
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effect of biomolecular corona on adsorption onto and desorption from a model lipid membrane†
Ceri J.
Richards
 ab,
Majid
Ahmadi
c,
Marc C. A.
Stuart
d,
Bart J.
Kooi
c,
Christoffer
Åberg
*b and
Wouter H.
Roos
*a
ab,
Majid
Ahmadi
c,
Marc C. A.
Stuart
d,
Bart J.
Kooi
c,
Christoffer
Åberg
*b and
Wouter H.
Roos
*a
aMolecular Biophysics, Zernike Institute for Advanced Materials, Rijksuniversiteit Groningen, 9747 AG Groningen, Netherlands. E-mail: w.h.roos@rug.nl
bPharmaceutical Analysis, Groningen Research Institute of Pharmacy, Rijksuniversiteit Groningen, 9713 AV Groningen, Netherlands. E-mail: christoffer.aberg@rug.nl
cNanostructure Materials and Interfaces, Zernike Institute for Advanced Materials, Rijksuniversiteit 9747 AG Groningen, Netherlands
dDepartment of Electron Microscopy, Groningen Biomolecular Sciences and Biotechnology Institute, Rijksuniversiteit Groningen, 9747 AG Groningen, Netherlands
First published on 6th December 2022
Abstract
The current lack of insight into nanoparticle–cell membrane interactions hampers smart design strategies and thereby the development of effective nanodrugs. Quantitative and methodical approaches utilizing cell membrane models offer an opportunity to unravel particle–membrane interactions in a detailed manner under well controlled conditions. Here we use total internal reflection microscopy for real-time studies of the non-specific interactions between nanoparticles and a model cell membrane at 50 ms temporal resolution over a time course of several minutes. Maintaining a simple lipid bilayer system across conditions, adsorption and desorption were quantified as a function of biomolecular corona, particle size and fluid flow. The presence of a biomolecular corona reduced both the particle adsorption rate onto the membrane and the duration of adhesion, compared to pristine particle conditions. Particle size, on the other hand, was only observed to affect the adsorption rate. The introduction of flow reduced the number of adsorption events, but increased the residence time. Lastly, altering the composition of the membrane itself resulted in a decreased number of adsorption events onto negatively charged bilayers compared to neutral bilayers. Overall, a model membrane system offers a facile platform for real-time imaging of individual adsorption–desorption processes, revealing complex adsorption kinetics, governed by particle surface energy, size dependent interaction forces, flow and membrane composition.
Introduction
From the perspective of developing smart nanomedicine design strategies, understanding the interactions of nanoparticles with biological interfaces is of great importance.1,2 However, there is still little known about how nanoparticle properties affect particle–cell binding.3–5 This is, in part, due to the heterogeneity and dynamic nature of the outer cell membrane, giving rise to complex interactions. Thus, at this interface both non-specific and specific interactions are present, potentially leading to a variety of particle–membrane binding and uptake mechanisms.2,3,6 Simplifying the system facilitates the study of these mechanisms in a structural manner. One example of such a simplification, and the approach taken here, is the replacement of the highly complex outer cell membrane with a model membrane.Membrane models, such as unilamellar vesicles and supported lipid bilayers, have various advantages compared to live cells, including: a known membrane composition, easy control over environmental factors, and compatibility with a variety of investigative techniques. Though model membrane systems comprised only of lipid species do not capture specific interactions such as those with receptors,7–9 these simplified membrane systems have been shown to exhibit analogous behaviour to live cells when exposed to nanoparticles.10–12 This highlights the importance of non-specific binding at the particle–cell membrane interface. Overall, model membranes offer a facile platform to characterize non-specific particle–membrane interactions in detail.10–17 This forms a first step towards understanding and disentangling the full landscape of particle–membrane interactions present in more complex systems.
The binding of nanoparticles to cell membranes or membrane models has been characterized via various qualitative and quantitative parameters. This includes studies on the amount of particles present (mass or fluorescence signal) on a membrane after a certain period of time;10–12,18–21 the adsorbing/desorbing mass across time;12,16 or the degree of membrane disruption10,11,21 It has been observed that properties such as particle charge,10,11,16,22 size,16,17,19,23 shape anisotropy,13,24,25 lipophilicity20,23 and surface features (porosity, spikes etc.)17,26 affect particle–membrane interactions. Furthermore, these interactions are strongly influenced by the presence of a biomolecular corona,10–12,21 that is, the layer(s) of biomolecules that associate with a particle surface when the latter enters biological fluids such as the blood stream (Fig. 1a).27 However, in addition to adsorption, also desorption of particles from the membrane should be considered.18,28 Furthermore, it has been speculated that, under biomolecular corona conditions, transient particle–membrane interactions are frequent, with long term adhesion occurring more rarely.1 This suggests that desorption is in fact very common and can occur quickly. However, dependent on the technique used, resolving these individual desorption processes may not be possible due to washing steps, a lack of temporal resolution,12,18 and/or only bulk data, lacking the necessary single particle information, being obtained.12,16 Fluorescence microscopy techniques, on the other hand, can be used to visualize single nanoparticles on membranes.15,29 Thus far, previous studies have mostly focused on final ‘snap shots’,10,11 rather than imaging the adsorption–desorption processes directly, or they have focussed on diffusion and transport processes of particles on membranes,7,30–32 as opposed to rates and duration of adsorption.
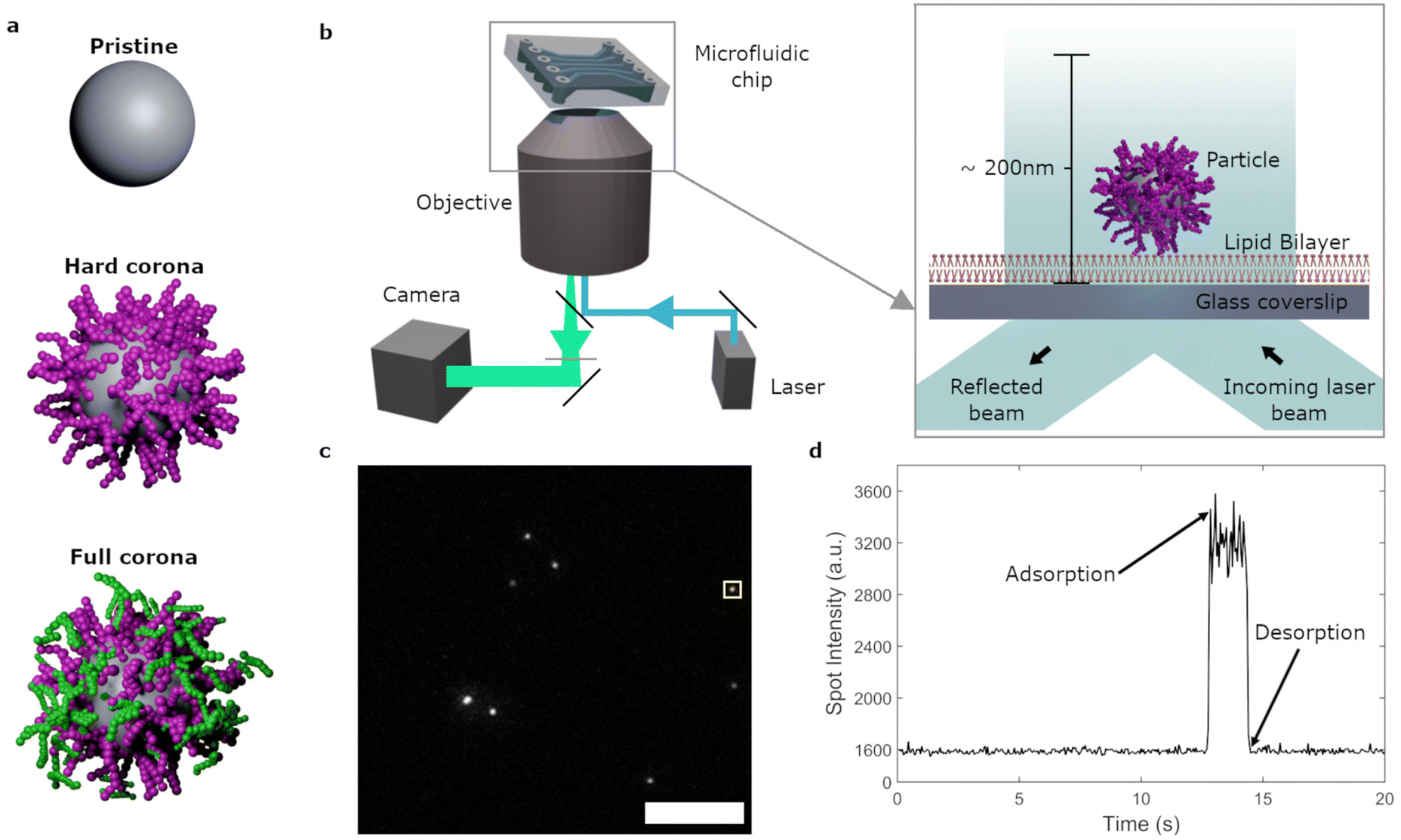 | ||
| Fig. 1 Experimental conditions and setup. a, Illustrations of particles under pristine, hard corona and full corona conditions, where biomolecules which are strongly bound to the particle are depicted in purple and loosely bound biomolecules are shown in green. Both the soft and hard corona consists of various biomolecular species. b, Schematic depiction of the TIRF microscopy set up. Bilayers were deposited within the channels of a microfluidic chip and fluorescent particles were added. The fluorescence signal was then imaged using the CCD camera. Inset: Laser excitation was used to visualize the particles within ∼200 nm of the glass surface using TIRF mode. c, Example of TIRF microscopy data where particles on/near the bilayer are seen as bright dots. Scale bar represents 20 μm. The yellow box denotes the object for which the intensity profile over time is given in d. d, Intensity profile over time of an object adsorbed to the bilayer. The adsorption time point is indicated by an increase in intensity from the baseline and desorption is signaled by a return to the background signal after 1.55 s (arrows). | ||
In the current study we focus on the non-specific interactions between nanoparticles and a membrane model, namely supported lipid bilayers. Using total internal reflection fluorescence (TIRF) microscopy, the interaction of individual nanoparticles within ∼200 nm of the membrane were observed with a 50 ms time resolution over timespans of minutes (Fig. 1b–d). Maintaining a simplistic membrane across experiments, we altered the particle and environmental conditions to investigate their effects on non-specific membrane adsorption. We studied the influence of the biomolecular corona, as well as the role of particle size and membrane composition, on these interactions. Additionally, flow conditions were introduced to mimic shear stresses present in the capillary environment in our simplified system. We quantified particle adsorption rates and the distributions of residence times on the membrane. Thereby, it was possible to distinguish the effect of each variable on transient as well as long lived events. The combination of event frequency and duration revealed how the biomolecular corona as well as flow conditions strongly affect non-specific interactions of the nanoparticles with the lipid bilayer.
Experimental
Nanoparticle preparation
100 nm, 40 nm and 20 nm yellow-green fluorescent carboxylated polystyrene nanoparticles (“FluoSpheres”) were purchased from Invitrogen (505/515 nm excitation/emission). Particles were dispersed in phosphate buffered saline (PBS; Gibco) to produce pristine particles. Dulbecco's minimal essential medium (Gibco) was supplemented with 10% foetal bovine serum (Gibco) to produce complete medium. To form full corona particles, the nanoparticles were dispersed in complete medium for 1 h on a nutator at room temperature. Hard corona particles were prepared by centrifugation of 100 nm full corona particles at 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Relative Centrifugal Force (RCF) at 4 °C for 75 min followed by removal of the resulting supernatant. PBS was added to the pellet and the particles were resuspended via bath sonication for 1 h.
000 Relative Centrifugal Force (RCF) at 4 °C for 75 min followed by removal of the resulting supernatant. PBS was added to the pellet and the particles were resuspended via bath sonication for 1 h.
The concentration of the particle dispersions was calculated using the nominal sizes and density of the particles. Dispersions were then diluted to a final concentration of 0.5 × 109 ml−1 in all cases except the 40 nm full corona particles which were diluted to a concentration of 2 × 109 ml−1. A correction for the concentration difference was applied for the comparison of the adsorption rates. Size characterization of the final dispersions was performed using a NanoSight LM14 and NTA 3.0 software (Malvern). Values for the particle hydrodynamic sizes obtained by NTA are reported as mean ± standard deviation. It should be noted that comparisons of particle concentrations measured by NTA suggested that the preparation procedure of the hard corona particles led to a reduced particle concentration compared to under the other two conditions. Therefore, we corrected the number of adsorption events by the concentration measured to determine the adsorption rate expected for a nominal number concentration of 0.5 × 109 ml−1.
Atomic force microscopy
Atomic force microscopy (AFM) of the particles was performed on a JPK NanoWizard (Bruker). Dispersions were prepared as above, using milli-q water instead of phosphate buffered saline. The full corona dispersions were diluted 20× with milli-q water after incubation in complete cell culture medium. Droplets of dispersion were placed on poly-L-lysine coated glass slides.33 After 10 min incubation, dispersions were removed without completely drying the substrate and then replaced with milli-q water. Imaging of the particles was performed in milli-q water in QI mode using the CB2 cantilever of the qp-BioAC-50 chip (NANOSENSORS) with a nominal spring constant of 0.06–0.18 N m−1. Analysis of the images was performed using JPK SPM Data Processing software. It should be noted that AFM typically displays broadening in the scanning plane and therefore only the heights are indicative of particle size. Heights are quoted as mean ± standard error of the mean.The arithmetic mean height of the poly-L-lysine substrate under the pristine condition was 0.6 ± 0.1 nm which increased to 1.2 ± 0.1 nm under the full corona conditions. This suggests biomolecules in solution did adsorb to the substrate. However, the influence of this on the measured heights of the particles was minimal, amounting to only ∼0.6 nm, if biomolecules adsorb in the region local to the particle.
Cryogenic transmission electron microscopy
Particle dispersions were prepared as above except that milli-q water replaced the usage of phosphate buffered saline. Full corona and hard corona samples were briefly centrifuged at 10000 RCF for 5 min after which 90% of the supernatant was removed to concentrate the dispersion. The loose pellet was resuspended in the remaining supernatant by pipetting. An aliquot (3 μL) of sample was deposited on glow-discharged holey carbon-coated grids (3.5/1 Quantifoil Micro Tools, Jena, Germany). After the excess liquid was blotted, the grids were vitrified in liquid ethane using a Vitrobot (FEI, Eindhoven, The Netherlands) and transferred to a FEI Tecnai T20 electron microscope equipped with a Gatan model 626 cryo-stage operating at 200 keV. Micrographs were recorded under low-dose conditions with a slow-scan CCD camera.
Liquid cell scanning transmission electron microscopy
Particle dispersions were prepared as above except that milli-q water replaced the usage of phosphate buffered saline. The dispersions were then diluted 500× in milli-q water. Graphene TEM grids were prepared by transferring graphene (mono and bi-layers) to the coated side of Au Quantifoil TEM grids. The grids were placed on graphene layers (deposited by chemical vapor deposition onto Cu foils) so that the coated side of the grids was in contact with the graphene. Droplets of isopropyl alcohol were placed onto the grids to improve adhesion to the graphene. After drying, the grids were transferred to FeCl3 solution (copper etchant, Sigma-Aldrich) for 30 min to etch away the Cu foil. The grids were washed several times with deionized water (see ESI Fig. S4† for a schematic of the grid preparation procedure). Droplets of particle dispersion were placed onto the grids and, after ambient drying, the graphene backed grids were sandwiched together to form ‘cells’ containing particle dispersion. The volumes of dispersion were isolated from the vacuum and therefore remained under ambient conditions whilst in the electron microscope. Images were taken with a double-corrected and monochromated Themis Z scanning transmission electron microscope (Thermo Fisher Scientific) operating at 300 kV through high-angle annular dark-field (HAADF) STEM mode. The beam convergence angle was measured ∼22.0 mrad. Data from the HAADF-STEM images were acquired from angles of about 31–186 mrad. Low dose STEM imaging was applied by using 10–30 pA e-beam current. Analysis of the images was performed using Velox software. Further details can be found in the ESI.†Bilayer preparation
Bilayers were produced using a 1:1:1:1.5:1.11 × 10−4 ratio of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), sphingomyelin, cholesterol and N-((6-(biotinoyl)amino)hexanoyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolaime (biotin-X DHPE). All lipids were purchased from Avanti Polar Lipids except biotin-X DHPE (Invitrogen). Octadecyl rhodamine B chloride (Invitrogen) was added to the lipid mixture at a 170 nM concentration as a lipophilic fluorescent label that binds to the lipid molecules. The dried lipid mixture was diluted with PBS to a 7 mM total lipid concentration. To obtain small unilamellar vesicles, several freeze–thaw cycles were performed on the subsequent liposome solution followed by sonication at temperatures above 55 °C for 30 min. HeLa mimicking bilayers34 were prepared as above using a 0.29:0.31:0.6 ratio of DOPC, DOPE, cholesterol and brain L-α-phosphatidylserine (PS) (Avanti Polar Lipids). After sonication, calcium chloride was added for a final concentration of 5 mM for both the lipids and CaCl. To form bilayers, liposome dispersions were deposited into a flow cell. The flow cell consisted of a polydimethylsiloxane (PDMS) microfluidic chip interfaced with 1.5 glass coverslips (VWR Scientific) to produce 5 separate 500 μm × 200 μm channels. The coverslips were cleaned via sonication in acetone, ethanol and 1 M KOH solution, rinsing with deionized water in between sonication steps. Prior to attachment to the microfluidic chip, coverslips were oxygen plasma cleaned for 30 min. After 1 h of incubation, the liposomes collapsed to produce bilayers within the channels. The bilayers were washed with PBS to remove excess lipid debris.15
Total internal fluorescence microscopy (TIRF)
Experiments were conducted on a home built TIRF microscope consisting of an IX-71 inverted microscope (Olympus), 60× oil immersion objective (NA 1.45, Olympus), EM-CCD camera (Hamamatsu) and 561 and 488 nm lasers (Coherent) for bilayer and nanoparticle fluorophore excitation, respectively.15 Fluorescence recovery after photobleaching (FRAP) was performed with the 561 nm laser to determine bilayer fluidity. Only bilayers for which recovery was observed were used for subsequent experiments. FRAP was also performed on bilayers that were incubated with complete cell culture medium for 1 h and then washed quickly with PBS. This was performed to ensure that this pre-incubation procedure (used in the pre-incubation experiments) did not disrupt or induce a fluid to gel phase transition within the lipid bilayer (ESI Fig. S6†). For the adsorption experiments under static conditions, particles were aspirated into the channel after which the flow was stopped once particles could be identified in the observation region. Any residual effects of the flow were allowed to dissipate before recording began. For the experiments under flow conditions, the flow used to aspirate particles into the channel was maintained for the entire observation period. Under both conditions, 10 min videos were recorded using a 50 ms exposure time and frame length (in total 12000 images per video). Experiments were repeated several times under the same conditions with each repeat performed on a new bilayer.
Analysis
The ImageJ/Fiji35,36 plugin TrackMate37 was used to identify particles. Bright spots, indicative of particles on or near the bilayer, were considered if the fluorescence intensity was above a given threshold. The threshold was chosen such that all (and only) particles observed by eye were identified. By linking the identified particles across time (frames), the first appearance (adsorption) and subsequent disappearance (desorption) of the particle were identified. Distinguishing between particles on and near the membrane using the intensity of the signal is challenging, given factors such as photobleaching, uneven illumination and the distribution of loaded fluorophores in the particles. Therefore, we chose to consider only stationary particles as adsorbed onto the bilayer, in other words, particles that did not move in the plane of the bilayer between the initial adsorption event and subsequent desorption from the bilayer. Therefore a 1 pixel interlinking distance was used. Intensity profiles of the particles across time were manually inspected to check that the outcome of the particle linking coincided with the peak in the intensity profile (Fig. 1d). In cases where a discrepancy was noted, the raw data was inspected to confirm the particle adsorption and desorption time points. This was necessary as both moving particles and those with intensities close to the detection threshold could be misidentified as multiple short events. Only events with minimally three frames were used, i.e., the first time point of entry, adherence (peak in intensity) and the last time point signalling the desorption process. The residence time was calculated as the time between attachment and detachment of the particle. It should be noted that our observations are limited by the temporal resolution of the experiments, implying that residence times shorter than 100 ms are not captured.To relate the length of the adsorption events to cell internalization, we applied a threshold to quantify the particles that, had they been adsorbed to a cell, would have remained sufficiently long, on average, to be internalized. Particles that remained adsorbed at the end of the measurement but had been adsorbed for minimally the threshold time were kept in the data set and all other particles that did not desorb were removed as they could not be ascribed a known residence time (more details on the analysis are provided in ESI Discussion “Analysis of microscopy videos”†). For choosing the threshold, we note that nanoparticles have been reported to be internalized by various mechanisms ranging from clathrin-mediated endocytosis to phagocytosis.38 These processes have typical time scales ranging from 40 s to several minutes.39–41 We defined 120 s as our threshold, but choosing other values for the threshold, for example 60 s and 180 s resulted in analogous outcomes for the analysis of the 100 nm pristine particles (ESI Fig. S7†).
Statistical testing of the residence time distributions was performed by a two sample Kolmogorov–Smirnov test at a 5% confidence interval with the null hypothesis that the two data sets compared came from the same continuous distribution. For the calculation of the average residence times (mean ± standard error of the mean) for the 100 nm and 40 nm full corona particles under static and flow conditions, the few events that were not observed to desorb were removed from the data set. Further details can be found in the ESI.†
Results and discussion
Particle characterization
We studied adsorption dynamics of carboxylated polystyrene particles with nominal diameters of 20 nm, 40 nm and 100 nm under pristine (untreated) and biomolecular corona conditions. The biomolecular corona can be divided into the hard corona, comprising tightly bound biomolecules, and the soft corona, which are molecules that interact transiently with the particle complex.42–44 Particles with both the hard and the soft corona are here termed full corona particles and we studied hard corona and full corona particles, next to pristine particles (Fig. 1a). Pristine nanoparticle dispersions were prepared simply by dispersion in buffer; full corona nanoparticles were prepared by dispersion in complete cell medium containing 10% foetal bovine serum; and hard corona covered nanoparticles were prepared in the same manner as the full corona particles, followed by a centrifugation and subsequent redispersion step in order to remove loosely bound biomolecules.42 Size characterization of the polystyrene particles was performed by atomic force microscopy (AFM) under all three conditions. The insets of Fig. 2a–c show AFM images of the 100 nm particles, which confirmed their spherical morphology (cross sections and larger images shown in ESI Fig. S1†). 40 nm and 20 nm particles both with and without corona coverage were also characterized by AFM (ESI Fig. S2†). Height determinations revealed an increase in size upon corona formation and indicated a 5 nm thick layer on all particles, irrespective of (pristine particle) size. This is in line with previous studies of biomolecule adsorption onto nanoparticles.43–45 The colloidal stability of the 100 nm particle dispersions was assessed using nanoparticle tracking analysis (NTA), which showed that all dispersions remained monodisperse (ESI Fig. S3†). Additionally, NTA yielded a diameter of 98 ± 12 nm for the pristine particles in line with the AFM measurements. Furthermore, NTA gave diameters of 131 ± 27 nm and 126 ± 16 nm for hard corona covered and full corona particles, respectively. There is a discrepancy between these measurements and those from AFM, which can be explained by the fact that NTA measures the hydrodynamic, rather than the ‘real’, particle size. | ||
| Fig. 2 Size characterization of 100 nm carboxylated polystyrene particles under various corona conditions via atomic force microscopy and cryogenic transmission electron microscopy. Height distributions obtained from cross sections of AFM images of a, pristine b, hard corona covered and c, full corona particles measured in liquid, yielding average particle sizes of 95 ± 1 nm, 104 ± 1 nm and 107 ± 2 nm, respectively. Insets show representative AFM images where color indicates height above the substrate. Scale bars 50 nm. Cryo EM images of d, pristine particles e, hard corona covered particles and f, full corona particles. Pristine particles show a well-defined particle edge, whereas the hard corona covered particles are covered by biomolecules (indicated by arrows). Under full corona conditions, not only the particles were covered with biomolecules, but biomolecules are also present in the background. The smaller particles are ice contaminants, as indicated. Scale bars 50 nm. | ||
Further characterization of the 100 nm particles was performed by cryogenic transmission electron microscopy (cryo-EM) and scanning transmission electron microscopy (STEM) to assess the degree of biomolecular coverage on particles (Fig. 2d–f). Cryo-EM images of pristine particles showed single spherical particles. Full corona particles were covered in material which could also be found in the surrounding dispersion. This material was more dense on and in the vicinity of the particles. Hard corona particles exhibited a coating which was clearly distinct from the less dense background (due to biomolecule removal in the centrifugation procedure). Therefore, we concluded that both full and hard corona particles were indeed completely coated by biomolecules. Liquid Cell STEM imaging confirmed these observations and, using this innovative technique, we could visualize biomolecules under ambient conditions (ESI Fig. S4†).
Pristine particle interactions with lipid bilayers
We next investigated the adsorption/desorption behaviour of individual 100 nm pristine particles on lipid bilayers under static conditions (that is, without flow). The lipid bilayer was composed of a 1:1:1:1.5:1.11 × 10−4 ratio of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), sphingomyelin, cholesterol and N-((6-(biotinoyl)amino)hexanoyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolaime (biotin-X DHPE). This model was chosen to mimic mammalian cell membranes which, in addition to phospholipids, include large proportions of sphingomyelin and cholesterol.46 A suspension of particles was exposed to the lipid bilayer and the nanoparticle–bilayer interactions were captured by TIRF.15 ESI Video S1† shows data from a typical experiment of 100 nm pristine particles interacting with the lipid bilayer. TIRF microscopy largely eliminates out-of-focus fluorescence by only visualizing up to ∼200 nm inside the flow cell (Fig. 1b). Furthermore, the closer to the lipid bilayer an object is, the stronger the fluorescence. Spots that suddenly appear and then grow in intensity thus represent nanoparticles that come into sight and subsequently approach the lipid bilayer and vice versa (Fig. 1c and d).
Particles were identified and linked across timeframes to obtain the nanoparticle residence times, that is, the time between adsorption and the frame prior to the desorption of each individual object. Fig. 3a shows the distribution of residence times, where the final bar of the histogram represents the proportion of events lasting 120 s or more (see also ESI Table S1†). We observed that the majority of adsorbing pristine particles (71%) resided on the bilayer for timespans of 120 s or more. The rest of the distribution was heavily skewed towards shorter residence times with only few events adsorbed for more than 2 s.
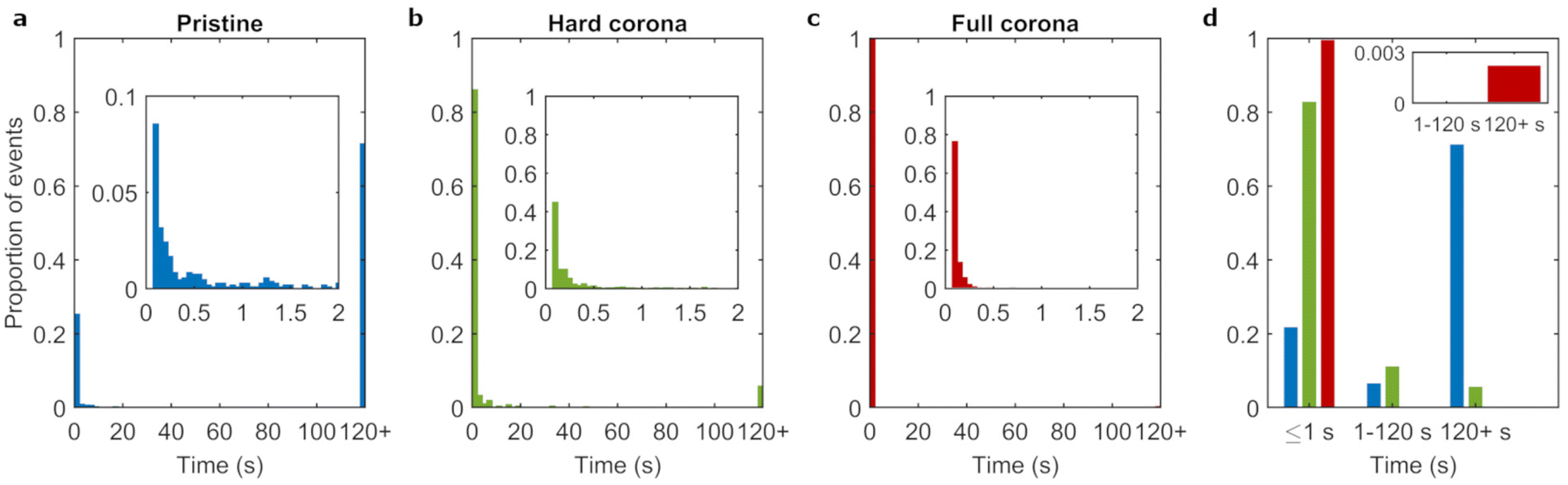 | ||
| Fig. 3 Residence times of the 100 nm particles at the lipid bilayer. Histograms of the pooled residence time distributions from repeat trials of a, pristine particles; b, hard corona covered particles; and c, full corona particles. Particles adsorbed to the lipid bilayer for 120 s or more are represented by the final bars (120+). The majority of the data is in the 120+ bin for pristine particles. Conversely, the largest proportion of the data is contained in the first histogram bars of the plots for the corona conditions. Insets show the sub 2 s residence time distributions with smaller (50 ms-sized) bins. Within this timescale, all three conditions show a skew to the shortest timespan measured. d, Comparative residence times distributions of pristine (blue), hard corona (green) and full corona (red) particles categorized into; transient (≤1 s), intermediate (1–120 s) and long lived events (≥120 s). Events of intermediate and long duration are present for the pristine and hard corona covered particle conditions, but are practically non-existent (see inset) for the full corona particle condition. | ||
In addition to the duration of the events, we also considered the actual number of adsorption events (Table 1 and ESI Fig. S5†) reflecting a combination of transport to the membrane and remaining there long enough to be detected due to interactions with the membrane. To compare quantities such as diffusion and sedimentation rates, we calculated the average adsorption rate. In this context, it is important to note that sedimentation effects are negligible for the light polystyrene nanoparticles we use (Table S3†). We therefore compare the experimentally measured adsorption rate with what is expected due to diffusion, that is, the number of 100 nm diameter particles colliding with a plane in the absence of interactions (Table 1; see ESI Discussion “Particle adsorption rates onto lipid bilayers”† for derivations). The experimentally measured particle adsorption rates were more than 5 orders of magnitude smaller than the expected number of particle–membrane collisions, indicating that particle adsorption onto the bilayer is rare.
| 100 nm | 100 nm | 100 nm | 40 nm | 20 nm | |
|---|---|---|---|---|---|
| Pristine | Hard corona | Full corona | Full corona | Full corona | |
| Measured adsorption rate (μm−2 s−1) | 6.3 × 10−5 | 1.1 × 10−5 | 1.9 × 10−5 | 0.6 × 10−5 | 0.2 × 10−5 |
| Calculated collision rate (μm−2 s−1) | 17.2 | 17.2 | 17.2 | 67.9 | 191.9 |
The biomolecular corona reduces particle adhesion
The experiments for 100 nm pristine particles discussed above were repeated for 100 nm corona-covered particles to assess the degree to which particle–membrane interactions are affected by adsorbed biomolecules. We investigated both hard corona covered (dispersed in buffer) and full corona (dispersed in complete medium) particles. We note that the hard and soft corona (and therefore by extension hard and full corona particles) are anticipated to differ not only in dynamics, but also in composition.47 A clear difference between the pristine condition (ESI Video S1†) and the two corona covered conditions (ESI Videos S2 and 3†) can already be discerned by eye. In the pristine condition, the majority of particles that can be seen to adsorb do so for long periods of time (on the order of seconds and longer), whereas in the corona conditions, many short lived events can be seen. This suggests a large influence of the biomolecular corona, where the presence of biomolecules visibly reduces the time span of particle adsorption to the membrane.We quantified the residence times of corona covered particles in the same manner as discussed above For the corona conditions, the fraction of events lasting 120 s or longer were 6% and 0.2% for hard and full corona particles, respectively (ESI Table S1†). This substantial decrease compared to the pristine condition indicates that the presence of a corona hinders strong particle–membrane binding. Previous experimental and simulation works have likewise shown decreased particle adhesion under biomolecular corona conditions, likely driven by surface energy effects (including from hydrophobicity/philicity of the particle surface).12,21,48,49
Fig. 3b and c show that, for both corona conditions, the most common residence time was the shortest time measured (100 ms) and few particles adsorbed for timespans longer than 1 s. Therefore, the majority of events for the corona conditions were transient (Fig. 3d). Furthermore, corona coverage also reduced the particle adsorption rate (Table 1 and ESI Fig. S5†). This is consistent with a previous study utilizing the same 100 nm polystyrene particles used here, where particles with a corona adhered far less to lipid bilayers compared to pristine particles (an outcome that was also replicated for cell membranes).12 We conclude that the corona acts to drastically reduce the affinity of the particles to the lipid bilayer, both in terms of duration and rate of adsorption events.
Hard corona particles reside longer than full corona particles on bilayers
Given the large difference between pristine and corona covered particles, it is interesting to observe more subtle differences between the two corona conditions. The minor fraction of events lasting more than 120 s increased more than 25 fold for hard corona particles compared to full corona particles. Similarly, the population that adsorbed for the shortest measured residence time (100 ms) constituted the majority under the full corona condition, whereas this value was below 50% in the hard corona condition. Furthermore, Fig. 3d shows that a population of particles with intermediate residence times was present in the hard corona condition (11% of events) but was not observed in the full corona condition (inset Fig. 3d). Statistical comparison of the residence time distributions confirmed that the distribution obtained from the hard corona covered particle data differed significantly from the full corona condition, and likewise both differed significantly from the pristine particle condition (ESI Table S2†). In summary, hard corona covered particles adsorbed for longer time spans compared to full corona conditions.The hard corona and full corona conditions differ in the absence and presence, respectively, of free (not irreversibly particle bound) biomolecules which contribute an additional transient layer of biomolecules to the particle surface (Fig. 1a). However, there is also a second difference, namely that under full corona conditions, biomolecules in the medium may also associate with the lipid bilayer, either by adsorbing or through more transient interactions. Both the presence of a soft corona and/or biomolecular association with the lipid bilayer could lead to the difference observed between hard and full corona conditions. We therefore investigated the effect of (potential) incorporation of biomolecules into the bilayer by incubating the bilayers with complete cell culture medium for 1 h, briefly rinsing with phosphate buffered saline, and finally adding hard corona particles as before. ESI Fig. S6† displays the distribution of residence times after this procedure, showing that the fraction of events lasting longer than 120 s increased from 6% to 20% due to the preincubation of the bilayer with complete medium. That is, the preincubation actually led to an increased difference between the hard corona and full corona conditions. This indicates that strongly bound biomolecules adsorbed onto the bilayer facilitate long term binding of hard corona particles. This implies that the decrease in long-lasting events under full corona conditions is not directly due to incorporation of biomolecules into the bilayer under these conditions. We therefore suggest that either transiently adsorbed biomolecules on the bilayer and/or the soft corona lead to the reduced residence times under full corona conditions compared to hard corona covered particles.
Particle size affects adsorption behaviour
It has been reported that uptake is dependent on particle diameter, with a maximum occurring for particles of ∼50 nm.25,50–53 To determine whether a likewise dependence occurs for residence time, the different sized nanoparticles were investigated under full corona conditions. Fig. 3c and 4a–b show the residence time distributions for the three particle diameters and ESI Table S3† lists the fraction of events lasting longer than 120 s. All particles mainly exhibited transient interactions with few events lasting for 120 s or more. Furthermore, on short timescales the three particle sizes showed a similar shape of the distribution, with a rapid decay of the number of events as a function of residence time. We conclude that the residence time distribution did not notably differ with particle size, though the (small) proportion of long-lasting events may still differ.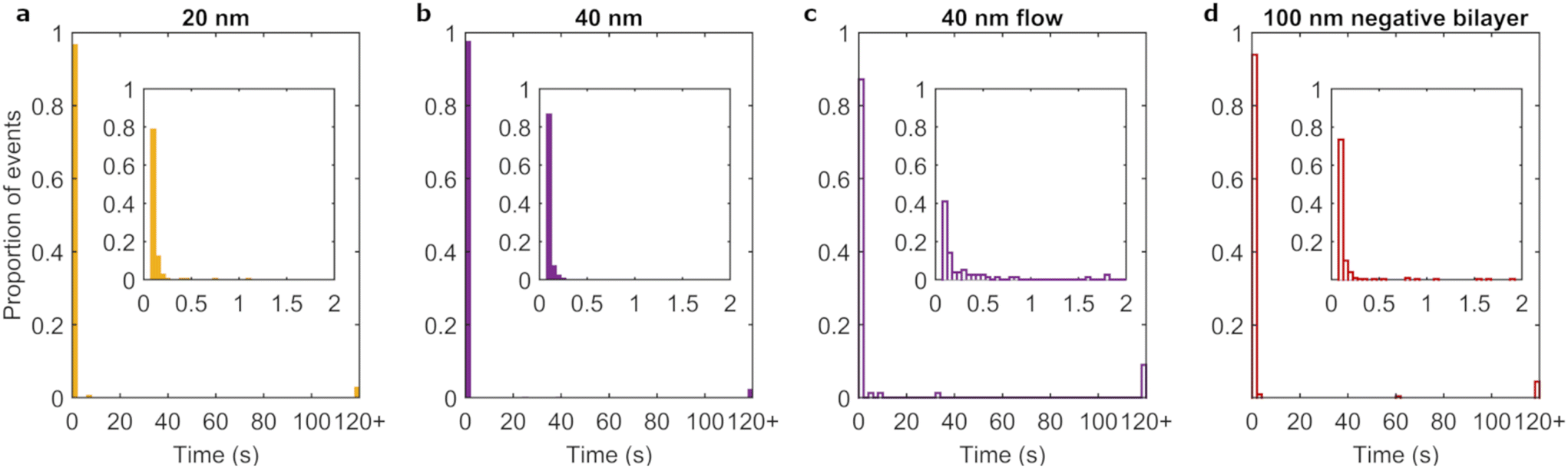 | ||
| Fig. 4 Size, flow and membrane composition effects on particle adsorption behavior. Histograms of the pooled residence time distributions of a, 20 nm full corona particles, b, 40 nm full corona particles, c, 40 nm full corona particles maintained under flow conditions and d, 100 nm full corona particles interacting with a HeLa cell mimicking negatively charged membrane under static conditions. The few particles that remained adsorbed to the lipid bilayer for 120 s or more are represented by the final bars (120+). Insets show the sub 2 s residence time distributions with smaller (50 ms-sized) bins, which accounts for the vast majority of the data regardless of condition. | ||
However, the adsorption rate was particle size dependent, with larger particles exhibiting higher rates (Table 1 and ESI Fig. S5†). In interpreting these results, we note that the number of events we observe is a combination of the nanoparticle first of all arriving to the bilayer and second of all remaining there (for long enough for us to observe it). To arrive to the membrane, sedimentation effects are essentially absent for polystyrene nanoparticles (ESI Table S3†), which have a density close to water. Furthermore, the particle impingement rate (see equation in ESI Discussion “Particle adsorption rates onto lipid bilayers”†) decreases with increasing particle size, and thus shows the opposite trend with particle size compared to what is empirically observed (Table 1). This hints at an effect in which adsorption rate is dependent on how long the particle remains close to the surface, rather than how often. Indeed, the impingement velocity goes down with increasing diameter and, related to that, the diffusion coefficient is smaller for larger particles (while the diffusion coefficient is also modulated close to a surface, the size dependence remains the same54). In addition, there is likely a size dependence of the particle–membrane interaction force. While the detailed functional form of the interaction force cannot be predicted from first principles, especially in the presence of the corona, it does not seem unreasonable that the interaction force is stronger the larger the particle. For example, this is the case for the van der Waals force of a particle near a plane.55,56 Ostensibly, the observation that the interaction is stronger for larger particles may seem inconsistent with the residence time largely being independent of size. However, both adsorption and desorption depend on the detailed form of the interaction force, certainly including the depth of the potential energy well, but also on the range of the interaction and eventual potential barriers.56 In addition, the lipid membrane may rearrange upon adsorption and thereby change the interaction at close range. Therefore the size dependence of adsorption rate and residence time may be decoupled. Overall, we thus interpret the size dependence of the adsorption rate to reflect a reduced diffusion close to the surface and/or a stronger interaction for the larger particles.
Flow conditions reduce the number of adsorbing particles
In nanomedicine approaches, particles are often exposed to membranes under flow conditions, e.g., in the blood stream. In order to mimic such scenarios, albeit to a limited extent, flow was introduced into the experimental design. We chose to match the shear stress (as opposed to flow velocity) at the bilayer surface to that at the endothelial cell layer within blood capillaries.57 Therefore, particles were aspirated through micro channels to achieve a shear stress of ∼0.5 N m−2. We investigated the flow effects on the adsorption of full corona particles as it most closely resembles the situation of particles in blood.The addition of flow reduced the average number of adsorption events 35-fold and 9-fold for 100 nm and 40 nm full corona particles, respectively. As a consequence, the number of events for the 100 nm full corona condition was insufficient to form a residence time distribution. However, we could calculate an average residence time from the pooled data set of observed adsorption events. The average residence time of 100 nm full corona particles was 1.05 ± 0.05 s in the presence of flow, whereas the same particles under static conditions had an average residence time of 0.12 ± 0.002 s. The residence time distribution of the 40 nm full corona particles under flow (Fig. 4c) was also different from the static condition (Fig. 4b). While in both cases most particles were adsorbed for less than 2 s, the distribution in the inset of Fig. 4c shows that the majority of events under flow did not fall within the first bin (100 ms), which was the case for the static scenario (inset Fig. 4b). This resulted in an increase in the average residence time from 0.17 ± 0.05 s under static conditions to 0.93 ± 0.5 s under flow. Therefore, for both particle sizes, the introduction of flow increased the average duration of adsorption events by up to an order of magnitude.
As already stated, sedimentation is negligible for these particles. Therefore, the reduced number of events is not due to the prevention of sedimentation caused by flow, as had been found in a previous study using (heavier) gold particles.58 Rather we interpret the difference as the introduction of flow being sufficient to overcome transient particle–membrane interactions. As transient interactions dominate in the full corona conditions, this could plausibly lead to the large reduction in the number of events observed. Likewise, the increase in average residence time for both particle sizes under flow conditions is consistent with a reduction of, in particular, the very short-lived adsorption events. That is, weakly interacting particles are typically washed away so we expect that the particles we do see adsorb are the rarer long adsorption events.
Membrane composition affects the number of adsorbing particles
Finally, we investigated the influence of membrane properties on the adsorption of 100 nm full corona particles under static conditions. We compared the neutral bilayers used throughout this work to HeLa cell mimicking bilayers34 which contain a small proportion (6%) of negatively charged lipid (brain PS). The majority of adsorption events were transient when this negatively charged bilayer were used (Fig. 4d), comparable to the earlier neutral bilayer (Fig. 3c). However, there were some subtle differences in the rest of the distribution, namely the presence of a few singular events with intermediate adsorption times and a small increase in the proportion of long term adsorption events.There was a more marked difference between the two membrane systems when comparing the number of adsorption events. The measured adsorption rate was approximately halved for the negatively charged membrane (8.6 × 10−6 μm−2 s−1) compared to the neutral membrane (Table 1). We exclude the possibility of decreased transport to the bilayer as the particles and environment are the same in both conditions. There is a difference in terms of the electrostatics, but electrostatics is strongly screened in physiological media59 and are therefore unlikely to result in decreased transport to the membrane. However, once the particles are in close vicinity to the bilayer (<1 nm),59 electrostatic interactions are influential. Therefore, similar to the size effects, we interpret the different adsorption rates to manifest from differences in how long particles remain once in the vicinity of the bilayer. In this case, we attribute the decreased duration to electrostatic interactions between the weakly negatively charged membrane and exposed negative domains of the biomolecular corona molecules.
Conclusions
In summary, the presence of a corona on the nanoparticle surface reduces both membrane adsorption rate and duration. This is consistent with the idea that surface energy effects drive both corona formation and membrane interactions.12,21,48 In the absence of a corona, the high surface energy of the bare nanoparticle surface makes the particle adhere strongly to the membrane. In the presence of a corona, on the other hand, the nanoparticle surface has already been passivated due to corona formation and subsequent particle–membrane interactions are weaker. Naturally, other interactions could modulate these effects, but they appear to be too weak to change the overall outcome. Aside from the large difference between the presence and absence of a corona, we also observed fewer long-lived adsorption events under full corona conditions compared to hard corona covered particles. Particle size, on the other hand, did not seem to affect the duration of binding, but it did alter the adsorption rate. We attribute this to either reduced dynamics at the surface and/or particle–membrane interactions. Under conditions mimicking the shear stress in blood capillaries due to flow, the number of events reduced substantially, but the average residence time increased. We interpret this as an inhibition of transient particle interactions under flow. Finally, a negatively charged HeLa cell membrane mimicking bilayer showed a decreased number of adsorption events compared to the neutral membrane. We attribute this to the presence of electrostatic interactions between the bilayer and molecules in the biomolecular corona once the particle complex is in the vicinity of the membrane (<1 nm).Non-specific lipid bilayer–particle interactions have been characterized as a first step towards understanding the more complex cellular membrane–particle interactions. The outcomes reported here are relevant for the interpretation of more biomimetic systems where both non-specific and specific interactions are present. As an example, linking to cellular uptake, it has been observed that a maximum occurs for nanoparticles of a size around 50 nm.25,50–53 Our results indicate that, in terms of non-specific interactions, the total number of adsorption events increases with particle size. Thus, the observation of a cell uptake maximum would have to be interpreted with this in mind. However, it remains to be determined to what degree the non-specific interactions investigated here are relevant when cell receptors are present. In addition to the introduction of specific interactions, it is also possible that the non-specific interactions are modulated by the presence of membrane proteins. Therefore, as a next step in understanding size dependent adhesion, we envision future experiments using membrane models containing specific receptors of interest, building towards the use of reconstituted cell membranes.7,10,60,61 We also note that for cell uptake, where full corona conditions are relevant, it is crucial for the nanoparticle to remain at the cell membrane long enough for an internalization mechanism to be concluded. Nanoparticles have been reported to be internalized by various mechanisms, ranging from clathrin-mediated endocytosis to phagocytosis with typical time scales ranging from 40 s to several minutes.39–41 Thus, the majority of the events we observe, as well as those too short for us to observe, are likely far too short to be relevant for cell uptake, putting the rare long-lived events in the spotlight. In this context it is interesting to note that we could not discern any particle size dependence in the proportion of such long-lived events.
Overall, our results underline the importance of characterizing the (bio)physical interactions of nano-sized objects with membranes if one wants to understand and optimize uptake. Together with a detailed characterization of the particle itself,62 this is likely to form a basis for a more biophysical approach to nanomedicine design.
Author contributions
CJR performed particle and lipid bilayer preparations, AFM, NTA and TIRF microscopy experimental work and analysis under the supervision of CÅ and WHR. MA performed STEM under the supervision of BJK. MCAS performed the cryo EM. All authors contributed to interpretation of data. The manuscript was written through contributions from CJR, MA, MCAS, CÅ and WHR.Conflicts of interest
There are no conflicts to declare.Acknowledgements
CJR was supported by a scholarship awarded under the Molecular Life and Health programme of the Faculty of Science and Engineering, Rijksuniversiteit Groningen. We would like to thank the Department of Pharmaceutical Technology and Biopharmacy of the Groningen Research Institute of Pharmacy for providing access to the NanoSight LM14 and NTA 3.0 software. We thank R. B. Lira of the Zernike Institute for Advanced Materials for suggestions on bilayer formation.References
- K. A. Dawson and Y. Yan, Nat. Nanotechnol., 2021, 1–14 Search PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- V. Francia, D. Montizaan and A. Salvati, Beilstein J. Nanotechnol., 2020, 11, 338–353 CrossRef CAS.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS.
- L. A. Lane, X. Qian, A. M. Smith and S. Nie, Annu. Rev. Phys. Chem., 2015, 66, 521–547 CrossRef CAS PubMed.
- P. Decuzzi and M. Ferrari, Biomaterials, 2007, 28, 2915–2922 CrossRef CAS.
- Y. Yu, Y. Gao and Y. Yu, ACS Nano, 2018, 12, 11871–11880 CrossRef CAS PubMed.
- G. Caracciolo, F. Cardarelli, D. Pozzi, F. Salomone, G. Maccari, G. Bardi, A. L. Capriotti, C. Cavaliere, M. Papi and A. Laganà, ACS Appl. Mater. Interfaces, 2013, 5, 13171–13179 CrossRef CAS.
- L. Digiacomo, F. Cardarelli, D. Pozzi, S. Palchetti, M. A. Digman, E. Gratton, A. L. Capriotti, M. Mahmoudi and G. Caracciolo, Nanoscale, 2017, 9, 17254–17262 RSC.
- L. Wang, N. Hartel, K. Ren, N. Alexander Graham and N. Malmstadt, Environ. Sci.: Nano, 2020, 7, 963–974 RSC.
- C. Montis, V. Generini, G. Boccalini, P. Bergese, D. Bani and D. Berti, J. Colloid Interface Sci., 2018, 516, 284–294 CrossRef CAS PubMed.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Åberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS.
- K. L. Chen and G. D. Bothun, Environ. Sci. Technol., 2014, 48, 873–880 CrossRef CAS.
- E. Rascol, J.-M. Devoisselle and J. Chopineau, Nanoscale, 2016, 8, 4780–4798 RSC.
- G. van der Borg, S. Braddock, J. S. Blijleven, A. M. van Oijen and W. H. Roos, J. Phys.: Condens. Matter, 2018, 30, 204005 CrossRef PubMed.
- X. Zhang and S. Yang, Langmuir, 2011, 27, 2528–2535 CrossRef CAS.
- Y. Roiter, M. Ornatska, A. R. Rammohan, J. Balakrishnan, D. R. Heine and S. Minko, Nano Lett., 2008, 8, 941–944 CrossRef CAS PubMed.
- C. Wilhelm, F. Gazeau, J. Roger, J. N. Pons and J.-C. Bacri, Langmuir, 2002, 18, 8148–8155 CrossRef CAS.
- A. L. Doiron, B. Clark and K. D. Rinker, Biotechnol. Bioeng., 2011, 108, 2988–2998 CrossRef CAS PubMed.
- C. A. Lochbaum, A. K. Chew, X. Zhang, V. Rotello, R. C. Van Lehn and J. A. Pedersen, ACS Nano, 2021, 15, 6562–6572 CrossRef CAS.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5857 CrossRef CAS PubMed.
- S. Ashraf, A. Hassan Said, R. Hartmann, M.-A. Assmann, N. Feliu, P. Lenz and W. J. Parak, Angew. Chem., Int. Ed., 2020, 59, 5438–5453 CrossRef CAS.
- P. Patel, K. P. Santo, S. Burgess, A. Vishnyakov and A. V. Neimark, ACS Nano, 2020, 14, 17273–17284 CrossRef CAS PubMed.
- L. Caselli, A. Ridolfi, G. Mangiapia, P. Maltoni, J.-F. Moulin, D. Berti, N.-J. Steinke, E. Gustafsson, T. Nylander and C. Montis, Phys. Chem. Chem. Phys., 2022, 24, 2762–2776 RSC.
- B. D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef CAS PubMed.
- S. M. Häffner, E. Parra-Ortiz, K. L. Browning, E. Jørgensen, M. W. A. Skoda, C. Montis, X. Li, D. Berti, D. Zhao and M. Malmsten, ACS Nano, 2021, 15, 6787–6800 CrossRef.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS.
- C. Åberg, Nanoscale Adv., 2021, 3, 2196–2212 RSC.
- N. Ruthardt, D. C. Lamb and C. Bräuchle, Mol. Ther., 2011, 19, 1199–1211 CrossRef CAS.
- C.-L. Hsieh, S. Spindler, J. Ehrig and V. Sandoghdar, J. Phys. Chem. B, 2014, 118, 1545–1554 CrossRef CAS PubMed.
- R. Bausinger, K. von Gersdorff, K. Braeckmans, M. Ogris, E. Wagner, C. Bräuchle and A. Zumbusch, Angew. Chem., 2006, 118, 1598–1602 CrossRef.
- S. Streck, S. S.-R. Bohr, D. Birch, T. Rades, N. S. Hatzakis, A. McDowell and H. Mørck Nielsen, ACS Appl. Bio Mater., 2021, 4, 3155–3165 CrossRef CAS.
- D. Vorselen, S. M. van Dommelen, R. Sorkin, M. C. Piontek, J. Schiller, S. T. Döpp, S. A. A. Kooijmans, B. A. van Oirschot, B. A. Versluijs, M. B. Bierings, R. van Wijk, R. M. Schiffelers, G. J. L. Wuite and W. H. Roos, Nat. Commun., 2018, 9, 4960 CrossRef.
- A. Botet-Carreras, M. T. Montero, J. Sot, Ò. Domènech and J. H. Borrell, Colloids Surf., A, 2021, 630, 127663 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J.-Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS.
- J.-Y. Tinevez, N. Perry, J. Schindelin, G. M. Hoopes, G. D. Reynolds, E. Laplantine, S. Y. Bednarek, S. L. Shorte and K. W. Eliceiri, Methods, 2017, 115, 80–90 CrossRef CAS.
- T.-G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- M. Ehrlich, W. Boll, A. van Oijen, R. Hariharan, K. Chandran, M. L. Nibert and T. Kirchhausen, Cell, 2004, 118, 591–605 CrossRef CAS.
- T. Kirchhausen, Trends Cell Biol., 2009, 19, 596–605 CrossRef CAS PubMed.
- D. Paul, S. Achouri, Y.-Z. Yoon, J. Herre, C. E. Bryant and P. Cicuta, Biophys. J., 2013, 105, 1143–1150 CrossRef CAS.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- C. Röcker, M. Pötzl, F. Zhang, W. J. Parak and G. U. Nienhaus, Nat. Nanotechnol., 2009, 4, 577–580 CrossRef.
- S. Milani, F. Baldelli Bombelli, A. S. Pitek, K. A. Dawson and J. Rädler, ACS Nano, 2012, 6, 2532–2541 CrossRef CAS.
- D. Walczyk, F. B. Bombelli, M. P. Monopoli, I. Lynch and K. A. Dawson, J. Am. Chem. Soc., 2010, 132, 5761–5768 CrossRef CAS.
- G. van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS PubMed.
- C. Weber, J. Simon, V. Mailänder, S. Morsbach and K. Landfester, Acta Biomater., 2018, 76, 217–224 CrossRef CAS.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS.
- H. Ding and Y. Ma, Biomaterials, 2014, 35, 8703–8710 CrossRef CAS PubMed.
- W. Jiang, B. Y. S. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- J. A. Varela, M. G. Bexiga, C. Åberg, J. C. Simpson and K. A. Dawson, J. Nanobiotechnol., 2012, 10, 39 CrossRef CAS PubMed.
- S. Zhang, J. Li, G. Lykotrafitis, G. Bao and S. Suresh, Adv. Mater., 2009, 21, 419–424 CrossRef CAS PubMed.
- F. Osaki, T. Kanamori, S. Sando, T. Sera and Y. Aoyama, J. Am. Chem. Soc., 2004, 126, 6520–6521 CrossRef CAS PubMed.
- H. Brenner, Chem. Eng. Sci., 1961, 16, 242–251 CrossRef CAS.
- J. N. Israelachvili, Intermolecular and surface forces, Elsevier, Amsterdam, 3rd edn, 2011 Search PubMed.
- D. Fennell Evans and H. Wennerström, in The Colloidal Domain: Where Physics, Chemistry, Biology, and Technology Meet; Advances in Interfacial Engineering, Wiley-VCH, New York, 1999, ch. 5, pp. 217–294 Search PubMed.
- A. G. Koutsiaris, S. V. Tachmitzi, N. Batis, M. G. Kotoula, C. H. Karabatsas, E. Tsironi and D. Z. Chatzoulis, Biorheology, 2007, 44, 375–386 Search PubMed.
- E. C. Cho, Q. Zhang and Y. Xia, Nat. Nanotechnol., 2011, 6, 385–391 CrossRef CAS PubMed.
- C.-H. Chu, I. Sarangadharan, A. Regmi, Y.-W. Chen, C.-P. Hsu, W.-H. Chang, G.-Y. Lee, J.-I. Chyi, C.-C. Chen, S.-C. Shiesh, G.-B. Lee and Y.-L. Wang, Sci. Rep., 2017, 7, 5256 CrossRef PubMed.
- U. Martens, U. Janke, S. Möller, D. Talbot, A. Abou-Hassan and M. Delcea, Nanoscale, 2020, 12, 19918–19930 RSC.
- Z. Li and A. A. Gorfe, Nanoscale, 2014, 7, 814–824 RSC.
- P. Delcanale, B. Miret-Ontiveros, M. Arista-Romero, S. Pujals and L. Albertazzi, ACS Nano, 2018, 12, 7629–7637 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05296b |
| This journal is © The Royal Society of Chemistry 2023 |