
High surface area siloxene for photothermal and electrochemical catalysis†
Yize
Su‡
 a,
Shenghua
Wang‡
a,
Liang
Ji‡
a,
Chengcheng
Zhang
a,
Haiting
Cai
b,
Hui
Zhang
a and
Wei
Sun
*a
a,
Shenghua
Wang‡
a,
Liang
Ji‡
a,
Chengcheng
Zhang
a,
Haiting
Cai
b,
Hui
Zhang
a and
Wei
Sun
*a
aState Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou, Zhejiang 310027, P. R. China. E-mail: sunnyway423@zju.edu.cn
bInstitute of Industrial Catalysis, College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China
First published on 29th November 2022
Abstract
Catalysis based on two-dimensional silicon has been under intense investigation recently. However, its substandard catalytic activity is far from industrialization. In this work, we demonstrate a new solution to this problem formulated on the batch synthesis of siloxene with an enhanced specific surface area (217.8 m2 g−1). A two-dimensional porous structure was prepared, enabling great support and dispersion of metal nanoparticles. Catalytic evaluations of such hybrid structures for the (photo)thermal CO2 hydrogenation reaction and the electrochemical hydrogen evolution reaction revealed a significant performance advantage over the benchmark two-dimensional silicon structures synthesized via the conventional method. This work may confer notable viability on two-dimensional silicon for advanced energy, catalytic, and environmental applications.
Introduction
Two-dimensional (2D) materials possess unique electronic, optical, and mechanical properties differing from their bulk form, and have been extensively investigated for catalysis.1 Abundant surface active sites, enhanced charge transfer, and the ability to confine metals between the layers have triggered new opportunities for 2D materials-based catalysis, especially in the field of conversion of small energy-related molecules like CO2, H2O, O2, CO, etc.1–5 Among the rising stars of 2D catalytic materials, 2D silicon has recently attracted considerable interest for heterogeneous catalysis,6 owing to the high Earth abundance and low toxicity of silicon, as well as some additional features beyond the common advantages of 2D materials.On the one hand, if synthesized with a widely applied route of exfoliating CaSi2 in HCl aqueous solution, 2D silicon with reductive Si–H bonds could be obtained, which enables spontaneous deposition of many catalytic metals on the surface of 2D silicon platforms (e.g. Pd, Cu, Ir, etc.).7–9 On the other hand, the space between the 2D silicon sheets and metal–O–Si linkages feasibly formed upon heating enables strong confinement of metal nanoparticles through the common impregnation method.10,11 These unique features have led to the development of relatively effective catalysts for various heterogeneous reactions exemplified by the CO2 reduction reaction, hydrogen evolution reaction, nitrogen oxide removal, dry reforming reaction, heavy metal ion removal, etc.9–16 However, despite various synthetic protocols having been explored,17–19 the surface area of 2D Silicon that can easily aggregate is still a major limiting factor for its catalytic performance. As one of the very few cases that investigated the influence of surface area on catalytic activity, Li and co-workers found the optimum reaction duration of HCl exfoliation for the highest photocatalytic reduction of Cr3+, but the highest surface area achieved was still below 50 m2 g−1.14 Qian and co-workers successfully deposited Pd nanoparticles onto 2DSi nanosheets through reductive Si–H bonds and demonstrated catalytic activity for the photothermal CO2 reduction, but the stabilized activity was only around 10 μmol g−1 h−1.7 Yan and co-workers discovered that impregnating Ni between the siloxene nanosheets could direct the selectivity of CO2 methanation, but at the cost of the total CO2 conversion due to the significant loss of exposed active sites and thereby the loss of production of CO.10 In this context, the 2DSi stacked structures still need further improvement in the surface area, and thereby the catalytic activity.
In this research, we demonstrate an optimized synthesis strategy to produce siloxene with a high specific surface area of 217.8 m2 g−1, which was much higher than that of 2D silicon obtained by the traditional HCl exfoliation methods (16.97 m2 g−1). Such siloxene obtained was further decorated with metal nanoparticles to achieve relatively high catalytic performances for CO2 hydrogenation and electrochemical hydrogen evolution. The results demonstrated the great viability and prospect of tuning the synthesis of siloxene and potentially other 2D materials for a variety of advanced catalytic systems.
Results and discussion
Characterization of the catalysts
We started with the commercial CaSi2 for the synthesis, but observed a substantial amount of bulk Si as the major impurity in this precursor (Fig. S1a†), which would be redundant for the synthesis and an impediment to further exfoliation; therefore, NaOH was employed to act as a corrodent to exclude the bulk Si impurity.20 The sample after etching in 5 M NaOH solution, denoted as CaSi2–NaOH, exhibited no bulk Si in the X-ray diffraction (XRD) pattern (Fig. S1a†). Further evidence of the erosion was provided by Raman spectra (Fig. S1b†). The symbolic peak of bulk Si (518 cm−1) vanished, and a new peak (382.5 cm−1) appeared after etching, indicating the etched CaSi2 surface and the exposure of its inner silicon domains.21–23 Surprisingly, we also observed the formation of a porous structure after NaOH treatment, as shown in scanning electron microscopy (SEM) images (Fig. S1c and d†). This was consistent with the erosion and exposure observed in the Raman spectra. A rough estimation of the ratio of Si/Ca obtained using an energy dispersive spectrometer (EDS) corresponded well to the conjecture mentioned above. The Si/Ca ratios were 2.38 for CaSi2 and 1.32 for CaSi2–NaOH, which confirmed the major consumption of silicon during the treatment. Next, the precursor needs exfoliation to acquire 2D structures. Previous synthesis strategies mostly include cooling the HCl solution to a temperature lower than 0 °C to reduce oxidation in the silicon backbone.24,25 However, in this research, a reaction temperature of 30 °C was adopted to separate silicon layers thoroughly (Fig. 1). Note that this temperature is more prone to industrial production regarding scalability and cost, compared with sub-zero temperatures. A sample prepared through the reported method by reacting CaSi2 with concentrated HCl at −15 °C for around a week (denoted as 2DSi) exhibited a low specific surface area of only 16.97 m2 g−1 (Table 1), whereas if we changed the reaction temperature to 30 °C but also used concentrated HCl, the resulting surface area of the sample (denoted as C-siloxene) rose sharply to 90.62 m2 g−1 (Table 1). Furthermore, we diluted the concentration of HCl to 1 M to mitigate the inter-layer condensation of Si–OH groups but obtain Kautsky's siloxene which incorporates O within the Si backbones,26 and the sample (denoted as siloxene (no NaOH)) exhibited an even increased surface area of 136.64 m2 g−1 (Table 1). Finally, we changed the precursor to our pre-treated CaSi2–NaOH, and mixed it with a diluted 1 M hydrochloride (HCl) solution at 30 °C. As the exfoliation proceeded, Ca ions between Si planes were gradually extracted with HCl, leaving the Si–H bonds like Si3![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–H (2110 cm−1) and O3Si–H (2250 cm−1),19,27 confirmed by Fourier-transform infrared (FTIR) spectroscopy (Fig. 2a and S2†). As schematized, the intensity of the Si–H region gradually increased to the maximum at 4.5 days, which might indicate thorough exfoliation. Afterwards, the Si–H intensity decreased, because prolonging the reaction time would promote oxidation and turn Si–H into Si–O–Si, which is detrimental to the reductive deposition of metals and may reduce the surface area through inter-layer crosslinking. Indeed, the siloxene obtained at 4.5 days exhibited a higher surface area (217.82 m2 g−1) than those obtained at 2.5 days (158.72 m2 g−1) and 5.5 days (205.04 m2 g−1). Therefore, the siloxene sample with the reaction time in HCl of 4.5 days was chosen for further study on catalysis, discussed later in this paper. In addition, it is noteworthy that it also has a much higher surface area than the siloxene (no NaOH)-4.5d (136.64 m2 g−1), as shown in Table 1.
Si–H (2110 cm−1) and O3Si–H (2250 cm−1),19,27 confirmed by Fourier-transform infrared (FTIR) spectroscopy (Fig. 2a and S2†). As schematized, the intensity of the Si–H region gradually increased to the maximum at 4.5 days, which might indicate thorough exfoliation. Afterwards, the Si–H intensity decreased, because prolonging the reaction time would promote oxidation and turn Si–H into Si–O–Si, which is detrimental to the reductive deposition of metals and may reduce the surface area through inter-layer crosslinking. Indeed, the siloxene obtained at 4.5 days exhibited a higher surface area (217.82 m2 g−1) than those obtained at 2.5 days (158.72 m2 g−1) and 5.5 days (205.04 m2 g−1). Therefore, the siloxene sample with the reaction time in HCl of 4.5 days was chosen for further study on catalysis, discussed later in this paper. In addition, it is noteworthy that it also has a much higher surface area than the siloxene (no NaOH)-4.5d (136.64 m2 g−1), as shown in Table 1.
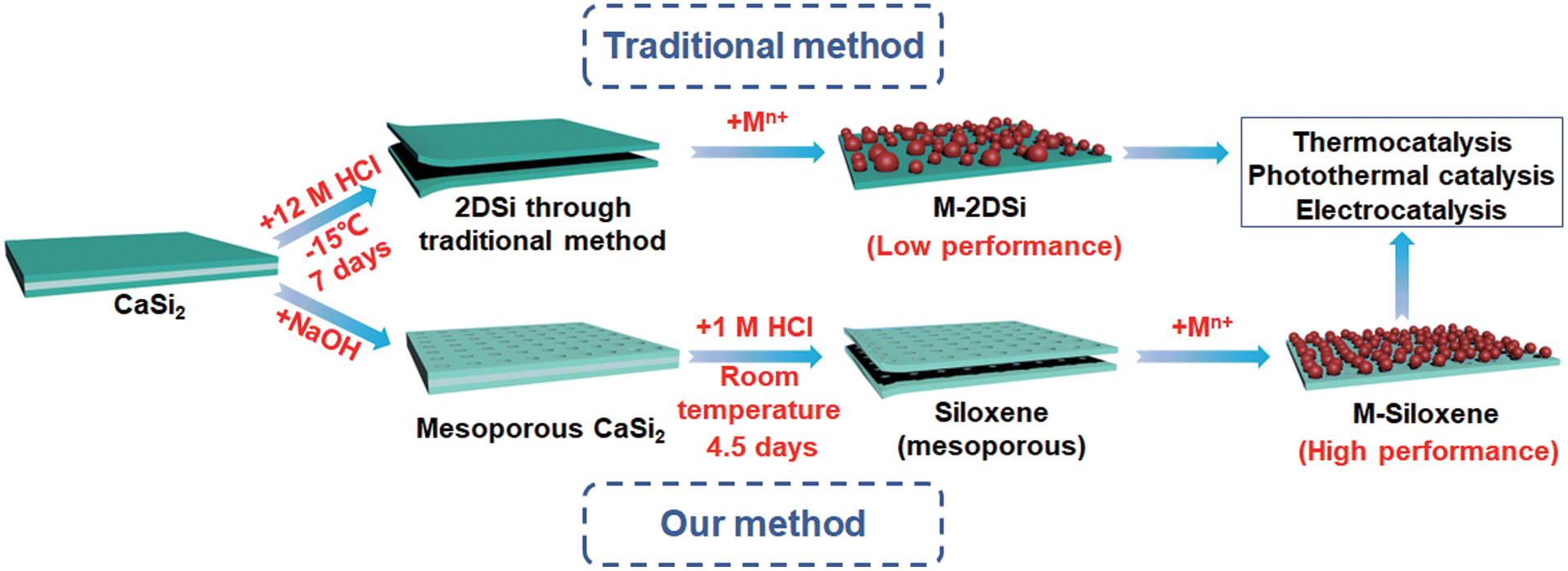 | ||
| Fig. 1 Schematic illustration of the formation of M-2DSi and M-siloxene (metallic nanoparticle-decorated 2DSi and siloxene). | ||
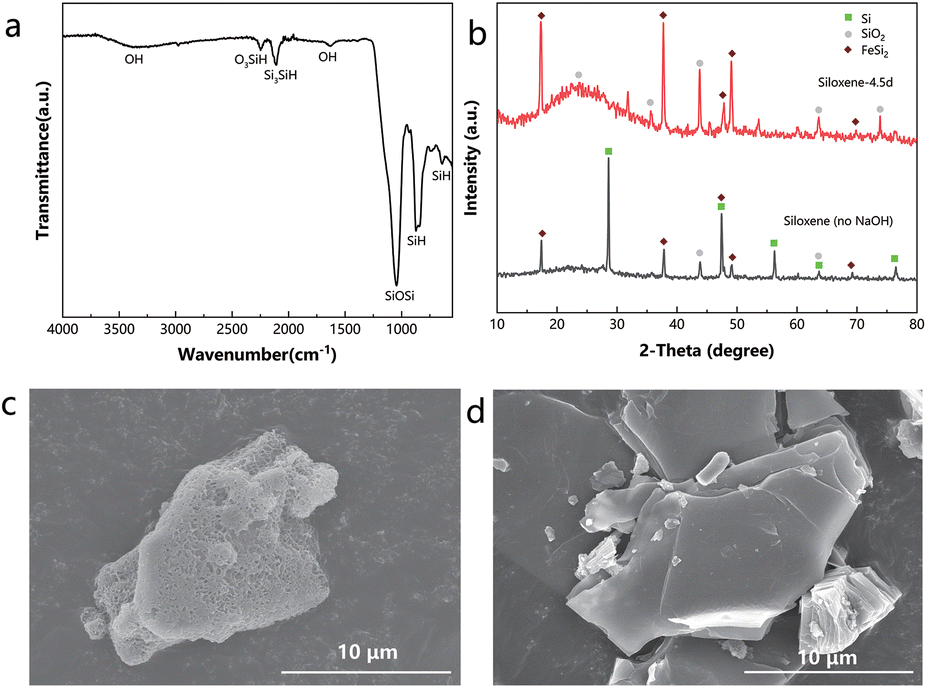 | ||
| Fig. 2 (a) FTIR spectrum of siloxene-4.5d. (b) XRD patterns of siloxene (no NaOH) and siloxene-4.5d, and the peaks are labelled according to the standard SiO2, the standard Si, and the standard FeSi2. SEM images of (c) siloxene-4.5d and (d) 2DSi. | ||
| Samples | CaSi2 | 2DSi | C-siloxene | Siloxene (no NaOH)-4.5d | Siloxene-2.5d | Siloxene-4.5d | Siloxene-5.5d |
|---|---|---|---|---|---|---|---|
| Specific surface area (m2 g−1) | 4.08 | 16.97 | 90.62 | 136.64 | 158.72 | 217.82 | 205.04 |
The XRD patterns show that a substantial amount of bulk Si remained in the siloxene (no NaOH)-4.5d, consistent with the XRD pattern of the original CaSi2, but in contrast to the siloxene-4.5d synthesized with the NaOH pre-treatment (Fig. 2b and Fig. S1a†). Moreover, siloxene (no NaOH) showed a much weaker signal of Si–H relative to Si–O–Si (494 cm−1) in the Raman spectra (Fig. S3†).20,23 These results demonstrate that the NaOH etching process also helped in delaminating CaSi2 and increasing the surface concentration of Si–H for siloxene.
To reiterate, by applying a NaOH pre-treatment of CaSi2, and altering the reaction temperature, HCl concentration, and reaction time, we successfully improved the surface area of the exfoliated 2D structure of silicon from 16.97 m2 g−1 to a significantly higher value of 217.82 m2 g−1, manifested through the Brunauer–Emmett–Teller (BET) measurements (Table 1 and Fig. S4†). Rather irregular patterns of pore size distribution and small pore volumes were seen for the materials prepared with the traditional methods (Fig. S5a–d†), but the pore sizes of clearly a few nanometres corresponding to large pore volumes were observed for the siloxene materials obtained with our optimized method (Fig. S5e–g†). With a larger specific surface area, the siloxene exhibited a larger pore volume per unit mass. SEM images clearly showed that the porous structure of CaSi2–NaOH was preserved in siloxene after exfoliation (Fig. 2c). Meanwhile, the 2DSi with the lowest surface area exhibited a rather smooth plane on its surface (Fig. 2d). The discrepancy in the surface morphology explained the huge difference in the surface area between siloxene and 2DSi.
In order to evaluate the effects of a large specific surface area on catalysis, siloxene-4.5d with the largest surface area and 2DSi with the lowest surface area were decorated with nanoparticles of Cu and Pd, which are widely used catalytic metals. Two different decoration methods were employed, in accordance with the recently reported catalytic hybrid structures derived from conventional 2D silicon precursors. The first method was the conventional impregnation method, including impregnating abundant metal precursors into the ensemble, followed by calcination in air and H2 in sequence.10 The other method utilized Si–H to reduce metal ions in the precursor solution and deposit nanoparticles on the surface.7,8
First, considering that Cu is a relatively cheap metal which is suitable for high loading amount, siloxene-4.5d decorated with Cu nanoparticles was prepared using an impregnation method, denoted as Cuim-siloxene-4.5d. XRD results suggested that Cu had successfully adhered to the surface of siloxene-4.5d, and also of the control sample prepared with 2DSi (Fig. S6a†). With a starting Cu amount of 25% for the preparation, the actual loading mass percentages of Cu for the resulting products prepared from siloxene-4.5d and 2DSi were similar: 18.3% and 17.7%, respectively, according to the inductively coupled plasma atomic emission spectroscopy (ICP-AES) measurements. The transmission electron microscopy (TEM) images of these samples showed that dark nanoparticles were distributed on the surface of thin 2DSi and siloxene-4.5d flakes, but more spatially segregated in the 18.3%Cuim-siloxene-4.5d sample (Fig. S7a and b†). The EDS mapping of 18.3%Cuim-siloxene-4.5d confirmed that such nanoparticles are Cu nanoparticles (Fig. S8a–d†). They have the size of tens of nanometers in diameter, which guaranteed a promising catalytic activity. A rough estimation using the Scherrer equation based on the XRD patterns indicated that 18.3%Cuim-siloxene-4.5d possessed a slightly smaller Cu crystallite size than 17.7%Cuim-2DSi (9 nm vs. 11 nm, Table S1†). The small discrepancy might originate from the high surface area of siloxene-4.5d, which limited the coagulation of Cu nanoparticles during the impregnation process.
Next, Pd as a noble metal and widely used electrochemical catalyst was also decorated on siloxene-4.5d and 2DSi for evaluation. PdH-siloxene-4.5d and PdH-2DSi were simply prepared by the method of reducing Na2PdCl4 with Si–H. The Pd nanoparticles were deposited on the surface of siloxene-4.5d and 2DSi successfully, as shown by XRD (Fig. S6b†). EDS mapping (Fig. S8e–h†) further confirmed the successful loading of Pd nanoparticles on siloxene, and the HRTEM images showed the (111) plane and the (200) plane of Pd (Fig. S9†). With the same starting amount of 5% of Pd for the preparation, the actual loading mass percentages were 3.8% for PdH-siloxene-4.5d and 2.4% for PdH-2DSi, respectively. The higher loading mass of PdH-siloxene-4.5d than that of PdH-2DSi revealed the fact that the high specific surface area of siloxene-4.5d helped in metal anchoring. In order to ensure fair comparison in terms of the loading amount for catalysis, PdH-2DSi with a loading mass of 3.6% was prepared by increasing the starting amount for preparation to 10%. Pd nanoparticles were scattered on the surface of siloxene-4.5d and 2DSi when the initial precursor feed amount of Pd was 5%, as shown in Fig. S7c and e.† As for 3.6%PdH-2DSi, an obvious agglomeration of Pd nanoparticles was observed (Fig. S7d†), and the Pd crystallite size was larger than those of both 3.8%PdH-siloxene-4.5d and 2.4%PdH-2DSi (Table S1†).
Catalytic performance
The improved specific surface area of the as-prepared metal-siloxene-4.5d hybrid could endow it with better catalytic activity in comparison with the samples obtained using the 2DSi prepared through the traditional method. To demonstrate this, we first explored their activities in the straightforward thermocatalytic CO2 hydrogenation reaction, since Cu is a well-known catalyst for the reverse water–gas shift (RWGS) reaction (CO2 + H2 → CO + H2O). As shown in Fig. S10,† 18.3%Cuim-siloxene-4.5d exhibited an obviously higher activity than 17.7%Cuim-2DSi under the same testing conditions in a flow reactor. At 400 °C, an enhancement in the activity of more than 100% was achieved, while at higher temperatures with a facilitated mass transfer, the enhancement was less pronounced, but still 18.3%Cuim-siloxene-4.5d exhibited a decent CO production rate of nearly 35 mmol g−1 h−1 at a high temperature of 500 °C, much higher than that of 17.7%Cuim-2DSi (around 20 mmol g−1 h−1). Bare 2DSi and siloxene-4.5d showed insignificant activities, suggesting that the Cu particles are the main active components. We also prepared a catalyst based on the commercial high-surface-area SiO2 support with a porous structure (Fig. S11†) to demonstrate the superiority of the siloxene. As illustrated in Fig. S12,† 18.3%Cuim-siloxene-4.5d showed higher catalytic activity and better stability than 17.1%Cuim-SiO2 at all test temperatures, which supports our rationale of employing the optimized siloxene as a promising catalyst support.Next, as the overall light reflections within the wavelength range between 200 nm and 2500 nm of 18.3%Cuim-siloxene-4.5d and 17.7%Cuim-2DSi were low (<21%, Fig. S13†), the advantage of the high specific surface area for siloxene-4.5d was further manifested with the photothermal catalytic CO2 hydrogenation reaction (Fig. 3), which could include the solar advantage for sustainability.28 Notably, the enhancement in the activity was even larger than that in the thermocatalytic tests. Compared with 17.7%Cuim-2DSi, an enhancement of 343% in the CO rate was seen for 18.3%Cuim-siloxene-4.5d under a focused light illumination of 34.3 suns (Fig. 3a), which can also be maintained over a 300 min continuous test (Fig. 3b). Under mild illumination, the contrast in the activities between the two samples was even more pronounced (Fig. 3a). There was barely any activity for 17.7%Cuim-2DSi under 20.0 suns. This might be ascribed to the lower overall temperatures of the catalysts in the photothermal tests as the reaction was driven by the local temperature of the active metal Cu. For example, 18.3%Cuim-siloxene-4.5d showed a similar CO rate in 400 °C-driven thermocatalysis (7.8 mmol g−1 h−1) and 34.3 suns-driven photothermal catalysis (8.4 mmol g−1 h−1). However, the temperature of 18.3%Cuim-siloxene-4.5d measured with a thermocouple under 34.3-suns illumination was only 331.3 °C (Fig. S14†). At a lower overall temperature in the photothermal system, only the high surface area sample 18.3%Cuim-siloxene-4.5d could ensure efficient mass transfer.
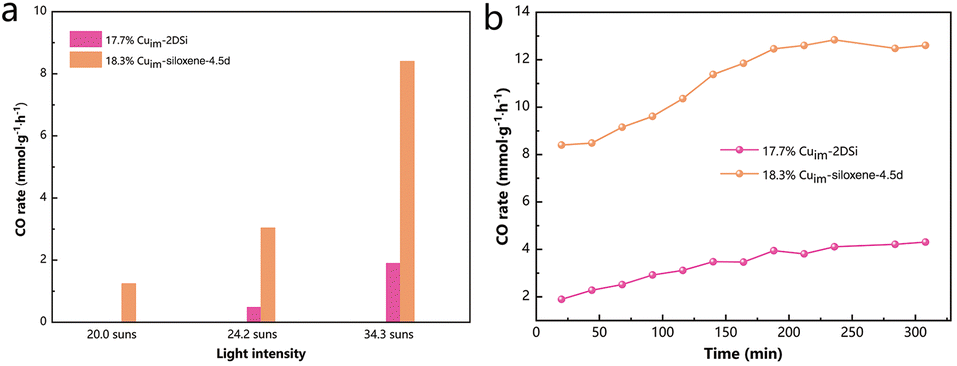 | ||
| Fig. 3 CO rates of different samples in photothermal catalytic reactions (testing conditions: 15 mg of the catalyst, CO2/H2/N2 = ∼2.5/10/2.5, and ambient pressure). | ||
In addition, we also tested the catalytic performance of siloxene (no NaOH), which has a surface area higher than 2DSi but lower than siloxene. The prepared sample was named 15.9%Cuim-siloxene (no NaOH). As expected, its overall activity was indeed between those of 17.7%Cuim-2DSi and 18.3%Cuim-siloxene, as shown in Table S2.† This also well confirmed our attribution of the activity to the surface area.
Finally, as Pd is a prominent metal for the electrochemical hydrogen evolution reaction (HER),29,30 which is considered as an efficient method to meet the growing demands of hydrogen production,31 we further illustrated the diverse advantages of the high specific surface area siloxene in this room-temperature reaction system in the liquid phase, distinct from the gas–solid reaction at elevated temperatures. HER tests were conducted in a 0.5 M H2SO4 solution. As shown in Fig. 4a and b, 3.8% PdH-siloxene-4.5d exhibited a small overpotential of 204 mV at a current density of 10 mA cm−2, which is much lower than that of 3.6% PdH-2DSi (356 mV). Moreover, the onset potential of 3.8% PdH-siloxene-4.5d (85 mV) was also significantly smaller than that of 3.6% PdH-2DSi (200 mV) as illustrated in Fig. 4b, indicating the facilitated catalytic behavior caused by the high specific surface area. Tafel plots were derived from the Linear Sweep Voltammetry (LSV) curves as shown in Fig. 4c, and 3.8% PdH-siloxene-4.5d exhibited a smaller Tafel slope of 96 mV dec−1vs. 145 mV dec−1 for 3.6% PdH-2DSi, suggesting a faster kinetics and the Volmer–Heyrovsky mechanism.32 To study the durability of the catalyst, chronoamperometry measurements were conducted, and as shown in Fig. 4d, 3.8% PdH-siloxene-4.5d can maintain over 50% activity after the 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 seconds durability test compared with the initial state. By comparison, 3.6% PdH-2DSi exhibited a prominent decay with only 10% maintained after 8000 seconds. In addition, the HER performance of the PdH-2DSi sample with a lower Pd loading of 2.4% was slightly better than that of 3.6% PdH-2DSi, but still much poorer than that of 3.8% PdH-siloxene-4.5d, as shown in Fig. S15.† Therefore, regardless of the Pd loading (and the extent of agglomeration of the Pd nanoparticles), the surface area was the main limiting factor for the electrochemical activity.
000 seconds durability test compared with the initial state. By comparison, 3.6% PdH-2DSi exhibited a prominent decay with only 10% maintained after 8000 seconds. In addition, the HER performance of the PdH-2DSi sample with a lower Pd loading of 2.4% was slightly better than that of 3.6% PdH-2DSi, but still much poorer than that of 3.8% PdH-siloxene-4.5d, as shown in Fig. S15.† Therefore, regardless of the Pd loading (and the extent of agglomeration of the Pd nanoparticles), the surface area was the main limiting factor for the electrochemical activity.
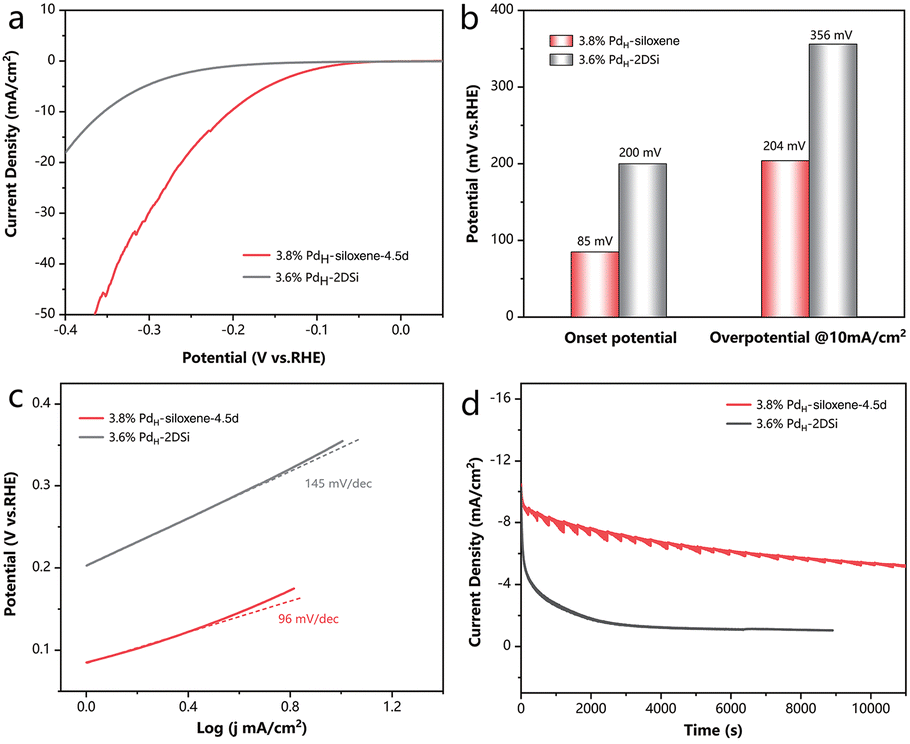 | ||
| Fig. 4 (a) Polarization curves of the 3.6% PdH-2DSi and 3.8% PdH-siloxene-4.5d catalysts. (b) Comparison of the overpotential at 10 mA cm−2 and the onset potential of the catalysts. (c) Tafel plots of the catalysts. (d) Chronoamperometry tests of the catalysts. | ||
Ruling out the effect of chemical states
To confirm that the high surface area is indeed responsible for the enhanced catalytic activities described above, the samples were further characterized by X-ray photoelectron spectroscopy (XPS) to investigate if there is discrepancy in their surface chemical states, a factor that often determines the reaction pathway. Only a small difference could be observed on comparing the XPS results between 17.7%Cuim-2DSi and 18.3% Cuim-siloxene-4.5d (Fig. 5). The peak positions of Cu 2p1/2 (952.4–952.5 eV) and Cu 2p3/2 (932.5–932.7 eV) were almost identical for the two samples (Fig. 5a).11,12 Moreover, X-ray induced Auger electron spectra (XAES) (Fig. 5b) revealed similar Cu(0)/Cu(+) ratios. In the the XPS patterns of Si 2p and O 1s, a similarity was consistently observed (Fig. 5c and d). These results indicate that after the impregnation procedure, the Cu sites in both the samples obtained the same chemical state and support environment, so the large discrepancy in the catalytic activity should have originated from the different surface areas.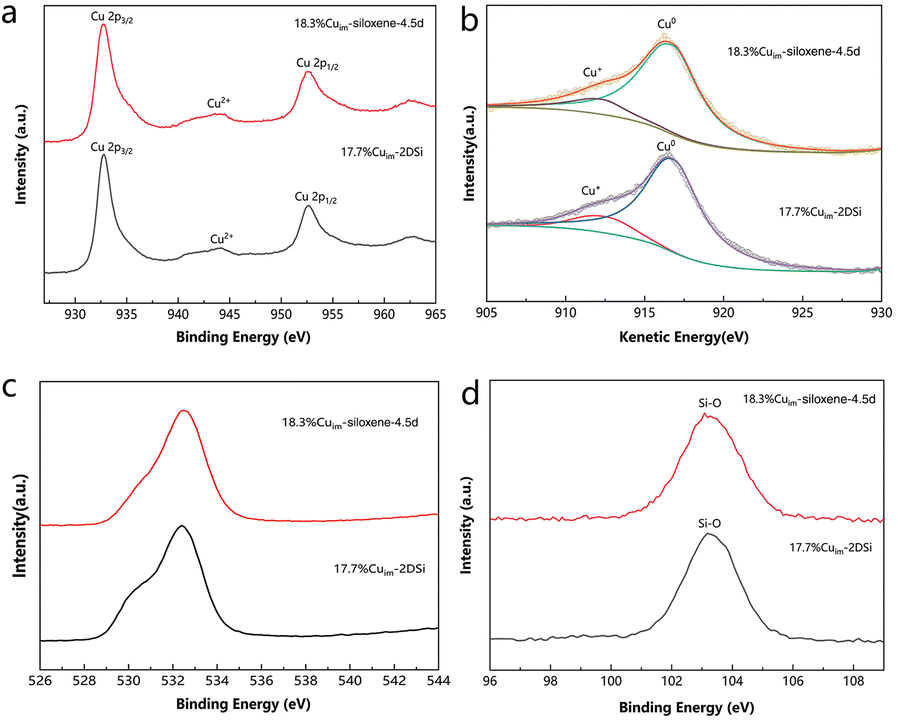 | ||
| Fig. 5 (a) Cu 2p, (c) O1s and (d) Si 2p XPS spectra of different Cu catalysts. (b) Cu LMM XAES spectra of different Cu catalysts. | ||
A similar conclusion can be drawn based on the XPS results obtained for the catalysts that contained Pd (Fig. S16†). Despite the Si 2p and O 1s peaks for 3.8% PdH-siloxene-4.5d shifting slightly to a higher binding energy compared with the 2DSi-based catalysts, due to the higher extent of the oxidation of siloxene induced by the preparation temperature (at 30 °C rather than at −15 °C), the Pd 3d patterns of all the three samples appeared the same: two peaks of 335.1 eV and 340.5 eV were observed and assigned to the Pd 3d5/2 and Pd 3d3/2 states, respectively.33,34 These results indicated that the active site Pd was successfully reduced by Si–H to the zerovalent metallic state in both kinds of the samples. Despite the oxidation of the siloxene support, its high surface area decisively enhanced the electrochemical activity compared with the catalysts based on 2DSi.
Experimental
Materials and chemicals
All the chemicals were used as received without further purification. Calcium silicide (CaSi2), sodium tetrachloropalladate (Na2PdCl4) and copper nitrate trihydrate (Cu(NO3)2·3H2O, 99.99%) were purchased from Sigma-Aldrich, Darui, and Macklin, respectively. A silicon oxide catalyst support with a high surface area (0.125 inch pellets, 250 m2 g−1) was purchased from Alfa Aesar. Sodium hydroxide (NaOH, AR), ethanol (GR, ≥99.8%), sulfuric acid (H2SO4) (AR, 98%) and hydrochloride acid (HCl) (36–38%, analytical reagent) were purchased from Sinopharm Chemical Reagent Co., Ltd. Milli-Q water (Millipore, 18.2 MΩ cm at 25 °C) was used in all experiments.Synthesis of siloxene materials
A typical synthesis of siloxene was started from the etching of CaSi2:NaOH dissolved in water to form a solution with a concentration of 5 M. A mixture of 2.0 g of CaSi2 and 200 ml of NaOH solution was then stirred at room temperature for 14 hours. After the reaction, the solid of the mixture, named CaSi2–NaOH, was collected by centrifugation, washed with water, and dried under vacuum sequentially. 1.0 g of CaSi2–NaOH was added into 100 ml of pre-diluted hydrochloride acid (1 M) in a round-bottom flask. The mixture was stirred at 30 °C for n (n varied from 0 to 7.5) days in N2. Then, the solvent was removed after centrifugation. The solid remaining was dried under vacuum after being washed with ethanol twice.
Siloxene (no NaOH) was prepared by a similar process over 4.5 days (n = 4.5) except for the substitution of CaSi2 for CaSi2–NaOH as the original precursor.
The preparation of C-siloxene was similar to that of siloxene (no NaOH) except for the substitution of 1 M hydrochloride acid with concentrated hydrochloride acid in the exfoliation procedure. The reaction lasted for 7 days.
Synthesis of 2DSi
In a typical synthesis of 2DSi, 1.0 g of CaSi2 was dispersed in 100 ml of concentrated hydrochloride acid, and it was precooled to −15 °C. The mixture was stirred at −15 °C under a nitrogen atmosphere for 7 days. A solid product was collected by centrifugation at room temperature, washed with ethanol, and dried under vacuum sequentially.Synthesis of Cuim-siloxene-4.5d, Cuim-2DSi, Cuim-siloxene (no NaOH) and Cuim-SiO2
380.2 mg of Cu(NO3)2 was added into 20 ml of ethanol under stirring. After complete dissolution, 300 mg of siloxene-4.5d was added to the solution followed by increasing the temperature to 80 °C. The solvent was evaporated at this temperature under stirring. The powder remaining was then calcined at 500 °C in air for two hours and reduced in H2 at 500 °C for another two hours. The product was denoted as Cuim-siloxene-4.5d.The production processes of Cuim-2DSi, Cuim-siloxene (no NaOH) and Cuim-SiO2 were identical to that of Cuim-siloxene-4.5d except for the substitution of 2DSi, siloxene (no NaOH) and SiO2 for siloxene-4.5d as the catalyst support, respectively.
Synthesis of PdH-siloxene-4.5d and PdH-2DSi
50 mg of siloxene-4.5d was added to the solution formed by dissolving 7.3 mg of Na2PdCl4 in 5 ml of water. Afterwards, a 3 min sonication was performed. The solid within the mixture was collected and washed with ethanol before drying under vacuum. The product obtained was denoted as 3.8%PdH-siloxene-4.5d.The production process of 2.4%PdH-2DSi was identical to that of 3.8%PdH-siloxene-4.5d except for the substitution of 2DSi for siloxene-4.5d as the catalyst support. 3.6%PdH-2DSi was prepared via the same method as that of 2.4%PdH-2DSi but the feed amount of Na2PdCl4 was doubled.
Thermocatalytic tests
Flow reactor studies for CO2 hydrogenation under atmospheric pressure were carried out in a quartz tube reactor. 30 mg of the catalyst (<80 mesh) was used for each test. Unless stated, the flow rates of CO2, H2, and N2 were set at ∼5, 20, and 5 mL min−1, respectively. The products at the reactor outlet were detected using an online gas chromatograph (Agilent 8860) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID).Photothermal catalytic tests
Photothermal tests were conducted similarly to thermocatalytic tests, except that a 300-W Xe arc lamp (PF300-T8E, BEIJING CHINA EDUCATION AU661 LIGHT CO., Ltd) rather than a heating modulate was used as the energy source. The mass of the catalyst in each test was 15 mg. The flow rates of CO2, H2, and N2 were set at ∼2.5, 10, and 2.5 mL min−1, respectively. The light intensity was measured using an optical power meter (PL-MW2000, Beijing Perfectlight Technology Co., Ltd).Electrochemical measurements
To prepare the catalyst ink, 10 mg of the sample was dispersed in a solution of DI water (4 ml) mixed with isopropanol (1 ml) and 5 wt% Nafion 117 solution (25 μl). An electrochemical workstation with a standard 3 electrode system was employed to further perform the electrochemical measurements. The ink (10 μl) was dropped on a glassy carbon electrode used as the working electrode, and Ag/AgCl and a graphite rod were used as the reference electrode and counter electrode, respectively. All of the measurements were carried out in Ar saturated 0.5 M H2SO4 at room temperature. Linear sweep voltammetry (LSV) was conducted within the range of −0.4–0.1 V (vs. RHE) at a scanning rate of 10 mV s−1. Chronoamperometry tests were carried out at the potential corresponding to an initial current density of 10 mA cm−2.Characterization
TEM and EDS mappings were performed on an FEI Talos F200X (200 kV) transmission electron microscope. SEM images were recorded using a HITACHI S4800 scanning electron microscope. A TriStar II 3020 specific surface area and pore structure analyzer was used for Brunauer–Emmett–Teller (BET) measurements. Raman spectroscopy was performed using a Bruker SENTERRA II Raman microscope. Fourier-transform infrared (FTIR) spectroscopy was performed using a Bruker Alpha FTIR spectrometer fitted with a universal attenuated total reflectance sampling accessory. X-ray photoelectron spectroscopy (XPS) and X-ray induced Auger electron spectroscopy (XAES) were performed using a Thermo Scientific K-Alpha scanning ESCA microprobe instrument (physical Electronics) equipped with an Al Kα X-ray source (hν = 1486.6 eV) and binding energies were referenced to the C 1s peak (284.8 eV). The metal contents of different samples were measured using an inductively coupled plasma optical emission spectrometer (ICP-OES) (i CAP Pro X, Thermo Fisher). X-ray diffraction (XRD) patterns were obtained using an XRD-700 X-ray diffractometer from Shimadzu. Raman spectra were obtained using a Raman spectrometer (Horiba LabRAM HR Evolution) equipped with a 532 nm laser excitation source.Conclusions
In the quest to improve the catalytic activity of 2D silicon based materials, we have proposed a novel synthesis strategy to obtain two-dimensional siloxene with a relatively high specific surface area. By applying NaOH treatment, and altering the time, temperature, and HCl concentration of the exfoliation procedure, we successfully increased the specific surface area from 16.97 m2 g−1 to 217.82 m2 g−1. Taking advantage of this high specific surface area, the catalysts with such siloxene as a support exhibited pronouncedly enhanced catalytic activity than the catalysts supported by conventional 2DSi. Inspiringly, such an improvement was seen in both (photo)thermal CO2 hydrogenation in the gas phase and electrochemical hydrogen evolution in the liquid phase. The room-temperature exfoliation, lower concentration of HCl, and compatibility with straightforward hybridization with catalytically active metals have endowed this strategy with potential for industrialization. This work takes a momentous step forward in facilitating the catalytic applications of two-dimensional silicon based materials, and may inspire more research with focus on the optimization of the synthesis of 2D materials for energy and environmental applications.Author contributions
Y. S., S. W., and L. J. contributed to this work equally as co-first authors. W. S., Y. S., S. W., and L. J. conceived and designed the experiments. Y. S. performed all the sample preparations and characterization studies. S. W. tested and analyzed the thermocatalytic and photothermal catalytic performances of the Cu-containing catalysts. L. J. tested and analyzed the electrocatalytic performances of the Pd-containing catalysts. C. Z. performed the TEM and diffuse reflection characterization studies. H. C. provided suggestions on the synthesis strategy and writing. H. Z. provided the electrocatalytic facilities and suggestions on the electrocatalytic experiments. Y. S. and W. S. wrote the paper. All authors commented on the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the National Key R&D Program of China (2021YFF0502000), the National Natural Science Foundation of China (51902287 and 61721005), the U of T-ZJU Joint Seed Fund, and the Fundamental Research Funds for the Central Universities (226-2022-00159 and 226-2022-00200). The authors are also grateful for the support from the State Key Laboratory of New Textile Materials and Advanced Processing Technologies (FZ2020020).References
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS.
- L. Tang, X. Meng, D. Deng and X. Bao, Adv. Mater., 2019, 31, 1901996 CrossRef CAS.
- L. X. Chen, Z. W. Chen, M. Jiang, Z. Lu, C. Gao, G. Cai and C. V. Singh, J. Mater. Chem. A, 2021, 9, 2018–2042 RSC.
- X. Chia and M. Pumera, Nat. Catal., 2018, 1, 909–921 CrossRef CAS.
- H. Jin, C. Guo, X. Liu, J. Liu, A. Vasileff, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem. Rev., 2018, 118, 6337–6408 CrossRef CAS.
- S. Wang, C. Wang, W. Pan, W. Sun and D. Yang, Sol. RRL, 2021, 5, 2000392 CrossRef CAS.
- C. Qian, W. Sun, D. L. H. Hung, C. Qiu, M. Makaremi, S. G. Hari Kumar, L. Wan, M. Ghoussoub, T. E. Wood, M. Xia, A. A. Tountas, Y. F. Li, L. Wang, Y. Dong, I. Gourevich, C. V. Singh and G. A. Ozin, Nat. Catal., 2019, 2, 46–54 CrossRef CAS.
- M. Ohashi, R. Yaokawa, Y. Takatani and H. Nakano, ChemNanoMat, 2017, 3, 534–537 CrossRef CAS.
- Q. Dai, Q. Meng, C. Du, F. Ding, J. Huang, J. Nie, X. Zhang and J. Chen, Chem. Commun., 2020, 56, 4824–4827 RSC.
- X. Yan, W. Sun, L. Fan, P. N. Duchesne, W. Wang, C. Kübel, D. Wang, S. G. H. Kumar, Y. F. Li, A. Tavasoli, T. E. Wood, D. L. H. Hung, L. Wan, L. Wang, R. Song, J. Guo, I. Gourevich, A. A. Jelle, J. Lu, R. Li, B. D. Hatton and G. A. Ozin, Nat. Commun., 2019, 10, 2608 CrossRef.
- S. Wang, K. Feng, D. Zhang, D. Yang, M. Xiao, C. Zhang, L. He, B. Yan, G. A. Ozin and W. Sun, Adv. Sci., 2022, 9, 2104972 CrossRef CAS.
- Z.-Q. Wang, Z.-N. Xu, S.-Y. Peng, M.-J. Zhang, G. Lu, Q.-S. Chen, Y. Chen and G.-C. Guo, ACS Catal., 2015, 5, 4255–4259 CrossRef CAS.
- W. Sun, X. Yan, C. Qian, P. N. Duchesne, S. G. Hari Kumar and G. A. Ozin, Faraday Discuss., 2020, 222, 424–432 RSC.
- M. Li, R. Ramachandran, T. Sakthivel, F. Wang and Z.-X. Xu, Chem. Eng. J., 2021, 421, 129728 CrossRef CAS.
- H. Imagawa, X. Wu, H. Itahara, S. Yin, K. Kojima, S. F. Chichibu and T. Sato, Dalton Trans., 2018, 47, 7070–7076 RSC.
- K. Krishnamoorthy, M. S. P. Sudhakaran, P. Pazhamalai, V. K. Mariappan, Y. S. Mok and S.-J. Kim, J. Mater. Chem. A, 2019, 7, 18950–18958 RSC.
- H. Nakano, M. Ishii and H. Nakamura, Chem. Commun., 2005, 23, 2945–2947 RSC.
- D. Karar, N. R. Bandyopadhyay, A. K. Pramanick, D. Acharyya, G. Conibeer, N. Banerjee, O. E. Kusmartseva and M. Ray, J. Phys. Chem. C, 2018, 122, 18912–18921 CrossRef CAS.
- B. J. Ryan, M. P. Hanrahan, Y. Wang, U. Ramesh, C. K. A. Nyamekye, R. D. Nelson, Z. Liu, C. Huang, B. Whitehead, J. Wang, L. T. Roling, E. A. Smith, A. J. Rossini and M. G. Panthani, Chem. Mater., 2020, 32, 795–804 CrossRef CAS.
- R. Fu, K. Zhang, R. P. Zaccaria, H. Huang, Y. Xia and Z. Liu, Nano Energy, 2017, 39, 546–553 CrossRef CAS.
- X. Xu, L. Zhou, D. Ding, Y. Wang, J. Huang, H. He and Z. Ye, ACS Appl. Nano Mater., 2020, 3, 538–546 CrossRef CAS.
- P. Pazhamalai, K. Krishnamoorthy, S. Sahoo, V. K. Mariappan and S.-J. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 624–633 CrossRef CAS.
- K. Krishnamoorthy, P. Pazhamalai and S.-J. Kim, Energy Environ. Sci., 2018, 11, 1595–1602 RSC.
- S. Mondal, T. K. Mondal, Y.-K. Su and S. K. Saha, J. Colloid Interface Sci., 2020, 562, 453–460 CrossRef CAS.
- R. Gonzalez-Rodriguez, R. M. del Castillo, E. Hathaway, Y. Lin, J. L. Coffer and J. Cui, ACS Appl. Nano Mater., 2022, 5, 4325–4335 CrossRef CAS.
- S. Yamanaka, H. Matsu-ura and M. Ishikawa, Mater. Res. Bull., 1996, 31, 307–316 CrossRef CAS.
- H. D. Fuchs, M. Stutzmann, M. S. Brandt, M. Rosenbauer, J. Weber, A. Breitschwerdt, P. Deák and M. Cardona, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 8172–8189 CrossRef CAS PubMed.
- S. Wang, A. A. Tountas, W. Pan, J. Zhao, L. He, W. Sun, D. Yang and G. A. Ozin, Small, 2021, 17, 2007025 CrossRef CAS PubMed.
- A. R. Poerwoprajitno, L. Gloag, S. Cheong, J. J. Gooding and R. D. Tilley, Nanoscale, 2019, 11, 18995–19011 RSC.
- S. Sarkar and S. C. Peter, Inorg. Chem. Front., 2018, 5, 2060–2080 RSC.
- C. Zhang, X. Liang, R. Xu, C. Dai, B. Wu, G. Yu, B. Chen, X. Wang and N. Liu, Adv. Funct. Mater., 2021, 31, 2008298 CrossRef CAS.
- D. C. Nguyen, T. L. Luyen Doan, S. Prabhakaran, D. T. Tran, D. H. Kim, J. H. Lee and N. H. Kim, Nano Energy, 2021, 82, 105750 CrossRef CAS.
- P. Xu, W. Lu, J. Zhang and L. Zhang, ACS Sustainable Chem. Eng., 2020, 8, 12366–12377 CrossRef CAS.
- C. Wang, Y. Li, C. Zhang, X. Chen, C. Liu, W. Weng, W. Shan and H. He, Appl. Catal., B, 2021, 282, 119540 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05140k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |