
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Communication of molecular fluorophores with other photoluminescence centres in carbon dots†
Michal
Langer
 ab,
Lukáš
Zdražil
a,
Miroslav
Medveď
*ac and
Michal
Otyepka
*ad
ab,
Lukáš
Zdražil
a,
Miroslav
Medveď
*ac and
Michal
Otyepka
*ad
aRegional Centre of Advanced Technologies and Materials, Czech Advanced Technology and Research Institute (CATRIN), Palacký University Olomouc, Šlechtitelů 241/27, 783 71 Olomouc, Czech Republic. E-mail: miroslav.medved@upol.cz; michal.otyepka@upol.cz
bChemical and Biological Systems Simulation Lab, Centre of New Technologies University of Warsaw, 2c Banacha Street, 02-097, Warszawa, Poland
cDepartment of Chemistry, Faculty of Natural Sciences, Matej Bel University, Tajovského 40, 974 01 Banská Bystrica, Slovak Republic
dIT4Innovations, VŠB-Technical University of Ostrava, 17. listopadu 2172/15, 708 00 Ostrava-Poruba, Czech Republic
First published on 31st January 2023
Abstract
The establishment of structure-photoluminescence (PL) relationships remains an ultimate challenge in the field of carbon dots (CDs). It is now commonly understood that various structural domains may evolve during the preparation of CDs; nonetheless, we are still far from capturing the specific features that determine the overall PL of CDs. Although the core, surface and molecular states are usually considered the three main sources of PL, it is not known to which extent they interact and/or affect one another. Expectedly, the communication between the different PL centres depends on the mutual arrangement and the type of linking. To gain insights into such a communication, time-dependent density functional theory (TD-DFT) calculations were performed for several (N-doped/O-functionalized) polyaromatic hydrocarbons (PAHs) as representative models for the core/surfaces PL states and the prototypical molecular fluorophore (MF) 5-oxo-1,2,3,5-tetrahydroimidazo-[1,2-α]-pyridine-7-carboxylic acid (IPCA), considering different interaction modes, namely hydrogen bonded and stacked complexes as well as covalently bonded and fused structures. Our results revealed that each of the studied arrangements in some way supported the communication between the PL centres. The deactivation pathways typically involve multiple charge and energy transfer events that can promote the formation of charge separated states and/or lead to the activation of other PL centres in CDs. Depending on the arrangement, the doping pattern and surface functionalization, both the CD core and the MF can act as an electron donor or acceptor, which could help to design CDs with desirable hole–electron surface/core characteristics.
Introduction
Carbon dots (CDs) represent a highly attractive class of zero-dimensional carbon nanoallotropes. Since their discovery by Xu et al.,1 the CDs have been praised as low-cost, biocompatible, chemically stable, water soluble nanosystems with outstanding optical and electrical properties, such as intense photoluminescence (PL), high resistance to photobleaching, high electron mobility, and photo-induced electron transfer.2,3 These properties endow CDs with applications ranging from photocatalysis, sensing, and imaging up to light-emitting diodes.4–9Generally, CDs are defined as quasi-spherical objects with a diameter below 10 nm possessing a multilayer graphite core and oxygen and/or nitrogen functional groups on the CD shell.10 Although CDs are often categorized into three types, i.e., graphene quantum dots (GQDs), carbon nanodots (CNDs), and carbonized polymeric dots (CPDs), their structural complexity is vast. This stems from miscellaneous synthetic procedures, as small changes in reaction conditions or precursors can lead to different CDs.11–14 To achieve better control over the properties of CDs for specific applications, the relationships between the structure and PL mechanisms of CDs need to be fully understood.
CDs exhibit multi-centre emission with three sources being used to explain the PL origins of CDs, i.e., core states due to carbon sp2 domains, surface states stemming from surface chemical groups, and molecular states due to the presence of molecular fluorophores (MFs).15–18 Various structural domains within CDs may contribute to the PL of CDs, and they can even co-operate and/or influence each other.19 Thus, many processes such as the charge transfer (CT), energy transfer (ET), radiationless de-excitations, (reverse) inter-system crossing (ISC), and involvement of trap states may be expected to occur upon photoexcitation of CDs (Fig. S5†). Nevertheless, despite all the efforts and acquired data, the clear link between a particular structural feature and the observed PL signal has not been established yet.
Computational chemistry tools are very helpful in elucidating relationships between the structure and PL properties of molecular systems. The theoretical studies focusing on the interplay between various PL centres in the context of CDs are, however, very limited.20,21 Concerning the molecular states, 5-oxo-1,2,3,5-tetrahydroimidazo-[1,2-α]-pyridine-7-carboxylic acid (IPCA) has been identified as a prototypical MF in CDs.22 The optical properties of IPCA monomer and dimer have been theoretically studied in gas and aqueous solvent,23 and also as a non-covalently bonded dimer in the model of CD by QM/MM.24 Nonetheless, neither experiments nor modelling really provided any direct insight into the communication of the molecular states stemming from the presence of IPCA with other structural motifs in CDs. Not fully resolved structural organization of MFs within CDs contributes to this conundrum. For example, citric-acid based fluorophore moieties have been reported as a free-floating by-product of CDs preparation,25 but also as being present in the CD interior26 as well as CD exterior,27 or even covalently bonded to the CD surface,28 and these possible scenarios very likely depend on synthetic conditions.29
Here, we describe the PL properties of a series of core/surface/MF models involving a prototypical MF IPCA in various chemical and structural context within CDs. In particular, our models cover all plausible types of structural arrangements (H-bonded, stacked, covalently bonded, and fused systems) of IPCA and a pyrene-like polyaromatic hydrocarbon (PAH) molecule representing the small-sized core of CDs. Our results show that the interplay between these PL centres may not only introduce new peaks in the absorption spectra of the complex (e.g., in covalently bonded systems), but many new dark states emerge, which can come to play during de-excitation processes. Also, depending on the structural organization of IPCA and PAH units, plausible CT and ET events between the molecular and core states along the internal inversion de-excitation cascade were identified, which can lead to the emission from a PL centre (e.g., core) different from the one photo-activated during absorption (e.g., IPCA).
Experimental
Models
Our studied structural domains can be sorted into four groups (Fig. 1), namely H-bonded and stacked complexes, covalently single-bonded systems, and fused systems. All model systems were constructed from one prototypical MF, IPCA, and one PAH molecule, which represents a CD core (Fig. 1, Fig. S1†). These models cover the main structural types, which can be present and co-operate in the overall PL of the CDs. Let us note that the fused structures can also be considered as models giving rise to the surface PL states. | ||
| Fig. 1 The schematic figure on how CDs (two models in the middle), can be formed from PAHs and different interaction systems of PAH/IPCA that were used as models for our calculations (corresponding chemical structures are shown in Fig. S1†). Colouring scheme: blue – nitrogen; red – oxygen; white – hydrogen; grey, green, orange, beige, cyan, violet, yellow – carbon. Different colours of carbons were chosen to clearly show their location in the models of CDs. | ||
Besides a parent pyrene molecule (model A; see Fig. S1a†), CD core models containing two inner-lattice graphitic nitrogens (graphitic-N-core models B, D, and F) and two graphitic-edge nitrogens (graphitic-N-edge models C and G) were considered (Fig. S1a†). As CDs usually possess oxygen-containing functional groups on their surface and/or edges, some PAHs were functionalized with either two oxo-groups (models D and E) or one carboxylic group (models F and G). In the model E, the graphitic-edge doping was replaced by two graphitic-core nitrogens in the same ring (graphitic-N-core2), as we only considered Kekulé structures. To assess the core-size effects, covalently bonded as well as stacked complexes containing an N-doped coronene moiety were also considered (Fig. S15†).
Methods
To examine the effects of various binding modes on the nature of electronic transitions in CD/MF systems, the lowest excited states of coupled systems were thoroughly analysed in terms of the vertical (de-)excitation energies and the corresponding oscillator strengths as well as in terms of electron density difference (EDD) plots and natural transition orbitals (NTOs).The density functional theory (DFT) and time-dependent density functional theory (TD-DFT)30 calculations were performed by applying the Coulomb-attenuating three-parameter Becke, Lee–Yang–Parr (CAM-B3LYP)31,32 functional including the D3 correction to account for dispersion interactions33 in combination with the def2-TZVP34 basis set. All calculations were performed for hydrated systems, employing the implicit universal solvation model based on solute electron density (SMD),35 and using the Gaussian 16 (revision B.01).36 The structures were rendered in PyMOL37 and Chemcraft.38
To simulate absorption spectra, the ground state (GS) geometry of all the models (Fig. S1†) was first optimized without any symmetry constraints. All the optimized geometries were verified to be true minima on the potential energy surface by the absence of imaginary frequencies in the harmonic vibrational analysis. The electronic vertical excitation energies (VEEs) were calculated within the TD-DFT framework using the linear response (LR) as well as corrected linear response (cLR) approaches,39–43 with the first thirty singlet states taken into consideration in the former. Furthermore, LR-TD-DFT calculations of ten lowest singlets and triplet excitations were performed, on the GS geometries, to judge on the feasibility of ISC in model structures. It should be noted that the evaluation of adiabatic singlet–triplet energy gaps (ΔEST) was not achievable for all systems due to convergence problems (using the applied method) during geometry optimizations of triplet excited state structures.
For the calculation of radiative de-excitations, Kasha emissions from the S1 state were considered. The S1 geometry was first optimized with the def2-SVP basis set, followed by calculation of emission energies with the def2-TZVP basis set applying the equilibrium solvation regime for the ES calculations and nonequilibrium solvation for the subsequent calculation of the GS.
CT and ET analyses were performed based on the NTOs that were rendered in Chemcraft (version 1.8) and electron density differences (EDDs) displayed in GaussView (version 5.0).44 The calculations of total coupling of electronic energy transfer (EET) were performed using TD-DFT as implemented in Gaussian 16, where solvent effects were introduced using the SMD cavity for the whole system;45,46 ES calculations were performed on each fragment, and all the couplings among all the resulting states were computed; and ONIOM-like47 link-atom input information to cap the fragments was used for our models bonded through a single covalent bond in EET calculations.
Results and discussion
First, we describe the changes in absorption and emission of CDs due to the formation of the PAH/IPCA complexes (Fig. 1). In the second part, we demonstrate how a particular bonding type and/or specific structural arrangement of IPCA and PAH can affect the de-excitation processes assuming the photo-activation of the system by a 350 nm source of light, i.e., in the region of the absorption maximum of IPCA. The analysis of the optical properties of isolated molecules from which we constructed our models to establish structure-PL relationships in CDs and trace the communication between the core/surface and molecular states is detailed in the ESI.†Changes in absorption spectra
In the H-bonded systems, the interaction of both IPCA and PAH units with light remains practically independent to each other. This means that the formation of H-bonded complexes does not give rise to any significant new peaks in the CDs absorption spectra (Fig. 2a and b), even though the frontier orbital analysis (Fig. S1b and S7†) suggests a possible CT excitation from the PAH unit to IPCA. Indeed, a CT π–π* transition (state S4, 401 nm) from PAH to IPCA with a rather small intensity (f = 0.026) due to a small overlap of the involved MOs was observed in our COOH_core model (Tables S11 and S12†). A similar, except being dark, CT state (S3, λmax = 371 nm, f = 0.002) was observed for the COOH_edge model.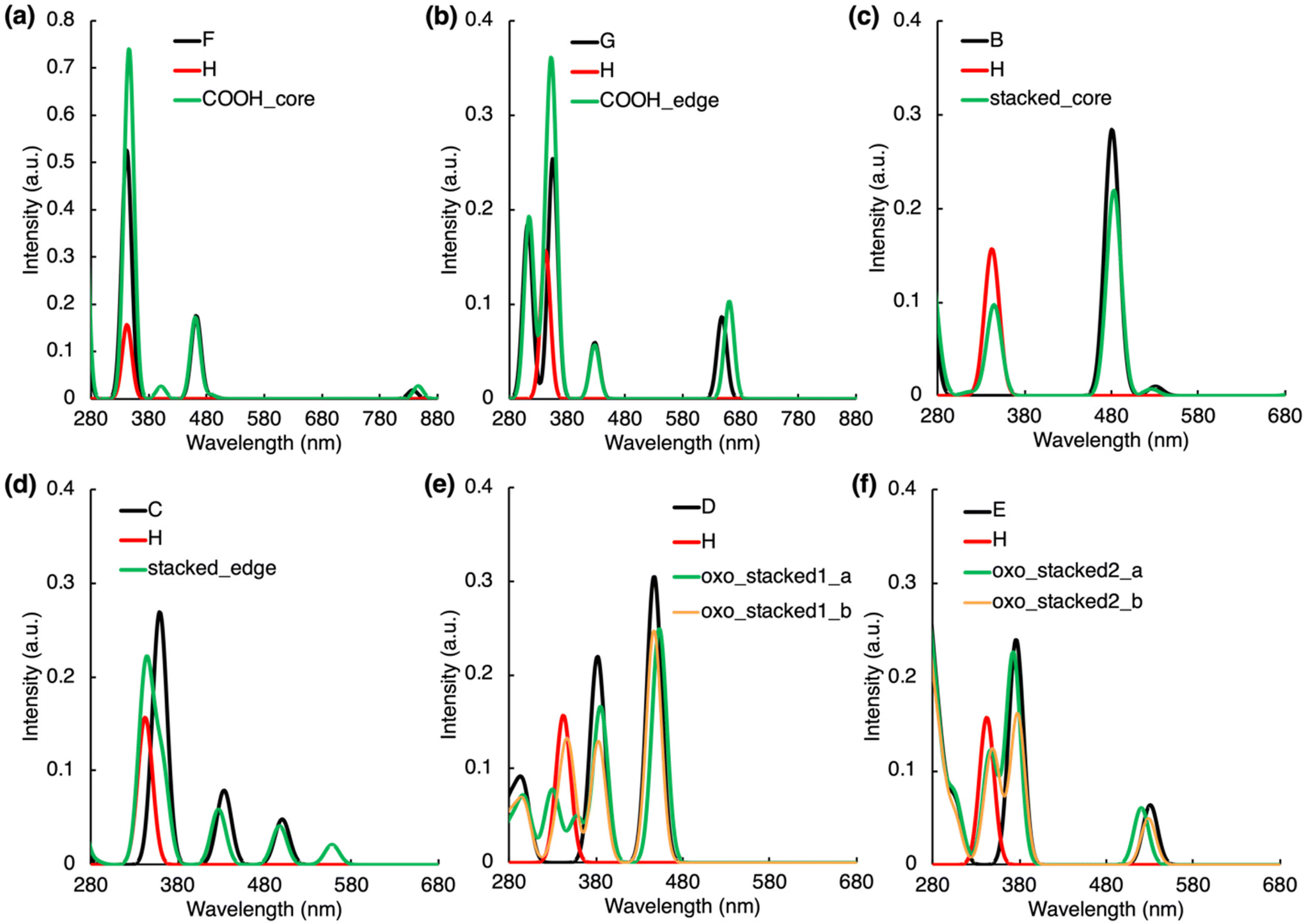 | ||
| Fig. 2 Absorption spectra of non-covalently bonded complexes along with those of the separated molecules which form the complexes (see molecules A–H in Fig. S1†). For each spectrum, the line spectra (considering 30 lowest singlet states) were convoluted by a Gaussian function assuming the inhomogeneous broadening of peaks with σ = 20 nm. | ||
The stacking arrangement of PAHs and IPCA opens new absorption channels in CDs. Notably higher energy of the highest occupied molecular orbital (HOMO) in N-doped PAH units and slightly lower energy of the lowest unoccupied molecular orbital (LUMO) in IPCA (Fig. S1†) indicate the plausibility of low-lying CT excitations from N-doped pyrene to IPCA, as demonstrated in stacked_core and stacked_edge models (Fig. 2c, d, S8; Tables S13 and S14†). However, in the model with a graphitic-N-edge unit (stacked_edge model), the S0 → S1 electronic excitation (λmax = 558 nm) exhibits much stronger CT character (D index 3.48 Å) from PAH to IPCA (Table S14†) compared to stacked_core complex (D index 1.33 Å). Owing to its non-zero oscillator strength (f = 0.021) a new small peak in the visible region can be observed (Fig. 2d). Nevertheless, the intense absorption bands of stacked_core and stacked_edge (above 300 nm) are due to LEs within the molecular components (Tables S13 and S14†), which is in line with a good overlap of the sum spectra with those of separate molecules albeit the intensity of peaks slightly decreases upon the complex formation (Fig. 2c and d).
Introducing O-functional groups in the stacked models (oxo_stacked1_a, oxo_stacked1_b) preserves the shape of the sum absorption spectra of components only above 380 nm, i.e., the main absorption bands are predominantly due to LE transitions in this region (Fig. 2e). The S0 → S1 transition is a bright LE in O-PAH (f = 0.250) peaking at around λmax ≈ 453 nm. Below 380 nm, new absorption peaks, relevant for photoexcitation using a ∼350 nm irradiation source, emerge upon the stacking. In oxo_stacked1_a, these peaks appear due to the hybridized local and charge-transfer (HLCT) S0 → S3 transition (λmax ≈ 360 nm, f = 0.049), where an electron is transferred from π-orbital delocalized over both components to a π* orbital localized only on IPCA (Table S16†), and S0 → S4,5 (λmax ≈ 330 nm, f = 0.04) transitions (Fig. 2e, Table S16†). In oxo_stacked1_b, a new absorption peak corresponding to S0 → S3 CT excitation (PAH → IPCA) arose at ∼350 nm (f = 0.054, Fig. 2e, Table S17†), overlapping with the LE peak of the IPCA molecule. Another albeit less bright CT (IPCA → PAH) excitation (λmax ≈ 335 nm, f = 0.027) can be detected in the region of spectrum, where typical excitation wavelengths are targeted for photoexcitation of CDs.
The change of the N-doping pattern in the O-functionalized stacked complexes can cause significant structural distortions (Tables S18 and S19, Fig. S17†). Whereas oxo_stacked2_b keeps a compact stacked arrangement, oxo_stacked2_a corresponds to a partially open stacked configuration stabilized by the formation of H-bonding (IPCA–N⋯O–PAH). Consequently, the absorption spectrum of oxo_stacked2_a copies to a large extent those of the isolated molecules. On the other hand, the compact arrangement of oxo_stacked2_b gives rise to slight modifications of the band shapes in the 350–400 nm region (Fig. 2f) resulting mainly from the S0 → S3 (λmax ≈ 352 nm, f = 0.075) and S0 → S5 (λmax ≈ 343 nm, f = 0.041) transitions exhibiting HLCT character, where the electron is transferred from π-orbital of IPCA to π*-orbital delocalized over the complex (Table S19†).
Absorption spectra of models when the core/molecular PL centres CDs are connected with an ester bond (referred as ester1_core and ester2_edge models) greatly overlap with the absorption spectra of the isolated molecular components (Fig. 3a and b). These structural motifs are also vital for understanding the CDs PL as they may occur during the synthesis of CDs before the carbonization process is completed. No bright excitations with CT character were observed in these models, which we attribute to the twisted mutual orientation of both units (Fig. S11a–h, Tables S20 and S21†) and to the character of the ester bond, which does not enable efficient electron delocalization.
 | ||
| Fig. 3 Absorption spectra of covalently bonded complexes along with those of the separated molecules which form the complexes (see molecules A–H in Fig. S1†). For each spectrum, the line spectra (considering 30 lowest singlet states) were convoluted by a Gaussian function assuming the inhomogeneous broadening of peaks with σ = 20 nm. | ||
On the other hand, an amide bond allows the whole complex being quasi-planar which can significantly affect the electronic energy levels of linked components. In the amide1_core model, the peak corresponding to LE on IPCA is blue-shifted by 28 nm compared to an isolated IPCA molecule, as the –COOH group of IPCA was replaced by –CONH– group (λmax ≈ 315 nm, f = 0.173, Fig. 3c). In amide1_edge, a small part of electron density is transferred from N-doped pyrene to a linking amide group and to the IPCA unit upon the S0 → S2 excitation (λmax ≈ 444 nm, f = 0.177), which indicates efficient interaction between the two PL centres. An additional small peak (f = 0.020) can be observed at 385 nm due to S0 → S3, which exhibits CT character from PAH to IPCA. Due to the replacement of –COOH by –CONH–, the LE on IPCA is also blue-shifted to 316 nm, giving rise to an additional peak in the absorption spectrum of amide1_edge model (Fig. 3c).
The position of an amide binding group covalently linking PL structural motifs inside CDs can also significantly affect the energy levels of the system. The observed changes are indirectly related to the dihedral angle between IPCA and pyrene units (Fig. S11†), as the quasi-planar orientation can lead to a more intense interaction of molecular orbitals of the components. Our results corroborate this hypothesis, as several CT excitations were identified in amide2 and amide3 positions for both N-doping patterns (Tables S22–S27†). Models with a graphitic-core unit, amide2_core and amide3_core, do not absorb light in the spectral region 330–400 nm, similarly to amide1_core model (Fig. 3d). The electron is transferred to the amide group and IPCA in S0 → S3 in amide2_core (Table S24†), causing a slight redshift of the absorption maximum. The change of the linking position also introduces the new peaks around 430 nm, which can be described as (weak) HLCT S0 → S4 excitation from PAH to IPCA. A striking difference due to different linking of –CONH– bond was noted for amide3_edge, in which the absorption peak for S0 → S1 HLCT transition from PAH to IPCA was blue-shifted to λmax ≈ 539 nm (Fig. 3e) and attained a notable oscillator strength (f = 0.122).
The formation of fused PAH/IPCA systems (fused, fused_core, fused_edge models) in CDs would produce many new peaks in their absorption spectra (Fig. 3f–h), which may be assigned to the extended delocalization effects and the presence of multiple bright CT excitations. Contrary to previously analysed systems, the frontier orbital analysis suggests that the fused systems should be considered more as core/shell systems because their HOMOs and LUMOs are delocalized over the entire model structures (Fig. S14†). In fused model, all ten lowest bright excitations have to some extent the π–π* character with the π-orbitals delocalized over the whole system (Table S28†), e.g., the S0 → S2 transition is a HLCT excitation, where the electron density predominantly transfers from the whole structure to a PAH part, however, it is less intense (f = 0.014). For the fused_core model, LE on IPCA is now the S0 → S4 transition with λmax ≈ 363 nm (Fig. 3g), i.e., red-shifted by 21 nm compared to IPCA, with an increased oscillator strength (f = 0.392) in comparison with an isolated IPCA molecule (Table S29†). There are several CT excitations from PAH to the whole system or mostly to IPCA unit for S0 → S3 (λmax ≈ 503 nm, f = 0.079). The S0 → S5 transition is a HLCT excitation from PAH to the whole system. The fact that it is the bright excitation lying in the visible part of electromagnetic spectrum (354 nm, f = 0.026) offers the interesting region to target the absorption in CDs. Concerning the fused_edge model, the S0 → S1 excitation (λmax ≈ 592 nm, f = 0.122) occurs on the whole model structure, dominantly on the PAH unit. Again, new excitations in comparison with IPCA and N-doped isolated molecules are present, which correspond to CT excitations delocalized over the whole system (Table S30†). These peaks positioned at 350, 558 and 511 nm make the fused_edge model plausible for the rationalization of typical photoexcitation wavelengths of CDs.
To address the impact of the core size on the absorption spectra of the studied systems, representative covalently bonded as well as stacked complexes containing an N-doped coronene moiety were investigated (Fig. S15†). In amide-linked models (amide1_cor and amide1_cor_edge), bright CT excitations from PAH to IPCA corresponding in both cases to S0 → S4 transition are centered around 460 nm (Fig. S16, Tables S31 and S32†), i.e., they are red-shifted (by ca. 14 and 72 nm, respectively) compared to pyrene-like analogs (Tables S22 and S23†). The S0 → S1 vertical excitation energy is even more red-shifted (by 171 nm and 415 nm) for amide1_cor and amide1_cor_edge, respectively, with respect to the smaller-core systems. For the stacked complex without O-functionalization (stacked_cor model), a brighter CT state was found for N-coronene/IPCA in comparison to the N-pyrene/IPCA counterpart, i.e., S0 → S7 transition (λmax ≈ 320 nm, f = 0.030) for N-coronene/IPCA vs. S0 → S6 transition (λmax ≈ 317 nm, f = 0.004) for N-pyrene/IPCA (Tables S13 and S33†). Again, the S0 → S1 maximum is red-shifted by 156 nm (λmax ≈ 938 nm) with respect to N-pyrene/IPCA. In stacked_oxo_cor model, the HLCT excitonic transition (S0 → S7; λmax ≈ 341 nm) has the oscillator strength with the value f = 0.044 (Table S34†), which is similar as in the pyrene counterpart (see oxo_stacked1_a, b). Similar CT states were found also in stacked_oxo2_cor model (S0 → S4; S0 → S6; S0 → S7; Table S35†) and in the stacked complex with different N-doping pattern (stacked_oxo_cor2; S0 → S4; S0 → S5; S0 → S7; Table S36†). Interestingly, the S0 → S1 excitation energy is red-shifted only by 38 nm (λmax ≈ 491 nm) for the N-coronene/IPCA stacked complex with oxo-functionalization (stacked_oxo_cor model) in comparison to the oxo_stacked1_a model (with pyrene-sized PAH). Despite a different N-doping pattern imprinted in the stacked_oxo_cor2 model, the absorption peak corresponding to S0 → S1 is not red-shifted outside the Vis region (λmax ≈ 518 nm, f = 0.051). In general, our results indicate that even in models with a larger core than pyrene, the overlap of molecular orbitals of the components remains efficient, and new CT states are present. Additionally, the absorption peaks are red-shifted for the larger core.
To sum up this part, in the 340–360 nm region, where the isolated IPCA and its dimers absorb light, other multiple absorption events can occur, thus photoactivating different PL centres. It was also shown that not only large sp2 carbon core domains but also the interaction between IPCA and PAH units in fused and amide-bonded systems are likely responsible for the absorption in the visible region. Moreover, it was demonstrated that the electronic excitations with CT character from IPCA to PAH only occurred in CD core models functionalized with oxo-groups due to the relative positions of the frontier orbitals isolated of IPCA and oxo-PAHs.
De-excitation pathways
The analysis of de-excitation pathways in the studied models of various interacting PAH/IPCA structural domains of CDs provides valuable insights into the PL deactivation processes occurring in real CD samples after irradiation. We chose to describe the sequential de-excitation cascades (Sn → S1) after the photoexcitation with a 350 nm energy source, which is the typical excitation wavelength presumably targeting the absorption maxima of MFs such as IPCA. Along the de-excitation pathways based on the NTO analysis, we identified CT/ET processes between the interacting CD components (Fig. S5†). Despite being dark, the CT and ET states can play an important role during de-excitation dynamic processes, leading to charge-separated excitons and/or activation of multiple PL centres in CDs. The here presented de-excitation cascades represent alternative relaxation channels to local de-activation of IPCA and its dimers leading to the emission from molecular states.23,24The formation of H-bonded complexes of IPCA and N-doped PAHs within CDs appears to promote radiationless channels and thus lowers the PL QY in CDs. The reason is that the de-excitation cascade brings the photoactivated H-bonded systems down to the S1 state which gets (after relaxation) too close to the GS (Fig. 4a and b). In particular, in the COOH_core model, the 350 nm excitation source causes population of S6 and S5 energy levels, which are LEs on IPCA and PAH, respectively. Sequential internal conversions of S5 to S3 involve the electron transfer from PAH to IPCA (S5 → S4) and reverse CT (S4 → S3) according to the NTO analysis (Table S11†). The EDD plot for S1* → S0* at the relaxed S1 geometry (inset in Fig. 4a) shows the localization of an exciton on PAH, and the proximity of S0 and S1 energy levels in this geometry suggests the non-radiative de-excitation. An analogous picture was drawn for the COOH_edge model, with one low-lying dark state missing in the deactivation cascade (Fig. 4b, Table S12†).
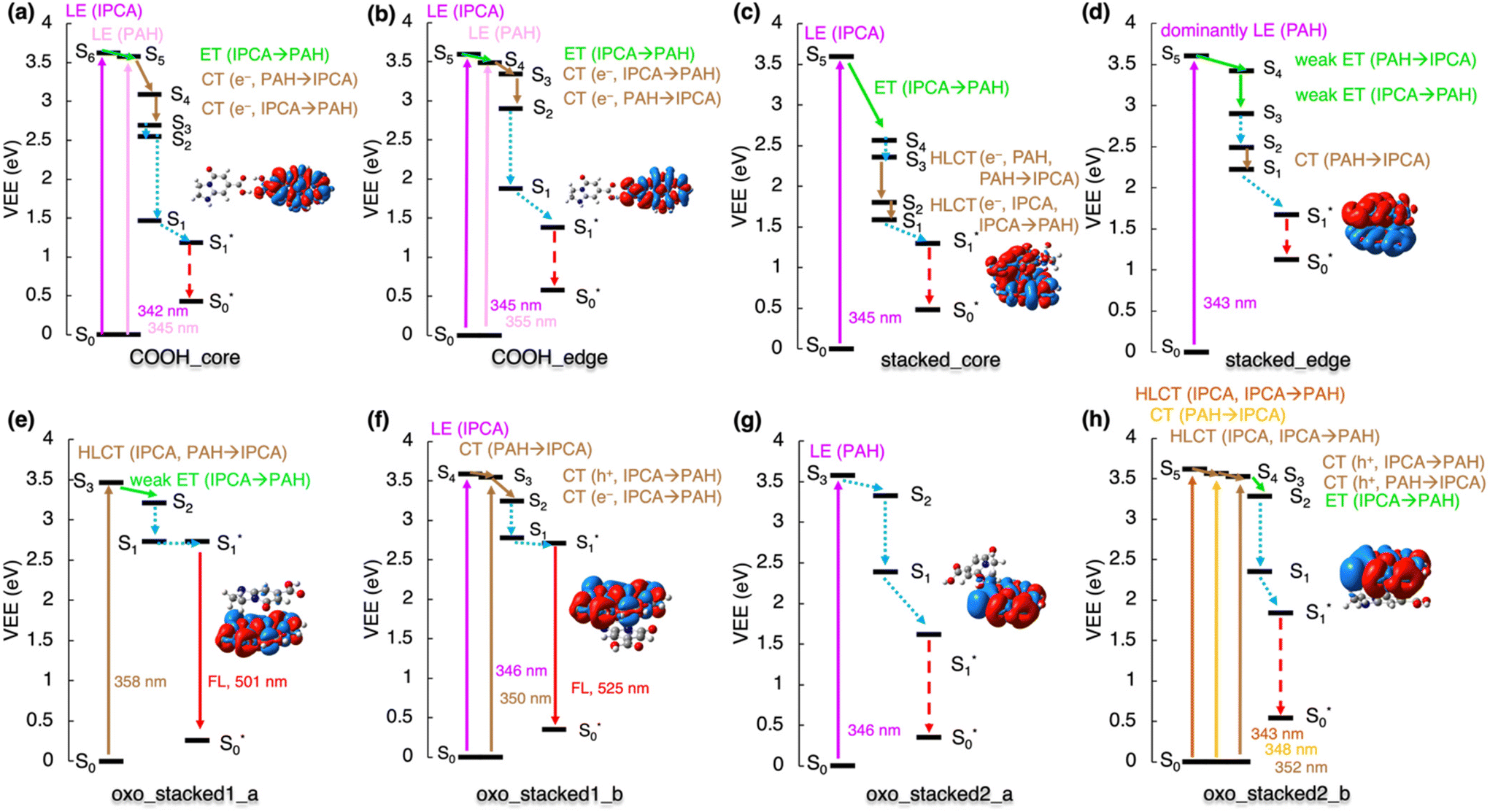 | ||
| Fig. 4 De-excitation cascade for the non-covalently bonded complexes (see Fig. 1 for system labeling) excited by the 350 nm energy source. LE represents local excitation, CT (e−/h+) charge (electron/hole) transfer, HLCT hybridized local and charge-transfer, ET energy transfer. Insets: EDD plots for the S0 → S1 transition (red/blue regions indicate increase/decrease of the electron density upon the excitation). Note: For stacked_edge structure, the S1* state was calculated with the LR approach (see ESI† for the explanation). | ||
Similarly, the stacked complexes of non-functionalized N-doped PAHs with IPCA endorse non-radiative channels in CDs, lowering the QY (Fig. 4c). There are several dark CT states (e.g., S2 and S6 in stacked_core model, and S6 in stacked_edge model) which cannot be identified in the absorption spectrum but are important in the de-excitation processes. After local excitation (S5) on IPCA at 345 nm in the stacked_core model, S5 → S4 ET to the PAH part of the complex can occur (see section 10 in ESI† for the evaluations of total couplings of the EET analysis) although the relatively large energy difference (∼1 eV) between the two states suggests a competitive pathway via direct emission from the S5 state localized on IPCA in some stacked arrangements.23,24 In the case of the S5 → S4 internal conversion, ET is followed by hybrid local charge-transfer (HLCT) to IPCA (S3 → S2) and HLCT to PAH (S2 → S1) and internal conversion to S1*. Thus, the emission takes place from this HLCT state, and it is significantly redshifted to 1512 nm. Moreover, these HLCT excitations have almost zero oscillator strengths, suggesting low efficiency of radiative deactivation. With the 350 nm source used for the photoexcitation, the formation of an exciton (S1 → S5) dominantly localized on PAH part of the stacked_edge model can be expected (Fig. 4d). The transition to from the S5 state to CT S1 state could be described as a weak ET from PAH to IPCA forth and back, with several NTOs involved in the transitions. Relaxing from S2 to S1, the electron is transferred to IPCA part. Here, the TD-DFT predicted that the S1 state in the relaxed geometry is energetically lower than GS, which is apparently an artefact of TD-DFT, which is not appropriate for describing quasi-degenerate states and suggests the occurrence of a conical intersection in this model.
The stacked complexes of IPCA with O-functionalized N-doped PAHs can lead to both radiative and radiationless de-excitation pathways in CDs depending on the doping pattern. The excitation to the S3 HLCT state in oxo_stacked1_a is followed by weak ET localizing the exciton on the PAH moiety (Fig. 4e). The S1 → S0 emission is bright (f = 0.558) with λmax ≈ 500 nm. If oxo_stacked1_b is photoexcited with the 350 nm source, S4 and S3 states become populated (Fig. 4f). The NTO analysis suggests that the S1 energy level is reached via a sequential ET process (from IPCA to PAH) through hole transfer (S4 → S3) followed by electron transfer (S3 → S2). This non-covalent structural domain similarly emits the light of green colour (525 nm, f = 0.464).
Altering the doping sites in O-functionalized N-doped stacked complexes can result in different de-excitation cascades lowering PL QY in CDs including possible occurrence of ISC (Fig. 4g and h). In oxo_stacked2_a, the 350 nm source would cause the populating of S3 localized on PAH, which can internally convert to S1via the S2 state, both keeping the character of LE on PAH. The LE character is also preserved during the relaxation of the S1 state, despite notable differences between the S0 and S1 structures (Fig. S17†). Besides the Kasha emission being in the IR region (λmax ≈ 976 nm, f = 0.093), a small singlet–triplet energy gap ΔEST = 1.1 kcal mol−1 (Fig. S6†) suggests the plausibility of ISC, thus opening a radiationless deactivation channel.
In the case of oxo_stacked2_b, the 350 nm excitation source would populate HLCT S5, CT S4 and HLCT S3 states. The transition from S5 to S1 involves hole transfer from IPCA to PAH and back, followed by ET from S3 to S2 and vibration relaxation to S1. As in oxo_stacked2_a, the Kasha emission occurs from S1 localized on PAH unit (see EDD plot in Fig. 4g) at 952 nm (f = 0.091), and the small singlet–triplet energy gap ΔEST = 0.4 kcal mol−1 (Fig. S6†) again suggests ISC as a possible radiationless decay channel.
The linking of IPCA with N-doped PAHs via a single covalent bond leads either to local emission from IPCA,23,24 or to a non-radiative de-excitation pathway lowering the PL QY of CDs, as the Kasha emission occurring on PAH is red-shifted to the red end of the visible range with low oscillator strength or even to the IR region. The involvement of low-lying dark CT states with a hole on PAH and an electron on IPCA in de-excitation processes in models with the ester bond is depicted in Fig. 5a and b. The S0 → S1 transition in ester1_core corresponding to a LE on PAH is dark and lies in the red spectral region (λmax ≈ 748 nm, f = 0.000), while the emission is far in the IR region (1370 nm, f = 0.001), suggesting a non-radiative decay. On the other hand, the S1 state in ester1_edge model with a graphitic-N-edge unit is the bright LE on pyrene peaking at 493 nm (f = 0.045), and the emission is shifted to the red spectral region (751 nm, f = 0.080) and it only slightly decreases the QY in comparison with the PL of isolated IPCA molecule. However, it should be noted that the S5 state in ester1_core is relatively well separated from S4, which suggests that a competitive pathway via direct emission from the S5 state localized on IPCA is plausible.
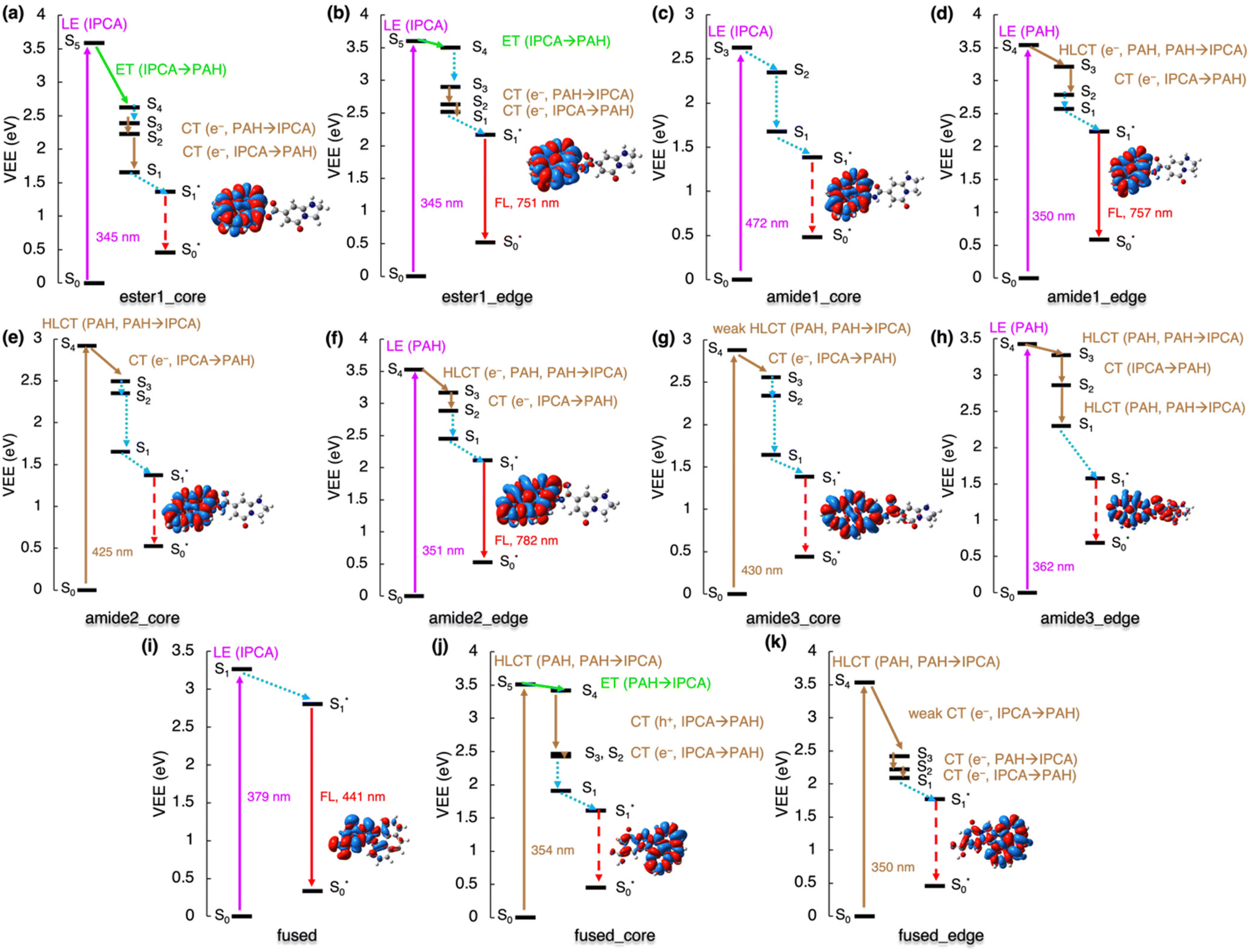 | ||
| Fig. 5 De-excitation cascade for the covalently bonded complexes excited by the 350 nm energy source. LE represents local excitation, CT (e−/h+) charge (electron/hole) transfer, HLCT hybridized local and charge-transfer, ET energy transfer. Insets: EDD plots for the S0 → S1 transition (red/blue regions indicate increase/decrease of the electron density upon the excitation). | ||
Contrary to other studied cases, the 350 nm energy source cannot activate PL channels of single-bonded complexes of IPCA/N-doped PAHs with graphitic-N-core doping pattern as there are no absorption peaks in this region. A lower energetic source could stimulate a LE on PAH (Fig. 5c) for amide1_core. Its de-excitation from this S3 state only includes the internal conversion to S1 and the calculated vertical emission energy in the IR region (λ = 1372 nm) suggests non-radiative deactivation (Fig. 5c). If the HLCT states amide2_core and amide3_core was targeted with the lower-energy excitation source, similar de-excitation pathways could be followed in both these models, starting with electron transfer from IPCA to PAH (S4 → S3) and sequential internal conversions and vibration relaxations to S1, where the exciton is localized on PAH (Fig. 5e and g). The existence of these complexes would, again, lower the QY of CDs emission as the emission was calculated to be in the IR region.
The amide-bonded complexes with graphitic-N-edge doping could be photoexcited with 350 nm laser. In amide1_edge, it would cause LE on PAH (S0 → S4) and the de-excitation would involve dark HLCT from PAH to IPCA (S4 → S3) and back transfer (S3 → S2), followed by vibration relaxation to S1 (Fig. 5d). Eventually, this radiative channel would lead to emission at 757 nm (f = 0.081). Photoexcitation with 350 nm energy source would also create an exciton pair on PAH in amide2_edge and amide3_edge complexes. Similar de-excitation pathways were identified for these two models, where transitions between bright HLCT and CT states could be tracked (Fig. 5f and h; Tables S25 and S27†). The amide2_edge model exhibited vertical emission at the end of the visible region, while HLCT character of S1 in amide3_edge resulted in a non-radiative de-excitation pathway, which could be related to smaller distortion of the two components in the S1 state (Fig. S18†). Moreover, ISC cannot be excluded from considerations as the closest singlet–triplet gap out of all single-bonded models is 1.9 kcal mol−1 for amide3_edge (Fig. S6†).
The formation of fused structures in CDs can open either radiative or radiationless channels depending on the number and topology of the graphitic nitrogens in their structure (Fig. 5i–k). In the fused model with two graphitic nitrogens being remnants of fusion with IPCA, the calculated Kasha emission was predicted at 441 nm (f = 0.834), which is very close to the IPCA emission band. If the HLCT state (354 nm) of fused_core was targeted with a 350 nm excitation source, two ET could be witnessed during deactivation processes to S1, i.e., internal conversion from S5 to S4, and a successive ET (S4 → S2) involving two-step hole and electron transfers (Fig. 5j). The S0 → S1 transition is not bright (λmax ≈ 651 nm, f = 0.003), and the corresponding Kasha emission is far in IR region at 1071 nm with the low oscillator strength (f = 0.005), suggesting non-radiative deactivations. Targeting S4 of fused_edge with the 350 nm source, electron CT from IPCA to PAH, back to IPCA and again to PAH are needed to reach the S1 level. The S1 → S0 emission is in the near IR region (λmax ≈ 945 nm, f = 0.023). Although the singlet–triplet gap for the S1 state of these fused structures is in the range of 4.3–15.8 kcal mol−1, thus discouraging the feasibility of the ISC pathway, it should be noted that the gap determined using the GS geometries can differ from that obtained from the adiabatic picture. The general features in our models representing interplay between core/surface and molecular states in possible structural domains occurring within CDs are summarized in Fig. 6.
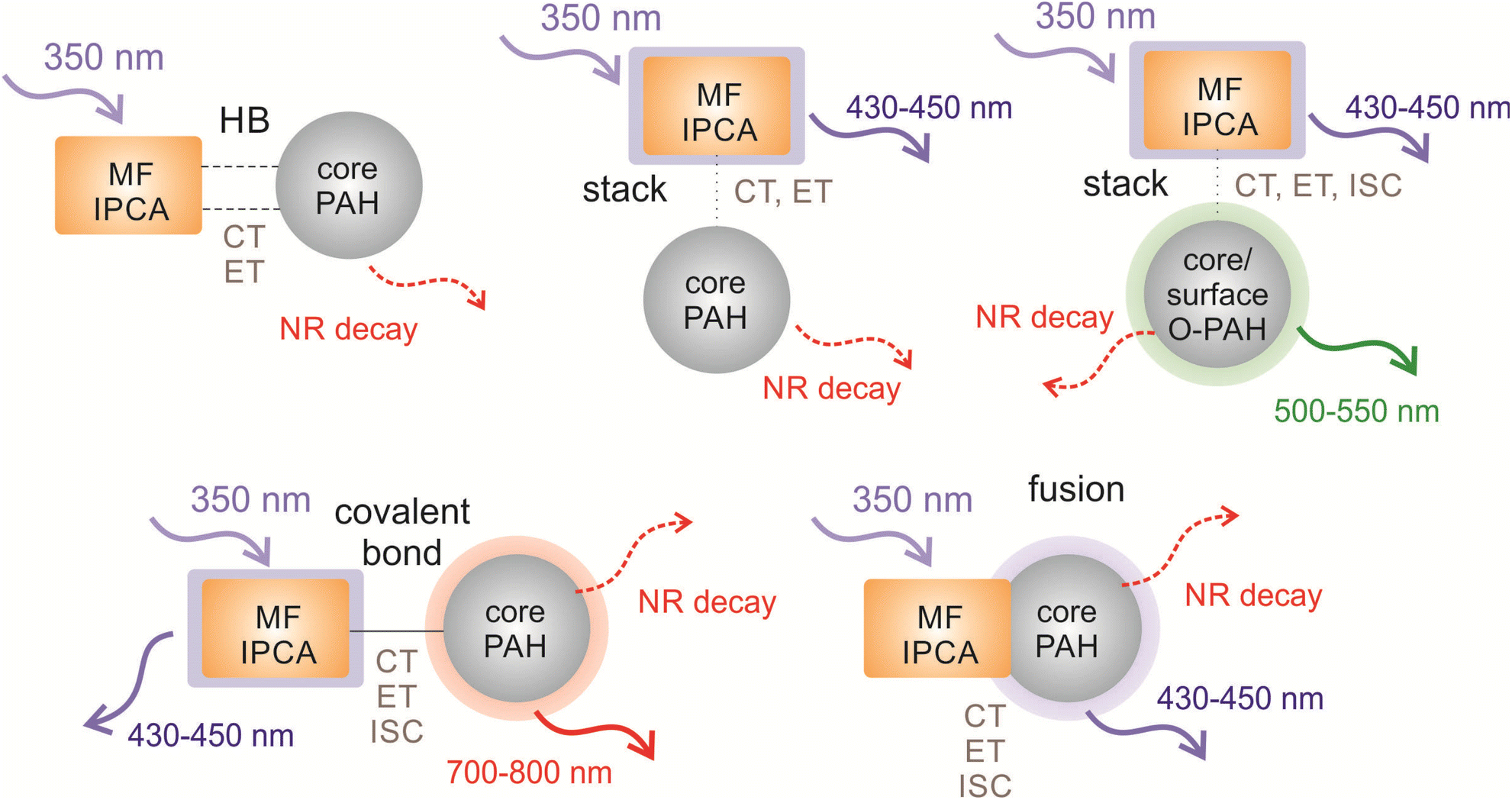 | ||
| Fig. 6 Overview of the processes occuring in the representative models of CD structural domains after the irradiation with a 350 nm energy source presumably targeting the absorption maximum of IPCA (displayed as incoming arrows). The outcoming arrows represent the PL (solid lines) from core and/or molecular states and dissipative NR channels (dashed lines). Notation: LE – local excitation, CT – charge transfer, ET – energy transfer, ISC – intersystem crossing, NR – non-radiative de-excitation, HB – hydrogen bond, MF – molecular fluorophore, PAH – polyaromatic hydrocarbon. | ||
Conclusions
We analysed absorption and emission properties of computationally feasible CD core/surface/molecular fluorophore systems, focusing on the possible interplay between different PL centres via charge and energy transfers that can occur during the de-excitation processes. By including both non-covalently and covalently bonded systems as well as fused structures, our models covered the main structural domain types and thus reflected the semi-local complexity of CDs. Our results revealed that all the studied arrangements supported, to some extent, the mutual communication between the core, surface, and molecular states. However, we showed that, in some cases (e.g., in stacking structures and covalent single-bonded systems), the direct emission from the photoactivated MF competed with an internal conversion de-excitation pathway, while in others (e.g., H-bonded, and fused systems), efficient CT and ET events led to either a long-wavelength emission from the core state or a non-radiative decay.The non-covalent interactions mediated by H-bonding did not significantly affect the optical properties of molecular components; however, the low-lying dark CT states were still able to assist non-radiative decay pathways. A more efficient overlap of frontier MOs in the stacked structures modified electronic excited levels, and new peaks corresponding to bright CT states emerged in their absorption spectra. Such changes in the absorption as well as emission spectra were enhanced in those types of covalent bonding, where steric factors allowed the molecular units to attain a quasi-planar arrangement, such as in systems linked via an amide bond. The most significant changes in the optical properties compared to the behaviour of separated molecules were found for fused structures formed via sharing one edge bond, where the CT character of excitations between the core and surface/molecular states was demonstrated.
The deactivation pathways in the studied systems typically involve multiple CT and/or ET events that can promote the formation of charge separated states and/or lead to the activation of other PL centres in CDs. In addition, it was shown that, depending on the arrangement, the doping pattern and surface functionalization, both the CD core and the MF can act as an electron donor or acceptor in the charge transfer processes, which could help to design CDs with desirable hole–electron surface/core characteristics. Our study represents a significant step towards deciphering the complexity of the PL mechanisms in CDs via establishing a link between their structure and the PL properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. M. acknowledges financial support of the Slovak Research and Development Agency (APVV-20-0098). This work was supported by the Ministry of Education, Youth and Sports of the Czech Republic via OP RDE of ERDF (CZ.02.1.01/0.0/0.0/16_019/0000754), e-INFRA CZ (ID: 90140), other computational resources were supplied, among others, by the project “e-Infrastruktura CZ” (e-INFRA LM2018140) provided within the program Projects of Large Research, Development and Innovations Infrastructures. The COST Action CA21101 is also acknowledged.References
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed
.
- T. Yuan, T. Meng, P. He, Y. Shi, Y. Li, X. Li, L. Fan and S. Yang, J. Mater. Chem. C, 2019, 7, 6820–6835 RSC
- Z. L. Wu, Z. X. Liu and Y. H. Yuan, J. Mater. Chem. B, 2017, 5, 3794–3809 RSC
- M. Li, T. Chen, J. J. Gooding and J. Liu, ACS Sens., 2019, 4, 1732–1748 CrossRef CAS PubMed
- H. Yu, R. Shi, Y. Zhao, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 9454–9477 CrossRef CAS PubMed
- S. Kalytchuk, L. Zdražil, Z. Bad'ura, M. Medved’, M. Langer, M. Paloncýová, G. Zoppellaro, S. V. Kershaw, A. L. Rogach, M. Otyepka and R. Zbořil, ACS Nano, 2021, 15, 6582–6593 CrossRef CAS PubMed
- B. Zhao, Z. Wang and Z. Tan, Nat. Photonics, 2020, 14, 130–131 CrossRef CAS
- X. Bao, Y. Yuan, J. Chen, B. Zhang, D. Li, D. Zhou, P. Jing, G. Xu, Y. Wang, K. Holá, D. Shen, C. Wu, L. Song, C. Liu, R. Zbořil and S. Qu, Light: Sci. Appl., 2018, 7, 91 CrossRef PubMed
- M. Semeniuk, Z. Yi, V. Poursorkhabi, J. Tjong, S. Jaffer, Z. H. Lu and M. Sain, ACS Nano, 2019, 13, 6224–6255 CrossRef CAS PubMed
- L. Cui, X. Ren, M. Sun, H. Liu and L. Xia, Nanomaterials, 2021, 11, 3419 CrossRef CAS PubMed
- S. Y. Lim, W. Shen and Z. Gao, Chem. Soc. Rev., 2014, 44, 362–381 RSC
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230 RSC
- P. Roy, P. C. Chen, A. P. Periasamy, Y. N. Chen and H. T. Chang, Mater. Today, 2015, 18, 447–458 CrossRef CAS
- H. Luo, N. Papaioannou, E. Salvadori, M. M. Roessler, G. Ploenes, E. R. H. Eck, L. C. Tanase, J. Feng, Y. Sun, Y. Yang, M. Danaie, A. B. Jorge, A. Sapelkin, J. Durrant, S. D. Dimitrov and M. Titirici, ChemSusChem, 2019, 12, 4432–4441 CrossRef CAS PubMed
- C. M. Carbonaro, D. Chiriu, L. Stagi, M. F. Casula, S. V. Thakkar, L. Malfatti, K. Suzuki, P. C. Ricci and R. Corpino, J. Phys. Chem. C, 2018, 122, 25638–25650 CrossRef CAS
- H. Ding, X. H. Li, X. B. Chen, J. S. Wei, X. B. Li and H. M. Xiong, J. Appl. Phys., 2020, 127, 231101 CrossRef CAS
- S. Mura, L. Stagi, L. Malfatti, C. M. Carbonaro, R. Ludmerczki and P. Innocenzi, J. Phys. Chem. A, 2020, 124, 197–203 CrossRef CAS PubMed
- W. Wang, B. Wang, H. Embrechts, C. Damm, A. Cadranel, V. Strauss, M. Distaso, V. Hinterberger, D. M. Guldi and W. Peukert, RSC Adv., 2017, 7, 24771–24780 RSC
- Z. Gan, H. Xu and Y. Hao, Nanoscale, 2016, 8, 7794–7807 RSC
- N. V. Tepliakov, E. V. Kundelev, P. D. Khavlyuk, Y. Xiong, M. Y. Leonov, W. Zhu, A. V. Baranov, A. V. Fedorov, A. L. Rogach and I. D. Rukhlenko, ACS Nano, 2019, 13, 10737–10744 CrossRef CAS PubMed
- F. Mocci, L. D. V. Engelbrecht, C. Olla, A. Cappai, M. F. Casula, C. Melis, L. Stagi, A. Laaksonen and C. M. Carbonaro, Chem. Rev., 2022, 122, 13709–13799 CrossRef CAS PubMed
- Y. Song, S. Zhu, S. Zhang, Y. Fu, L. Wang, X. Zhao and B. Yang, J. Mater. Chem. C, 2015, 3, 5976–5984 RSC
- F. Siddique, M. Langer, M. Paloncýová, M. Medved, M. Otyepka, D. Nachtigallova, H. Lischka and A. J. A. Aquino, J. Phys. Chem. C, 2020, 124, 14327–14337 CrossRef CAS
- M. Langer, T. Hrivnák, M. Medved’ and M. Otyepka, J. Phys. Chem. C, 2021, 125, 12140–12148 CrossRef CAS
- M. Righetto, A. Privitera, I. Fortunati, D. Mosconi, M. Zerbetto, M. L. Curri, M. Corricelli, A. Moretto, S. Agnoli, L. Franco, R. Bozio and C. Ferrante, J. Phys. Chem. Lett., 2017, 8, 2236–2242 CrossRef CAS PubMed
- R. Bhuyan, K. Bramhaiah and S. Bhattacharyya, J. Colloid Interface Sci., 2022, 605, 364–372 CrossRef CAS PubMed
- M. Langer, M. Paloncýová, M. Medved’ and M. Otyepka, J. Phys. Chem. Lett., 2020, 11, 8252–8258 CrossRef CAS PubMed
- F. Meierhofer, F. Dissinger, F. Weigert, J. Jungclaus, K. Müller-Caspary, S. R. Waldvogel, U. Resch-Genger and T. Voss, J. Phys. Chem. C, 2020, 124, 8894–8904 CrossRef CAS
- A. Das, D. Roy, C. K. De and P. K. Mandal, Phys. Chem. Chem. Phys., 2018, 20, 2251–2259 RSC
- F. Furche and R. Ahlrichs, J. Chem. Phys., 2002, 117, 7433–7447 CrossRef CAS
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, et al., Gaussian16 Revision B.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed
-
The PyMOL Molecular Graphics System, Version 1.7, Schrodinger, LLC Search PubMed
-
G. A. Zhurko and D. A. Zhurko, Chemcraft. Version 1.8, https://www.chemcraftprog.com (accessed July 17, 2022) Search PubMed
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC
- M. Caricato, B. Mennucci, J. Tomasi, F. Ingrosso, R. Cammi, S. Corni and G. Scalmani, J. Chem. Phys., 2006, 124, 124520 CrossRef PubMed
- R. Cammi, S. Corni, B. Mennucci and J. Tomasi, J. Chem. Phys., 2005, 122, 104513 CrossRef CAS PubMed
- A. Bussy and J. Hutter, J. Chem. Phys., 2021, 155, 034108 CrossRef CAS PubMed
-
R. Dennington, T. Keith and J. Millam, Gauss View, Version 5, Semichem Inc., Shawnee Mission, 2009 Search PubMed
- C. Curutchet and B. Mennucci, J. Am. Chem. Soc., 2005, 127, 16733–16744 CrossRef CAS PubMed
- V. Russo, C. Curutchet and B. Mennucci, J. Phys. Chem. B, 2007, 111, 853–863 CrossRef CAS PubMed
- M. Svensson, S. Humbel, R. D. J. Froese, T. Matsubara, S. Sieber and K. Morokuma, J. Phys. Chem., 1996, 100, 19357–19363 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr05114a |
| This journal is © The Royal Society of Chemistry 2023 |