
Synthesis of a bilayered SDS/ionic liquid stabilized ferrofluid and magnetic cubic mesoporous silica using a long chain ionic liquid†
Jing
Shen
 *a,
Dongxiu
Wang
a,
Li
Yang
a,
Tongwen
Wang
a,
Qianmo
Yang
b and
Jun
Wang
*a
*a,
Dongxiu
Wang
a,
Li
Yang
a,
Tongwen
Wang
a,
Qianmo
Yang
b and
Jun
Wang
*a
aCollege of Chemistry and Chemical Engineering, Yunnan Normal University, Kunming 650092, China. E-mail: shenjingbox0225@hotmail.com; 4478@ynnu.edu.cn
bKunming World Youth Academy, 901 Longxiang Street, Kunming, 650000, China
First published on 15th November 2022
Abstract
Bilayered sodium dodecyl sulfate (SDS)/ionic liquids (alkyl imidazolium type ionic liquid Cnmim+, n = 10, 12, 14, and 16) were first employed to modify the surface of Fe3O4 nanoparticles for the formation of stable aqueous ferrofluids. Subsequently, in the synthesized bilayered SDS/C16mim+-stabilized ferrofluid (Fe3O4/SDS/C16mim+), magnetic mesoporous silica with a cubic (Ia3d) mesostructure (Fe3O4@MCM-48) has been synthesized using tetraethylorthosilicate as the silicon source and the long chain C16mim+ as a template again. The research results showed that the combination of SDS and long chain Cnmim+ (such as C16mim+) is superior to that of SDS and short chain Cnmim+ (such as C10mim+) for the production of stable ferrofluids in water. Moreover, Fe3O4@MCM-48 (0.1 g) with Fe3O4 particles embedded in the network of cubic mesoporous silica showed decolorization rates of up to ∼90% in 25 min for four dye (methylene blue, rhodamine B, malachite green, and methyl violet) solutions (50 mL, 100 mg L−1). In addition, the malachite green-adsorbed Fe3O4@MCM-48 can be separated and recovered under an external magnetic field. This method in which the long chain ionic liquid C16mim+ can be manipulated in turn not only as the modifier but also as the mesoporous templating agent provides important guidelines for the multifunctional applications of C16mim+ in the synthesis of various nanostructured materials.
1. Introduction
Surfactant-stabilized magnetic particles have been extensively studied for the stabilization of magnetic fluid suspensions by steric repulsion of surfactants.1 The electrostatic repulsion occurring in the magnetic particles can be also produced by the introduction of a surfactant-coated bilayer as proposed by Shimoiizaka et al. in the 1980s.2 The assembly of bilayered surfactants onto the magnetic particles was achieved by first coating the surface of the preformed magnetic particles with the primary surfactant, followed by coating the surface of the primary surfactant-coated particles with a second surfactant. For example, Wooding et al. employed saturated and unsaturated fatty acids as primary and secondary surfactants to construct stable magnetic fluids in water.3 Based on these results, Laibinis and Hatton group further demonstrated and quantified surfactant bilayered structures using iron oxide nanoparticles.4 Subsequently, various surfactant bilayered structures were developed to stabilize the magnetic fluids, such as some combinations of sodium oleate and PEG-type,5 oleic acid and polyisobutylene succinimide,6 oleic acid and SDS,7 and so on. Despite this demonstrated progress, exploiting new species for surfactant bilayered coating and enhancing the stabilization of aqueous magnetic fluids continue to be a research issue.Recently, the application of ionic liquids has opened a new route in the fields of synthesis and creation of new and interesting materials.8–10 Particularly, long-chain ionic liquids with imidazolium head groups and long alkyl chains (more than 10 carbon atoms) can show both the surface activity of common surfactants and the special properties of the short-chain ionic liquids such as the properties of low melting temperature, strong solubility, increasing polarity in water, and so on. Numerous studies were performed on the self-organization properties of long-chain ionic liquids in aqueous solutions.11–13 For example, Zheng and his group contributed much effort to study the micelle properties of ionic liquids with long alkyl chains in water via monitoring their conductivity characterization, fluorescence properties, surface tension changes, etc.14–16 These results showed that the activity of long-chain ionic liquids is slightly higher than that of cationic surfactants with a quaternary ammonium type structure in aqueous solutions. Besides, long-chain ionic liquids were proved to exhibit the properties of both thermotropic and lyotropic liquid crystals.17,18 The various features of long-chain ionic liquids have also been utilized to establish various composite nanomaterials.19 For instance, 1-hexadecyl-3-methylimidazole ions (abbr. Cnmim+, n refers to the number of carbon atoms in the alkyl chain on the imidazolium group) were reported to exhibit unique templating behavior in the preparation of silica with a hexagonal mesoporous structure (MCM-41-type).20 Zhou et al. synthesized supermicroporous lamellar silica using the long-chain C16mim+ as the template.21 After that, it was found that silica with cubic and hexagonal mesoporous structures can be also prepared using the long-chain C16mim+ as the template in our earlier research work.22 More recently, we have reported self-assembly bilayer structures of the Cnmim+/Cnmim+ onto magnetic Fe3O4 nanoparticles. Moreover, we found that in the bilayer C16mimCl+-based ferrofluid (Fe3O4/C16mim+/C16mim+), hexagonal magnetic mesoporous silica with Fe3O4 NPs embedded into the porous silica framework (Fe3O4@MCM-41 type) can be prepared using the C16mim+ as the template again.23 However, unexpectedly, although we have made many attempts to investigate the effects of varied synthesized proportions and parameters on the formation of ordered cubic magnetic mesoporous silica (Fe3O4@MCM-48 type) in the ferrofluid (Fe3O4/C16mim+/C16mim+), we still cannot obtain the cubic magnetic mesoporous silica Fe3O4@MCM-48. This may be due to the narrow phase area of the cubic phase in the liquid crystal phase diagram, or the mismatching ratio of the Fe3O4/C16mim+/C16mim+ particles, silicon precursor (TEOS) and the C16mim+ template agent.
In this study, first, we prepared a self-assembly bilayer of SDS/Cnmim+ (n = 10, 12, 14, and 16) to modify magnetic Fe3O4 NPs for the production of stable ferrofluids. Second, based on the bilayered SDS/C16mim+-based ferrofluid, magnetic mesoporous silica with a cubic mesoporous structure and Fe3O4 NPs embedded into the cubic mesoporous network was synthesized using TEOS as a silicon source and the C16mim+ as a mesoporous templating agent. Furthermore, the adsorption properties of the obtained cubic magnetic mesoporous silica (Fe3O4@MCM-48) were also investigated.
