
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterisation of group 11 metal complexes with a guanidine-tagged triphenylphosphine and evaluation of the isolated Au(I) complexes in gold-mediated organic reactions†
Zdeněk
Leitner
,
Ivana
Císařová
and
Petr
Štěpnička
 *
*
Department of Inorganic Chemistry, Faculty of Science, Hlavova, 2030, 128 40 Prague, Czech Republic. E-mail: petr.stepnicka@natur.cuni.cz
First published on 16th February 2023
Abstract
Phosphines bearing guanidine substituents at the backbone are attractive hybrid ligands that have not yet received adequate attention. This paper describes group 11 metal complexes of a guanidine-substituted triphenylphosphine, viz., N′′-[2-(diphenylphosphino)phenyl]-N,N′-diisopropylguanidine (1). Reactions of 1 with Cu(I) and Ag(I) precursors yielded the P,N-chelate complexes [M(1-κ2P,N)2]X, where M/X = Cu/BF4, Cu/Br, Ag/SbF6 and Ag/Br. Conversely, reacting 1 and the hydrochloride 1·HCl with [AuCl(SMe2)] produced the corresponding phosphine complexes [AuCl(1-κP)] and [AuCl(1H-κP)]Cl, which were further converted into [{μ(P,N)-1}2Au2][SbF6]2 and [AuCl(1H-κP)][SbF6], respectively, by reacting with Ag[SbF6]. These compounds and the bis-phosphine complex [Au(1-κP)2][SbF6] were studied as precatalysts in the Au-mediated cyclisation of N-propargylbenzamide and the addition of benzoic acid across terminal alkynes. Of the Au(I)-1 complexes studied, the complex [{μ(P,N)-1}2Au2][SbF6]2 was particularly attractive as a stable and well-defined, silver-free precatalyst, which can be conveniently activated in situ by the addition of a protic acid (either as an additive or a substrate).
Introduction
A particular combination of functional groups, whose properties can be widely varied through substituents, makes guanidine-substituted phosphines attractive hybrid donors. For instance, triphenylphosphine derivative A (Scheme 1) was studied as a supporting ligand in Re(I) complexes used to investigate the elementary steps of CO hydrogenation.1 More recently, compound B was employed in platinum metal complexes, which were subsequently utilised in small molecule activation and indole nitroethylation.2 Analogous phosphines bearing cyclic guanidine moieties, C and D, were evaluated as substrate-directing ligands in hydroformylation of β,γ-unsaturated carboxylic acids3 and found to form highly active H/D-exchange iridium catalysts (type E).4 | ||
| Scheme 1 Examples of phosphinoguanidine donors (Tol = 4-tolyl, Cy = cyclohexyl, Xyl = 2,6-dimethylphenyl). | ||
Recently, we prepared a series of ferrocene-based phoshinoguanidines F (Scheme 1) and studied their coordination behaviour,5 demonstrating that the ligating properties of these donors can be substantially modified by changing the protonation state of their guanidine moiety.6 We also focused on the analogous triphenylphosphine derivative 1 and described the protonation-dependent coordination properties of this compound in Pd(II) complexes.7 Here, we extend our studies focused on compound 1 further towards complexes with group 11 metals and further explore the catalytic properties of the prepared Au(I)-1 complexes in gold-mediated organic transformations.
Results and discussion
Cu(I) and Ag(I) complexes
Initially, compound 1 was allowed to react with Cu(I) and Ag(I) precursors with weakly coordinating anions, such as [Cu(MeCN)4][BF4] and Ag[SbF6] (Scheme 2). These reactions, performed at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 metal-to-ligand ratio in dichloromethane, proceeded similarly to produce the respective bis-chelate complexes 2a and 3a, which were isolated as air-stable, colourless solids with good yields (93% for 2a isolated by precipitation and 70% for the crystallised complex 3a). Analogous reactions with CuBr and AgBr proceeded similarly to give complexes 2b and 3b. In this case, the addition of ligand 1 to a suspension of the bromide salt resulted in the dissolution of the metal precursor and the formation of a colourless solution. The products were isolated by precipitation or crystallisation. As solids, however, the bromide salts were practically insoluble in common organic solvents, which precluded any analysis by common solution methods (e.g., NMR spectroscopy). Notably, analogous reactions employing equimolar amounts of the starting materials yielded the same, [M(1)2]+-type products.
:2 metal-to-ligand ratio in dichloromethane, proceeded similarly to produce the respective bis-chelate complexes 2a and 3a, which were isolated as air-stable, colourless solids with good yields (93% for 2a isolated by precipitation and 70% for the crystallised complex 3a). Analogous reactions with CuBr and AgBr proceeded similarly to give complexes 2b and 3b. In this case, the addition of ligand 1 to a suspension of the bromide salt resulted in the dissolution of the metal precursor and the formation of a colourless solution. The products were isolated by precipitation or crystallisation. As solids, however, the bromide salts were practically insoluble in common organic solvents, which precluded any analysis by common solution methods (e.g., NMR spectroscopy). Notably, analogous reactions employing equimolar amounts of the starting materials yielded the same, [M(1)2]+-type products.
 | ||
| Scheme 2 Synthesis of Cu(I) and Ag(I) complexes with ligand 1. | ||
Complex formation was revealed in the 31P NMR spectra. For 2a, a broadened 31P NMR signal was observed at δP −17.8, which is upfield from the free ligand (δP −13.8), while complex 3a displayed a resonance at δP −5.5 that was split into a double doublet due to interactions with 107Ag and 109Ag (1JAgP = 436 and 503 Hz, respectively). The 1H NMR and 13C NMR spectra displayed all expected signals. However, the 13C NMR signals of the 31P-coupled carbons were observed as virtual triplets due to virtual coupling in the 13C–31P–Pd–31P–12C AA′X-type spin systems8 (in the case of 3a, further splitting due to C–Ag interactions was observed). The presence of cations [M(1)2]+ (in solution) was verified by mass spectrometry using soft ionisation techniques (ESI or MALDI), and the sample purity was corroborated by elemental analysis. The latter methods were also used to characterise the insoluble compounds 2b and 3b.
The crystal structures of 2a·C2H4Cl2, 2b·CH2Cl2 and 3a were determined using X-ray diffraction analysis. Compound 3b yielded only poor-quality crystals: the collected data allowed us to confirm the structure but not a satisfactory refinement. The complex cations in the structures of 2a·C2H4Cl2, 2b·CH2Cl2 and 3a are generally similar. A structure diagram of the representative compound 2b is shown in Fig. 1, and the remaining structures are presented in the ESI.† The geometric data are summarised in Table 1.
 | ||
| Fig. 1 View of the complex cation in the structure of 2b·CH2Cl2. | ||
| Parametera | 2a·C2H4Cl2b | 2b·CH2Cl2 | 3a |
|---|---|---|---|
| a P–M–N is the angle pertaining to the chelating phosphinoguanidine ligand (bite angle), whereas P–M–N′ is the corresponding “open” angle, where the P and N atoms belong to the different ligands. b The molecule resides on the crystallographic two-fold axis, and hence, only its half is structurally independent. | |||
| M | Cu | Cu | Ag |
| M–P | 2.240(1) | 2.2392(7)/2.2366(7) | 2.401(1)/2.418(1) |
| M–N | 2.121(2) | 2.126(2)/2.130(2) | 2.599(2)/2.476(2) |
| P–M–N | 83.13(7) | 82.82(5)/83.33(5) | 71.95(6)/74.50(6) |
| P–M–P′ | 125.12(4) | 124.23(3) | 139.41(2) |
| N–M–N′ | 111.48(9) | 111.97(7) | 101.98(8) |
| P–M–N′ | 129.68(7) | 127.53(6)/132.49(6) | 139.65(6)/132.25(5) |
| τ 4 | 0.71 | 0.71 | 0.57 |
| N–C2–C3–P | 7.1(4) | −4.2(3)/−7.9(3) | −4.9(3)/−9.8(3) |
| C1–N1 | 1.332(4) | 1.330(3)/1.332(3) | 1.310(4)/1.307(4) |
| C1–N2 | 1.353(4) | 1.350(3)/1.349(3) | 1.357(4)/1.353(4) |
| C1–N3 | 1.346(5) | 1.357(3)/1.354(3) | 1.369(4)/1.378(4) |
The coordination spheres in 2a·C2H4Cl2, 2b·CH2Cl2 and 3a are distorted not only due to varying M-donor distances but also severely twisted. While the M–P bond lengths do not change much in the entire series, the M–N distances vary more, presumably due to steric factors and a lower covalence of the M–N dative bonds.9
The P–M–N angles associated with the chelate rings are significantly narrower (72–83° in the series) than the remaining interligand angles, reflecting the size and rigidity of the 1,2-phenylene backbone. Only the N–M–N′ angles remain close to the ideal tetrahedral values (102–112°), while the P–M–P′ and nonchelate P–M–N angles are significantly widened. The distortion of the coordination sphere is more pronounced in the Ag(I) complex than in its Cu(I) analogues that, in turn, differ only marginally. This can be illustrated by the τ4 index,10 which is 0.71 for both Cu(I) complexes and 0.57 for 3a (N.B. ideal tetrahedral and planar coordination would yield τ4 = 1 and 0, respectively) and by the angle subtended by the {M,P,N} planes of the two chelate rings, which is 78.5(1)° in 2a, 78.23(8)° in 2b, and 67.22(9)° in 3a (in a regular tetrahedron, these planes would be perpendicular).
No significant torsion is observed at the central benzene ring, as evidenced by the N–C2–C3–P torsion angles (Table 1). The guanidine moieties are planar and partly delocalised: the C–N1 distance involving the coordinated nitrogen atom is consistently shorter than the remaining C–N bonds. The NH groups in 2a and 2b are syn and directed away from the central atom, while those in 3a assume mutually anti-positions. These differences can be ascribed to hydrogen-bond interactions (see the ESI†).
Au(I) complexes
Au(I) complexes were obtained through reactions of 1 with Au(I) precursors with labile sulfide ligands (Scheme 3). Thus, the replacement of dimethylsulfide in [AuCl(SMe2)] with 1 (Au:1 = 1:1) produced the chlorogold(I) complex [AuCl(1-κP)] (4), whereas the reaction with [Au(tht)2][SbF6] (Au:1 = 1:2, tht = tetrahydrothiophene) gave the bis-phosphine complex [Au(1-κP)2][SbF6] (5). Subsequent halogen removal using Ag[SbF6] in MeCN cleanly converted the former compound into the dinuclear complex [{μ(P,N)-1}2Au2][SbF6]2 (6) rather than a solvent complex (e.g., [Au(1-κP)(MeCN)][SbF6]). The preferred formation of 6 can be explained by a stabilising effect of the aurophilic interaction11 in its structure (vide infra).
 | ||
| Scheme 3 Synthesis of Au(I) complexes 6–10 (tht = tetrahydrothiophene). | ||
In view of subsequent catalytic testing, Au(I) complexes were also prepared using the protonated ligand 1·HCl. The protonation expectedly prevented the coordination of the guanidine moiety: the reaction of [AuCl(SMe2)] with 1·HCl yielded the phosphine complex [AuCl(1H-κP)]Cl (7a), which reacted with Ag[SbF6] (1 equiv.) under anion exchange to give [AuCl(1H-κP)][SbF6] (7b).
Complexes 4–7 were characterised using NMR spectroscopy, ESI MS and elemental analysis. The 1H NMR and 13C NMR spectra displayed the expected signals but are difficult to compare because different solvents had to be used to dissolve the compounds. The 31P NMR signals appeared downfield compared to the free ligand and their position depends on the other ligand in gold (δP ≈ 24–25 for 4, 6 and 7a/b, and δP ≈ 37 for 5; in different solvents). The ESI MS of 4 and 7a/b displayed ions attributable to AuCl(1H)+ (m/z 636), while for 5 and 6, signals due to Au(1)2+ were observed at m/z 1003.
The molecular structure of 4 is shown in Fig. 2 (the structures of solvated 7a and 7a are presented in the ESI†); Table 2 contains the relevant structural parameters.
 | ||
| Fig. 2 The molecular structure of 4. | ||
| Parameter | 4 | 7a·H2O·CH2Cl2 | 7b·½C2H4Cl2 |
|---|---|---|---|
| a The guanidine moiety is disordered, which particularly affects these distances. | |||
| Au–P | 2.2287(5) | 2.2276(7) | 2.2279(8) |
| Au–Cl | 2.2960(6) | 2.2821(7) | 2.2968(9) |
| P–Au–Cl | 178.84(2) | 176.35(3) | 177.38(3) |
| C1–N1 | 1.301(2) | 1.358(3) | 1.355(4) |
| C1–N2/N3 | 1.356(2)/1.366(2) | 1.335(3)/1.321(3) | —a |
| N–C2–C3–P | −11.7(2) | −3.6(3) | −4.8(4) |
| C2–C3–P–Au | 50.1(1) | 59.4(2) | 57.9(3) |
The Au(I) ions in 4 and solvated 7a and 7b present typical linear coordination with parameters similar to those of [AuCl(PPh3)].12 In all structures, the linear P–Au–Cl moieties are bent out of the plane of the ligand's 1,2-phenylene backbone (cf. C2–C3–P–Au angles in Table 2), which is somewhat twisted in 4 (N.B.7a and 7b show smaller N–C2–C3–P torsion angles). The guanidine moieties in 4 and 7a are essentially planar with anti-positioned NH moieties and show differentiated C–N bonds: in 4, the C1–N1 bond is shorter than the C1–N2/3 bonds, while the opposite is observed for 7a with a protonated guanidine moiety. The orientation of the guanidine moiety towards the phenylene plane changes with intermolecular interactions, the lack of which can result in disorders such as in 7b (see the ESI;† the dihedral angles of the C(2–7) and {C1,N1,N2,N3} planes are 67.97(9)° in 4 and 84.7(1)° in 7a).
The gold atom in the structure of 5 also shows linear coordination (Fig. 3). The Au–P bond lengths are similar to those in [Au(PPh3)2]X (X = SbF6, PF6 and BF4)13 and are elongated with respect to 4 due to a strong trans influence of the phosphine ligands,14 which destabilise each other.15 Steric factors may also play some role, as the ligands are oriented so that the P-bound phenyl groups and guanidine moieties from the two ligands are oriented towards each other (syn).
 | ||
| Fig. 3 View of the complex cation in the structure of 5. The selected distances and angles for ligand 1 [ligand 2] (in Å and deg): Au–P 2.3007(6) [2.3035(6)], P–Au–P 175.84(3), C1–N1 1.315(2) [1.304(2)], C1–N1 1.353(2) [1.357(2)], C1–N3 1.367(3) [1.357(2)], N1–C1–N2 118.9(2) [119.6(2)], N1–C1–N3 125.4(2) [124.2(2)], N2–C1–N3 115.6(2) [116.2(2)], N1–C2–C3–P 2.6(2). | ||
Complex 6 (Fig. 4) crystallised as a dichloromethane solvate, 6·2CH2Cl2, with the [{μ(P,N)-1}2Au2]2+ cations residing over the crystallographic inversion centres (space group P21/n). The structure of the complex cation is stabilised by intramolecular aurophilic interactions (Au–Au′ ≈ 2.90 Å), resulting in a slight inclination of the gold centres and bending of the P–Au–N moiety (175°). The Au–P bond length is similar to the value determined for 4 (trans influences of Cl− and N-donors are similar), and the Au–N distance compares to the values reported for similar cationic dimers obtained from 2-(diphenylphosphino)-1-methylimidazole16 and 2-(diphenylphosphino)-1-methylbenzimidazole.17 The guanidine moiety is planar (within 0.002 Å) and the guanidine C–N bonds differ less than in the previously discussed structures (both NH groups are oriented towards the pivotal atom N1). The guanidine plane is twisted by 70.9(2)° from the plane of the 1,2-phenylene ring C(2–7), which diverts by 47.9(1)° from the plane of the central {Au2P2N2} moiety. Due to the imposed symmetry, the complex cation adopts an anti-arrangement, such that the phenylene ring from one ligand and the guanidine unit and one P-bound phenyl ring from the other are located on one side of the {Au2P2N2} ring.
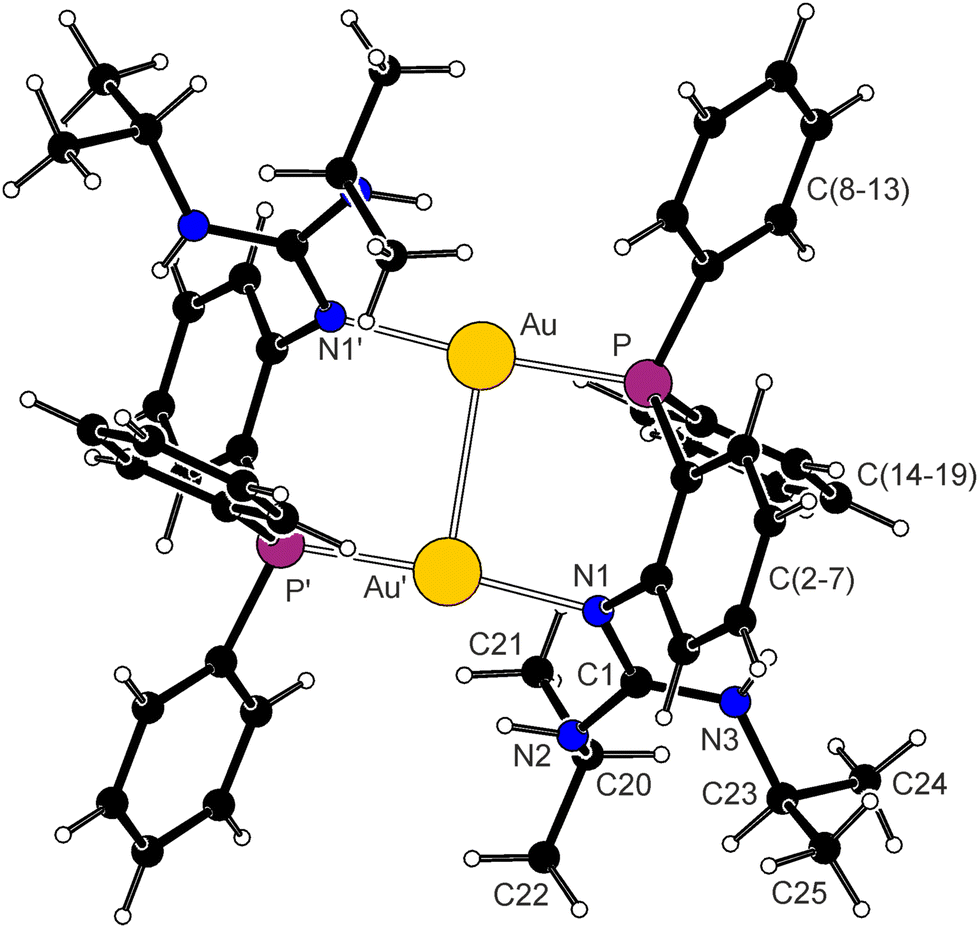 | ||
| Fig. 4 View of the complex cation in the structure of 6·2CH2Cl2. Selected distances and angles (in Å and deg): Au–Au′ 2.9003(4), Au–P 2.2491(8), Au–N′ 2.072(2), P–Au–N′ = 174.77(7), C1–N1 1.340(3), C1–N2 1.358(3), C1–N3 1.336(3), N1–C2–C3–P 1.9(3). | ||
Catalysis
The catalytic properties of the prepared gold(I) complexes were first evaluated using gold-mediated cyclisation of N-propargylbenzamide (8) into 5-methylene-2-phenyl-4,5-dihydrooxazole (9; Scheme 4).18 The reaction, performed19 in CD2Cl2 at 25 °C using 1 mol% of the gold catalyst, proceeded selectively, producing 9 as the only detectable product. As such, it was easily followed in situ by 1H NMR spectroscopy. | ||
| Scheme 4 Au-catalysed cyclisation of N-propargylbenzamide (8). | ||
The results collected in Table 3 illustrate the markedly different catalytic activities of complexes 4–7, which were used without any promotor (e.g., silver salt to abstract the chloride ligand). Compounds 4, 5 and 7b did not show any appreciable catalytic activity (<1% yield after 6 h), and complexes 6 and 7a reacted only very slowly. Eventually, a fast-reacting catalyst was obtained upon adding bis(trifluoromethanesulfonyl)imide (HNTf2)20 to complex 6. One equiv. of HNTf2 per gold atom markedly accelerated the reaction but better yields at shorter reaction times were obtained with 2 equiv. of the acid. Addition of more than 2 equiv. of HNTf2 per the gold atom had no beneficial effect.
| Catalyst | Yield of 9 [%] | ||
|---|---|---|---|
| 1 h | 3 h | 6 h | |
| a The reaction was performed in CD2Cl2 (c(8) = 0.25 M) at 25 °C using 1 mol% Au. HNTf2 was added in the form of a freshly prepared stock solution in CD2Cl2. The amount of HNTf2 is given in molar equivalents per gold atom. | |||
| 4 | 0 | 0 | 0 |
| 5 | 0 | 0 | 0 |
| 6 | 4 | 6 | 10 |
| 6 + HNTf2 | 44 | 89 | 100 |
| 6 + 2HNTf2 | 48 | 99 | 100 |
| 6 + 3HNTf2 | 43 | 99 | 100 |
| 6 + 3HNTf2 | 40 | 99 | 100 |
| 7a | 3 | 8 | 14 |
| 7b | 0 | 0 | 0 |
In contrast to analogous complexes with P,N-bridging phosphinonitrile ligands,21 complex 6 does not dissociate spontaneously under the reaction conditions. However, the addition of HNTf2 to 6 leads to the protonation of the strongly basic guanidine moiety and, consequently, the cleavage of the dimeric structure into coordinatively unsaturated and catalytically active species Au(1H)+ or their anion-stabilised form [Au(1H)(NTf2)]. The activity of the formed species is comparable to that of species resulting from the spontaneous dissociation of [Au2L2]2+ cations, which contain bridging ferrocene-based phosphinonitrile ligands and the prototypical gold(I) catalyst [Au(PPh3)(MeCN)][SbF6].19
Catalyst activation was further followed by NMR titration: the addition of HNTf2 to a solution of 6 in acetone-d6 resulted in the gradual appearance of another set of signals (illustrated in Fig. 5 for the CHMe2 signals and the 31P NMR resonances), which cleanly replaced the signals due to 6. The 31P NMR signal of the new species shifted slightly upfield (δP ≈ 20).
 | ||
| Fig. 5 Changes in the 31P{1H} and 1H NMR spectra (region of CHMe2 protons) of complex 6 after adding various amounts of HNTf2 (recorded in CDCl3 at 25 °C). The amount of HNTf2 is given in molar equivalents per one gold atom. | ||
The easy activation and high reactivity of the formed species led us to further consider complex 6 as an instant, silver-free gold precatalyst for reactions with acidic substrates that may activate the gold complex in situ. To demonstrate such applications, we performed additional reaction tests including the challenging gold-catalysed addition of benzoic acids across terminal alkynes to produce enol esters22 (Scheme 5, results in Table 4).
 | ||
| Scheme 5 Au-catalysed addition of benzoic acids across 1-octyne. | ||
| Acid | Yield of 12 [%] | ||
|---|---|---|---|
| 3 h | 6 h | 18 h | |
| a The reaction was performed in 1,2-dichloroethane at 80 °C (c(10) = c(11) = 0.3 M) using 5 mol% of the gold catalyst (i.e., 2.5 mol% of 6). | |||
| 10a | 13 | 26 | 40 |
| 10b | 13 | 16 | 25 |
| 10c | 23 | 33 | 45 |
Indeed, the model reaction between benzoic acid (10a) and 1-octyne (11) in 1,2-dichlorethane at 80 °C in the presence of 5 mol% of complex 6 proceeded selectively and gave a 40% yield of the Markovnikov addition product 12a (with no other isomers detected). In contrast, no reaction occurred when using compounds 4, 5, 7a and 7b under similar conditions and even with complex 6 when the reaction solvent was changed to MeCN and 1,4-dioxane.
The relatively weaker acid 10b gave a lower yield of the respective addition product (12b: 25%), while a slight improvement was noted when using the stronger acid 10c (12c: 45%). Such results correspond with the presumed mode of catalyst activation, for which stronger acids are expected to be more efficient.
Conclusion
In summary, the results reported here indicate a similar behaviour of Cu(I) and Ag(I) ions in reactions with ligand 1. Both ions favour the formation of tetracoordinate bis-chelate cations of the type [M(1-κ2P,N)2]+, which were isolated from experiments performed at the 1:1 and 1:2 metal-to-ligand ratios and obtained from precursors containing both coordinating and weakly coordinating anions. In contrast, the tested gold(I) precursors afford discrete mononuclear Au(I)–1 complexes with linearly coordinated gold centres. Nevertheless, removal of the halide ligand from [AuCl(1-κP)] with Ag[SbF6] results in the formation of a dimeric cation [{μ(P,N)-1}2Au2]2+, stabilised by intramolecular aurophilic interactions. This cation can be cleaved through the action of Brønsted acids that protonate the guanidine moiety, thereby preventing its coordination. The resulting species, presumably Au(1H)+, are highly catalytically active. As such, complex 6 represents an attractive silver-free gold catalyst23 that eliminates the possible interference of silver species arising from silver salts typically used to activate LAuCl-type precatalysts, which may also be catalytically active or can hamper the reaction, e.g., by forming Ag–Au species.24 The protonated guanidine moiety can further increase the affinity of the Au(1H)+ species towards substrates via charge-supported hydrogen bond interactions.
Experimental
Materials and methods
All syntheses were performed under a dinitrogen atmosphere using conventional Schlenk techniques. Compounds 17 and 819 were prepared according to the literature procedure. Other starting materials were purchased from commercial suppliers (Sigma-Aldrich, Alfa-Aesar, TCI) and used without additional purification. Dry and deoxygenated dichloromethane was obtained using a PureSolv MD5 solvent purification system (Innovative Technology). Solvents used for chromatography and crystallisation were used as received (analytical grade, Lach-Ner, Czech Republic).NMR spectra were recorded on a Varian UNITY Inova 400 spectrometer at 25 °C unless stated otherwise. Chemical shifts (δ in ppm) are expressed relative to internal SiMe4 (1H and 13C) and to external 85% aqueous H3PO4 (31P), all set to 0 ppm. Electrospray ionisation (ESI) mass spectra were measured on a Bruker Compact Q-TOF instrument using samples dissolved in HPLC-grade methanol. MALDI TOF mass spectra were obtained on a Bruker MALDI TOF/TOF Ultraflex instrument. Elemental analyses were performed on a PerkinElmer PE 2400 Series II CHNS/O Elemental Analyser. The amount of clathrated solvent was determined by 1H NMR analysis.
Syntheses
1H NMR (400 MHz, CD2Cl2): δ 0.72 (d, 3JHH = 6.4 Hz, 24 H, CHMe2), 3.55 (vq, J = 6.4 Hz, 4 H, CHMe2), 4.86 (d, 3JHH = 8.3 Hz, 4 H, NH), 6.85–6.91 (m, 2 H, C6H4), 7.05 (dtdd, J = 8.2, 2.9, 1.1, 0.5 Hz, 2 H, C6H4), 7.19–7.40 (m, 24 H, 4 C6H4 + PPh2). 13C{1H} NMR (101 MHz, CD2Cl2): δ 22.58 (s, 8 C, CHMe2), 44.81 (s, 4 C, CHMe2), 121.26 (vt, JPC = 2 Hz, 2 C, CH C6H4), 121.50 (s, 2 C, CH C6H4), 125.07 (vt, JPC = 20 Hz, 2 C, C–P C6H4), 129.07 (vt, JPC = 5 Hz, 8 C, CH PPh2), 130.16 (s, 4 C, CHpara PPh2), 131.92 (s, 2 C, CH C6H4), 133.73–133.88 (m, 10 C, 2 CH C6H4 + 8 CH PPh2), 155.66 (vt, JPC = 12 Hz, 2 C, C–N C6H4), 160.55 (s, 2 C, Cipso guanidine). 31P{1H} NMR (162 MHz, CD2Cl2): δ −17.8 (br s). ESI+ MS: m/z 869 ([M – BF4]+), 420 ([1O + H]+), 404 ([1 + H]+). Anal. calcd for C50H60BCuF4N6P2 (957.37): C 62.73, H 6.32, N 8.78%. Found: C 62.63, H 6.15, N 8.41%.
1H NMR (400 MHz, CD2Cl2): δ 0.73 (d, 3JHH = 6.3 Hz, 24 H, CHMe2), 3.60 (d of sept, 3JHH = 7.8, 6.5 Hz, 4 H, CHMe2), 3.74 (d, 3JHH = 8.1 Hz, 4 H, NH), 6.95–7.02 (m, 6 H, C6H4), 7.38–7.52 (m, 22 H, 2 C6H4 + PPh2). 13C{1H} NMR (101 MHz, CD2Cl2): δ 22.05 (s, 8 C, CHMe2), 44.09 (s, 4 C, CHMe2), 122.27 (vt, JPC = 3 Hz, 2 C, CH C6H4), 123.64 (vt, JPC = 2 Hz, 2 C, CH C6H4), 125.05 (vt, JPC = 22 Hz, 2 C, C–P C6H4), 129.55 (vt, JPC = 5 Hz, 8 C, CH PPh2), 131.24 (s, 4 C, CHpara PPh2), 131.80 (vt of doublets, JPC = 16 Hz, 2JAgC = 5 Hz, 4 C, Cipso PPh2), 132.53 (s, 2 C, CH C6H4), 133.56 (vt of doublets, JPC ≈ JAgC ≈ 1.5 Hz, 2 C, CH C6H4), 134.42 (vt of doublets, JPC = 9 Hz, 3JAgC = 2 Hz, 8 C, CHortho PPh2), 153.50 (vt of doublets, JPC = 8 Hz, 2JAgC = 1 Hz, 2 C, C–N C6H4), 154.93 (s, 2 C, Cipso guanidine). 31P{1H} NMR (162 MHz. CD2Cl2): δ −5.5 (pair of concentric doublets, 1JAgP = 503 (109Ag), 436 (107Ag) Hz). MALDI TOF: m/z 913 ([M – SbF6]+), 510 ([M – 1 – SbF6]+). Anal. calcd for C50H60AgF6N6P2Sb (1150.64): C 52.19, H 5.26, N 7.30%. Found: C 52.18, H 5.09, N 7.36%.
ESI+ MS: m/z 869 ([M – Br]+), 466 ([M – 1 – Br]+), 420 ([1O + H])+, 404 ([1 + H])+. Anal. calcd for C50H68BrCuN6P2·0.2CH2Cl2 (975.50): C 62.32, H 6.29, N 8.69%. Found: C 62.41, H 6.62, N 8.23%. The NMR spectra could not be acquired because the compound is insoluble in nonpolar and moderately polar deuterated solvents and decomposes in strongly polar solvents.
ESI+ MS: m/z 995 ([M + H]+), 915 ([M – Br]+), 404 ([L + H]+). Anal. calcd for C50H60AgBrN6P2·C6H12 (1078.95): C 62.34, H 6.73, N 7.79%. Found: C 61.78, H 6.39, N 7.63%. The compound is virtually insoluble in common deuterated solvents, which precluded recording the NMR spectra.
:1, 25 mL). The resultant mixture was stored at 4 °C for 3 h, whereupon it deposited a crystalline solid, which was isolated by suction filtration, washed with pentane and dried under vacuum. Yield: 564 mg (88%), colourless, needle-like crystals.
1H NMR (400 MHz, CDCl3): δ 1.04 (d, 3JHH = 6.4 Hz, 12 H, CHMe2), 3.57 (d of sept, 3JHH = 7.7, 6.5 Hz, 2 H, CHMe2), 3.82 (d, 3JHH = 7.4 Hz, 2 H, NH), 6.65 (ddd, J = 11.8, 7.8, 1.4 Hz, 1 H, C6H4), 6.82 (ddt, J = 7.7, 2.4, 1.1 Hz, 1 H, C6H4), 6.96 (ddd, J = 8, 5.2, 0.7 Hz, 1 H, C6H4), 7.36–7.61 (m, 11 H, 1 C6H4 + PPh2). 13C{1H} NMR (101 MHz, CDCl3): δ 23.61 (s, 4 C, CHMe2), 43.12 (s, 2 C, CHMe2), 120.26 (d, JPC = 10 Hz, 1 C, CH C6H4), 121.63 (d, 1JPC = 71 Hz, 1 C, C–P C6H4), 121.95 (d, JPC = 6 Hz, 1 C, CH C6H4), 128.62 (d, JPC = 12 Hz, 4 C, CH PPh2), 130.16 (d, 1JPC = 63 Hz, 2 C, Cipso PPh2), 130.95 (d, 4JPC = 2 Hz, 2 C, CHpara PPh2), 132.77 (d, JPC = 2 Hz, 1 C, CH C6H4), 133.70 (d, JPC = 8 Hz, 1 C, CH C6H4), 149.13 (s, 1 C, Cipso guanidine), 153.95 (d, 2JPC = 8 Hz, 1 C, C–N C6H4). 31P{1H} NMR (161.90 MHz, CDCl3): δ 26.2 (s). ESI+ MS: m/z 636 ([M + H]+). Anal. calcd for C25H30AuClN3P (635.93): C 47.22, H 4.76, N 6.61%. Found: C 47.29, H 4.58, N 6.90%.
1H NMR (400 MHz, CDCl3): δ 0.75 (d, 3JHH = 6.3 Hz, 24 H, CHMe2), 3.59 (sept, 3JHH = 6.9 Hz, 4 H, CHMe2), 3.73 (d, 3JHH = 7.9 Hz, 4 H, NH), 6.71 (dtd, J = 7.2, 5.6, 1.5 Hz, 2 H, C6H4), 6.87 (dtd, J = 7.6, 2.5, 1.3 Hz, 2 H, C6H4), 7.03 (dtd, J = 8.2, 2.8, 0.6 Hz, 2 H, C6H4), 7.42–7.60 (m, 22 H, 2 C6H4 + PPh2). 13C{1H} NMR (101 MHz, CDCl3): δ 23.05 (s, 8 C, CHMe2), 42.84 (s, 4 C, CHMe2), 120.57 (vt, JPC = 5 Hz, 2 C, CH C6H4), 121.61 (broad t, 2 C, CH C6H4), 121.69 (vt, JPC = 34 Hz, C–P C6H4), 129.15 (vt, JPC = 6 Hz, 8 C, CH PPh2), 129.83 (vt, JPC = 30 Hz, 4 C, Cipso PPh2), 131.58 (s, 4 C, CHpara PPh2), 133.30 (s, 2 C, CH C6H4), 133.76 (vt, JPC = 4 Hz, CH C6H4), 133.99 (vt, JPC = 8 Hz, 8 C, CH PPh2), 150.12 (s, 2 C, Cipso guanidine), 153.69 (vt, JPC = 5 Hz, 2 C, C–N C6H4). 31P{1H} NMR (161.90 MHz, CDCl3): δ 37.4 (s). ESI+ MS: m/z 1003 ([M – SbF6]+). Anal. calcd for C50H60AuF6N6P2Sb (1239.72): C 48.44, H 4.88, N 6.78%. Found: C 48.58, H 4.91, N 6.68%.
:1, 25 mL), whereupon a white solid was deposited. The mixture was stored at 4 °C for 18 h before the solid was collected on a glass frit and dried under vacuum to give 6 as a white solid. Yield: 285 mg (68%).
1H NMR (400 MHz, acetone-d6): δ 1.12 (d, 3JHH = 6.4 Hz, 24 H, CHMe2), 4.16 (br s, 4 H, CHMe2), 5.53 (br s, 4 H, NH), 6.86 (ddd, J = 12.8, 7.9, 1.5 Hz, 2 H, C6H4), 7.36 (dddd, J = 15.3 ≈ 15.3, 1.5 ≈ 1.4 Hz, 2 H, C6H4), 7.61 (ddd, J = 8.0, 5.0, 1.2 Hz, 2 H, C6H4), 7.65–7.88 (m, 22 H, 2 HC6H4 + 20 H, PPh2). 13C{1H} NMR (101 MHz, acetone-d6): δ 23.37 (s, 8 C, CHMe2), 47.09; 47.18 (d, JPC = 9 Hz, CHMe2), 125.65 (d, 1JPC = 65 Hz, 2 C, C–P C6H4), 127.84 (d, JPC = 10 Hz, 2 C, CH C6H4), 128.06 (d, 1JPC = 66 Hz, 4 C, Cipso PPh2), 130.29 (filled d, J = 6 Hz, 2 C, CH C6H4), 130.83 (filled d, J = 13 Hz, 8 C, CH PPh2), 133.88 (vt, J′ = 1 Hz, CHpara PPh2), 135.37 (filled d, J = 15 Hz, 8 C, CH PPh2), 135.44 (s, 2 C, CH C6H4), 135.74 (d, JPC = 7 Hz, 2 C, CH C6H4), 148.67 (d, 2JPC = 5 Hz, C–N C6H4), 158.76 (d, 4JPC = 5 Hz, Cipso guanidine). 31P{1H} NMR (162 MHz, acetone-d6): δ 23.84 (s). ESI+ MS: m/z 1003 (Au(1)2+). Anal. calcd for: C50H60Au2F12N6P2Sb2 (1672.45): C 35.91, H 3.62, N 5.03%. Found: C 35.78, H 3.45, N 4.77%.
1H NMR (400 MHz, acetone-d6): δ 1.18 (d, 3JHH = 6.4 Hz, 12 H, CHMe2), 4.03 (br s, 2 H, CHMe2), 7.06 (dd, J = 11.5, 7.8 Hz, 1 H, C6H4), 7.50 (br t, J = 7.4 Hz, 1 H, C6H4), 7.56–7.68 (m, 11 H, 1 C6H4 + 10 PPh2), 7.76 (br t, J = 7.4 Hz, 1 H, C6H4), 8.03 (br s, 2 H, NH), 9.97 (br s, 1 H, NH+). 13C{1H} NMR (101 MHz, acetone-d6): δ 23.18 (s, 4 C, CHMe2), 46.38 (d, JPC = 9 Hz, 2 C, CHMe2), 129.25 (d, 1JPC = 64 Hz, 2 C, Cipso PPh2), 129.39 (d, JPC = 10 Hz, 1 C, C6H4), 130.56 (d, JPC = 12 Hz, 4 C, CH PPh2), 133.16 (d, JPC = 3 Hz, 2 C, CHpara PPh2), 134.68 (d, JPC =2 Hz, 1 C, C6H4), 135.33 (d, JPC = 14 Hz, 4 C, CH PPh2), 135.87 (d, JPC = 6 Hz, 1 C, C–N C6H4), 140.84 (s, 1 C, C6H4), 154.99 (s, 1 C, Cipso guanidine). The signal due to C–P C6H4 was not observed. 31P{1H} NMR (162 MHz, acetone-d6): δ 24.72 (s). ESI+ MS: m/z 636 ([M – Cl]+). Anal. calcd for C25H31AuCl2N3P·H2O·C2H4Cl2 (789.35): C 41.08, H 4.72, N 5.32%. Found: C 40.73, H 4.27, N 5.21%.
:1, 20 mL). After the separated solid was aged at 4 °C for 18 h, the solvent was poured away (decantation), and the residue was dried under vacuum to give 7b as a white powdery solid. Yield: 84 mg (96%).
1H NMR (400 MHz, CD2Cl2): δ 1.21 (d, 3JHH = 6.2 Hz, 12 H, CHMe2), 3.63 (vq, J = 6.2 Hz, 2 H, CHMe2), 5.25 (br, 2 H, NH), 6.73 (br, 1 H, NH+), 7.00 (dd, J = 11.4, 7.6 Hz, 1 H, C6H4), 7.53 (vt, J = 7.4 Hz, 1 C, C6H4), 7.56–7.70 (m, 11 H, C6H4 + 10 PPh2), 7.80 (vt, J = 7.4 Hz, 1 H, C6H4). 13C{1H} NMR (101 MHz, CD2Cl2): δ 22.79 (s, 4 C, CHMe2), 46.23 (s, 2 C, CHMe2), 126.32 (d, 1JPC = 65 Hz, 2 C, Cipso PPh2), 130.55 (d and br s, JPC = 12 Hz, 5 C, CH C6H4 + 4 CH PPh2), 133.66 (d, 4JPC = 2 Hz, 2 C, CHpara PPh2), 134.84 (d, JPC = 14 Hz, 4 C, CH PPh2), 135.06 (s, 1 C, CH C6H4), 135.50 (d, JPC = 6 Hz, 1 C, CH C6H4), 139.29 (br s, 1 C, C–N C6H4), 152.10 (s, 1 C, Cipso guanidine). The signals due to CH and C–P of C6H4 were not observed, presumably due to overlaps. 31P{1H} NMR (162 MHz, CD2Cl2): δ 24.21 (s). ESI+ MS: m/z 636 ([M – SbF6]+). Anal. calcd for C25H31AuClF6N3PSb (872.68): C 34.41, H 3.58, N 4.82%. Found: C 34.56, H 3.39, N 4.55%.
Catalytic experiments
The reaction mixture was transferred to an NMR tube and analysed by 1H NMR spectroscopy at 25 °C after 1, 3 and 6 h of mixing. The conversion was determined by the integration of the NCH2 signals due to substrate 8 (δH 4.21) and the cyclisation product 5-methylene-2-phenyl-4,5-dihydrooxazole (9; δH 4.63).19 The reaction proceeded selectively; no other compounds were detected in the NMR spectra.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)25 and the formyl group of the standard (δP 9.87, singlet). Reactions with different benzoic acids and in different solvents were performed similarly.
CH2)25 and the formyl group of the standard (δP 9.87, singlet). Reactions with different benzoic acids and in different solvents were performed similarly.
X-ray crystallography
The full-sphere diffraction data were recorded at 120 or 150 K using a Bruker D8 VENTURE Kappa Duo diffractometer equipped with a PHOTON III detector and a Cryostream Cooler (Oxford Cryosystems). Mo Kα radiation (λ = 0.71073 Å) was used in all cases. The structures were solved by direct methods (SHELXT-2014/201826) and refined by a full-matrix least-squares routine based on F2 (SHELXL-201727). All nonhydrogen atoms were refined with anisotropic displacement parameters. The NH hydrogen atoms were located on the difference electron density map and refined as riding atoms with Uiso(H) = 1.2Ueq(N). Hydrogen atoms in the CHn groups were placed in their theoretical positions and refined similarly using the standard parameters in SHELXL.Compound 2a·C2H4Cl2 crystallised as a racemic twin (space group C2, refined contributions of the two enantiomeric domains were ≈94:6). One of the phenyl substituents had to be refined over two positions as an ideal hexagon due to disorder (occupancies: 71:29). In addition, the solvent in this structure was severely disordered within structural voids and, hence, was treated as a diffuse electron density using PLATON SQUEEZE.28 A similar situation was encountered in 7a·H2O·CH2Cl2 and 7b·½C2H4Cl2. In the former compound, not only were the solvent molecules disordered but also the chloride anion and water molecule occupied the same positions with equal abundance. For the latter compound, the disorder also affected the guanidine moiety, which had to be refined over two positions.
Relevant crystallographic data and refinement parameters are available in the ESI† (Table S1). All geometric data and structural diagrams were obtained using a recent version of the PLATON program.29 The numerical values were rounded to one decimal place with respect to their estimated standard deviations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Charles University Research Centre program (project no. UNCE/SCI/014).Notes and references
- T. S. Teets, J. A. Labinger and J. E. Bercaw, Organometallics, 2013, 32, 5530 CrossRef CAS.
- (a) M. Carmona, J. Ferrer, R. Rodríguez, V. Passarelli, F. J. Lahoz, P. García-Orduña, L. Cañadillas-Delgado and D. Carmona, Chem. – Eur. J., 2019, 25, 13665 CrossRef CAS PubMed; (b) A. Parker, P. Lamata, F. Viguri, R. Rodríguez, J. A. López, F. J. Lahoz, P. García-Orduña and D. Carmona, Dalton Trans., 2020, 49, 13601 RSC; (c) M. Carmona, R. Pérez, J. Ferrer, R. Rodríguez, V. Passarelli, F. J. Lahoz, P. García-Orduña and D. Carmona, Inorg. Chem., 2022, 61, 13149 CrossRef CAS PubMed.
- T. Šmejkal, D. Gribkov, J. Geier, M. Keller and B. Breit, Chem. – Eur. J., 2010, 16, 2470 CrossRef PubMed.
- (a) K. Jess, V. Derdau, R. Weck, J. Atzrodt, M. Freytag, P. G. Jones and M. Tamm, Adv. Synth. Catal., 2017, 359, 629 CrossRef CAS; (b) M. Valero, D. Becker, K. Jess, R. Weck, J. Atzrodt, T. Bannenberg, V. Derdau and M. Tamm, Chem. – Eur. J., 2019, 25, 6517 CrossRef CAS PubMed.
- O. Bárta, R. Gyepes, I. Císařová, A. Alemayehu and P. Štěpnička, Dalton Trans., 2020, 49, 4225 RSC.
- O. Bárta, I. Císařová and P. Štěpnička, Dalton Trans., 2021, 50, 14662 RSC.
- Z. Leitner, I. Císařová and P. Štěpnička, New J. Chem., 2022, 46, 1060 RSC.
- W. H. Hersh, J. Chem. Educ., 1997, 74, 1485 CrossRef CAS.
- The metal-donor distances are comparable with those determined for phosphinoamine and phosphinooxazoline Cu(I) and Ag(I) complexes. For selected examples, see: (a) E. W. Ainscough, A. M. Brodie, S. L. Ingham and J. M. Waters, Inorg. Chim. Acta, 1994, 215, 191 CrossRef; (b) P. Papathanasiou, G. Salem, P. Waring and A. C. Willis, J. Chem. Soc., Dalton Trans., 1997, 3435 RSC; (c) W. Kaim, M. Wanner, A. Knodler and S. Zalis, Inorg. Chim. Acta, 2002, 337, 163 CrossRef CAS; (d) O. Bárta, M. Drusan, I. Císařová, R. Šebesta and P. Štěpnička, New J. Chem., 2018, 42, 11450 RSC; (e) R. Giereth, A. K. Mengele, W. Frey, M. Kloss, A. Steffen, M. Karnahl and S. Tschierlei, Chem. – Eur. J., 2020, 26, 2675 CrossRef CAS PubMed.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955 RSC.
- (a) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931 RSC; (b) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- S. P. C. Dunstan, P. C. Healy, A. N. Sobolev, E. R. T. T. iekink, A. H. White and M. L. Williams, J. Mol. Struct., 2014, 1072, 253 CrossRef CAS.
- (a) F. Inagaki, C. Matsumoto, Y. Okada, N. Maruyama and C. Mukai, Angew. Chem., Int. Ed., 2015, 54, 818 CrossRef CAS PubMed; (b) R. J. Staples, C. King, M. N. I. Khan, R. E. P. Winpenny and J. P. Fackler, Jr., Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 472 CrossRef; (c) J.-C. Wang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 53, 611 CrossRef.
- (a) T. G. Appleton, H. C. Clark and L. E. Manzer, Coord. Chem. Rev., 1973, 10, 335 CrossRef CAS; (b) F. R. Hartley, Chem. Soc. Rev., 1973, 2, 163 RSC.
- (a) R. G. Pearson, Inorg. Chem., 1973, 12, 712 CrossRef CAS; (b) J. Vicente, A. Arcas, D. Bautista and P. G. Jones, Organometallics, 1997, 16, 2127 CrossRef CAS.
- V. J. Catalano and S. J. Horner, Inorg. Chem., 2003, 42, 8430 CrossRef CAS PubMed.
- D. E. Jenkins and Z. Assefa, J. Mol. Struct., 2017, 1133, 374 CrossRef CAS.
- (a) A. S. K. Hasmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391 CrossRef PubMed; (b) J. P. Weyrauch, A. S. K. Hashmi, A. Schuster, T. Hengst, S. Schetter, A. Littmann, M. Rudolph, M. Hamzic, J. Visus, F. Rominger, W. Frei and J. W. Bats, Chem. – Eur. J., 2010, 16, 956 CrossRef CAS PubMed.
- O. Bárta, I. Císařová, J. Schulz and P. Štěpnička, New J. Chem., 2019, 43, 11258 RSC.
- W. Zhao and J. Sun, Chem. Rev., 2018, 118, 10349 CrossRef CAS PubMed.
- K. Škoch, I. Císařová and P. Štěpnička, Chem. – Eur. J., 2015, 21, 15998 CrossRef PubMed.
- (a) V. Cadierno, Catalysts, 2017, 7, 328 CrossRef; (b) V. Cadierno, Catalysts, 2020, 10, 1206 CrossRef CAS.
- H. Schmidbaur and A. Schier, Z. Naturforsch., 2011, 66b, 329 CrossRef.
- For selected examples, see: (a) D. Weber and M. R. Gagné, Org. Lett., 2009, 11, 4962 CrossRef CAS PubMed; (b) D. Wang, R. Cai, S. Sharma, J. Jirak, S. K. Thummanapelli, N. G. Akhmedov, H. Zhang, X. Liu, J. L. Petersen and X. Shi, J. Am. Chem. Soc., 2012, 134, 9012 CrossRef CAS PubMed; (c) Z. Lu, J. Han, G. B. Hammond and B. Xu, Org. Lett., 2015, 17, 4534 CrossRef CAS PubMed; (d) A. Franchino, M. Montesinos-Magraner and A. M. Echavarren, Bull. Chem. Soc. Jpn., 2021, 94, 732 CrossRef.
- (a) C. D. Yi and R. Gao, Organometallics, 2009, 28, 6585 CrossRef CAS PubMed; (b) B. C. Chary and S. Kim, J. Org. Chem., 2010, 75, 7928 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct, Chem., 2015, 71, 3 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct, Chem., 2015, 71, 9 CrossRef CAS PubMed.
- (a) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS; (b) A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structure diagrams, summary of relevant crystallographic parameters and copies of the NMR spectra. CCDC 2237665–2237672. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj00451a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |