
High surface area siloxene for photothermal and electrochemical catalysis†

Abstract
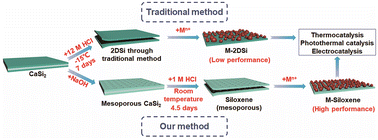
Catalysis based on two-dimensional silicon has been under intense investigation recently. However, its substandard catalytic activity is far from industrialization. In this work, we demonstrate a new solution to this problem formulated on the batch synthesis of siloxene with an enhanced specific surface area (217.8 m2 g−1). A two-dimensional porous structure was prepared, enabling great support and dispersion of metal nanoparticles. Catalytic evaluations of such hybrid structures for the (photo)thermal CO2 hydrogenation reaction and the electrochemical hydrogen evolution reaction revealed a significant performance advantage over the benchmark two-dimensional silicon structures synthesized via the conventional method. This work may confer notable viability on two-dimensional silicon for advanced energy, catalytic, and environmental applications.
- This article is part of the themed collections: 2024 Lunar New Year Collection and CO2 capture and conversion
 Please wait while we load your content...
Please wait while we load your content...