In our design, the primary goal is to construct a bimolecular modification layer SDS/Cnmim+ onto Fe3O4 NPs. It can be expected that the cationic imidazole head-group on the Fe3O4 NPs surface extends to the solution, which may lead to a high charge density and strong hydrophilicity on the particle surface. This special surface structure should be able to form a stable aqueous ferrofluid. Our second goal is to confirm the magical templating behavior of the C16mim+ in the synthesis of cubic mesoporous silica with magnetism in the obtained Fe3O4/SDS/C16mim+ ferrofluid. In order to prove the adsorption performance of this magnetic cubic mesoporous silica, four dyes, methylene blue, rhodamine B, malachite green, and methyl violet, were selected as adsorption models. Furthermore, the adsorbed amount of the magnetic cubic mesoporous silica Fe3O4@MCM-48 for malachite green solutions with different initial concentrations was investigated, also including its magnetic separation properties and reuse efficiency. We expect that the long-chain ionic liquid Cnmim+ should be manipulated in a synthetic system in turn not only as the outer layer modifier of Fe3O4/SDS/Cnmim+ particles but also as the mesoporous templating agent in the formation of magnetic cubic mesoporous silica.
2. Experimental section
2.1 Materials
1-Chlorodecane, 1-chlorododecane, 1-chlorotetradecane, 1-chlorodexadecane, 1-chlorooctadecane and 1-methylimidazole (analytical reagent, AR) were purchased from Acrös Organics. Sodium dodecyl sulfate (SDS) (AR) was purchased from Tianjin Zhiyuan Chemical Reagent Co., Ltd China. FeCl2·4H2O and FeCl3·6H2O (AR) were obtained from Chengdu Jinshan Chemical Reagent Co., Ltd, China. Tetraethylorthosilicate (TEOS) (AR) was purchased from Shanghai Aladdin Bio-Chem Technology Co., Ltd, China. All the chemicals were used as received.2.2 Preparation of SDS-coated Fe3O4 particles
Sodium dodecyl sulfate (SDS)-coated Fe3O4 particles were first synthesized by the chemical coprecipitation of Fe2+ and Fe3+ from ammonia water in a molar ratio of 1/2 M with a little amount of SDS, followed by a modifying process of SDS. In a typical preparation step, SDS (0.10 g), FeCl2·4H2O (0.86 g), and FeCl3·6H2O (2.35 g) were dissolved in deionized water (50 mL) by mechanical stirring under N2 purging. After mixing for 5 minutes, the temperature of the mixture was increased to 353 K, followed by adjusting the pH value of the mixture to 8 with ammonia (25% w/w). Then, SDS was further added in five 0.1 g amounts over 5 min until the amount of total SDS reached 0.6 g. After 30 minutes, the reaction mixture was cooled slowly. Through a magnetic decantation method, the resultant reaction solid can be obtained from the solution, followed by washing using deionized water and ethanol. This washing-magnetic decantation steps were repeated three times. The resultant solid product is denoted as Fe3O4/SDS.2.3 Preparation of bilayered SDS/Cnmim+-stabilized ferrofluids
Long-chain alkyl imidazolium ionic liquids Cnmim+, (n = 10, 12, 14, 16, and 18, respectively) were synthesized and identified according to the earlier report.23 In order to synthesize stable aqueous ferrofluids, the Fe3O4/SDS nanoparticles were further coated with a series of Cnmim+. In a typical synthesis procedure of Fe3O4/SDS/C16mim+ ferrofluid, C16mim+ solution (1 mL, 40 g L−1) was added to 20 mL of deionized water. Then, the pH value of the solution was adjusted to 3 by dropping a certain amount of dilute hydrochloric acid, followed by adding Fe3O4/SDS (0.5 g) to the solution under ultrasound. After mixing for 15 minutes, the C16mim+ solution with a concentration of 40 g L−1 was added drop by drop to the mixture again in 1 mL amounts over 5 min with mechanical stirring. This process was continued until no obvious precipitate and phase separation phenomenon were observed when a small permanent magnet was placed at the bottom of the beaker containing the mixture for 10 min. The resultant solution is denoted as the Fe3O4/SDS/C16mim+ ferrofluid. The preparation procedures of other SDS/Cnmim+ bilayered structures onto the Fe3O4 particles were accomplished by repeating the above operation steps using the corresponding combination of SDS and Cnmim+ (n = 10, 12, and 14, respectively) as inner and outer layer modifiers, respectively. For subsequent detection, a small portion of dried Fe3O4/SDS/Cnmim+ particles were separated out of the obtained ferrofluids using the decantation method with a small permanent magnet placed at the bottom of a beaker to provide a magnetic field of about 0.3 T over three weeks.2.4 Preparation of magnetic cubic mesoporous silica
In the obtained Fe3O4/SDS/C16mim+ ferrofluid, magnetic cubic mesoporous silica was prepared using the hydrothermal synthesis method in alkaline medium using C16mim+ as the templating agent and TEOS as the silicon source. Typically, NaOH and C16mim+ were dispersed in the Fe3O4/SDS/C16mim+ ferrofluid with mechanical stirring. Then, TEOS was added in drops to the mixture. The starting molar composition was: 1.0 TEOS:xFe3O4/SDS/C16mim+:0.2 C16mim+:0.5 NaOH:70 H2O (x = 0.050, 0.075, 0.10, respectively). After 1 hour of the reaction, the reaction mixture was transferred to a sealed Teflon-lined autoclave. The hydrothermal reaction in the autoclave was carried out for 3 days in an oven at 373 K. The resultant white powder was further filtered and washed and then calcined at 823 K for 5 h to remove the C16mim+ templating agent. The final product is denoted as Fe3O4@MCM-48.2.5 Characterization
Transmission electron microscopy (TEM) was carried out on a JEM-2100 electron microscope with an acceleration voltage of 200 kV. The samples were prepared by dipping a carbon-coated copper TEM grid (300 meshes) into the sample dispersions and dried at room temperature. The structural analysis of the samples was performed on an X-ray diffractometer (TTR III powder X-ray diffractometer) with Cu Kα (λ = 0.1542 nm) radiation. The XRD was operated at 40 KV and 30 mA over a 2θ range of 1–8° and 20–80°, respectively. The electrophoresis values of the colloidal suspensions were measured using an electrophoresis apparatus (Nanjing Sangli DYL-3). The magnetization experiments of the samples were conducted on a vibrating sample magnetometer (VSM 7407). The thermal properties of samples were investigated using thermogravimetric analysis (TGA) and differential thermal analysis (DTA), respectively, on a ZRY-1P thermal analysis apparatus. The N2 adsorption–desorption isotherms for the magnetic cubic mesoporous silica were obtained at 77 k on a Tristar 3000 automated gas adsorption analyser. Before the sorption experiments, a Micromeritics VacPrep 061 degasser was used to remove the gas of the samples overnight at 423 K under 100 μTorr pressure. The BJH (Barrett–Joyner–Halenda) method was employed to analyze the obtained data. The BET value was determined at a range of P/P0 = 0.05–0.35, and pore size distribution curves were obtained from the desorption branch of the isotherm. The Fourier transform infrared (FT-IR) spectra of samples were characterized on a TENSOR 27 FTIR spectrometer. The contents of iron in samples were determined by a well-known o-phenanthroline spectrophotometer, and then, the content of the modified double-layer was obtained by subtracting the corresponding amount of iron oxide from the total amount of the sample.2.6 Adsorption experiment of magnetic cubic mesoporous silica
Four dye solutions, methylene blue, rhodamine B, malachite green, and methyl violet solutions, were selected as absorbed objects to study the adsorption and separation properties of the magnetic cubic mesoporous Fe3O4@MCM-48. In a typical adsorption procedure, the synthesized Fe3O4@MCM-48 (0.1 g) was added to 50 mL of the dye aqueous solutions (100 mg L−1). At several fixed time intervals, the resultant sediments were separated from the adsorbed suspensions using a dumping method under magnetic induction, and then, the absorption spectra of the remaining solutions were recorded on an UV-vis spectrograph (SHIMADZU UV-1780) at 664, 554, 617, and 580 nm for methylene blue, rhodamine B, malachite green, and methyl violet solutions, respectively. The decolorization rates of the resultant solutions were calculated according to the following equation:Decolorization rate:
 | (1) |
Moreover, the adsorption and separation characteristics of Fe3O4@MCM-48 in the malachite green solutions with different initial concentrations of 25, 50, 100, 200, and 300 mg L−1 were also investigated, respectively. The concentrations of the malachite green solutions after adsorption at several fixed time intervals can be given according to the standard working curves of malachite green solutions. The adsorption quantity of Fe3O4@MCM-48, qt, (mg g−1) was determined from the following equation:
 | (2) |
The Freundlich isotherm model is the most important adsorption model for multilayered adsorbent surfaces. The linear form of the Freundlich isotherm model can be represented as
 | (3) |
In addition, the malachite green-adsorbed Fe3O4@MCM-48 can be calcined continuously at 823 K for 5 h to remove the adsorbed malachite green. By tracing the UV-vis absorption spectra at different cycle times, the recycling rate of Fe3O4@MCM-48 can be investigated. In contrast, the adsorption experiment of malachite green on the pure cubic mesoporous silica (MCM-48) prepared using the same C16mim+ templating agent,22 without adding Fe3O4 NPs, was carried out. At the same time, the adsorption experiment of malachite green on the bare Fe3O4 NPs prepared by the same chemical coprecipitation method without adding C16mim+ as the modifier was also performed.
3. Results and discussion
3.1 Synthesis and structure of bilayer SDS/Cnmim+ on Fe3O4 NPs
Our strategy to prepare bilayer SDS/Cnmim+ on Fe3O4 NPs includes first synthesizing Fe3O4 NPs and then immobilizing the bilayer SDS/Cnmim+ on Fe3O4 NPs. The well-established chemical coprecipitation technique of Fe2+ and Fe3+ in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 from ammonia water was used to synthesize the Fe3O4 NPs.24 By adding NH4OH (25% w/w), the pH of the reaction system was adjusted to 8, which led to the formation of positively charged Fe3O4 particles.25,26 Subsequently, the obtained Fe3O4 NPs were coated with SDS physisorbed to the surface of Fe3O4 NPs by electrostatic action between the positively charged Fe3O4 particle surface and the negatively charged SDS head-group, as shown in Fig. 1. It was easy to observe that the SDS-coated Fe3O4 particles (Fe3O4/SDS) settled down from the solution within a few minutes (Fig. S1A, ESI†). The reason of this unstable appearance may be due to the alkyl chain of SDS on the Fe3O4 particle surface extending to the water. The black Fe3O4/SDS particles can be attracted by placing an experimental small magnet next to the beaker (Fig. S1B, ESI†). This simple experiment demonstrated that the Fe3O4/SDS particles are magnetic. Fig. S2(a) and (b) (ESI†) show the wide-angle XRD patterns of the pure Fe3O4 particles and the Fe3O4/SDS particles, respectively. All characteristic diffraction peak positions are consistent with those of the crystalline Fe3O4 with the face-centered cubic (fcc) structure according to JCPDS card, No. 00-001-1111. The peak intensity of Fe3O4/SDS particles is slightly lower than that of pure Fe3O4 particles. The average grain sizes were calculated from the Scherrer formula to be 9.52 nm for Fe3O4/SDS particles and 10.20 nm for pure Fe3O4 particles, respectively. These results indicated that SDS modification onto Fe3O4 particles limited the growth of Fe3O4 particles in part.
:2 from ammonia water was used to synthesize the Fe3O4 NPs.24 By adding NH4OH (25% w/w), the pH of the reaction system was adjusted to 8, which led to the formation of positively charged Fe3O4 particles.25,26 Subsequently, the obtained Fe3O4 NPs were coated with SDS physisorbed to the surface of Fe3O4 NPs by electrostatic action between the positively charged Fe3O4 particle surface and the negatively charged SDS head-group, as shown in Fig. 1. It was easy to observe that the SDS-coated Fe3O4 particles (Fe3O4/SDS) settled down from the solution within a few minutes (Fig. S1A, ESI†). The reason of this unstable appearance may be due to the alkyl chain of SDS on the Fe3O4 particle surface extending to the water. The black Fe3O4/SDS particles can be attracted by placing an experimental small magnet next to the beaker (Fig. S1B, ESI†). This simple experiment demonstrated that the Fe3O4/SDS particles are magnetic. Fig. S2(a) and (b) (ESI†) show the wide-angle XRD patterns of the pure Fe3O4 particles and the Fe3O4/SDS particles, respectively. All characteristic diffraction peak positions are consistent with those of the crystalline Fe3O4 with the face-centered cubic (fcc) structure according to JCPDS card, No. 00-001-1111. The peak intensity of Fe3O4/SDS particles is slightly lower than that of pure Fe3O4 particles. The average grain sizes were calculated from the Scherrer formula to be 9.52 nm for Fe3O4/SDS particles and 10.20 nm for pure Fe3O4 particles, respectively. These results indicated that SDS modification onto Fe3O4 particles limited the growth of Fe3O4 particles in part.
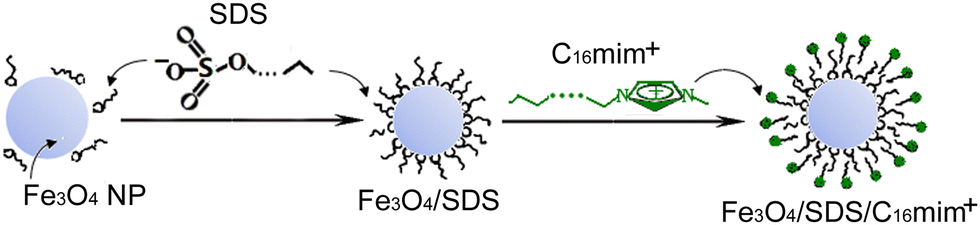 | ||
| Fig. 1 Schematic illustration of the surface modification procedure of Fe3O4/SDS/C16mim+ particles. | ||
In order to synthesize stable aqueous ferrofluids, the Fe3O4/SDS particles were coated with Cnmim+ (n is 10, 12, 14, 16, respectively) as outer layer. It was found that the Cnmim+-coated Fe3O4/SDS particles (Fe3O4/SDS/Cnmim+) exhibited a remarkable stability in the aqueous colloidal suspensions. No sediment was found for more than 6 months (e.g. for Fe3O4/SDS/C16mim+, Fig. S3, ESI†). Approximately +35 mV of Zeta potential (ζ) value was shown for the Fe3O4/SDS/C16mim+ colloidal suspension in the corresponding electrophoresis experiment. These results suggest a hypothetical structure of the Fe3O4/SDS/Cnmim+ particles, that is, the positively charged imidazole head-groups in the outer layer of the particles extend to the aqueous solution, while the alkyl chain of imidazole is towards the Fe3O4/SDS particles or partially inserted into the alkyl chain of SDS coated on the particles due to hydrophobic interaction, as shown in Fig. 1. The hypothetical structure that the positively charged imidazole head-groups on the outer layer of the particles provided a very high charge density and hydrophilic particle surface, resulting in the formation of an aqueous stable ferrofluid.
Fig. 2(A) and (B) show the typical TEM image of the Fe3O4/SDS/C16mim+ particles and its corresponding particle size histogram, respectively. It is clear that the Fe3O4/SDS/C16mim+ particles exhibited the characteristics of polydispersed spherical particles with a clear interface and an average diameter of approximately 10.15 nm (Fig. 2(B)). Electron diffraction in the corresponding region is shown in the insets in Fig. 2(A). The diffraction image consists of a series of rings that can be indexed to a magnetite structure, which shows the same result as that of the wide-angle XRD pattern shown in Fig. S2 (ESI†). Fig. 2(C) shows the high-resolution TEM (HRTEM) image of a randomly oriented nanocrystallite, which is approximately 10 nm in diameter and possesses clearly resolved lattice fringes, indicating the high crystallinity of the sample particle. It should be noted that no double-layered structure of SDS/C16mim+ was observed in the TEM image, which may be because of the low resolution of the molecular arrangement of double-layered SDS/C16mim+ onto the Fe3O4 particles.
 | ||
| Fig. 2 TEM image of (A) Fe3O4/SDS/C16mim+ particles and electron diffraction image of a large zone in the inset, (B) corresponding particle size histogram and (C) HRTEM image of a unique single-crystal particle. | ||
Fig. 3 shows the magnetization curves of pure Fe3O4, Fe3O4/SDS, and Fe3O4/SDS/C10mim+ particles at room temperature, respectively. These curves exhibited typical magnetization “S-shaped” curves, and no obvious hysteresis loops were observed, that is, there were no significant reduced remanence and intrinsic coercivity in these samples, indicating the superparamagnetic feature of these samples. The saturation magnetizations (Ms) of pure Fe3O4, Fe3O4/SDS, and Fe3O4/SDS/C10mim+ particles were determined to be 62.61, 60.10 and 53.90 emu g−1, respectively. It is mentionable that the gradual reducing change of Ms(Fe3O4) > Ms(Fe3O4/SDS) > Ms(Fe3O4/SDS/C10mim+) can be observed with the increase of the number of modification layers. This phenomenon may be due to a separation of dipole coupling, which originated from the nonmagnetic species (such as SDS and Cnmim+) on the Fe3O4 particle surface.27,28
 | ||
| Fig. 3 Magnetization curves of (a) pure Fe3O4 particles, (b) Fe3O4/SDS particles, and (c) Fe3O4/SDS/C10mim+ particles. | ||
Fig. 4 exhibits the typical thermogravimetric analysis (TG) (left axis) and differential thermal analysis (DTA) (right axis) curves of the Fe3O4/SDS/C16mim+ particles, respectively. For the TG curve, when the temperature was below 120 °C, a small thermal weightlessness of about 2.64% can be observed, which should be due to the removal of adsorbed water from the sample. A more significant percentage weight loss of ∼10.24% appeared between 210 and 480 °C, which can be attributed to the combustion removal of the bilayered SDS/C16mim+ molecules. The DTA curve of the sample exhibited an intense exothermic peak at 321 °C and a weak exothermic peak at 389 °C, which may be due to the thermal decomposition of the outer C16mim+ and the inner layer SDS, respectively. These results can be easily explained by the differences in molecular weights and boiling point between C16mim+ and SDS molecules. Furthermore, these two exothermic peaks appeared at different temperature positions, which may be due to the difference between the two interactions, such as hydrophobic interaction between SDS and C16mim+ and electrostatic attraction between SDS and Fe3O4 particles. These results revealed the coexistence of two different kinds of molecules (SDS and C16mim+) onto Fe3O4 particles. Similar to our results, the obvious difference in the weight loss between the bilayer fatty acid surfactants coated on Fe3O4 particles has also been reported by Shen et al.4
 | ||
| Fig. 4 TG and DTA curves of Fe3O4/SDS/C16mim+ particles. | ||
The multiple coverage parameters for inner layer SDS and outer layer Cnmim+ onto Fe3O4 NPs can be further calculated. In this calculation, the content of iron in samples was determined by well-known o-phenanthroline spectrophotometry. Then, the content of the modifier (SDS and Cnmim+) was obtained by subtracting the corresponding amount of iron oxide from the total amount of the sample. In addition, the Fe3O4 particle was regarded as a sphere with an average diameter of 10 nm according to the results of TEM (Fig. 2). These multiple coverage parameters of the bilayer SDS/Cnmim+ assembled onto Fe3O4 NPs are listed in Table 1. Two characteristics should be noted. It is first clear that the weight percents of the Fe3O4/SDS/Cnmim+ (n = 10, 12, 14, 16) particles are larger than those of the Fe3O4/SDS particles. The difference can be attributed to the outer layer Cnmim+. For example, the weight percents of the bilayer SDS/Cnmim+ were estimated onto Fe3O4 particles to be 10.46 wt% (SDS/C10mim+), 12.50 wt% (SDS/C12mim+), 17.42 wt% (SDS/C14mim+) and 21.98 wt% (SDS/C16mim+), respectively, corresponding to the monolayer SDS of 6.01 wt% (third column in Table 1). This result proved again the existence of the bilayer SDS/Cnmim+ molecular structures onto the Fe3O4 NPs. Secondly, it was found that with the increasing n value (the number of carbon atoms in the alkyl chain of Cnmim+) from 10 to 16, the weight percent of outer layer Cnmim+ increased from 4.45 wt% to 15.97 wt% (fourth column in Table 1), and the number of outer layer Cnmim+ on each Fe3O4/SDS particle increased from 316 to 1006 (sixth column in Table 1) (in the experiment, the modifying amount of adding outer Cnmim+ was the same). This C16mim+ molecule number of 1006 on each Fe3O4/SDS particle is close to that of the saturated fatty acid modifiers (such as myristic acid) coated onto Fe3O4 particles with a densely packed structure pattern.3 In the experiment, we found that the long-chain C16mim+ displayed a stronger assembly tendency (it was easier to form stable ferrofluid) than that of the short-chain ionic liquid (e.g. C10mim+). The reason is that during the modification processes of outer layer Cnmim+, there is a competition of two interactions. One is a hydrophilic interaction between imidazole head-groups of Cnmim+ and water molecules. Another is a hydrophobic interaction between hydrocarbon tails of outer layer Cnmim+ and inner layer SDS. It is well known that positively charged ionic liquids, such as imidazolium salts, have strong hydrophilicity.19 The shorter the hydrocarbon chain of the Cnmim+, the stronger the hydrophilic tendency of the Cnmim+. When the short-chain Cnmim+ (such as C10mim+) was employed as the outer layer modifier, the short-chain C10mim+ might tend to stay in water because of its strong hydrophilic interaction with water molecules, which causes only a small amount of C10mim+ to adhere to the particles, forming a loose arrangement in the particle surface. In the experiments, it can be observed that when n ≤ 10, the SDS/Cnmim+-coated Fe3O4 particle exhibited an unstable ferrofluid state, that is, the Fe3O4/SDS/C10mim+ particles settled down within minutes. However, when the n value was increased to 18, the solubility of this C18mim+ in water deceased markedly and an insoluble floating substance (C18mim+) similar to sponge appeared, which indicated that it was impossible to produce an effective bilayer SDS/C18mim+ structure due to the strong hydrophobicity of the C18mim+. When n was 16, the hydrophobic and hydrophilic interactions of the C16mim+ were found to be matching, resulting in a significant increase in the assembly capability. Thus, we believe that the Fe3O4/SDS/C16mim+ particles can provide a strongly hydrophilic and highly charge density surface because of a closely packed structure of C16mim+ molecules exposing their imidazole headgroups to the aqueous solution. The special particle surface structure can prevent the particles not only to agglomerate together but also to oxidize between the Fe3O4/SDS/C16mim+ NPs due to electrostatic and steric repulsions, leading to the production of stable aqueous ferrofluids.
| Sample | Wt%a (Fe3O4) | Wt% (modifier onto Fe3O4 NPs) | Wt% (outer layer modifier) | Wtoutb (outer layer Cnmim+ on per g Fe3O4) (g) | N out (No. of molecules of outer layer Cnmim+ on each particle) |
|---|---|---|---|---|---|
| a The mass percentage of Fe3O4 determined by the analysis of iron in samples using o-phenanthroline spectrophotometry. b Wtout = wt%(out)/wt%(Fe3O4), where the masspercentage of Fe3O4 was calculated by subtracting the masspercentage of Cnmim+ and corresponding adsorbed water. c N out = N1/N2, where N1 = Wtout × NA/Mout is the number of Cnmim+ molecules per g Fe3O4, with NA being the Avogadro constant and Mout being the molar mass of outer layer Cnmim+, and N2 = V1/V2 is the number of Fe3O4 particles per g Fe3O4, with V1 = 1/ρ and V2 = 4πR3/3 being the volume per g Fe3O4 and the volume per particle, respectively, where ρ is the density of Fe3O4 (5.18 g cm−3) and R is the radius of the Fe3O4 particle. | |||||
| Fe3O4/SDS | 93.99 | 6.01 | 6.01 | — | — |
| Fe3O4/SDS/C10mim+ | 89.54 | 10.46 | 4.45 | 0.05 | 316 |
| Fe3O4/SDS/C12mim+ | 87.50 | 12.50 | 6.49 | 0.07 | 401 |
| Fe3O4/SDS/C14mim+ | 82.58 | 17.42 | 11.41 | 0.14 | 730 |
| Fe3O4/SDS/C16mim+ | 78.02 | 21.98 | 15.97 | 0.21 | 1006 |
3.2 Characterization of magnetic cubic mesoporous silica
The schematic diagram of the synthesized procedure of the cubic magnetic mesoporous silica is shown in Fig. 5. In this preparation procedure, Fe3O4/SDS/C16mim+ particles, TEOS and C16mim+ template were used as precursors, and subsequently, the procedure included a hydrothermal treatment and finally the calcination step. | ||
| Fig. 5 Schematic illustration of the synthesized procedure of magnetic cubic mesoporous silica prepared using C16mim+ as the template in the Fe3O4/SDS/C16mim+ ferrofluid. | ||
Three samples were synthesized with initial molar ratios of n(Fe3O4/SDS/C16mim+)/n(TEOS) of 0.050, 0.075 and 0.10, respectively, using C16mim+ as the templating agent and TEOS as the silica source in the aqueous Fe3O4/SDS/C16mim+ ferrofluid. The low-angle XRD patterns of the calcined samples (Fig. 6) show well-resolved reflections with d211-spacings of 3.91, 3.79, and 3.87 nm, indicating a cubic gyroid phase (Ia3d) structure, which is consistent with the low-angle XRD patterns of the siliceous MCM-48 prepared using a quaternary ammonium type surfactant as the template.29 The unit-cell dimensions a0 of the cubic mesoporous structures were calculated to be 9.58, 9.29, and 9.48 nm for the three samples with the initial molar ratio n(Fe3O4/SDS/C16mim+)/n(TEOS) of 0.050, 0.075 and 0.10, respectively (the unit-cell dimension a0 assuming space group Ia3d was calculated according to the XRD data using a0 = d211√6). The unit-cell dimension a0 values of the three samples are slightly larger than those of pure cubic mesoporous silica prepared with the same C16mim+ as the template in our earlier work,22 which may be due to the introduction of the magnetic particles. The inset in Fig. 6 is an enlarged view of Fig. 6(b) in the range of 2θ = 3.5–5° (multiple peaks), further confirming the existence of the cubic phase structure. Compared with the low-angle XRD patterns of the pure cubic mesoporous silica prepared using C16mim+ as the template,22 we found that the presence of the Fe3O4/SDS/C16mim+ particles in the defined concentration range did not destroy the cubic gyroid phase structure of silica. If we compare the distinguishability of the diffraction peaks shown in Fig. 6(b) and (c), we can find that the resolution of the peaks in Fig. 6(b) is higher than that in Fig. 6(c), revealing that with an increasing initial molar ratio n(Fe3O4/SDS/C16mim+)/n(TEOS) from 0.075 to 0.100, the order degree of the cubic magnetic mesopores would decrease. This result may be due to excessive addition of Fe3O4/SDS/C16mim+ particles, which leads to the decrease of the matching degree of charge density between the silicon species and C16mim+ template. This phenomenon can be observed when the guest species was loaded into the ordered mesoporous structure and the distinguishability of their XRD lines would be reduced.30
 | ||
| Fig. 6 Low-angle XRD patterns of the three samples prepared with the initial molar ratio n(Fe3O4/SDS/C16mim+)/n(TEOS) of (a) 0.050, (b) 0.075 and (c) 0.10 using C16mim+ as the templating agent in the aqueous Fe3O4/SDS/C16mim+ ferrofluid. | ||
The TEM images of the calcined sample prepared with the initial molar ratio n(Fe3O4/SDS/C16mim+)/n(TEOS) of 0.075 using C16mim+ as the template are shown in Fig. 7. These TEM images taken along the [111] (Fig. 7(A)), [311] (Fig. 7(B)) and [110] (Fig. 7(C)) zone axes revealed that the sample exhibited an ordered mesoporous structure with cubic symmetry, which is in agreement with the TEM analysis on the pure cubic mesoporous silica (MCM-48) templated by the quaternary ammonium ion surfactant agent.31,32 Simultaneously, in the TEM images (Fig. 7(A)–(C)), some dark quasi spherical particles can be observed. The dark particles should be Fe3O4 NPs, evidenced by some well-resolved Bragg peaks of XRD shown in Fig. S4A (ESI†), which was indexed to a crystalline ferromagnetic structure according to JCPDS card no. 28-0491. From the TEM images, we also found that the dark magnetite particles were randomly embedded into the channel network of the cubic ordered mesoporous silica. Therefore, these TEM images revealed the coexistence of the arrangement of cubic mesopore channels and quasi spherical Fe3O4 NPs. Fig. S4B (ESI†) further displays a magnetization “S” curve of this sample. No obvious hysteresis loop was observed, that is, there were no remarkable remanence and coercivity in the sample, demonstrating the superparamagnetic properties of the magnetic cubic mesoporous silica.
