (Fig. 5B & D, ESI: Fig. S9, S10 and S12†). Here, the final mesoporous population at t = 5500 s of heated Ni-struvite with a mean pore diameter of 14 ± 4 nm is much broader and larger than the narrow mesoporous population of Mg-struvite of 8 ± 1 nm. The first two maxima (1st, 2nd) represent the pores in the mesoscale, while the last one appearing at high q (>5 nm−1) originates from the micropores (1st*). In addition, Mg-struvite exhibits a higher volume of mesopores in relation to the volume of micropores compared to Ni-struvite (approximately for Mg: φmicro ≈ 83% to φmeso ≈ 17% and for Ni: φmicro ≈ 60% to φmeso ≈ 40%, ESI: Table S5†).
 | ||
| Fig. 5 In situ SAXS data and fits of isothermally treated (A) Mg- and (B) Ni-struvite at T = 90 °C with a fixed local volume fraction ν = 40%; resulting pore size distribution of heated (C) Mg- and (D) Ni-struvite with relative and cumulative (final time) volume fraction of pores. | ||
Based on the relative volume fraction of meso- and micropores from SAXS, the final heated amorphous magnesium phosphate phase exhibits 2–2.5 higher volume of mesopores than the final heated Ni–PO4 phase (Mg: φmeso = 40% vs. Ni: φmeso = 17%).
Although a pure Co-struvite precursor did not evolve mesopores in the final stage, a detailed view on in situ data could reveal transitional pores formed in the heating process, which eventually disappeared. In fact, we could observe short-lived micro- to mesopores of an average size of 2–3 nm at around t = 150 s (Fig. 4E). They evolved during a thermal treatment due to the simultaneous decomposition/degassing of Co-struvite. After this time, all released gases, mainly H2O, had evaporated and Co-dittmarite recrystallized immediately, resulting in a progressive decline of the mesopore signal until its disappearance at around t = 700 s. This points out again to a sharp crystalline–crystalline phase transition between Co-struvite and Co-dittmarite. Interestingly, permanent well-defined narrow peaks at 5.1 and 5.5 nm−1 developed rapidly already within the first 150 s and remained present in the scattering pattern. Such peaks did not appear either in the Mg or Ni system (Fig. 4C). These narrow and sharp reflections are probably associated with the crystal structure changes rather than with an actual evolving microporosity.
In the context of stark differences between Ni- and Co-struvite, we also investigated the evolution of the NixCo1−x-struvites with various metal contents. Namely, for comparison against the pure Ni- and Co-endmembers, ex situ combined SAXS/WAXS measurements were performed on three isothermally heated NixCo1−x-struvite solid solutions with Ni# = 0.8, Ni# = 0.5 and Ni# = 0.2 and fitted with the same model (ESI: Fig. S13, Table S6†). As indicated by the BET surface measurements, the amount of mesopores decreased with lowering Ni content. Based on SAXS/WAXS, it was further evident that also the pore structure itself changed with different Ni# in the material (ESI: Fig. S13, Table S6†). The Ni-rich composition with Ni# = 0.8 exhibited a micro-to-mesoporous population with minor macropores as in the Ni endmember. In contrast, the intermediate NixCo1−x-mixtures showed a broad pore size distribution with a significant amount of macropores and a lower amount of mesopores. Finally, the Co-rich NixCo1−x-mixture demonstrated no significant mesoporosity (ESI: Fig. S13, Table S6†) in correlation with the results from the other methods. Cobalt-richer compositions of the heated NixCo1−x-struvite shift the pore distribution towards larger pore sizes visible in a scattering signal at lower q and in agreement with the BET data (ESI: Fig. S6, Table S4†). Reiterating in the context of our previous results, the amorphicity of the material is necessary to evolve any significant mesoporosity.
Coordination environment of amorphous phases and the T-effect on crystalline phases
Based on our SAXS/WAXS results, we observed a significant change in the pore size during heating (Fig. 5), which is counterintuitive considering just simple evaporation of ammonia and water. This finding suggests a potential condensation or a change of the amorphous structure with extended heating times, resulting in a decrease of the pore sizes. Therefore, to characterize the amorphous structure and explain its correlation with the occurrence of the mesoporous framework, X-ray absorption spectroscopy (XAS) measurements were performed. The obtained spectra of the time series of the heated Ni-struvite are displayed in Fig. 6. They correlate the evolution of mesoporosity with the change in the coordination environment. In addition, the selected NixCo1−x-struvites and their heated products at T = 90 °C were measured for the Ni- and Co-edge to compare their distortion of the M2+-octahedron in a solid solution. In particular, the intensity of the pre-peak (XANES region) originating from the 1s–3d transitions expresses the degree of centrosymmetry or distortion in the vicinity of the considered ions (inset of Fig. 6A, ESI Note 2†). As only 3d–4p hybridization enables parity allowed transitions, the extent of those is strongly correlated with the coordination geometry. Near-ideal centrosymmetric coordinations show very low pre-peak intensities, while more distorted geometries exhibit higher intensities. As X-ray spectroscopy considers primarily the shell of the absorber atom and is therfore element specific, an analysis of crystalline and amorphous samples is possible. In general, highly crystalline phases demonstrate narrower and more intense peaks in the R-space compared to amorphous phases because more ions have similar bond distances to the metal cation due to a regular periodic structure (towards a delta-function-like distribution). Conversely, as amorphous phases do not display an established long-range order, they show broader, less intense, and less-pronounced peaks in the R-space mostly in the low R-region due to an irregular arrangement of the lattice ions. | ||
| Fig. 6 (A) Normalized XAS spectra of the crystalline precursor Ni-struvite (NIS), time series of thermally treated Ni-struvite and hydrothermally synthesized crystalline Ni-dittmarite (NID) compared with multiple M2+ (M = Ni2+, Co2+) standards (only the Ni-foil is shown for clarity) with detailed view of the pre-peak region; (B) Fourier-transformed Ni spectra plotted in the R-space; (C) (010) crystal plane of Ni-struvite structure space group Pmn21 with a black dashed line of potential near spherical micropores inside the structure and amorphous phase. | ||
The XANES region (Fig. 6A) clearly proves the exclusive presence of bivalent Ni2+ and/or Co2+ species in the materials. No indications of oxidation to Ni3+or Co3+ were observed either in the pure endmember or in the bicationic solid solutions. Quantitative pre-peak integration results App (area of pre-peak) were calculated for comparison of the distortion in the first coordination sphere (ESI: Fig. S14, Tables S7–S9†). Ni exhibits a slightly higher degree of distortion in the amorphous phase than in the precursor Ni-struvite, which is visible through higher pre-peak intensities (Ni-struvite: App (t = 0 min) = 0.0250(1), App (t = 150 min) = 0.0298(1), ESI: Fig. S14, Table S7†).
Looking at the pre-peak region of NixCo1−x-struvite mixtures and their heated mixtures, Ni2+ forms a near-ideal octahedral coordination in the crystalline NixCo1−x-struvite structure and in the heated products regardless of the Ni#, as is indicated by a low pre-peak intensity (ESI: Fig. S14, Tables S8 and S9†). In contrast, Co2+ shows a slightly higher degree of distortion with increasing Ni content in the struvite structure (precursor Ni0.8Co0.2-struvite and App = 0.032(2) vs. Ni0.2Co0.8-struvite and App = 0.025(1)). Similarly, the degree of distortion increases with higher Ni# in the heated products (heated Ni0.8Co0.2-struvite App = 0.046(2) vs. heated Ni0.2Co0.8-struvite App = 0.034(2)). Based on the XRD results (Fig. 2), NixCo1−x-struvites decompose to the amorphous phase, as a single phase in Ni-rich compositions above Ni# > 0.5 and as a secondary phase at values of 0.2 < Ni# ≤ 0.5. Following on that, Co-dittmarite recrystallizes at Ni# ≥ 0.5 as a primary phase and it is the only occurring phase below Ni# < 0.2 (ESI: Tables S8 and S9†). Hence, Ni2+ tends to be incorporated in the amorphous phase, while Co2+ favors the crystalline M-dittmarite structure. Surprisingly, the distortion degree of Ni2+ barely changes in all observed structures, in the precursor M-struvite, M-dittmarite or in the amorphous phase. In contrast, Co2+ forms a more distorted non-centrosymmetric coordination environment than Ni2+ in the pure compounds and the distortion increases even more in the mixed NixCo1−x- ones with higher Ni content (ESI: Tables S8 and S9†).
Based on the comparison of the distortion (Fig. 6, ESI: Fig. S14, Tables S7–S9†) and on fitting of the spectra of the Ni time series (ESI: Fig. S15 and S16, Tables S10–S18, ESI Note 2†), the structure of the amorphous Ni phases formed at 90 °C showed a similarity to that of Ni-dittmarite, albeit with a less pronounced long-range order. After a 40 min-long heating time, the transformation was completed, visible in the pre-peak area integration results and calculated bond distances (ESI: Fig. S16, Tables S10–S18†).
Discussion
Phase transitions and correlated amorphicity/crystallinity
In the thermal treatment, metal struvites underwent degassing of ammonia and water resulting in amorphization, with an associated development of meso- to microporous frameworks, or in a simple recrystallization. Based on our results, we suggest that the preservation of the mesoporous structures is directly correlated with the stability of the amorphous phase.The occurring phase transitions in heated M-struvites (i.e. amorphous–amorphous, crystalline–amorphous or crystalline–crystalline) are highly dependent on humidity, temperature, heating rate, and on the metal cation in the precursor struvite. From the same isostructural M-struvite precursor, various amorphous and/or transition metal derivatives with different pore structures can be obtained. The main phase transition is assumed to be driven by the removal of close to five water molecules per formula unit at around 70–90 °C. Mg and Ni-struvite decompose to the amorphous phase at TMg ≈ TNi ≈ 85 °C, while Co-struvite transforms through a crystalline–crystalline phase transition to Co-dittmarite already at TCo ≈ 72 °C. Based on the chemical composition of Co-dittmarite and thermal decomposition studies of Mg-struvite,35 probably only minor amounts of ammonia are removed in this step. Both amorphous phases of Mg and Ni exhibit mesoporous networks although their evolution and final pore structure differ significantly. The amorphous phase of Ni locally resembles the Ni-dittmarite structure, but is less ordered, at larger distances of R > 4 Å. Interestingly, similar effects of humidity were observed in iron phosphate minerals.53,54 In contrast, Co-struvite decomposed to Co-dittmarite already at room temperature after several days when exposed to air (ESI: Fig. S1D,† more details in the literature20). When heated, this process is accelerated. In both systems, the phase transitions mimic pseudomorphism where the amorphous phase/the crystalline Co-dittmarite follows after Ni/Co-struvite similar to other dehydrating compounds such as calcite after ikaite.55
Looking at the NixCo1−x-struvite precursors, even low amounts of Ni could prevent the decomposition to the M-dittmarite compound at room temperature. The heated products of the NixCo1−x-solid solutions transform to a single amorphous phase, to single crystalline M-dittmarite or a two-phase mixture of both depending on the Ni# (ESI: Fig. S1B†). High Ni content is favorable for the formation of the amorphous phase, while high Co content leads to the recrystallization of M-dittmarite. Polyamorphism, the occurrence of multiple phases with a different amorphous structure, cannot be completely excluded, especially in the heated NixCo1−x-mixtures, based on the XAS signal. But even if several amorphous phases are present in considerable amounts, they appear to have a similar short-range order. In this regard, the Fourier transformed spectra in R-space demonstrate no peak splitting.
Temporal formation of mesoporosity and structural reconfiguration
As the mesoporosity is directly dependent on the change of the amorphous structure, heated Mg- and Ni-struvite differ significantly in their absolute and temporal evolution of mesoporous frameworks. Primarily, isolated pores form due to the removal of mainly water and, to a minor extent, ammonia. In the initial degassing step, barely any reconfiguration occurs in the compound, which results in a polydisperse population of larger mesopores (>10 nm for Mg, >20 nm for Ni) and micropores. Upon further heating, the building units, PO4 and MO6 begin probably to bind to each other causing a potential shrinkage of the pores (disappearance of larger mesopores), as implied by the R-spectra in XAS and partially resembling shrinkage mechanisms in consolidated xerogels.56 Moreover, the isolated ammonium unit decomposes partly to ammonia and released protons could form, in combination with oxygen, free water as a condensation product. As the bond distances were just evaluated ex situ with EXAFS, the dynamic reconfiguration process of the compound is likely to be more far more complex. At final t = 5500 s, Mg- and Ni-struvite exhibit bimodal distributions of pores: a single microporous population of around 1 nm, one other mesoporous population at around 8 ± 1 nm for Mg and 14 ± 4 nm for Ni. We assume that the isolated micropores begin to partly coalesce to mesopores 5–15 nm in size, and we speculate that channel/worm-like, elongated geometries (5–15 nm) of pores also evolve. Such a geometry would explain the pore interconnectivity and therefore the accessibility for N2 gas in the BET measurement. The shape of pores has to be further confirmed, and this geometry is not explicitly included in our scattering model due to its mathematical constraints. However, in Mg-struvite these channel-like/elongated pores, if actually present, seem to form fast and gradually increase with higher heating time (progressively increasing intensity of the main peak at around q = 0.7–0.8 nm−1). Therefore, the relative volume fraction of mesopores is much higher than that of micropores (approximately for Mg: φmicropores ≈ 83% to φmesopores ≈ 17%). Uniform and narrow pore size distributions of 8 ± 1 nm and 1.0 ± 0.5 nm develop through time. In contrast, Ni-struvite decomposes much slower than Mg-struvite to the amorphous phase and preserves a significant fraction of isolated micropores. Therefore, we assume that Ni-struvite forms only a limited amount of interconnected pore frameworks which would explain the significant lower BET surface area compared to a heated Mg-struvite (Mg: 240 m2 g−1vs. Ni: 90 m2 g−1) and the higher amount of micropores compared to mesopores (approximately for Ni: φmicropores ≈ 60% to φmesopores ≈ 40%). The ratio of the absolute surface areas, pore volumes of heated Mg- (240 m2 g−1, 0.31 cm3 g−1) and Ni-struvite (90 m2 g−1, 0.13 cm3 g−1) and the average pore sizes obtained from BET N2 gas sorption are mostly in the same order of magnitude. Small discrepancies of a few nanometers in the calculated BJH pore sizes and the SAXS pore sizes could be originated in the BJH method as it always includes desorption artefacts.Within the NixCo1−x-struvite mixtures, a higher Co2+ content leads to a higher polydispersity and higher average pore size (ESI: Fig. S13, Table S6†). Coincidentally, the absolute BET surface area and pore volume decrease significantly at Ni# < 0.5. After thermal treatment, only a limited amount of Co2+ can be probably incorporated into the amorphous phase, since most of Co tends to form its own crystalline phase, namely Co-dittmarite (ESI: Fig. S1†). This results in a binary phase mixture: amorphous and crystalline. Based on the EXAFS analysis, the structure of the amorphous phases resembles the local structure of crystalline Ni-dittmarite NH4NiPO4·H2O with a less pronounced long-range order. An indicator for this assumption is the linking of the phosphate units with the metal octahedron associated with the characteristic appearance of R-distances of around 2.7 Å (Fig. 6B). Interestingly, a high Ni content in the precursor struvite and heated products leads to a significant distortion of Co2+ in its octahedral coordination. Ni2+ shows barely any correlation with increasing Co content. Co2+ with a d7 electron configuration demonstrates a strong Jahn–Teller effect in a high-spin octahedral coordination (H2O acts as a weak field ligand) due to more unpaired electrons compared to Ni2+ as a d8-ion. Based on the pre-peak integration results (ESI: Fig. S14, Tables S7–S9†), a mutual influence of both 3d metals in the shared solid solution on the coordination sphere is assumed, whether in the precursor M-struvite, in the amorphous phase or M-dittmarite structure. The significant correlation of the distortion degree of Co2+ in NixCo1−x-mixtures is in agreement with electron spin resonance spectroscopy.57 As stated also in our previous study,20 we assume a strong dependency between the stabilities of the different transition metal struvites and their electronical d-configuration.
Regardless of their individual differences in the pore size evolution and absolute surface area, heated Mg-, Ni- and Ni-rich NixCo1−x-struvites preserve mesoporous frameworks. On the other hand, Co- and Co-rich NixCo1−x struvite eliminate any potential mesoporous structures completely due to their fast reconfiguration to crystalline M-dittmarite NH4MPO4·H2O. The amorphization to a stable non-crystalline phase during degassing seems to be necessary to obtain any mesoporous system and probably prevents the collapse of mesopores due to the absence of a crystalline reconfiguration. The high stability of an occurring amorphous phase could have stopped further diffusion of volatiles out of the compound. Since a particular mesoporous framework configuration seems to be directly linked to an element specific amorphous phase and cannot be predicted a priori from the precursor material, we suggest that the small-angle scattering signal itself might constitute a fingerprint for a specific amorphous phase. Porous amorphous phases could be classified in such a way. The major differences in the mesoporosity of the heated Mg- and Ni-struvite, which are directly linked to the stability and chemical composition of the amorphous phases, support this hypothesis. However, this idea would need to be verified with other porous materials.
By adjusting the reaction conditions (Ni/Co ratio in precursor M-struvite, temperature, humidity), mesoporous materials with different pore structures can be obtained. In such a way, an engineering of the mesoporous framework can be performed for a distinct applicational use, e.g. in electrocatalysis. The surface area values of the mesoporous struvite-derived phosphate materials are comparable to other mesoporous phosphates and their related phosphonates, synthesized in much more complex chemical procedures involving templates.58–62 Transition metal phosphates are frequently used in electrodes or supercapacitors;3,40,41,63 and the same mesoporous analogues would potentially exhibit a higher performance due to enhanced reactivity. The mesoporous ammonium transition metal phosphates could also be further converted by ion exchange into lithium/sodium metal phosphates, which are critical for some types of batteries.5,8 A coated or doped amorphous mesoporous transition metal phosphate material might potentially improve properties e.g. high electrochemical activity in the water splitting reaction or ionic conductivity. However, this aspect needs to be further tested. As we suggested in a previous study,20 M-struvite could be precipitated out of industrial or mining wastewaters. Due to its fast precipitation and low solubility product Ksp, it is also occurring as a biomineral in specialized bacteria species.64,65 Through a simple thermal treatment, these compounds containing critical raw materials such as phosphate or cobalt could be re- and upcycled for industrial purposes.
In summary, we observed three major effects in M-struvite-derived materials which are visualized in Fig. 7: (a) the amorphization and consolidation of Mg- and Ni-struvite mainly through linking of the PO4 units with the MO6 units due to the loss of coordinating H2O together with minor NH4, and (b) their formation of isolated micropores and the connection to larger potentially elongated/worm-like mesoporous frameworks. In (c) a complete reconfiguration of X-shaped Co-struvite occurs to crystalline 2D sheet-shaped Co-dittmarite, without the evolution of significant porosity.
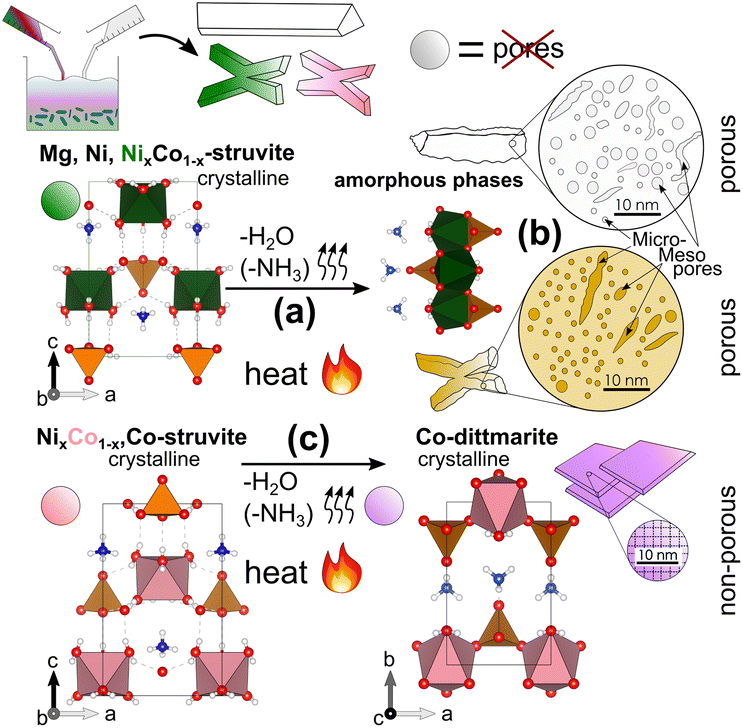 | ||
| Fig. 7 Summary sketch including the three major processes in the samples: (a) the amorphization of the crystalline M-struvite, (b) the formation of isolated micropores and the connection to (potentially) elongated mesoporous frameworks, the condensation of the material majorly through linking the PO4 units with the MO6 units due to loss of coordinating water and minorly of ammonia and (c) a crystalline–crystalline phase transition of X-shaped Co-struvite to 2D plate Co-dittmarite with no significant mesoporosity. | ||
Conclusions
Through a simple thermal treatment of precursor M-phosphate compounds such as M-struvites NH4MPO4·6H2O (M = Mg, Ni, Ni-rich, NixCo1−x), simultaneously mesoporous and amorphous metal phosphate materials were synthesized. The temporal evolution of mesopores responsible for the high surface area was accompanied by a crystalline–amorphous phase transformation during degassing of volatile compounds, NH3 and H2O. Such thermally induced phase transformation was linked to the response in the coordination environment of Ni2+ and Co2+ and their structural reconfiguration in the solid compound. Mg-, Ni- and Ni-rich NixCo1−x-struvites form amorphous phases during thermal treatment under dry conditions and evolve stable mesoporous frameworks. At first, near spherical micropores form and coalesce later to possibly more elongated mesopores. The condensation degree to mesoporous structures associated with surface area, pore size and pore shape is directly dependent on the complex amorphous structure. Here, we report a low temperature of 90 °C, at least two hours of duration time and low humidity for optimal evolution of mesoporosity. No significant formation of mesopores occurred in Co- and Co-rich NixCo1−x-struvite after thermal treatment as these compounds exhibit several crystalline–crystalline phase transitions (to Co-dittmarite). Therefore, the relative content of Ni and Co determines the transformation pathways of the TMPs: amorphous/mesoporous for Ni-rich compositions, crystalline for Co-rich compositions. This result is beneficial for structure engineering, because a high Ni-content (x ≫ 0.5) in the mixed NixCo1-xMPs ensures the mesoporosity of structures which still contain considerable amounts of Co, without “locking” Co into a crystalline dittmarite. Ni2+ demonstrated a near-ideal centrosymmetric octahedral coordination sphere in the precursor and heated material compared to a heavily distorted one of Co2+ in the processed compounds. The obtained mesoporous transition metal phosphate materials could potentially be used as OER catalysts, supercapacitors or in proton exchange membranes due to their enhanced reactivity accompanied by their increased surface area.Data availability statement
The datasets supporting this article have been uploaded as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Dr Daniel Hermida Merino for his support at the Dutch-Belgian Beamline (DUBBLE) BM26 beamline, and Dr Martin Radtke for his help at the BAMline beamline of BESSY II. We acknowledge the Dutch-Belgian Beamline (DUBBLE) BM26 for providing us with the beamtime at the ESRF (Proposal A26-02-928). We thank BAM and Helmholtz-Zentrum Berlin (HZB) for providing us with the beamtime at BESSY II. We thank Ines Feldmann for the electron microscopy measurements.References
- H. Zhao and Z. Yuan, Insights into Transition Metal Phosphate Materials for Efficient Electrocatalysis, ChemCatChem, 2020, 12(15), 3797–3810 CrossRef CAS.
- R. Lin and Y. Ding, A Review on the Synthesis and Applications of Mesostructured Transition Metal Phosphates, Materials, 2013, 6(1), 217–243 CrossRef CAS PubMed.
- X. Xu, P. Du, T. Guo, B. Zhao, H. Wang and M. Huang, In situ Grown Ni phosphate@Ni12P5 Nanorod Arrays as a Unique Core–Shell Architecture: Competitive Bifunctional Electrocatalysts for Urea Electrolysis at Large Current Densities, ACS Sustainable Chem. Eng., 2020, 8(19), 7463–7471 CrossRef CAS.
- A. G. Meguerdichian, T. Jafari, M. R. Shakil, R. Miao, L. A. Achola, J. Macharia, A. Shirazi-Amin and S. L. Suib, Synthesis and Electrocatalytic Activity of Ammonium Nickel Phosphate, [NH4]NiPO4.6H2O, and beta-Nickel Pyrophosphate, beta-Ni2P2O7: Catalysts for Electrocatalytic Decomposition of Urea, Inorg. Chem., 2018, 57(4), 1815–1823 CrossRef CAS PubMed.
- V. G. Koleva, T. J. Boyadzhieva and R. K. Stoyanova, Crystal and Morphology Design of Dittmarite-Type Ammonium Iron–Manganese Phosphates, NH4Mn1−xFexPO4·H2O, as Precursors for Phospho-olivine Electrodes, Cryst. Growth Des., 2019, 19(7), 3744–3754 CrossRef CAS.
- Z.-H. Guo, J.-Y. Lin, P.-R. Chen, K.-Q. Ou, X.-Y. Xu, J.-K. Li, S. Huang, X.-X. Sang, J.-C. Liu and J.-L. Song, Fast and scale-up synthesis of amorphous C,N co-doped mesoporous Co-based phosphates as advanced electrodes for supercapacitors and water oxidation, Sustainable Energy Fuels, 2021, 5(22), 5741–5747 RSC.
- X. Guo, N. Li, Y. Cheng, G. Wang, Y. Zhang and H. Pang, General synthesis of nitrogen-doped metal (M = Co2+, Mn2+, Ni2+, or Cu2+) phosphates, Chem. Eng. J., 2021, 411, 128544 CrossRef CAS.
- K. Raju, Ammonium metal phosphates: Emerging materials for energy storage, Curr. Opin. Electrochem., 2020, 21, 351–357 CrossRef CAS.
- J. Pandey, M. M. Seepana and A. Shukla, Zirconium phosphate based proton conducting membrane for DMFC application, Int. J. Hydrogen Energy, 2015, 40(30), 9410–9421 CrossRef CAS.
- R. N. Bulakhe, J. Lee, C. V. Tran and J. B. In, Mesoporous nanohybrids of 2D layered Cu–Cr phosphate and rGO for high-performance asymmetric hybrid supercapacitors, J. Alloys Compd., 2022, 926, 166864 CrossRef CAS.
- N. L. W. Septiani, Y. V. Kaneti, K. B. Fathoni, Y. Guo, Y. Ide, B. Yuliarto, X. Jiang, N. Nugraha, H. K. Dipojono, D. Golberg and Y. Yamauchi, Tailorable nanoarchitecturing of bimetallic nickel–cobalt hydrogen phosphate via the self-weaving of nanotubes for efficient oxygen evolution, J. Mater. Chem. A, 2020, 8(6), 3035–3047 RSC.
- X. Li, P. Xu, M. Chen, G. Zeng, D. Wang, F. Chen, W. Tang, C. Chen, C. Zhang and X. Tan, Application of silver phosphate-based photocatalysts: Barriers and solutions, Chem. Eng. J., 2019, 366, 339–357 CrossRef CAS.
- F. Pang, X. Liu, M. He and J. Ge, Ag3PO4 colloidal nanocrystal clusters with controllable shape and superior photocatalytic activity, Nano Res., 2014, 8(1), 106–116 CrossRef.
- W. Zhang, P. Oulego, S. K. Sharma, X. L. Yang, L. J. Li, G. Rothenberg and N. R. Shiju, Self-Exfoliated Synthesis of Transition Metal Phosphate Nanolayers for Selective Aerobic Oxidation of Ethyl Lactate to Ethyl Pyruvate, ACS Catal., 2020, 10(7), 3958–3967 CrossRef CAS PubMed.
- C. Zhang, W. Zhou, M. M. Ehteshami, Y. Wang and S. H. Chan, Determination of the optimal operating temperature range for high temperature PEM fuel cell considering its performance, CO tolerance and degradation, Energy Convers. Manage., 2015, 105, 433–441 CrossRef CAS.
- T. Anfimova, Metal Phosphates as Proton Conducting Materials for Intermediate Temperature Fuel Cell and Electrolyser Applications, Technical University of Denmark, 2014 Search PubMed.
- Y. Huang, Q. Li, A. H. Jensen, M. Yin, J. O. Jensen, E. Christensen, C. Pan, N. J. Bjerrum and W. Xing, Niobium phosphates as an intermediate temperature proton conducting electrolyte for fuel cells, J. Mater. Chem., 2012, 22(42), 22452–22452 RSC.
- W. Huang, S. Komarneni, Y. D. Noh, J. Ma, K. Chen, D. Xue, X. Xue and B. Jiang, Novel inorganic tin phosphate gel: multifunctional material, Chem. Commun., 2018, 54(21), 2682–2685 RSC.
- L. Mathur, I.-H. Kim, A. Bhardwaj, B. Singh, J.-Y. Park and S.-J. Song, Structural and electrical properties of novel phosphate based composite electrolyte for low-temperature fuel cells, Composites, Part B, 2020, 202 CrossRef CAS.
- S. Karafiludis, A. G. Buzanich, Z. Kochovski, I. Feldmann, F. Emmerling and T. M. Stawski, Ni- and Co-Struvites: Revealing Crystallization Mechanisms and Crystal Engineering toward Applicational Use of Transition Metal Phosphates, Cryst. Growth Des., 2022, 22(7), 4305–4315 CrossRef CAS.
- S. Bach, E. Visnow, M. Panthöfer, T. Gorelik, A. G. Buzanich, A. Gurlo, U. Kolb, F. Emmerling, C. Lind and W. Tremel, Hydrate Networks under Mechanical Stress - A Case Study for Co3(PO4)2·8H2O, Eur. J. Inorg. Chem., 2016, 2016(13–14), 2072–2081 CrossRef CAS.
- J. Fan, T. Wang, Y. Yuan, C.-L. Do-Thanh, X. Suo, Z. Yang, H. Chen and S. Dai, Mechanochemically Assisted Synthesis of High-Entropy Layer-Structured Dittmarite Analogues, ACS Appl. Energy Mater., 2022, 5(3), 3290–3297 CrossRef CAS.
- N. Gupta, R. K. Sahu, T. Mishra and P. Bhattacharya, Microwave-assisted rapid synthesis of titanium phosphate free phosphorus doped Ti3C2 MXene with boosted pseudocapacitance, J. Mater. Chem. A, 2022, 10(29), 15794–15810 RSC.
- Y. Zhao, Z. Chen, D. B. Xiong, Y. Qiao, Y. Tang and F. Gao, Hybridized Phosphate with Ultrathin Nanoslices and Single Crystal Microplatelets for High Performance Supercapacitors, Sci. Rep., 2016, 6, 17613 CrossRef CAS PubMed.
- V. A. Volkovich, T. R. Griffiths and R. C. Thied, Formation of lanthanide phosphates in molten salts and evaluation for nuclear waste treatment, Phys. Chem. Chem. Phys., 2003, 5(14), 3053–3060 RSC.
- P. P. Prosini, L. Cianchi, G. Spina, M. Lisi, S. Scaccia, M. Carewska, C. Minarini and M. Pasquali, Synthesis and Characterization of Amorphous 3Fe2O3·2P2O5·10H2O and Its Electrode Performance in Lithium Batteries, J. Electrochem. Soc., 2001, 148(10), A1125 CrossRef CAS.
- O. Delmer, P. Balaya, L. Kienle and J. Maier, Enhanced Potential of Amorphous Electrode Materials: Case Study of RuO2, Adv. Mater., 2008, 20(3), 501–505 CrossRef CAS.
- A. Ziaee, S. O'Dea, A. Howard-Hildige, L. Padrela, C. Potter, J. Iqbal, A. B. Albadarin, G. Walker and E. J. O'Reilly, Amorphous solid dispersion of ibuprofen: A comparative study on the effect of solution based techniques, Int. J. Pharm., 2019, 572, 118816 CrossRef CAS PubMed.
- V. Mathew, S. Kim, J. Kang, J. Gim, J. Song, J. P. Baboo, W. Park, D. Ahn, J. Han, L. Gu, Y. Wang, Y.-S. Hu, Y.-K. Sun and J. Kim, Amorphous iron phosphate: potential host for various charge carrier ions, NPG Asia Mater., 2014, 6(10), e138–e138 CrossRef CAS.
- J. Rouquerol, D. Avnir, C. W. Fairbridge, D. H. Everett, J. H. Haynes, N. Pernicone, J. D. F. Ramsay, K. S. W. Sing and K. K. Unger, Recommendations for the characterization of porous solids, International Union of Pure and Applied Chemistry (IUPAC), 1994, pp. 1739–1758 Search PubMed.
- J. Feng, Z. Wang, B. Shen, L. Zhang, X. Yang and N. He, Effects of template removal on both morphology of mesoporous silica-coated gold nanorod and its biomedical application, RSC Adv., 2014, 4(54), 28683–28690 RSC.
- M. Barczak, Template removal from mesoporous silicas using different methods as a tool for adjusting their properties, New J. Chem., 2018, 42(6), 4182–4191 RSC.
- R. A. Lorenzo, A. M. Carro, C. Alvarez-Lorenzo and A. Concheiro, To remove or not to remove? The challenge of extracting the template to make the cavities available in Molecularly Imprinted Polymers (MIPs), Int. J. Mol. Sci., 2011, 12(7), 4327–4347 CrossRef CAS PubMed.
- T. Qin, Y. Han, P. Zhang, I. H. Wani, F. Nikolajeff, K. Leifer and H. Engqvist, Template-free synthesis of phosphate-based spheres via modified supersaturated phosphate buffer solutions, J. Mater. Sci. Mater. Med., 2017, 28(7), 99 CrossRef PubMed.
- J. Hövelmann, T. M. Stawski, R. Besselink, H. M. Freeman, K. M. Dietmann, S. Mayanna, B. R. Pauw and L. G. Benning, A template-free and low temperature method for the synthesis of mesoporous magnesium phosphate with uniform pore structure and high surface area, Nanoscale, 2019, 11(14), 6939–6951 RSC.
- K. S. Le Corre, E. Valsami-Jones, P. Hobbs and S. A. Parsons, Phosphorus Recovery from Wastewater by Struvite Crystallization: A Review, Crit. Rev. Environ. Sci. Technol., 2009, 39(6), 433–477 CrossRef CAS.
- A. Uysal, Y. D. Yilmazel and G. N. Demirer, The determination of fertilizer quality of the formed struvite from effluent of a sewage sludge anaerobic digester, J. Hazard. Mater., 2010, 181(1–3), 248–254 CrossRef CAS PubMed.
- X. Hao, C. Wang, M. C. van Loosdrecht and Y. Hu, Looking beyond struvite for P-recovery, Environ. Sci. Technol., 2013, 47(10), 6–4965 CrossRef PubMed.
- M. Sena, M. Seib, D. R. Noguera and A. Hicks, Environmental impacts of phosphorus recovery through struvite precipitation in wastewater treatment, J. Cleaner Prod., 2021, 280(1), 124222 CrossRef CAS.
- J. Zhao, H. Pang, J. Deng, Y. Ma, B. Yan, X. Li, S. Li, J. Chen and W. Wang, Mesoporous uniform ammonium nickel phosphate hydrate nanostructures as high performance electrode materials for supercapacitors, CrystEngComm, 2013, 15(30), 5950–5955 RSC.
- Y. Liu, X. Zhai, K. Yang, F. Wang, H. Wei, W. Zhang, F. Ren and H. Pang, Mesoporous NH4NiPO4.H2O for High-Performance Flexible All-Solid-State Asymmetric Supercapacitors, Front. Chem., 2019, 7, 118 CrossRef CAS PubMed.
- W. Bras, I. P. Dolbnya, D. Detollenaere, R. van Tol, M. Malfois, G. N. Greaves, A. J. Ryan and E. Heeley, Recent experiments on a combined small–angle/wide–angle X–ray scattering beam line at the ESRF, J. Appl. Crystallogr., 2003, 36, 791–794 CrossRef CAS.
- V. Dyadkin, P. Pattison, V. Dmitriev and D. Chernyshov, A new multipurpose diffractometer PILATUS@SNBL, J. Synchrotron Radiat., 2016, 23(Pt 3), 825–829 CrossRef CAS PubMed.
- G. J. Smales and B. R. Pauw, The MOUSE project: a meticulous approach for obtaining traceable, wide-range X-ray scattering information, J. Instrum., 2021, 16(06), P06034 CrossRef CAS.
- B. R. Pauw, A. J. Smith, T. Snow, N. J. Terrill and A. F. Thunemann, The modular small-angle X-ray scattering data correction sequence, J. Appl. Crystallogr., 2017, 50(Pt 6), 1800–1811 CrossRef CAS PubMed.
- I. Bressler, B. R. Pauw and A. Thunemann, McSAS: software for the retrieval of model parameter distributions from scattering patterns, J. Appl. Crystallogr., 2015, 48(Pt 3), 962–969 CrossRef CAS PubMed.
- S. Visser and S. Cooper, Analysis of Small-Angle X-ray Scattering Data for Model Polyurethane Ionomers: Evaluation of Hard-Sphere Models, Macromolecules, 1991, 24, 2584–2593 CrossRef CAS.
- D. J. Kinning and E. L. Thomas, Hard-Sphere Interactions between Spherical Domains in Diblock Copolymers, Macromolecules, 1984, 17, 1712–1718 CrossRef CAS.
- A. G. Baldwin, Y. Yang, N. J. Bridges and J. C. Braley, Tributyl Phosphate Aggregation in the Presence of Metals: An Assessment Using Diffusion NMR Spectroscopy, J. Phys. Chem. B, 2016, 120(47), 12184–12192 CrossRef CAS PubMed.
- O. Lehmann, H. Meyssamy, K. Kömpe, H. Schnablegger and M. Haase, Synthesis, Growth, and Er3+ Luminescence of Lanthanide Phosphate Nanoparticles, J. Phys. Chem. B, 2003, 107, 7449–7453 CrossRef CAS.
- W. Görner, M. P. Hentschel, B. R. Müller, H. Riesemeier, M. Krumrey, G. Ulm, W. Diete, U. Klein and R. Frahm, BAMline: the first hard X-ray beamline at BESSY II, Nucl. Instrum. Methods Phys. Res., Sect. A, 2001, 467–468, 703–706 CrossRef.
- B. Ravel and M. Newville, ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT, J. Synchrotron Radiat., 2005, 12(Pt 4), 537–541 CrossRef CAS PubMed.
- G. Pratesi, C. Cipriani, G. Giuli and W. D. Birch, Santabarbaraite: a new amorphous phosphate mineral, Eur. J. Mineral., 2003, 15(1), 185–192 CrossRef CAS.
- B. Jin, Z. Liu, C. Shao, J. Chen, L. Liu, R. Tang and J. J. De Yoreo, Phase Transformation Mechanism of Amorphous Calcium Phosphate to Hydroxyapatite Investigated by Liquid-Cell Transmission Electron Microscopy, Cryst. Growth Des., 2021, 21(9), 5126–5134 CrossRef CAS.
- N. Sánchez-Pastor, M. Oehlerich, J. M. Astilleros, M. Kaliwoda, C. C. Mayr, L. Fernández-Díaz and W. W. Schmahl, Crystallization of ikaite and its pseudomorphic transformation into calcite: Raman spectroscopy evidence, Geochim. Cosmochim. Acta, 2016, 175, 271–281 CrossRef.
- C. J. Brinker and G. W. Scherer, CHAPTER 9 - Structural Evolution during Consolidation, in Sol-Gel Science, ed. C. J. Brinker and G. W. Scherer, Academic Press, San Diego, 1990, pp. 514–615 Search PubMed.
- M. B. G. Foglio, Study of Co2+ in Different Crystal Field Environments, Braz. J. Phys., 2005, 36, 40–54 Search PubMed.
- A. Styskalik, D. Skoda, Z. Moravec, P. Roupcova, C. E. Barnes and J. Pinkas, Non-aqueous template-assisted synthesis of mesoporous nanocrystalline silicon orthophosphate, RSC Adv., 2015, 5(90), 73670–73676 RSC.
- E. Kim, M. G. Kim, Y. Kim and J. Cho, Effect of Pore Size and Pore Wall Thickness of Mesoporous Phase in Tin Phosphate Composite on Electrochemical Cycling, Electrochem. Solid-State Lett., 2005, 8(9), A452 CrossRef CAS.
- J. Lu, Y. Li and C. Deng, Facile synthesis of zirconium phosphonate-functionalized magnetic mesoporous silica microspheres designed for highly selective enrichment of phosphopeptides, Nanoscale, 2011, 3(3), 1225–1233 RSC.
- M. Pramanik, J. Lee, S. Tominaka, Y. Ide, J. H. Kim and Y. Yamauchi, Unique nanocrystalline frameworks in mesoporous tin phosphate prepared through a hydrofluoric acid assisted chemical reaction, J. Mater. Chem. A, 2016, 4(46), 18091–18099 RSC.
- M. Pramanik, C. Li, M. Imura, V. Malgras, Y. M. Kang and Y. Yamauchi, Ordered Mesoporous Cobalt Phosphate with Crystallized Walls toward Highly Active Water Oxidation Electrocatalysts, Small, 2016, 12(13), 1709–1715 CrossRef CAS PubMed.
- W.-X. Lu, B. Wang, W.-J. Chen, J.-L. Xie, Z.-Q. Huang, W. Jin and J.-L. Song, Nanosheet-like Co3(OH)2(HPO4)2 as a Highly Efficient and Stable Electrocatalyst for Oxygen Evolution Reaction, ACS Sustainable Chem. Eng., 2019, 7(3), 3083–3091 CrossRef CAS.
- F. S. Costa, F. Langenhorst and E. Kothe, Biomineralization of Nickel Struvite Linked to Metal Resistance in Streptomyces mirabilis, Molecules, 2022, 27(10), 3061 CrossRef CAS PubMed.
- G. Haferburg, G. Kloess, W. Schmitz and E. Kothe, “Ni-struvite” - a new biomineral formed by a nickel resistant Streptomyces acidiscabies, Chemosphere, 2008, 72(3), 517–523 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Note 1: Critical analysis of SAXS and WAXS processing and of the applied fitting model, Supplementary Note 2: description of K-edge EXAFS spectra in R-space, Fig. S1: XRD diagrams and TGA/DSC measurements of NixCo1−x-struvites, Fig. S2: crystal structure and XRD patterns of T-series of Ni- and Co-struvite from 25 °C – 800 °C, Fig. S3: selected EDX mappings of NixCo1−x-struvite crystals, Fig. S4: SE images of Mg-, Ni – and Co -struvite and their thermally processed derivatives, Fig. S5: N2 adsorption/desorption measurements of thermally treated Mg and Ni-struvite, Fig. S6: N2 adsorption/desorption measurements of thermally treated NixCo1−x-struvite, Fig. S7: McSAS fits of Mg-struvite isothermally heated at 90 °C, Fig. S8: pore size distribution histograms of McSAS fits of Mg-struvite isothermally heated at 90 °C, Fig. S9: McSAS fits of Ni-struvite isothermally heated at 90 °C, Fig. S10: pore size distribution histograms of McSAS fits of Ni-struvite isothermally heated at 90 °C, Fig. S11: McSAS fits of different local volume fractions (ν = 35%, 40% and 50%) and pore size distributions (relative volume fraction of pores) of Mg-struvite isothermally heated at 90 °C, Fig. S12: McSAS fits of different local volume fractions (ν = 35%, 40% and 50%) and pore size distributions (relative volume fraction of pores) of Ni-struvite isothermally heated at 90 °C, Fig. S13: ex situ combined SAXS/WAXS measurements and associated McSAS fits of isothermally heated NixCo1−x-struvite with x = 0.2, 0.5, 0.8 at T = 90 °C for t = 24 h, Fig. S14: Ni- and Co-K-edge XAS spectra of Ni-, Co- and NixCo1−x-struvites, Fig. S15: fits of Ni–K-edge EXAFS data of Ni time series, Fig. S16: Ni K-edge EXAFS data of Ni time series with the corresponding bond distances, eqn (S1): full explanation of the Percus-Yevick structure factor, eqn (S2): calculation of the approximated volume fraction of pores in Mg-struvite from BET N2 adsorption/desorption data, Table S1: DSC integration results of M-struvite, Table S2: summarized EDS data for all NixCo1−x-mixtures (x = 0.1–0.95), Table S3: BET N2 gas sorption results for the pure Mg-, Ni- and Co-struvites, Table S4: BET N2 gas sorption results for the NixCo1−x-mixtures, Table S5: calculated McSAS average pore sizes of all pure M-struvites, Table S6: calculated McSAS average pore sizes of heated NixCo1−x-struvite, Table S7: XANES Ni–K (8333 eV) pre-peak integration results of heated Ni-struvite t-series, Table S8: XANES Ni–K (8333 eV) and Co–K (7709 eV) pre-peak integration results of selected NixCo1−x-struvite precursors and heated products at T = 90 °C and t = 1 d, Table S9: summarized Ni- and Co–K-pre-peak area integration results and phase composition of the NixCo1−x-mixtures, Table S10: fit parameter for Ni-struvite R-factor = 0.005, Table S11: fit parameter for Ni-struvite for t = 10 min, R-factor = 0.008, Table S12: fit parameter for Ni-struvite for t = 20 min, R-factor = 0.009, Table S13: fit parameter for Ni-struvite for t = 40 min, R-factor = 0.011, Table S14: fit parameter for Ni-struvite for t = 60 min, R-factor = 0.013, Table S15: fit parameter for Ni-struvite for t = 90 min, R-factor = 0.008, Table S16: fit parameter for Ni-struvite for t = 120 min, R-factor = 0.010, Table S17: fit parameter for Ni-struvite for t = 150 min, R-factor = 0.009 and Table S18: fit parameter for Ni-monohydrate synthesized in a hydrothermal route, R-factor = 0.012. See DOI: https://doi.org/10.1039/d2nr05630e |
| This journal is © The Royal Society of Chemistry 2023 |