
First representatives of C-glycosyl 1,2,4,5-tetrazines: synthesis of 3-β-D-glucopyranosyl 1,2,4,5-tetrazines and their transformation into 3-β-D-glucopyranosyl pyridazines†
Éva
Bokor
 *,
Dóra T.
Kecskés
,
Ferenc
Gombás
,
Alexandra
Fehér
,
Eszter
Kardos
,
Akram
Dabian
,
Zsófia
Vonza
,
Eszter
Szennyes
and
László
Somsák
*
*,
Dóra T.
Kecskés
,
Ferenc
Gombás
,
Alexandra
Fehér
,
Eszter
Kardos
,
Akram
Dabian
,
Zsófia
Vonza
,
Eszter
Szennyes
and
László
Somsák
*
Department of Organic Chemistry, University of Debrecen, H-4002 Debrecen, POB 400, Hungary. E-mail: bokor.eva@science.unideb.hu; somsak.laszlo@science.unideb.hu
First published on 7th November 2022
Abstract
1,2,4,5-Tetrazines (s-tetrazines) are a long known class of compounds with many applications e.g. in heterocyclic syntheses and recently in bioorthogonal chemistry. C-Glycopyranosyl tetrazines are unknown in the literature, therefore, we have started to study their synthesis. In this paper ring closing reactions leading to s-tetrazines have been investigated with suitable β-D-glucopyranosyl precursors and the feasible transformations have been identified. In addition, the obtained C-glucopyranosyl tetrazines’ basic protecting group compatibility and their utility in inverse electron demand Diels–Alder cycloadditions towards a variety of C-glucopyranosyl pyridazines have been demonstrated.
Introduction
1,2,4,5-Tetrazines, also called s-tetrazines, have been known since the pioneering synthetic work of Adolf Pinner described more than a hundred years ago.1 For the preparation of 1,2,4,5-tetrazine derivatives (Scheme 1, I) basically two main synthetic approaches exist.2–6 One of them aims at the construction of the 1,2,4,5-tetrazine ring itself via the formation of a 1,4-dihydrotetrazine intermediate obtained by cyclocondensation of electrophilic carbon precursors (e.g. nitriles, iminoesters, carboxamidines, 1,2-bis-chloroalkylidene-hydrazines) with hydrazine, followed by an oxidative aromatization (Type A). The other approach is focused on various replacements of substituents of pre-formed s-tetrazines (Type B). These transformations are based on nucleophilic substitutions or metal-catalyzed cross-couplings (e.g. Sonogashira, Negishi, Suzuki, Liebeskind–Srogl), wherein s-tetrazines having an exchangeable group (e.g. chloride, alkylsulfanyl) are functionalized with a desired substituent to reach higher structural diversity.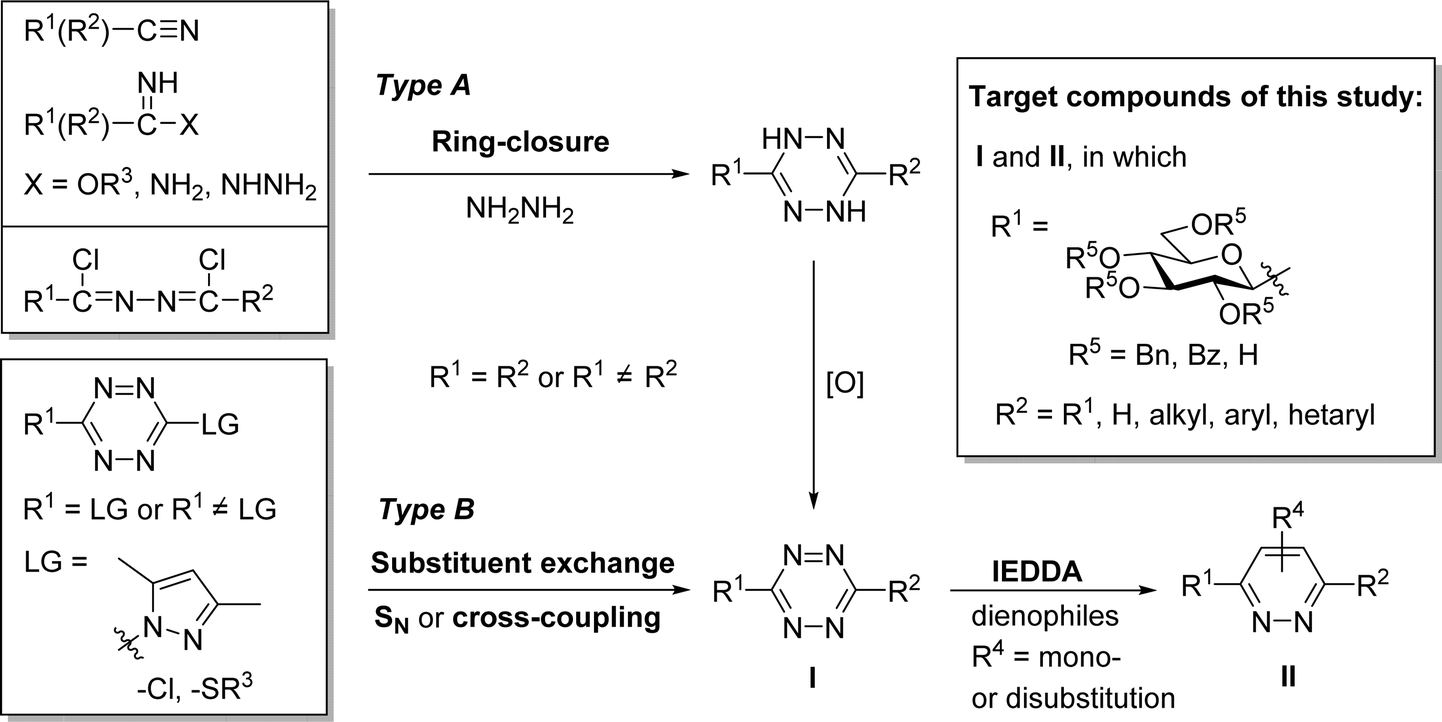 | ||
| Scheme 1 Synthetic methods for the preparation of s-tetrazines and target compounds of this work. | ||
The synthetic importance of these heterocyclic compounds lies in their applicability in ring-transformation reactions. Possessing a –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NC– structural motif within the ring they are able to react as azadienes both in [4+1] and [4+2] cycloadditions, thereby providing access to further N-heterocycles, such as pyrazoles, 1,2,4-triazines and pyridazines.7–10
N–NC– structural motif within the ring they are able to react as azadienes both in [4+1] and [4+2] cycloadditions, thereby providing access to further N-heterocycles, such as pyrazoles, 1,2,4-triazines and pyridazines.7–10
A special type of [4+2] cycloadditions, the strain-promoted variant of the inverse electron-demand Diels–Alder reactions (IEDDA) of 1,2,4,5-tetrazines has received the greatest attention in recent years. Due to the extremely fast catalyst-free kinetics and biocompatibility supplemented with outstanding selectivity, such transformations have emerged as the most powerful bioorthogonal reactions.11–15 The bioorthogonal manipulations in general,13,16–18 involving IEDDAs in particular14,19–21 appear to be new tools for mapping out physiological or pathological events by labeling of biomolecules. Since carbohydrates play pivotal roles in numerous metabolic and recognition processes, the labeling of proteins and glycans or their conjugates (glycoproteins, glycolipids etc.) via sugar-derived precursors has become a reasonable concept in this line.14,20,22–24 The IEDDA-assisted carbohydrate labeling is usually based on the use of a sugar-derived chemical reporter, functionalized with a dienophile (e.g. terminal alkene, cyclopropene, norbornene, cyclooctene, bicyclononyne), and its subsequent visualization triggered by the conjugation with an s-tetrazine suitable for imaging.14,20 As an alternative, the interchangeability of the reactive functional groups has also been demonstrated in a few cases. Thus, the introduction of a tetrazine moiety into carbohydrate building blocks via various linkers was performed, and subsequent IEDDA with appropriate dienophiles led to the formation of the desired sugar conjugates.25–27
Despite the growing interest in variously functionalized s-tetrazines for bioorthogonal11–15,20,21 and other applications2,3,6 there is no example in the literature for the preparation of C-glycosyl 1,2,4,5-tetrazines. Therefore, we set out to investigate the possibilities for the synthesis of this hitherto unknown compound class.
Previously, we prepared several 2,6-anhydro-aldonic acid derivatives (e.g. esters, nitriles, amidines) and used them as starting materials for the synthesis of a variety of C-glycopyranosyl azoles28–30 and pyrimidines.31–33
In the present study, a series of investigations to get C-glucopyranosyl 1,2,4,5-tetrazines (I in Scheme 1) by ring-closing reactions of the above type glucose-based precursors has been envisaged. The main goals of the work have been (i) to evaluate the applicability of several s-tetrazine ring forming reactions with glucose derived starting compounds, and (ii) to demonstrate the tolerance towards basic O-protection/deprotection reactions, an important issue in carbohydrate chemistry, with ether and ester protected as well as unprotected glucopyranosyl precursors. In addition, some IEDDA reactions of the obtained 1,2,4,5-tetrazines to explore their synthetic potential for the synthesis of barely represented 3-C-glucopyranosylated pyridazines (II) have also been examined.
Results and discussion
For the preparation of a 3-monosubstituted 1,2,4,5-tetrazine the Pinner type cyclocondensation of a mixture of a nitrile and formamidine with hydrazine, followed by the oxidation of the resulting 1,4-dihydrotetrazine is one of the most common methods. In such reactions sulfur is an often used additive to facilitate the ring-formation by increasing the nucleophilicity of the hydrazine.3Thus, O-perbenzylated β-D-glucopyranosyl cyanide341 and formamidine hydrochloride were treated with an excess of hydrazine monohydrate in the presence of S8 at rt in DMF to get intermediate 2, whose oxidation by in situ generated nitrous acid furnished the O-perbenzylated 3-(β-D-glucopyranosyl)-1,2,4,5-tetrazine 3 in high yield (Scheme 2). A similar reaction with benzamidine hydrochloride to get 3-glucosyl-6-phenyl-1,2,4,5-tetrazine resulted in a mixture of each possible symmetric and asymmetric s-tetrazines and other unknown by-products, from which the target heterocycle could not be obtained in satisfactory purity and yield.
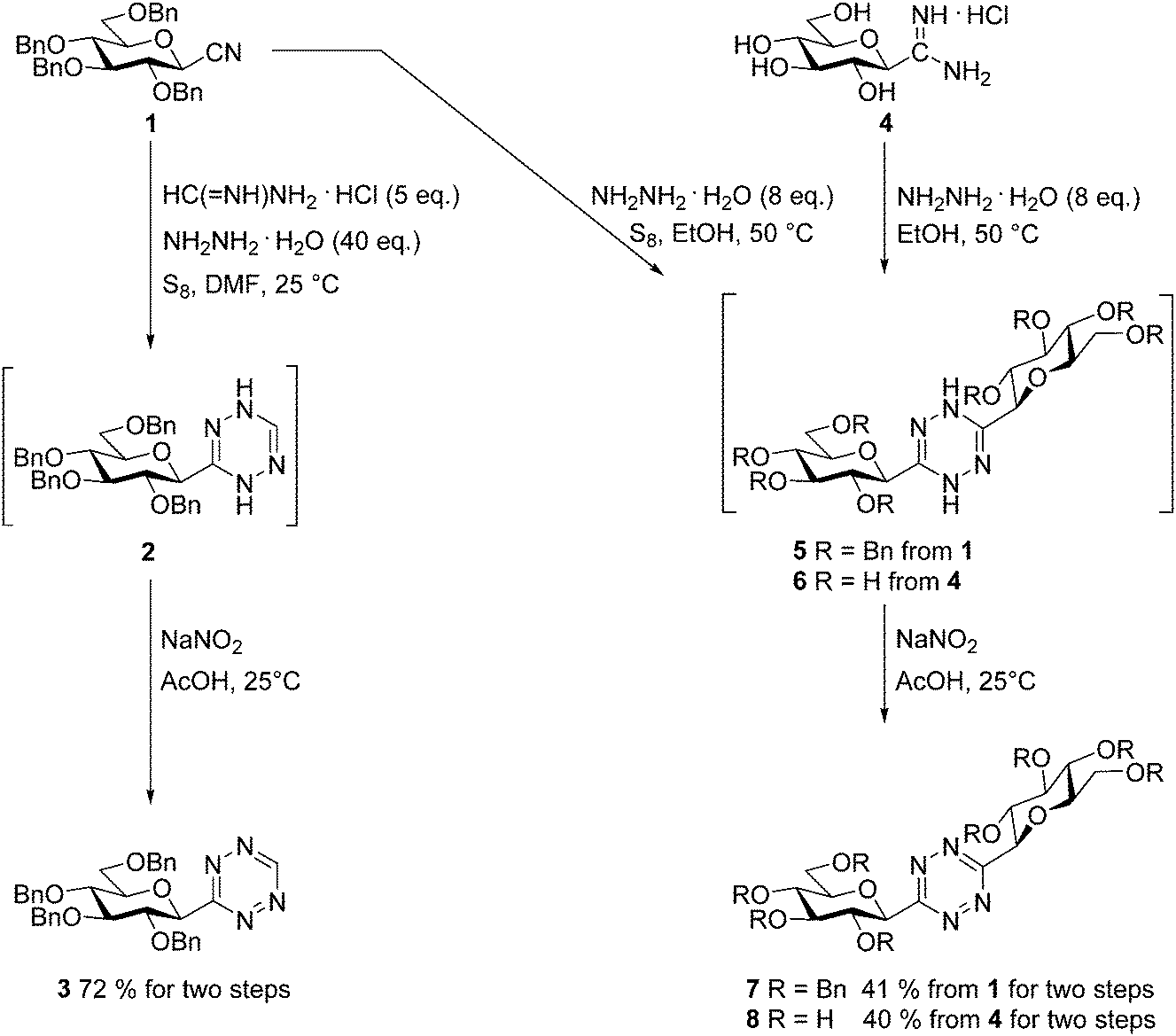 | ||
| Scheme 2 Synthesis of 3-mono- and 3,6-bis-glucopyranosylated 1,2,4,5-tetrazines. | ||
To get 3,6-bis-glucosyl-1,2,4,5-tetrazines we also turned to Pinner type transformations (Scheme 2). Thermal reaction of cyanide 1 with an excess of hydrazine monohydrate assisted by S8 in EtOH and subsequent oxidation of the resulting dihydrotetrazine 5 gave the desired symmetrically diglucopyranosylated 1,2,4,5-tetrazine 7 in O-perbenzylated form. The unprotected derivative 8 was also achieved from the hydrochloride salt of C-β-D-glucopyranosyl formamidine314via the corresponding dihydrotetrazine 6 in good yield, without the need of using sulfur.