 | ||
| Fig. 7 TEM images of the sample prepared with the initial molar ratio n(Fe3O4/SDS/C16mim+)/n(TEOS) of 0.075 using C16mim+ as the templating agent in the Fe3O4/SDS/C16mim+ aqueous ferrofluid, and the images were taken along (A) [111], (B) [311], and (C) [110] axes’ directions. | ||
A nitrogen physisorption isotherm (Fig. 8(A)) was recorded to estimate the textural properties of this calcined sample (n(Fe3O4/SDS/C16mim+)/n(TEOS) of 0.075). The isotherm is of type IV with a distinct hysteresis loop in the relative pressure 0.35–0.70, indicating a narrow pore size distribution in the mesoporous range.33 The pore size distribution (Fig. 8(B)) is mainly located around 2.3–4 nm, and the average pore size calculated from the BJH model using the desorption branch was determined to be about 2.35 nm, which is consistent with the observation of the TEM images (Fig. 7). The BTE surface area and pore volume for the sample were determined to be 952 m2 g−1 and 0.66 cm3 g−1, respectively. It can be found that this BET surface area of the magnetic cubic mesoporous sample is lower than that of the pure cubic mesoporous silica synthesized using C16mim+ as the template (∼1250–1300 m2 g−1),22 which may be attributed to the introduction of the magnetic particles into the cubic mesoporous silica channel framework.
 | ||
| Fig. 8 (A) N2 adsorption–desorption isotherm of the sample prepared with the initial molar ratio n(Fe3O4/SDS/C16mim+)/n(TEOS) of 0.075 using C16mim+ as the templating agent in the Fe3O4/SDS/C16mim+ aqueous ferrofluid, and (B) its BJH pore size distribution calculated from the desorption branch using the BJH method. | ||
In the classical preparation of mesoporous silica materials, surfactant chemistry is the key to the synthesis of solid mesophases.34 It was reported that the synthesis of the cubic mesoporous phase (Ia3d) is more difficult than that of the hexagonal mesoporous phase. This reason may be due to the narrow phase area of the cubic phase in the liquid crystal phase diagram and the relatively harsh synthesis conditions for the synthesis of the cubic phase.35 At the same time, the introduction of the modified Fe3O4 NPs would interfere with the synthesis of the composite mesoporous phase and increase the difficulty of the synthesis of the cubic phase. In our early experiments, we found that magnetic Fe3O4@MCM-41 with a coexistence structure of a hexagonal mesoporous silica array and Fe3O4 NPs can be obtained in a magnetic Fe3O4/C16mim+/C16mim+ fluid.23 However, in the bilayer C16mim+-based magnetic fluid, although we have made many attempts to study the effects of n(Fe3O4/C16mim+/C16mim+)/n(TEOS), n(TEOS)/n(C16mim+) and n(TEOS)/n(H2O) on the formation of the magnetic cubic mesoporous Fe3O4@MCM-48, we still cannot obtain Fe3O4@MCM-48. In this article, SDS and C16mim+ were used as the modifiers for the inner and outer layers to construct modified bilayers onto the Fe3O4 particles, respectively. We found that in the Fe3O4/SDS/C16mim+ ferrofluid, cubic magnetic mesoporous Fe3O4@MCM-48 can be formed. The results may be explained by two aspects. One is that the anion headgroup of SDS is smaller than the cationic imidazole headgroup of C16mim+. When SDS is used as the modified inner layer of the modified double layer on the surface of Fe3O4 particles, smaller Fe3O4/SDS/C16mim+ particles with higher radii of curvature can be obtained compared with the Fe3O4/C16mim+/C16mim+ particles. The existence of such small particles has little interference on the production of magnetic cubic mesoporous Fe3O4@MCM-48. Another reason may be due to the use of rational proportions of the n(Fe3O4/SDS/C16mim+)/n(TEOS), n(TEOS)/n(C16mim+) and n(TEOS)/n(H2O) in the initial synthesis compositions. Based on our earlier research, we found that in the concentrated initial mixture solution, it is advantageous to synthesize cubic mesoporous silica by using C16mim+ as the template.22 Therefore, on the other hand, the addition of the magnetic particles may increase the concentration of the initial mixture component, leading to the formation of the magnetic cubic mesoporous phase. The result also shows that it is possible to prepare the magnetic cubic mesoporous silica by using C16mim+ as the template in the aqueous Fe3O4/SDS/C16mim+ ferrofluid.
3.3 Adsorption properties of magnetic cubic mesoporous Fe3O4@MCM-48
To assess the adsorption ability of the synthesized Fe3O4@MCM-48, four dyes, methylene blue, rhodamine B, malachite green, and methyl violet, were selected as adsorption models, respectively. Fig. 9 shows the decolorization rates of the four dye solutions (100 mg L−1, 50 mL) after adsorption using the synthesized Fe3O4@MCM-48 (0.1 g) as the adsorbent at different time intervals. As shown in Fig. 9, within 25 min, the decolorization rates of the four dye solutions increased rapidly and reached a plateau (∼90%), indicating the strong adsorption capacity of Fe3O4@MCM-48. Fig. 10 further shows decolorization rates of malachite green solutions and adsorbed amount of Fe3O4@MCM-48. Obviously, when the initial concentration of malachite green solution (50 mL) is less than 100 mg L−1 (Fig. 10(A)), the decolorization rate of the malachite green solution increased faster in the first 5 min, and then the value reached a platform (95%) in the next 15 min, indicating that the adsorption equilibrium was reached. However, when the initial concentrations of the malachite green solutions were increased to 200 or 400 mg L−1, the decolorization rates decreased to 84% or 66% at 15 min, respectively (Fig. 10(A)). These results indicated that there is a saturation value for the adsorption of Fe3O4@MCM-48. The adsorption capacity of Fe3O4@MCM-48 at equilibrium (30 min) was found to be 11.5, 15.0, 36.5, 85.5 and 161 mg g−1 for the initial malachite green concentrations of 25, 50, 100, 200 and 400 mg L−1, respectively (Fig. 10(B)). If we compare the adsorption capacities of the Fe3O4@MCM-48 and pure mesoporous silica MCM-48 prepared by the same C16mim+ template at equilibrium (30 min),22 it was found that the adsorption quantity of Fe3O4@MCM-48 (161 mg g−1) was lower than pure MCM-48 (376 mg g−1). This phenomenon may be due to the fact that the BET surface area of Fe3O4@MCM-48 (952 m2 g−1) is lower than that of the pure mesoporous silica MCM-48 (∼1250–1300 m2 g−1).22 However, Fe3O4@MCM-48 was easily recovered from the adsorbed solution by magnetic separation technology, while the pure cubic mesoporous silica MCM-48 can only be retrieved by centrifugation. Another notable problem was that the bare Fe3O4 particles had an adsorption quantity of 10.5 mg g−1, which is lower than that of Fe3O4@MCM-48. The reason is probably that Fe3O4@MCM-48 had adequate mesoporous channels and highly adsorption capacity compared with the bare Fe3O4 particles. | ||
| Fig. 9 Decolorization rates of (a) methylene blue, (b) rhodamine B, (c) malachite green, and (d) methyl violet solutions after adsorption using Fe3O4/MCM-48 as the adsorbent and magnetic separation at different time intervals. The initial concentrations of the four dye solutions were all 100 mg L−1. | ||
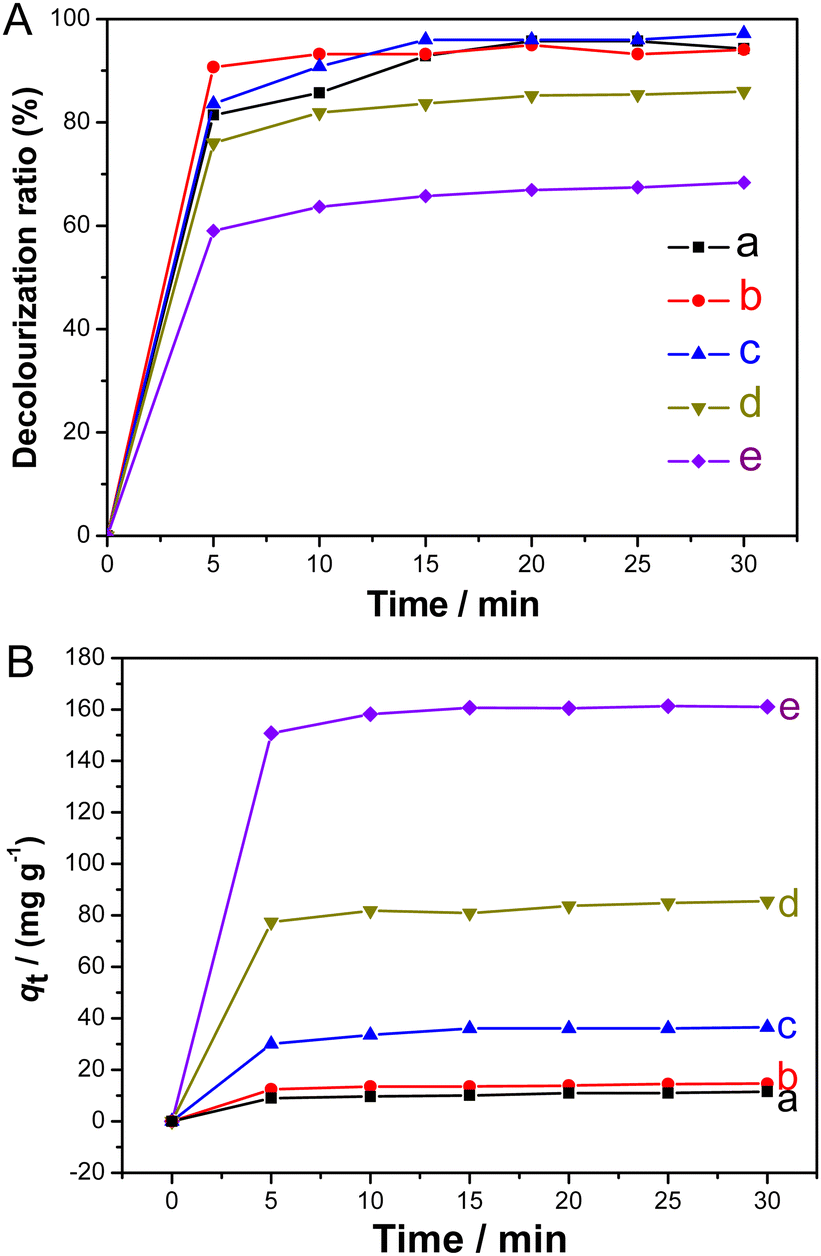 | ||
| Fig. 10 (A) Decolorization rates of malachite green solutions and (B) adsorbed quantity of Fe3O4@MCM-48 at different time intervals. The initial concentrations of malachite green solutions are (a) 25, (b) 50, (c) 100, (d) 200 and (e) 400 mg L−1, respectively. | ||
For rough surfaces, the Freundlich isotherm model is considered as the most important multilayer adsorption model. For the adsorption of malachite green solutions at different initial concentrations (25, 50, 100, 200 and 400 mg L−1), the equilibrium adsorption quantity (qe) of Fe3O4@MCM-48 and the equilibrium concentration (Ce) of malachite green solutions at 30 min can be treated according to the Freundlich isotherm model. A well-defined linear relation was obtained (see Fig. S5, ESI†). From the linear relation, we can calculate that values of KF and 1/n are 18 mg g−1 and 0.53, respectively. This result (especially n is greater than 1) indicated that the synthesized Fe3O4@MCM-48 possessed spacious porous channels and special high surface area, resulting in relatively strong adsorption capacity. Therefore, in some liquid-phase adsorption processes, the synthesized Fe3O4@MCM-48 can be potentially used as an adsorbent. The synthesized Fe3O4@MCM-48 has generous surface silicon hydroxyl groups with negative charge in the mesoporous channels, which can adsorb positively charged cationic dyes, for example, methylene blue, malachite green, rhodamine B and methyl violet. Therefore, we believe that the physical adsorption caused by the electrostatic interaction between the cationic dyes and the silicon hydroxyls of negatively charged mesoporous channels may be the main reason, that is, the adsorption is more beneficial to cationic dyes.
If a small laboratory magnet was placed next to the small beaker containing the mixture, the dye-adsorbed Fe3O4@MCM-48 was recovered simply by decanting clear solution or removing clear solution using a pipette. From one side, this simple experiment also showed that Fe3O4@MCM-48 is magnetic. In other words, Fe3O4@MCM-48 can be considered as a magnetic adsorbent to remove some positively charged cationic dyes from the corresponding liquid-phase. We can also find that after the adsorption process, Fe3O4@MCM-48 was recovered by magnetic separation, dried at room temperature and calcined at 823K for 2 h to remove the adsorbed dyes. The reclaimed Fe3O4@MCM-48 can be used as an adsorbent again. It was found that after three successive cycle uses of Fe3O4@MCM-48, the decolorization rates of malachite green solutions decreased to 94, 91, and 85 mg L−1, respectively. It is worth noting that the decolorization rate was decreased to about 74% after the fourth cycle, which suggests that its adsorption performance was reduced. This result may be attributed to the partial collapse of the cubic mesoporous channels of Fe3O4@MCM-48 due to the multiple recycled adsorptions and calcinations.
4. Conclusions
Stable aqueous ferrofluids were first synthesized by using bilayer SDS/ionic liquid (alkyl imidazolium ionic liquid Cnmim+, n = 10, 12, 14, and 16, respectively) as modifiers onto Fe3O4 NPs. It was found that the long chain ionic liquid (such as C16mim+) is superior to the short chain ionic liquid (such as C10mim+) in the construction of bilayer modifiers. The SDS/C16mim+-based Fe3O4 particles can offer the hydrophilic and positively charged outer surface, resulting in the formation of a stable aqueous ferrofluid. Furthermore, in the aqueous SDS/C16mim+-based ferrofluid, the magnetic cubic mesoporous silica framework (Fe3O4@MCM-48) can be synthesized by using C16mim+ as the template again. The synthesized Fe3O4@MCM-48 with a specific surface area of up to 952 m2 g−1 showed high decolorization rates of up to ∼90% in 25 min for four dye solutions (methylene blue, rhodamine B, malachite green, and methyl violet solutions). In addition, it was found that the malachite green-adsorbed Fe3O4@MCM-48 can be separated and recovered from the corresponding solution under an external magnetic field. It was found that for successive three cycles, Fe3O4@MCM-48 enabled the decolorization rates of the malachite green solution to reach 94, 91, and 85 mg L−1, respectively. The application of C16mim+ as the modifier of the outer layer of Fe3O4/SDS/C16mim+ particles and cubic mesoporous templating agent in turn may provide some information for the multifunctional applications of C16mim+ in the synthesis of various nanomaterials.Author contributions
Jing Shen: resources, funding acquisition, data analysis, and writing-original draft. Dongxiu Wang: investigation, data analysis and resources. Li Yang: investigation and resources. Tong Wang: funding acquisition. Qianmo Yang: investigation. Jun Wang: funding acquisition, writing – review and editing, and supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (No. 21564018, 21063017, 21363029 and 22165034).Notes and references
- B. M. Berkovsky, V. F. Medvedev and M. S. Karkov, Magnetic Fluids: Engineering Applications, Oxford University Press, New York, 1993 Search PubMed.
- J. Shimoiizaka, K. Nakatsuka, T. Fujita and A. Kounosu, IEEE Trans. Magn., 1980, 16, 368–371 CrossRef.
- A. Wooding, M. Kilner and D. B. Lambrick, J. Colloid Interface Sci., 1991, 144, 236–242 CrossRef CAS.
- L. F. Shen, P. E. Laibinis and T. A. Hatton, Langmuir, 1999, 15, 447–453 CrossRef CAS.
- R. Y. Hong, S. Z. Zhang, Y. P. Han, H. Z. Li, J. Ding and Y. Zheng, Powder Technol., 2006, 170, 1–11 CrossRef CAS.
- B. Bateer, Y. Qu, X. Meng, C. Tian, S. Du, R. Wang, K. Pan and H. Fu, J. Magn. Magn. Mater., 2013, 332, 151–156 CrossRef CAS.
- M. Soleymani and M. Edrissi, J. Dispersion Sci. Technol., 2016, 37, 693–698 CrossRef CAS.
- B. Xin and J. Hao, Chem. Soc. Rev., 2014, 43, 7171–7187 RSC.
- X. Zhao, L. Guo, T. Xu, H. Wang, R. Zheng and Z. Jiang, New J. Chem., 2022, 46, 15901–15910 RSC.
- Y. Tian, C. Xing, W. Wang, S. Zhang and Y. Zhang, New J. Chem., 2022, 46, 8855–8862 RSC.
- M. Blesic, M. H. Marques, N. V. Plechkova, K. R. Seddon, L. P. N. Rebelo and A. Lopes, Green Chem., 2007, 9, 481–490 RSC.
- J. Łuczak, J. Hupka, J. Thöming and C. Jungnickel, Colloids Surf., A, 2008, 329, 125–133 CrossRef.
- H. Cao, Y. Hu, W. Xu, Y. Wang and X. Guo, J. Mol. Liq., 2020, 319, 114354 CrossRef CAS.
- B. Dong, N. Li, L. Zheng, L. Yu and T. Inoue, Langmuir, 2007, 23, 4178–4182 CrossRef CAS.
- T. Inoue, H. Ebina, B. Dong and L. Zheng, J. Colloid Interface Sci., 2007, 314, 236–241 CrossRef CAS PubMed.
- B. Dong, X. Zhao, L. Zheng, J. Zhang, N. Li and T. Inoue, Colloids Surf., A, 2008, 317, 666–672 CrossRef CAS.
- T. Bleasdale, G. Tiddy and E. Wyn-Jones, J. Phys. Chem., 1991, 14, 5385–5386 CrossRef.
- F. Neve, O. Francescangeli and A. Crispini, Inorg. Chim. Acta, 2002, 338, 51–58 CrossRef CAS.
- M. Antonietti, D. B. Kuang, B. M. Smarsly and Z. Yong, Angew. Chem., Int. Ed., 2004, 43, 4988–4992 CrossRef CAS.
- C. J. Adams, A. E. Bradley and K. R. Seddon, Aust. J. Chem., 2001, 54, 679–681 CrossRef CAS.
- Y. Zhou and M. Antonietti, Adv. Mater., 2003, 15, 1452–1455 CrossRef CAS.
- T. Wang, H. Kaper, M. Antonietti and B. M. Smarsly, Langmuir, 2007, 23, 1489–1495 CrossRef CAS PubMed.
- J. Shen, W. He and T. Wang, RSC Adv., 2019, 9, 3504–3513 RSC.
- R. Massart and V. Cabuil, J. Chem. Phys., 1987, 84, 967–973 CAS.
- S. E. Khalafalla and G. W. Reimers, IEEE Trans. Magn., 1980, 16, 178–183 CrossRef.
- E. Dubois, J. Chevalet and R. Massart, J. Mol. Liq., 1999, 83, 243–254 CrossRef CAS.
- J. Wang, Q. Chen, C. Zeng and B. Hou, Adv. Mater., 2004, 16, 137–140 CrossRef CAS.
- D. Wang, C. Cao, S. Xue and H. Zhu, J. Cryst. Growth, 2005, 277, 238–245 CrossRef CAS.
- Q. Huo, D. I. Margolese and G. D. Stucky, Chem. Mater., 1996, 8, 1147–1160 CrossRef CAS.
- C. K. Krishnan, T. Hayashi and M. Ogura, Adv. Mater., 2008, 20, 2131–2136 CrossRef.
- V. Alfredsson and M. W. Anderson, Chem. Mater., 1996, 8, 1141–1146 CrossRef CAS.
- A. A. Romero, M. D. Alba, W. Zhou and J. Klinowski, J. Phys. Chem., 1997, 101, 5294–5300 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- J. N. Israelachvili, D. J. Mitchell and B. W. Ninham, J. Chem. Soc., Faraday Trans., 1976, 272, 1525–1568 RSC.
- J. C. Vartuli, K. D. Schmitt, C. T. Kresge, W. J. Roth, M. E. Leonowicz, S. B. McCullen, S. D. Hellring, J. S. Beck, J. L. Schlenker, D. H. Olson and E. W. Sheppard, Chem. Mater., 1994, 6, 2317–2326 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj04958a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |