
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of tosyl starch in eco-friendly media†
Phitawat
Namnouad‡
a,
Manisa
Kongkaew
b,
Suttiporn
Pikulthong
a,
Rungtiwa
Wongsagonsup
c,
Taweechai
Amornsakchai
ad,
Siwaporn Meejoo
Smith
 *ad and
Thanthapatra
Bunchuay‡
a
*ad and
Thanthapatra
Bunchuay‡
a
aDepartment of Chemistry, Faculty of Science, Mahidol University, Rama VI Rd, Bangkok 10400, Thailand. E-mail: siwaporn.smi@mahidol.edu
bDepartment of Science and Technology, Faculty of Science, Pibulsongkram Rajabhat University, Phlai Chumphon, Mueang Phitsanulok District, Phitsanulok 65000, Thailand
cFood Technology Division, School of Interdisciplinary Studies, Mahidol University, Kanchanaburi Campus, Kanchanaburi, 71150, Thailand
dCenter of Sustainable Energy and Green Materials, Faculty of Science, Mahidol University, Salaya, Nakorn Pathom 73170, Thailand
First published on 21st July 2023
Abstract
A green strategy for efficient tosylation of starch was demonstrated in the eco-friendly solvent NaOH–urea in the presence of surfactants. The influence of surfactant nature, surfactant amounts, and the molar ratio between p-toluenesulfonyl chloride (TsCl) and the anhydroglucose unit (AGU) on the degree of substitution (DS) was studied. The tosyl products were characterised by means of CHNS elemental analysis and spectroscopy including FTIR, 1H-NMR and 13C-NMR. The results demonstrate that this methodology allows a range of degrees of substitution (DS) to be observed depending on the surfactants and prove that neutral surfactants are not necessary to obtain high DS of tosyl groups as in the case with cellulose starting materials previously reported. Subsequent nucleophilic displacement (SN) reactions of the tosyl groups in this medium were also investigated.
Starch is a polysaccharide containing anhydroglucose monomeric units linked via glycosidic bonds. They are composed of two main components with different physicochemical properties: amylose, a linear polymer with α-(1 → 4) glycosidic linkages, and amylopectin, a highly branched polymer with lots of short chains due to additional α-(1 → 6) glycosidic linkages.1,2 Starch from different botanical resources has varying percentages of amylose and amylopectin, and hence diverse physicochemical properties for varied areas of applications.3,4 Starch has outstanding characteristic properties including natural abundancy, cheapness, renewability, non-toxicity, and biodegradability. Blending starch with other materials can, therefore, provide materials with a variety of physicochemical benefits. Corn and potato starch, for example, were incorporated into tissue scaffold matrices to improve their mechanical properties and biocompatibility.5–7 Starch was also used in a controlled-release drug delivery system to enhance drug's efficiency and reduce drug's toxicity in the pharmacological area.8,9
Enhancing materials properties using native starch is, however, proved challenging as there are inherent limitations of native starch to overcome such as its poor solubility, low mechanical strength, tendency to retrograde, and the difficulty to process.10,11 Chemical modifications, in this regard, provide an approach to eliminate these shortcomings and to achieve desired properties for specific applications.12 The multifunctional structure of starch allows three readily available hydroxyl groups per anhydroglucose unit (AGU) repeating unit to be chemically derivatised with different functional groups and various functionalisation patterns, unfolding endless possibilities to diversify its properties. Most common chemical modifications of starch include esterification,13–15 etherification,16,17 carbonisation,18,19 crosslinking, and oxidation.20–22 Esterification, for example, was used to reduce starch's hygroscopicity by converting hydrophilic hydroxyl groups to hydrophobic ester groups so that the modified-starch can be used as a renewable alternative to fossil-based feedstock.23–25 Etherification, as a step towards cationic starch derivatives, on the other hand, plays an important role in wastewater treatment as this can improve starch adsorptive capacity for the removal of anionic dyestuffs from the textile industries.26,27
The discovery of an easily prepared reactive starch intermediate whose functional groups can be more diversely transformed will enable the fabrication of a larger library of starch derivatives. In the past few decades, tosyl starch, derived from the reaction of starch with toluene sulfonyl chloride, represents the most promising candidate in this regard due to the ease of subsequent nucleophilic displacements. The major problem towards the manufacture is, however, the immiscible nature of starch in aqueous solution as well as most common organic solvents. Starch tosylation in pyridine was initially studied but only under heterogenous course of reaction.28,29 Even when tosylation is carried out in high-polar solvents such as dimethyl sulfoxide (DMSO) and N,N-dimethyl formamide (DMF), only a small degree of substitution (DSTos) is observed due to phase separation of the reaction products. A combination of N,N-dimethylacetamide (DMA) and lithium chloride offers an efficient homogenous synthetic path towards products with a wide range of DSTos depending on the conditions.30,31 However, there comes serious health and environmental concerns associated with the use of these solvents as they are flammable, toxic and hazardous. Thus, the discovery of green solvents is a long-term endeavour that would impact a wide range of research as this would open a new paradigm for homogeneous starch modification on both the lab and commercialisation scale.
In the past few decades, much research has been devoted to ionic liquids and supercritical carbon dioxide as green solvents for starch modification.32–35 However, because of the cost and difficulty to handle, they are deemed unsuitable for large scale processing. New aqueous solvents seem to be a better alternative but most of them suffer from the hydrophobicity of the reaction products which leads to phase separation and hence small DSTos due to the insufficient reactivity.36 Recently, T. Heinze and co-workers reported the multigram scale synthesis of cellulose tosylates of high DSTos with the use of environmentally friendly aqueous NaOH–urea media with the addition of surfactants.37 They found that this solvent system shows predominant functionalisation at C6 and high DSTos products were observed with the use of a non-ionic surfactant such as imbentin. Inspired by this work, we envision the possibility of using an analogous system for starch tosylation.
Herein we describe a synthetic methodology for the functionalisation of tosyl groups into the starch scaffold by using eco-friendly aqueous NaOH–urea media with a range of surfactants, and investigate the relationship between the surfactant nature, DSTos values, and functionalisation pattern of the obtained products. Importantly, we also highlight the fact that neutral surfactants are not necessary for obtaining high DSTos in starch as in the case of cellulose tosylation reported previously, but ionic surfactants could also give satisfactory DSTos. The tosylated samples were characterised by elemental analysis and spectroscopic methods including FTIR, 13C-NMR and 1H-NMR. Starch tosylation in this media was found to occur predominantly at C2 as evidenced by 13C-NMR and 1H-NMR spectra, and this was shown to be in contrast with the previously reported analogous reaction with cellulose whose functionalisation occurs mainly at C6.
Experiments
Chemicals and materials
Cassava starch was purchased from a local supermarket in Bangkok and used without further purifications, while pineapple starch was prepared in-house using the procedure reported by our research group.38p-Toluenesulfonyl chloride (TsCl), Tween®-20, Tween®-20, pyridine, and SDS were purchased from Tokyo Chemical Industry (TCI). Sodium hydroxide (NaOH, 97%), Brij®30 and TritonTMX 100 were purchased from Sigma-Aldrich. All solvents were purchased from RCI Labscan. Deionised (DI) water from a Milli-Q Advantage A10 Water Purification System with an electronic resistance ≥18.2 MΩ cm at 25 °C was used in all experiments.Characterisation
Fourier transform infrared (FT-IR) spectra were recorded using the attenuated total reflection technique (Bruker, Alpha) 4000–500 cm−1 to confirm successful functionalisation of all modified starch. Elemental analysis was carried out to determine the percentage content of carbon, hydrogen, nitrogen, and sulfur in all starch samples using a Thermo Scientific FlashSmart Elemental Analyzer (PS42267-EN 1116). Thermogravimetric analyses (TGA) were performed on a SDT 2960 SDT V3.0F in the temperature range of 40–1000 °C under air flow with a heating rate of 10 °C min−1. The samples for NMR analysis were prepared as d6-DMSO solutions. Their 1H NMR spectra were recorded using a Bruker AVANCE 400 spectrometer (400 MHz), a Bruker AVANCE 500 spectrometer (500 MHz), and a Bruker AVANCE 600 spectrometer (600 MHz), while 13C NMR spectra were recorded on the same machine (100 MHz) at 298 K.Aqueous tosylation of starch in NaOH–urea/surfactant
Oven-dried starch (1.0 g, 6.2 mmol) was dissolved in distilled water (30 ml) followed by the addition of NaOH (1.8 g, 44 mmol) and urea (3.0 g, 50 mmol) with stirring to obtain a clear solution. TsCl (4.7 g, 25 mmol, 4.0 equivalents of AGU) and Brij® 30 (1.0 ml, 2.62 mmol) were then added to the mixture. After the reaction solution was stirred vigorously at 25 °C for 24 h, the mixture was precipitated into 200 ml ethanol to obtain a white precipitate which was subsequently filtered off, and washed with ethyl acetate. The white solid was resuspended in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) mixture of ethanol–water (15 ml), and then filtered off. The obtained white precipitate was ground using a pestle and mortar, washed with ethanol, and filtered off. The grinding and washing steps were performed twice to remove the remaining surfactants. The collected product was then dried in an oven at 60 °C for 24 h. Brij could be replaced with other surfactants including neutral surfactants (Tween®20, Tween®80, and Triton™X-100), and ionic surfactants (CTAB and SDS). The effect of of surfactants and TsCl per AGU molar ratio was also studied by using the same conditions, but the amount of surfactants and TsCl was subsequently changed according to Table 2.
:1 (v/v) mixture of ethanol–water (15 ml), and then filtered off. The obtained white precipitate was ground using a pestle and mortar, washed with ethanol, and filtered off. The grinding and washing steps were performed twice to remove the remaining surfactants. The collected product was then dried in an oven at 60 °C for 24 h. Brij could be replaced with other surfactants including neutral surfactants (Tween®20, Tween®80, and Triton™X-100), and ionic surfactants (CTAB and SDS). The effect of of surfactants and TsCl per AGU molar ratio was also studied by using the same conditions, but the amount of surfactants and TsCl was subsequently changed according to Table 2.
Tosylation of starch in DMA/LiCl
Tosylation of starch in DMA/LiCl media was modified from the synthetic procedure reported by Heinze et al.31 An oven-dried starch (5 g, 31 mmol) was suspended in DMA (100 ml) and stirred at 130 °C for 1 h. After the slurry had been allowed to cool to 100 °C, anhydrous LiCl (10 g, 202 mmol) was added portion wise. During cooling to room temperature with stirring, the starch suddenly turned from slurry to a clear-viscous solution. A solution of triethylamine (14.8 ml, 107 mmol) in DMA (10 ml) was added. The solution was further cooled down to about 8 °C and TsCl (23.6 g, 123 mmol) in DMA (30 ml) was added dropwise with stirring. The reaction mixture was additionally stirred for 24 h at room temperature and then slowly poured into 500 ml of ice water. The precipitate was filtered off, washed with ethanol, and the product was dried at 60 °C for 24 h.Tosylation of starch in DMSO/LiCl
Oven-dried starch (1.0 g, 6.2 mmol) was dissolved in DMSO (20 ml). Anhydrous LiCl (2.0 g, 40 mmol) was added, followed by the addition of triethylamine (2.9 ml, 21.4 mmol) and TsCl (2.4 g, 12 mmol) portion wise. After stirring at room temperature for 24 h, the reaction mixture was poured into ice-water (150 ml). The precipitate was filtered off, washed with ethanol, and the product was dried at 60 °C for 24 h.Tosylation of starch in DMF/LiCl
The reaction was carried out using the same procedure as aforementioned for tosylation in DMSO/LiCl but DMSO was replaced by DMF with the same volume.Tosylation of starch in anhydrous pyridine
Oven-dried starch (1.0 g, 6.2 mmol) was dissolved in anhydrous pyridine (25 ml) and stirred at room temperature for 16 h. Subsequently, TsCl (3.5 g, 18 mmol) was added portion wise. The reaction mixture was kept stirring and heated at 60 °C for 2 h. After the reaction mixture was cooled down to room temperature, the mixture was poured into a 4:1 (v/v) mixture of methanol–water (250 ml). The solid product was then filtered off, washed with water, and dried to give a white powder.
Nucleophilic displacement reactions of starch
The oven-dried tosyl sample prepared in Brij® 30 (DSTos = 0.65; 0.2 g, 0.68 mmol) was dissolved in distilled water (6 ml) followed by the addition of NaOH (0.3 g, 7.33 mmol) and urea (0.5 g, 8.33 mmol) with stirring to obtain a clear solution. Sodium azide (57.9 mg, 0.89 mmol, 1.5 equivalents of DSTos) and Brij® 30 (1.0 ml, 2.62 mmol) were then added to the mixture. After the reaction mixture was stirred vigorously at 80 °C for 24 h, the mixture was precipitated into 40 ml ethanol to obtain a white precipitate which was subsequently filtered off and washed with ethanol. The product was dried in a vacuum for 8 h. Sodium azide could be replaced with potassium phthalimide (164.8 mg, 0.89 mmol, 1.5 equivalents of DSTos) under the same conditions. Similar nucleophilic displacement reactions were also performed in DMSO (6 ml).Results and discussion
Tosylation reaction of starch was carried out by reacting TsCl with dried cassava starch in different solvent systems and the amount of TsCl was initially kept constant at a 4:1 molar ratio of TsCl per anhydroglucose unit (AGU) (Scheme 1). The resulting products were investigated after the successful tosylation by determination of the degree of tosyl substitution (DSTos) and percentage of sulfur (%S) in each sample via1H NMR spectral analysis and CHNS analysis, respectively. Selections of reaction media played key roles in controlling the DSTos and %S presented in each sample (Table 1). The reaction carried out in a mixture of the DMA/LiCl solvent system gave a homogeneous solution and produced the tosyl starch product with DSTos of 0.75% and %S of 9.26 (Table 1, entry 1). Replacement of DMA with other polar aprotic solvents including DMF and DMSO significantly reduced the solubility of reaction products as observed by the formations of the heterogeneous clumps through the course of reactions, resulting in a negligible DSTos (Table 1, entries 2 and 3). A negligible DSTos was also observed in the tosylation reaction carried out in DMSO and DMF without the addition of LiCl (Table 1, entries 4 and 5). Interestingly, tosylation of starch in anhydrous pyridine followed the method reported by Hirase et al. and Horton et al. gave negligible DSTos (Table 1, entry 6) and %S. According to the original reports, a nucleophilic substitution reaction of tosyl groups with sodium iodide in 2,5-hexane-dione was employed to indirectly confirm that tosylation occurred predominantly at C6 of AGU in modified starch.28,29 However, 1H NMR spectral evidence was not used as a main characterisation technique in these reports.
 | ||
| Scheme 1 Tosylation reaction of starch in aqueous media. | ||
| Entry | Solvent | Additives | Surfactant | %Sa | DSTosb |
|---|---|---|---|---|---|
| a %S is percentage of sulfur determined by elemental analysis. b DSTos was calculated from the sulfur content of the product according to the equation: DSTos = 162 × S (%)/[3200 (%) − 155 × S (%)]. c %S = 0 and the resulting product did not show 1H-NMR signal of tosyl moieties. d A negligible DSTos and 1H-NMR spectrum revealed proton signals of tosyl moieties. | |||||
| 1 | DMA | LiCl | — | 9.26 | 0.75 |
| 2 | DMSO | LiCl | — | —c | —c |
| 3 | DMF | LiCl | — | —c | —c |
| 4 | DMSO | — | — | —c | —c |
| 5 | DMF | — | — | —c | —c |
| 6 | Pyridine | — | — | —d | —d |
| 7 | Water | NaOH, urea | — | 0.21 | 0.01 |
| 8 | Brij® 30 | 7.88 | 0.65 | ||
| 9 | Tween®20 | 3.38 | 0.20 | ||
| 10 | Tween®80 | 3.08 | 0.18 | ||
| 11 | Triton™X-100 | 7.15 | 0.55 | ||
| 12 | SDS | 6.91 | 0.53 | ||
| 13 | CTAB | 4.42 | 0.28 | ||
Our effort moved towards tosylation in eco-friendly aqueous media. Starch was insoluble in water and the dissolution can be facilitated by the addition of NaOH and urea (Fig. S1, ESI†) as a result of dissociation of a double-helical structure of the native starch and crystal to amorphous structural transformation under these conditions.39 However, an aqueous NaOH–urea system still afforded only small DSTos due to the immiscibility of reagents such as TsCl over the course of reaction (Table 1, entry 7). Enhanced solubility of starch and TsCl can be achieved by the addition of surfactants. In this work, we used two types of surfactants including commercial non-ionic and ionic surfactants (Scheme 1). In the presence of surfactants, tosyl starch products were obtained with the DSTos in the range of 0.20–0.65, and showed no distinct correlation between the DSTos of the products and the types of the surfactants added (ionic or non-ionic surfactants). With a variety of surfactants used under investigation, our results are in stark contrast with the previously reported analogous cellulose tosylation reaction where Imbentin, a neutral surfactant, could give a higher DSTos in comparison to the conditions using SDS. In our cases, neutral surfactants are not always champion in the tosylation of starch as evidenced by lower DSTos values observed in Tween®20 and Tween®80 (Table 1, entries 9 and 10, DSTos = 0.20 and 0.18, respectively) than the values obtained from SDS and CTAB (Table 1 entries 12 and 13, DSTos = 0.53 and 0.28, respectively). These results suggested that suitable surfactants can facilitate tosylation reactions in aqueous media with satisfactory DSTos values.
In order to investigate the effect of surfactant amount and TsCl per AGU molar ratio on the DSTos in aqueous tosylation reaction, we chose Brij® 30 as a model in this study due to Brij® 30 giving the highest DSTos amongst the surfactants studied (Table 1, entry 8). Brij® 30 detergents are non-ionic surfactants derived from ethoxylating natural alcohols, consisting of alkyl and polyoxyethylene (POE) ether components, and used in a variety of applications including liquid crystals and protein solubilisation by forming micellar structures upon adding it above the critical micelle concentration (CMC).40,41 In this study, we set a concentration range of Brij® 30 above its CMC (7.0–14.5 mg L−1) to ensure that micelles are formed in the mixture. We found that, at a constant TsCl per AGU molar ratio (4:1), DSTos increases with the amount of Brij® 30 added (Table 2, entries 1–8). This is postulated by the fact that a higher amount of surfactant in the system implies a higher ability to form more micelles in the solution, and this helps with the homogeneous dissolution of higher DSTos products during the course of the reaction. Correspondingly, at the same molar ratio of Brij® 30 per AGU, the increasing molar ratio of tosyl chloride per AGU yields products with higher DSTos (Table 2, entries 8–11).
| Entry | Molar ratio | %Sb | DSTosc | |
|---|---|---|---|---|
| Brij® 30:AGU |
TsCl:AGU |
|||
| a The reactions were carried out in an aqueous NaOH–urea system. b %S is the percentage of sulfur determined by elemental analysis. c DSTos was calculated from the sulfur content of the product according to the equation: DSTos = 162 × S (%)/[3200 (%) − 155 × S (%)]. | ||||
| 1 | 0.016 | 4 | 1.07 | 0.06 |
| 2 | 0.026 | 4 | 0.75 | 0.04 |
| 3 | 0.034 | 4 | 0.68 | 0.04 |
| 4 | 0.042 | 4 | 2.04 | 0.12 |
| 5 | 0.084 | 4 | 4.95 | 0.33 |
| 6 | 0.170 | 4 | 7.39 | 0.62 |
| 7 | 0.254 | 4 | 6.99 | 0.54 |
| 8 | 0.424 | 4 | 7.88 | 0.65 |
| 9 | 0.424 | 3 | 6.26 | 0.45 |
| 10 | 0.424 | 2 | 5.94 | 0.42 |
| 11 | 0.424 | 1 | 2.65 | 0.15 |
The structures of pure starch and tosyl starch samples obtained under different solvent media were investigated and compared by means of FTIR spectroscopy (Fig. 1). The starch FTIR spectrum shows broad absorption at 3600–3000 cm−1 belonging to multiple O–H groups in the polysaccharide backbone. The tosyl starch samples obtained from the DMA/LiCl system, however, reveals a significant decrease in O–H absorption with the appearance of characteristic absorption peaks at 1353 and 1173 cm−1 belonging to –SO2 symmetric and asymmetric stretches, and the appearance of signals at 1597 cm−1 due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching and at 810 and 665 cm−1 due to CC bending of the benzene ring. Tosyl starch samples obtained under anhydrous pyridine shows similar absorption to pure starch due to negligible substitution under this condition while tosyl samples obtained with Brij® 30 shows a similar FTIR spectrum to that of DMA/LiCl. The FTIR spectra of the tosyl samples with other surfactants were also compared and they are all representative of tosyl starch (Fig. S2, ESI†).
C stretching and at 810 and 665 cm−1 due to CC bending of the benzene ring. Tosyl starch samples obtained under anhydrous pyridine shows similar absorption to pure starch due to negligible substitution under this condition while tosyl samples obtained with Brij® 30 shows a similar FTIR spectrum to that of DMA/LiCl. The FTIR spectra of the tosyl samples with other surfactants were also compared and they are all representative of tosyl starch (Fig. S2, ESI†).
 | ||
| Fig. 1 FTIR spectra of tosyl starch samples obtained in different solvent systems. | ||
Insight details were further studied by 1H-NMR and 13C-NMR spectral analysis of the tosylated products in d6-DMSO. First, the product obtained using DMA/LiCl was studied. The 1H-NMR spectrum of the product obtained from a combination of DMA/LiCl revealed that tosylation occurred predominantly at the secondary hydroxyl group of C2 in accordance with the literature (Fig. S5, ESI†).31 The spectrum shows characteristic tosyl moiety signals at 7.80, 7.39 and 2.37 ppm corresponding to aromatic protons Ha, Hb and methyl proton Hc, respectively. Despite tosylation in anhydrous pyridine showing a negligible DSTos, the 1H-NMR spectrum of the tosyl starch synthesised under this condition shows two sets of aromatic protons (Fig. S4, ESI†). The first set of aromatic proton signals appeared at 7.46 and 7.11 ppm belonging to Ha and Hb tosyl protons, respectively, while the second set of aromatic proton signals appeared at further downfield resonances 8.66, 8.01 and 7.57 ppm belonging to pyridinium moieties. These result suggest that displacement of tosyl groups by the pyridine solvent occurred and thus significantly reduced %S as well as DSTos to negligible values (Table 1, entry 6). Therefore, only CHNS elemental analysis could result in misinterpretation and emphasise the need for NMR spectroscopic techniques for insight studies. By using our NMR spectral results together with analysis results from previous studies, we can use the 1H-NMR spectra of the tosyl starch prepared from DMA/LiCl to represent the predominant C2 functionalisation and from pyridine to represent the predominant C6 functionalisation albeit partial pyridine substitutions observed (Fig. 2).
 | ||
| Fig. 2 1H-NMR spectra of tosyl starch prepared under the conditions using (a) DMA/LiCl, (b) NaOH–urea/Brij® 30, and (c) pyridine solvent systems, recorded in d6-DMSO at 25 °C. | ||
The 1H-NMR spectrum of the tosyl starch product obtained with NaOH–urea-Brij®30 system (DSTos = 0.65) recorded in d6-DMSO reveals that the tosylation reaction occurs at hydroxyl positions C2 and C6 (Fig. 2b). There are two sets of tosyl protons in both aromatic and methyl regions assigned to be tosylations of hydroxyl groups at positions 2 and 6 of AGU. The aromatic region shows signals at δ = 7.68 ppm and δ = 7.35 ppm belonging to Ha and Hb of the tosyl groups at C2 and signals at δ = 7.48–7.46 ppm and δ = 7.12–7.10 ppm (d, J = 7.9 Hz) belonging to Ha and Hb of the tosyl groups at C6. The tosyl proton signals at C2 positions become slightly upfield shifted and broad in comparison with the chemical shifts of tosyl signals observed in the spectrum of the tosyl starch prepared from DMA/LiCl. Peak broadening and slight chemical shift changes are presumably caused by the variations in non-covalent interactions of C2-tosyl groups when there are high levels of C6-tosyl groups present in the polymeric chain. In addition, the methyl signals at δ = 2.34 and 2.28 ppm are assigned to Hc of the tosyl moieties at C2, and C6, respectively. The 1H-NMR spectrum of the tosyl products obtained from other surfactants also show similar spectral patterns, suggesting that hydroxyl groups at positions C2 and C6 in the AGU are made available for tosylation reaction under the influence of surfactants (Fig. S13–S17, ESI†). Note that there might be marginal tosyl substitutions at C3 but their spectral features are unresolved.
The percent distribution of functional groups of the obtained products at different DSTos prepared with Brij® 30 was also determined by the integration method (Table 3, see spectra in the ESI,† Fig. S6–S12). Here we demonstrated that starch tosylation with the NaOH–urea-surfactant favourably occurs at position C2 followed by C6 with a substitution ratio of 9:1. T. Heinze and co-workers used chemical shifts and splitting patterns in the 13C-NMR spectra of tosyl cellulose at different DSTos to determine the functionalisation pattern of cellulose tosylation in the NaOH–urea system. In this work, the 13C-NMR spectra of tosyl starch samples in NaOH–urea-Brij® 30 with DSTos of 0.33, 0.54 and 0.65 were therefore recorded in d6-DMSO at 25 °C for a similar purpose (Fig. 3). Carbon chemical shifts of aromatic tosyl moieties (C7, C8, C9 and C10) and tosyl methyl signal (C11) can be observed at 128–146 and 22 ppm, respectively. The signals on the carbohydrate backbone (C1–6) were assigned in accordance with the literature. It is noteworthy that, at higher DSTos, a stark splitting of the C1 signal is observed due to significant tosylation of starch at position 2 while the intensity of the C6 signal appreciably remains, suggesting a small degree of substitution at position 6. There appears no new signal of C4 influenced by C3-tosylation which would normally be observed at δ = 73.1 ppm, suggesting insignificant C3-tosylation. Therefore, the tosylation reaction of starch in NaOH–urea-Brij® 30 was further confirmed to occur predominantly at position 2 in accordance with 1H-NMR integration data previously discussed.
| Entry | DSTos | Percentage distribution of tosyl groups | |
|---|---|---|---|
| C2 (%) | C6 (%) | ||
| 1 | 0.15 | 88.50 | 11.50 |
| 2 | 0.33 | 93.46 | 6.54 |
| 3 | 0.45 | 87.72 | 12.28 |
| 4 | 0.54 | 93.46 | 6.54 |
| 5 | 0.65 | 88.89 | 11.11 |
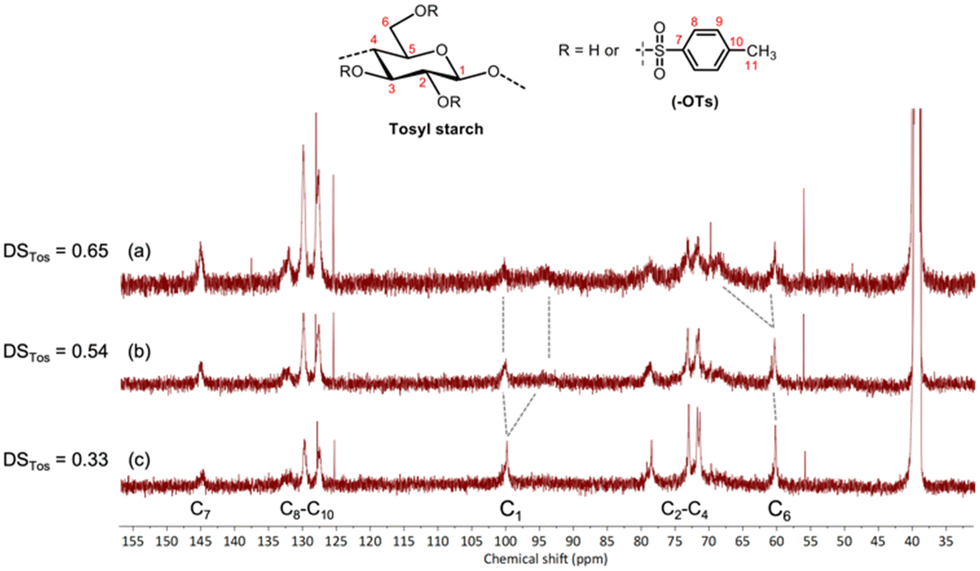 | ||
| Fig. 3 13C-NMR spectra of tosyl starch (a) with a degree of substitution (DSTos) = 0.65 (b) DSTos = 0.54, and (c) DSTos = 0.33, recorded in d6-DMSO at 25 °C. | ||
The thermal properties of tosylated starch samples were also investigated via thermogravimetric (TGA) analysis under an N2 atmosphere. Native starch showed an initial weight loss of water adsorbed in the sample around 23% of the initial mass while the main weight loss of 77% occurred at the decomposition temperature around 338 °C. Functionalisations of native starch with tosyl groups enhanced the hydrophobicity of the samples as observed by a significantly reduced amount of water adsorbed in the samples (from 23% to the range of 4–6%). Introduction of multiple tosyl groups into the anhydroglucose units interrupted formations of intra- and inter-chain hydrogen bonding of native starch and shifted the decomposition temperature to a lower temperature range (193–210 °C) with higher percentages of char yields in comparison to the native starch (Fig. S26–S30, ESI†).
Tosyl starch has been a key intermediate in the preparation of starch derivatives. The tosyl groups can be simply substituted by subsequent nucleophilic substitution reactions. In this work, the transformations of tosyl starch with sodium azide and potassium phthalimide in DMSO and aqueous NaOH–urea-Brij® 30 were studied. The products were characterised by spectroscopic techniques including FTIR and 1H-NMR (Fig. S18–S25 and Tables S2–S5, ESI†). The 1H-NMR spectra of both obtained products carried out in DMSO medium show variations of aromatic and methyl proton signals belonging to tosyl groups at C2 (δ of Ha, Hb = 7.84–7.41 ppm, and δ of Hc = 2.42–2.34 ppm), with insignificant signal for tosyl protons at C6 (Fig. S18 and S19, ESI†). The FTIR spectra confirm the presence of the azide and phthalimide groups showing characteristic absorptions at 2103 cm−1 belonging to the NNN stretching of the azide group and at 1773 and 1713 cm−1 indicative of a doubly carbonyl stretch of a cyclic imide in phthalimide (Fig. S22, S23 and Tables S2, S3, ESI†). These results indicate that the substitution successfully occurs mainly at the least steric position C6. The 1H-NMR spectra of the obtained products obtained for a mixture of NaOH–urea-Brij® 30 show a large presence of the broad aromatic and methyl proton downfield-shifted signals belonging to tosyl groups at C2 (δ of Ha = 7.73 ppm, δ of Hb = 7.38 ppm, and δ of Hc = 2.34 ppm), with only a slight remaining signal for tosyl protons at C6 (δ of Ha = 7.47 ppm, δ of Hb = 7.11 ppm, and δ of Hc = 2.28 ppm) (Fig. S20 and S21, ESI†). The FTIR spectra, however, reveal negligible signals of azide and phthalimide groups, suggesting that hydrolysis occurs instead of substitution reactions under this condition (Fig. S24, S25 and Tables S4, S5, ESI†). Notably, from the results, position C2 is less susceptible to undergo hydrolysis than position C6. This insight presents a novel green strategy towards dominant tosyl functionalisation of starch at position C2. By exploiting NaOH–urea-surfactant media for starch tosylation followed by hydrolysis reactions, a C2-preferred functionalised tosyl starch can be yielded.
Conclusions
Tosyl starch was successfully synthesised in an eco-friendly NaOH–urea solvent with the use of surfactants for achieving higher DSTos. Elemental analysis and spectroscopic techniques including FTIR, 1H-NMR and 13C-NMR were used for the product characterisation. The 1H-NMR spectra of the tosyl products prepared from the NaOH–urea-surfactant solvent reveal tosylation at hydroxyl positions C2 and C6 with predominant functionalisation at position C2. The nucleophilic displacement (SN) reactions of the tosyl groups in DMSO and this eco-friendly medium were also studied and the substitutions were shown to be successful in DMSO with predominant displacement of tosyl groups at the least hindered site C6. However, in the NaOH–urea-surfactant solvent, hydrolysis alternatively occurs mainly at position C6, leaving a significant number of remaining tosyl groups at position C2. This notably demonstrates a green strategy for efficient C2-regioselective starch modification by using these aqueous media for tosylation with subsequent hydrolysis. With the growing health and environmental concerns on using toxic and hazardous chemicals, further research is still needed to develop environmentally friendly techniques to accommodate the increasing demands on future manufacturing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for the financial support from Mahidol University (Basic Research Fund: fiscal year 2022; Grant no. BRF1-046/2565).References
- E. Ogunsona, E. Ojogbo and T. Mekonnen, Eur. Polym. J., 2018, 108, 570–581 CrossRef CAS.
- Z. Shokri, F. Seidi, M. R. Saeb, Y. Jin, C. Li and H. Xiao, Mater. Today Chem., 2022, 24, 100780 CrossRef CAS.
- D. Seung, New Phytol., 2020, 228, 1490–1504 CrossRef CAS PubMed.
- P. A. Magallanes-Cruz, P. C. Flores-Silva and L. A. Bello-Perez, J. Food Sci., 2017, 82, 2016–2023 CrossRef CAS PubMed.
- H. Gujral, A. Sinhmar, M. Nehra, V. Nain, R. Thory, A. K. Pathera and P. Chavan, Int. J. Biol. Macromol., 2021, 186, 155–162 CrossRef CAS PubMed.
- M. R. Mohd Roslan, N. L. Mohd Kamal, M. F. Abdul Khalid, N. F. Mohd Nasir, E. M. Cheng, C. Y. Beh, J. S. Tan and M. S. Mohamed, Materials, 2021, 14, 1960 CrossRef PubMed.
- K. Wang, W. Wang, R. Ye, A. Liu, J. Xiao, Y. Liu and Y. Zhao, Food Chem., 2017, 216, 209–216 CrossRef CAS PubMed.
- N. Salah, L. Dubuquoy, R. Carpentier and D. Betbeder, Int. J. Pharm., 2022, 4, 100114 CAS.
- M. T. Soe, T. Pongjanyakul, E. Limpongsa and N. Jaipakdee, Carbohydr. Polym., 2020, 245, 116556 CrossRef CAS PubMed.
- J. Gao, Z.-G. Luo and F.-X. Luo, Carbohydr. Polym., 2012, 89, 1215–1221 CrossRef CAS PubMed.
- C. Cao, B. Nian, Y. Li, S. Wu and Y. Liu, J. Agric. Food Chem., 2019, 67, 12366–12373 CrossRef CAS PubMed.
- Y. Fan and F. Picchioni, Carbohydr. Polym., 2020, 241, 116350 CrossRef CAS PubMed.
- A. Zarski, S. Ptak, P. Siemion and J. Kapusniak, Carbohydr. Polym., 2016, 137, 657–663 CrossRef CAS PubMed.
- B. J. Orzolek, M. A. Rahman and P. M. Iovine, ACS Sustainable Chem. Eng., 2018, 6, 13562–13569 CrossRef CAS.
- A. Lehmann and B. Volkert, Carbohydr. Polym., 2011, 83, 1529–1533 CrossRef CAS.
- K. Petzold, A. Koschella, D. Klemm and B. Heublein, Cellulose, 2003, 10, 251–269 CrossRef CAS.
- R. W. Kerr and K. C. Hobbs, Ind. Eng. Chem., 1953, 45, 2542–2544 CrossRef CAS.
- Y. Wang and W. Xie, Carbohydr. Polym., 2010, 80, 1172–1177 CrossRef CAS.
- K. Li, H. Liu, W. Gao, M. Chen, Y. Zeng, J. Liu, L. Xu and D. Wu, Biomaterials, 2015, 39, 131–144 CrossRef CAS PubMed.
- P. L. Bragd, A. C. Besemer and H. van Bekkum, Carbohydr. Res., 2000, 328, 355–363 CrossRef CAS PubMed.
- P. S. Chang and J. F. Robyt, J. Carbohydr. Chem., 1996, 15, 819–830 CrossRef CAS.
- S. Borsacchi, L. Calucci, M. Geppi, F. La Terra, C. Pinzino and M. Bertoldo, Carbohydr. Polym., 2014, 112, 245–254 CrossRef CAS PubMed.
- M. Ruhul Amin, F. R. Anannya, M. A. Mahmud and S. Raian, J. Polym. Res., 2019, 27, 3 CrossRef.
- C. Rivard, L. Moens, K. Roberts, J. Brigham and S. Kelley, Enzyme Microb. Technol., 1995, 17, 848–852 CrossRef CAS.
- M. A. Otache, R. U. Duru, O. Achugasim and O. J. Abayeh, J. Polym. Environ., 2021, 29, 1365–1379 CrossRef CAS.
- Q. Chen, H. Yu, L. Wang, Z. ul Abdin, Y. Chen, J. Wang, W. Zhou, X. Yang, R. U. Khan, H. Zhang and X. Chen, RSC Adv., 2015, 5, 67459–67474 RSC.
- R. Cheng, B. Xiang, Y. Li and M. Zhang, J. Hazard. Mater., 2011, 188, 254–260 CrossRef CAS PubMed.
- R. L. Whistler and S. Hirase, J. Org. Chem., 1961, 26, 4600–4605 CrossRef CAS.
- D. M. Clode and D. Horton, Carbohydr. Res., 1971, 17, 365–373 CrossRef CAS PubMed.
- A. S. Gross, A. T. Bell and J.-W. Chu, J. Phys. Chem. B, 2013, 117, 3280–3286 CrossRef CAS PubMed.
- R. Dicke, K. Rahn, V. Haack and T. Heinze, Carbohydr. Polym., 2001, 45, 43–51 CrossRef CAS.
- F. Ren, J. Wang, F. Xie, K. Zan, S. Wang and S. Wang, Green Chem., 2020, 22, 2162–2183 RSC.
- H. Muljana, F. Picchioni, H. J. Heeres and L. P. B. M. Janssen, Carbohydr. Polym., 2009, 78, 511–519 CrossRef CAS.
- H. Muljana, F. Picchioni, F. Picchioni, Z. Knez, H. J. Heeres and L. P. B. M. Janssen, Carbohydr. Res., 2011, 346, 1224–1231 CrossRef CAS PubMed.
- H. Muljana, S. van der Knoop, D. Keijzer, F. Picchioni, L. P. B. M. Janssen and H. J. Heeres, Carbohydr. Polym., 2010, 82, 346–354 CrossRef CAS.
- P.-H. Elchinger, P.-A. Faugeras, C. Zerrouki, D. Montplaisir, F. Brouillette and R. Zerrouki, Green Chem., 2012, 14, 3126–3131 RSC.
- S. Schmidt, T. Liebert and T. Heinze, Green Chem., 2014, 16, 1941–1946 RSC.
- N. Nakthong, R. Wongsagonsup and T. Amornsakchai, Ind. Crops Prod., 2017, 105, 74–82 CrossRef CAS.
- J. Hu, F. Cheng, Y. Lin, K. Zhao and P. Zhu, J. Appl. Polym. Sci., 2016, 133, 43390 CrossRef.
- W. C. Chan and H. You, Afr. J. Biotechnol., 2010, 9, 5914–5921 CAS.
- D. J. Kerr, T. E. Wheldon, J. G. Russell, H. R. Maurer, A. T. Florence, G. W. Halbert, R. I. Freshney and S. B. Kaye, Eur. J. Cancer Clin. Oncol., 1987, 23, 1315–1322 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nj00830d |
| ‡ P. N. and T. B. contributed equally to this research article. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |