
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnusually cyclized triterpenoids: occurrence, biosynthesis and chemical synthesis
Hidayat
Hussain
 *a,
Jianbo
Xiao
bc,
Akbar
Ali
d,
Ivan R.
Green
e and
Bernhard
Westermann
*a
*a,
Jianbo
Xiao
bc,
Akbar
Ali
d,
Ivan R.
Green
e and
Bernhard
Westermann
*a
aDepartment of Bioorganic Chemistry, Leibniz Institute of Plant Biochemistry, Weinberg 3, Halle (Saale) D-06120, Germany. E-mail: Hidayat.Hussain@ipb-halle.de; Bernhard.Westermann@ipb-halle.de
bInternational Research Center for Food Nutrition and Safety, Jiangsu University, Zhenjiang 212013, China
cDepartment of Analytical Chemistry and Food Science, Faculty of Food Science and Technology, University of Vigo – Ourense Campus, Ourense, E-32004, Spain
dDepartment of Chemistry, Government College University Faisalabad, Faisalabad 38000, Pakistan
eDepartment of Chemistry and Polymer Science, University of Stellenbosch, Private Bag X1, Matieland, Stellenbosch 7600, South Africa
First published on 2nd December 2022
Abstract
Covering: 2009 to 2021
Biosynthetically, most of the syntheses of triterpenes follow the cascade cyclization and rearrangement of the acyclic precursors viz., squalene (S) and 2,3-oxidosqualene (OS), which lead to the very well known tetra- and pentacyclic triterpene skeletons. Aside from these, numerous other triterpenoid molecules are also reported from various natural sources and their structures are derived from “S” and “OS” via some unusual cyclization operations which are different from the usual tetra- and pentacyclic frameworks. Numerous compelling advances have been made and reported in the identification of these unusual cyclized mono-, di-, tri- and tetracyclic triterpenes between 2009 and 2021. Besides a dramatic increase in the newly isolated uncommon cyclized triterpenoids, substantial progress in the (bio)-synthesis of these triterpenes has been published along with significant progress in their biological effects. In this review, 180 new unusual cyclized triterpenoids together with their demonstrated biogenetic pathways, syntheses and biological effects will be categorized and discussed.
 Hidayat Hussain | Hidayat Hussain was Post Doc (2004–2007), senior scientist (2009–2010) in University of Paderborn Germany, and Post Doc fellow (2007–2008) at University of Maine France. He worked at Uni. Nizwa Oman as Associate Professor (2011–2017) and AvH guest Professor (2018–2019) at Leibniz Institute of Plant Biochemistry (IPB), Germany. He was also a Visiting Professor at Scripps Institution of Oceanography in 2017. Currently Dr Hidayat is associated with IPB, Germany and has 350 publications with impact factor over 1070. According to the Stanford University database, his name is included in the top 2% of the most-cited scientists for 2020, 2021 and 2022. |
 Jianbo Xiao | Dr. Jianbo Xiao is current a full professor of Jiangsu University (China) and a distinguished researcher of University of Vigo (Spain). He has worked as a Postdoc supported by AvH foundation at University of Würzbug, Germany (2013–2015) and then worked as an assistant professor in University of Macau (2015–2020). His research is focusing on chemistry and function of natural products. Dr Xiao has been selected as Clarivate Analytics Highly Cited Researchers for 5 times since 2016. He has accepted and published more than 300 peer reviewed papers including Nature Reviews Drug Discovery, Seminars in Cancer Biology, and so on. |
 Akbar Ali | Dr. Akbar Ali is currently working as assistant professor at the department of chemistry, G. C. University Faisalabad. Before, he worked for 3 years as assistant professor at institute of Chemistry, University of Sargodha. Dr Akbar Ali has completed his PhD in the area of synthetic organic chemistry from Federal University of Sao Carlos, Brazil and Leibniz Institute of Plant Biochemistry (IPB), Germany. To date he has more than 70 publications in reputed international journals with around 250 impact factor, 1102 citations, 20 h-index and 34 i10-index. His interests include organic synthesis, click chemistry, green and photo chemistry and asymmetric organocatalysis. |
 Ivan R. Green | Ivan Green graduated with a PhD in Synthetic Organic Chemistry in 1972 from the University of Cape Town. He started his career as Lecturer in 1973 and was made a full Professor in 1986 and later a Senior Professor in 1990 at the University of the Western Cape where he lectured for 39 years until his retirement in July of 2011. Upon retirement, he was invited to join the University of Stellenbosch as a Research Associate where he is involved in doing research in various topics. He has over 200 publications, and has supervised 24 PhD students. |
 Bernhard Westermann | Prof. Dr Bernhard Westermann received his PhD in 1989, Profs. W. Sucrow and H.-J. Altenbach at the University of Paderborn, Germany. After a postdoctoral stay at the University of Toronto, Canada (Prof. J. B. Jones) from 1990 to 1991, he started his independent scientific career at the University of Paderborn. After his habilitation in 1998, he stayed as an associate professor. In 2004 he moved to the Leibniz-Institute of Plant Biochemistry as head of Biofunctional Synthesis, where he continues to work on topics dealing with natural product synthesis, ligation strategies with multicomponent reactions, ligation strategies, and plant hormones. |
1 Introduction
Triterpenes represent a versatile group of terpenes comprised of six isoprene subunits and to date over 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 triterpenes have been identified from various natural sources.1 Moreover, around 200 different triterpene skeletons have been reported to be derived from cyclization products of squalene (S), oxidosqualene (OS), or bis-oxidosqualene (BOS) catalysed by tripterpene synthases.2 Apart from these, constantly new enzymes are identified which arrange precursors to these skeletons. Very recently, Tao et al.3 described the non-squalene-dependent triterpene biosynthesis and discovered two chimeric fungal class I triterpene synthases (TrTSs) for the conversion and production of hexaprenyl diphosphate into triterpenes.
000 triterpenes have been identified from various natural sources.1 Moreover, around 200 different triterpene skeletons have been reported to be derived from cyclization products of squalene (S), oxidosqualene (OS), or bis-oxidosqualene (BOS) catalysed by tripterpene synthases.2 Apart from these, constantly new enzymes are identified which arrange precursors to these skeletons. Very recently, Tao et al.3 described the non-squalene-dependent triterpene biosynthesis and discovered two chimeric fungal class I triterpene synthases (TrTSs) for the conversion and production of hexaprenyl diphosphate into triterpenes.
Triterpenes are naturally present in fruit, vegetables and medicinal plants and are hence part of the daily human diet. A driving force behind triterpene research is the wide range of biological and pharmacological effects including cytotoxicity, inhibition of pathogenic microbes, anti-inflammatory, antiprotozoal, antitubercular, and antifeedant effects to name but a few. In addition, some triterpenes have illustrated enzymatic effects and have been shown to modulate specific receptor functions.4–7
The large majority of triterpenoids are 6-6-6-5 tetra-cycles, 6-6-6-6-5 pentacycles, or 6-6-6-6-6 pentacycles and these compounds are biosynthetically formed via cascade cyclizations and rearrangements of either squalene (S) or 2,3-oxidosqualene (OS). Although the majority of the triterpenes contain tetra- or pentacyclic skeletons, together with these triterpenes derived from cyclization processes there are a number that deviate from these mainstream polycyclization of S and OS to tetra- or pentacyclic compounds which have been reported from Nature. These triterpene structures, referred to as “unusually cyclized triterpenoids” are generated from unusual cyclizations of squalene (S) and 2,3-oxidosqualene (OS).2,8 These “unusually cyclized triterpenes” are biosynthesized via: (i) incomplete cyclization of the S-derived molecules whose cyclization processes start from the epoxide or terminal isopropylidene core and generate mono-, bi- and tricyclic triterpenoid skeletons. (ii) Cyclization of S or OS to generate polycyclic triterpenoid skeletons followed by cleavage of the already established ring systems. (iii) Two independent cyclizations of the S- or OS-derived molecule.8
This review provides a comprehensive and critical coverage of 180 new unusual cyclized triterpenoids reported from January 2009 through to December 2021, and serves to complement the previous review of unusual cyclized triterpenoids published in Natural Product Reports.8 In addition, this review includes a few previously omitted articles published between 1998 and 2008. Notably, included are nor-iridal and nor-isomalabaricane terpenes which are biosynthetically derived from iridal and isomalabaricane-type triterpenoids. In much the same way as the previous review, the format focuses on the discovery of new unusual cyclized triterpenoids from natural sources along with their synthesis and biological effects. Additionally, recent studies of the fascinating biosynthesis of these unusual cyclized triterpenoids are covered in this review. There have been numerous synthetic strategies towards forming these unusual cyclized triterpenoids as end products and these studies are also covered in this review. Notably, characteristic features of unusual cyclized triterpenoids with their high degree of skeletal and structural diversification are also discussed.
2 Iridal triterpenes
2.1 Monocyclic iridals
The basic iridal nucleus is depicted in Fig. 1 and this skeleton forms from squalene epoxide. In addition, the majority of the iridal natural products are typified by a C-1 carbonyl function (mostly aldehyde), and an ester, or alcohol at C-3. Four monocyclic iridal triterpenes viz., 8-hydroxylisoiridogermanal (1), belamcandane A (2), 3-O-acetyliridobelamal A (3), and belamcandal A (4) (Fig. 1) have been isolated from the rhizomes of Belamcanda chinensis collected from China. Iridals 1–3 feature a hydroxyl group at C-16 of the polyene side chain while triterpene 4 has a hydroxyl group at C-17.9 In addition, compounds 1 and 4 have an aldehyde function at C-1 and C-25, respectively. On the other hand, compound 3 is a rare iridal terpenoid which is lacking a carbonyl function at C-1 or the C-25 position. Bioassays for compounds 1–4 revealed inactivities towards various human cancer cells.9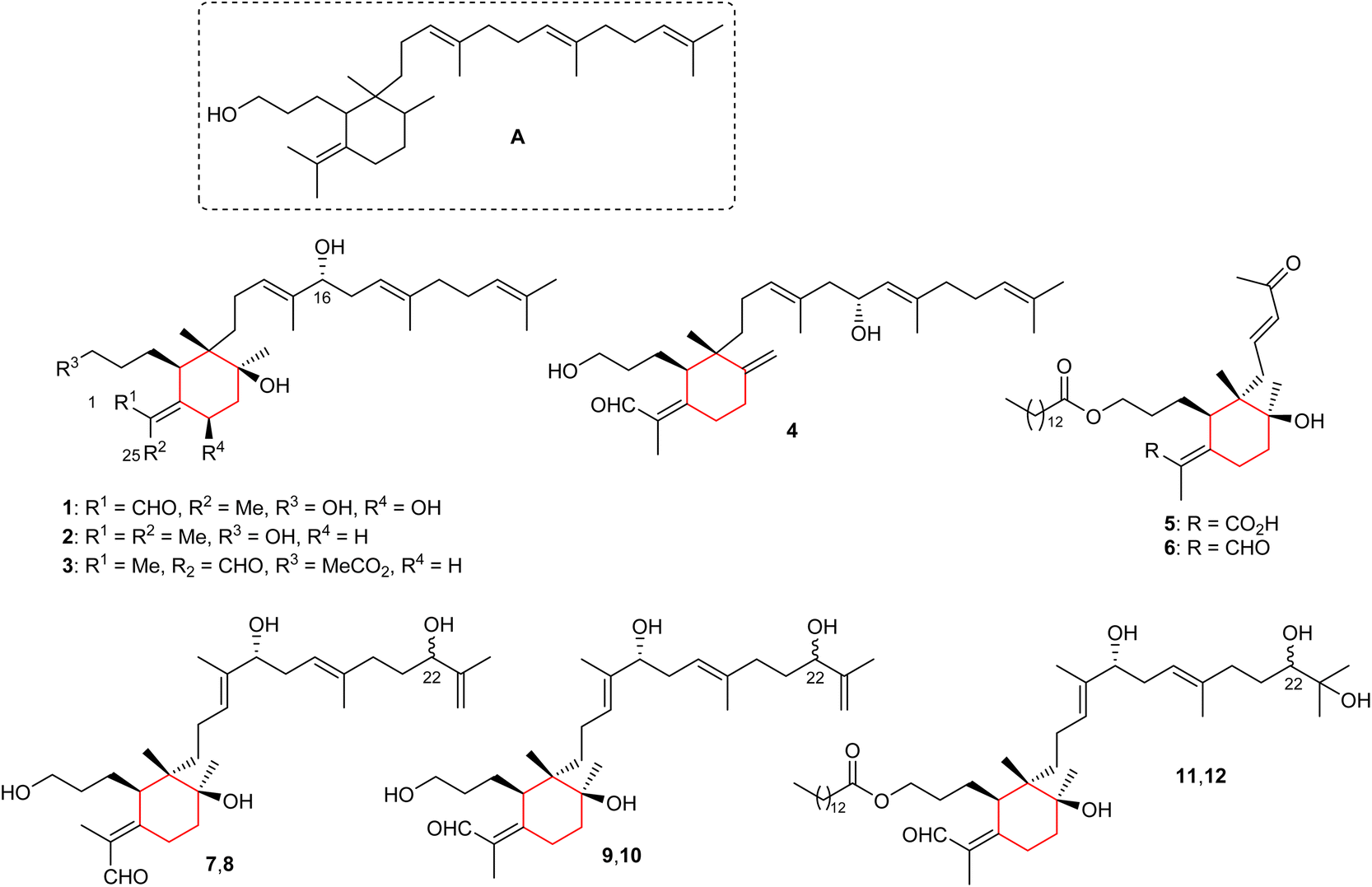 | ||
| Fig. 1 Structures of monoclic iridals 1–12. | ||
Iriwattins A (5) and B (6) (Fig. 1), two unusual sixteen carbon comprising nor-iridal triterpenes were isolated from the flower Iris wattii. Furthermore, compounds 5 and 6 illustrated notable protein-tyrosine phosphatase 1B (PTP1B) enzyme inhibition of 52% and 14%, respectively.10 Iritectol D (7), 22-epiiritectol D (8), iritectol E (9), 22-epiiritectol E (10), iritectol F (11), and 22-epiiritectol F (12) were produced by Iris tectorum. All of them were isolated as pairs of C-22 epimers and all possess a hydroxyl group at C-16 of the polyene side chain while metabolites 11/12 have a C-14-ester group at the C-3 hydroxyl group of the side chain of the ring along with an additional hydroxyl moiety at C-23.11
Three monocyclic iridal-type triterpenoids 13–15 (Fig. 2) were isolated from Iris delavayi. Iridal 13 is the geometrical isomer of 15, with respect to the aldehyde group at C-1. In addition, metabolites 13–15 are rare iridals missing the C-10 tertiary hydroxyl group.12 A literature survey revealed that only a few iridals described earlier are missing the C-10 tertiary hydroxyl group, such as marneral,13 marnerol,13 and 22,23-epoxy-10-deoxy-21-hydroxyiridal.14 Furthermore, compounds 13 and 15 contain an epoxide function at the C-18/C-19 position of the side chain.12
 | ||
| Fig. 2 Structures of monocyclic iridals 13–19. | ||
In another report, two iridal-type triterpenoids, named forrestins A (16) and B (17), were reported from I. forrestii. Both compounds are geometrical isomers of each other with respect to the aldehyde group. Moreover, compounds 16 and 17 have a tertiary hydroxyl group at C-19 of which the stereochemistry has not be established.15 Shi et al.16 reported iridojaponal C (18) from I. japonica and this compound illustrated moderate hepatoprotective effects. Moreover, iridal 18 has three hydroxyl groups in the polyene side chain. In 2018, Chen et al.17 reported the isolation of 17-hydroxyl-27-ene-iridal (19) from I. confusa. It features an exocyclic double bond between C-10/C-27 which is interestingly, also present in belamcandal A (4). Compounds having an exocyclic double at C-10/C-27 are very rare and were not reported in the previous review.8
2.2 Bicyclic iridals
Spirioiridoconfal C (20) along with isobelamcandal (21) (Fig. 3) were reported from related I. confusa. Metabolites 20 and 21 are spirioiridoconfals bearing C-19 and C-29 based exocyclic double bonds while compound 20 has an additional exocyclic bond at C-15 and C-28. Metabolite 20 (IC50: 85 μM; SI = 2.1) illustrated anti-HBV pronounced effects towards HBV DNA replication.17 A literature survey revealed that the earlier described iridotectoral B18 and hoogianal19 have a similar cyclopentane based spirocyclic core and were reported from the Iris species. Belamcanda chinensis rhizomes, collected from the Hebei province in China, produced two unusual iridal triterpenes named belamcanolide A (22), and belamcanoxide A (23). The latter compound demonstrated good cytotoxic effects towards colon (HCT-116), liver (HepG2), gastric (BGC-823), and lung (NCI-H1650) cancer cells with IC50 ranging from 3.2 to 7.5 μM.20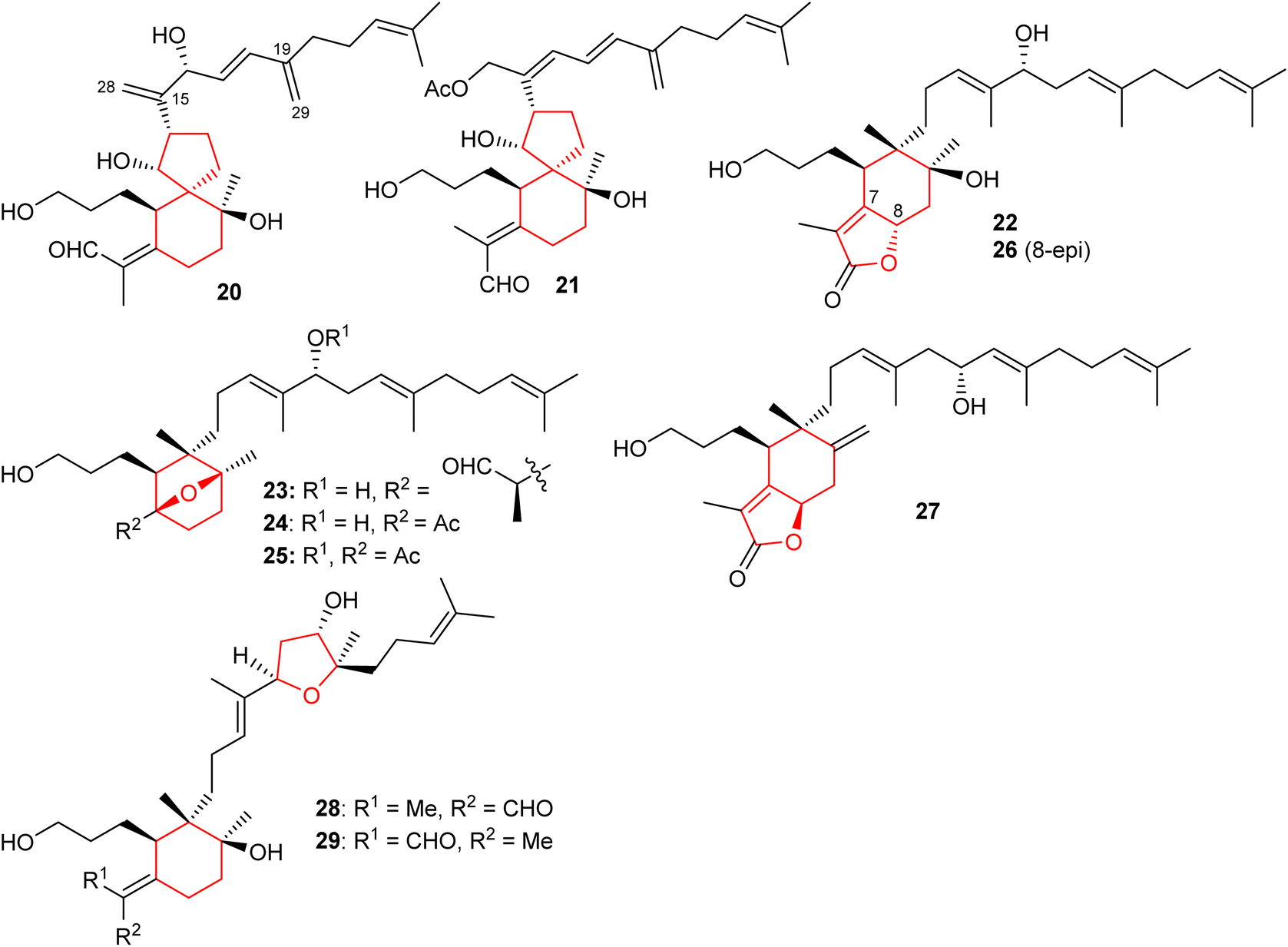 | ||
| Fig. 3 Structures of bicyclic iridals 20–29. | ||
Two nor-iridals named belamcanoxide B (24) and 16-O-acetylbelamcanoxide B (25) were isolated from Belamcanda chinensis. Furthermore, belamcanoxide B (24) illustrated good cytotoxic effects towards MCF-7 and HCT-116 cell lines with IC50: 3.4 and 5.6 μM respectively.9 In addition, this plant produces two further iridal lactones viz., belamcanolides B (26), and C (27).9 Compounds 22, 26, and 27 are rare examples of a 5/6-fused bicyclic iridal lactone, in which these metabolites bear a furanone ring (lactone ring) attached between C-7 and C-8. Interestingly, compounds 23–25 feature an ether linkage between C-7 and C-10 of the 6-membered ring. In addition, molecule 23 is a structurally unique iridal having an isolated aldehyde in place of the α,β-unsaturated aldehyde moiety which is common for iridal-type triterpenoids. It is interesting to note that compounds 22 and 26 are C-8 epimers while iridals 22, 23, and 26 feature a hydroxyl group at C-16.9
Both geometrical isomers viz., iritectols C (28)11 and G (29)21 (Fig. 3), include the tetrahydrofuran moiety between C-16/C-19 in the polyene side chain, and are produced by Iris tectorum. Iritectol 28 illustrated good cytotoxic effects towards liver cancer (HePG2) with IC50: 4.1 μM. On the other hand, it also possessed moderate effects towards ovarian cancer (A2780: IC50: 6.5 μM), colon cancer (HCT-16: IC50: 9.7 μM), and lung cancer (NCI-H1650: IC50: 9.8 μM).21 Additionally, iritectol G (29) illustrated anti-epileptic effects via inhibiting spontaneous (IC50: 8.2 μM) and 4-aminopyridine-evoked calcium oscillations (IC50: 12.5 μM).21
2.3 Tricyclic iridals
Li et al.22 reported four iridal-type triterpenoids named polycycloiridals P-T (30–34) (Fig. 4) from Belamcanda chinensis, which is an ornamental plant of the Iridaceae family. These bicyclic metabolites have an additional cyclopentane core unit resulting from a C-18 and C-22 cyclization of the homofarnesyl chain. Furthermore, iridals 30/31 and the corresponding 32/33 are pairs of Δ2(7) olefinic geometric isomers. Moreover, compound 34 has an ethoxy group at C-12 and based on extraction protocols, the authors proposed that this compound might be an artifact, although as yet, not proven. | ||
| Fig. 4 Structures of tricyclic iridals 30–36. | ||
Investigations of B. chinensis extracts yielded two unusual tricyclic-iridal triterpenoids named belamcandanes A (35) and B (36) (Fig. 4). It was found that these metabolites carry a rare α-terpineol moiety in their polyene side chain. Both compounds possessed moderate hepatoprotective effects while they were not active towards NCI-H1650, HepG2, BGC 823, HCT-116, and A2780 cancer cells.23 Iridals 30–34 feature a spiro furan ring formed via the C-13 hydroxyl group attack to a C-26 aldehyde group. A literature survey revealed that iridotectoral A,18 (13R)-iridotectoral C,24 and D,18 have a similar furan based spiro cyclic core reported from other Iris species.
A further three iridal-type triterpenoids, iritectol G (37), iritectol H (38) and 19-epi-iritectol H (39) (Fig. 5) bearing a rearranged homofarnesyl side chain and a 6/7/5 core system, were also reported from I. tectorum. Moreover, compound 37 is the second compound having been assigned the same name viz., iritectol G because compound 29 has already been reported with the same name. Iridals 38 and 39 are C-19 epimers and this diastereomeric mixture was shown to possess moderate neuroprotective effects.25
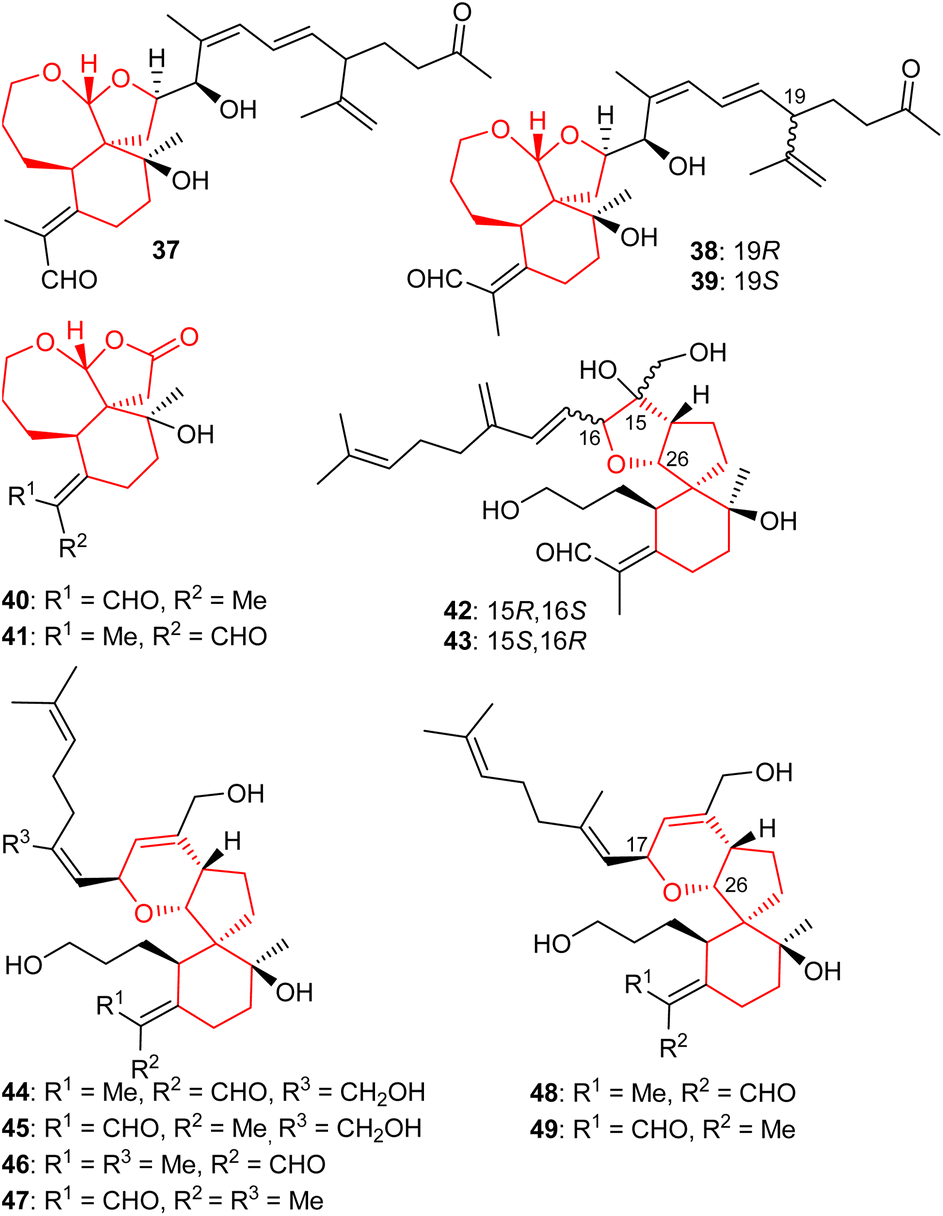 | ||
| Fig. 5 Structures of tricyclic iridals 37–49. | ||
In another report, iridojaponal A (40) and B (41) were produced by Iris japonica. Notably, iridojaponal A (40) and B (41) are sixteen carbon comprising degraded iridals. Iridals 40 and 41 demonstrated moderate hepatoprotective effects.16 Chen et al.17 reported that the spirioiridoconfals A (42) B (43) were isolated from I. confusa. Metabolites 42 and 43 are spiro-iridals bearing a C-16 and C-26 based tetrahydrofuran ring.
Six iridal-type triterpenoids, named spirioiridotectals A−F (44–49) representing three pairs of geometrical isomers (with respect to aldehyde group), were produced by I. tectorum. Interestingly, compounds 44–49 feature a 3,6-dihydro-2H-pyran core between C-17 and C-26. Moreover, iridals 44 and 45 are geometrical isomers of compounds 46 and 48 with respect to the exo-C-18/19-double bond only. Furthermore, metabolites 44, 45 and 49 possess moderate neuroprotective effects towards PC12 cell.26 In addition, spirioiridotectal D (45) suppressed NO production with an IC50: 0.54 μM.27
2.4 Tetracyclic iridals
Four new iridal polycycloiridals A−D (50–53) (Fig. 6), having an unusual α-terpineol group were isolated from I. tectorum. Iridals 50 and 52 have a distinct absolute configuration at the α-terpineol group. Moreover, compounds 50–53 illustrate hepatoprotective effects towards D-galactosamine-induced toxicity with activity ranging between 65 to 96%.28 In another report, polycycloiridals E–J (54–59) were reported from I. tectorum. These compounds possess a 6/5/7 tricyclic core, as observed earlier. In addition, metabolites 54–59 possess a cyclopentane ring which is suggested to be formed via cyclization between C-18 and C-22 of the homofarnesyl chain. Furthermore, these compounds contain two E configured double bonds at Δ14(15) and Δ16(17).27 Li et al.22 reported five tricyclic iridal-type triterpenoid analogs bearing the 6/5/7-core named polycycloiridals K–O (60–64) from Belamcanda chinensis and these metabolites possess a cyclopentane core resulting via cyclization between C-18 and C-22 of the homofarnesyl chain. Moreover, these compounds illustrated weak PTP1B inhibitory effects.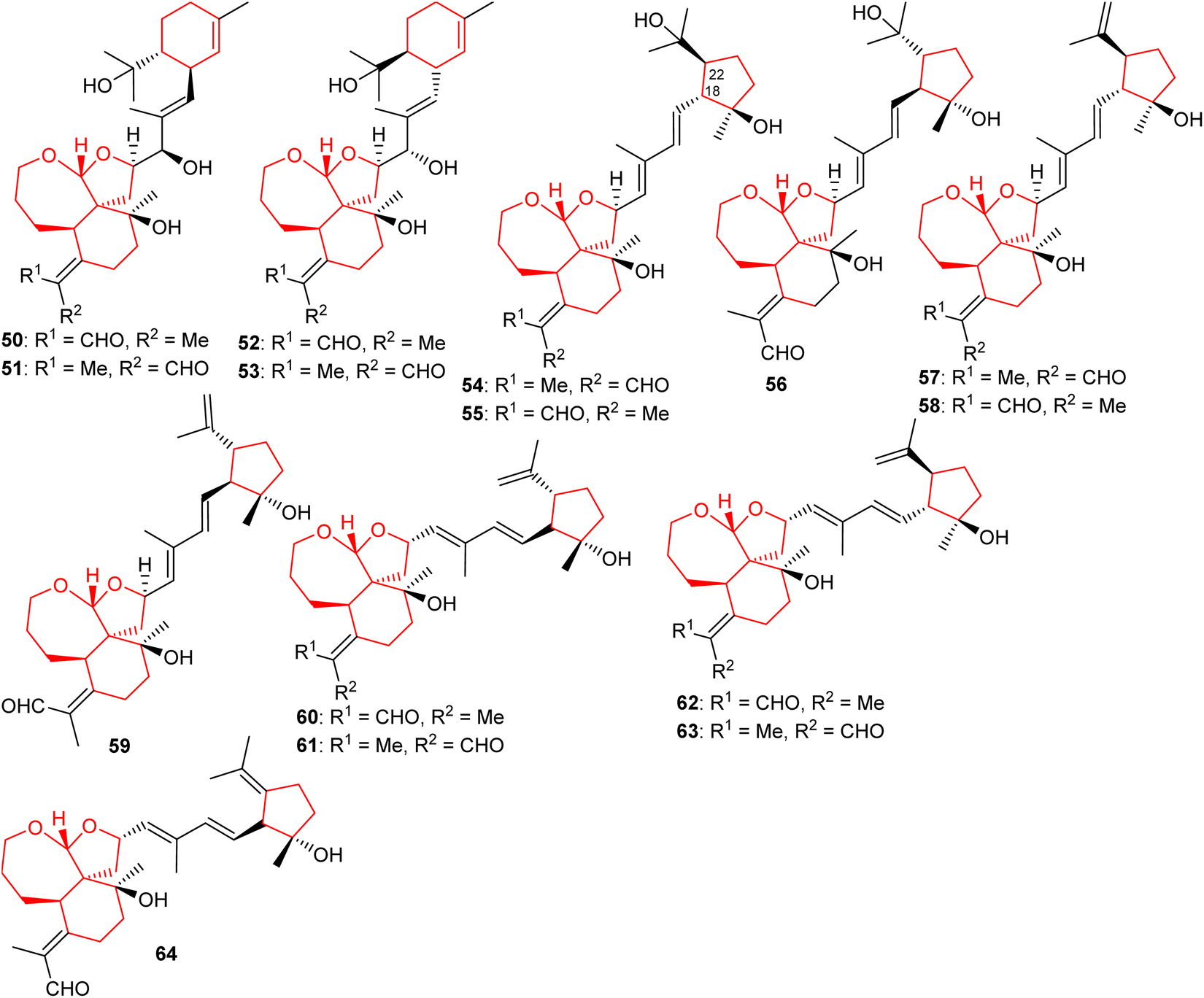 | ||
| Fig. 6 Structures of tetracyclic iridals 50–64. | ||
2.5 Miscellaneous iridal triterpenoids
Three spiroiridal-isoflavone hybrid compounds named belamcandanins A–C (65–67) (Fig. 7), have been isolated from B. chinensis. Metabolites 65–67 represent rare iridal-flavone hybrid compounds. Biological investigation of these compounds demonstrated that they were not active towards lung cancer (NCI-H1650), hapetocelluar cancer (HepG2), stomach cancer (BGC 823), colon cancer (HCT-116), and breast cancer (MCF-7).20 An unusual dimeric iridal-type triterpenoid, named dibelamcandal A (68) having two iridal type triterpenoid units and linked via a cyclohexene ring (gives rise to dimeriziaton via a [4 + 2]-cycloaddition), was reported from B. chinensis. Furthermore, this compound illustrates promising molluscicide effects towards Pomacea canaliculate with an LC50: 1.3 μg mL−1.29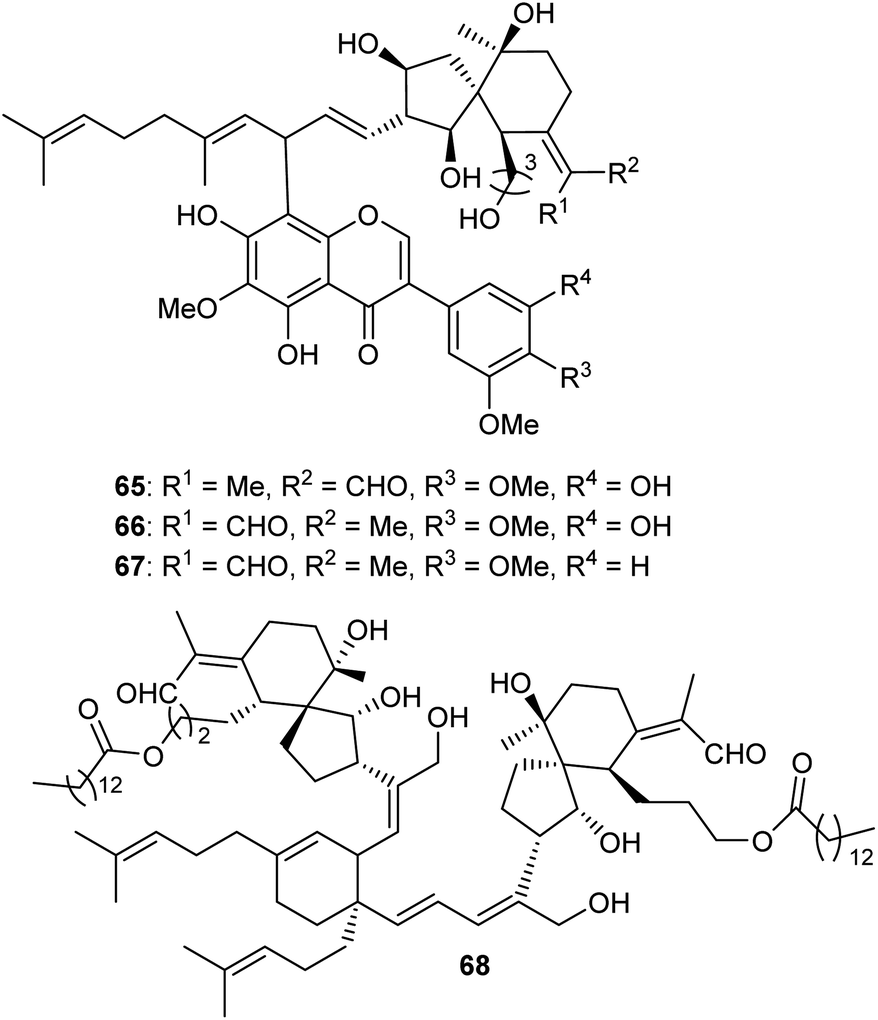 | ||
| Fig. 7 Structures of miscellaneous iridals 65–68. | ||
3 Biosynthesis
3.1 Biosynthesis of monocyclic iridals
Oxidosqualene cyclases are key enzymes which are responsible for the intriguing structural diversity of triterpenes derived from oxidosqualene (69)30,31 (Scheme 1). Over 100 triterpene skeletons have been reported from oxidosqualene (69) via catalytic variability in cation–π annulation, ring expansion, hydride or 1,2-methyl shifts, and deprotonation. In 2006, Xiong et al.30 experimentally proved that oxidosqualene (69) is converted into bicyclic 72via cyclization, hydride and 1,2-methyl shifts, which subsequently undergoes a Grob-type fragmentation to achieve monocyclic iridal skeleton 73. The latter compound could be reduced or oxidized in order to achieve iridals 74a,b (see also Fig. 1). The study further demonstrated that the Arabidopsis thaliana gene At5g42600 is responsible to encode an oxidosqualene cyclase acting as a catalyst for the Grob-type fragmentation. In addition, further hydroxylation and dehydration, and dehydrogenation in the polyene side chains provided iridals 1–19.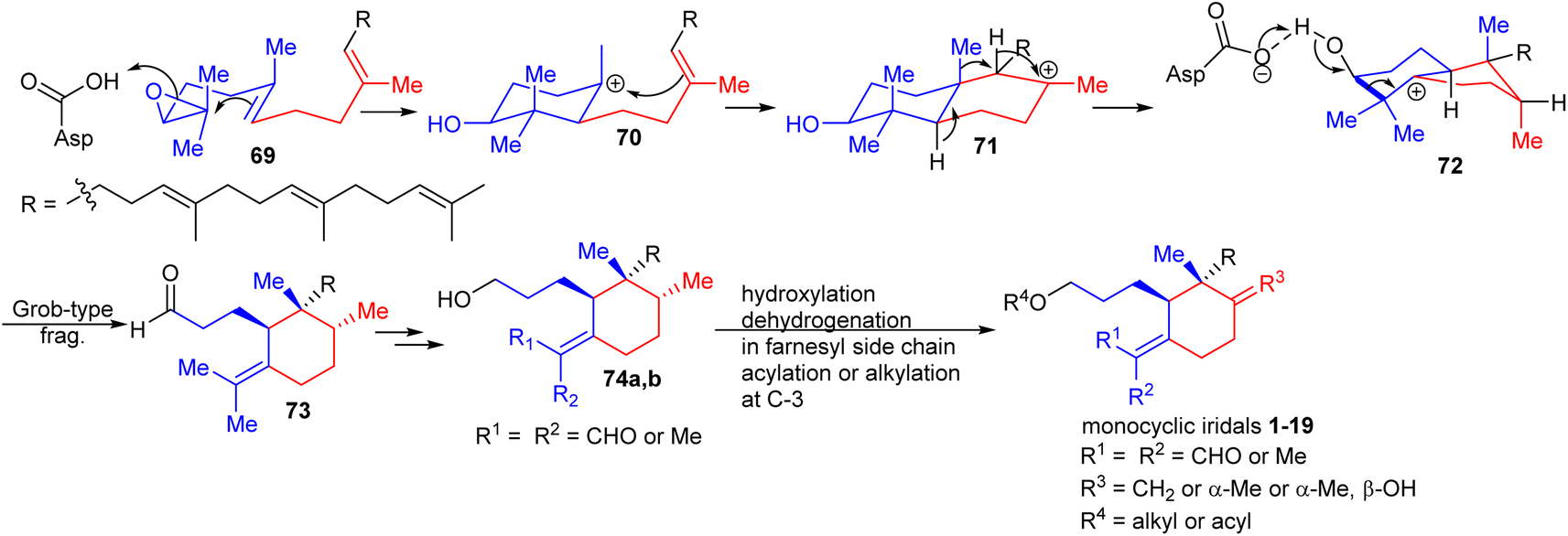 | ||
| Scheme 1 Biosynthesis of monocyclic iridals 1–19. Reproduced from ref. 30 and 31 with permission from the John Wiley and Sons. | ||
3.2 Biosynthesis of bicyclic iridals
Plausible biosynthetic pathways for some bicyclic iridals were proposed as shown in Schemes 2 and 3.23 Upon oxidation of the Me-26 groups in iridal 75a and its geometric isomer 75b,23,32,33 nucleophilic attack of the C-14 double bond to the C-26 aldehyde carbonyl (76a,b) forms the 6/5 spiro bicyclic skeletons 77a,b in an ene-type reaction. Subsequently, further functionalization would be possible at the unsaturated polyene side chain via hydroxylation, dehydrogenation, and acetylation and thus could provides iridals 20 and 21. | ||
| Scheme 2 Hypothetical biosynthesis of bicyclic iridals 20 and 21. Reproduced from ref. 23 with permission from the Elsevier. | ||
 | ||
| Scheme 3 Plausible biosynthesis of bicyclic iridals 22, 24, 25, 28, and 29. Reproduced from ref. 11 and 20 with permission from the Elsevier and Royal Society of Chemistry respectively. | ||
After oxidation of the C-25 aldehyde moiety in iridobelamal A (77) along with C-8 hydroxylation, the subsequent lactonization between the CO2H group at C-25 and the C-8 hydroxyl group to belamcanolide A (22) is initiated (Scheme 3). Moreover, belamcanoxide B (24) and 16-O-acetylbelamcanoxide B (25) are suggested to be derived from belamcanoxide A (79), which would involve an oxa-Michael addition, and acetylation to lead to the target compounds 24 and 25.20 On the other hand, iritectols C (28) and G (29) could biosynthetically be derived from iridobelamal A (77) and its isomeric isoiridogermanal (79), which after epoxidation at C-18/C-19 following by nucleophilic attack of the C-16 hydroxyl group to C-19 is formed.11
3.3 Biosynthesis of tri- and tetracyclic iridals
The biosynthesis of tri- and tetracyclic iridals 30–33 is initiated by C-13 hydroxylation in compound 76 to yield intermediate 82a,b, followed by nucleophilic attack of the C-13 OH group to the C-26 aldehyde carbonyl to achieve the 6/5 spiro bicyclic hemiacetals 83a,b (Scheme 4). Subsequently, epoxidation at C-18/C-19 which is consequently opened by hydroxyl attack at C-23 is followed by the cascade reaction resulting in attack of the resulting C-22 nucleophile at C-18 of the epoxide ring leading to the C-22/C-18 cyclopentane ring systems (85a,b). The latter compound could then undergo a dehydration of the tertiary alcohol to produce iridals 30–33. On the other hand the C-22/C-23 double in the side chain could be epoxidized to form 86a,b and the iridals 35/36 could then be formed through cyclization of the unsaturated side chain via nucleophilic attack of the C-16/C-17 double to C-22 followed by C-13 hydroxylation.23 | ||
| Scheme 4 Plausible biosynthesis of tricyclic iridals 30–33, 35, and 36. Reproduced from ref. 23 with permission from the Elsevier. | ||
The biosynthesis of iridals 37–39 and 54–63 is initiated by acetal formation between the C-3 hydroxyl group and C-26 hemiacetal in 87a,b affording ketal 88a,b which is followed by epoxidation at C-18/C-19 to epoxide 89a,b (Scheme 5).25 Further conversion steps viz., nucleophilic addition of an OH at C-23, nucleophilic attack of the double bond to C-18, and dehydration lead to iridals 54–63. The final formation to iridals 37–39 could be considered by an additional epoxidation at C-15/C-16 to form 90a,b followed by attack at the epoxide ring and cleavage of the cyclopentane ring leading to the ring-opened hydroxy ketones 37–39.25
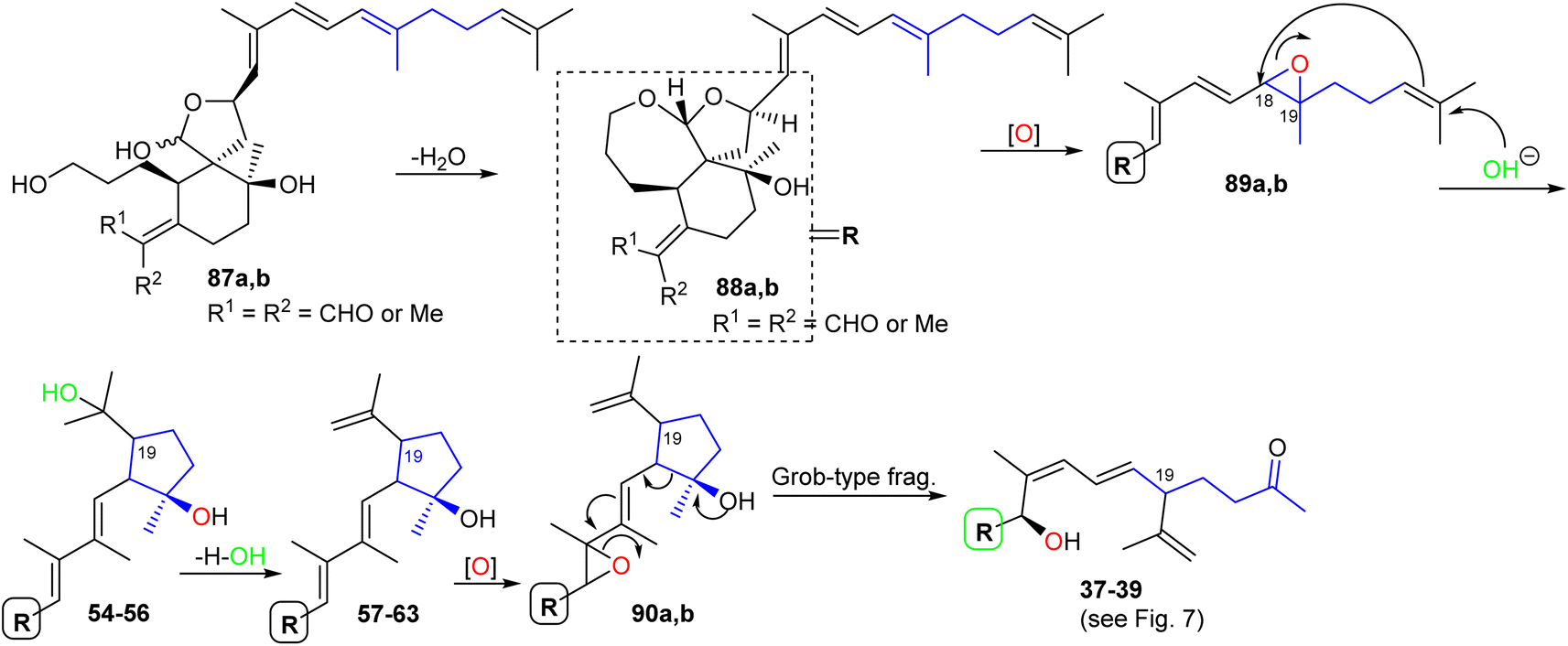 | ||
| Scheme 5 Hypothetical biosynthesis of tricyclic iridals 37–39 and 54–63. Reproduced from ref. 25 with permission from the Elsevier. | ||
For the formation of iridals 40–43, 48–53, spirocyclic 77a,b (Scheme 6) undergoes a 5-exo trig cyclization followed by C-15/C-28 epoxidation and then resulting epoxide hydrolysis could provide iridals 42 and 43. Similarly, the same intermediate might also undergo 6-endo trig cyclization followed by C-15/C-28 epoxidation and dehydration to produce iridals 48 and 49. According to Baldwin's rule, 5-exo trig and 6-endo trig cyclizations are favored but enzymes can possibly catalyze the chemically disfavoured pathways as well.34 Moreover, the biosynthetic precursor of iridals 50–53 might possibly be traced back to either epi-anhydrobelachinal or iso-anhydrobelachinal (91a,b),28,35 which could undergo a series of conversions such as epoxidation at C-22/C-23, nucleophilic hydroxyl addition at C-14, and intramolecular cyclization to form 50–53 (Scheme 6). The biosynthetic route of iridals 40 and 41 could commence from iridals 50–53via acid catalysis to produce the key intermediates 94a,b. Subsequently, intermediates 94a,b might undergo C-13/C-14 bond oxidative cleavage to afford 95a,b. Finally, dehydration of the later compound could form the degraded iridals, iridojaponals A (40) and B (41).16
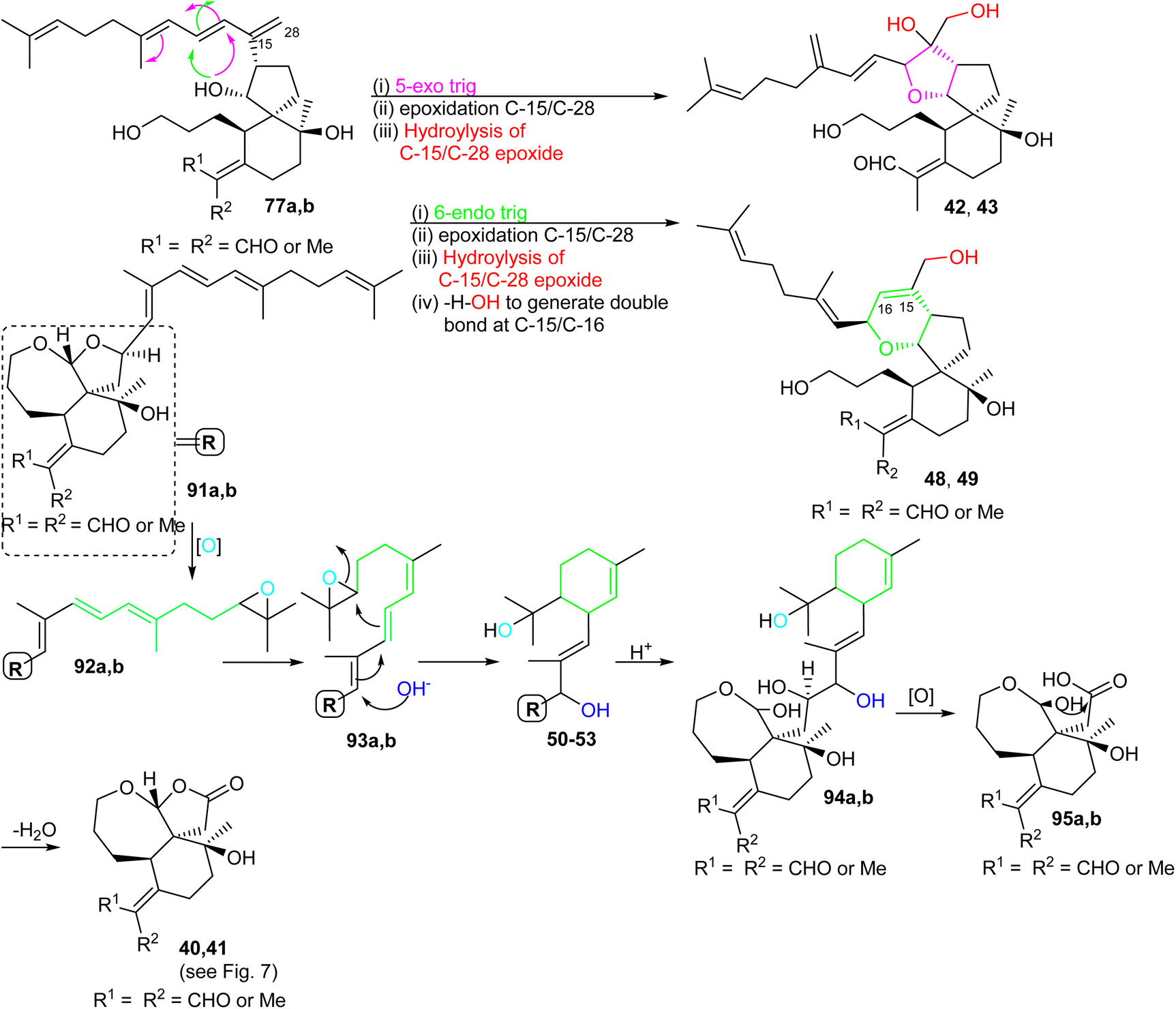 | ||
| Scheme 6 Hypothetical biosynthesis of tricyclic iridals 40–43 and 48–53. Reproduced from ref. 16 with permission from the Elsevier. | ||
3.4 Biosynthesis of miscellaneous iridals
Belamcandal (96) or its geometrical isomer could initially undergo deacetylation, double bond isomerization, and C-17 hydroxylation. Moreover chlorination of the resulting hydroxyl group could afford the chlorinated product 98, which might undergo dechlorination to provide cation 99 (Scheme 7). Subsequent, Friedel–Crafts alkylation between later cation 99 with isoflavone 100via typical Friedel–Crafts alkylation mechanism would produce belamcandanins A-C (65–67).20 On the other hand, dibelamcandal A (68) could be biosynthesized through the [4 + 2]π Diels–Alder reaction at C-29/C-29′ and C-17/C-19′ between the two monomers 101 and 102 (Scheme 7).29 | ||
| Scheme 7 Plausible biosynthesis of miscellaneous iridals 65–68. Reproduced (with slight modification from 97 to 65–67) from ref. 20 and 29 with permission from the Royal Society of Chemistry and American Chemical Society respectively. | ||
4 Tricyclic 6-6-5 triterpenes
4.1 Isomalabaricanes-type terpenes
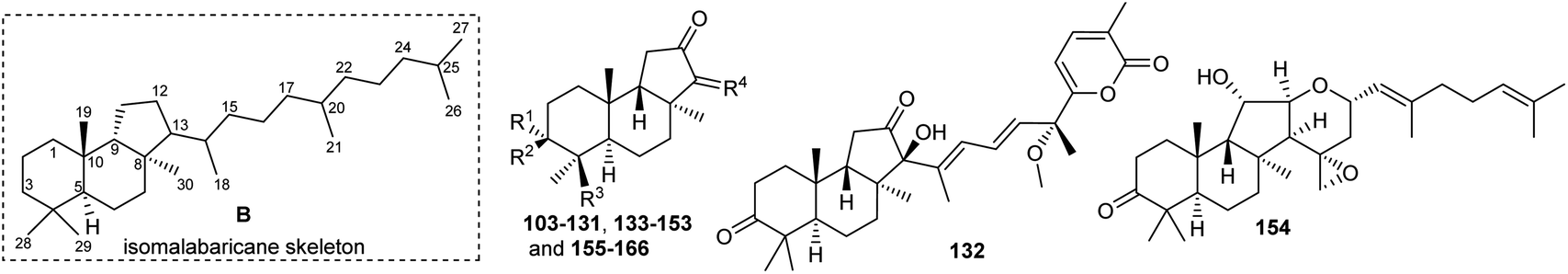
Literature revealed that the genus Rhabdastrella, which can be found in tropical oceans36 produced various isomalabaricane triterpenes with intriguing chemical diversity and demonstrating a wide range of biological effects.37–39 Isomalabaricanes are a group of tricyclic triterpenes bearing a 6/6/5 trans-syn-trans tricyclic core attached to a polyene side chain (Table 1, structure “B”). The difference between isomalabaricanes and malabaricanes is in the stereochemistry of the B–C ring junction: trans-syn-trans (iso) and trans-anti-trans, respectively. The overwhelming number of structural features distinguishing these metabolites can be found in the terpenoid side chain, which consists of acyclic isoprene-units up to sesquiterpenes. The side chains remain acyclic, or depending on the oxidation state, have been cyclized into gamma and delta lactones. Carbocyclic moieties within the side-chains most likely result from ene- or aldol reactions.|
|
|||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | Source |
| Rhabdastrellin A (103) | H | OH | CH2OH |
|
Rhabdastrella aff. distincta36 |
| Rhabdastrellin B (104) | H | OH | CH2OH |

|
Rhabdastrella aff. distincta36 |
| Rhabdastrellin C (105) | H | OH | CH2OH |
|
Rhabdastrella aff. distincta36 |
| Rhabdastrellin D (106) | H | OH | CH2OH |

|
Rhabdastrella aff. distincta36 |
| Rhabdastrellin E (107) | H | OH | Me |
|
Rhabdastrella aff. distincta36 |
| Rhabdastrellin F (108) | H | OH | Me |
|
Rhabdastrella aff. distincta36 |
| Rhabdastrellin G (109) | O | — | Me |

|
Rhabdastrella providentiae 41 |
| Rhabdastrellin H (110) | O | — | Me |
|
Rhabdastrella providentiae 41 |
| Rhabdastrellin I (111) | O | — | Me |

|
Rhabdastrella providentiae 41 |
| Rhabdastrellin J (112) | H | OAc | Me |

|
Rhabdastrella providentiae 41 |
| Rhabdastrellin K (113) | H | OAc | Me |
|
Rhabdastrella providentiae 41 |
| Globostelletin B (114) | O | — | Me |

|
Rhabdastrella globostellata 42 |
| Globostelletin C (115) | O | — | Me |

|
Rhabdastrella globostellata 42 |
| Globostelletin D (116) | O | — | Me |

|
Rhabdastrella globostellata 42 |
| Globostelletin E (117) | O | — | Me |

|
Rhabdastrella globostellata 42 |
| Globostelletin F (118) | O | — | Me |

|
Rhabdastrella globostellata 42 |
| Globostelletin G (119) | O | — | Me |

|
Rhabdastrella globostellata 42 |
| Globostelletin H (120) | O | — | Me |

|
Rhabdastrella globostellata 42 |
| Globostelletin I (121) | O | — | Me |
|
Rhabdastrella globostellata 42 |
| Globostelletin J (122) | O | — | Me |
|
Rhabdastrella globostellata 43 |
| Globostelletin K (123) | O | — | Me |
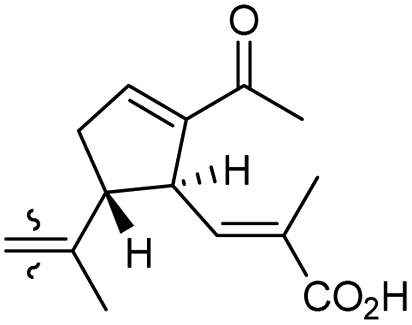
|
Rhabdastrella globostellata 43 |
| Globostelletin L (124) | O | — | Me |
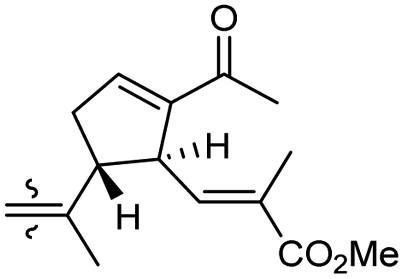
|
Rhabdastrella globostellata 43 |
| Globostelletin M (125) | O | — | Me |
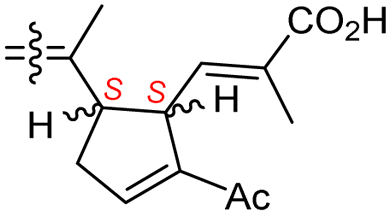
|
Rhabdastrella globostellata 43 |
| Globostelletin N (126) | O | — | Me |

|
Rhabdastrella globostellata 43 |
| Globostelletin O (127) | H | OH | CH2OH |
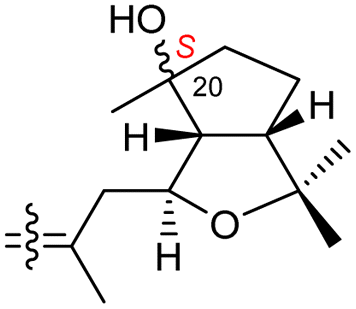
|
Rhabdastrella globostellata 43 |
| Globostelletin P (128) | H | OH | CH2OH |

|
Rhabdastrella globostellata 43 |
| Globostelletin Q (129) | H | OH | CH2OH |
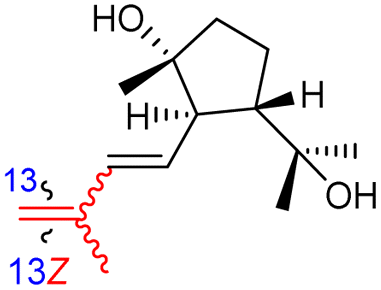
|
Rhabdastrella globostellata 43 |
| Globostelletin R (130) | H | OH | CH2OH |
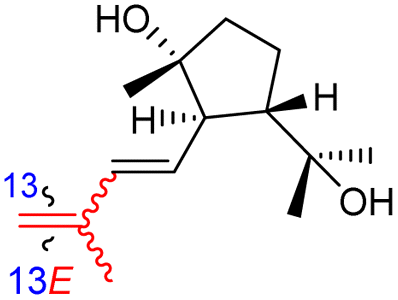
|
Rhabdastrella globostellata 43 |
| Stellettin N (131) | H | OAc | Me |
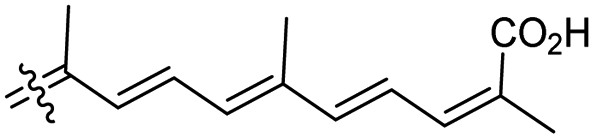
|
Stelletta sp.44 |
| Stellettin O (133) | O | — | Me |

|
Stelletta tenuis 45 |
| Stellettin P (134) | O | — | Me |
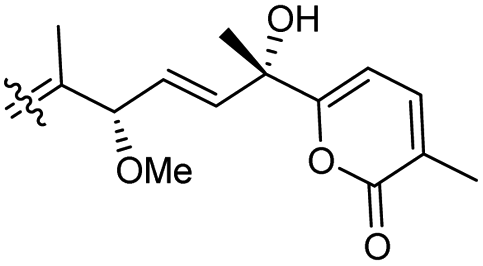
|
Stelletta tenuis 45 |
| Stellettin Q (135) | H | OAc | Me |

|
Stelletta sp.47 |
| Stellettin R (136) | H | OAc | Me |

|
Stelletta sp47 |
| Auroral 137 | OH | H | CH2OH |
|
Rhabdastrella globostellata 48 |
| Auroral 138 | OH | H | CH2OH |

|
Rhabdastrella globostellata 48 |
| Auroral 139 | OH | H | CH2OH |
|
Rhabdastrella globostellata 48 |
| Auroral 140 | OH | H | CH2OH |
|
Rhabdastrella globostellata 48 |
| Jaspiferin C (141) | O | — | Me |
|
Jaspis stellifera 49 |
| Jaspiferin D (142) | H | OH | CH2OAc |

|
Jaspis stellifera 49 |
| Jaspiferin E (143) | H | OH | CH2OAc |
|
Jaspis stellifera 49 |
| Jaspiferin F (144) | H | OH | Me |
|
Jaspis stellifera 49 |
| Jaspiferin G (145) | H | OH | CH2OH |

|
Jaspis stellifera 50 |
| Jaspiferin H (146) | O | — | Me |

|
Jaspis stellifera 51 |
| Jaspiferin I (147) | H | OH | Me |
|
Jaspis stellifera 51 |
| Jaspiferin J (148) | H | OH | Me |

|
Jaspis stellifera 51 |
| Rhabdaprovidine A (149) | H | OAc | Me |

|
Rhabdastrella providentiae 52 |
| Rhabdaprovidine B (150) | H | OAc | Me |
|
Rhabdastrella providentiae 52 |
| Rhabdaprovidine C (151) | H | OAc | Me |
|
Rhabdastrella providentiae 52 |
| Rhabdaprovidine D (152) | O | — | Me |

|
Rhabdastrella providentiae 53 |
| Rhabdaprovidine E (153) | O | — | Me |

|
Rhabdastrella providentiae 53 |
| Stelliferin J (155) | H | OAc | Me |
|
Rhabdastrella globostellata 46 |
| Stelliferin K (156) | H | OAc | Me |

|
Rhabdastrella globostellata 46 |
| Stelliferin L (157) | H | OAc | Me |

|
Rhabdastrella globostellata 46 |
| Stelliferin M (158) | H | OAc | Me |
|
Rhabdastrella globostellata 46 |
| Stelliferin N (159) | H | OAc | Me |
|
Rhabdastrella globostellata 46 |
| Rhabdastin A (160) | H | OAc | Me |

|
Rhabdastrella globostellata 54 |
| Rhabdastin B (161) | H | OH | Me |

|
Rhabdastrella globostellata 54 |
| Rhabdastin C (162) | H | OAc | CO2Me |

|
Rhabdastrella globostellata 54 |
| Rhabdastin D (163) | H | OAc | Me |
|
Rhabdastrella globostellata 54 |
| Rhabdastin E (164) | H | OAc | Me |
|
Rhabdastrella globostellata 54 |
| Rhabdastin F (165) | H | OAc | Me |

|
Rhabdastrella globostellata 54 |
| Rhabdastin G (166) | H | OAc | Me |
|
Rhabdastrella globostellata 54 |
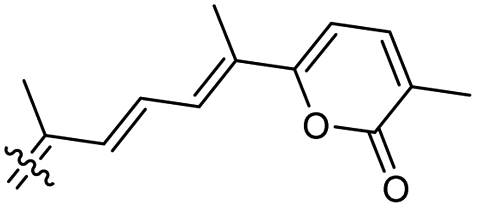
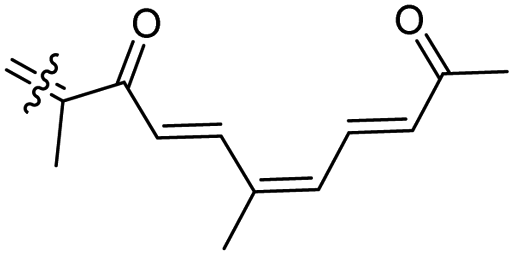
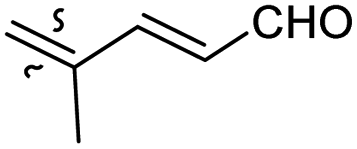
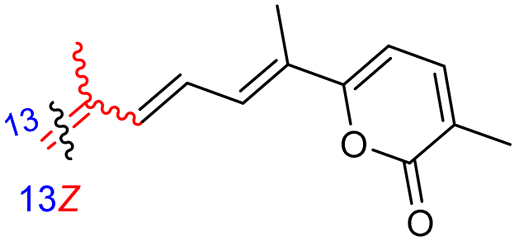
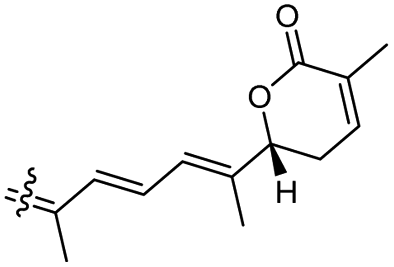
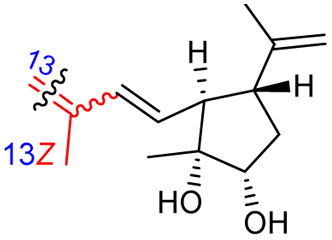
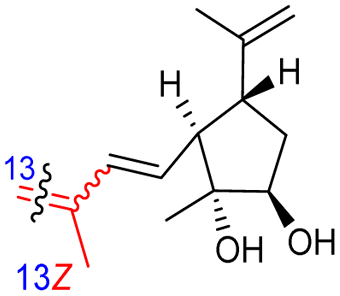
Among the isomalabaricanes isolated from R. aff. distincta (103–108), only rhabdastrellin A (103) showed moderate cytotoxic effects toward leukemia (HL-60: IC50: 4.2 μg mL−1), which may be related to the 2-pyranone modified side chain and which closely resembles previously reported Jaspolides A and B.40 Rhabdastrellins B–F (104–108) contain only acyclic side chains altering in their oxidized positions, although the stereochemistry of the C-24 hydroxyl group of the side chain in Rhabdastrellin B (104) could not be unambiguously established. For the relative stereoconfiguration of isomalabaricanes, it was demonstrated during isolation, that these triterpenoids are isomerized at C-13 via exposure to light and thus, it was suggested that the 13-Z isomers might be artefacts rather than new entities.36 In another report, Kiem et al.41 reported isomalabaricane derived nor-terpenoids named rhabdastrellins G–K (109–113) from the sponge R. providentiae. Compounds 109–111 possessed cytotoxic effects towards HepG2, MCF-7, LU-1, SK-Mel2, and HL-60 with IC50 ranging from 11 to 85 μM. Notably, the 20(22)-Z isomer 110 illustrated much better cytotoxic effects (IC50: from 11 to 16 μM) in comparison with the 20(22)-E isomer 109 (IC50: from 56 to 87 μM).
The marine sponge R. globostellata produced the side-chain shortened isomalabaricane triterpenes globostelletins B–I (114–121) (Table 1).42 Compounds 114–121 were tested against various cancer cell lines with only moderate activities. Again, among the 13E/13-Z isomers, globostelletins H (120) and I (121), isomer 121 (13-Z) illustrated better inhibitory effects towards A2780 (IC50 = 7.6 μM) than its E isomer viz., compound 120 (13-E) (A2780: IC50 = 8.1 μM).
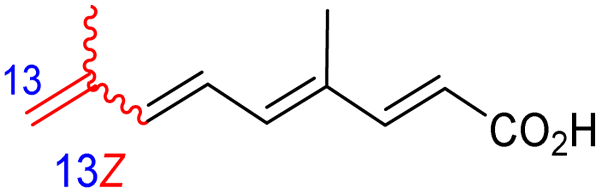
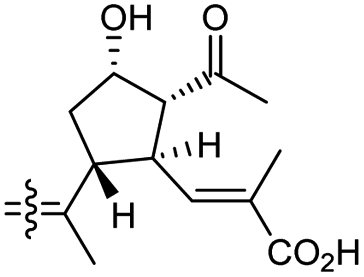
The same organism also produced globostelletins J–R (122–130) bearing cyclopentane modified side chains. Natural products 122–126 have the five-membered cyclopentane core formed between C-15 and C-23 in the polyene side chain while compounds 127 and 128 (Table 1) feature a 25-dimethyltetrahydrofuran moiety, which was joined to another cyclopentane core such as 20-hydroxy-20-methylcyclopentane at the C-17 and C-24 positions.43 Moreover, compounds 129 and 130 bearing a substituted cyclopentane core feature a hydroxyl group at C-22. Globostelletins K (123) and L (124) illustrated moderate inhibition towards focal adhesion kinase (FAK), protein kinases (ALK), Aurora-B, and VEGF-R2. On the other hand, the remaining compounds were not active towards sixteen tumor-related protein kinase enzymes.43
In another report the sponge Stelletta sp. collected from Hainan, China, produced the isomalabaricane triterpene stellettin N (131).44 This single member was associated with a natural product family resulting from isolation studies done on St. tenuis. In this regard Li et al.45 isolated isomalabaricane triterpenes viz., stellettins N–P (132–134) from this marine sponge. Unfortunately, one of the compounds viz., 132 was again published as “stellettin N′′ because metabolite 131 was already named as “stellettin N”.44 Some features of compound 132 are unique such as the C-13 β-hydroxyl group as well as the saturated C-13/C-14 bond. The vast majority of this family have fully conjugated polyene side chains. Very few isomalabaricane triterpenes have been reported which have only one or two of the normally unsaturated bonds saturated viz., stelliferins J–N and globostelletins J–P.43,46 Compounds 132–134 were evaluated against lung cancer (A549), gastric cancer (AGS), and glioblastoma (U-251MG) where it was found that these compounds were only active towards AGS with IC50: in the range from 4.5 to 9.6 μM.45 Stellettins Q (135) and R (136) were isolated from Stelletta sp. and both compounds have a cyclopentane ring in the polyene side chain formed between C-15 and C-22. Moreover, isomalabaricane triterpenes 135 and 136 (Table 1) are a 13-Z/E geometric pair of the same 15R,23S isomers.47
Isomalabaricanes such aurorals 137–140 are produced by the sponge R. globostellata. Biological studies revealed that aurorals 137 and 138 possessed good cytotoxic effects towards epidermoid carcinoma (KB cells) with ID50: 0.2 μg mL−1. On the other hand, compounds 139 and 140 displayed moderate effects with an ID50: 8 μg mL−1.48
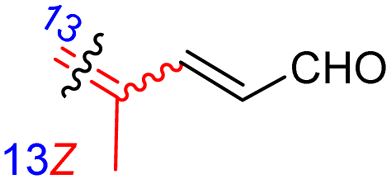
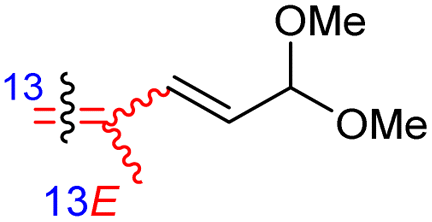
Jaspiferins C–F (141–144) were isolated from the marine sponge Jaspis stellifera and jaspiferin C (141) has a six-membered carbon core formed between C-15 and C-23 which is rather rare among isomalabaricane triterpenes.49 An isomalabaricane triterpenoid, jaspiferin G (145), comprising a cyclopentane ring in the pendant polyene side chain was produced by the sponge Jaspis stellifera.50 Jaspiferins H-J (146–148) were obtained from J. stellifera, among which Jaspiferin H (146) has a five membered lactone ring (γ-lacone).51 From the sponge R. providentiae Dung et al.52,53 reported rhabdaprovidines A–E (149–153) and G (154). The latter one, rhabdaprovidine G (154) has an additional pyran ring formed between C-12 and C-15. Notably, this metabolite possesses a novel chemical structure of the 6-6-5-6-tetracyclic core along with nine chiral carbon centres which is quite unique among isomalabaricane skeletons.53 Bioactivity studies revealed that rhabdaprovidines A–C (149–151) inhibited NO production in LPS in which the IC50: ranged from 17.5 to 46.8 μM with 150 (Table 1) being the most potent IC50: 17.5 μM.52
Stelliferins J–N (155–159) were isolated from the sponge R. globostellata collected from Hainan Island, China. The configuration of the stereogenic centres in triterpenoids 155–159 were determined via CD and NMR measurements using Mosher's method. All these natural products possess a diol (secondary/tertiary hydroxyl group) in the polyene side chains with terpenes 155 and 156 featuring a acyclic side chain and 157–159 a cyclopentane diol unit. Metabolites 157 and 159 illustrated antimicrobial effects towards Bacillus subtilis with IC50 8 μg mL−1.46
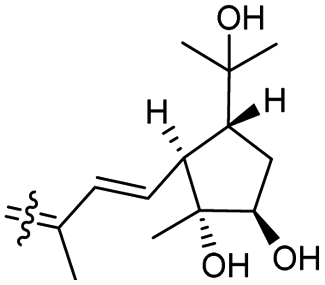
Rhabdastins A–G (160–166, Table 1) were produced by the sponge R. globostellata. Furthermore, compounds 160–166 have a cyclopentane group generated in the conjugated polyene side chains.54 Compounds 163, 164, and 166 demonstrated modest cytotoxicity on leukemia cells (HL-60) with IC50: 21, 29, and 44 μM, respectively. Isomalabaricanes 163 and 164 have been studied in detail revealing to cause caspase 3 cleavage in HL-60 cells at 10 μM without effecting expression of Bcl-2 in HL-60 cells.54 Among the latest natural products of this class, Lai et al.55 isolated rhabdastins H (167) and (168) (Fig. 8) from the sponge Rhabdastrella sp. These isolates demonstrate anti-proliferative effects towards leukemic cells (Molt4 and K562) with IC50 values ranging from 9.8 to 16.5 μM. Jaspolides G (169) and H (170), two unusual bisisomalabaricanes isomers were reported from the marine sponge Jaspis sp.56 The intriguing structures not only involve two isomalabaricane moieties, they also possess 2-pyrone heterocycles, which makes the C60 natural product even more impressive.
 | ||
| Fig. 8 Structures of isomalabaricane-type triterpenoids 167,168 and dimeric isomalabaricane 169,170. | ||
4.2 Isomalabaricane-derived nor-terpenoids
Two C-19-nor isomalabaricane triterpenes named cyclobutastellettolides A (171) and B (172) were produced by Stelletta sp. (Fig. 9). The bioassay-guided isolation revealed them to significantly enhance reactive oxygen species (ROS) generation in murine macrophages. Notably, these two compounds comprise four fused rings, of which one is a cyclobutanone and this is indeed a rare feature in isomalabaricane triterpenes.57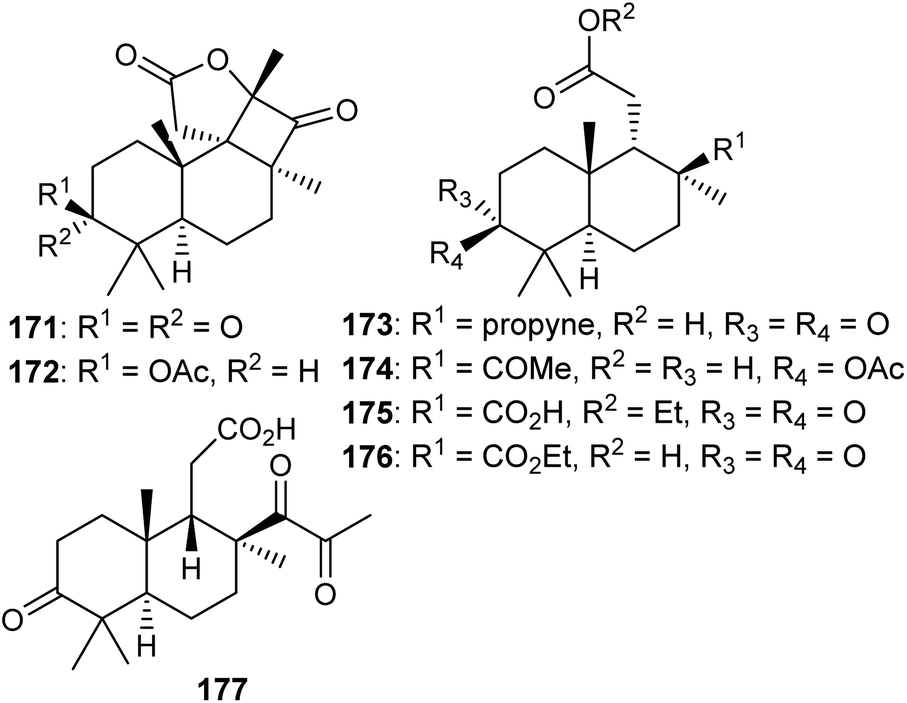 | ||
| Fig. 9 Structures of isomalabaricane-derived nor-terpenoids 171–177. | ||
In another report, isomalabaricane-derived nor-terpenoids, stellettins S–V (173–176) were isolated from the marine sponge Stelletta sp. Stellettin S (173) represents an intriguing example because this compound has an alkyne group and this is the first example of an isomalabaricane triterpene which features this rare functionality as a substituent.47 The marine sponge R. globostellata produces nor-isomalabaricanes triterpene globostelletin A (177) which has a rather unique structure.42
4.3 Malabaricane type triterpenes
Malabaricanes are a group of tricyclic triterpenes bearing a 6-6-5 trans-anti-trans tricyclic core attached to a polyene side chain (Fig. 10, structure “C”). Six malabaricane type triterpenes 178, 179 with cyclobutane modified side chains, and 182–185 (Fig. 10) were reported from the oleoresin of Ailanthus malabarica.58 During the time covered by this report, Duan et al.59 demonstrated that triterpenes 178 and 179 with the suggested unusual cyclobutane modifications have the same NMR data (in particular 13C NMR data) as the dammarane-type triterpenenes ocotillone (180) and gardaubryone C (181), respectively, leading to a revision of the formerly proposed structures. Of note, their structural reassignment was established by further using DFT chemical shift analysis and the CASE algorithm. Bio-guided isolation studies on Commiphora africana provided the malabaricane triterpene commafric A (186) bearing a methyl group at C-13 instead of C-8. It illustrated significant cytotoxic effects towards lung cancer lines (A549: IC50: 4.52 μg mL−1) along with moderate effects towards ovarian cancer (A2780), stomach cancer (SNU638), and pancreatic cancer (MIA-PaCa-2), with IC50 ∼ 10 μg mL−1.60 | ||
| Fig. 10 Structures of malabaricane-type triterpenoids 178, 179, 182–190. | ||
Modified side chains have been identified in ailanthusins F (187) and G (188) (Fig. 10), which are produced by the stem of Ailanthus triphysa, a tree commonly found in Thailand and Australia. Moreover, these metabolites were not active in cytotoxicity screening towards MOLT-3, HepG2, MDA-MB-321, HuCCA-1, T47-D, A549, HeLa, and MRC-5.61 In another report, Messina et al.62 isolated malabaricatrienone (189) and malabaricatrienol (190) from the methanol extract of the resin of Bursera microphylla which is called “elephant tree” and is widely present in Mexico. In addition, these metabolites were not active against various murine and human cancer cells.
5 Biosynthesis of isomalabaricane and malabaricane trietrpenoids
Isomalabaricanes and malabaricanes feature 6-6-5 trans-syn-trans and trans-anti-trans tricyclic cores while both skeletons have different stereochemistry at the C-8 and C-9 positions (Table 1 and Fig. 10). The basic core skeleton of isomalabaricane 195 is thought to be biosynthetically generated through cyclizations of 2,3-oxidosqualene (69) via chair−boat−chair (191) transition states (Scheme 8).57,63,64 Similarly, it has been suggested that the biosynthesis of the basic core skeleton of malabaricane triterpenoids also proceeds through cyclizations of 2,3-oxidosqualene (69) via a chair–chair–chair (193) transition state57,63,64 and triterpene synthases are responsible to generate the malabaricane basic skeleton 196 (Scheme 8).64,65 | ||
| Scheme 8 Hypothetical biosynthesis of isomalabaricane (195) malabaricane (196) basic skeletons. Reproduced from ref. 64 and 65 with permission from the American Chemical Society. | ||
It has been reported that two Arabidopsis thaliana genes including At5g4810 and At4g15340 encode OSCs which furnish arabidiol and thalianol.8,66,67 In addition, numerous malabaricanes have been obtained via site-directed mutagenesis of A. acidocaldarius squalene hopene cyclase which in turn comprise point mutants of Tyr420, Phe601 and Ile261.8,68,69 This suggests that the malabaricanes are produced in nature with custom tailored enzymes. Lodeiro et al.65 demonstrated that the isomalabaricane core arises from a lanosterol cyclase mutant (Tyr510) while the native enzyme has yet to be discovered. Stelletin E (197) (Scheme 9) could be the plausible precursor to generate globostelletins B (114), D (116), F (118), and I (121) via oxidative degradation reactions.42,57,70
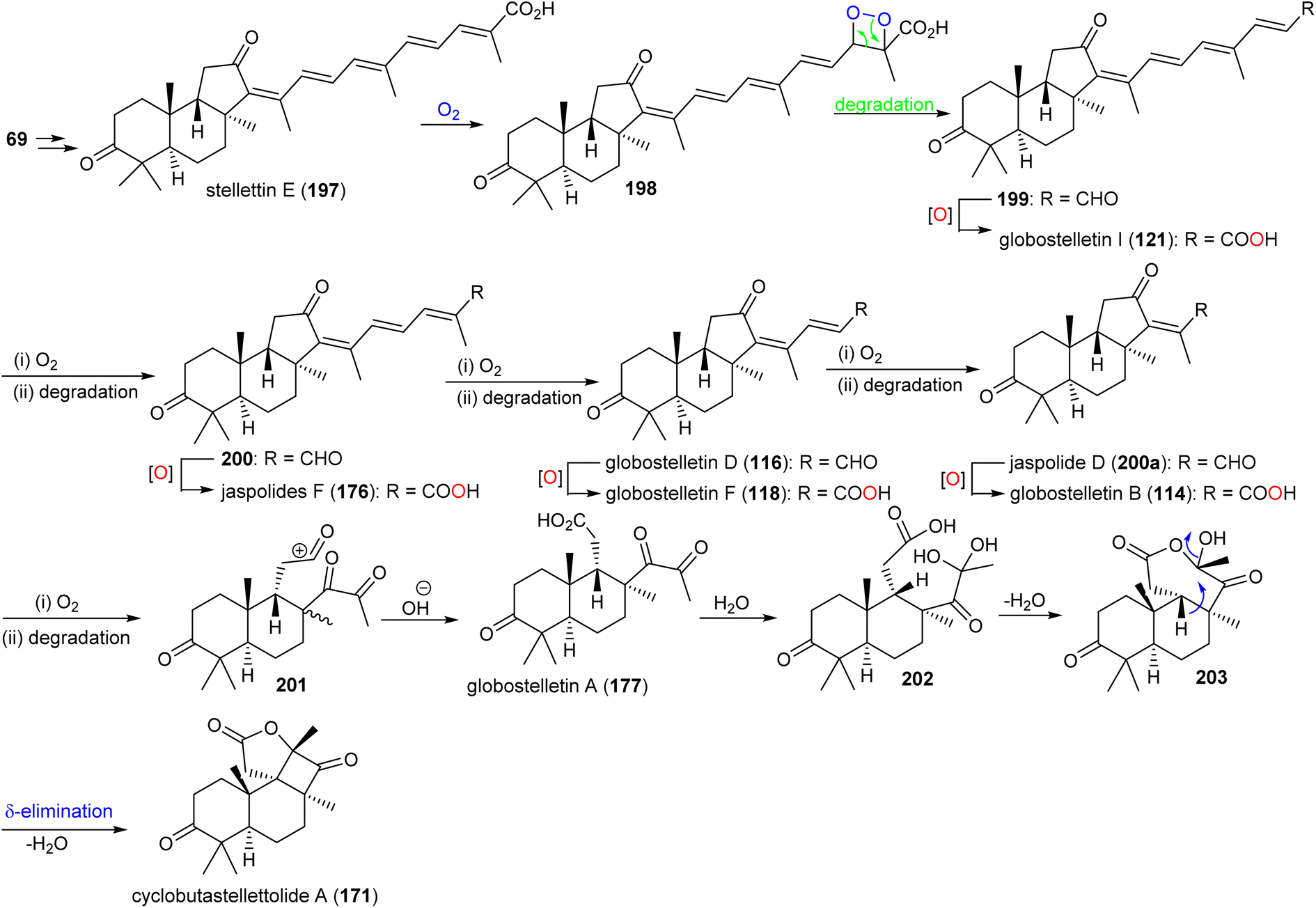 | ||
| Scheme 9 Hypothetical biosynthesis of isomalabaricane triterpenoids 114, 116, 118, 121, 171 and 177. Reproduced from ref. 57 with permission from the American Chemical Society. | ||
Globostelletin A (177) is assumed to be generated by oxidative degradation (loss of CO2) from globostelletin B (114) and therefore it is also considered to be a member of the isomalabaricane class42 (Scheme 9). Moreover, globostelletin A (177) is considered as the putative biosynthetic precursor of cyclobutastellettolide A (171) (Scheme 9). Globostelletin A (177), or its hydrate 202 may be converted into cyclobutastellettolides A (171) via an intramolecular dehydration followed by δ-elimination. The ΔG calculation results for possible biosynthetic intermediates indicated that the seven-membered ring formation in tricyclic 171 is preferred to happen prior to the formation of the fused γ-lactone, in contrast to the simultaneous generation of the cyclobutane-fused lactone.57 Cyclobutastellettolide B (172) was presumed to be generated by the same protocol from stellettin H or I (3-OAc derivatives of stellettin E).57,71
Stellettin E (196), which co-existed in the same specimen (R. globostellata), is thought to be considered as the putative precursor for globostelletins J–L (122–124), which bear an unusual cyclopentane derived side chain (Scheme 10).43 Formation of this unit might be initiated via a 15,23-electrocyclization followed by 20,22-epoxidation to afford the cyclohexane unit 204. Moreover, the later intermediate could then undergo a semi-pinacol rearrangement, 17-hydroxylation, dehydration and methylation to generate the globostelletine triterpenes 122–124. On the other hand, 3-epi-stelletin J (206) could be derived from stellettin E (196) and possibly be the precursor for the biosynthetic formation of globostelletins O–Q (127–129). Considering the very likely epoxidation at C-24/C-25 to form epoxide 207 followed by attack by the nucleophilic Δ17(20) double bond and hydroxylation of the resulting tertiary carbocation at C-20 (208), this will eventually provide globostelletin Q (129). Globostelletins O (127) and P (128) are more than likely to be derived from 129 through nucleophilic attack of the C-25 hydroxyl group at the C-16 double bond to form the pyran ring.
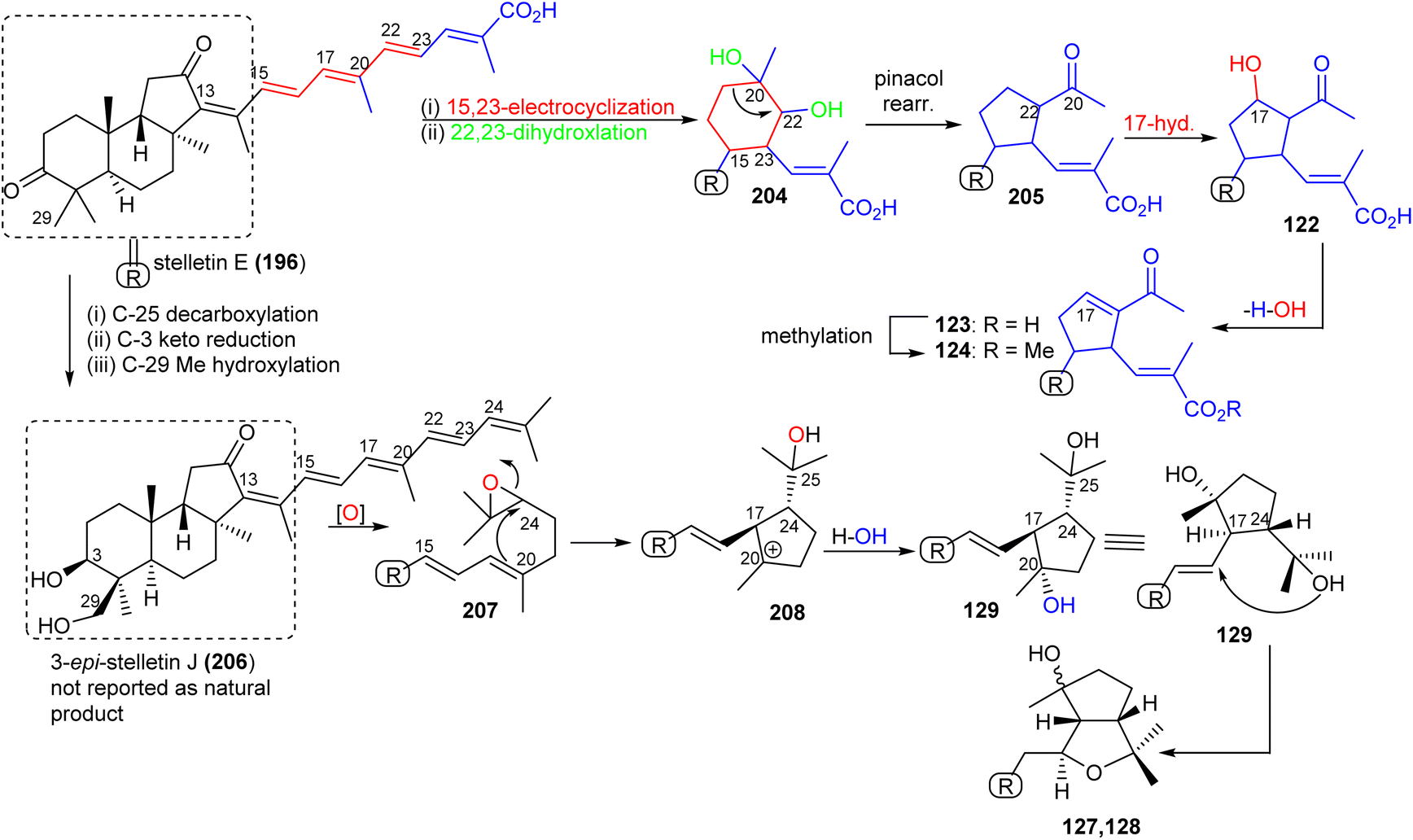 | ||
| Scheme 10 Hypothetical biosynthesis of isomalabaricane triterpenoids 122–124 and 127–129. Reproduced from ref. 43 with permission from Elsevier. | ||
Stelliferins can be considered to be starting materials for another modified isomalabaricane, Jaspiferin C (141). It contains a cyclohexenyl moiety in the side chain which is most likely derived from stellettin E (196). Its biosynthesis could initially be formed by lactonization between the C-25 carboxylic acid group attacking the C-24 double bond followed by dehydrogenation to afford 2-pyrone-modified stelletin B (209) (Scheme 11). The resulting lactone ring in the stelletin B (209) could reasonably be ring-opened via an hydroxyl attack to generate intermediate 210. The later intermediate features an enolate tautomer. Finally, the cyclohexenyl moiety may be generated between C-15 and C-23 to form the resulting cyclohexenone core unit in the polyene side chain of jaspiferin C (141).49
 | ||
| Scheme 11 Plausible biosynthesis of isomalabaricane triterpenoid 141. Reproduced from ref. 49 with permission from the Taylor & Francis. | ||
Rhabdastins A–C (160–162) are thought to be biogenetically generated via oxidative cleavage of stelliferin derivatives as described for globostelletin B (114) (Scheme 9). A proposed biogenetic route for rhabdastins D–G (163–166) via oxidation and cyclization of stelliferins is depicted in Scheme 12.54 Stelliferin D (212) formation could proceed by initial C-3 acetylation and C-21 hydroxylation followed by epoxidation of the double bond at C-24 in stelliferin A (213)72 to form epoxide 214 at C-24/C-25. Thereafter, attack by the Δ17(20) double bond in later compound may cleave the epoxide and provide a tertiary carbocation at C-20 (215).
 | ||
| Scheme 12 Hypothetical biosynthesis of isomalabaricane triterpenoid 163–166. Reproduced from ref. 54 with permission from the American Chemical Society. | ||
This could then be followed by nucleophilic attack by water from the α-face to produce rhabdastin G (166) which in turn could be followed by the C-25 OH group being lost via a dehydration to give rhabdastin E (164). On the other hand, rhabdastin F (165) may be a photoisomer of 164, because this later compound was slowly transformed into its C-13 isomer 165 under room light conditions. In addition, rhabdastin D (163) may presumably be formed through a similar route from stelliferin B (218)72 including epoxidation, epoxide ring-opening, α-face nucleophilic attack by water as described for compounds 164–166. Additionally, alternative biogenetic routes for compounds 163–166via formation of stelliferin C (216) from stelliferin A (213)72 are also depicted in Scheme 12.
Jaspolides G (169) and H (170) are the first examples of isomalabaricane based dimers and these are geometrical isomers with respect to the C-13 double bond.56 The authors postulated that the structure of jaspolide G (169) could be biosynthetically formed between stellettin A (221)73 and isogeoditin A (223)74 which are representative isomalabaricane type triterpenes and a nor-triterpenes, respectively56 (Scheme 13). Initially, hydroxylation might occur at C-13 of stellettin A (221) which would afford the intermediate 222 with a concomitant migration of the double bond at C-14/C-18. A double bond between C-23/C-24 of geoditin A (223) and the conjugated double bonds at C-15/C-16 and C-14/C-18 of the intermediate 222 might then undergo a [4 + 2]π cycloaddition to afford the corresponding dimer containing the newly formed cyclohexene ring. The C-13 isomerization viz., Z-form (169) to E-form (170) is frequently present in isomalabaricanes due to their thermal sensitivity.
 | ||
| Scheme 13 Hypothetical biosynthesis of isomalabaricane triterpenoids 169 and 170. Reproduced from ref. 56 with permission from the Elsevier. | ||
5 Tetranordammarane and polypodane triterpenoids
Three unusual triterpene saponins named nototronesides A–C (224–226) (Fig. 11), that feature a rare 6/6/9 tricyclic tetranordammarane core, are produced by Panax notoginseng, commonly referred Chinese ginseng. They were screened for their neuroprotective effects where it was found that metabolite 225 enhanced the cell viability remarkably by 79%.75Commiphora mukul gum resin, also known as guggul, has been employed to treat obesity, inflammation, and lipid disorders in Ayurvedic medicine. Myrrhanol C (227) and its corresponding keto analog myrrhanone C (228), were reported from this guggul gum resin.76,77 Furthermore, myrrhanol C (227) was reported from Pistacia lentiscus resin and found to possess potent effects towards prostate cancer cells.78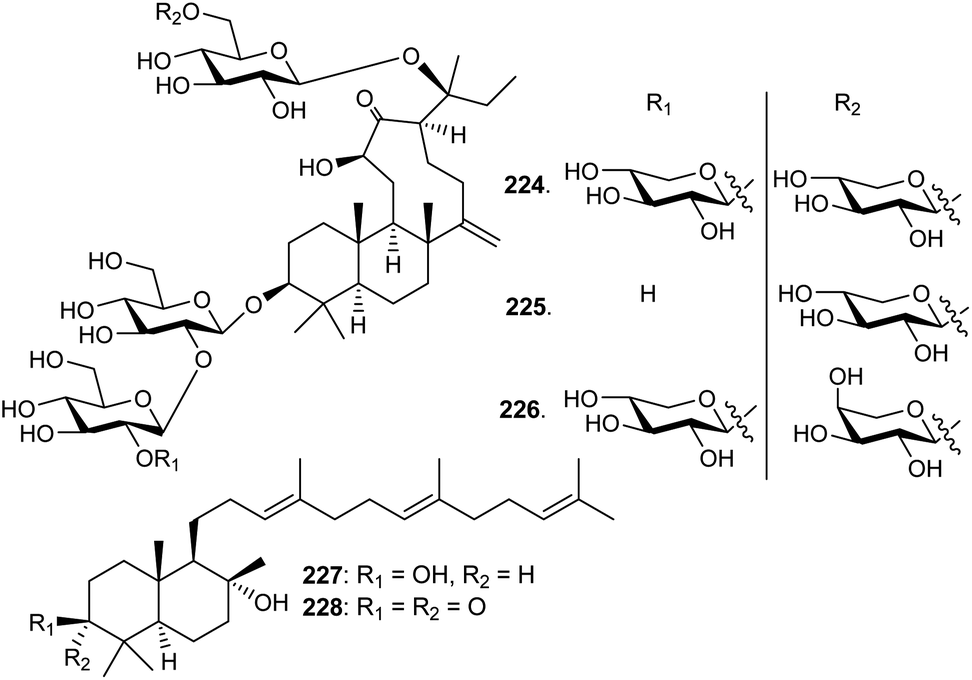 | ||
| Fig. 11 Structures of triterpenes 224–228. | ||
6 Biosynthesis of tetranordammaranes
The aglycon of nototroneside A (224) (nototrone A, 238) represents the first example of an unusual tricyclic dammarane-type triterpene.75 A proposed biosynthetic pathway of nototrone A (238) is depicted in Scheme 14.75 In this scheme the authors proposed that tricyclic (238) is biosynthetically derived from protopanaxadiol (229), which is a reported aglycon of numerous saponins from Panax plants.75,79,80 Initially, dehydration of the C-12 hydroxyl group of protopanaxadiol (229) would generate a double bond at the Δ12,13 position, which could subsequently undergo epoxidation via an enzyme-catalyzed epoxidation to afford the corresponding oxidized analog 231. This latter derivative could then undergo a fragmentation of the highly strained epoxide intermediate by cleavage of the C-13/C-14 bond to produce a nine-membered enol 232.81 The intermediate enol 232 is thought to then undergo keto–enol a plausible tautomerisation which would lead to the generation the keto anion 233 and upon a deprotonation lead to the formation of tricyclic triterpene 234. Enzymatic oxidation of 234 to acid 235, its decarboxylation and C-13 oxidation would ultimately lead to nototrone A (238).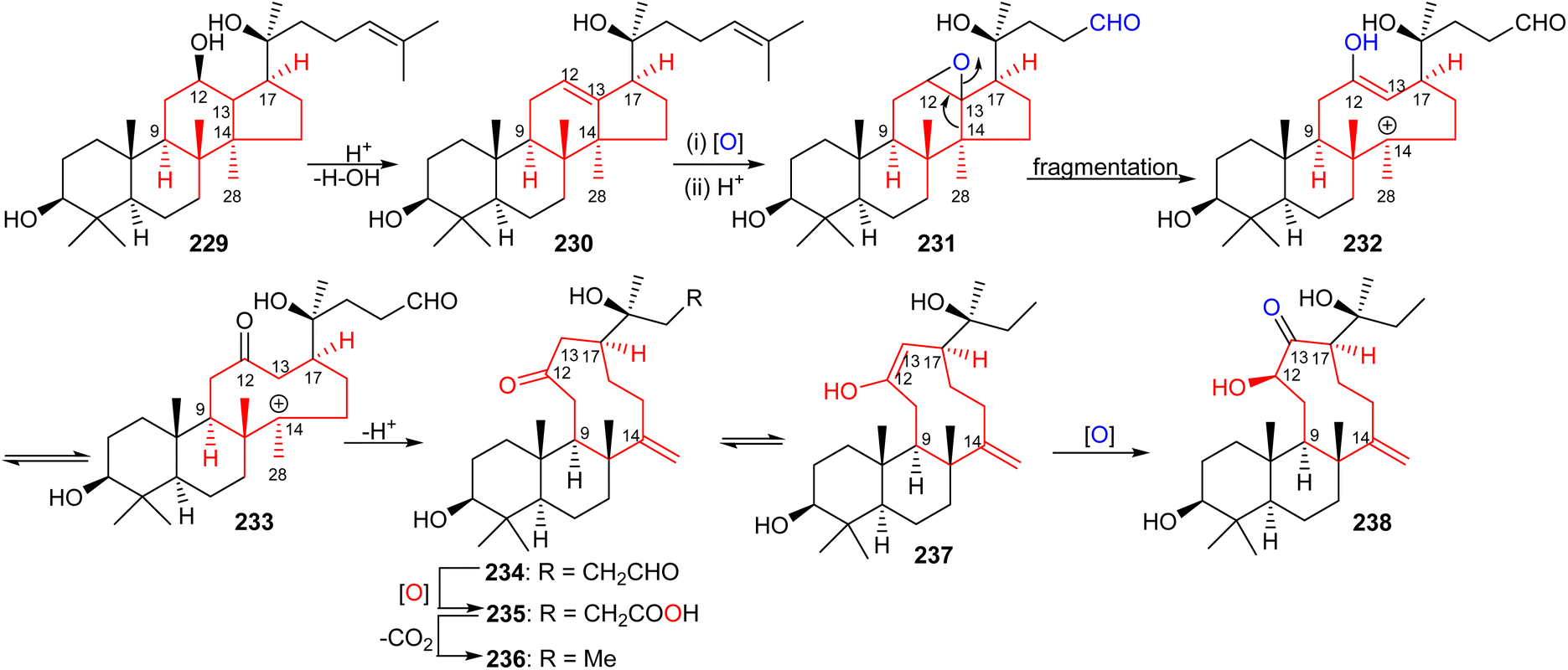 | ||
| Scheme 14 Hypothetical biosynthesis of nototroneside A (224) sapogenin nototrone A (238). Reproduced from ref. 75 with permission from the American Chemical Society. | ||
7 Schiglautone and volvalerane type triterpenes
Chemical investigation of the evergreen shrubs Schisandra glaucescens (Schisandraceae) provided the schiglautane-type triterpene named schiglautone A (239) (Fig. 12), which feature an unusual 6/7/9-fused tricyclic core. The structure of this unusual scaffold was confirmed via spectroscopic as well as X-ray analysis. Compound 239 with an unusual 7,9-membered bicyclic moiety seems to have no precedent among reported secondary metabolites or synthetic compounds and is clearly different from numerous skeletons reported from the Schisandraceae family. Moreover, it illustrated weak cytotoxic effects towards HeLa, Hep G2, and SGC-7901.82 Volvalerenol A (240), which features a 7/12/7 tricyclic ring system, was obtained from the EtOH extracts of the roots of Valeriana hardwickii.83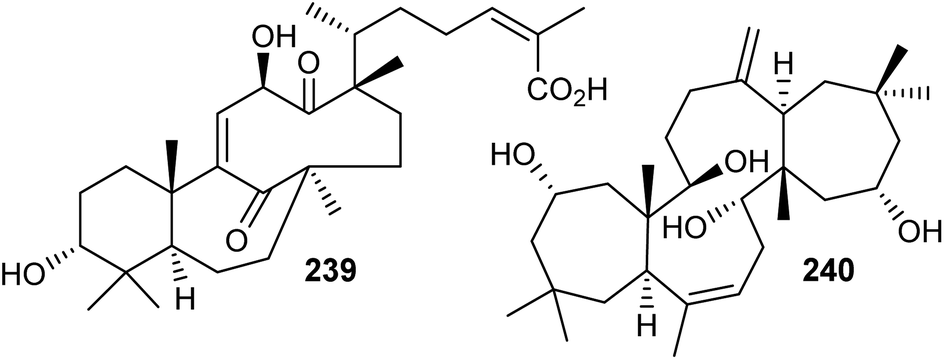 | ||
| Fig. 12 Structures of triterpenes 239 and 240. | ||
8 Biosynthesis of schiglautone and volvalerane type triterpenes
As could be expected, the biosynthesis of these very unusual triterpenes was of tremendous interest. The starting point for schiglautone A (239) is thought to be derived from anwuweizic acid (241) (Scheme 15),82 which was also reported from the same plant species in large quantities. The authors postulated that triterpene 239 might conceivably be generated from 241via enzymatic conversion reactions which include oxidation, acidification hydroxylation, and most importantly ring expansion. The key biosynthetic conversions include two ring expansions i.e. degradation of one isoprene unit in the purple color in intermediate 243 to form a nine-membered ring and ring expansion via the shifting of one carbon unit of the isoprene unit in green color (245) to form a seven member ring (246).82 On the other hand, the 7/12/7 tricyclic comprising triterpene volvalerenol A (240) could be biogenetically derived from two units of FPP (farnesyl pyrophosphate, 250). Initially FPP 250 could undergo head-to-tail condensation followed by a series of enzymatic reactions including epoxidation, dehydrogenation, electrophilic addition, and oxidation reactions (Scheme 15).83 | ||
| Scheme 15 Hypothetical biosynthesis for schiglautone A (239) and volvalerenol A (240). Reproduced from ref. 82 and 83 with permission from the American Chemical Society. | ||
9 Unusual tricyclic seco/nor-lanostane
Chemical investigation of traditionally used Kadsura coccinea, which was collected from China, yielded one unusual tricyclic triterpenoid, kadcotrione A (255) (Fig. 13) bearing an 18(13 → 12)-abeo-13,17-seco-14β-lanostane skeleton.84 Notably, metabolite 255 bears a 6/6/6-fused skeleton having a β-oriented C-14 methyl group which is not consistent with the methyl group at the C-28 position present in lanostane-type triterpenoids. In addition, this compound also features a C11 side chain attached to C-14. This side chain comprises a keto, methyl, and carboxylic group. Furthermore, its absolute configuration was established via both the calculated and experimental ECD spectra.84 | ||
| Fig. 13 Structures of triterpenes 255–257. | ||
The norlanostane triterpenoid named kadcotrione B (256), having a 14(13 → 12)-abeo-13,18-dinorlanostane core was also produced by K. coccinea. Additionally, this plant produced kadcotrione C (257) which features a 14(13 → 12)-abeo-12,13-secolanostane core system. Both triterpenes 256 and 257 have a 6/6/5 ring scaffold system which is configurationally similar to the isomalabaricane skeleton but is different from that of isomalabaricane, because of a C13 or C11 side chain along with a Me group at the 28 position. Furthermore, only compound 256 illustrated weak anti-HIV-1 effects with EC50: 30.2 μM. In addition, compounds 255 and 256 were not active towards SMMC-7721, A-549, MCF-7, and SW480.84
10 Biosynthesis of seco/nor-lanostane
The structures of kadcotriones A-C (255–257) possess the same ring A-B trans-relationship to that of 12β-hydroxycoccinic acid (258), a lanostane triterpene which was also isolated from K. coccinea together with triterpenes 255–257 (Scheme 16).84 Based on their structural similarities, the triterpene 258 could be considered to be the biosynthetic precursor and thus might undergo a series of enzymatic conversions including dehydration, 1,2-migration of a methyl group, and oxidation/oxidative degradations.84 Furthermore, the key step in the biosynthesis of kadcotriones A–C (255–257) is a fragmentation of the C–D ring in intermediate 261, and oxidative cleavage of the C-13/C-17 double bond in 265 (to generate compound 255), and, similarly, tricyclic 267 in order to biosynthesize triterpenes 256 and 257.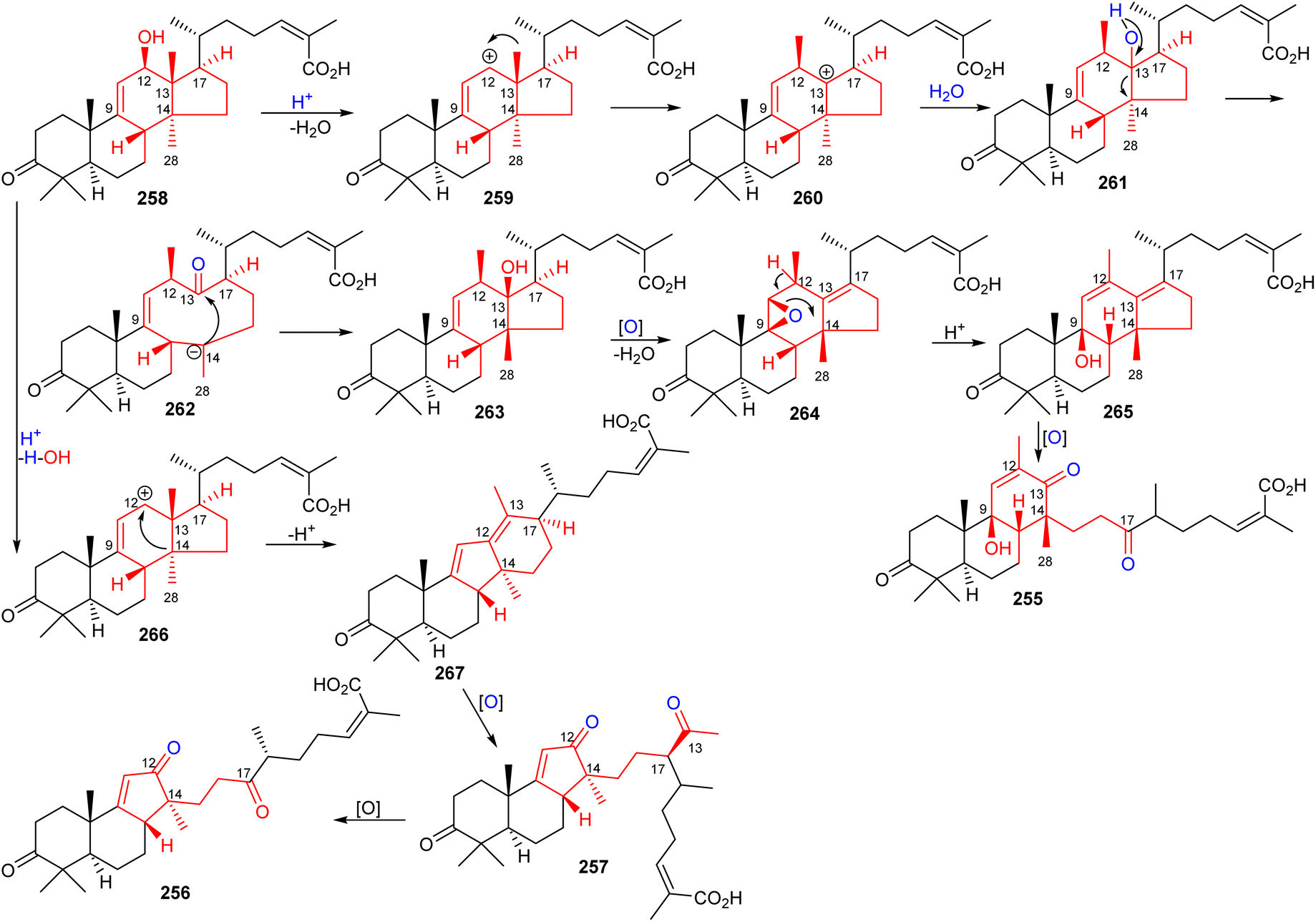 | ||
| Scheme 16 Hypothetical biosynthesis of kadcotriones A-C (255–257). Reproduced from ref. 84 with permission from the American Chemical Society. | ||
11 Sipholanes and siphonellines
The sipholane skeleton may be described as a cis-fused cyclopentane-cycloheptane ethylene linked hexane-oxepane.85 Investigations of the sponge Siphonochalina siphonella yielded the two benzoxepine-azulene linked sipholane type triterpenes, sipholenols N (268) and O (269) (Fig. 14).86 On the other hand sipholane type triterpenoids also include the octahydro-chromene-azulene system linked87via a two carbon bridge. Jain et al. reported sipholane 270 from Siphonochalina siphonella, which possessed no cytotoxic effects toward epidermoid cancer cells such as KB-C2 and KB-3-1.87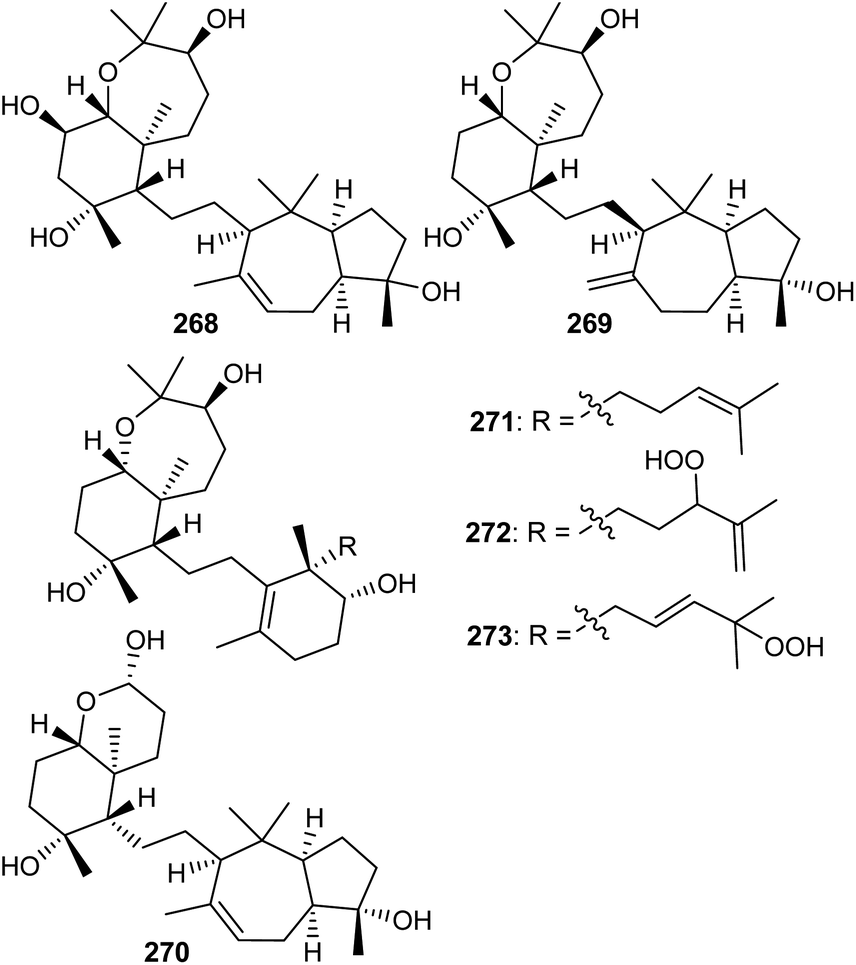 | ||
| Fig. 14 Structures of sipholanes and siphonellines 268–273. | ||
Siphonellines comprising of decahydro tetramethylbenzooxepine ring systems attached to cyclohexenol analogs via an ethylene bridge are also known. Callyspongia sp. produced siphonelline triterpenes named siphonellinol D (271), E (272), and siphonellinol C-23-hydroperoxide (273) (Fig. 14) and these triterpenes were inactive toward the two epidermoid cancer cells (KB-C2 and KB-3-1).87
12 Sodwanones and neviotines
Sodwanones have structural similarities to the sipholanes as well as possess a hexane-oxepane subunit attached to a variety of decalins. Chemical examination of the red sea sponge S. siphonella yielded five sodwanones 274–278 (Fig. 15) and these triterpenes proved to be inactive toward the two epidermoid cancer cell lines KB-C2 and KB-3-1.87 Neviotine type triterpenoids are comprised of a pentacyclic core system including a bicyclic (octahydro-1H-indene) ethylene linked with a tricyclic (dodecahydro-2H-indeno[5,4-b]oxepine). Siphonochalina siphonella produced the two neviotine triterpenes named neviotines C (279)88 and D (280).86 Moreover, compound 280 illustrated significant inhibition of RANKL induced osteoclastogenesis with IC50: 12.8 μM.86 On the other hand, neviotines C (279) illustrated moderate cytotoxic effects towards MCF-7, PC-3 and A549.88 | ||
| Fig. 15 Structures of sodwanones and neviotines 274–280. | ||
13 Biosynthesis of sipholanes, siphonellines, sodwanones, and neviotines
Sipholanes, siphonellines, sodwanones, and neviotines could feasibly be biosynthesized via two separate cascade cyclizations starting from the multiple-oxidized di- (282) or triepoxysqualenes (283). In all triterpene skeletons, cyclization of the doubly epoxidized half could be initiated by acid catalyzed enzymatic opening of the epoxide moiety in 282 and 283 (Scheme 17).89 On the other hand, for the siphonellinoles the western part appears to follow a cascade-cyclization protocol via an electrophilic protonation of a double bond (Scheme 17), while the western part in sodwanones could be biosynthesized after protonation of the western half carrying a mono-epoxidized terminal moiety.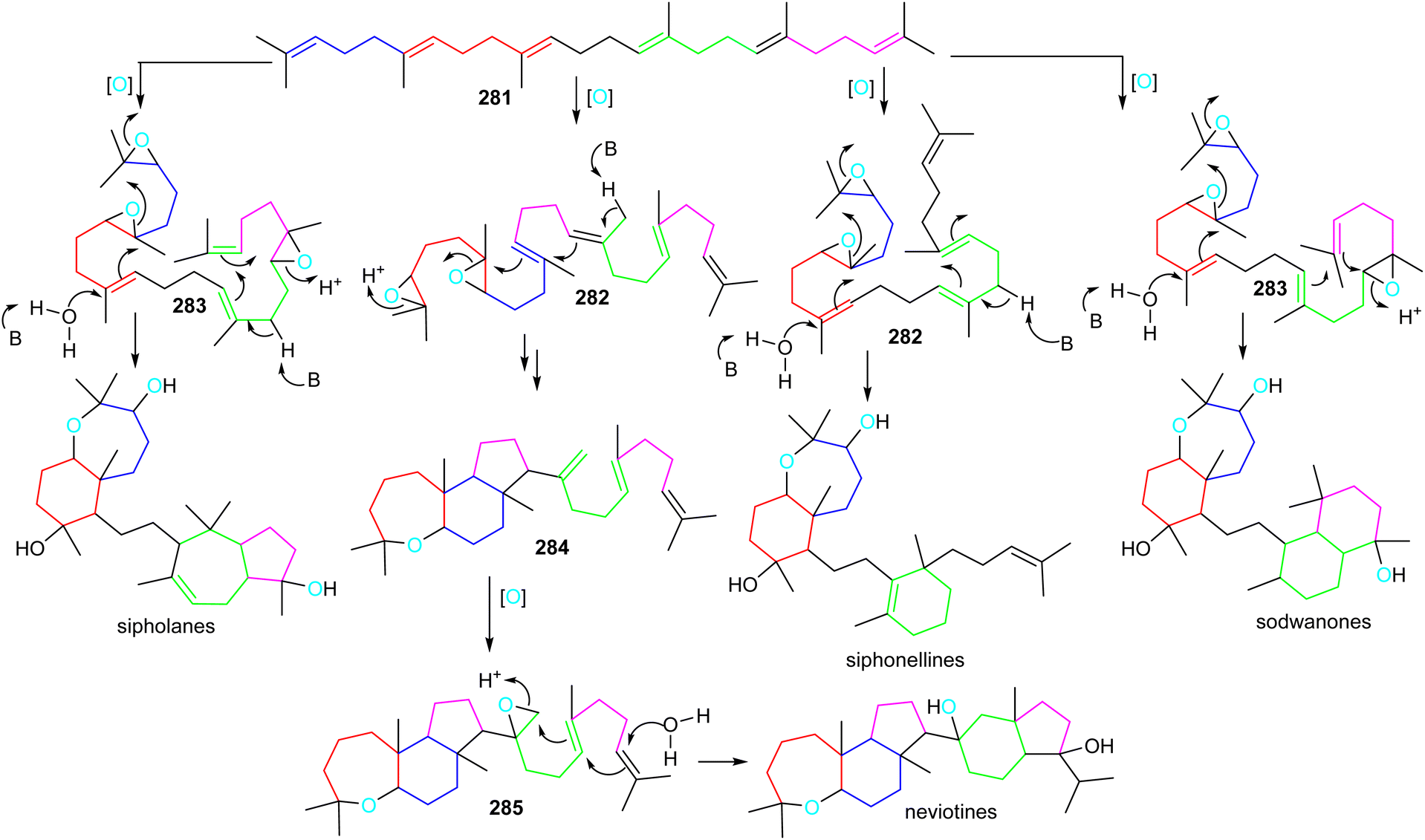 | ||
| Scheme 17 Hypothetical biosynthesis of sipholane, siphonelline, sodwanone and neviotine skeletons. Reproduced from ref. 89 and 90 with permission from the American Chemical Society and Springer Nature respectively. | ||
A more different and complex biosynthetic route could involve the biogenesis of neviotine type triterpenes (Scheme 17).89,90 The starting di-epoxide in 282 could be the same as for the biosynthesis of sipholanes, and sodwanones, with an interrupted cyclization cascade, delivering the tricyclic core structure 284. Subsequently, the eastern half cyclization could be initiated via late-stage epoxidation of the C-15 methylene group to form epoxide 285, thus setting up the second cascade cyclization to generate the neviotane skeleton.
14 Onocerane and belamchinane-type triterpenoids
Two onocerane triterpenoids named reinereins A (286) and B (287) (Fig. 16), were isolated via a bioassay guided chromatographic protocol from the bark of Reinwardtiodendron cinereum, found in Malaysia and Indonesia. Triterpene 286 illustrated moderate cytotoxic effects towards HL-60, A549, MCF7, and HepG2 cancer cells with IC50 ranging from 11.5 to 27.8 μM. On the other hand, compound 287 was only active towards HL-60 and A549 with IC50: 27.8 and 29.7 μM, respectively.91 | ||
| Fig. 16 Structures of onocerane and belamchinane-type triterpenoids 286–288. | ||
In another report, the belamchinane-type triterpenoid named belamchinenin A (288), exhibiting an unusual 6/5/6-fused tricyclic skeleton has been reported from Belamcanda chinensis, whose root extract is widely used in China and found successful in curing various diseases including cancer. Notably, metabolite 288 bears a half-caged tricyclic core along with a flexible geranyl group. As could be expected by the activity guided fractionation studies, compound 288 demonstrated cytotoxic effects towards NCI-H1650, HepG2, BGC 823, HCT-116, and MCF-7 cancer cells in which the IC50: ranged from 2.29 to 4.47 μM.92
15 Biosynthesis of onocerane and belamchinane-type triterpenoids
Members of the natural product family of onoceranes have been isolated previously, e.g. α-onocerin (291) was discovered in 1855 and its complete structure was established in 1955.93 Later, its biosynthetic route was postulated to involve two cyclizations starting from 2,3,22,23-dioxidosqualene (289).94,95 Despite the fact that these findings on α-onocerin (291) were established over six decades ago, the nature of the enzymes or genes that catalyze the two right and left cyclizations remained elusive. In 2016, Kushiro and his team96,97 investigated the biosynthesis of α-onocerin (291) (Scheme 18) and identified the two elusive genes from the fern Lycopodium clavatum, LCC and LCD of the oxidosqualene cyclase (OSC) family. The findings demonstrated that dioxidosqualene cyclase LCC catalyzed the first cyclization to get a decalin-intermediate (ring A,B) pre-α-onocerin (290) and subsequently onocerin synthase LCD catalyzed the second cyclization to afford α-onocerin (291). Moreover, reinereins A (286) and B (287) could be biosynthetically derived from α-onocerin (291) through biosynthetic conversions including oxidative ring A and D cleavage, oxidation and double bond isomerization. | ||
| Scheme 18 Hypothetical biosynthesis of onoceranes 286 and 287. Reproduced from ref. 96 and 97 with permission from the John Wiley and Sons and American Chemical Society respectively. | ||
Most of the iridal-type triterpenoids have the same absolute configurations at C-6, C-10, and C-11 (e.g. iridobelamal A (77), Scheme 19).92,98 Furthermore, belamchinenin A (288), which feature a novel 6/5/6-tricyclic core system, has stereochemistry at C-6, C-10, and C-11 being the same as for iridal-type triterpenoids, indicating that triterpene 288 could likely be derived from iridal-type triterpenoids. The iridal-type triterpenoid iridobelamal A (77) could be considered to be the biogenetic precursor, which after dehydratation to 293 might be converted into tricyclic 294 through intramolecular [4 + 2] cyclisation followed by oxidative and dehydrogenative steps to afford the scaffold of belamchinenin A (288).92
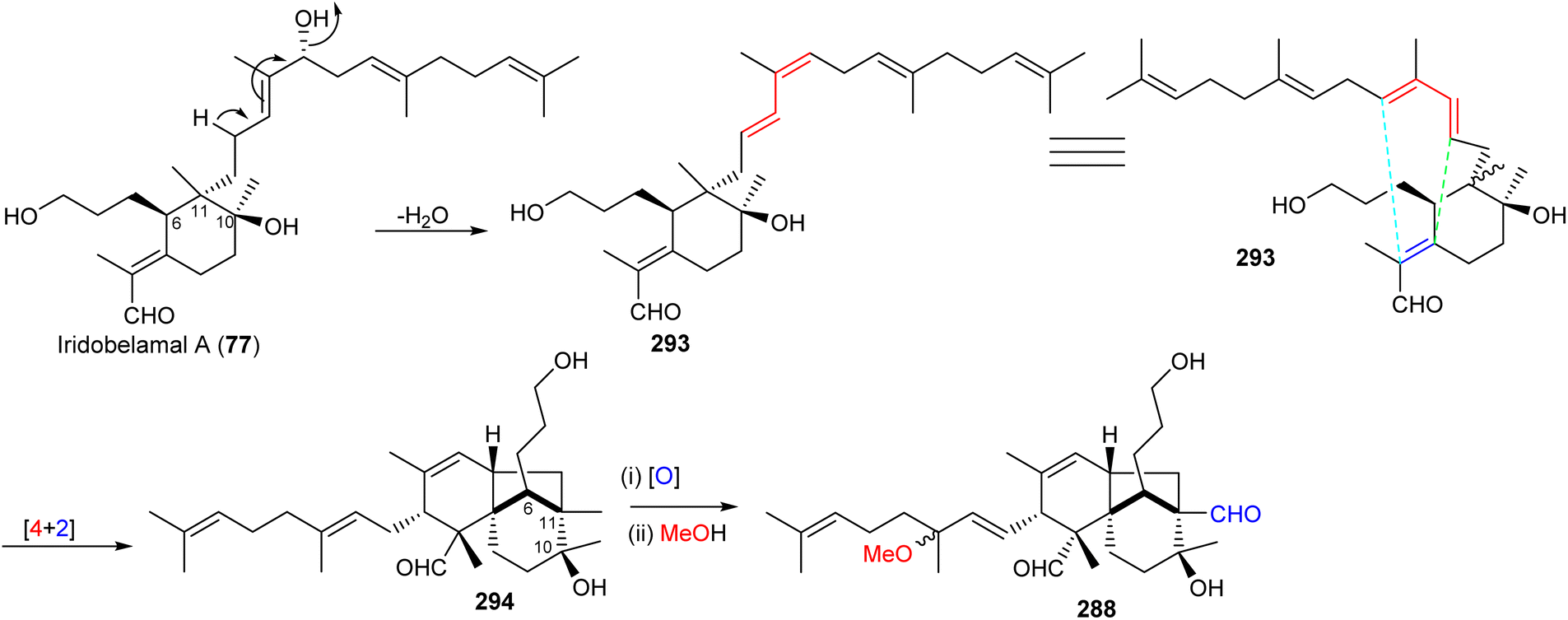 | ||
| Scheme 19 Biosynthesis of belamchinenin A (288). Reproduced from ref. 92 with permission from the Royal Society of Chemistry. | ||
16 Chemical synthesis of unusual cyclized triterpenoids
16.1 Isomalabaricane-type triterpenoids
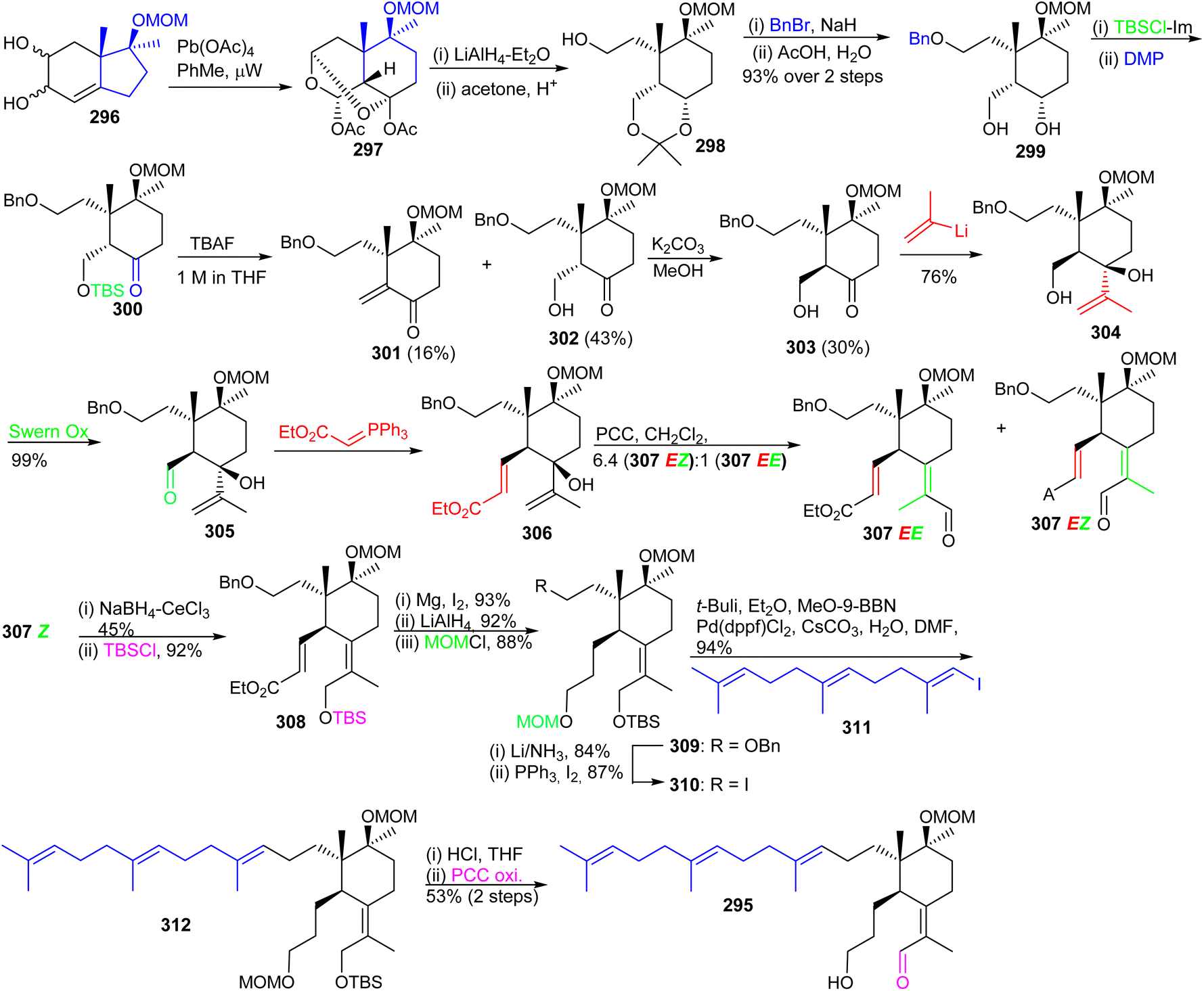 | ||
| Scheme 20 Diastereoselective synthesis of 21-deoxy-iridogermanal (295). | ||
Precursor 303 on reaction with (2-propenyl)lithium furnished the corresponding carbinol 304 by Swern oxidation of the primary hydroxl group at C-5 provided aldehyde 305. The latter aldehyde was subjected to a Wittig olefination to afford the α,β-unsaturated ester 306 which upon a Dauben–Michno oxidative rearrangement102 gave a mixture of the aldehydes 307EZ and 307EE. Reduction of aldehyde 307EZ under Luche reduction conditions and a subsequent silylation of the resulting primary hydroxyl group afforded 308. Next, selective Mg/MeOH mediated reduction of the double bond of the acrylate moiety of 308103 followed by LiAlH4 mediated reduction of the ester group and subsequent MOM protection of the resulting alcohol at C-3 provided the advanced intermediate 309.
Debenzylation of 309 with Li/NH3 afforded the resulting C-13 alcohol which was subsequently transformed to iodide 310. Moreover, E-vinyl iodide 311 which was produced from geranyl acetone according to a published two step procedure104 and using the Marshall protocol105 for a Suzuki–Miyaura reaction.106 The latter compound was coupled to the key iodide 310 to produce the sp2–sp3 coupled 312 which contains the complete iridal triterpene skeleton. Synchronized acidic deprotection of all protecting groups and then PCC-mediated selective allylic oxidation provided the desired (+)-iridal 295 in 53% isolated yield. Throughout the synthesis, the chiral quaternary center at C-4a was evident in governing the diastereoselective synthesis.
16.2 Isomarabarican-type triterpenes
:1 epimeric mixture at C-8. The bicyclic ketone 316 was then converted to the key alkyne precursor 317 by virtue of a two-step protocol including conversion of the ketone functionality to an α,β-unsaturated aldehyde by employing dichloromethyllithium followed by lithium acetylide addition and an in situ pivaloate protection.
 | ||
| Scheme 21 Preparation of (±)-stelletin E (196) and (±)-rhabdastrellic acid A (313). | ||
The α-fluoro hydrazone 318 was prepared via an efficient annulation and functionalization protocol by reacting 317 with a cationic gold(I) catalyst in the presence of Selectfluor to give the C-ring α-fluoro enone as a single diastereomer. Notably, in situ p-toluenesulfonyl hydrazone formation facilitates construction of the α-fluoro hydrazone 318. The key trans-syn-trans intermediate 319 was obtained from 318 first by treating the latter with triethylamine in methanol to promote a conjugate addition of the methanol solvent and produce an azoalkene followed by stereospecific addition of hydrogen via a retro-ene rearrangement of the allylic diazene. Next, 319 was transformed into the enone 320 by employing reductive zirconation and copper-catalyzed cross-coupling with acetyl chloride as C-2 unit. The triketone skeleton 321 was synthesized by the relay hydroboration of the double bond of the ketone 320 followed by an in situ silyl group deprotection and IBX oxidation. The key tricyclic intermediate 322 was synthesized as a single isomer from ketone 321 by bromination using a Vilsmeier reagent. The next core skeleton to be synthesized was the polyene side chain 326. In order to prepare polyene side chain 326, initially, stannane dienal 324 was synthesized from 3-picoline (323) according to a published procedure.111
Compound 324 was coupled with phosphonate 325 (Scheme 21) under typical Horner–Wadsworth–Emmons olefination conditions to furnish the polyene coupling partner 326. Coupling between the tricyclic vinyl bromide 322 and 326 was achieved to produce the methyl ester of rhabdastrellic acid A which upon saponification produces the (±)-rhabdastrellic acid A (313). The geometric isomer stelletin E (196) was prepared upon irradiation of (±)-rhabdastrellic acid A (313).
In order to prepare stelletin A (327),112 initially, gibepyrone C (330) was prepared for the pyrone side-chain in both target molecules (Scheme 22). Dimerization of tiglic acid (328)113 provided lactone 329 which was subsequently converted into gibepyrone C (330) through a Johnson–Lemieux oxidation to afford a ketone. This is followed by ethoxy vinyl zinc addition and hydrolysis to afford the target aldehyde 330. This in turn was subjected to a Boron–Wittig reaction114 to furnish the pyrone coupling partner 331. Finally, Suzuki coupling of diketone 321 and the pyrone derivative 331 gave stelletin A (327). Unfortunately, stelletin A (327) could not been converted into stelletin A (209) via the photoisomerization protocol due to significant decomposition under UV light.112
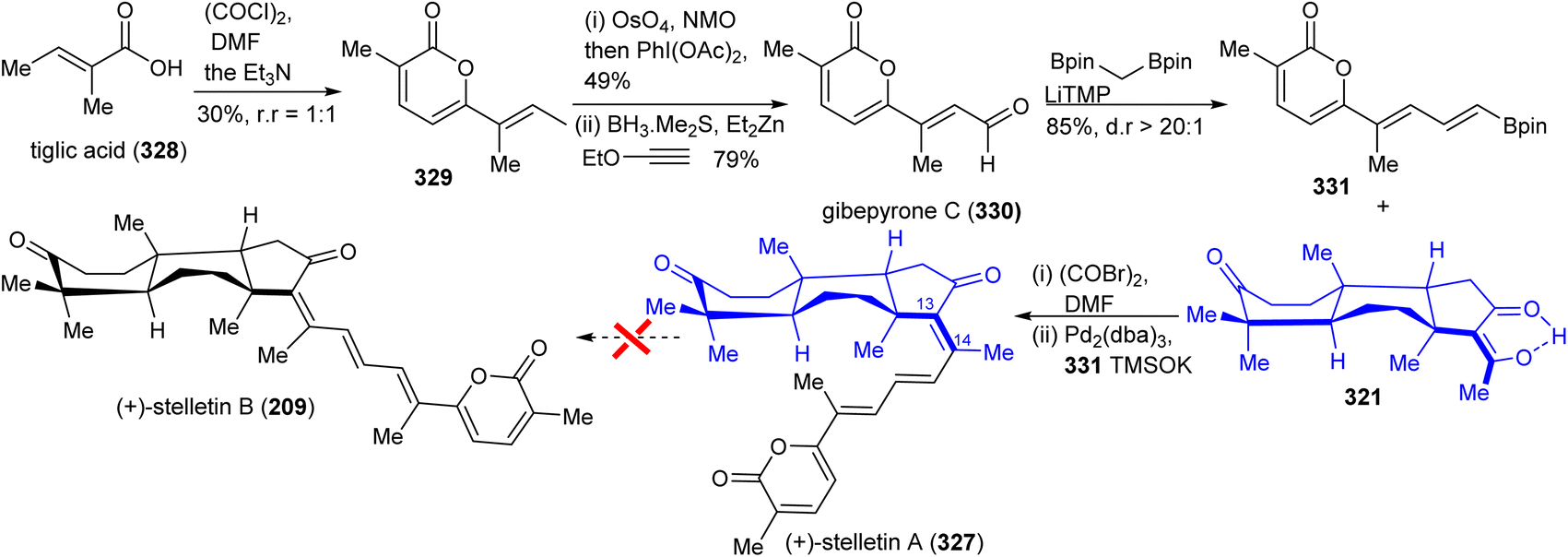 | ||
| Scheme 22 Synthesis of (±)-stelletin A (327). | ||
 | ||
| Scheme 23 Synthesis of 13E,17E-globostellatic acid X methyl ester mimic 332. | ||
An alternative route was therefore developed. Thus, reduction of the ester group in 336via DIBAL-H provided methacrolein 340 which was further successfully converted into the vinyl iodide 341via a Takai olefination. Notably, the keto group in ring C of the latter compound was generated by using CrO3/3,5-dimethylpyrazole and this was followed by ketal deprotection to afford the coupling fragment 342. The aldehyde 349 which was required for installation of the polyene side chain, was prepared in six steps from methylmalonate (343) (Scheme 23). Wittig reaction between aldehyde 349 and tributylphosphonium salt 350 established the side chain precursor 351. Lastly, the desired BC-ring 332 was achieved via Stille cross-coupling between fragments 342 and 351.116
16.3 Polypodanes
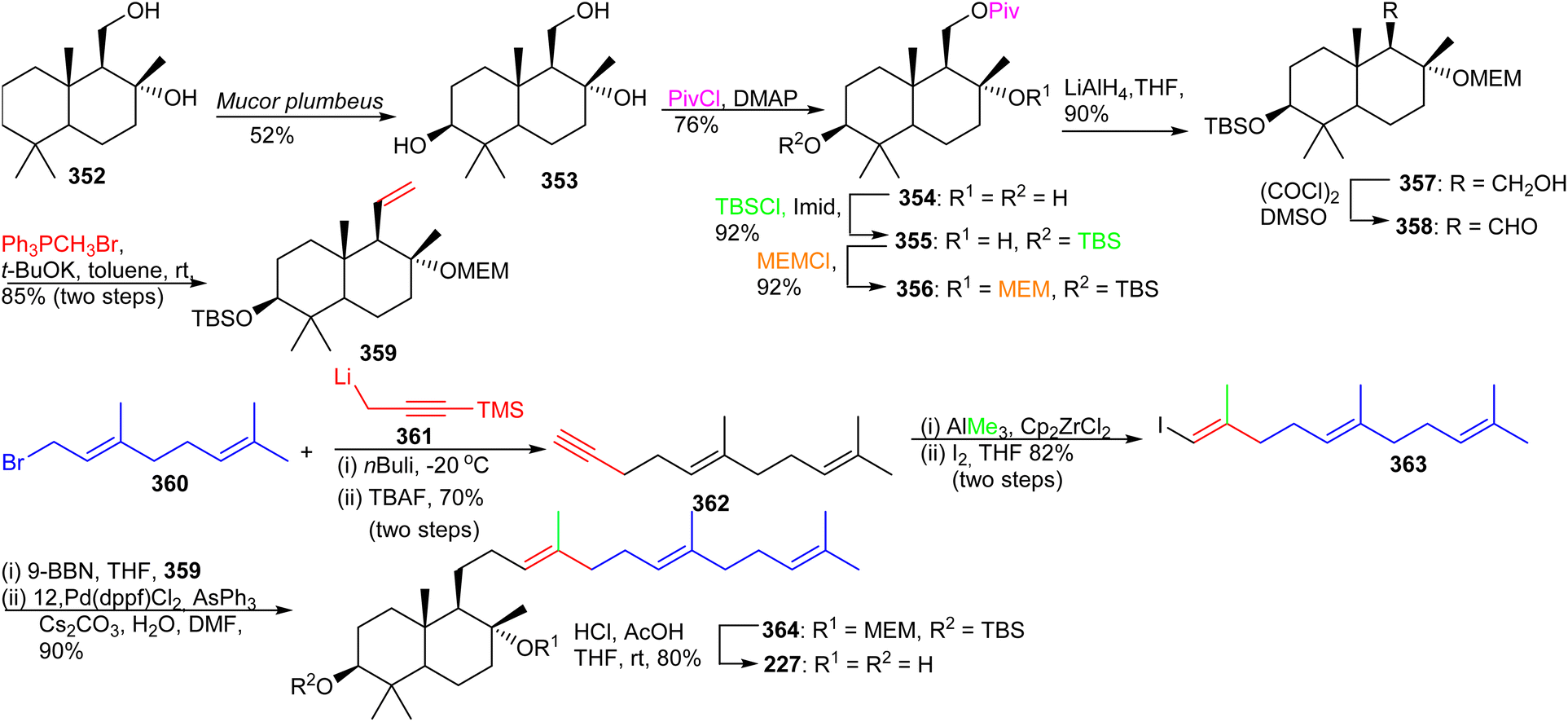 | ||
| Scheme 24 Total synthesis of (+)-myrrhanol C (227). | ||
The advanced intermediate 359 was synthesized by via primary alcohol 357, formed by reduction of the pivaloyl ester of 356 and Swern oxidation to the corresponding aldehyde 358. This latter compound was subjected to a Wittig olefination employing methyltriphenylphosphonium bromide to afford the bicyclic fragment 359. Vinyl halide 363 was prepared by coupling geranyl bromide 360 and 361 followed by Zirconium-catalyzed carboalumination of the acetylene group and iodination. Suzuki–Miyaura coupling of precursor 359 and iodide 363 provided bicyclic 364 and lastly, acid hydrolysis furnished myrrhanol C (227).
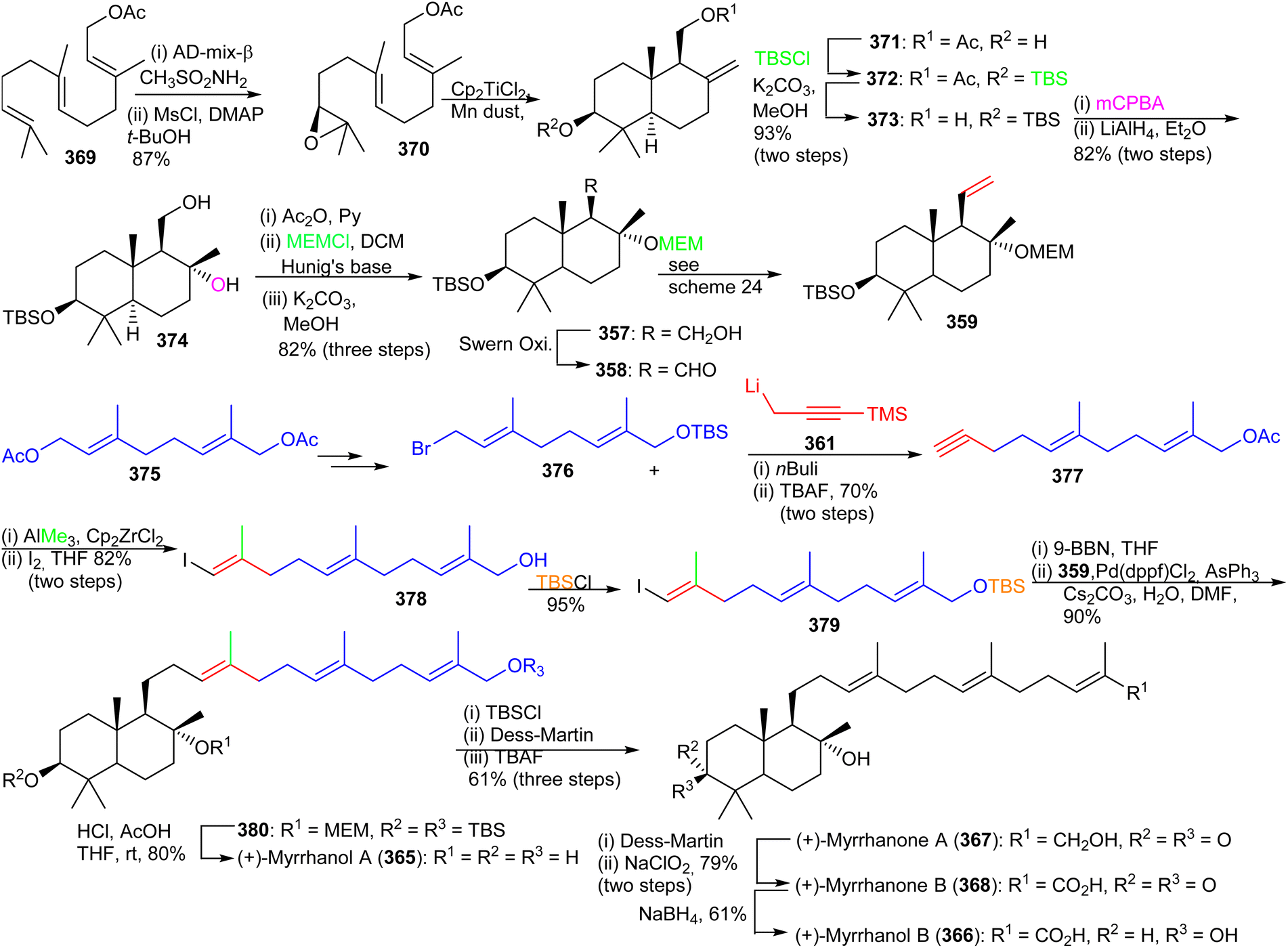 | ||
| Scheme 25 Total synthesis of (+)-myrrhanols A (365), B (366), (+)-myrrhanones A (367), and (+)-B (368). | ||
Precursor 357 was transformed to the bicyclic synthon 359 by Swern oxidation of the primary alcohol followed by a Wittig olefination of with resulting aldehyde. Preparation of the coupling partner, vinyl iodide 379, was accomplished using geranyl acetate and employing the same synthetic protocol described for iodide 363. The key intermediates, bicyclic synthon 359 and vinyl iodide 379 were coupled using the Suzuki–Miyaura protocol to produce 380 which was finally converted to the target compound, (+)-myrrhanol A (365) upon deprotection of the three protecting groups. Following on to this, (+)-myrrhanol A (365) was converted to (+)-myrrhanone A (367) employing a Dess–Martin oxidation of the C-3 hydroxyl group. The (+)-myrrhanone A (367) could be converted to (+)-myrrhanone B (368) upon further Dess–Martin oxidation to produce firstly the corresponding aldehyde and after further oxidation, the corresponding carboxylic acid. Formation of the final target (+)-myrrhanol B (366) was achieved by chemo- and stereoselective reduction of (+)-myrrhanone B (368) with NaBH4.121
16.4 Schiglautanes
 | ||
| Scheme 26 Synthesis of tricyclic core 392. | ||
The lactone 387 was constructed from 385 by employing the Gansäuer protocol,125 which upon reduction, Swern oxidation, subsequent chemoselective Grignard addition and silylation, provided bicyclic ketone 388. The O-allylated 389 was generated after treatment of 388 with LiHMDS/allyl bromide and this was followed by a Claisen rearrangement to construct diene 390. This rearrangement afforded the C-1 bridging unit. The tricyclic carbon skeleton 391 was prepared from diene 390via a Grubbs II catalyst based ring closing metathesis, yielding a cis-configured double bond. The correctly folded and substituted 392 was generated from ketone 391 through a Jones oxidation along with an addition of methyl lithium and a formal allylic ([3,3]-rearrangement) oxidation via the Dauben–Michno protocol.
Michael addition of the CuBr-modified Grignard reagent 393 to 392 (Scheme 27) followed by a subsequent Rubottom oxidation with DMDO resulted in formation of the hydroxylated and epoxidized 394. Treatment of the epoxide of 394 with the strong oxidant PCC afforded the ‘diol’-cleaved aldehyde 395. Horner–Wadsworth–Emmons reaction of the latter aldehyde with 396 provided the Z-configured 397 selectively upon addition of crown ethers. This triketone was first treated with methyl acrylate to give the correct side-chain moiety and then with DMDO to afford the ene-dione 398 in a one-pot procedure. After cleavage of the C-3 benzyl group, reduction of the C-12 ketone was achieved via Luche conditions and delivered the major diastereomer 399 along with hemiketal 400 and aldehyde 401. Lastly, desilylation of 399 with TBAF afforded diastereoselectively 402. Surprisingly, NMR data of 402 did not accurately resemble that of natural schiglautone A (239)82 and based on NMR and X-ray data the structure of the synthesized compound 402 was established as a diastereomeric atropisomer of 239.
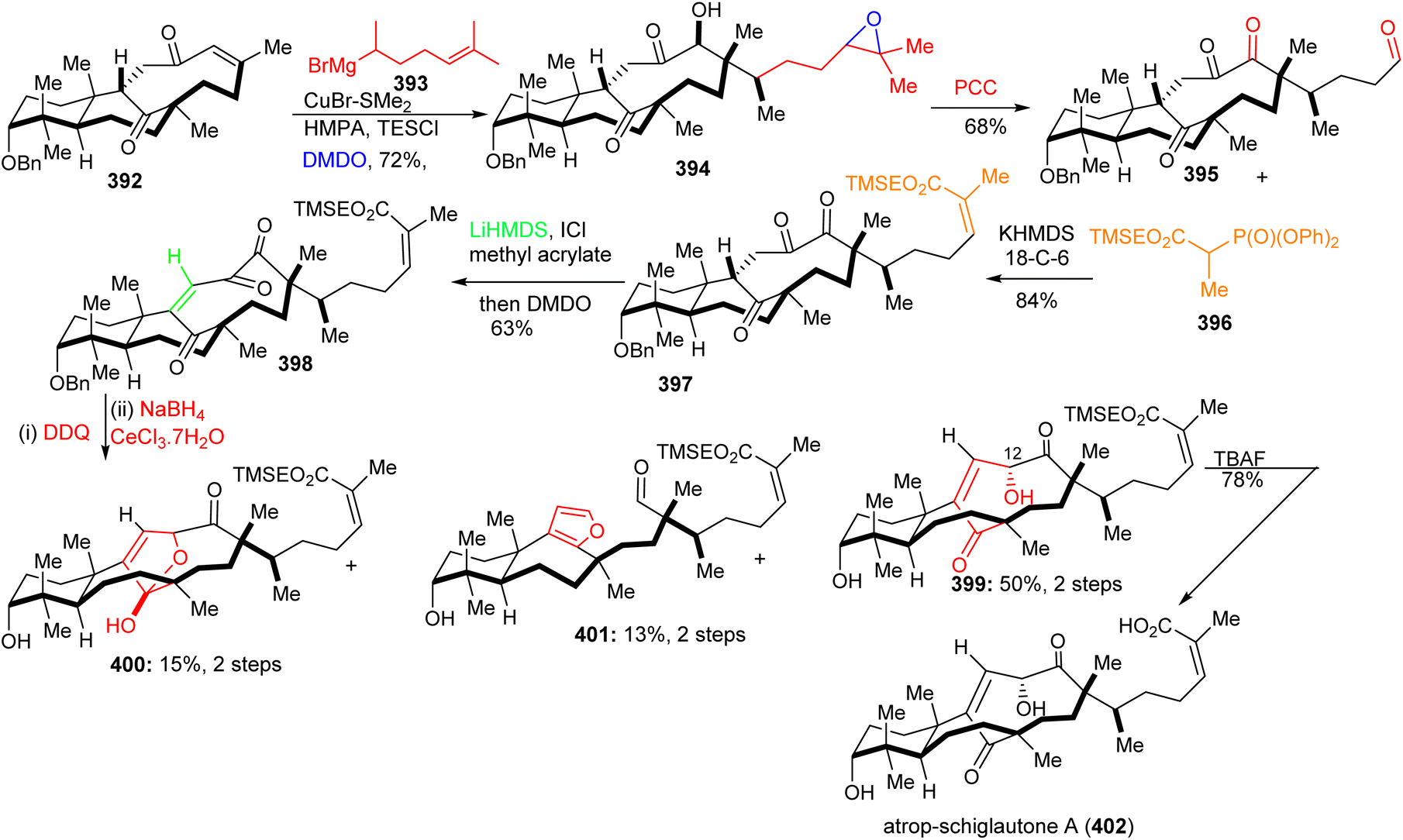 | ||
| Scheme 27 Synthesis of atrop-schiglautone A (402). | ||
 | ||
| Scheme 28 Synthesis of fragment 409. | ||
The preparation of ketone 419 commenced from (−)-citronellene (410), which was initially transformed to aldehyde 411 in two steps128 (Scheme 29). The latter molecule was subjected to a Grignard addition (derived from 5-bromo-1-pentene) to afford alcohol 412 and this step was followed by oxidation to the ketone and then followed by a second Grignard addition to furnish triene 413. Grubbs I catalyst based ring-closing metathesis between the terminal primary alkene groups delivered allylic alcohol 414 and oxidative transposition provided enone 415. Luche reduction of enone 415 provided allylic racemic alcohol 416. This mixture was subjected to enzymatic resolution to afford allylic alcohol 417.
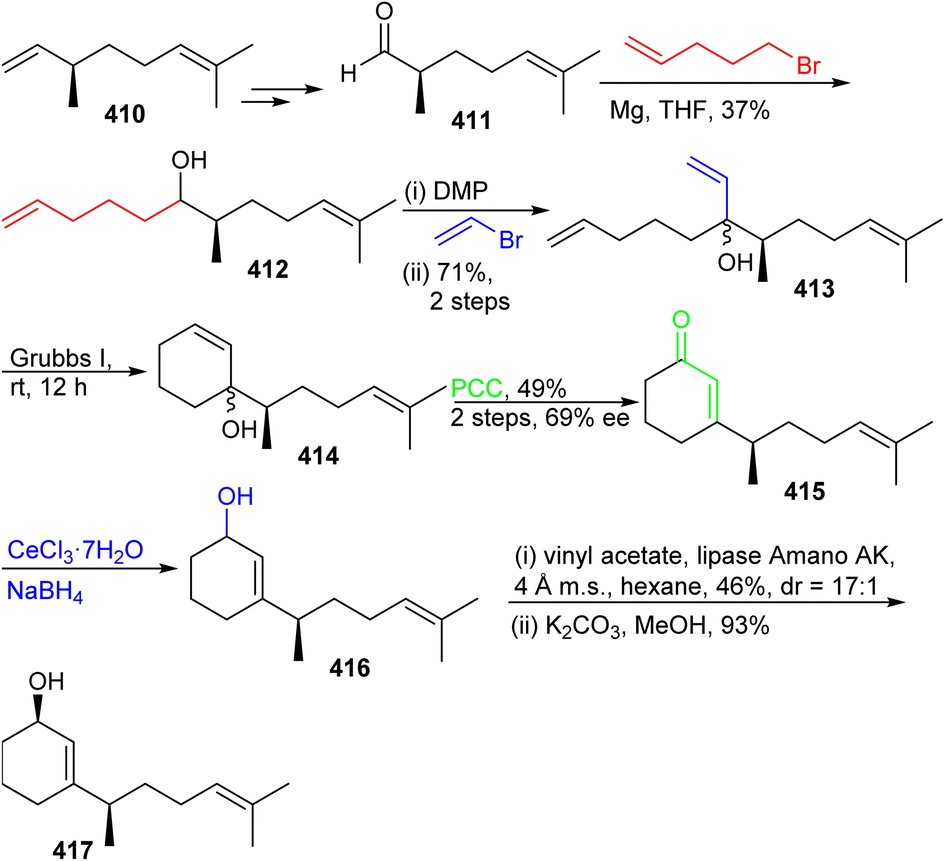 | ||
| Scheme 29 Synthesis of fragment 417. | ||
The precursor 417 was subjected to chemoselective cyclopropanation to furnish alcohol 418 (Scheme 30) and PCC mediated oxidation to the ketone along with Birch reduction mediated cyclopropane ring opening provided ketone 419. Aldol addition between aldehyde 409 and ketone 419 provided enone 420 which was elaborated into ketone 421 through conjugate reduction with LAH/CuI. The thermodynamically stable silyl enol ether of 421 was constructed and subjected to Furukawa's cyclopropanation reagent129 to provide the desired product 422 together with side products 423–425. Lastly, the desired precursor 422 was transformed into ketone 403 through cyclopropane ring opening with NaOH/EtOH.
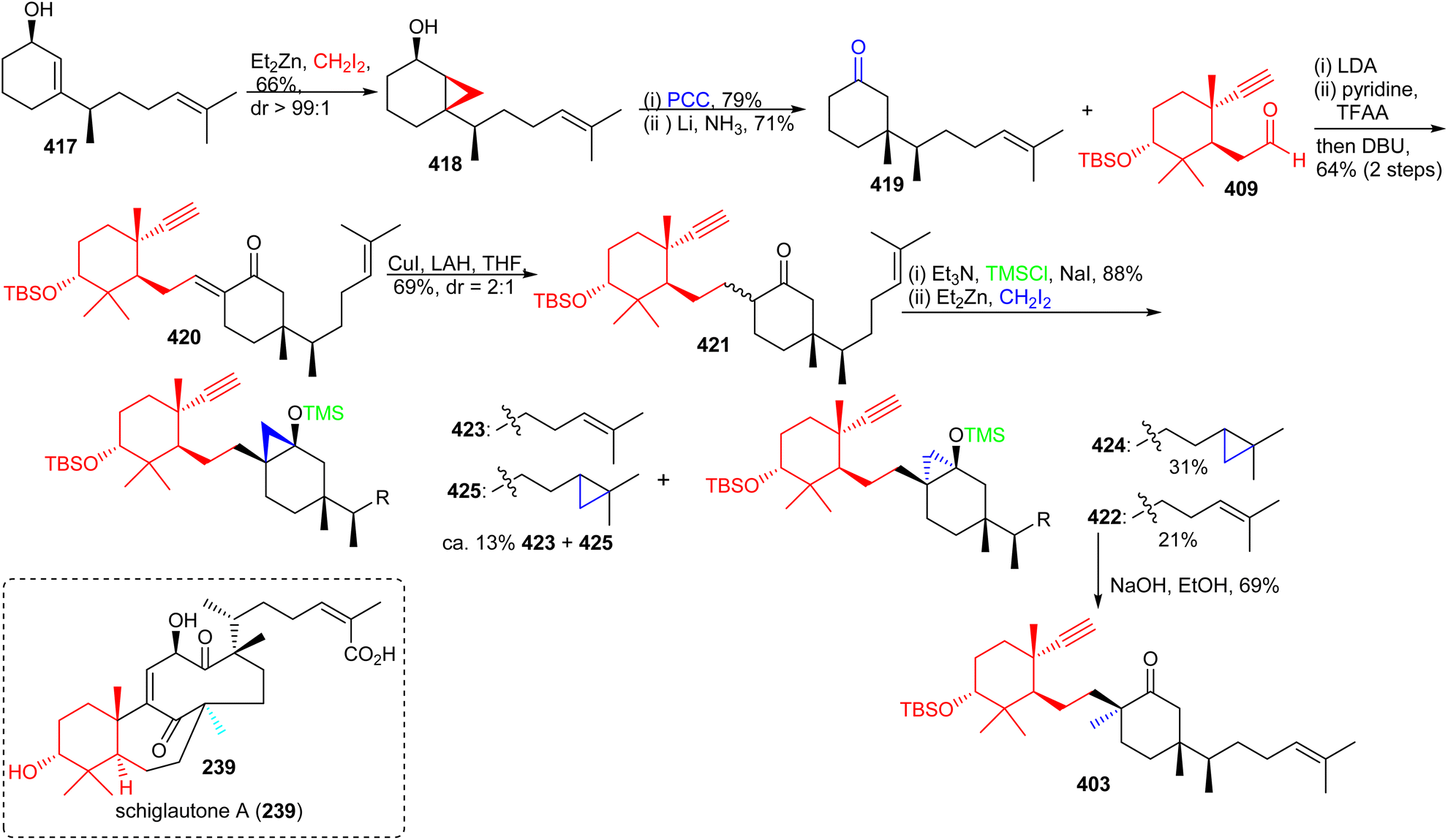 | ||
| Scheme 30 Synthesis of advanced intermediate 403. | ||
16.5 Unusual tricyclic seco/nor-lanostane
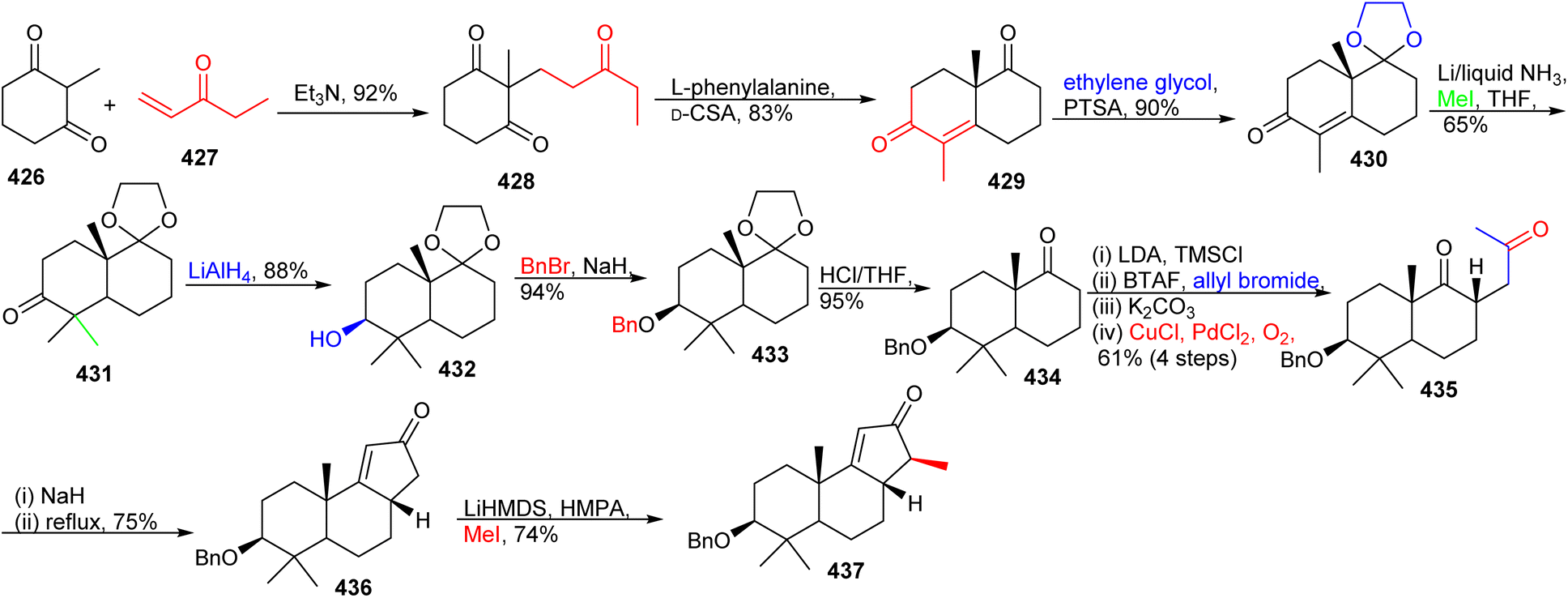 | ||
| Scheme 31 Synthesis of tricyclic fragment 437. | ||
The synthesis of aliphatic fragment 448, as depicted in Scheme 32, commenced with 1,5-pentanediol (438) via the protected aldehyde 439 by following the Tsuji protocol.133 This latter compound was elaborated into the Z-methacrylate derivative 441 with the Still–Gennari phosphonate 440 under the Horner–Wadsworth–Emmons reaction protocol. The ester group in 441 was reduced to the primary alcohol via DIBAL-H to afford alcohol 442 which was followed by protection of the resulting hydroxyl group as its benzyl ether and then cleavage of the TBS group afforded 443. Furthermore, the primary alcohol group in the latter molecule was transformed into the corresponding carboxylic acid through two oxidation procedures viz., Swern and Pinnick oxidations to afford 444.134
 | ||
| Scheme 32 Synthesis of fragment 448. | ||
Precursor 444 was next transformed into amide 446via a coupling reaction with (R)-4-benzyloxazolidin-2-one (445) and diastereoselective methylation of 446 was achieved by following the Evans protocol135 to deliver molecule 447. The amide 447 was converted into the main fragment 448 through a three step procedure including chiral auxiliary cleavage, transformation into a Weinreb amide and treatment with vinylmagnesium bromide. With the key fragments 448 and 437 in hand, initial attempts to combine them through Michael addition and further elaboration into Kadcotrione B (256) via a two-step sequence including the deprotection of the benzyl group, and oxidation of primary and secondary alcohols in a single step were investigated but proved inadequate to deliver the desired 449. The various reaction conditions that were investigated (NaOEt, t-BuOK, LDA, KHMDS, NaHMDS, LiHMDS) for the crucial Michael addition between 437 and 448 resulted in the decomposition of starting materials.
16.6 Abudinols
 | ||
| Scheme 33 Total syntheses ent-abudinol B (ent-460). | ||
Furthermore, ketone 455 was elaborated into diepoxy-enolsilane 456 through two steps including addition of vinylmagnesium chloride to the acylsilane 455 and Brook rearrangement. The TMSOTf mediated regio- and stereoselective tandem tricyclization of 456 provided the trans-anti-trans-fused tricyclic core 457 which upon treatment with the classical Wittig reagent, transformed it into the disubstituted alkene 458. Diepoxide ent-459α was subsequently prepared from alkene 458 through regio- and stereoselective double epoxidation. Lastly, TMSOTf catalyzed cyclization of diepoxide ent-459α produce ent-abudinol B (ent-460) in low yield (15%).136
16.7 Muzitones
 | ||
| Scheme 34 Total synthesis of muzitone (ent-472) and ent-muzitone (ent-470). | ||
Protection of the two free hydroxyl groups as their acetates in the latter compound and migration of the double bond from the C-13/C-14 into the C-14/C-25 position was accomplished after treatment with HI to afford isomer 469. The compound ent-470, which is considered as “pre muzitone” was achieved by deacetylation of 469 employing K2CO3 in methanol. Finally, ent-muzitone (ent-472) was constructed via the oxidative cleavage of the tetrasubstituted alkene in ent-470 with O3/Me2S or indirectly by from 469 by producing diol 471 initially through osmylation followed by reduction and then deacetylation along with Pb(OAc)4 mediated oxidative cleavage which produces the same desired ent-muzitone (ent-472). In addition, X-ray analysis of bis-p-nitrobenzoate analog ent-472 established the ent-472 is truly ent-muzitone. Unfortunately, Boone et al.136 observed some discrepancies in their NMR data for ent-472 and the original natural muzitone.137 Based on this synthetic investigation, the structure of muzitone assigned by Kashman group is consequently incorrect.
17 Conclusions
This review provides the second comprehensive overview on some intriguing unusual cyclized triterpenes. Marine organisms, in particular sponges, produce unusual cyclized triterpenoids with remarkable chemical diversity. Advanced scientific studies into these unusually cyclized triterpenes establishes a sound strategy in the importance of these molecules. These triterpenoids have shown a broad spectrum of biological effects including anti-epileptic, cytotoxic, hepatoprotective, neuroprotective, anti-inflammatory, and antidiabetic effects.The majority of these unusual triterpenes include malabaricane, isomalabaricane, and iridal-type triterpenoids. Overall, isomalabaricanes were reported from four genera of sponges including Rhabdastrella, Jaspis, Geodia, and Stelletta. However, taxonomic identification of these sponges remains questionable.139,140 On the other hand, malabaricane triterpenoids were present in higher plants, fungi, ferns, and bottom sediments.140 Moreover, iridals were found in the Iridaceae family, mainly in the genus Iris. However only a few were reported from the genera Tigridia and Belamcanda. Furthermore, the unusual and quite fascinating structures have challenged the esteemed synthetic chemists to construct these demanding molecules with the correct absolute configuration and which has required them establishing new synthetic methodologies.
There is no doubt that nature has established fascinating pathways to construct natural products which has unfortunately resulted in less effort by scientists being focused on investigating biosynthetic pathways of unusual cyclized triterpenoids and also understanding the structures along with mechanisms of SC and OSC enzymes which generate these molecules. On the positive side, Xiong et al.30 investigated how oxidosqualene is converted into the monocyclic iridal skeleton. In addition, two Arabidopsis thaliana genes encode OSCs which furnish the malabaricane skeleton8,66–69 Lodeiro et al.65 demonstrated that the isomalabaricane core arises from a lanosterol cyclase mutant (Tyr510) while the native enzyme has yet to be discovered. Moreover, Kushiro and his team96,97 investigated the biosynthesis of the α-onocerin skeleton and successfully identified the two genes from the fern Lycopodium clavatum, LCC and LCD of the oxidosqualene cyclase (OSC) family.
Thus, more intensive investigations on cyclizing enzymes which generate these triterpenes, will be an intriguing and challenging research field for future natural product research. In addition, there are still numerous fundamental questions about enzyme mechanisms which still need to be fully investigated. Notably, the relationship between active-site structure and cyclization mechanism of the numerous SC and OSC enzymes presents a formidable task. Further studies are required for the enzyme proteins structure-based engineering along with manipulating substrates. Finally, future research needs to focus on the genomics and synthetic biology for identifying and diversifying these unusual triterpenoid molecules because we believe that this will facilitate remarkable progress in finding new lead compounds for potential biomedical applications.
18 Author contributions
H. H. and B. W.: conceptualized, supervised, and edited the review process. J. X. and A. A. wrote the manuscript and drew the figures/schemes. I. R. G. wrote and edited the manuscript.19 Conflicts of interest
There are no conflicts to declare.20 Acknowledgments
H Hussain is thankful to the Alexander von Humboldt Foundation for its generous support in providing the opportunity to do work in Germany which facilitated the writing of this review.21 Notes and references
- K. Muffler, D. Leipold, M. C. Scheller, C. Haas, J. Steingroewer, T. Bley, H. E. Neuhaus, M. A. Mirata, J. Schrader and R. Ulber, Process Biochem., 2011, 46, 1 CrossRef CAS.
- R. Xu, G. C. Fazio and S. P. T. Matsuda, Phytochemistry, 2004, 65, 261 CrossRef CAS PubMed.
- H. Tao, L. Lauterbach, G. Bian, R. Chen, A. Hou, T. Mori, S. Cheng, B. Hu, L. Lu, X. Mu, M. Li, N. Adachi, M. Kawasaki, T. Moriya, T. Senda, X. Wang, Z. Deng, I. Abe, J. S. Dickschat and T. Liu, Nature, 2022, 606, 414 CrossRef CAS.
- H. N. Sultani, I. Morgan, H. Hussain, A. H. Roos, H. H. Haeri, G. N. Kaluđerović, D. Hinderberger and B. Westermann, Int. J. Mol. Sci., 2021, 22, 7125 CrossRef CAS PubMed.
- B. A. Shah, G. N. Qazi and S. C. Taneja, Nat. Prod. Rep., 2009, 26, 72 RSC.
- H. Hussain, I. Ali, D. Wang, F. L. Hakkim, B. Westermann, L. Rashan, I. Ahmed and I. R. Green, Expert Opin. Drug Discovery, 2021, 16, 851–867 CrossRef CAS PubMed.
- H. Tenga, B. Yuan, S. Gothai, P. Arulselvan, X. Song and L. Chen, Trends Food Sci. Technol., 2018, 72, 34 CrossRef.
- V. Domingo, J. F. Arteaga, J. F. Q. del Moral and A. F. Barrero, Nat. Prod. Rep., 2009, 26, 115 RSC.
- J. Li, G. Ni, L. Li, Y. Liu, Z. Mai, R. Wang and D. Yu, Bioorg. Chem., 2019, 83, 20 CrossRef CAS PubMed.
- G. Ni, J. Y. Li and D. Q. Yu, Phytochem. Lett., 2019, 31, 43 CrossRef CAS.
- C. L. Zhang, Y. Wang, Y. F. Liu, D. Liang, Z. Y. Hao, H. Luo, Q. J. Zhang, G. R. Shi, R. Y. Chen, Z. Y. Cao and D. Q. Yu, Tetrahedron, 2015, 71, 5579 CrossRef CAS.
- Y. Hasegawa, X. Gong and C. Kurod, Nat. Prod. Commun., 2011, 6, 789 CAS.
- Q. Xiong, W. K. Wilson and S. P. Matsuda, Angew. Chem., Int. Ed., 2006, 45, 1285 CrossRef CAS PubMed.
- L. Taillet, J. Bonfils, F. Marner and Y. Sauvaire, Phytochemistry, 1999, 52, 1597 CrossRef CAS.
- G. Ni, J. Y. Li and Y. De-Quan, J. Asian Nat. Prod. Res., 2019, 21, 881–886 CrossRef CAS PubMed.
- G. R. Shi, X. Wang, Y. F. Liu, C. L. Zhang, G. Ni, R. Y. Chen and D. Q. Yu, Tetrahedron Lett., 2016, 57, 5761 CrossRef CAS.
- X. Chen, X. Zhang, Y. Ma, Z. Deng, C. Geng and J. Chen, Fitoterapia, 2018, 129, 126 CrossRef CAS.
- K. Takahashi, Y. Hoshino, S. Suzuki, Y. Hano and T. Nomura, Phytochemistry, 2000, 53, 925 CrossRef CAS PubMed.
- F. J. Marner and B. Hanisch, Helv. Chim. Acta, 2001, 84, 933 CrossRef CAS.
- G. Ni, G. R. Shi, J. Y. Li and D. Q. Yu, RSC Adv., 2017, 7, 20160 RSC.
- C. Zhang, J. Chen, F. Zhao, R. Chen, D. Yu and Z. Cao, Fitoterapia, 2017, 122, 20–25 CrossRef CAS PubMed.
- J. Li, G. Ni, Y. Liu, Z. Mai, R. Wang and D. Yu, J. Nat. Prod., 2019, 82, 1759 CrossRef CAS.
- G. Ni, J. Y. Li, Z. P. Mai and D. Q. Yu, Tetrahedron Lett., 2018, 59, 151 CrossRef CAS.
- K. Takahashi, S. Suzuki, Y. Hano and T. Nomura, Biol. Pharm. Bull., 2002, 25, 432 CrossRef CAS PubMed.
- C. L. Zhang, Y. Wang, F. Zhao, Y. F. Liu, G. R. Shi, R. Y. Chen, D. Q. Yu and Z. Y. Cao, Fitoterapia, 2019, 137, 104193 CrossRef CAS PubMed.
- C. L. Zhang, Y. Wang, Y. F. Liu, G. Ni, D. Liang, H. Luo, X. Y. Song, W. Q. Zhang, R. Y. Chen, N. H. Chen and D. Q. Yu, J. Nat. Prod., 2014, 77, 411 CrossRef CAS PubMed.
- C. L. Zhang, Z. Y. Hao, Y. F. Liu, Y. Wang, G. R. Shi, Z. B. Jiang, R. Y. Chen, Z. Y. Cao and D. Q. Yu, J. Nat. Prod., 2017, 80, 156 CrossRef CAS PubMed.
- C. L. Zhang, Y. F. Liu, Y. Wang, D. Liang, Z. B. Jiang, L. Li, Z. Y. Hao, H. Luo, G. R. Shi, R. Y. Chen, Z. Y. Cao and Y. De-Quan, Org. Lett., 2015, 17, 5686 CrossRef CAS PubMed.
- Z. J. Song, X. M. Xu, W. L. Deng, S. L. Peng, L. S. Ding and H. H. Xu, Org. Lett., 2011, 13, 462–465 CrossRef CAS PubMed.
- Q. Xiong, W. K. Wilson and S. P. T. Matsuda, Angew. Chem., 2006, 118, 1307 CrossRef.
- C. I. Lin, R. M. McCarty and H. W. Liu, Angew. Chem., Int. Ed., 2017, 56, 3446 CrossRef CAS.
- F. J. Marner, D. Gladtke and L. Jaenicke, Helv. Chim. Acta, 1988, 71, 1331 CrossRef CAS.
- F. J. Marner, K. N. Yasmin and L. Jaenicke, Helv. Chim. Acta, 1990, 73, 433 CrossRef CAS.
- J. A. Piccirilli, Chem. Biol., 1999, 6, R59 CrossRef CAS.
- L. Zhang, K. Wei, J. Xu, D. Yang, C. Zhang, Z. Wang and M. Li, J. Ethnopharmacol., 2016, 186, 1–13 CrossRef CAS PubMed.
- F. Lv, M. Xu, Z. Deng, N. J. de Voogd, R. W. M. van Soest, P. Proksch and W. Lin, J. Nat. Prod., 2008, 71, 1738 CrossRef CAS.
- J. W. Blunt, B. R. Copp, W.-P. Hu, M. H. G. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2007, 24, 31 RSC.
- J.-F. Guo, J.-M. Zhou, Y. Zhang, R. Deng, J.-N. Liu, G.-K. Feng, Z.-C. Liu, D.-J. Xiao, S.-Z. Deng and X.-D. Zhu, Cell Biol. Int., 2008, 32, 48 CrossRef.
- J. A. Clement, M. Li, S. M. Hecht and D. G. I. Kingston, J. Nat. Prod., 2006, 69, 373 CrossRef CAS.
- S. Tang, Y. Pei, H. Fu, Z. Deng, J. Li, P. Proksch and W. Lin, Chem. Pharm. Bull., 2006, 54, 4 CrossRef CAS.
- P. V. Kiem, D. T. Dung, P. H. Yen, N. X. Nhiem, T. H. Quang, B. H. Tai and C. V. Minh, Phytochem. Lett., 2018, 26, 199 CrossRef CAS.
- J. Li, B. Xua, J. Cui, Z. Deng, N. J. de Voogd, P. Proksch and W. Lin, Bioorg. Med. Chem., 2010, 18, 4639 CrossRef CAS.
- J. Li, H. Zhu, J. Ren, Z. Deng, N. J. de Voogd, P. Proksch and W. Lin, Tetrahedron, 2012, 68, 559 CrossRef CAS.
- D. Q. Xue, S. C. Mao, X. Q. Yu and Y. W. Guo, Biochem. Syst. Ecol., 2013, 49, 101 CrossRef CAS.
- Y. Li, H. Tang, X. Tian, H. Lin, M. Wang and M. Yao, Fitoterapia, 2015, 106, 226 CrossRef CAS.
- N. Tanaka, R. Momose, A. Shibazaki, T. Gonoi, J. Fromont and J. Kobayashi, Tetrahedron, 2011, 67, 6689 CrossRef CAS.
- S. A. Kolesnikova, E. G. Lyakhova, A. B. Kozhushnaya, A. I. Kalinovsky, D. V. Berdyshev, R. S. Popov and V. A. Stonik, Molecules, 2021, 26, 678 CrossRef CAS PubMed.
- M. L. Bourguet-Kondracki, A. Longeon, C. Debitus and M. Guyot, Tetrahedron Lett., 2000, 41, 3087 CrossRef CAS.
- D. J. Jin, S. A. Tang, G. S. Xing, W. J. Zhao, C. Zhao, H. Q. Duan and W. Han, J. Asian Nat. Prod. Res., 2014, 16, 427 CrossRef CAS.
- W. G. Xu, J. Wang, G. S. Xing, J. J. Xu, W. Qiao, C. Zhao and S. A. Tang, Z. Naturforsch., C: J. Biosci., 2016, 71, 111 CrossRef CAS PubMed.
- W. G. Xu, J. Wang, W. Qiao, C. Zhao and S. A. Tang, Chem. Nat. Compd., 2018, 54, 84 CrossRef CAS.
- D. T. Dung, P. H. Yen, N. X. Nhiem, T. H. Quang, B. H. Taia, C. V. Minh, D. C. Kim, H. Oh, Y. C. Kim and P. V. Kiem, Nat. Prod. Commun., 2018, 13, 661 Search PubMed.
- D. T. Dung, D. T. T. Hang, N. Nhiem, T. H. Quang, B. H. Tai, P. H. Yen, N. T. Hoai, D. C. Thung, C. V. Minh and P. V. Kiem, Nat. Prod. Commun., 2018, 13, 1251 Search PubMed.
- M. Hirashima, K. Tsuda, T. Hamada, H. Okamura, T. Furukawa, S. I. Akiyama, Y. Tajitsu, R. Ikeda, M. Komatsu, M. Doe, Y. Morimoto, M. Shiro, R. W. M. van Soest, K. Takemura and T. Iwagawa, J. Nat. Prod., 2010, 73, 1512 CrossRef CAS.
- K. H. Lai, Z. H. Huang, M. El-Shazly, B. R. Peng, W. C. Wei and J. H. Su, Mar. Drugs, 2021, 19, 206 CrossRef CAS PubMed.
- S. Tang, Z. Deng, P. Prokschc and W. Lin, Tetrahedron Lett., 2007, 48, 5443 CrossRef CAS.
- S. A. Kolesnikova, E. G. Lyakhova, A. I. Kalinovsky, D. V. Berdyshev, E. A. Pislyagin, R. S. Popov, B. B. Grebnev, T. N. Makarieva, C. V. Minh and V. A. Stonik, J. Nat. Prod., 2019, 82, 3196 CrossRef CAS.
- P. S. Achanta, R. K. Gattu, A. R. V. Belvotagi, R. R. Akkinepally, R. K. Bobbala and A. R. V. N. Achanta, Fitoterapia, 2015, 100, 166 CrossRef CAS PubMed.
- Z. K. Duan, T. M. Lv, G. S. Song, Y. X. Wang, B. Lin and X. X. Huang, Nat. Prod. Res., 2020, 36, 229 CrossRef PubMed.
- W. Dinku, J. Isaksson, F. G. Rylandsholm, P. Bouř, E. Brichtová, S. U. Choi, S. H. Lee, Y. S. Jung, Z. S. No, J. S. M. Svendsen, A. J. Aasen and A. Dekebo, Appl. Biol. Chem., 2020, 63, 16 CrossRef CAS.
- S. Thongnest, J. Boonsombat, H. Prawat, C. Mahidol and S. Ruchirawat, Phytochemistry, 2017, 134, 98 CrossRef CAS PubMed.
- F. Messina, M. Curini, C. Di Sano, C. Zadra, G. Gigliarelli, L. A. Rascón-Valenzuela, R. E. R. Zepeda and M. C. Marcotullio, J. Nat. Prod., 2015, 78, 1184 CrossRef CAS.
- Q. Xiong, F. Rocco, W. K. Wilson, R. Xu, M. Ceruti and S. P. T. Matsuda, J. Org. Chem., 2005, 70, 5362 CrossRef CAS.
- C. J. Huck, Y. D. Boyko and D. Sarlah, Acc. Chem. Res., 2021, 54, 1597 CrossRef CAS PubMed.
- S. Lodeiro, W. K. Wilson, H. Shan and S. P. T. Matsuda, Org. Lett., 2006, 8, 439 CrossRef CAS.
- T. Xiang, M. Shibuya, Y. Katsube, T. Tsutsumi, M. Otsuka, H. Zhang, K. Masuda and Y. Ebizuka, Org. Lett., 2006, 8, 2835 CrossRef CAS PubMed.
- G. C. Fazio, R. Xu and S. P. T. Matsuda, J. Am. Chem. Soc., 2004, 126, 5678 CrossRef CAS.
- T. Hoshino and T. Sato, Chem. Commun., 2002, 291 RSC.
- I. Abe, Nat. Prod. Rep., 2007, 24, 1311 RSC.
- G. Klaus and G. Axel, Tetrahedron, 1984, 40, 3235 CrossRef.
- D. Tasdemir, G. C. Mangalindan, G. P. Concepcion, S. M. Verbitski, S. Rabindran, M. Miranda, M. Greenstein, J. N. A. Hooper, M. K. Harper and C. M. Ireland, J. Nat. Prod., 2002, 65, 210 CrossRef CAS.
- M. Tsuda, M. Ishibashi, K. Ageta, T. Sasaki and J. Kobayashi, Tetrahedron, 1991, 47, 2181 CrossRef CAS.
- J. Su, Y. Meng, L. Zeng, X. Fu and F. J. Schmitz, J. Nat. Prod., 1994, 57, 1450 CrossRef CAS PubMed.
- F. Lv, Z. Deng, J. Li, H. Fu, R. W. M. van Soest, P. Proksch and W. Lin, J. Nat. Prod., 2004, 67, 2033–2036 CrossRef.
- X. Y. Liu, C. J. Li, F. Y. Chen, J. Ma, S. Wang, Y. H. Yuan, L. Li and D. M. Zhang, Org. Lett., 2018, 20, 4549 CrossRef CAS PubMed.
- J. A. Francis, S. N. Raja and M. G. Nair, Chem. Biodiversity, 2004, 1, 1842 CrossRef CAS.
- L. O. Hanus, T. Rezanka, V. M. Dembitsky and A. Moussaieff, Biomed. Pap., 2005, 149, 3 CrossRef CAS.
- S. A. F. Morad, C. Schmidt, Be. Buechele, B. Schneider, M. Wenzler, T. Syrovets and T. Simmet, J. Nat. Prod., 2011, 74, 1731 CrossRef CAS.
- H. Z. Li, Y. J. Zhang and C. R. Yang, Nat. Prod. Res., 2006, 18, 549 CAS.
- M. Yoshikawa, T. Morikawa, Y. Kashima, K. Ninomiya and H. Matsuda, J. Nat. Prod., 2003, 66, 922 CrossRef CAS.
- K. Prantz and J. Mulzer, Chem. Rev., 2010, 110, 3741 CrossRef CAS.
- F. Y. Meng, J. X. Sun, X. Li, H. Y. Yu, S. M. Li and H. L. Ruan, Org. Lett., 2011, 13, 1502 CrossRef CAS PubMed.
- P. C. Wang, X. H. Ran, H. R. Luo, Q. Y. Ma, Y. Q. Liu, H. F. Dai, J. Zhou and Y. X. Zhao, Org. Lett., 2013, 15, 2898 CrossRef CAS PubMed.
- C. Q. Liang, Y. M. Shi, X. Y. Li, R. H. Luo, Y. Li, Y. T. Zheng, H. B. Zhang, W. L. Xiao and H. D. Sun, J. Nat. Prod., 2013, 76, 2350 CrossRef CAS.
- U. Shmueli, S. Carmely, A. Groweiss and Y. Kashman, Tetrahedron Lett., 1981, 22, 709 CrossRef CAS.
- A. A. Beih, A. H. El-Desoky, M. A. Al-Hammady, A. I. Elshamy, M. E. F. Hegazy, H. Kato and S. Tsukamoto, Fitoterapia, 2018, 128, 43 CrossRef PubMed.
- S. Jain, I. Abraham, P. Carvalho, Y. H. Kuang, L. A. Shaala, D. T. A. Youssef, M. A. Avery, Z. S. Chen and K. A. El Sayed, J. Nat. Prod., 2009, 72, 1291 CrossRef CAS.
- R. F. Angawi, E. Saqer, A. Abdel-Lateff, F. A. Badria and S. E. N. Ayyad, Pharmacogn. Mag., 2014, 10, S334–S341 CrossRef.
- Y. Kashman, T. Yosief and S. Carmeli, J. Nat. Prod., 2001, 64, 175 CrossRef CAS.
- Y. Kashman and A. Rudi, Phytochem. Rev., 2004, 3, 309 CrossRef CAS.
- A. E. Nugroho, D. Inoue, C. P. Wong, Y. H. Irasawa, T. Kaneda, O. Shirota, A. H. A. Hadi and H. Morita, J. Nat. Rem., 2018, 72, 588 CAS.
- G. Ni, J. Y. Li and D. Q. Yu, Org. Biomol. Chem., 2018, 16, 3754 RSC.
- D. H. R. Barton and K. H. Overton, J. Chem. Soc., 1955, 2639 RSC.
- M. G. Rowan, P. D. G. Dean and T. W. Goodwin, FEBS Lett., 1971, 12, 229 CrossRef CAS PubMed.
- M. G. Rowan and P. D. G. Dean, Phytochemistry, 1972, 11, 3111 CrossRef CAS.
- T. Araki, Y. Saga, M. Marugami, J. Otaka, H. Araya, K. Saito, M. Yamazaki, H. Suzuki and T. Kushiro, ChemBioChem, 2016, 17, 288 CrossRef CAS PubMed.
- Y. Saga, T. Araki, H. Araya, K. Saito, M. Yamazaki, H. Suzuki and T. Kushiro, Org. Lett., 2017, 19, 496 CrossRef CAS PubMed.
- W. Krick, F. J. Marner and M. L. Jaenicke, Helv. Chim. Acta, 1984, 67, 318 CrossRef CAS.
- W. Krick, F. J. Marner and L. Z. Jaenicke, Naturforsch, 1983, C38, 179 CrossRef.
- A. Corbu, M. Aquino, T. V. Pratap, P. Retailleau and S. Arseniyadis, Org. Lett., 2008, 10, 1787 CrossRef CAS PubMed.
- A. Corbu, G. Gauron, J. M. Castro, M. Dakir and S. Arseniyadis, Org. Lett., 2007, 9, 4745 CrossRef CAS.
- W. G. Dauben and D. M. Michno, J. Org. Chem., 1977, 42, 643 CrossRef.
- J. A. Profitt, D. S. Watt and E. J. Corey, J. Org. Chem., 1975, 40, 127 CrossRef CAS.
- E. Negishi, A. O. King and W. L. Klima, J. Org. Chem., 1980, 45, 2526 CrossRef CAS.
- J. Marshall and G. M. Schaaf, J. Org. Chem., 2003, 68, 7428 CrossRef CAS.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- Y. D. Boyko, C. J. Huck and D. Sarlah, J. Am. Chem. Soc., 2019, 141, 14131 CrossRef CAS PubMed.
- O. H. Oldenziel, D. Van Leusen and A. M. Van Leusen, J. Org. Chem., 1977, 42, 3114 CrossRef CAS.
- C. P. Ting, G. Xu, X. Zeng and T. J. Maimone, J. Am. Chem. Soc., 2016, 138, 14868 CrossRef CAS.
- A. Fernandez-Mateos, H. P. Teijon, R. R. Clemente, R. R. Gonzalez and F. S. Gonzalez, Synlett, 2007, 17, 2718 CrossRef.
- T. D. Michels, J. U. Rhee and C. D. Vanderwal, Org. Lett., 2008, 10, 4787 CrossRef CAS.
- Y. D. Boyko, C. J. Huck, S. Ning, A. S. Shved, C. Yang, T. Chu, J. Tonogai, P. J. Hergenrother and D. Sarlah, J. Am. Chem. Soc., 2021, 143, 2138 CrossRef CAS PubMed.
- A. F. Barrero, J. E. Oltra, M. M. Herrador, E. Cabrera, J. F. Sanchez, J. F. Quilez, F. J. Rojas and J. F. Reyes, Tetrahedron, 1993, 49, 141 CrossRef CAS.
- J. R. Coombs, L. Zhang and J. P. Morken, Org. Lett., 2015, 17, 1708 CrossRef CAS PubMed.
- N. Kotoku, N. Tamada, A. Hayashi and M. Kobayashi, Bioorg. Med. Chem. Lett., 2008, 18, 3532 CrossRef CAS.
- S. Aoki, M. Sanagawa, Y. Watanabe, A. Setiawan, M. Arai and M. Kobayashi, Bioorg. Med. Chem., 2007, 15, 4818 CrossRef CAS PubMed.
- V. Domingo, L. Lorenzo, J. F. Quilez del Moral and A. F. Barrero, Org. Biomol. Chem., 2013, 11, 559 RSC.
- A. F. Barrero, E. A. Manzaneda, J. Altarejos, S. Salido, J. M. Ramos, M. S. Simmonds and W. M. Blaney, Tetrahedron, 1995, 51, 7435 CrossRef CAS.
- J. H. George, J. E. Baldwin and R. M. Adlington, Org. Lett., 2010, 12, 2394 CrossRef CAS PubMed.
- H. Tanimoto and T. Oritani, Tetrahedron: Asymmetry, 1996, 7, 1695 CrossRef CAS.
- V. Domingo, L. Silva, H. R. Dieguez, J. F. Arteaga, J. F. Quılez del Moral and A. F. Barrero, J. Org. Chem., 2009, 74, 6151 CrossRef CAS.
- A. F. Barrero, M. M. Herrador, J. F. Quilez delMoral, P. Arteaga, J. F. Arteaga, M. Piedra and E. M. Sanchez, Org. Lett., 2005, 7, 2301 CrossRef CAS.
- B. Ma, Y. Zhao, C. He and H. Ding, Angew. Chem., 2018, 130, 15793 CrossRef.
- A. F. Barrero, J. M. Cuerva, M. M. Herrador and M. V. Valdivia, J. Org. Chem., 2001, 66, 4074 CrossRef CAS.
- A. Gansäuer, M. Pierobon and H. Bluhm, Angew. Chem., Int. Ed., 1998, 37, 101 CrossRef.
- C. L. Chapelain, Org. Biomol. Chem., 2017, 15, 6242 RSC.
- J. C. Gilbert and U. Weerasooriya, J. Org. Chem., 1982, 47, 1837 CrossRef CAS.
- B. V. Burger, W. G. B. Petersen, W. G. Weber and Z. M. Munro, J. Chem. Ecol., 2002, 28, 2527 CrossRef CAS.
- A. Gris, N. Cabedo, I. Navarro, I. de Alfonso, C. Agullo and A. Abad-Somovilla, J. Org. Chem., 2012, 77, 5664 CrossRef CAS.
- J. S. Reddy, M. Gangababu, P. Manimala, A. Rammohan and J. S. Yadav, Synthesis, 2020, 52, 735 CrossRef CAS.
- H. Hagiwara and H. Uda, J. Org. Chem., 1988, 53, 2308 CrossRef CAS.
- A. B. Smith and R. Mewshaw, J. Org. Chem., 1984, 49, 3685 CrossRef CAS.
- J. Tsuji, I. Shimizu and K. Yamamoto, Tetrahedron Lett., 1976, 34, 2975 CrossRef.
- B. S. Bal, W. E. Childers Jr and H. W. Pinnick, Tetrahedron, 1981, 37, 2091 CrossRef CAS.
- D. A. Evans, M. D. Ennis and D. J. Mathre, J. Am. Chem. Soc., 1982, 104, 1737 CrossRef CAS.
- M. A. Boone, R. Tong, F. E. McDonald, S. Lense, R. Cao and K. I. Hardcastle, J. Am. Chem. Soc., 2010, 132, 5300 CrossRef CAS PubMed.
- A. Rudi, T. Yosief, M. Schleyer and Y. Kashman, Tetrahedron, 1999, 55, 5555 CrossRef CAS.
- A. N. L. Batista, B. R. P. Angrisani, M. E. D. Lima, S. M. P. da Silva, V. H. Schettini, H. A. Chagas, F. M. dos Santos Jr, J. M. Batista Jr and A. L. Valverde, J. Braz. Chem. Soc., 2021, 32, 1499 CAS.
- P. Cárdenas, J. Gamage, C. M. Hettiarachchi and S. Gunasekera, Mar. Drugs, 2022, 20, 190 CrossRef PubMed.
- V. A. Stonik and S. A. Kolesnikova, Mar. Drugs, 2021, 19, 327 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |