
Enhanced chemodynamic and photoluminescence efficiencies of Fe–O4 coordinated carbon dots via the core–shell synergistic effect†
Xiaoyan
Wu
a,
Feng
Yu
b,
Yifei
Han
 c,
Lei
Jiang
b,
Zijian
Li
*a,
Junfa
Zhu
d,
Qian
Xu
*d,
Antonio Claudio
Tedesco
be,
Jiangwei
Zhang
f and
Hong
Bi
*a
c,
Lei
Jiang
b,
Zijian
Li
*a,
Junfa
Zhu
d,
Qian
Xu
*d,
Antonio Claudio
Tedesco
be,
Jiangwei
Zhang
f and
Hong
Bi
*a
aSchool of Materials Science and Engineering, Anhui University, Hefei 230601, China. E-mail: bihong@ahu.edu.cn; 22018@ahu.edu.cn
bSchool of Chemistry and Chemical Engineering, Anhui University, Hefei 230601, China
cCAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Anhui 230026, China
dNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230029, China. E-mail: qianxu@ustc.edu.cn
eDepartment of Chemistry, Center of Nanotechnology and Tissue Engineering-Photobiology and Photomedicine Re-search Group, Faculty of Philosophy, Sciences and Letters of Ribeirão Preto, University of São Paulo, Ri-beirão Preto, São Paulo 14040-901, Brazil
fCollege of Chemistry and Chemical Engineering, Inner Mongolia University, Hohhot 010021, China
First published on 24th November 2022
Abstract
In natural systems like photosynthetic organisms and photo-active enzymes, the spatial organization of chromophores is critical for efficient light harvesting and bio-catalysis. Inspired by nature, a novel modular nanoplatform with both biological imaging and therapeutic functions is constructed by taking advantage of the intrinsic core–shell structure of Fe-decorated carbon dots. Light-harvesting chromophores with deep-red photoluminescence are densely packed into the carbon core. Simultaneously, the atomically dispersed Fe3+ catalytic sites accounting for efficient conversion of H2O2 to ˙OH are discretely distributed on the shell. Precise control over their spatial distribution leads to the elegant integration and exciting interplay of the functional moieties. On the one hand, incorporating a catalysis shell enhances the emission of chromophores via synergistic shielding and rigidifying effects. On the other hand, visible light excitation of the chromophores significantly increases the catalytic activity and cytotoxicity against cancer cells, ascribed to the promotion of the charge transfer process. This nanoplatform exhibits excellent biocompatibility, bright red fluorescence, and light-regulated cytotoxicity for anti-cancer treatment, promising its applications in smart nanocatalytic medicines and efficient chemodynamic therapy.
Introduction
Chemodynamic therapy (CDT) is emerging as a highly effective therapeutic modality for cancer treatment due to its high logicality and selectivity derived from internal triggers.1–3 Particularly, the catalytic effect observed in CDT can convert hydrogen peroxide (H2O2), an over-expressed species in the tumor microenvironment, into harmful reactive oxygen species (ROS) such as hydroxyl radicals (˙OH), thereby inducing tumor cell death by apoptosis and necrosis pathways. Along with the fast evolution of nanotechnology, many nanocatalysts, also termed nanocatalytic medicine, have been applied to initiate catalytic reactions for generating therapeutic effects.4–7 Nevertheless, most current nanocatalytic medicines are inorganic metal oxides (e.g., Fe3O4, TiO2, CeO2).4,8,9 They decrease the utilization efficiency of metal atoms and may suffer from the issue of low biodegradability. Moreover, the mechanisms of nanocatalysts in therapy are still challenging to clarify,8 which requires the integration of diagnostic and therapeutic functions into a single nanoparticle formulation.Carbon dots (CDs) are core–shell structured nanoparticles10,11 that have recently attracted considerable attention for biomedical applications due to their excellent biocompatibility, high stability, and good water solubility.12–14 More importantly, the unique and tunable photophysical and photochemical properties endow CDs with great application potential in diagnostic imaging protocols, photodynamic therapy (PDT), and CDT.15–19 Besides, metal (e.g., Fe, Cu) doping could endow CDs with catalytic activity and thus enable their use as nanocatalytic medicines.20,21 Among the various metal ions, iron doping represents one of the most explored and efficient strategies for enhancing the CDT effect due to the facilitation of in situ ˙OH generation by an iron mediated Fenton reaction and the Haber–Weiss reaction.22–24 Unfortunately, the incorporation of Fe inevitably quenches the photoluminescence (PL) of CDs, hampering the construction of multifunctional nanomaterials. In addition, the catalytic activity of Fe-doped CDs is still insufficient and needs further improvement to optimize the therapeutic outcomes.
Photochemical regulation of the catalytic behavior is highly desirable owing to the high spatiotemporal resolution of light.25–28 Recently, several groups have reported the light-enhanced or regulated catalytic efficiency of nanocatalytic medicines.29–32 However, their photoactivation mechanisms are attributed to their simple photothermal and/or photocatalysis effects. The situation is different from natural photo-active enzymes,33,34 in which light-harvesting chromophores and catalytic centers are discretely distributed while spatially organized, facilitating the harnessing of light energy to power the catalytic process by either an electron or charge transfer pathway. Learning from the sophisticated structure of enzymes from nature to construct artificial nanomaterials is challenging yet very interesting for both fundamental and practical research.
Herein, a novel modular nanoplatform with both imaging and catalytic functions was constructed by taking advantage of the intrinsic core–shell structure of EDTA-CDs (Scheme 1). Specifically, the light-harvesting chromophores are densely packed into the carbon core, responsible for visible light absorption and bright red fluorescence. On the other hand, the surface was decorated with abundant carboxyl moieties, which serve as anchor sites for the deposition of atomically dispersed Fe3+ (Scheme 1a). The incorporation of Fe3+ not only endows Fe@EDTA-CDs with the catalytic ability of converting H2O2 to ˙OH, but also, to our gratifying surprise, increases the emission efficiency of chromophores via synergistic shielding and rigidifying effects (Scheme 1b). More interestingly, visible light excitation of the chromophores in the carbon core enhances the catalytic activity of the Fe coordination sites on the surface, ascribed to the light-promoted charge transfer process (Scheme 1b). Fe@EDTA-CDs not only feature excellent biocompatibility and bright red fluorescence, but also possess a high ROS generation rate and light-regulated anticancer efficiency, which promise their application in smart nanocatalytic medicines.
 | ||
| Scheme 1 Schematic diagram of the construction of an Fe@EDTA-CD nanoplatform and the core–shell interaction. Schematic representation for (a) the synthesis process of Fe@EDTA-CDs and (b) the construction of an Fe@EDTA-CD nanoplatform with bioimaging and biomedicine functions by the core–shell synergistic effect. The balance and harmony between “Yin” and “Yang” in Daoism is an analogy of the interplay and synergistic effect of the carbon core and Fe–O4 coordinated shell of Fe@EDTA-CDs here. | ||
Results and discussion
EDTA-CDs were synthesized by a one-step hydrothermal method using o-phenylenediamine (o-PD) and ethylene diamine tetra-acetic acid (EDTA) as precursors (Scheme 1a and Fig. 1a). In our modular design, o-PD was employed to produce the structural skeleton and chromophores of CDs, and the EDTA was used to introduce carboxyl groups onto the surface of CDs to provide coordinate sites for Fe3+ (Fig. 1a). Characterization of the as-prepared EDTA-CDs by transmission electron microscopy (TEM) shows monodisperse and spherical particles with an average size of 6.80 nm (Fig. 1b). High-resolution TEM images reveal that EDTA-CDs have a lattice spacing of 0.24 nm (Fig. 1c and S1a†), consistent with the (100) plane of graphite carbon.35 According to the X-ray powder diffraction (XRD) profiles (Fig. 1d), a broad (002) peak centered at 22° was detected. Notably, both the TEM and XRD results indicate the formation of a graphite carbon core, which is typical for phenylenediamine-derived CDs (Fig. 1a). Besides, the Raman spectrum shows two peaks at 1409 (crystalline D band) and 1579 cm−1 (disordered G band), with a D to G intensity ratio (ID/IG) of 1.77 (Fig. 1e). This demonstrates the coexistence of graphitic and amorphous carbons in EDTA-CDs.36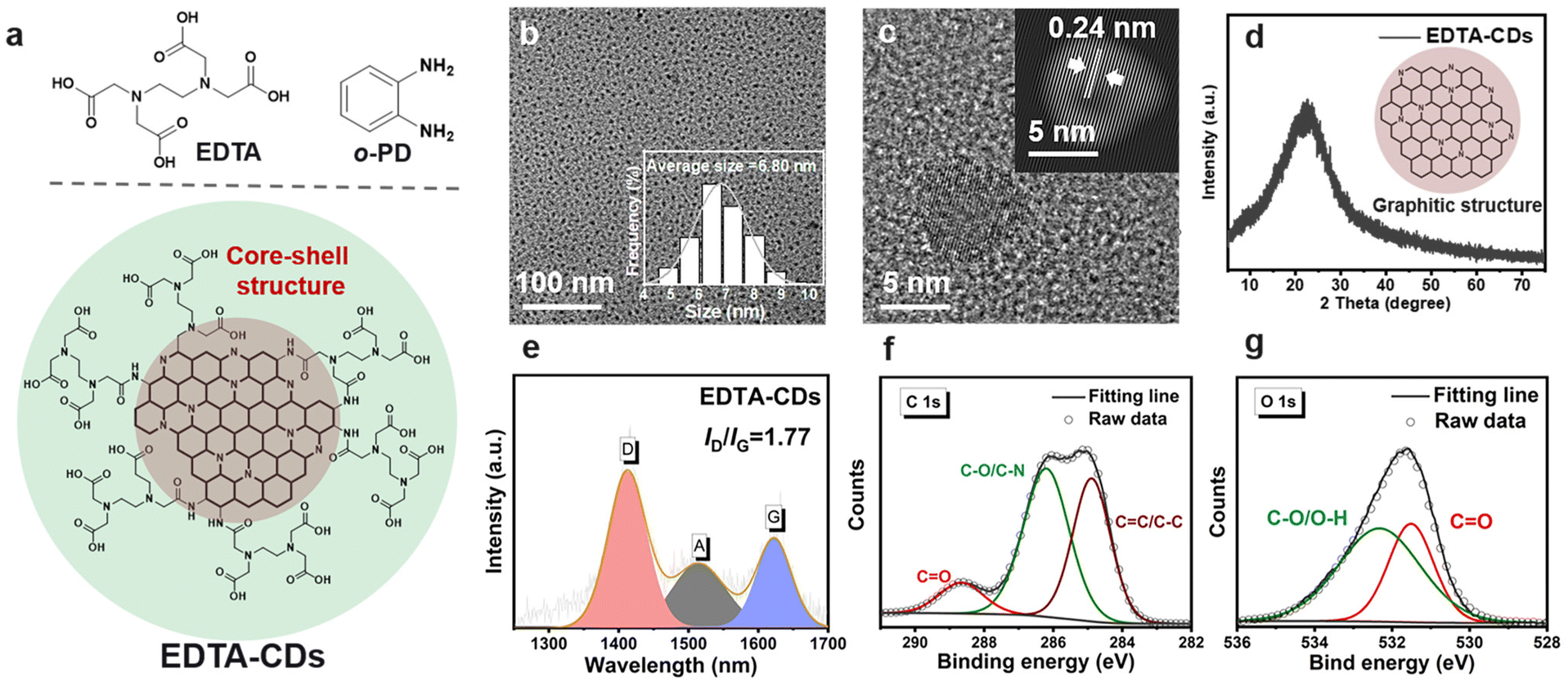 | ||
| Fig. 1 Structural characterization of EDTA-CDs. (a) Chemical structures of the precursors and graphical representation for the core–shell structure of EDTA-CDs. (b) TEM image and the inset of (b) shows the size distribution of EDTA-CDs. (c) High-resolution TEM image of EDTA-CDs (the inset shows the corresponding specially filtered image). (d) XRD pattern, (e) Raman spectrum (λex = 532 nm), and (f) high-resolution C 1s and (g) high-resolution O 1s XPS spectra of EDTA-CDs. Here filtration means a sequence of 2D-operators (FFT, masking, and inverse FFT) applied to the high-resolution TEM image. | ||
The compositions of EDTA-CDs were then analyzed by X-ray photoelectron spectroscopy (XPS). Screening the full spectra identifies three peaks at 285, 400, and 532 eV, which are attributed to C 1s (65.54%), N 1s (13.73%), and O 1s (19.60%), respectively (Fig. S2a†). The C 1s band can be deconvoluted into three components corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C (284.85 eV), C–O/C–N (286.20 eV), and CO (288.65 eV) (Fig. 1f), while pyridinic N (398.95 eV), pyrrolic N (400.05 eV), and graphitic N (400.85 eV) species are present in the N 1s band (Fig. S2b†). These phenomena suggest that the carbon core of CDs is mainly composed of N-doped polyaromatic structures, which originate from the condensation of o-PD precursors.37,38 Different from o-PD, which is primarily responsible for forming the carbon core, another precursor EDTA contributes more to the surface state of CDs. Specifically, C–O/O–H and CO species were found in the O 1s (532.32 and 531.53 eV) XPS spectra, suggesting the abundance of carboxyl groups on the surface of CDs (Fig. 1g).
C/C–C (284.85 eV), C–O/C–N (286.20 eV), and CO (288.65 eV) (Fig. 1f), while pyridinic N (398.95 eV), pyrrolic N (400.05 eV), and graphitic N (400.85 eV) species are present in the N 1s band (Fig. S2b†). These phenomena suggest that the carbon core of CDs is mainly composed of N-doped polyaromatic structures, which originate from the condensation of o-PD precursors.37,38 Different from o-PD, which is primarily responsible for forming the carbon core, another precursor EDTA contributes more to the surface state of CDs. Specifically, C–O/O–H and CO species were found in the O 1s (532.32 and 531.53 eV) XPS spectra, suggesting the abundance of carboxyl groups on the surface of CDs (Fig. 1g).
This is further evidenced by the Fourier transform infrared (FT-IR) spectra (Fig. S3†), where the stretching vibration bands of CO and O–H were observed at 1628 and 3400 cm−1, respectively. According to these results, we speculated that EDTA-CDs have a core–shell structure, where the carbon core is mainly composed of o-PD derived π-conjugated domains and the shell is decorated with abundant carboxyl moieties left by EDTA (Fig. 1a).
Due to the interplay of chromophores in the carbon core and functional groups in the shell (Scheme 1), EDTA-CDs exhibit extensive absorption from UV to the red-light region in water (Fig. 2a). The UV absorption band centered at 270 nm has a high extinction coefficient and could be assigned to the n → π* transition of CO moieties. In addition, an absorption tail with a relatively weak intensity could be found between 400 and 700 nm, which almost covers the whole visible spectrum (Fig. 2a, inset). Visible light excitation of EDTA-CDs shows strong red fluorescence (Fig. 2b, inset). The peak maximum (λmax = 642 nm) and band shape are independent of the excitation wavelength (λex = 505–590 nm), indicative of its origin from a single emission center (Fig. 2b). It is also noted that the visible absorption and red emission band are vibrational structured.39 It reveals progressional spacings of 1295 and 1331 cm−1, in accordance with the skeletal vibrational modes of aromatic rings (Fig. 2b). This suggests that they originate from the band-gap transitions of polyaromatic structures in the carbon core.40 Additional evidence was obtained from the PL excitation spectrum, which agrees well with the absorption profile in the visible range (Fig. 2b). Meanwhile, minor contributions come from UV absorption, demonstrating its quite different origin. Considering the core–shell structure of EDTA-CDs and the carbon core responsible for its visible-light response, the absorption in the UV region should be originated from the carboxyl decorated shell.
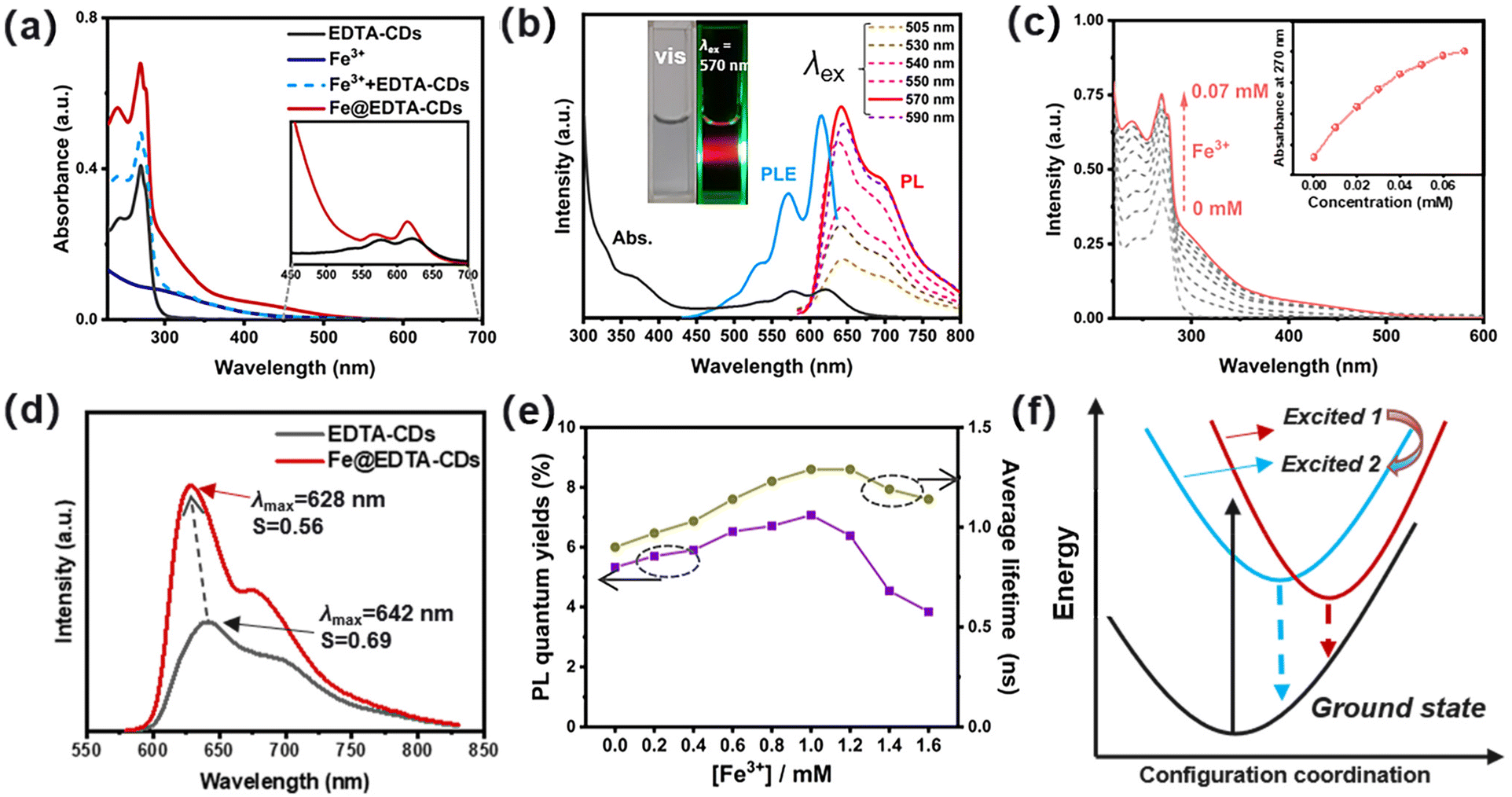 | ||
| Fig. 2 Optical properties of EDTA-CDs and Fe@EDTA-CDs. (a) UV-vis spectra of EDTA-CDs, Fe3+, Fe3+ + EDTA-CDs and Fe@EDTA-CDs. (b) Absorption, photoluminescence (PL) and photoluminescence excitation (PLE) spectra of EDTA-CDs. Inset: images of an aqueous solution of EDTA-CDs under ambient light (left) and 570 nm laser irradiation (right). (c) UV-vis spectra and the inset of (c) shows the intensity changes of absorbance at 270 nm of Fe@EDTA-CDs (20 μg mL−1 in water) with different concentrations of Fe3+ (from 0 to 0.07 mM). (d) PL spectra (λex = 570 nm) of EDTA-CDs and Fe@EDTA-CDs in an aqueous medium. (e) Absolute fluorescence quantum yields and the average lifetime of Fe@EDTA-CDs in an aqueous medium with different concentrations of Fe3+. (f) Graphical representation for the rigidifying effect from EDTA-CDs to Fe@EDTA-CDs. | ||
The carboxyl groups on the surface of EDTA-CDs may serve as potential sites for complexation with metal ions such as Fe3+ (Scheme 1). As shown in Fig. 2a and c, the addition of FeCl3 significantly strengthens the absorption of EDTA-CDs (20 μg mL−1 in water) in the UV region (λ = 240–380 nm). In contrast, the visible absorption is much less affected (Fig. 2a). This demonstrates that it is the carboxyl groups on the shell, not the carbon core, that complex with Fe3+. Furthermore, monitoring the absorption intensity at 270 nm gives a titration curve, where the slope value decreases as the concentration of Fe3+ increases (Fig. 2c, inset). This is a typical characteristic of the saturation curve, suggesting the spectroscopic changes originating from the metal–ligand coordination.
Emission change upon Fe3+/EDTA-CD complexation was then investigated. Fe3+ is known as an emission quencher due to its weak ligand field splitting.41 However, with the addition of Fe3+ (1.0 mM) to EDTA-CDs (1 mg mL−1), no obvious emission quenching or color change occurs for the mixed aqueous solution (Fig. 2d and S4a, b†). It is also worth noting that both EDTA-CDs and Fe@EDTA-CDs exhibited good dispersity and high stability in physiological media, including water, phosphate-buffered solution (PBS), and Dulbecco's modified Eagle's medium (DMEM) (Fig. S4c and S4d†) The emission quantum yield (Φem) of Fe@EDTA-CDs was determined to be 7.07% (λex = 570 nm), which, to our surprise, surpasses that of EDTA-CDs (Φem = 5.33%) (Table 1 and Fig. 2e). Simultaneously, the excited-state lifetime (τ) is also prolonged (τ0: 0.90 and 1.29 ns for EDTA-CDs and Fe@EDTA-CDs, respectively) (Table 1; Fig. 2e and S5†). Radiative (kr) and non-radiative (knr) decay rates were obtained (Table 1). The kr value of EDTA-CDs (5.92 × 107 s−1) is comparable to that of Fe@EDTA-CDs (5.48 × 107 s−1). It suggests negligible participation of Fe3+ in the electronic excited states. On the other hand, the knr value decreases by 31.4% from EDTA-CDs (1.05 × 109 s−1) to Fe@EDTA-CDs (0.72 × 109 s−1). Evidently, the reduced knr value is primarily responsible for the emission enhancement of Fe@EDTA-CDs.
| [Fe3+] (mM) | λ max (nm) |
Φ
em![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (%) a (%) |
τ
0b (ns) |
k
rc (107 s−1) |
k
nrc (109 s−1) |
|---|---|---|---|---|---|
| a Absolute fluorescence quantum yield. b Average fluorescent lifetime. c Obtained from kr = Φem/τ0 and knr = 1/τ0 − kr. | |||||
| 0 | 642 | 5.33 | 0.90 | 5.92 | 1.05 |
| 0.2 | 638 | 5.70 | 0.97 | 5.88 | 0.97 |
| 0.4 | 635 | 5.90 | 1.03 | 5.73 | 0.91 |
| 0.6 | 632 | 6.52 | 1.14 | 5.72 | 0.82 |
| 0.8 | 630 | 6.71 | 1.23 | 5.46 | 0.76 |
| 1.0 | 628 | 7.07 | 1.29 | 5.48 | 0.72 |
| 1.2 | 629 | 6.38 | 1.29 | 4.95 | 0.73 |
| 1.4 | 630 | 4.54 | 1.19 | 3.82 | 0.80 |
| 1.6 | 632 | 3.84 | 1.14 | 3.37 | 0.84 |
Taken together, the mechanism of PL enhancement was further elucidated. The pH perturbation caused by the addition of Fe3+ was first excluded (Fig. S6†), since the fluorescence of EDTA-CDs is stable in such pH ranges. We presume that both the shielding and rigidifying effects contribute to this intriguing phenomenon.42 For one, when added to EDTA-CDs, Fe3+ tends to be anchored on the surface of CDs via coordinating with the carboxyl groups. It effectively shields the chromophores from Fe3+ as reflected by the invariable kr values (Table 1), which avoids their direct contact and thereby minimizes the emission quenching.43 This could also explain why adding excess Fe3+ would further exert negative effects on PL efficiency (Fig. S4†). Specifically, continuously adding FeCl3 after the carboxyl groups are coordinatively saturated produces large amounts of free Fe3+ ions, which might interact with the chromophores and the PL intensity decreases. For another, the complexation between Fe3+ and carboxyl groups rigidifies the shell. It provides a conformationally restricted environment for the chromophores and thus blocks the non-radiative relaxation channels (Fig. 2f).
To obtain further information on the EDTA-CDs and Fe coordination, we performed X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements. A comparison of the Fe K-edge XANES spectral features of Fe@EDTA-CDs with those of Fe foil, FeO, Fe2O3, and FePc samples reveals that the Fe species in Fe@EDTA-CDs is close to +3 valence (Fig. 3a). This is corroborated by the Fe 2p XPS analysis, where Fe3+ 2p3/2 and Fe3+ 2p1/2 are present at 711.12 and 724.42 eV, respectively (Fig. S7†).44 The Fourier transform of the EXAFS spectra of Fe@EDTA-CDs shows main peaks at 1.79 and 2.64 Å attributed to the Fe–O and Fe–O–C coordinations, respectively (Fig. 3b). It could also be reflected by the 3D contour Wavelet transform extended EXAFS map, where the scattering path signals of Fe–O and Fe–O–C were found at [3.86, 1.79] and [2.84, 2.64] (Fig. 3c). Simultaneously, no scattering path signal of Fe–Fe bonds was detected. This phenomenon, together with the absence of diffraction peaks from iron oxides in XRD (Fig. S8†), suggests that all Fe species are atomically dispersed. C, N, and O K-edge near-edge X-ray absorption fine structure (NEXAFS) analyses were further performed. First, the coordination between Fe and N was excluded as there is no appreciable difference between the N K-edge NEXAFS spectra of EDTA-CDs and Fe@EDTA-CDs (Fig. S9†). In contrast, their C and O K-edge NEXAFS spectra are distinct. Specifically, the incorporation of Fe3+ lowers the signal of C![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) species in O K-edge NEXAFS spectrum notably (π* C at 530.59 eV, Fig. 3d) while enhancing the intensity of
species in O K-edge NEXAFS spectrum notably (π* C at 530.59 eV, Fig. 3d) while enhancing the intensity of ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O in the C K-edge NEXAFS spectrum (π* O at 286.68 eV, Fig. 3e). These results demonstrate that the Fe atoms are coordinated by carboxyl groups, consistent with the observations and analysis of the XANES spectra. In addition, a FT-IR signal attributed to Fe–O coordination emerged at 684 cm−1 for Fe@EDTA-CDs, which is absence for EDTA-CDs and thus provides additional evidence for the existence of Fe–O (Fig. S3†).
O in the C K-edge NEXAFS spectrum (π* O at 286.68 eV, Fig. 3e). These results demonstrate that the Fe atoms are coordinated by carboxyl groups, consistent with the observations and analysis of the XANES spectra. In addition, a FT-IR signal attributed to Fe–O coordination emerged at 684 cm−1 for Fe@EDTA-CDs, which is absence for EDTA-CDs and thus provides additional evidence for the existence of Fe–O (Fig. S3†).
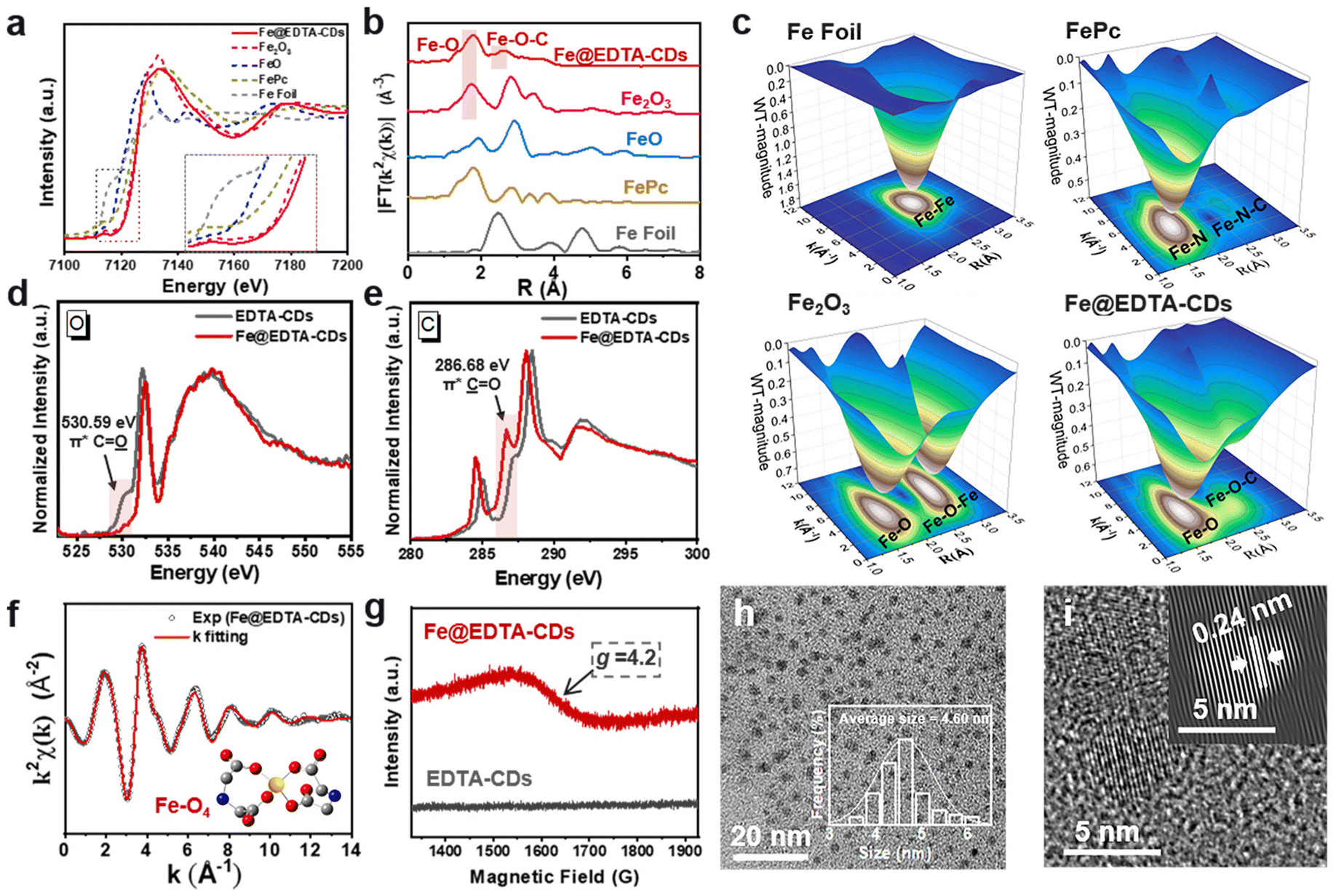 | ||
| Fig. 3 Structural analysis of Fe@EDTA-CDs. (a) Normalized Fe K-edge XANES spectra and (b) radial distance χ(R) spectra of Fe foil, FePc, FeO, Fe2O3, and Fe@EDTA-CDs. (c) 3D contour wavelet transform extended EXAFS maps with the 2D projection of Fe foil, Fe2O3, FePc, and Fe@EDTA-CDs. (d) O K-edge XANES spectra and (e) C K-edge XANES spectra of EDTA-CDs and Fe@EDTA-CDs. (f) Fitting curves of the EXAFS of Fe@EDTA-CDs in the K space. Inset: a model for the surface of Fe@EDTA-CDs. (C, N, O, and Fe are presented by gray, blue, red, and yellow atoms.) (g) EPR spectra of Fe@EDTA-CDs in the solid state. (h) TEM image and the inset of (h) shows the size distribution of Fe@EDTA-CDs. (i) High-resolution TEM image and the inset of (i) shows the corresponding specially filtered image of Fe@EDTA-CDs. Here filtration means a sequence of 2D-operators (FFT, masking, and inverse FFT) applied to the high-resolution TEM image. | ||
The coordination mode between Fe3+ and carboxyl groups was further investigated. As shown in Fig. 3f and S10, ESI,† the good fitting results of χ(R) and χ(k) space spectra with reasonable R-factor (Table S1†) support the coordination structure of Fe–O4 in Fe@EDTA-CDs. In addition, Fe@EDTA-CDs exhibit broad resonance signals at g ≈ 4.2 in the electron paramagnetic resonance (EPR) spectrum (Fig. 3g). According to previous studies,45 this signal is attributed to tetrahedral coordinated Fe3+ with rhombic symmetry sites. These results imply that one Fe3+ ion is chelated by two carboxyl groups (Fig. 3f, inset), which rigidifies the shell and thus leads to a slight decrease in the average diameter from EDTA-CDs (6.80 nm) (Fig. 1b) to Fe@EDTA-CDs (4.60 nm) (Fig. 3h and i and S1b†). According to the ICP-MS and XPS results, the mass and atomic ratios of Fe in Fe@EDTA-CDs are 4.80% and 1.13%, respectively (Fig. S7a†). In addition, the zeta potential of EDTA-CDs was measured to be −18.23 ± 0.2 mV. It shifts to almost neutral (−3.20 ± 0.2 mV) for Fe@EDTA-CDs due to the consumption of carboxyl groups via Fe coordination (Fig. S11†).
The successful doping of atomically dispersed Fe3+ into Fe@EDTA-CDs has prompted us to explore their potential in the catalysis of H2O2 to produce hydroxyl radicals (˙OH). Upon adding a trace amount of Fe@EDTA-CDs (20 μg mL−1) to an aqueous solution of H2O2 and 5,5-dimethyl-1-pyrroline N-oxide (DMPO, a radical scavenger), four lines with an intensity ratio of 1:2:2:1 were observed in the EPR spectrum (Fig. 4a). This strong signal is ascribed to the capture of ˙OH generated in situ by DMPO, demonstrating the H2O2 catalytic activity of Fe@EDTA-CDs. When Fe@EDTA-CDs is absent or replaced by EDTA-CDs, no EPR signals could be detected under the same conditions. This suggests that the coordination Fe sites play an essential role in the catalytic activity, which allows us to calculate the catalyst loading capacity of Fe@EDTA-CD (0.86 mmol mg−1) from the ICP-MS results.
 | ||
| Fig. 4 Photo-enhanced catalytic properties of Fe@EDTA-CDs. (a) EPR spectra of DMPO in different groups. (b) UV-vis absorption spectra of TMB in different groups. Inset: the corresponding images of TMB in the presence of H2O2 (left), EDTA-CDs + H2O2 (middle) and Fe@EDTA-CDs + H2O2 (right). (c) Lineweaver–Burk plot of Fe@EDTA-CDs. (d) UV-vis absorption spectra of TMB in the presence of H2O2, H2O2(+), Fe@EDTA-CDs + H2O2, and Fe@EDTA-CDs + H2O2(+). (e) Normalized absorbance of methylene blue (A/A0) at 665 nm in the presence of Fe@EDTA-CDs + H2O2 and Fe@EDTA-CDs + H2O2(+). (f) CV curves of an Fe@EDTA-CDs/glassy carbon electrode in the dark and light treatment (10 min) (LED lamp, 400–500 nm, 100 mW cm−2). (g) The transient photocurrent response of EDTA-CDs and Fe@EDTA-CDs. (h) Possible mechanism of the photo-enhanced H2O2 catalytic activity of Fe@EDTA-CDs. The concentrations of EDTA-CDs, Fe@EDTA-CDs and H2O2 are 20 μg mL−1, 20 μg mL−1, and 1 mM, respectively. (+) in the related panels refers to light treatment (LED lamp, 400–500 nm, 100 mW cm−2), and the light irradiation time in Fig. 4a and d is 2 min. | ||
A quantitative evaluation of the H2O2 catalytic efficiency was carried out by employing 3,3′,5,5′-tetramethyl-benzidine (TMB) as a substrate. As TMB is rapidly consumed by ˙OH to afford bluish oxidized TMB (oxTMB), a strong absorption band centered at 652 nm emerges for the Fe@EDTA-CDs/H2O2 system (Fig. 4b). It is worth noting that the H2O2 catalytic activity reaches its maximum at pH = 4, and this pH was adopted in the following experiments (Fig. S12†). On this basis, the kinetics parameters were obtained for the H2O2 substrate. Specifically, the maximum initial velocity (vmax) and Michaelis–Menten constant (Km) of Fe@EDTA-CDs were determined to be 6.63 × 10−4 mM s−1 and 1.47 mM at 25 °C, respectively, which are comparable to those of the well-studied Fe3O4 nanozyme or natural horseradish peroxidase (HPR) (Fig. 4c and Table S2†).46 Moreover, the turnover number (TON) value of Fe@EDTA-CDs was calculated to be 3.87 × 10−2 s−1, which is among the highest in developed artificial H2O2 catalysts (Table S3†). This suggests that Fe@EDTA-CDs catalyze H2O2 with a high catalytic efficiency. In addition, we found that Fe@EDTA-CDs also exhibited high activity for the decomposition of glutathione (GSH), an antioxidant agent that exists in most cells (Fig. S13†). This indicates that Fe@EDTA-CDs can reduce or even deplete the cellular GSH, facilitating the accumulation of reactive oxygen species (ROS) in tumor cells and thereby enhancing oxidative damage.2
The broad absorption of Fe@EDTA-CDs in the visible region further inspired us to explore the influence of light on its catalytic activity. When exposed to mild visible light (LED lamp, 400–500 nm, 100 mW cm−2), the EPR signal arising from ˙OH enhances notably, suggesting that Fe@EDTA-CDs catalyze H2O2 more efficiently than in the dark (Fig. 4a). It is corroborated by the TMB colorimetric reaction, where the amount of generated ˙OH increases by 89.50% after light irradiation (denoted as the photo-enhanced catalytic efficiency, PCE) (Fig. 4d). The PCE effect was also reflected by the light-boosted degradation rate of methylene blue (Fig. 4e and S14†). This phenomenon is very interesting considering the chromophores (which absorbs visible light) and catalysis center (which catalyzes H2O2) in Fe@EDTA-CDs are located at different sections (Scheme 1). It is worth noting that the PCE is not likely caused by simple photocatalysis of the chromophores, since no ˙OH was produced from EDTA-CDs under the same irradiation conditions (Fig. 4a). Moreover, the temperature variation of the EDTA-CD and Fe@EDTA-CD solutions is negligible after 8 minutes of irradiation, and their time-dependent temperature curves resemble that of pure water (Fig. S15†). Hence, the photothermal effect, which is employed to account for the recently reported PCE behaviors, is also ruled out in our cases.32
In order to study the origin of PCE, the catalytic activity of Fe@EDTA-CDs with different [Fe3+] was evaluated. As shown in Fig. S16 and S17,† the PCE effect first enhances, reaches the maximum at an [Fe] of 0.8 mol L−1, then decreases significantly with further increase of [Fe]. This trend, to our interest, is somewhat resembling that of the PL lifetime. Given that the lifetime is closely related to the stability of excitons, an increase in the lifetime generally means that the excitons generated in the chromophores can interact with surrounding species (i.e., catalytic sites on the surface) more efficiently. This may result in enhanced charge separation and promotes the catalysis performance accordingly. To support this assumption, cyclic voltammetry (CV) and transient photocurrent response measurements were conducted. As shown in Fig. 4f, the CV curve of Fe@EDTA-CDs under dark conditions shows single electron oxidation and reduction peaks attributable to the redox of Fe catalytic sites (Fig. S18†). Upon irradiation, the oxidation potential shows a slight negative shift, accompanied by a significant increase in the oxidation current. This phenomenon suggests that photoexcitation of the chromophores could reduce the interfacial resistance and thereby boost the charge transfer efficiency of the catalysis center. This result is further supported by the stable and reversible photocurrent response of Fe@EDTA-CDs (Fig. 4g), which confirms the crucial role of photo-induced charge separation in the PCE effect (Fig. 4h).
Density functional theory (DFT) calculations were performed to achieve deeper insights into the photo-induced charge separation process. A simplified molecular model consists of a chromophore and a catalytic unit was first constructed for Fe@EDTA-CDs (Fig. 5a). It is well documented that 2,3-diaminophenazine (DPA) (or its derivatives) is responsible for the red emission of o-PD derived CDs, and thus is chosen as the chromophore. Meanwhile, a Fe-EDTA coordination structure, which serves as the catalytic unit, was covalently attached to DPA via amide bonds. The optimized geometry for the Fe@EDTA-CDs model reveals the tetrahedral coordination mode between the Fe3+ and carboxyl groups on EDTA, consistent with the aforementioned EXAFS and EPR results (Fig. 3f and g). Additionally, according to time-dependent DFT calculations (Fig. 5b), the theoretical simulated spectrum exhibits absorption from 350 to 700 nm (Fig. 5c), which agrees well with the broad visible-light absorption in the experimental profile. Carefully screening of the calculated transitions with major contributions (oscillator strengths > 0.01) to the absorption identifies excited states 15, 24, 36, and 39 (Fig. 5c). These four transitions have charge transfer characters, which involve the molecular orbitals (MOs) of DPA and Fe-EDTA moieties. In particular, for excited state 15 with an excitation wavelength of 590 nm, the electron densities are mainly distributed over the π-conjugated moieties of DPA (MO-259A, Fig. 5d), while it migrates to the Fe-EDTA coordinated unit upon photoexcitation (MO-262A, Fig. 5d). Such photo-induced electron injection would potentially increase the electron densities of the Fe-EDTA unit and thereby enhance the catalytic activity.47
 | ||
| Fig. 5 Photo-induced charge separation process. (a) Optimized ground state geometry and (b) energy level diagram for the α and β MOs of Fe@EDTA-CDs. (c) Simulated absorption spectrum and (d) calculated MOs diagrams (MO-259A and MO-262A) for the molecular model of Fe@EDTA-CDs. | ||
As confirmed above, Fe@EDTA-CDs show bright red fluorescence and intriguing photoenhanced catalytic activity, which promises their uses for synergistic imaging and cancer therapy. The in vitro biocompatibility was first assessed. The cell electron microscopy analysis reveals that the amount of autophagosomes (highlighted by red arrows), an indicator of the level of autophagic activity running, in the Fe@EDTA-CD group is much less than that of EDTA-CDs (Fig. 6a). This suggests that the incorporation of Fe improves the biocompatibility of Fe@EDTA-CDs. CCK-8 results show that the cell viability exceeds 80% even at a high Fe@EDTA-CD concentration of 100 μg mL−1 (Fig. 6b), notably higher than that of the EDTA-CD group (Fig. S19†). Similar results were obtained from lactate dehydrogenase (LDH) assays, where HepG2 cells exposed to Fe@EDTA-CDs for 24 h led to a lower LDH leakage than those exposed to EDTA-CDs (Fig. S19b and c†). The lowered cytotoxicity of Fe@EDTA-CDs may arise from the reduction of metal chelating ability and/or abundant negative charges of the carboxyl groups by Fe complexation (Fig. S11†). Additionally, Fe@EDTA-CDs exhibit negligible toxicity to red blood cells (RBCs) as revealed by hemolysis assays,48 which show a low hemolysis rate of 4.67% at a concentration of 100 μg mL−1. These results indicate the excellent biocompatibility of Fe@EDTA-CDs (Fig. S19d†).
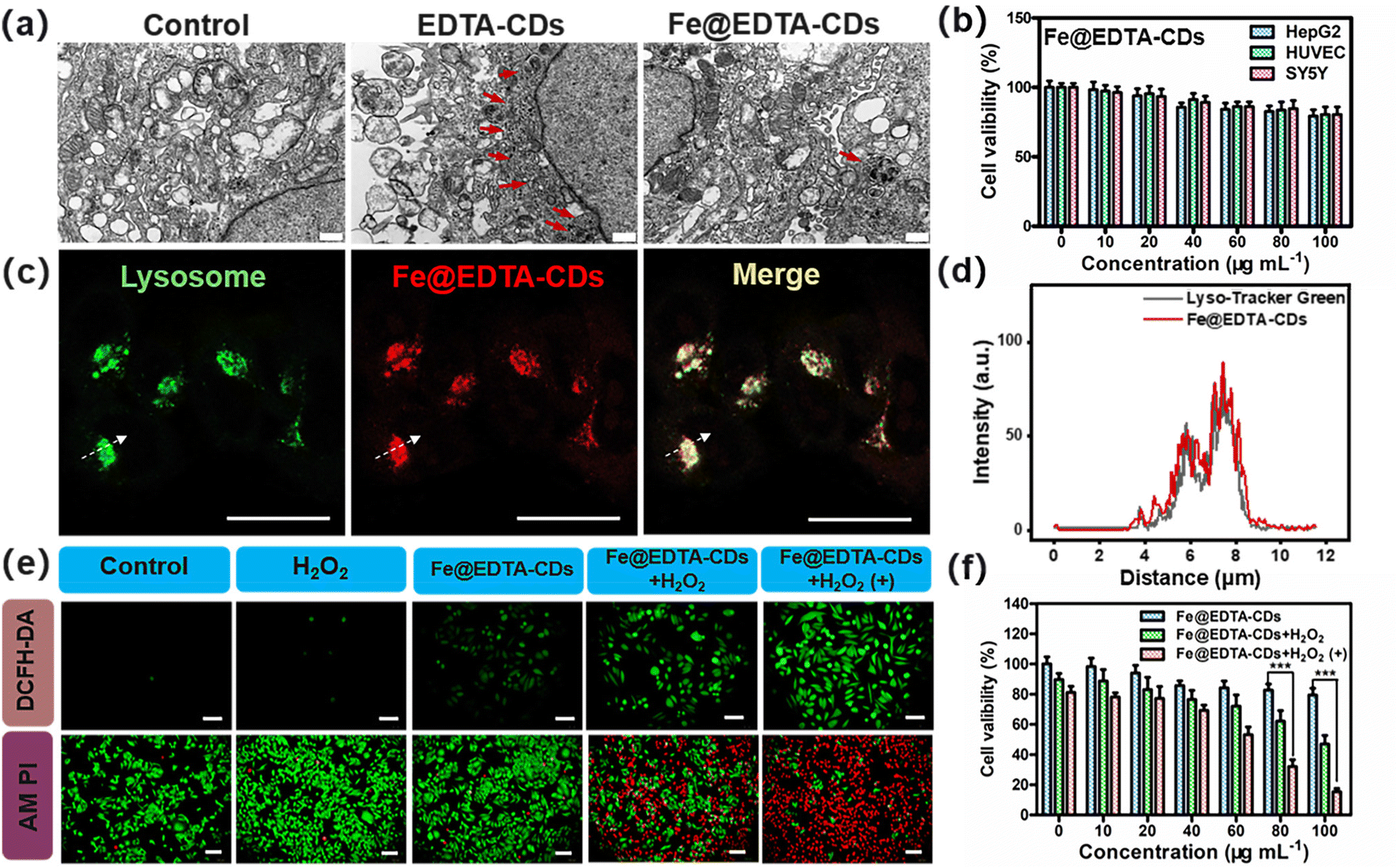 | ||
| Fig. 6 Intracellular anti-cancer therapy. (a) Typical bio-TEM images of HepG2 cells treated with EDTA-CDs (80 μg mL−1) and Fe@EDTA-CDs (80 μg mL−1). Scale bar: 500 nm. (b) Cell viability of different cells incubated with Fe@EDTA-CDs at different concentrations. (c) CLSM images of HepG2 cells incubated with Fe@EDTA-CDs (80 μg mL−1) and LysoTracker Green (100 nM). Scale bar: 20 μm. (d) The fluorescence intensity curves between the green and red channels in HepG2 cells. (e) Fluorescence images of HepG2 cells with DCFH-DA and calcein-AM/PI staining under different treatments for 4 h. (f) Cancer cell viability of HepG2 cells for Fe@EDTA-CDs, Fe@EDTA-CDs + H2O2, and Fe@EDTA-CDs + H2O2(+) at different concentrations of Fe@EDTA-CDs (H2O2: 100 μM L−1). *p < 0.05, **p < 0.01, and ***p < 0.001 (n = 6). (+) in the related panels refers to light treatment (LED lamp, 400–500 nm, 100 mW cm−2, 12 min). | ||
On this basis, we explored Fe@EDTA-CDs as luminescent agents for bioimaging. As shown in confocal laser scanning microscopy (CLSM) (Fig. S20 and S21†), HepG2 cells incubated with EDTA-CDs exhibit red PL with weak intensity. In contrast, intense fluorescence signals with a high signal-to-background ratio were observed for the Fe@EDTA-CD group (Fig. S20†). This demonstrates the improved bioimaging capability of Fe@EDTA-CDs, which should be derived from the enhanced emission efficiency upon Fe complexation. Experiments with a commercial lysosome probe (Lyso-Tracker Green) further reveal the lysosomes-targeting capability of Fe@EDTA-CDs, as evidenced by the excellent overlay (Pearson's coefficient = 0.88) between the fluorescence signals from Fe@EDTA-CDs (red channel) and LysoTracker Green (green channel) (Fig. 6c and d and Fig. S21c, d†). It is also noted that the localization of Fe@EDTA-CDs in lysosomes (pH range 4.5–5.5) is favorable for their ˙OH generation since the H2O2 activity of Fe@EDTA-CDs is boosted in an acid environment (Fig. S12†).
Furthermore, the in vitro anticancer efficiency of Fe@EDTA-CDs was evaluated using HepG2 cells. For simulating the H2O2-rich tumor microenvironment, H2O2 was added to the 2D cells with a concentration of 50 × 10−6 M. Considering the PCE properties of Fe@EDTA-CDs, the influence of light irradiation on the anticancer activity was further tested. When the Fe@EDTA-CDs group was exposed to mild visible light (400–500 nm, 100 mW cm−2, 12 min), the cell viability decreases sharply to 15% as compared with that in the dark (79%) (Fig. 6f), which demonstrates the light enhanced cytotoxicity of Fe@EDTA-CDs. The enhanced cytotoxicity is attributed to an increase in the intracellular ROS level revealed by the 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA) staining experiment (Fig. 6e). This result is also reflected by calcein AM (green emission) and propidium iodide (PI, red emission) staining assays. The coexistence of green and red emissions suggests that only partial cells are killed for the Fe@EDTA-CDs + H2O2 group in the dark. Gratifying, the proportion of death cells increases dramatically upon visible light irradiation, as revealed by the homogeneous red fluorescence. This result shows that the light-enhanced anticancer efficiency of Fe@EDTA-CDs is essentially derived from the PCE effect.
For the phenotypic analysis of the antitumor activity of Fe@EDTA-CDs, an SY5Y 3D multicellular spheroid (3D MC) model, a typical model which mimics the tumor microenvironment, was established using SY5Y cells (Fig. S22†). As expected, the size of 3D MCs increases rapidly over time in the absence of Fe@EDTA-CDs, indicative of no antitumor activity by merely using H2O2 and/or light. With the incubation of Fe@EDTA-CDs, the growth of 3D MCs was inhibited as no appreciable difference in the size was observed with time. Moreover, the 3D MCs incubated with Fe@EDTA-CDs and H2O2 show a pronounced decrease in the size as a function of incubation time, suggesting their good antitumor activity. The antitumor efficiency was further improved when light was applied, as evidenced by the accelerated diminishing process of 3D MCs. After 7 days of incubation, the 3D MCs with 12 min day−1 of light irradiation was almost eliminated. These phenomena reveal that light, H2O2 and Fe@EDTA-CDs have a synergistic effect on the killing of tumor cells in the three-dimensional microenvironment.
Bacterial infection can cause major diseases, such as tuberculosis, plague, syphilis, and cholera, which is a severe global health concern.49,50 In view of the excellent ROS generation efficiency, we further explored the possibility of using Fe@EDTA-CDs as antibacterial agents. As can be seen from Fig. S23,† the number of Fe@EDTA-CD bacterial plaques decreased significantly with the increase of Fe@EDTA-CD concentration upon light exposure, reaching an antibacterial rate of 86% against Staphylococcus aureus and 62% against E. coli at 100 μg mL−1. This demonstrates the good antibacterial capability of Fe@EDTA-CDs. In contrast, the antibacterial efficacy is notably compromised in the absence of light or using the EDTA-CDs as antibacterial agents.
Conclusions
In summary, inspired by the spatial organization in natural photosynthesis and photoactive organisms, we have presented a simple and versatile strategy for developing a nanocatalytic platform with the integration of imaging and therapy functions. The spatial arrangement of light-harvesting chromophores and catalytic sites in Fe@EDTA-CDs is discretely addressable by core–shell compartmentalization, giving rise to the intriguing interplay and mutual promotion between each function. Specifically, not only the red emission quantum yield increases due to the synergistic shielding and rigidifying effects rendered by Fe coordination, but also the catalytic activity can be notably enhanced by the visible light-promoted charge transfer process. Because of these combined attributes, Fe@EDTA-CDs are endowed with a high ROS generation rate and light-enhanced anticancer efficiency, suggesting their promising application in CDT. More importantly, this work may provide a new perspective for designing smart nanocatalytic medicines featuring full modularity and facile integration.Experimental section
Materials and methods, synthetic procedures and characterization are available in the ESI.†Author contributions
X. W., Z. L. and H. B. designed all of the experiments; X. W. and Z. L. performed the experiments and wrote the manuscript; F. Y. helped X. W. in the cell experiment; Y. H. performed the density functional theory calculation. L. J. helped in analyzing the data; J. Z. and Q. X. performed the NEXAFS and EXAFS measurements of all samples and helped X. W. and H. B. in the data analysis; J. Z. helped in analyzing the data of NEXAFS and EXAFS measurements; A. C. T. and Z. L. revised the manuscript; H. B. supervised the whole work and revised the manuscript. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundations of China (Grant No. 52172033, 51772001, 22005280, 21872131) and the Anhui Province Key Research and Development Plan Project International Science and Technology Cooperation Special Project (No. 202004b11020015). We gratefully acknowledge the financial support from the National Key R&D Program of China (Grant No. 2021YFA1600202). We acknowledge the support of the Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Anhui University, Hefei, Anhui 230601, China and the Key Laboratory of Functional Inorganic Material Chemistry of Anhui Province, Anhui University, Hefei 230601, P. R. China. We also acknowledge the support of the Key Laboratory of Environment-Friendly Polymer Materials of Anhui Province, Hefei, China. We acknowledge the 1W1B beamline of Beijing's Synchrotron Radiation Facility (BSRF) and the BL11U beamline of National Synchrotron Radiation Laboratory. We would also like to thank Dr Xiaojie Wang from the School of Life Sciences of Anhui University for helping us in performing the antibacterial experiment.References
- L. Lin, J. Song, L. Song, K. Ke, Y. Liu, Z. Zhou, Z. Shen, J. Li, Z. Yang, W. Tang, G. Niu, H. Yang and X. Chen, Angew. Chem., Int. Ed., 2018, 57, 4902–4906 CrossRef CAS PubMed.
- Y. Sang, F. Cao, W. Li, L. Zhang, Y. You, Q. Deng, K. Dong, J. Ren and X. Qu, J. Am. Chem. Soc., 2020, 142, 5177–5183 CrossRef CAS PubMed.
- J. J. Chen, Y. F. Zhu, C. T. Wu and J. L. Shi, Chem. Soc. Rev., 2020, 49, 9057–9094 RSC.
- B. Yang, Y. Chen and J. Shi, Adv. Mater., 2019, 31, 1901778 CrossRef PubMed.
- H. Lin, Y. Chen and J. Shi, Chem. Soc. Rev., 2018, 47, 1938–1958 RSC.
- M. Liang and X. Yan, Acc. Chem. Res., 2019, 52, 2190–2200 CrossRef CAS PubMed.
- X. Qin, C. Wu, D. Niu, L. Qin, X. Wang, G. Wang and Y. Li, Nat. Commun., 2021, 12, 5243 CrossRef CAS.
- H. Hu, W. Feng, X. Qian, L. Yu, Y. Chen and Y. Li, Adv. Mater., 2021, 33, 2005062 CrossRef CAS PubMed.
- A. Robert and B. Meunier, ACS Nano, 2022, 16, 6956–6959 CrossRef CAS PubMed.
- F. Rigodanza, M. Burian, F. Arcudi, L. Dordevic, H. Amenitsch and M. Prato, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- F. Arcudi, L. Dordevic and M. Prato, Acc. Chem. Res., 2019, 52, 2070–2079 CrossRef CAS PubMed.
- Q. Jia, J. Ge, W. Liu, X. Zheng, S. Chen, Y. Wen, H. Zhang and P. Wang, Adv. Mater., 2018, 30, 1706090 CrossRef.
- J. Liu, R. Li and B. Yang, ACS Cent. Sci., 2020, 6, 2179–2195 CrossRef CAS.
- C. Dong, M. Xu, S. Wang, M. Ma, O. U. Akakuru, H. Ding, A. Wu, Z. Zha, X. Wang and H. Bi, J. Nanobiotechnol., 2021, 19, 1–8 CrossRef.
- B. L. Li, S. J. Zhao, L. Huang, Q. Wang, J. F. Xiao and M. H. Lan, Chem. Eng. J., 2021, 408, 127245 CrossRef CAS.
- C. Y. Jia, Y. X. Guo and F. G. Wu, Small, 2022, 18, 2103868 CrossRef CAS PubMed.
- Y. Chung, J. Kim and C. B. Park, ACS Nano, 2020, 14, 6470–6497 CrossRef CAS PubMed.
- X. Xu, L. Mo, Y. Li, X. Pan, G. Hu, B. Lei, X. Zhang, M. Zheng, J. Zhuang, Y. Liu and C. Hu, Adv. Mater., 2021, 33, 2104872 CrossRef CAS.
- D. Li, E. V. Ushakova, A. L. Rogach and S. Qu, Small, 2021, 17, 2102325 CrossRef CAS PubMed.
- X. Li, Y. Fu, S. Zhao, J. Xiao, M. Lan, B. Wang, K. Zhang, X. Song and L. Zeng, Chem. Eng. J., 2022, 430, 133101 CrossRef CAS.
- P. Muhammad, S. Hanif, J. Li, A. Guller, F. U. Rehman, M. Ismail, D. Zhang, X. Yan, K. Fan and B. Shi, Nano Today, 2022, 45, 101530 CrossRef CAS.
- B. Yu, W. Wang, W. Sun, C. Jiang and L. Lu, J. Am. Chem. Soc., 2021, 143, 8855–8865 CrossRef CAS PubMed.
- C. Zhang, W. Bu, D. Ni, S. Zhang, Q. Li, Z. Yao, J. Zhang, H. Yao, Z. Wang and J. Shi, Angew. Chem., 2016, 128, 2141–2146 CrossRef.
- L. Huang, J. Chen, L. Gan, J. Wang and S. Dong, Sci. Adv., 2019, 5, eaav5490 CrossRef CAS PubMed.
- J. Zhang and J. Liu, Nanoscale, 2020, 12, 2914–2923 RSC.
- J. Lin, W. Tian, Z. Guan, H. Zhang, X. Duan, H. Wang, H. Sun, Y. Fang, Y. Huang and S. Wang, Adv. Funct. Mater., 2022, 32, 2201743 CrossRef CAS.
- H. Zhu, W. Zan, W. Chen, W. Jiang, X. Ding, B. L. Li, Y. Mu, L. Wang, S. Garaj and D. T. Leong, Adv. Mater., 2022, 34, 2200004 CrossRef CAS PubMed.
- A. Call, C. Casadevall, F. Acuña-Parés, A. Casitas and J. Lloret-Fillol, Chem. Sci., 2017, 8, 4739–4749 RSC.
- Y. Zhu, Z. Wang, R. Zhao, Y. Zhou, L. Feng, S. Gai and P. Yang, ACS Nano, 2022, 16, 3105–3118 CrossRef CAS PubMed.
- L. Huang, D. W. Sun and H. Pu, Small, 2022, 18, 2200178 CrossRef CAS PubMed.
- G. Zhou and M. Li, Adv. Mater., 2022, 2200871 CrossRef CAS PubMed.
- Y. Lin, X. Liu, Z. Liu and Y. Xu, Small, 2021, 17, 2103348 CrossRef CAS PubMed.
- D. Sorigué, B. Légeret, S. Cuiné, S. Blangy, S. Moulin, E. Billon, P. Richaud, S. Brugière, Y. Couté and D. Nurizzo, Science, 2017, 357, 903–907 CrossRef PubMed.
- S. Zhang, D. J. Heyes, L. Feng, W. Sun, L. O. Johannissen, H. Liu, C. W. Levy, X. Li, J. Yang and X. Yu, Nature, 2019, 574, 722–725 CrossRef CAS PubMed.
- X. T. Tian and X. B. Yin, Small, 2019, 15, 1901803 CrossRef CAS PubMed.
- F. Yuan, Z. Wang, X. Li, Y. Li, Z. A. Tan, L. Fan and S. Yang, Adv. Mater., 2017, 29, 1604436 CrossRef PubMed.
- B. Wang, Z. Wei, L. Sui, J. Yu, B. Zhang, X. Wang, S. Feng, H. Song, X. Yong and Y. Tian, Light: Sci. Appl., 2022, 11, 1–14 CrossRef PubMed.
- X. Zhang, G. Li, J. Wang, J. Chu, F. Wang, Z. Hu and Z. Song, ACS Appl. Mater. Interfaces, 2022, 14, 27968–27978 CrossRef CAS PubMed.
- M.-C. Tang, C.-H. Lee, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 9341–9349 CrossRef CAS PubMed.
- J. Xu, Q. Liang, Z. Li, V. Y. Osipov, Y. Lin, B. Ge, Q. Xu, J. Zhu and H. Bi, Adv. Mater., 2022, 34, 2200011 CrossRef CAS PubMed.
- J. D. Braun, I. B. Lozada, C. Kolodziej, C. Burda, K. M. Newman, J. van Lierop, R. L. Davis and D. E. Herbert, Nat. Chem., 2019, 11, 1144–1150 CrossRef CAS PubMed.
- K. Li, G. S. M. Tong, Q. Wan, G. Cheng, W.-Y. Tong, W.-H. Ang, W.-L. Kwong and C.-M. Che, Chem. Sci., 2016, 7, 1653–1673 RSC.
- S. Kuila, K. V. Rao, S. Garain, P. K. Samanta, S. Das, S. K. Pati, M. Eswaramoorthy and S. J. George, Angew. Chem., 2018, 130, 17361–17365 CrossRef.
- C. Gao, Y. Su, X. Quan, V. K. Sharma, S. Chen, H. Yu, Y. Zhang and J. Niu, Appl. Catal., B, 2020, 276, 119016 CrossRef CAS.
- R. Naik, S. Prashantha, H. Nagabhushana and K. Girish, J. Lumin., 2018, 197, 233–241 CrossRef CAS.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang and S. Perrett, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- J. Zhang, T. S. Wu, H. V. Thang, K. Y. Tseng, X. Hao, B. Xu, H. Y. T. Chen and Y. K. Peng, Small, 2022, 18, 2104844 CrossRef CAS PubMed.
- G. Gao, Y.-W. Jiang, H.-R. Jia, J. Yang and F.-G. Wu, Carbon, 2018, 134, 232–243 CrossRef CAS.
- J. J. Yang, G. Gao, X. D. Zhang, Y. H. Ma, X. K. Chen and F. G. Wu, Carbon, 2019, 146, 827–839 CrossRef CAS.
- X. Y. Wu, K. Abbas, Y. X. Yang, Z. J. Li, A. C. Tedesco and H. Bi, Pharmaceuticals, 2022, 15, 487 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Other experimental procedures, characterization, other supplementary figures and author contributions. See DOI: https://doi.org/10.1039/d2nr05281d |
| This journal is © The Royal Society of Chemistry 2023 |