The Pinner type reactions of nitriles were primarily restricted for the preparation of aromatic mono- and symmetrically substituted s-tetrazines for a long time. One reason for this is that aliphatic nitriles are generally not reactive enough for such transformations. On the other hand, the good-yielding synthesis of unsymmetrical 3,6-disubstituted s-tetrazines from two different nitriles is hampered by the unavoidable formation of symmetrically disubstituted s-tetrazines.4,5
To overcome these issues, Devaraj and co-workers introduced the use of Lewis acid catalysts (Zn(II) and Ni(II) triflates) for the activation of the electrophilic carbon centre of the nitriles towards nucleophilic attack of the hydrazine.35 With this modification and the use of anhydrous hydrazine the preparation of a series of symmetric 3,6-dialkyl- (24–95% yields) and 3-methyl-6-substituted-1,2,4,5-tetrazines (36–70%, with a 10-fold excess of acetonitrile in the latter cases) was carried out. 3-Benzyl-6-pentyl-1,2,4,5-tetrazine, the only example with a higher homologous alkyl group, was isolated in 12% yield only.35
Based on these preliminaries, we have investigated similar reactions for the preparation of 3-(β-D-glucopyranosyl)-6-substituted-1,2,4,5-tetrazines. First, the reaction of glucosyl cyanide 1 with acetonitrile was examined (Table 1, entry 1). An important modification of the original procedure has been the use of the less hazardous hydrazine monohydrate instead of anhydrous hydrazine. In addition, a higher excess of acetonitrile was used to completely avoid the formation of 3,6-bis-glucosyl-1,2,4,5-tetrazine consuming the glucosyl cyanide. These alterations in the cyclisation step proved to be efficient and furnished a high-yield of the desired heterocycle 19 after the oxidative treatment.
|
|
|||
|---|---|---|---|
| Entry | R | Product | Yield for two steps from 1 (%) |
| Conditions: (i) Compound 1 (0.36 mmol), 2 mL of R-CN, NH2NH2·H2O (25 eq.), Zn(OTf)2 (0.25 eq.), 70 °C, Ar atm.; (ii) NaNO2, H2O, AcOH, 0 °C. | |||
| 1 | –CH3 | 19 | 62 |
| 2 | –CH2CH3 | 20 | 61 |
| 3 |
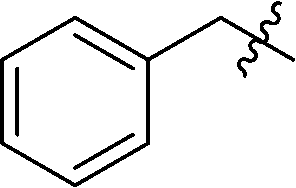
|
21 | 53 |
| 4 |
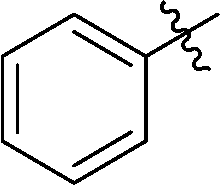
|
22 | 51 |
| 5 |
|
23 | 46 |
| 6 |
|
24 | 42 |
| 7 |
|
25 | 45 |
| 8 |
|
26 | 27 |
| 9 |
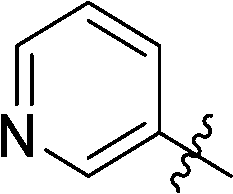
|
27 | 25 |
| 10 |
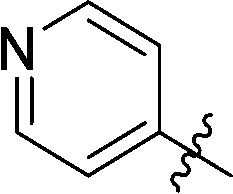
|
28 | 18 |
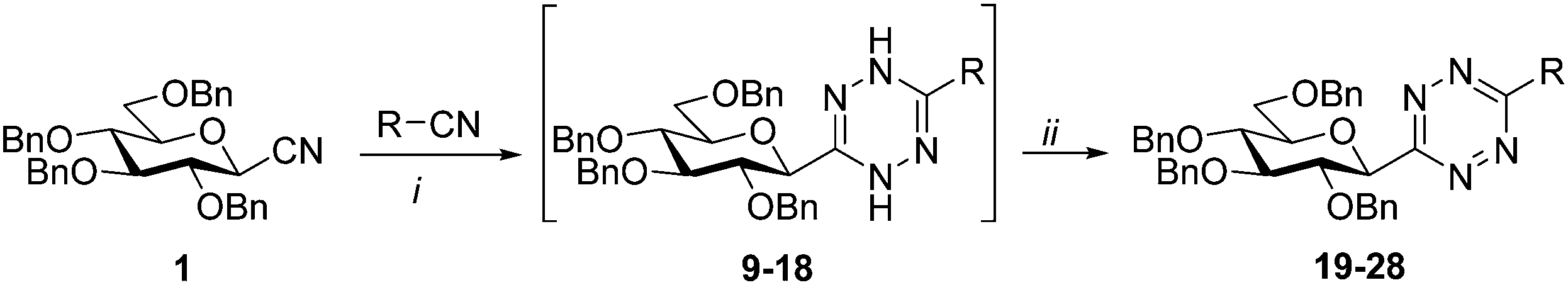
Next, the applicability of the above method for the synthesis of variously substituted 3-glucosyl 1,2,4,5-tetrazines was studied, which proved suitable for the formation of a set of compounds constructed from 1 and liquid or low melting nitriles (Table 1). The two-step procedures starting from cyanide 1 and aliphatic (entries 1–3) and aromatic nitriles (entries 4–7) were smoothly accomplished providing the target unsymmetrical s-tetrazines 20–25 in good yields. Pyridyl-substituted 3-glucosyl-1,2,4,5-tetrazines (26–28) could also be obtained, albeit in lower yields (entries 8–10). The symmetric 3,6-disubstituted-1,2,4,5-tetrazines derived from the reactant nitriles were formed in each reaction as expected, could, however, be separated from the target glucose derivatives by crystallization and/or column chromatography. Fortunately, under the applied reaction conditions, the undesirable transformation of cyanide 1 into 3,6-bis-glucosyl-1,2,4,5-tetrazine 7 could be completely avoided or highly suppressed (in some cases traces were detectable but could not be isolated). Such reactions with chloroacetonitrile, trichloroacetonitrile, pentafluorobenzonitrile as well as 2-cyanopyrimidine were also attempted, however, the formation of the desired 3-glucosyl-6-substituted-1,2,4,5-tetrazines was observed in neither case.
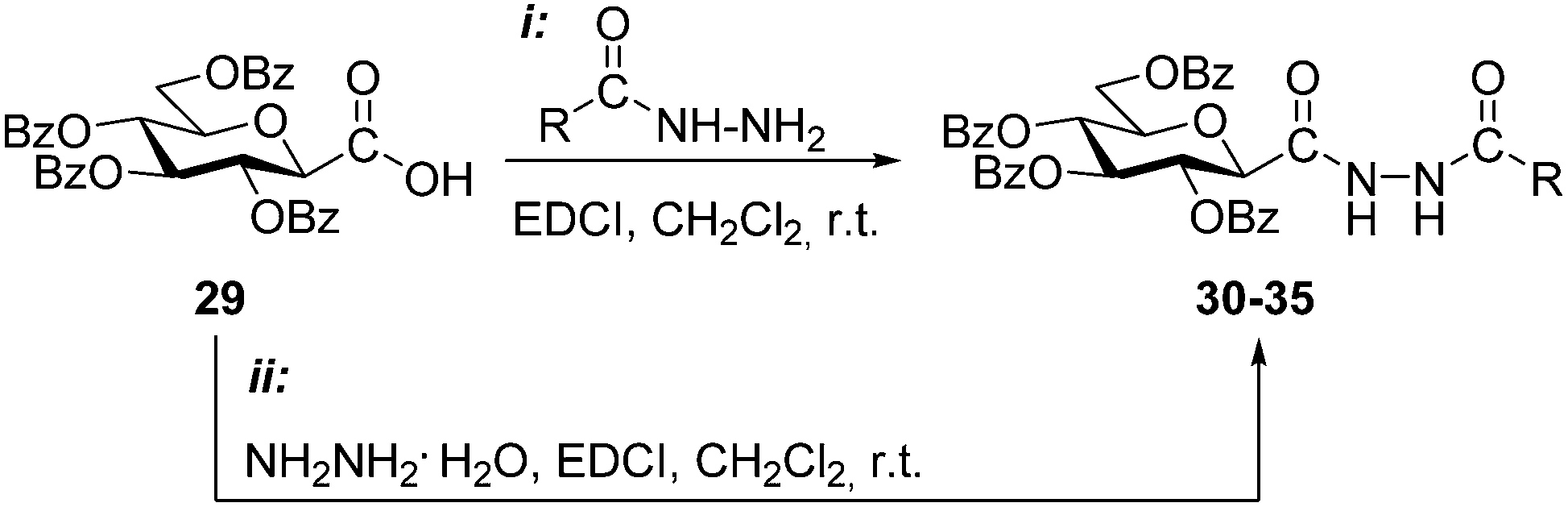
Among synthetic possibilities based on ring-formation, the cyclisation of 1,2-bis-chloroalkylidene-hydrazines with hydrazine provides another alternative to access unsymmetrically disubstituted 1,4-dihydrotetrazine intermediates.4,5 In order to study this approach a set of O-perbenzoylated N-(β-D-glucopyranosylcarbonyl)-N′-acyl hydrazines as precursors was first prepared (Table 2). The 2,6-anhydro-aldonic acid 29 was treated with carboxylic acid hydrazides in the presence of EDCI resulting in the N,N′-diacyl hydrazines 30–34 in moderate to high yields. In addition, N,N′-bis(β-D-glucopyranosylcarbonyl)hydrazine 35 was also obtained by the reaction of the same acid 29 with hydrazine monohydrate.
|
|
|||
|---|---|---|---|
| Conditions | R | Product | Yield (%) |
| i | –H | 30 | 71 |
| i | –CH3 | 31 | 65 |
| i | –CF3 | 32 | 51 |
| i |
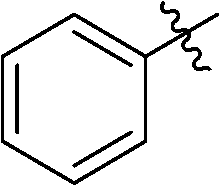
|
33 | 85 |
| i |
|
34 | 85 |
| ii |
|
35 | 43 |
By the adaptation of a literature procedure36 the transformation of the synthesized N,N′-diacyl hydrazines 30–35 into C-glucosyl 1,2,4,5-tetrazines via the corresponding 1,2-bis-chloroalkylidene-hydrazines was then investigated (Scheme 3). Chlorination of the N′-benzoyl hydrazine 33 with PCl5 was smoothly effected and the desired bis-imidoyl chloride 36 could be isolated in excellent yield. Treatment of 36 with hydrazine monohydrate and subsequent oxidation of the resulting 1,4-dihydrotetrazine 37 by nitrosation furnished the desired 6-phenyl substituted s-tetrazine 38 in high overall yield (72% for three steps). The same chlorination of N′-picolinyl hydrazine 34 provided a less stable intermediate 39, therefore, this was subjected to the subsequent cyclisation–oxidation sequence without isolation. The target 6-(2-pyridyl)-1,2,4,5-tetrazine 41 was formed accompanied with the difficult-to-separate 2-(2′,3′,4′,6′-tetra-O-benzoyl-β-D-glucopyranosyl)-5-(2-pyridyl)-1,3,4-oxadiazole37 (40). In case of the chlorination of the N-trifluoroacetyl hydrazine derivative 32 the formation of the expected 1,2-bis-chloroalkylidene-hydrazines was not observed, this reaction led directly and exclusively to the formation of the 5-trifluoromethyl-1,3,4-oxadiazole3842. Our attempt to convert this oxadiazole to the desired s-tetrazine by a ring-transformation10 also failed, as heating of 42 with 20 equiv. of hydrazine monohydrate in DMF, followed by oxidation resulted in a multicomponent reaction mixture. In addition, the chlorination–cyclisation–oxidation sequence was also tried with other N,N′-diacyl hydrazines (30, 31 and 35), but none of the target 6-substituted-1,2,4,5-tetrazines could be identified in satisfactory amount in the resulting multicomponent reaction mixtures.
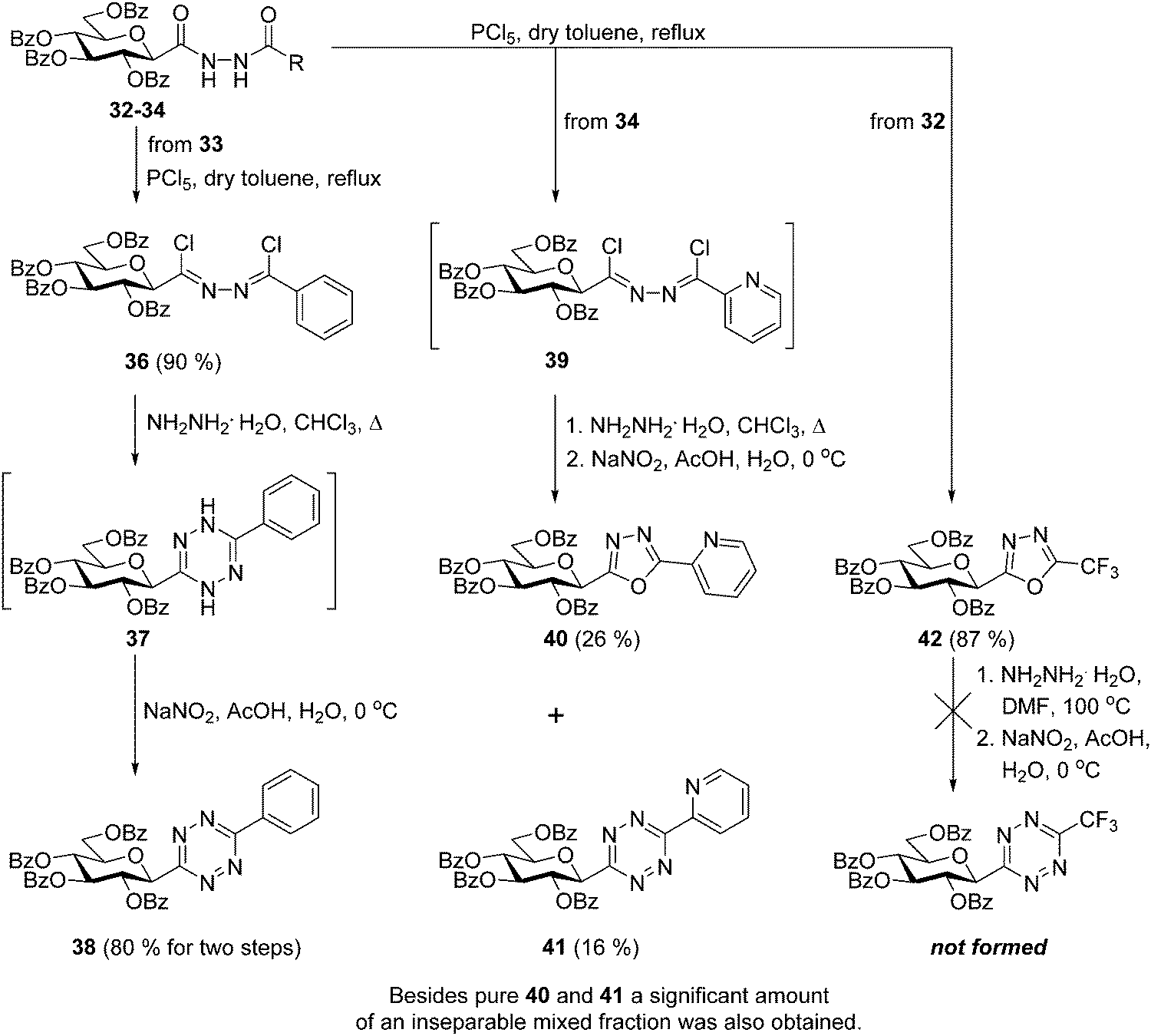 | ||
| Scheme 3 Investigation of the synthesis of O-perbenzoylated 3-(β-D-glucopyranosyl)-6-substituted-1,2,4,5-tetrazines starting from N-(β-D-glucopyranosylcarbonyl)-N′-acyl hydrazines. | ||
We have also investigated the stability of the heteroaromatic moiety of the newly synthesized C-glucosyl 1,2,4,5-tetrazines under standard O-protection–deprotection conditions often used in carbohydrate chemistry.39 Therefore, a set of experiments in this direction was also performed (Scheme 4). Lewis acid mediated O-debenzylation of tetrazines 3 and 19 gave the corresponding unprotected glucosyl s-tetrazines 43 and 44, respectively, in moderate to high yields. O-Debenzoylation of tetrazine 38 under Zemplén conditions resulted in the corresponding O-deprotected derivative 45 in good yield. A direct exchange of the benzyl protecting groups to benzoyl groups was also carried out by a Lewis acid mediated reaction40 of 19 with benzoyl chloride, affording the O-perbenzoylated analog 46 in moderate yield. The latter compound was also achieved in a two-step procedure via the unprotected derivative 44 and this route (19 → 44 → 46) proved to be more efficient resulting in a 77% combined yield.
 | ||
| Scheme 4 O-Protections-deprotections of the synthesized C-glucopyranosyl 1,2,4,5-tetrazines. | ||
To demonstrate the synthetic utility of the prepared 3-glucopyranosyl-1,2,4,5-tetrazines, we have carried out their transformations into 3-glucopyranosyl pyridazines. C-Glycosyl pyridazines are known in the literature, nevertheless, mostly with a glycofuranosyl sugar part and, to the best of our knowledge, only a single 3-glucopyranosyl pyridazine has been described as a candidate SGLT2 inhibitor.41 This latter compound was obtained in a multistep procedure via O-perbenzylated glucopyranosyl ethene obtained from the corresponding gluconolactone.41 Among C-glycofuranosyl pyridazines the 3-glycosyl derivatives were prepared from 2-glycosyl furanes, which were oxidatively transformed to a 1,4-dicarbonyl compound42 or its equivalents43–46 to be ring-closed by hydrazine to the corresponding pyridazine. A 3-glycofuranosyl isoxazoline was also used as the starting material for a similar synthesis of 3-glycosyl pyridazines.47 The more frequent 4-glycosyl pyridazines were mostly obtained by cycloadditions of glycofuranosyl ethynes with 3,6-disubstituted-1,2,4,5-tetrazines,48–53 and similar reactions of glycopyranosyl ethynes are also known.52 In addition, a 3-glycofuranosyl furane → 1,4-dicarbonyl compound → ring closure by hydrazine route towards 4-glycosyl pyridazines was also described.54,55
For the preparation of 3-glucopyranosyl-5-substituted pyridazines inverse electron-demand Diels–Alder reactions (IEDDA) of 3-glucopyranosyl-1,2,4,5-tetrazine 3 with terminal alkynes was investigated first (Table 3). The ring-transformation of 3 with propargyl acetate, N-Boc-propargylamine, phenylacetylene and 2-ethynylpyridine in boiling m-xylene took place regioselectively, resulting in the expected 5-substituted pyridazines 47–50, respectively, in good to excellent yields. The position of the R substituent in 47–50 was determined by 1H NMR. In the spectra of 47–50 a characteristic doublet for H-6 of the pyridazine moiety appeared in the range of 9.75–9.00 ppm with a small coupling constant (J = 1.6–2.3 Hz) indicating the meta position of the pyridazine protons. Preferred formation of the 3-glucosyl-5-substituted-1,2,4,5-tetrazines over the isomeric 3-glucosyl-4-substituted derivatives may be explained by a sterically less crowded [4+2] cycloaddition transition state of lower energy.
|
|
|||
|---|---|---|---|
| R | Reaction time (h) | Product | Yield (%) |
| –CH2OAc | 12 | 47 | 89 |
| –CH2NHBoc | 10 | 48 | 81 |
|
24 | 49 | 60 |
|
10 | 50 | 97 |
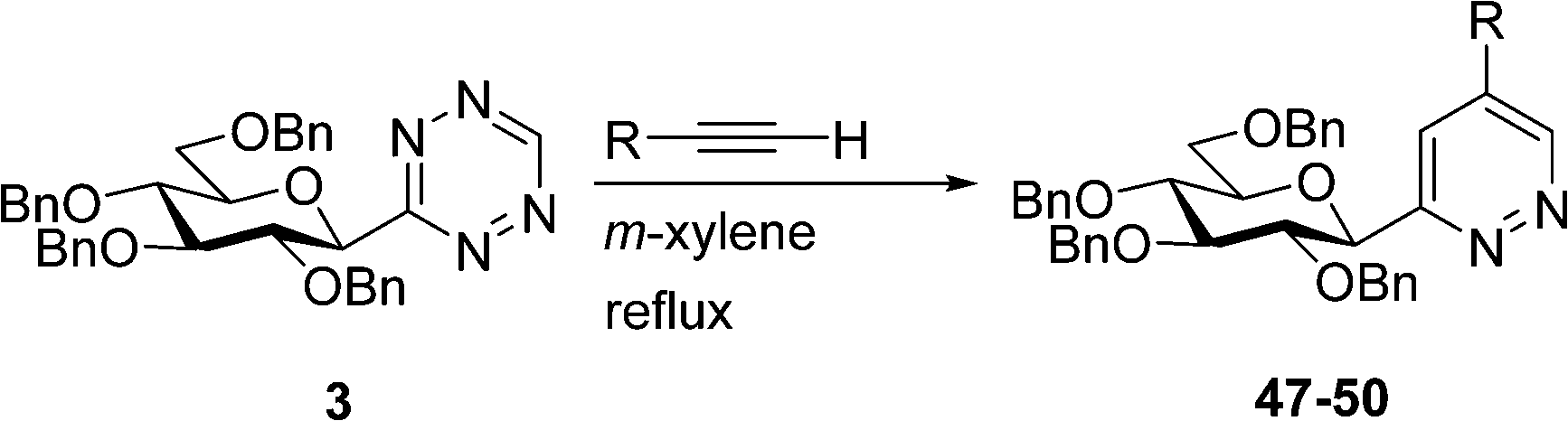
A high-yielding synthesis of two annelated 3-glucosyl pyridazines (51 and 52) was also performed by IEDDA reactions of tetrazine 3 with 1-pyrrolidino-1-cycloalkenes in m-xylene (Scheme 5) to demonstrate the applicability of other electron rich dienophiles.
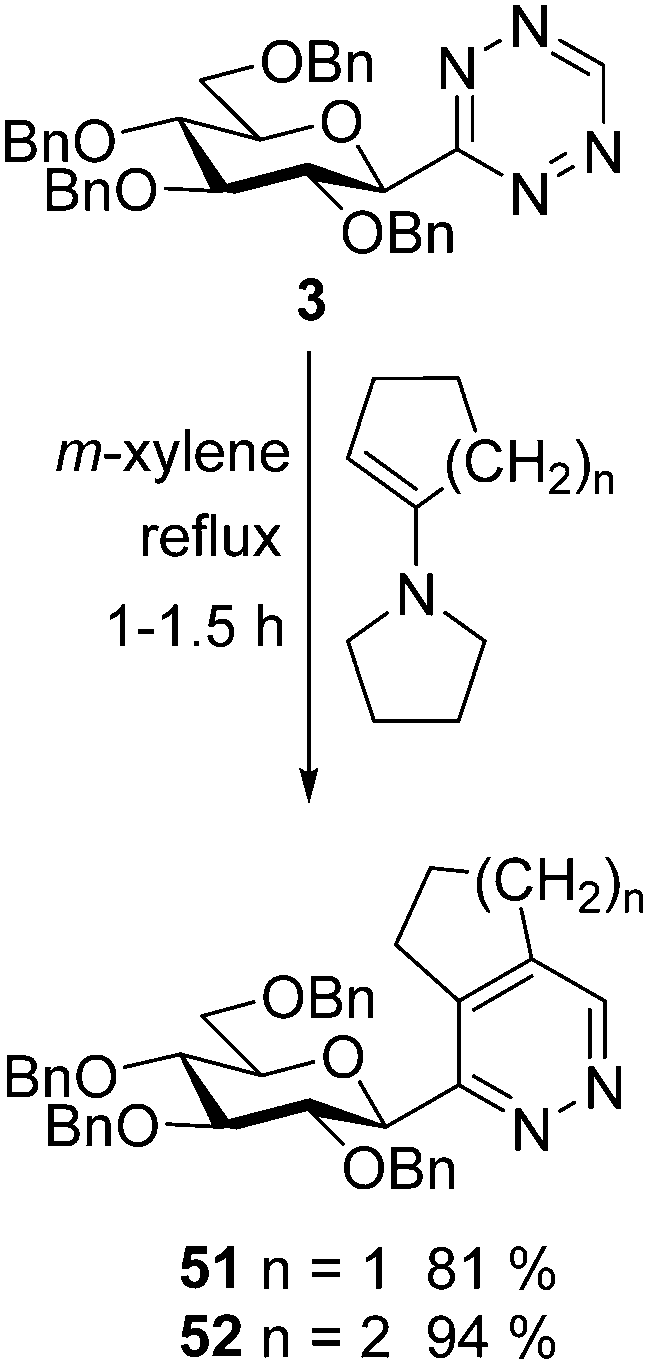 | ||
| Scheme 5 Synthesis of annelated 3-β-D-glucopyranosyl pyridazines. | ||
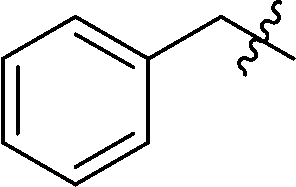
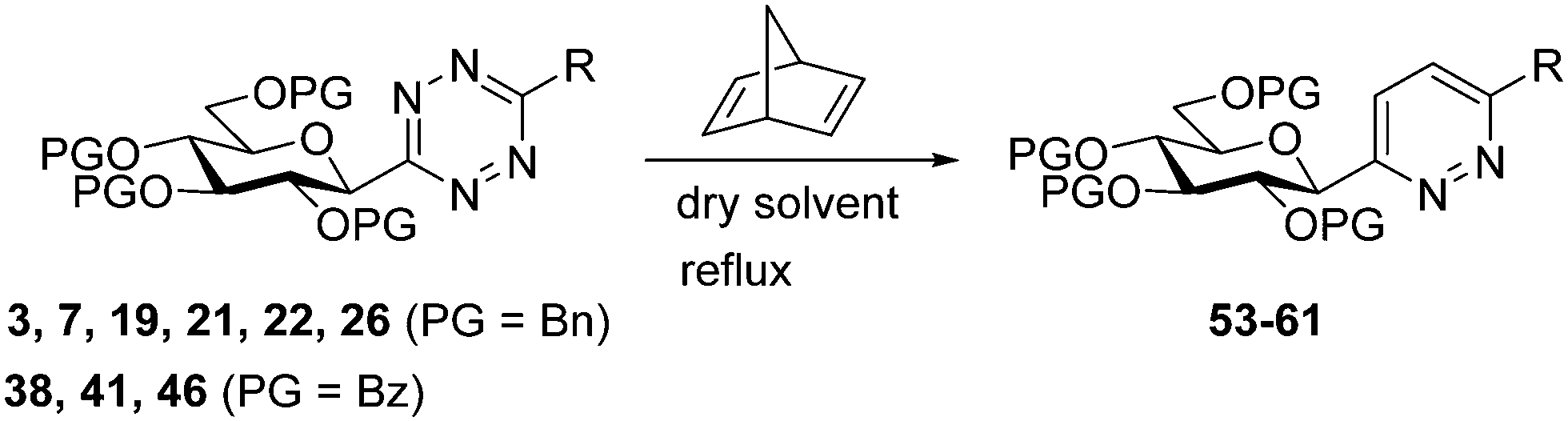
In addition, several O-protected 3-glucopyranosyl-6-substituted pyridazines were also obtained by the reaction of selected 3-glucopyranosyl-6-substituted-1,2,4,5-tetrazines (3, 7, 19, 21, 22, 26, 38, 41, 46) with 2,5-norbornadiene (Table 4). The conversion of these s-tetrazines was carried out in boiling toluene, or in some cases in dichloromethane. Under the applied reaction conditions, the [4+2] cycloadditions were accompanied by the elimination of cyclopenta-1,3-diene, and the desired pyridazines 53–61 were formed in high yields (Table 4). Among the O-perbenzylated 3-glucosyl s-tetrazines (3, 7, 19, 21, 22, 26), the sterically least crowded 3-glucosyl-1,2,4,5-tetrazine 3 and the most electron deficient 6-(2-pyridyl)-1,2,4,5-tetrazine 26 proved to be the most reactive in toluene (entries 2, 9 vs. entries 3, 5, 6, 11), and could also be converted into the appropriate pyridazines 53 and 59, respectively, in dichloromethane (entries 1 and 8). A comparison of the reaction times of the O-perbenzylated and O-perbenzoylated 3-glucosyl-1,2,4,5-tetrazine analogs (19vs.46 in entries 3 and 4, and 22vs.38 in entries 6 and 7) shows that the electron withdrawing ester protection of the sugar part accelerates the reaction. In the whole series, tetrazine 41 with an O-perbenzoylated sugar part and a 2-pyridyl substituent proved to be the most reactive, whose transformation into pyridazine 60 could be achieved even in dichloromethane in a very short reaction time (entry 10).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Tetrazine | PG | R | Solvent | R. time | Product | Yield (%) |
| 1 | 3 | Bn | –H | CH2Cl2 | 6 h | 53 | 72 |
| 2 | Toluene | 15 min | 72 | ||||
| 3 | 19 | Bn | –CH3 | Toluene | 3 h | 54 | 85 |
| 4 | 46 | Bz | –CH3 | Toluene | 30 min | 55 | 90 |
| 5 | 21 | Bn |
|
Toluene | 6 h | 56 | 83 |
| 6 | 22 | Bn |
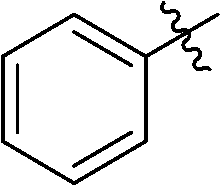
|
Toluene | 4 h | 57 | 90 |
| 7 | 38 | Bz |
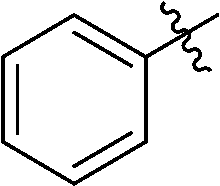
|
Toluene | 2 h | 58 | 70 |
| 8 | 26 | Bn |
|
CH2Cl2 | 1.5 h | 59 | 85 |
| 9 | Toluene | 10 min | 90 | ||||
| 10 | 41 | Bz |
|
CH2Cl2 | 30 min | 60 | 94 |
| 11 | 7 | Bn |
|
Toluene | 1.5 h | 61 | 83 |
Conclusion
In this work we have studied s-tetrazine syntheses to establish feasible methods to get C-glucopyranosyl 1,2,4,5-tetrazines for the first time. The transformation of the precursors gave the corresponding 1,4-dihydro-1,2,4,5-tetrazines as intermediates, which were not isolated but immediately oxidized to the target tetrazines, yields thus refer to these two-step sequences. O-Perbenzylated β-D-glucopyranosyl cyanide and O-unprotected C-(β-D-glucopyranosyl)formamidine were converted to the corresponding 3-mono- and 3,6-bis-glucopyranosylated 1,2,4,5-tetrazines, respectively, by hydrazine monohydrate in the presence of elemental sulfur with good to excellent yields. The O-perbenzylated β-D-glucopyranosyl cyanide gave the expected unsymmetrical 3-(β-D-glucopyranosyl)-6-substituted-1,2,4,5-tetrazines in good yields by Lewis acid catalyzed Pinner reactions with several liquid and low-melting nitriles. A series of N-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosylcarbonyl)-N′-acyl hydrazines were converted to the corresponding 1,2-bis-chloroalkylidene-hydrazines which were subsequently treated by hydrazine monohydrate. From these reactions only the 3-(β-D-glucopyranosyl)-6-phenyl-1,2,4,5-tetrazine could be isolated in excellent yield, however, all other reactions failed by resulting in 1,3,4-oxadiazole type by-products or inseparable product mixtures. The obtained C-glucopyranosyl tetrazines are compatible with basic protecting group manipulations (O-debenzylation, O-debenzoylation and O-benzoylation). Reactions of the C-glucopyranosyl tetrazines with terminal alkynes, norbornadiene and 1-pyrrolidino-1-cycloalkenes gave the corresponding 3-(β-D-glucopyranosyl)-5- and 6-substituted pyridazine and 4,5-alkylene pyridazine derivatives, respectively. Thus, the synthesis of the first representatives of hitherto unknown C-glycopyranosyl 1,2,4,5-tetrazines and their synthetic utility has been demonstrated. This can pave the way towards a new type of carbohydrate labeling of biomolecules, and in this direction further studies to increase the diversity of C-glycopyranosyl 1,2,4,5-tetrazines and their utility as bioorthogonal labeling agents are in progress and will be published in due course.Experimental
General methods
The melting points were measured on a Kofler hot-stage instrument and are uncorrected. Optical rotations were measured by a Jasco P-2000 or a PerkinElmer 241 polarimeter at r.t. NMR spectra were recorded with Bruker DRX360 (360/90 MHz for 1H/13C) and DRX400 (400/100 MHz for 1H/13C) spectrometers. Chemical shifts were referenced to internal Me4Si (1H-NMR) or to the residual solvent signals (13C-NMR). HRMS data were measured by a Bruker maXis II instrument in positive ionization mode. TLC analysis was performed on DC Kieselgel 60 F254 plates (Sigma-Aldrich). The spots were checked under UV light and were developed by gentle heating. For column chromatographic purifications Kieselgel 60 silica gel (Molar Chemicals, particle size 0.063–0.2 mm) was applied. Anhydrous EtOH and pyridine were purchased from VWR Chemicals. Other anhydrous solvents were freshly prepared following standard distillation procedures: CH2Cl2, CHCl3, toluene and m-xylene were obtained by distillation from P4O10 and stored then over 4 Å molecular sieves (CH2Cl2, CHCl3) or sodium wires (toluene, m-xylene). MeOH was distilled over Mg turnings and iodine. 2,3,4,6-Tetra-O-benzyl-β-D-glucopyranosyl cyanide34 (1), C-(β-D-glucopyranosyl)formamidine hydrochloride31 (4) and C-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)formic acid56 (29) were prepared following procedures worked out in our laboratory.General procedure I for the synthesis of 6-substituted-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-1,2,4,5-tetrazines (19–28) from 2,3,4,6-tetra-O-benzyl-β-D-glucopyranosyl cyanide 1
In an ace pressure tube, 2,3,4,6-tetra-O-benzyl-β-D-glucopyranosyl cyanide34 (1, 0.2 g, 0.36 mmol), the corresponding nitrile (2 mL) and Zn(OTf)2 (0.09 mmol, 0.25 equiv.) were mixed and hydrazine monohydrate (0.44 mL, 91 mmol, 25 equiv.) was added. The tube was filled with Ar and closed with a screw cap. The reaction mixture was stirred at 70 °C for 10 hours, then at rt overnight. The complete conversion of cyanide 1 was judged by TLC (hexane–EtOAc 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The reaction mixture was then diluted with glacial acetic acid (3 mL) and cooled down in an ice bath. An aqueous solution of NaNO2 (0.5 g in 4 mL of water) was added in two portions in 10 minutes. Gas evolution and change of the colour of the mixture to dark pink was observed. After completion of the oxidation (20–30 min) monitored by TLC (hexane–EtOAc 5:1) the mixture was diluted with CHCl3 (50 mL) and extracted with water (30 mL), saturated aqueous solution of NaHCO3 (2 × 30 mL) and water (30 mL), respectively. The separated organic phase was dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. Column chromatographic purification yielded the target 1,2,4,5-tetrazine.
:1). The reaction mixture was then diluted with glacial acetic acid (3 mL) and cooled down in an ice bath. An aqueous solution of NaNO2 (0.5 g in 4 mL of water) was added in two portions in 10 minutes. Gas evolution and change of the colour of the mixture to dark pink was observed. After completion of the oxidation (20–30 min) monitored by TLC (hexane–EtOAc 5:1) the mixture was diluted with CHCl3 (50 mL) and extracted with water (30 mL), saturated aqueous solution of NaHCO3 (2 × 30 mL) and water (30 mL), respectively. The separated organic phase was dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. Column chromatographic purification yielded the target 1,2,4,5-tetrazine.
General procedure II for the synthesis of N-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosylcarbonyl)-N′-acylhydrazines (30–34)
To a solution of C-(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosyl)formic acid56 (29) in dry CH2Cl2 (3 mL/100 mg acid) the appropriate carboxylic acid hydrazide (1 equiv.) and EDCI (1.2 equiv.) were added. The reaction mixture was stirred at rt and the transformation was monitored by TLC (hexane–EtOAc 2:1). After total conversion of the acid, the mixture was diluted with CH2Cl2 (20 mL) and extracted with brine (2 × 20 mL). The separated organic layer was dried over MgSO4, filtered and the solvent was evaporated in vacuo. The residue was purified by column chromatography.
General procedure III for the synthesis of 5-substituted-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)pyridazines (47–50)
To a solution of tetrazine 3 in m-xylene (3 mL/100 mg tetrazine) the corresponding alkyne (1 equiv.) was added. The reaction mixture was heated at reflux temperature until the TLC (hexane–EtOAc 3:1) showed complete conversion of the tetrazine. After completion of the reaction the solvent was removed under diminished pressure and the crude compound was purified by column chromatography.
General procedure IV for the synthesis of O-protected 3-(β-D-glucopyranosyl)-6-substituted pyridazines (53–61)
To a solution of the corresponding 3-glucopyranosyl-s-tetrazine in dry toluene (1 mL/20 mg tetrazine) 2,5-norbornadiene (2 equiv.) was added. The reaction mixture was refluxed until the TLC (hexane–EtOAc 5:1) showed complete disappearance of the tetrazine. After completion of the reaction the solvent was removed under diminished pressure and the crude compound was purified by column chromatography.
3-(2′,3′,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (3)
2,3,4,6-Tetra-O-benzyl-β-D-glucopyranosyl cyanide34 (1, 2.00 g, 3.64 mmol), formamidine hydrochloride (1.47 g, 18.19 mmol, 5 equiv.) and sulfur (116 mg, 3.64 mmol) were suspended in DMF (10 mL) and hydrazine monohydrate (7.1 mL, 0.146 mol, 40 equiv.) was added. The mixture was stirred at rt and the reaction was monitored by TLC (hexane–EtOAc 3:1). After complete disappearance of the cyanide (1 day) water (20 mL) and 2 M aq. solution of HCl (4 mL) were added to the reaction mixture. The precipitated white solid was filtered off (intermediate 2), then suspended in glacial acetic acid (10 mL). This mixture was placed in an ice bath and NaNO2 (0.50 g, 7.28 mmol) was added. The color of the mixture turned to pink. After 30 minutes the solvent was removed under reduced pressure, then the residue was dissolved in EtOAc (50 mL). The solution was extracted with water (3 × 30 mL), dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. The resulting pink oil was purified by column chromatography (hexane–EtOAc 3:1) to give 1.60 g (72%) pink oil, which could be crystallized from EtOH yielding pink solid. Rf = 0.42 (hexane–EtOAc 3:1). 1H NMR (360 MHz, CDCl3) δ (ppm): 10.14 (1H, s, H-6), 7.38–7.10 (18H, m, Ar), 6.82 (2H, dd, J = 8.0, 1.8 Hz, Ar), 4.99, 4.95 (2 × 1H, 2 d, J = 11.0 Hz in each, PhCH2), 4.92 (1H, d, J = 9.8 Hz, H-1′), 4.88, 4.61 (2 × 1H, 2 d, J = 10.7 Hz in each, PhCH2), 4.74, 4.33 (2 × 1H, 2 d, J = 11.6 Hz in each, PhCH2), 4.55, 4.48 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.31 (1H, pt, J = 9.9, 9.0 Hz, H-2′ or H-3′ or H-4′), 3.97 (1H, pt, J = 9.0, 8.7 Hz, H-2′ or H-3′ or H-4′), 3.83 (1H, pt, J = 9.3, 8.7 Hz, H-2′ or H-3′ or H-4′), 3.78–3.73 (3H, m, H-5′, H-6′a, H-6′b). 13C NMR (90 MHz, CDCl3) δ (ppm): 168.6 (C-3), 159.0 (C-6), 138.4, 138.0, 138.0, 137.5, 128.6–127.8 (Ar), 87.2, 80.5, 80.2, 79.8, 78.1 (C-1′–C-5′), 75.9, 75.4, 74.8, 73.6 (4 × PhCH2), 68.9 (C-6′). ESI-HRMS positive mode (m/z): calcd for C36H37N4O5+ [M + H]+ 605.2758; C36H36N4NaO5+ [M + Na]+ 627.2578. Found: [M + H]+ 605.2752; [M + Na]+ 627.2570.
3,6-Bis-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (7)
In an ace pressure tube, 2,3,4,6-tetra-O-benzyl-β-D-glucopyranosyl cyanide34 (1, 0.20 g, 0.36 mmol) and sulfur (23 mg, 0.73 mmol) were suspended in dry EtOH (3 mL) and hydrazine monohydrate (142 μL, 2.91 mmol, 8 equiv.) was added. The sealed tube was heated at 50 °C for 1 day. The complete transformation of the cyanide was judged by TLC (hexane–EtOAc 4:1). The resulting yellow heterogeneous mixture, which contained intermediate 5, was diluted with CH2Cl2 (3 mL) and cooled down in an ice bath. To this mixture a solution of NaNO2 (0.25 g, 3.64 mmol, 10 equiv.) in glacial acetic acid (1.25 mL) was added under stirring. The color of the mixture turned to pink. After completion of the oxidation monitored by TLC (10 min, hexane–EtOAc 4:1) the reaction mixture was diluted with CH2Cl2 (20 mL) and extracted with water (2 × 10 mL). The organic phase was dried over MgSO4, filtered and the solvent was evaporated under reduced pressure. The residue was purified by column chromatography (hexane–EtOAc 4:1). Yield: 84 mg (41%), pink oil. Rf = 0.18 (hexane–EtOAc 4:1). 1H NMR (360 MHz, CDCl3) δ (ppm): 7.36–6.77 (2 × 20H, m, Ar), 4.99, 4.94 (2 × (2 × 1H), 2 d, J = 11.1 Hz in each, PhCH2), 4.95 (2 × 1H, d, J = 9.7 Hz, H-1′), 4.88, 4.64 (2 × (2 × 1H), 2 d, J = 10.7 Hz in each, PhCH2), 4.70, 4.34 (2 × (2 × 1H), 2 d, J = 11.2 Hz in each, PhCH2), 4.54, 4.46 (2 × (2 × 1H), 2 d, J = 12.1 Hz in each, PhCH2), 4.38 (2 × 1H, pt, J = 9.7, 9.3 Hz, H-2′ or H-3′ or H-4′), 3.98 (2 × 1H, pt, J = 8.9, 8.9 Hz, H-2′ or H-3′ or H-4′), 3.86 (2 × 1H, pt, J = 9.0, 8.9 Hz, H-2′ or H-3′ or H-4′), 3.79–3.73 (2 × 3H, m, H-5′, H-6′a, H-6′b). 13C NMR (90 MHz, CDCl3) δ (ppm): 167.0 (C-3, C-6), 138.3, 137.9, 137.1, 128.4–127.6 (Ar), 87.1, 80.3, 79.8, 79.0, 78.0 (C-1′–C-5′), 75.7, 75.2, 74.6, 73.5 (4 × PhCH2), 68.8 (C-6′). ESI-HRMS positive mode (m/z): calcd for C70H71N4O10+ [M + H]+: 1127.5165; C70H70N4NaO10+ [M + Na]+: 1149.4984. Found: [M + H]+: 1127.5167; [M + Na]+: 1149.4983.
3,6-Bis-(β-D-glucopyranosyl)-1,2,4,5-tetrazine (8)
In an ace pressure tube, C-(β-D-glucopyranosyl)formamidine hydrochloride31 (4, 0.10 g, 0.41 mmol) was suspended in dry EtOH (2 mL) and hydrazine monohydrate (161 μL, 3.30 mmol, 8 equiv.) was added. The sealed tube was heated at 50 °C for one day. The complete transformation of the amidine was checked by TLC (CHCl3–MeOH 2:1). The reaction mixture was cooled down in an ice bath and a solution of NaNO2 (0.29 g, 4.13 mmol, 10 equiv.) in glacial acetic acid (1.5 mL) was added. The color of the mixture turned to pink. The completion of the oxidation step (10 min) was judged by TLC (CHCl3–MeOH 3:2). After completion of the transformation the solvent was evaporated under diminished pressure. The residual pink syrup was purified by column chromatography (CHCl3–MeOH 3:2). Yield: 34 mg (40.5%), pink syrup. Rf = 0.22 (CHCl3–MeOH 3:2). 1H NMR (360 MHz, CD3OD) δ (ppm): 4.91 (2 × 1H, d, J = 9.8 Hz, H-1′), 4.05 (2 × 1H, pt, J = 9.5, 9.2 Hz H-2′ or H-3′ or H-4′), 3.91 (2 × 1H, dd, J = 12.1, 1.3 Hz, H-6′a), 3.74 (2 × 1H, dd, J = 12.1, 5.2 Hz, H-6′b), 3.67 (2 × 1H, pt, J = 9.0, 8.6, H-2′ or H-3′ or H-4′), 3.62–3.58 (2 × 1H, m, H-5′), 3.55 (2 × 1H, pt, J = 9.5, 8.6 Hz, H-2′ or H-3′ or H-4′); 13C NMR (90 MHz, CD3OD) δ (ppm): 169.2 (C-3, C-6), 83.2, 82.0, 79.3, 74.2, 71.6 (C-1′–C-5′), 62.9 (C-6′). ESI-HRMS positive mode (m/z): calcd for C14H22N4NaO10+ [M + Na]+: 429.1228. Found: 429.1228.
6-Methyl-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (19)
Prepared from cyanide 1 (0.2 g, 0.36 mmol) and acetonitrile (2 mL) according to General procedure I. Purified by column chromatography (hexane–EtOAc 5:1) to give 137 mg pink oil (62%). Trituation of the pure product with EtOH yielded 127 mg pink solid. Rf = 0.5 (hexane–EtOAc 5:2). 1H NMR (360 MHz, CDCl3) δ (ppm): 7.37–7.08 (18H, m, Ar), 6.84 (2H, d, J = 6.8 Hz, Ar), 4.99, 4.95 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.90 (1H, d, J = 9.7 Hz, H-1′), 4.88, 4.62 (2 × 1H, 2 d, J = 10.7 Hz in each, PhCH2), 4.75, 4.35 (2 × 1H, 2 d, J = 11.7 Hz in each, PhCH2), 4.55, 4.47 (2 × 1H, 2 d, J = 12.1 Hz in each, PhCH2), 4.34 (1H, pt, J = 8.6 Hz, H-2′ or H-3′ or H-4′), 3.97 (1H, pt, J = 8.9, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.83 (1H, pt, J = 8.8, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.76–3.73 (3H, m, H-5′, H-6′a, H-6′b), 2.99 (3H, s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 168.5, 165.6 (C-3, C-6), 138.3, 137.9, 137.8, 137.5, 128.4–127.3 (Ar), 87.1, 80.2, 79.8, 79.8, 78.0 (C-1′–C-5′), 75.7, 75.1, 74.6, 73.4 (4 × PhCH2), 68.8 (C-6′), 21.3 (CH3). ESI-HRMS positive mode (m/z): calcd for C37H39N4O5+ [M + H]+ 619.2915. Found: 619.2910.
6-Ethyl-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (20)
Prepared from cyanide 1 (0.2 g, 0.36 mmol) and propionitrile (2 mL) according to General procedure I. Purified by column chromatography (hexane–EtOAc 9:1) to give 140 mg pink oil (61%). Rf = 0.38 (hexane–EtOAc 5:1). 1H NMR (360 MHz, CDCl3) δ (ppm): 7.37–7.08 (18H, m, Ar), 6.85–6.82 (2H, m, Ar), 4.99, 4.96 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.91 (1H, d, J = 9.9 Hz, H-1′), 4.88, 4.62 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.76, 4.34 (2 × 1H, 2 d, J = 11.6 Hz in each, PhCH2), 4.56, 4.47 (2 × 1H, 2 d, J = 12.1 Hz in each, PhCH2), 4.35 (1H, pt, J = 9.0, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.95 (1H, pt, J = 8.9, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.83 (1H, pt, J = 8.9, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.78–3.74 (3H, m, H-5′, H-6′a, H-6′b), 3.30 (2H, q, J = 7.6 Hz, CH2), 1.49 (3H, t, J = 7.6 Hz, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 172.0, 165.8 (C-3, C-6), 138.3, 137.9, 137.8, 137.6, 128.4–127.4 (Ar), 87.1, 80.2, 79.9, 79.8, 78.0 (C-1′–C-5′), 75.7, 75.1, 74.7, 73.4 (4 × PhCH2), 68.8 (C-6′), 28.5 (CH2), 12.1 (CH3). ESI-HRMS positive mode (m/z): calcd for C38H40N4NaO5+ [M + Na]+: 655.2891. Found: 655.2887.
6-Benzyl-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (21)
Prepared from cyanide 1 (0.2 g, 0.36 mmol) and benzyl cyanide (2 mL) according to General procedure I. Purified by column chromatography (hexane–EtOAc 7:1) to give 134 mg pink oil (53%). Rf = 0.27 (hexane–EtOAc 5:1). 1H NMR (360 MHz, CDCl3) δ (ppm): 7.40–7.00 (23H, m, Ar), 6.77–6.75 (2H, m, Ar), 4.98, 4.95 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.90 (1H, d, J = 9.9 Hz, H-1′), 4.87, 4.61 (2 × 1H, 2 d, J = 10.9 Hz in each, PhCH2), 4.73, 4.30 (2 × 1H, 2 d, J = 11.2 Hz, PhCH2), 4.57 (2H, s, CH2), 4.53, 4.46 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.34 (1H, pt, J = 9.2, 9.1 Hz, H-2′ or H-3′ or H-4′), 3.95, 3.82 (2 × 1H, 2 pt, J = 8.9, 8.9 Hz in each, H-2′ and/or H-3′ and/or H-4′), 3.79–3.72 (3H, m, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 170.1, 165.8 (C-3, C-6), 138.2, 137.8, 137.3, 135.5, 129.2–127.4 (Ar), 87.0, 80.1, 79.8, 79.7, 77.9 (C-1′–C-5′), 75.7, 75.1, 74.6, 73.4 (4 × PhCH2), 68.6 (C-6′), 41.3 (CH2). ESI-HRMS positive mode (m/z): calcd for C43H42N4NaO5+ [M + Na]+: 717.3047. Found: 717.3049.
6-Phenyl-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (22)
Prepared from cyanide 1 (0.2 g, 0.36 mmol) and benzonitrile (2 mL) according to General procedure I. Before column chromatographic purification the crude mixture was treated with EtOH, the precipitation (by-product 3,6-diphenyl-1,2,4,5-tetrazine) was filtered off, and the filtrate was evaporated under reduced pressure. The residue was purified by column chromatography (hexane–EtOAc 9:1) to give the target compound (127 mg, 51%) as a pink oil. Rf = 0.43 (hexane–EtOAc 7:1). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.58–8.56 (2H, m, Ar), 7.65–7.57 (3H, m, Ar), 7.39–7.19 (15H, m, Ar), 7.02–6.94 (3H, m, Ar), 6.85–6.84 (2H, m, Ar), 5.01, 4.98 (2 × 1H, 2 d, J = 11.4 Hz in each, PhCH2), 4.96 (1H, d, J = 9.8 Hz, H-1′), 4.90, 4.64 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.77, 4.41 (2 × 1H, 2 d, J = 11.2 Hz, PhCH2), 4.56, 4.48 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.38 (1H, pt, J = 9.5, 9.3 Hz, H-2′ or H-3′ or H-4′), 4.00 (1H, pt, J = 8.9, 8.9 Hz, H-2′ or H-3′ or H-4′), 3.85 (1H, pt, J = 9.2, 8.9 Hz, H-2′, H-3′ or H-4′), 3.80–3.75 (3H, m, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 165.6, 164.9 (C-3, C-6), 138.3, 137.9, 137.8, 137.4, 132.9, 131.6, 129.2–127.4 (Ar), 87.2, 80.2, 79.8, 79.7, 78.0 (C-1′–C-5′), 75.8, 75.2, 74.7, 73.4 (4 × PhCH2), 68.8 (C-6′). ESI-HRMS positive mode (m/z): calcd for C42H40N4NaO5+ [M + Na]+: 703.2891. Found: 703.2891.
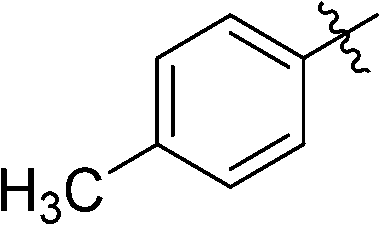
3-(2′,3′,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)-6-(p-tolyl)-1,2,4,5-tetrazine (23)
Prepared from cyanide 1 (0.2 g, 0.36 mmol) and p-tolunitrile (1.96 g, melted in a 50 °C water bath before using) according to General procedure I. Before column chromatographic purification the crude mixture was treated with EtOH, the precipitation (by-product 3,6-di-p-tolyl-1,2,4,5-tetrazine) was filtered off, and the filtrate was evaporated under reduced pressure. The residue was purified by column chromatography (hexane–EtOAc 9:1) to give the target compound (116 mg, 46%) as a pink oil. Rf = 0.43 (hexane–EtOAc 7:1). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.46 (2H, d, J = 8.3 Hz, Ar), 7.41–7.18 (17H, m, Ar), 7.03–6.97 (3H, m, Ar), 6.86–6.83 (2H, m, Ar), 5.01, 4.97 (2 × 1H, 2 d, J = 11.7 Hz in each, PhCH2), 4.94 (1H, d, J = 9.8 Hz, H-1′), 4.90, 4.64 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.76, 4.41 (2 × 1H, 2 d, J = 11.6 Hz in each, PhCH2), 4.56, 4.48 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.37 (1H, pt, J = 9.4, 9.3 Hz, H-2′ or H-3′ or H-4′), 3.99, 3.84 (2 × 1H, 2 pt, J = 8.9, 8.8 Hz in each, H-2′ and/or H-3′ and/or H-4′), 3.80–3.75 (3H, m, H-5′, H-6′a, H-6′b), 2.48 (3H, s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 165.4, 164.9 (C-3, C-6), 143.7, 138.3, 138.0, 137.9, 137.4, 130.0, 128.9, 128.5–127.5 (Ar), 87.2, 80.2, 79.9, 79.8, 78.1 (C-1′–C-5′), 75.8, 75.2, 74.7, 73.5 (4 × PhCH2), 68.9 (C-6′), 21.7 (CH3). ESI-HRMS positive mode (m/z): calcd for C43H42N4NaO5+ [M + Na]+: 717.3047. Found: 717.3047.
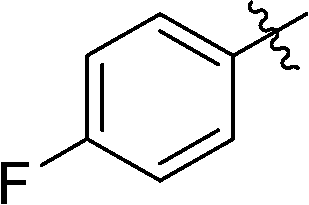
6-(4-Fluorophenyl)-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (24)
Prepared from cyanide 1 (0.2 g, 0.36 mmol) and 4-fluorobenzonitrile (2.22 g, melted in a 50 °C water bath before using) according to General procedure I. Before column chromatographic purification the crude mixture was treated with EtOH, the precipitation (by-product 3,6-bis(4-fluorophenyl)-1,2,4,5-tetrazine) was filtered off, and the filtrate was evaporated under reduced pressure. The residue was purified by column chromatography (hexane–EtOAc 9:1) to give the target compound (106 mg, 42%) as a pink oil. Rf = 0.31 (hexane–EtOAc 7:1). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.61–8.57 (2H, m, Ar), 7.39–7.19, 7.02–6.96, 6.85–6.83 (22H, m, Ar), 5.03–4.95 (2H, m, PhCH2), 4.94 (1H, d, J = 9.8 Hz, H-1′), 4.90, 4.64 (2 × 1H, 2 d, J = 10.7 Hz in each, PhCH2), 4.77, 4.41 (2 × 1H, 2 d, J = 11.8 Hz in each, PhCH2), 4.56, 4.48 (2 × 1H, 2 d, J = 12.1 Hz in each, PhCH2), 4.36 (1H, pt, J = 9.7, 9.2 Hz, H-2′ or H-3′ or H-4′), 3.99, 3.84 (2 × 1H, 2 pt, J = 8.9, 8.8 Hz in each, H-2′ and/or H-3′ and/or H-4′), 3.80–3.75 (3H, m, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 165.9 (d, J1C–F = 254.6, ArCq–F), 165.6, 164.0 (C-3, C-6), 138.3, 137.9, 137.9, 137.5 (Ar), 130.7 (d, J3C–F = 9.2 Hz, ArCH), 128.5–127.4 (Ar), 116.5 (d, J2C–F = 22.1 Hz, ArCH), 87.2, 80.2, 79.9, 79.8, 78.0 (C-1′–C-5′), 75.8, 75.2, 74.7, 73.5 (4 × PhCH2), 68.9 (C-6′). ESI-HRMS positive mode (m/z): calcd for C42H39FN4NaO5+ [M + Na]+: 721.2797. Found: 721.2797.
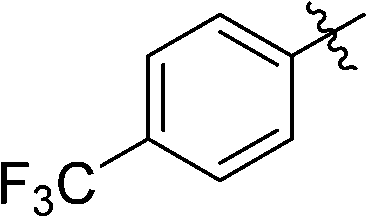
3-(2′,3′,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)-6-(4-trifluoromethylphenyl)-1,2,4,5-tetrazine (25)
Prepared from cyanide 1 (0.2 g, 0.36 mmol) and 4-(trifluoromethyl)benzonitrile (2.56 g, melted in a 50 °C water bath before using) according to General procedure I. Before column chromatographic purification the crude mixture was treated with EtOH, the precipitation (by-product 3,6-bis[4-(trifluoromethyl)phenyl]-1,2,4,5-tetrazine) was filtered off, and the filtrate was evaporated under reduced pressure. The residue was purified by column chromatography (hexane–EtOAc 9:1) to give the target compound (123 mg, 45%) as a pink oil. Rf = 0.44 (hexane–EtOAc 5:1). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.70, 7.88 (2 × 2H, 2 d, J = 8.2 Hz in each, Ar), 7.40–7.19, 7.00–6.94, 6.84–6.82 (2H, m, Ar), 5.03–4.97 (2H, m, PhCH2), 4.97 (1H, d, J = 9.7 Hz, H-1′), 4.90, 4.64 (2 × 1H, 2 d, J = 10.7 Hz in each, PhCH2), 4.77, 4.42 (2 × 1H, 2 d, J = 11.7 Hz in each, PhCH2), 4.56, 4.48 (2 × 1H, 2 d, J = 12.3 Hz in each, PhCH2), 4.36 (1H, pt, J = 9.7, 9.1 Hz, H-2′ or H-3′ or H-4′), 4.00 (1H, pt, J = 9.0, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.85 (1H, pt, J = 9.3, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.80–3.75 (3H, m, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.1, 163.9 (C-3, C-6), 138.3, 137.9, 137.8, 137.4 (Ar), 134.4 (q, J2C–F = 33.2 Hz, ArCq), 128.6–127.4, 126.2–125.6 (Ar), 123.7 (q, J1C–F = 272.7 Hz, CF3), 87.2, 80.2, 79.8, 79.7, 78.0 (C-1′–C-5′), 75.9, 75.2, 74.7, 73.5 (4 × PhCH2), 68.9 (C-6′). ESI-HRMS positive mode (m/z): calcd for C43H39F3N4NaO5+ [M + Na]+: 771.2765. Found: 771.2766.
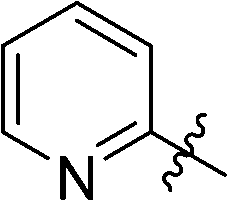
6-(2-Pyridyl)-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (26)
Prepared from cyanide 1 (0.2 g, 0.36 mmol) and 2-cyanopyridine (2.16 g, melted in a 50 °C water bath before using) according to General procedure I. Before column chromatographic purification the crude mixture was treated with EtOH, the precipitation (by-product 3,6-di(pyridin-2-yl)-1,2,4,5-tetrazine) was filtered off, and the filtrate was evaporated under reduced pressure. The residue was purified by column chromatography (hexane–EtOAc 2:1) to give the target compound (66 mg, 27%) as a pink oil. Rf = 0.45 (hexane–EtOAc 1:1). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.97 (1H, d, J = 4.6 Hz, Py-H-6), 8.62 (1H, d, J = 7.9 Hz, Py-H-3), 7.98 (1H, dt, J = 7.7, 1,7 Hz, Py-H-4), 7.56 (1H, ddd, J = 7.6, 4.6, 1.7 Hz, Py-H-5), 7.38–7.20 (15H, m, Ar), 7.04–6.93 (3H, m, Ar), 6.86–6.84 (2H, m, Ar), 5.04 (1H, d, J = 9.8 Hz, H-1′), 5.01, 4.98 (2 × 1H, 2 d, J = 11.2 Hz in each, PhCH2), 4.90, 4.64 (2 × 1H, 2 d, J = 10.7 Hz in each, PhCH2), 4.78, 4.42 (2 × 1H, 2 d, J = 11.6 Hz in each, PhCH2), 4.58, 4.49 (2 × 1H, 2 d, J = 12.1 Hz in each, PhCH2), 4.43 (1H, pt, J = 8.6, 8.6 Hz, H-2′ or H-3′ or H-4′), 4.01, 3.88 (2 × 1H, 2 pt, J = 8.9, 8.9 Hz in each, H-2′ and/or H-3′ and/or H-4′), 3.80–3.77 (3H, m, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.3, 164.2 (C-3, C-6), 150.9 (Py-C-6), 150.0 (Py-C-2), 138.3, 137.9, 137.8, 137.3 (Ar), 137.4 (Py-C-4), 128.4–127.4 (Ar), 126.5, 124.3 (Py-C-3, Py-C-5), 87.1, 80.3, 79.8, 79.7, 78.0 (C-1′–C-5′), 75.7, 75.2, 74.7, 73.5 (4 × PhCH2), 68.8 (C-6′). ESI-HRMS positive mode (m/z): calcd for C41H39N5NaO5+ [M + Na]+: 704.2843. Found: 704.2842.
6-(3-Pyridyl)-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (27)
Prepared from cyanide 1 (0.2 g, 0.36 mmol) and 3-cyanopyridine (2.32 g, melted in a 50 °C water bath before using) according to General procedure I. Before column chromatographic purification the crude mixture was treated with EtOH, the precipitation (by-product 3,6-di(pyridin-3-yl)-1,2,4,5-tetrazine) was filtered off, and the filtrate was evaporated under reduced pressure. The residue was purified by column chromatography (hexane–EtOAc 7:2) to give the target compound (61 mg, 25%) as a pink oil. Rf = 0.25 (hexane–EtOAc 7:3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.77 (1H, d, J = 1.5 Hz, Py-H-2), 8.90 (1H, dd, J = 4.8, 1.5 Hz, Py-H-6), 8.81 (1H, dt, J = 8.1, 4.8, 1.8 Hz, Py-H-4), 7.57 (1H, dd, J = 8.1, 4.8 Hz, Py-H-5), 7.40–7.19 (15H, m, Ar), 7.00–6.95 (3H, m, Ar), 6.85–6.83 (2H, m, Ar), 5.02, 4.98 (2 × 1H, 2 d, J = 11.0 Hz in each, PhCH2), 4.97 (1H, d, J = 9.8 Hz, H-1′), 4.90, 4.64 (2 × 1H, 2 d, J = 10.7 Hz in each, PhCH2), 4.77, 4.42 (2 × 1H, 2 d, J = 11.9 Hz in each, PhCH2), 4.56, 4.48 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.36 (1H, pt, J = 9.5, 9.4 Hz, H-2′ or H-3′ or H-4′), 4.00 (1H, pt, J = 8.9, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.85 (1H, pt, J = 9.4, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.81–3.75 (3H, m, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.2, 163.8 (C-3, C-6), 153.4, 149.6 (Py-C-2, Py-C-6), 138.2, 137.9, 137.8, 137.4 (Ar), 135.4 (Py-C-4), 128.5–127.4 (Ar), 127.7 (Py-C-3), 123.9 (Py-C-5), 87.2, 80.2, 79.8 (2), 78.0 (C-1′–C-5′), 75.8, 75.2, 74.7, 73.5 (4 × PhCH2), 68.9 (C-6′). ESI-HRMS positive mode (m/z): calcd for C41H39N5NaO5+ [M + Na]+: 704.2843. Found: 704.2844.
6-(4-Pyridyl)-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (28)
Prepared from cyanide 1 (0.2 g, 0.36 mmol) and 4-cyanopyridine (2.22 g, melted in a 100 °C oil bath before using) according to General procedure I. Before column chromatographic purification the crude mixture was treated with EtOH, the precipitation (by-product 3,6-di(pyridin-4-yl)-1,2,4,5-tetrazine) was filtered off, and the filtrate was evaporated under reduced pressure. The residue was purified by column chromatography (hexane–EtOAc 7:2) to give the target compound (45 mg, 18%) as a pink oil. Rf = 0.36 (hexane–EtOAc 3:2). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.94 (2H, d, J = 5.8 Hz, Py-H-2, Py-H-6), 8.45 (2H, d, J = 5.9 Hz, Py-H-3, Py-H-5), 7.40–7.19 (15H, m, Ar), 6.99–6.92 (3H, m, Ar), 6.83–6.81 (2H, m, Ar), 5.02, 4.98 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.97 (1H, d, J = 9.8 Hz, H-1′), 4.91, 4.64 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.77, 4.41 (2 × 1H, 2 d, J = 11.9 Hz in each, PhCH2), 4.56, 4.48 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.34 (1H, pt, J = 9.6, 9.2 Hz, H-2′ or H-3′ or H-4′), 4.01 (1H, pt, J = 8.9, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.85 (1H, pt, J = 9.4, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.81–3.74 (3H, m, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.7, 163.7 (C-3, C-6), 151.1 (Py-C-2, Py-C-6), 139.0, 138.2, 137.9, 137.8, 137.4 (Ar, Py-C-4), 128.5–127.4 (Ar), 121.5 (Py-C-3, Py-C-5), 87.2, 80.3, 79.8, 79.7, 78.0 (C-1′–C-5′), 75.9, 75.2, 74.7, 73.5 (4 × PhCH2), 68.8 (C-6′). ESI-HRMS positive mode (m/z): calcd for C41H39N5NaO5+ [M + Na]+: 704.2843. Found: 704.2846.
N-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosylcarbonyl)-N′-formyl hydrazine (30)
Prepared from acid 29 (2.00 g, 3.2 mmol) and formic acid hydrazide (0.19 g, 3.2 mmol) according to General procedure II. Reaction time: 5 h. Purified by column chromatography (hexane–EtOAc 1:2) to give 1.51 g (71%) pale yellow solid. Rf = 0.39 (hexane–EtOAc 1:3); mp = 138–139 °C; [α]D = +39 (c 0.83, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.00, 8.64 (2 × 1H, 2 d, J = 4.2 Hz in each, NH), 8.05–7.84, 7.53–7.25 (21H, m, Ar, CHO), 5.97 (1H, pt, J = 9.3, 9.3 Hz, H-2 or H-3 or H-4), 5.80 (1H, pt, J = 9.6, 9.5 Hz, H-2 or H-3 or H-4), 5.74 (1H, pt, J = 9.7, 9.5 Hz, H-2 or H-3 or H-4), 4.68 (1H, dd, J = 12.5, 2.2 Hz, H-6a), 4.52 (1H, dd, J = 12.5, 4.8 Hz, H-6b), 4.46 (1H, d, J = 9.6 Hz, H-1), 4.21 (1H, ddd, J = 9.5, 4.8, 2.5 Hz, H-5); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.3, 165.7, 165.4, 165.0, 164.4 (–O–CO, –NH–CO), 157.9 (H–CO), 133.5, 133.4, 133.3, 133.2, 129.8–128.3 (Ar), 76.3, 75.6, 73.5, 69.9, 68.9 (C-1–C-5), 62.7 (C-6). ESI-HRMS positive mode (m/z): calcd for C36H30N2O11+ [M + H]+ 667.1922; C36H30N2NaO11+ [M + Na]+ 689.1742. Found: [M + H]+ 667.1923; [M + Na]+ 689.1741.
N-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosylcarbonyl)-N′-acetyl hydrazine (31)
Prepared from acid 29 (1.0 g, 1.6 mmol) and acethydrazide (0.12 g, 1.6 mmol) according to General procedure II. Reaction time: 4 h. Purified by column chromatography (hexane–EtOAc 1:2) to give 0.71 g (65%) white solid. Rf = 0.36 (hexane–EtOAc 1:3); mp = 197 °C; [α]D = +38 (c 0.88, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.01, 8.89 (2 × 1H, 2d, J =4.0 Hz in each, NH), 8.03–7.86 (8H, m, Ar), 7.52–7.25 (12H, m, Ar), 6.00 (1H, pt, J = 9.4, 9.4 Hz, H-2 or H-3 or H-4), 5.86 (1H, pt, J = 9.6, 9.5 Hz, H-2 or H-3 or H-4), 5.78 (1H, pt, J = 9.6, 9.6 Hz, H-2 or H-3 or H-4), 4.67 (1H, dd, J = 12.5, 2.5 Hz, H-6a), 4.53 (1H, dd, J = 12.5, 4.6 Hz, H-6b), 4.47 (1H, d, J = 9.6 Hz, H-1), 4.23 (1H, ddd, J = 9.5, 4.6, 2.5 Hz, H-5), 1.80 (3H, s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 167.6, 166.2, 165.7, 165.3, 165.0, 164.2 (–O–CO, –NH–CO), 133.5, 133.3 (2), 133.1, 129.8–128.3 (Ar), 76.2, 75.6, 73.6, 70.0, 68.9 (C-1–C-5), 62.7 (C-6), 20.3 (CH3). ESI-HRMS positive mode (m/z): calcd for C37H33N2O11+ [M + H]+ 681.2079; C37H32N2NaO11+ [M + Na]+ 703.1898. Found: [M + H]+ 681.2077; [M + Na]+ 703.1894.
N-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosylcarbonyl)-N′-trifluoroacetyl hydrazine (32)
Prepared from acid 29 (0.50 g, 0.8 mmol) and 2,2,2-trifluoroacetic acid hydrazide (0.21 g, 1.6 mmol) according to General procedure II. Reaction time: 15 h. Purified by column chromatography (hexane–EtOAc 2:1) to give 0.30 g (51%) white amorphous solid. Rf = 0.45 (hexane–EtOAc 1:1); [α]D = +32 (c 0.68, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.78 (2H, br s, NH), 8.03–7.91 (8H, m, Ar), 7.55–7.23 (12H, m, Ar), 6.05 (1H, pt, J = 9.7, 9.1 Hz, H-2 or H-3 or H-4), 6.00 (1H, pt, J = 9.9, 9.1 Hz, H-2 or H-3 or H-4), 5.82 (1H, pt, J = 9.3, 9.2 Hz, H-2 or H-3 or H-4), 4.70–4.60 (2H, m, H-6a, H-6b), 4.49 (1H, d, J = 9.1 Hz, H-1), 4.25 (1H, m, H-5); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.5, 165.8, 165.6, 165.3, 165.1 (–O–CO, –NH–CO), 154.8 (d, J2C–F = 38.9 Hz, OC–CF3), 133.6,133.5, 133.4, 133.3, 129.9–128.4 (Ar), 115.2 (q, J1C–F = 287.2 Hz, CF3), 76.5, 75.4, 73.6, 69.7, 68.9 (C-1–C-5), 62.8 (C-6). ESI-HRMS positive mode (m/z): calcd for C37H30F3N2O11+ [M + H]+ 735.1796; C37H29F3N2NaO11+ [M + Na]+ 757.1616. Found: [M + H]+ 735.1796; [M + Na]+ 757.1617.
N-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosylcarbonyl)-N′-benzoyl hydrazine (33)
Prepared from acid 29 (2.0 g, 3.2 mmol) and benzhydrazide (0.44 g, 3.2 mmol) according to General procedure II. Reaction time: 3 days. Purified by column chromatography (hexane–EtOAc 3:2) to give 2.03 g (85%) white solid. Rf = 0.46 (hexane–EtOAc 1:1); mp = 211–212 °C; [α]D = +15 (c 0.64, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.43, 9.04 (2 × 1H, 2 d, J = 3.7 Hz in each, NH), 8.03–7.88 (8H, m, Ar), 7.63–7.17 (17H, m, Ar), 6.09 (1H, pt, J = 9.5, 9.4 Hz, H-2 or H-3 or H-4), 5.99 (1H, pt, J = 9.3, 9.3 Hz, H-2 or H-3 or H-4), 5.86 (1H, pt, J = 9.7, 9.4 Hz, H-2 or H-3 or H-4), 4.71 (1H, dd, J = 12.5, 2.2 Hz, H-6a), 4.57 (1H, dd, J = 12.5, 4.6 Hz, H-6b), 4.45 (1H, d, J = 9.7 Hz, H-1), 4.25 (1H, ddd, J = 9.5, 4.6, 2.2 Hz, H-5); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.3, 165.8, 165.4, 165.0, 164.8, 164.8 (–O–CO, –NH–CO), 133.5, 133.3, 133.2, 133.0, 132.1, 131.0, 129.8–129.7, 129.4–128.2, 127.3 (Ar), 76.3, 75.7, 73.9, 69.9, 69.0 (C-1–C-5), 62.9 (C-6). ESI-HRMS positive mode (m/z): calcd for C42H35N2O11+ [M + H]+ 743.2235; C42H34N2NaO11+ [M + Na]+ 765.2055. Found: [M + H]+ 743.2235; [M + Na]+ 765.2053.
N-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosylcarbonyl)-N′-(pyridine-2-carbonyl)hydrazine (34)
Prepared from acid 29 (3.0 g, 4.8 mmol) and picolinohydrazide (0.66 g, 4.8 mmol) according to General procedure II. Reaction time: 3 days. Purified by column chromatography (hexane–EtOAc 1:4) to give 3.02 g (85%) white solid. Rf = 0.48 (EtOAc); mp = 217–217 °C; [α]D = −0.7 (c 1.0, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 10.09, 9.30 (2 × 1H, 2 br s, NH), 8.47 (1H, d, J = 4.1 Hz, Py-H-6), 8.12–7.76, 7.56–7.22 (23H, m, Ar, Py-H-3–Py-H-5), 5.99 (1H, pt, J = 9.3, 9.3 Hz, H-2 or H-3 or H-4), 5.85 (1H, pt, J = 9.5, 9.5 Hz, H-2 or H-3 or H-4), 5.76 (1H, pt, J = 9.7, 9.5 Hz, H-2 or H-3 or H-4), 4.73 (1H, dd, J = 12.3, 2.7 Hz, H-6a), 4.55 (1H, dd, J = 12.3, 4.6 Hz, H-6b), 4.54 (1H, d, J = 9.6 Hz, H-1), 4.25 (1H, ddd, J = 9.5, 4.6, 2.7 Hz, H-5); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.2, 165.6, 165.2, 165.0 (–O–CO), 163.4, 160.9 (–NH–CO), 148.3 (Py-C-6), 148.0 (Py-C-2), 137.2 (Py-C-4), 133.5, 133.2, 133.1 (2), 129.9–129.7, 129.4, 129.1, 128.7, 128.6, 128.4–128.2 (Ar), 126.7 (Py-C-3), 122.4 (Py-C-5), 76.4, 75.9, 73.6, 69.9, 68.9 (C-1–C-5), 62.7 (C-6). ESI-HRMS positive mode (m/z): calcd for C41H34N3O11+ [M + H]+ 744.2188; C41H33N3NaO11+ [M + Na]+ 766.2007. Found: [M + H]+ 744.2187; [M + Na]+ 766.2009.
N,N′-Bis(2,3,4,6-tetra-O-benzoyl-β-D-glucopyranosylcarbonyl)hydrazine (35)
To a solution of acid 29 (0.6 g, 0.96 mmol) in dry CH2Cl2 (15 mL) EDCI (0.20 g, 1.1 equiv.) and hydrazine monohydrate (23 μL, 0.48 mmol, 0.5 equiv.) were added. The reaction was stirred at rt and monitored by TLC (hexane–EtOAc 1:2). After total transformation of the acid (18 h) the reaction mixture was diluted with CH2Cl2 (35 mL) and extracted with brine (2 × 30 mL). The separated organic phase was dried over MgSO4, filtered and evaporated. Column chromatographic purification of the residue (hexane–EtOAc = 3:2) gave 0.26 g (43%) white solid. Rf = 0.34 (hexane–EtOAc 1:1); mp = 140–142 °C; [α]D = +25 (c 0.58, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.75 (2H, s, NH), 8.02, 7.93, 7.90, 7.83 (4 × 2 × 2H, 4 d, J = 7.5 Hz in each, Ar), 7.51–7.25 (24H, m, Ar), 5.92 (2H, pt, J = 9.4, 9.4 Hz, 2 × (H-2 or H-3 or H-4)), 5.75 (2H, pt, J = 9.6, 9.6 Hz, 2 × (H-2 or H-3 or H-4)), 5.69 (2H, pt, J = 9.7, 9.6 Hz, 2 × (H-2 or H-3 or H-4)), 4.65 (2H, dd, J = 12.5, 1.9 Hz, 2 × H-6a), 4.46 (2H, dd, J = 12.5, 4.6 Hz, 2 × H-6b), 4.41 (2H, d, J = 9.7 Hz, 2 × H-1), 4.13 (2H, ddd, J = 9.3, 4.6, 1.9 Hz, 2 × H-5); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.2, 165.7, 165.2, 165.0, 164.2 (2 × (4 × –O–CO), 2 × –NH–CO)), 133.5, 133.2, 133.2, 133.1, 129.9–128.3 (Ar), 76.3, 75.7, 73.6, 69.7, 68.8 (2 × (C-1–C-5)), 62.6 (2 × C-6). ESI-HRMS positive mode (m/z): calcd for C70H56N2NaO20+ [M + Na]+ 1267.3319. Found: 1267.3311.
6-Phenyl-3-(2′,3′,4′,6′-tetra-O-benzoyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (38)
N-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosylcarbonyl)-N′-benzoyl hydrazine (33, 1.5 g, 2.02 mmol) and phosphorus pentachloride (2.1 g, 10.1 mmol, 5 equiv.) were suspended in dry toluene (40 mL). The reaction mixture was refluxed and the transformation was monitored by TLC (hexane–EtOAc 1:1). After completion of the reaction (5 h) the solvent was removed under reduced pressure and the residue was purified by column chromatography to give 1.42 g intermediate 36 (90%) as a white foam.
O), 140.3, 140.8 (2 × CN), 133.5, 133.3, 133.2, 133.1, 132.9, 131.9, 129.9–128.3 (Ar), 80.1, 76.3, 73.9, 69.7, 69.2 (C-1–C-5), 63.0 (C-6).
Intermediate 36 (1.42 g, 1.82 mmol) was dissolved in CHCl3 (30 mL) and the solution was cooled down in an ice bath. Hydrazine monohydrate (133 μL, 2.73 mmol, 1.5 equiv.) was added and after 10 min the reaction mixture was started to be boiled. After 1.5 h the TLC (hexane–EtOAc 2:1) showed incomplete conversion of intermediate 36, therefore an additional portion of hydrazine monohydrate (133 μL, 2.73 mmol, 1.5 equiv.) was added to the reaction mixture. After 30 min the total transformation of 36 was observed, thus, the solvent was removed under reduced pressure. The resulting yellow oil containing intermediate 37 was dissolved in glacial acetic acid (60 mL) and the solution was cooled down in an ice bath. A solution of NaNO2 (0.77 g, 9.11 mmol, 5 equiv.) in water (2 mL) was added to the mixture and the completion of the oxidation (15 min) was judged by TLC (hexane–EtOAc 2:1). The reaction mixture was diluted with CHCl3 (200 mL) and extracted with water (100 mL), sat. aq. solution of NaHCO3 (2 × 100 mL) and water (100 mL). The separated organic phase was dried over MgSO4, filtered and the solvent was removed under reduced pressure. The residue was purified by column chromatography (hexane–EtOAc 7:2) yielding 1.07 g of 38 (80% for two steps). Pink oil, Rf = 0.41 (hexane–EtOAc 2:1).
O), 133.5, 133.4, 133.3, 133.2, 133.1, 131.2, 129.8–128.3 (Ar), 78.9, 77.2, 74.0, 71.6, 69.3 (C-1′–C-5′), 63.2 (C-6′). ESI-HRMS positive mode (m/z): calcd for C42H33N4O9+ [M + H]+ 737.2242; C42H32N4NaO9+ [M + Na]+ 759.2061. Found: [M + H]+ 737.2243; [M + Na]+ 759.2058.
6-(2-Pyridyl)-3-(2′,3’,4′,6′-tetra-O-benzoyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (41)
N-(2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosylcarbonyl)-N′-(pyridine-2-carbonyl)hydrazine (34, 1.0 g, 1.3 mmol) and phosphorus pentachloride (1.4 g, 6.7 mmol, 5 equiv.) were suspended in dry toluene (20 mL). The reaction mixture was refluxed and the transformation was monitored by TLC (hexane–EtOAc 1:1). After completion of the reaction (5 h) the solvent was removed under reduced pressure and the resulting intermediate 39 was used in the next step without further purification.
Intermediate 39 was dissolved in CHCl3 (20 mL) and the solution was cooled down in an ice bath. Hydrazine monohydrate (130 μL, 2.6 mmol) was added and after 10 min the reaction mixture was started to be boiled. After 1.5 h, an additional portion of hydrazine monohydrate (130 μL, 2.6 mmol) was added to the reaction mixture. After 2 hours the the solvent was removed under reduced pressure. The residue was dissolved in glacial acetic acid (30 mL) and the solution was cooled down in an ice bath. A solution of NaNO2 (0.46 g) in water (2 mL) was added to the mixture and the completion of the oxidation (15 min) was judged by TLC (hexane–EtOAc 1:1). The reaction mixture was diluted with CHCl3 (150 mL) and extracted with water (80 mL), sat. aq. solution of NaHCO3 (2 × 80 mL) and water (80 mL). The separated organic phase was dried over MgSO4, filtered and the solvent was removed under reduced pressure. The column chromatographic purification (hexane–EtOAc 3:2) gave 257 mg of oxadiazole 40 (26%) as the first fraction (Rf = 0.22, hexane–EtOAc 3:2), 325 mg of a mixed fraction containing 40 and 41, and 161 mg of tetrazine 41 (16% for three step) as the third fraction (Rf = 0.18, hexane–EtOAc 3:2). NMR data of compound 40 corresponds to the reported ones.37
O), 150.9 (Py-C-6), 149.6 (Py-C-2), 137.4 (Py-C-4), 133.4, 133.3, 133.2, 133.0, 133.1, 131.2, 129.8–128.2 (Ar), 126.7, 124.6 (Py-C-3, Py-C-5), 78.8, 77.2, 74.0, 71.5, 69.2 (C-1′–C-5′), 63.1 (C-6′). ESI-HRMS positive mode (m/z): calcd for C41H32N5O9+ [M + H]+ 738.2195; C41H31N5NaO9+ [M + Na]+ 760.2014. Found: [M + H]+ 738.2195; [M + Na]+ 760.2014.
3-(β-D-Glucopyranosyl)-1,2,4,5-tetrazine (43)
A stirred solution of tetrazine 3 (300 mg; 0.50 mmol) in dry CH2Cl2 (15 mL) was cooled down to −60 °C and a 1 M solution of BCl3 in CH2Cl2 (2.5 mL, 2.5 mmol, 5 equiv.) was added. The completion of the reaction was judged by TLC (CHCl3–MeOH 4:1). After 1.5 hours, dry MeOH (20 mL) was added to the reaction mixture and the solvents were removed under diminished pressure. The resulting syrup was purified by column chromatography (CHCl3–MeOH 9:1). Yield: 55 mg (45%), pink oil. 1H NMR (360 MHz, CD3OD) δ (ppm): 10.44 (1H, s, H-6), 4.82 (1H, H-1′), 3.99 (1H, pt, J = 9.4, 9.3 Hz, H-2′ or H-3′ or H-4′), 3.87 (1H, dd, J = 12.0, 1.5 Hz, H-6′a), 3.68 (1H, dd, J = 12.0, 5.6 Hz, H-6′b), 3.60 (1H, pt, J = 9.0, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.55 (1H, ddd, J = 9.0, 5.6, 1.5 Hz, H-5′), 3.48 (1H, pt, J = 9.2, 9.0 Hz, H-2′ or H-3′ or H-4′). 13C NMR (90 MHz, CD3OD) δ (ppm): 170.2 (C-3), 160.6 (C-6), 83.1, 82.1, 79.3, 74.1, 71.5 (C-1′–C-5′), 62.9 (C-6′). ESI-HRMS positive mode (m/z): calcd for C8H12N4NaO5+ [M + Na]+ 267.0700. Found: 267.0698.
3-(β-D-Glucopyranosyl)-6-methyl-1,2,4,5-tetrazine (44)
A stirred solution of tetrazine 19 (300 mg; 0.50 mmol) in dry CH2Cl2 (15 mL) was cooled down to −60 °C and and a 1 M solution of BCl3 in CH2Cl2 (2.5 mL, 2.5 mmol, 5 equiv.) was added. The completion of the reaction was judged by TLC (CHCl3–MeOH 8:1). After three hours, dry MeOH (20 mL) was added to the reaction mixture and the solvents were removed under diminished pressure. The resulting syrup was purified by column chromatography (CHCl3–MeOH 8:1, then 4:1). Yield: 108 mg (86%), pink oil. Rf = 0.24 (CHCl3–MeOH 4:1). 1H NMR (360 MHz, CD3OD) δ (ppm): 4.83 (1H, d, J = 9.7 Hz, H-1′), 4.00 (1H, pt, J = 9.6, 9.1 Hz, H-2′ or H-3′ or H-4′), 3.90 (1H, dd, J = 12.1, 2.0 Hz, H-6′a), 3.72 (1H, dd, J = 12.1, 5.4 Hz, H-6′b), 3.63 (1H, pt, J = 9.0, 8.7 Hz, H-2′ or H-3′ or H-4′), 3.58 (1H, ddd, J = 9.6, 5.4, 2.0 Hz, H-5′), 3.52 (1H, pt, J = 9.5, 8.6 Hz, H-2′ or H-3′ or H-4′), 3.06 (3H, s, CH3); 13C NMR (90 MHz, CD3OD) δ (ppm): 170.4, 167.4 (C-3, C-6), 83.0, 81.7, 79.3, 74.0, 71.5 (C-1′–C-5′), 62.8 (C-6′), 21.3 (CH3). ESI-HRMS positive mode (m/z): calcd for C9H15N4O5+ [M + H]+ 259.1037; C9H14N4NaO5+ [M + Na]+ 281.0856. Found: [M + H]+ 259.1037; [M + Na]+ 281.0856.
3-(β-D-Glucopyranosyl)-6-phenyl-1,2,4,5-tetrazine (45)
To a solution of tetrazine 38 (0.24 g, 0.33 mmol) in a solvent mixture of dry MeOH (4 mL) and dry CHCl3 (1 mL) a catalytic amount of a NaOMe solution (1 M in MeOH) was added. The reaction mixture was kept at rt and the transformation was monitored by TLC (hexane–EtOAc 2:1 and CHCl3–MeOH 4:1). When the reaction was complete, the mixture was neutralized by the addition of acetic acid and then the solvents were removed under diminished pressure. Purification of the residue by column chromatography (CHCl3–MeOH 9:1) gave 60 mg (58%) pink amorphous solid. Rf = 0.47 (CHCl3–MeOH 4:1). 1H NMR (360 MHz, CD3OD) δ (ppm): 8.62–8.59 (2H, m, Ph), 7.71–7.62 (3H, m, Ph), 4.91 (1H, H-1′), 4.07 (1H, pt, J = 9.5, 9.2 Hz, H-2′ or H-3′ or H-4′), 3.93 (1H, dd, J = 12.1, 1.9 Hz, H-6′a), 3.74 (1H, dd, J = 12.1, 5.6 Hz, H-6′b), 3.65 (1H, pt, J = 8.9, 8.7 Hz, H-2′ or H-3′ or H-4′), 3.59 (1H, ddd, J = 9.5, 5.6, 1.9 Hz, H-5′), 3.54 (1H, pt, J = 9.4, 8.7 Hz, H-2′ or H-3′ or H-4′); 13C NMR (90 MHz, CD3OD) δ (ppm): 167.7, 166.7 (C-3, C-6), 134.1, 133.2, 130.5 (2), 129.3(2) (Ph), 83.2, 81.79, 79.4, 74.2, 71.7 (C-1′–C-5′), 63.0 (C-6′).
6-Methyl-3-(2′,3′,4′,6′-tetra-O-benzoyl-β-D-glucopyranosyl)-1,2,4,5-tetrazine (46)
:2). After completion of the reaction (5 d) the mixture was diluted with CH2Cl2 (20 mL) and extracted with water (20 mL), sat. aq. NaHCO3 (3 × 15 mL) and with water (20 mL). The organic phase was dried over MgSO4, filtered and the solvent was removed under reduced pressure. Column chromatographic purification of the residue (hexane–EtOAc 3:1) gave 23 mg (54%) pink oil.
:1 and hexane–EtOAc 1:1) showed incomplete conversion of 44, therefore an additional portion of benzoyl chloride (108 μL, 0.93 mmol) was added to the mixture. After completion of the reaction (4 h) the solvent was removed under diminished pressure. The residue was dissolved in CHCl3 (20 mL) and extracted with sat. aq. NaHCO3 (2 × 15 mL) then with water (15 mL). The organic phase was dried over MgSO4, filtered and the solvent was removed under reduced pressure. The crude product was purified by column chromatography (hexane–EtOAc 3:1, then 7:3). Yield: 118 mg (90%), pink oil. Rf = 0.31 (hexane–EtOAc 5:2). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.00, 7.94, 7.85, 7.76 (4 × 2H, 4 d, J = 7.3 Hz in each, Ar), 7.55–7.26 (12H, m, Ar), 6.20–6.14 (2H, m, H-2′ and/or H-3′ and/or H-4′), 5.92 (1H, pt, J = 9.6, 9.6 Hz, H-2′ or H-3′ or H-4′), 5.46 (1H, d, J = 9.3 Hz, H-1′), 4.68 (1H, dd, J = 12.4, 2.8 Hz, H-6′a), 4.56 (1H, dd, J = 12.4, 5.3 Hz, H-6′b), 4.45 (1H, ddd, J = 9.6, 5.3, 2.8 Hz, H-5′), 3.07 (3H, s, CH3); 13C NMR (100 MHz, CDCl3) δ (ppm): 169.1, 166.1, 165.8, 165.1, 164.7, 164.6 (C-3, C-6, 4 × CO), 133.5, 133.4, 133.3, 133.1, 129.8–128.3 (Ar), 78.9, 77.2, 74.0, 71.6, 69.3 (C-1′–C-5′), 63.2 (C-6′), 21.3 (CH3). ESI-HRMS positive mode (m/z): calcd for C37H31N4O9+ [M + H]+ 675.2086; C37H30N4NaO9+ [M + Na]+ 697.1905. Found: [M + H]+ 675.2087; [M + Na]+ 697.1907.
5-Acetoxymethyl-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)pyridazine (47)
Prepared from tetrazine 3 (0.1 g, 0.17 mmol) and propargyl acetate (16 μL, 0.17 mmol) according to General procedure III. Reaction time: 12 h. Purified by column chromatography (hexane–EtOAc 1:1) to give 99 mg (89%) pale yellow oil. Rf = 0.47 (hexane–EtOAc 1:1), [α]D = −96 (c 0.21, CH2Cl2). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.07 (1H, d, J = 1.6 Hz, H-6), 7.35–7.10, 6.88–6.86 (21H, m, Ar, H-4), 5.02–4.95 (2 × 2H, m, PhCH2, CH2), 4.89, 4.63 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.70 (1H, d, J = 9.4 Hz, H-1′), 4.57, 4.51 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.56, 4.14 (2 × 1H, 2 d, J = 11.2 Hz in each, PhCH2), 3.93, 3.83, 3.80 (3 × 1H, 3 pt, J = 9.2, 9.0 Hz in each, H-2′, H-3′, H-4′), 3.74–3.68 (3H, m, H-5′, H-6′a, H-6′b), 2.12 (3H, s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 170.1 (CO), 160.0 (C-3), 149.8 (C-6), 138.4, 137.9, 137.8, 137.3 (Ar), 135.9 (C-5), 128.4–127.5 (Ar), 123.8 (C-4), 86.8, 81.1, 80.8, 79.5, 78.0 (C-1′–C-5′), 75.6, 75.1, 74.4, 73.3 (4 × PhCH2), 68.9 (C-6′), 61.9 (CH2), 20.6 (CH3). ESI-HRMS positive mode (m/z): calcd for C41H43N2O7+ [M + H]+: 675.3065. Found: 675.3067.
5-(tert-Butoxycarbamoyl)methyl-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)pyridazine (48)
Prepared from tetrazine 3 (0.1 g, 0.17 mmol) and N-Boc-propargylamine (26 mg, 0.17 mmol) according to General procedure III. Reaction time: 10 h. Purified by column chromatography (hexane–EtOAc 3:2) to give 96 mg (81%) pale yellow oil. Rf = 0.23 (hexane–EtOAc 1:1); [α]D = −24 (c 0.97, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.02 (1H, d, J = 1.8 Hz, H-6), 7.36–7.10, 6.88–6.86 (21H, m, Ar, H-4), 5.04, 4.20 (2 × 1H, 2 dd, J = 10.7, 5.3 Hz in each, CH2), 4.96, 4.93 (2 × 1H, 2 d, J = 11.2 Hz in each, PhCH2), 4.88, 4.61 (2 × 1H, 2 d, J = 10.5 Hz in each, PhCH2), 4.67 (1H, d, J = 9.5 Hz, H-1′), 4.56, 4.50 (2 × 1H, 2 d, J = 12.3 Hz in each, PhCH2), 4.53, 4.12 (2 × 1H, 2 d, J = 10.9 Hz in each, PhCH2), 3.92, 3.81, 3.77 (3 × 1H, 3 pt, J = 9.1, 9.1 Hz in each, H-2′, H-3′, H-4′), 3.72–3.67 (3H, m, H-5′, H-6′a, H-6′b), 1.45 (9H, s, C(CH3)3); 13C NMR (90 MHz, CDCl3) δ (ppm): 159.8, 155.6 (CO, C-3), 150.5 (C-6), 139.0, 138.4, 137.9 (2), 137.4 (Ar, C-5), 128.3–127.4 (Ar), 123.9 (C-4), 86.8, 81.2, 80.8, 79.4, 78.0 (C-1′–C-5′), 80.2 (C(CH3)3), 75.6, 75.0, 74.4, 73.3 (4 × PhCH2), 69.0 (C-6′), 41.4 (CH2), 28.2 (C(CH3)3). ESI-HRMS positive mode (m/z): calcd for C44H50N3O7+ [M + H]+ 732.3643; C44H49N3NaO7+ [M + Na]+ 754.3463. Found: [M + H]+ 732.3640; [M + Na]+ 754.3469.
5-Phenyl-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)pyridazine (49)
Prepared from tetrazine 3 (0.1 g, 0.17 mmol) and phenylacetylene (18 μL, 0.17 mmol) according to General procedure III. Reaction time: 1 d. Purified by column chromatography (hexane–EtOAc 1:1) to give 67 mg (60%) pale yellow oil. Rf = 0.38 (hexane–EtOAc 1:1); [α]D = −38 (c 0.97, CHCl3). 1H NMR (400 MHz, CDCl3) δ (ppm): 9.39 (1H, d, J = 2.3 Hz, H-6), 7.60–6.88 (26H, m, Ar, H-4), 4.99, 4.95 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.89, 4.63 (2 × 1H, 2 d, J = 10.7 Hz in each, PhCH2), 4.74 (1H, d, J = 9.3 Hz, H-1′), 4.58, 4.53 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.59, 4.55 (2 × 1H, 2 d, J = 11.0 Hz in each, PhCH2), 3.95, 3.90, 3.80 (3 × 1H, 3 pt, J = 9.2, 9.0 Hz in each, H-2′, H-3′, H-4′), 3.75–3.69 (3H, m, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 160.2 (C-3), 149.4 (C-6), 138.8, 138.5, 138.1, 138.0, 137.3 (Ar), 134.5 (C-5), 130.0–127.2 (Ar), 122.7 (C-4), 86.9, 81.3, 81.0, 79.6, 78.2 (C-1′–C-5′), 75.6, 75.1, 74.5, 73.4 (4 × PhCH2), 69.0 (C-6′). ESI-HRMS positive mode (m/z): calcd for C44H43N2O5+ [M + H]+ 679.3166; C44H42N2NaO5+ [M + Na]+ 701.2986. Found: [M + H]+ 679.3169; [M + Na]+ 701.2983.
5-(2-Pyridyl)-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)pyridazine (50)
Prepared from tetrazine 3 (0.1 g, 0.17 mmol) and 2-ethynylpyridine (17 μL, 0.17 mmol) according to General procedure III. Reaction time: 10 h. Purified by column chromatography (hexane–EtOAc 5:4) to give 109 mg (97%) colourless oil. Rf = 0.33 (hexane–EtOAc 1:1); [α]D = −38 (c 1.0, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.76 (1H, d, J = 2.1 Hz, H-6), 8.76 (1H, d, J = 4.7 Hz, Py-H-6), 8.03 (1H, d, J = 2.1 Hz, H-4), 7.80 (1H, dt, J = 7.9, 1.6 Hz, Py-H-4), 7.74 (1H, d, J = 7.8 Hz, Py-H-3), 7.38–7.19, 7.05–6.89 (21H, m, Ar, Py-H-5), 4.98, 4.95 (2 × 1H, 2 d, J = 11.2 Hz in each, PhCH2), 4.90, 4.64 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.79 (1H, d, J = 9.0 Hz, H-1′), 4.59, 4.52 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.54, 4.18 (2 × 1H, 2 d, J = 11.0 Hz in each, PhCH2), 3.96, 3.93, 3.82 (3 × 1H, 3 pt, J = 9.3, 8.6 Hz in each, H-2′, H-3′, H-4′), 3.78–3.72 (3H, m, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 160.4 (C-3), 151.6 (Py-C-2), 150.3, 148.7 (C-6, Py-C-6), 138.5, 138.1, 138.0, 137.3, 136.7 (Ar, C-5), 137.0 (Py-C-4), 128.3–127.4 (Ar), 124.5, 122.3, 121.1 (C-4, Py-C-3, Py-C-5), 86.9, 81.3, 81.0, 79.6, 78.1 (C-1′–C-5′), 75.5, 75.0, 74.4, 73.3 (4 × PhCH2), 69.0 (C-6′). ESI-HRMS positive mode (m/z): calcd for C43H42N3O5+ [M + H]+ 680.3119; C43H41N3NaO5+ [M + Na]+ 702.2938. Found: [M + H]+ 680.3118; [M + Na]+ 702.2933.
1-(2′,3’,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)-6,7-dihydro-5H-cyclopenta[d]pyridazine (51)
A solution of tetrazine 3 (50 mg, 0.083 mmol) and 1-pyrrolidino-1-cyclopentene (12 μL, 0.083 mmol) in dry m-xylene (2 mL) was refluxed and the transformation was monitored by TLC (hexane–EtOAc 3:1). After completion of the reaction (1.5 h) the solvent was removed under reduced pressure. The residue was purified by column chromatography (hexane–EtOAc 1:1) to get 43 mg (81%) pale yellow syrup. Rf = 0.34 (hexane–EtOAc 1:1); [α]D = −11 (c 0.21, CH2Cl2). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.01 (1H, s, H-4), 7.38–7.08, 6.85–6.83 (20H, m, Ar), 4.99, 4.95 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.88, 4.62 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.70 (1H, d, J = 9.7 Hz, H-1′), 4.60, 4.26 (2 × 1H, 2 d, J = 11.2 Hz in each, PhCH2), 4.54, 4.49 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.05 (1H, pt, J = 9.4, 9.2 Hz, H-2′ or H-3′ or H-4′) 3.91 (1H, pt, J = 9.2, 9.0 Hz, H-2′ or H-3′ or H-4′), 3.76 (1H, pt, J = 9.4, 9.2 Hz, H-2′ or H-3′ or H-4′), 3.73–3.65 (3H, m, H-5′, H-6′a, H-6′b), 3.05–2.97, 2.87–2.82, 2.78–2.69, 1.99–1.90 (6H, m, 3 × CH2); 13C NMR (90 MHz, CDCl3) δ (ppm): 156.6 (C-1), 148.6 (C-4), 145.1, 144.2 (C-4a, C-7a), 138.4, 138.0, 137.9, 137.5, 128.4–127.4 (Ar), 87.2, 80.2, 79.4, 79.3, 78.0 (C-1′–C-5′), 75.6, 75.1, 74.3, 73.2 (4 × PhCH2), 69.0 (C-6′), 30.4, 30.3, 23.4 (3 × CH2). ESI-HRMS positive mode (m/z): calcd for C41H43N2O5+ [M + H]+ 643.3166; C41H42N2NaO5+ [M + Na]+ 665.2986. Found: [M + H]+ 643.3162; [M + Na]+ 665.2983.
1-(2′,3′,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)-5,6,7,8-tetrahydrophthalazine (52)
A solution of tetrazine 3 (50 mg, 0.083 mmol) and 1-pyrrolidino-1-cyclohexene (13 μL, 0.083 mmol) in dry m-xylene (2 mL) was refluxed and the transformation was monitored by TLC (hexane–EtOAc 1:1). After completion of the reaction (1 h) the solvent was removed under reduced pressure. The residue was purified by column chromatography (hexane–EtOAc 1:1) to get 51 mg (94%) pale yellow syrup. Rf = 0.40 (hexane–EtOAc 1:1); [α]D = +36 (c 0.21, CH2Cl2). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.77 (1H, s, H-4), 7.38–7.14, 6.92–6.89 (20H, m, Ar), 5.00, 4.96 (2 × 1H, 2 d, J = 11.5 Hz in each, PhCH2), 4.87, 4.59 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.75 (1H, d, J = 10.8 Hz, PhCH2), 4.66 (1H, d, J = 9.6 Hz, H-1′), 4.55–4.48 (3H, m, PhCH2), 4.45 (1H, pt, J = 9.2, 9.0 Hz, H-2′ or H-3′ or H-4′), 3.89 (1H, pt, J = 9.1, 9.0 Hz, H-2′ or H-3′ or H-4′), 3.76–3.65 (4H, m, H-2′ or H-3′ or H-4′, H-5′, H-6′a, H-6′b), 2.75–2.66 (4H, m, H-5, H-8), 1.73 (4H, br s, H-6, H-7); 13C NMR (90 MHz, CDCl3) δ (ppm): 157.1 (C-1), 152.0 (C-4), 138.6, 138.2, 138.1, 138.0 (Ar), 137.7, 137.2 (C-4a, C-8a), 128.4–127.4 (Ar), 87.6, 80.1, 79.8, 78.3, 77.7 (C-1′–C-5′), 75.6, 75.1, 74.8, 73.3 (4 × PhCH2), 69.3 (C-6′), 26.1, 23.6, 21.6, 21.0 (4 × CH2). ESI-HRMS positive mode (m/z): calcd for C42H45N2O5+ [M + H]+ 657.3323; C42H44N2NaO5+ [M + Na]+ 679.3142. Found: [M + H]+ 657.3322; [M + Na]+ 679.3140.
3-(2′,3′,4′,6′-Tetra-O-benzyl-β-D-glucopyranosyl)pyridazine (53)
Prepared from tetrazine 3 (50 mg, 0.08 mmol) and 2,5-norbornadiene (17 μL, 0.17 mmol) in toluene according to General procedure IV. Reaction time: 15 min. Purified by column chromatography (hexane–EtOAc 3:2) to give 36 mg (72%) colourless syrup. Rf = 0.38 (hexane–EtOAc 1:1); [α]D = −49 (c 0.20, CH2Cl2). 1H NMR (360 MHz, CDCl3) δ (ppm): 9.13 (1H, dd, J = 4.9, 1.7 Hz, H-6), 7.46 (1H, dd, J = 8.5, 1.7 Hz, H-4), 7.36–7.12, 6.90–6.88 (21H, m, Ar, H-5), 4.97, 4.93 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.88, 4.61 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.69 (1H, d, J = 9.4 Hz, H-1′), 4.57, 4.53 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.51, 4.10 (2 × 1H, 2 d, J = 11.0 Hz in each, PhCH2), 3.93 (1H, pt, J = 8.8, 8.7 Hz, H-2′ or H-3′ or H-4′), 3.84 (1H, pt, J = 9.5, 9.3 Hz, H-2′ or H-3′ or H-4′), 3.76 (1H, pt, J = 9.8, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.74–3.67 (3H, m, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 160.3 (C-3), 151.1 (C-6), 138.4, 138.1, 138.0, 137.4, 128.4–127.5 (Ar), 126.6, 125.8 (C-4, C-5), 86.8, 81.5, 81.0, 79.6, 78.1 (C-1′–C-5′), 75.6, 75.1, 74.5, 73.4 (4 × PhCH2), 69.0 (C-6′). ESI-HRMS positive mode (m/z): calcd for C38H39N2O5+ [M + H]+ 603.2853; C38H38N2NaO5+ [M + Na]+ 625.2673. Found: [M + H]+ 603.2851; [M + Na]+ 625.2675.
6-Methyl-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)pyridazine (54)
Prepared from tetrazine 19 (20 mg, 0.03 mmol) and 2,5-norbornadiene (7 μL, 0.06 mmol) in toluene according to General procedure IV. Reaction time: 3 h. Purified by column chromatography (hexane–EtOAc 1:1) to give 17 mg (85%) white solid. Rf = 0.29 (hexane–EtOAc 1:1); [α]D = −14 (c 0.52, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 7.34–7.25, 7.21–7.11, 6.93–6.90 (22H, m, Ar, H-4, H-5), 4.95, 4.92 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.88, 4.62 (2 × 1H, 2 d, J = 10.7 Hz in each, PhCH2), 4.66 (1H, d, J = 9.4 Hz, H-1′), 4.59–4.49 (3H, m, PhCH2), 4.15 (1H, d, J = 11.0 Hz, PhCH2), 3.92 (1H, pt, J = 8.8, 8.7 Hz, H-2′ or H-3′ or H-4′), 3.85 (1H, pt, J = 9.2, 8.9 Hz, H-2′ or H-3′ or H-4′), 3.81 (1H, pt, J = 9.4, 8.5 Hz, H-2′ or H-3′ or H-4′), 3.75–3.65 (3H, m, H-5′, H-6′a,b), 2.71 (3H, s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 159.7, 157.9 (C-3, C-6), 138.5, 138.2, 138.0, 137.6, 128.4–127.0 (Ar, C-4 or C-5), 125.7 (C-4 or C-5), 86.9, 81.5, 80.8, 79.5, 78.2 (C-1′–C-5′), 75.7, 75.1, 74.5, 73.4 (PhCH2), 69.1 (C-6′), 22.2 (CH3). ESI-HRMS positive mode (m/z): calcd for C39H41N2O5+ [M + H]+ 617.3010; C39H40N2NaO5+ [M + Na]+ 639.2829. Found: [M + H]+ 617.3012; [M + Na]+ 639.2831.
6-Methyl-3-(2′,3′,4′,6′-tetra-O-benzoyl-β-D-glucopyranosyl)pyridazine (55)
Prepared from tetrazine 46 (20 mg, 0.03 mmol) and 2,5-norbornadiene (6 μL, 0.06 mmol) in toluene according to General procedure IV. Reaction time: 30 min. Purified by column chromatography (hexane–EtOAc 1:1) to give 18 mg (90%) white solid. Rf = 0.21 (hexane–EtOAc 1:1); [α]D = −17 (c 0.51, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.04, 7.95, 7.82, 7.79 (4 × 2H, 4 dd, J = 7.8, 1.3 Hz in each, Ar), 7.69 (1H, d, J = 8.7 Hz, H-4 or H-5), 7.58–7.25 (13H, m, Ar, H-4 or H-5), 6.13 (1H, pt, J = 9.6, 9.6 Hz, H-2′ or H-3′ or H-4′), 5.85 (1H, pt, J = 9.8, 9.8 Hz, H-2′ or H-3′ or H-4′), 5.71 (1H, pt, J = 9.8, 9.8 Hz, H-2′ or H-3′ or H-4′), 5.25 (1H, d, J = 9.8 Hz, H-1′), 4.68 (1H, dd, J = 12.3, 2.6 Hz, H-6′a), 4.52 (1H, dd, J = 12.3, 5.1 Hz, H-6′b), 4.36 (1H, ddd, J = 9.8, 5.1, 2.6 Hz, H-5′), 2.65 (3H, s, CH3); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.1, 165.7, 165.3, 165.2 (4 × CO), 160.4, 156.5 (C-3, C-6), 133.5, 133.3, 133.2, 133.1, 129.9–128.3 (Ar), 127.7, 125.0 (C-4, C-5), 79.9, 76.7, 74.0, 72.2, 69.5 (C-1′–C-5′), 63.1 (C-6′), 22.1 (CH3). ESI-HRMS positive mode (m/z): calcd for C39H32N2NaO9+ [M + Na]+ 695.2000. Found: 695.2000.
6-Benzyl-3-(2′,3’,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)pyridazine (56)
Prepared from tetrazine 21 (30 mg, 0.04 mmol) and 2,5-norbornadiene (9 μL, 0.09 mmol) in toluene according to General procedure IV. Reaction time: 6 h. Purified by column chromatography (hexane–EtOAc 7:2) to give 25 mg (83%) white solid. Rf = 0.31 (hexane–EtOAc 2:1); [α]D = −28 (c 0.95, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 7.35–7.05, 6.87–6.85 (27H, m, Ar, H-4, H-5), 4.96, 4.92 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.87, 4.62 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.65 (1H, d, J = 9.0 Hz, H-1′), 4.55, 4.48 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.53, 4.12 (2 × 1H, 2 d, J = 10.9 Hz in each, PhCH2), 4.38, 4.33 (2 × 1H, 2 d, J = 15.0 Hz in each, PhCH2), 3.91, 3.86, 3.76 (3 × 1H, 3 pt, J = 9.0, 8.5 Hz in each, H-2′, H-3′, H-4′), 3.73–3.64 (3H, m, H-5′, H-6′a,b); 13C NMR (90 MHz, CDCl3) δ (ppm): 162.5, 158.3 (C-3, C-6), 138.5, 138.2, 138.1, 138.0, 137.6, 129.1–126.2 (Ar, C-4, C-5), 86.9, 81.5, 81.0, 79.6, 78.2 (C-1′–C-5′), 75.6, 75.1, 74.5, 73.4 (PhCH2), 69.1 (C-6′), 42.4 (PhCH2). ESI-HRMS positive mode (m/z): calcd for C45H45N2O5+ [M + H]+ 693.3323; C45H44N2NaO5+ [M + Na]+ 715.3142. Found: [M + H]+ 693.3319; [M + Na]+ 715.3135.
6-Phenyl-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)pyridazine (57)
Prepared from tetrazine 22 (30 mg, 0.04 mmol) and 2,5-norbornadiene (9 μL, 0.09 mmol) in toluene according to General procedure IV. Reaction time: 4 h. Purified by column chromatography (hexane–EtOAc 5:1) to give 27 mg (90%) white amorphous solid. Rf = 0.56 (hexane–EtOAc 2:1); [α]D = −23 (c 0.44, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.10–8.08 (2H, m, Ar), 7.68 (1H, d, J = 8.8 Hz, H-4 or H-5), 7.56–7.50 (3H, m, Ar), 7.45 (1H, d, J = 8.8 Hz, H-4 or H-5), 7.36–7.19, 7.10–7.04, 6.92–6.89 (20H, m, Ar), 4.97, 4.94 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.90, 4.63 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.72 (1H, d, J = 9.1 Hz, H-1′), 4.58, 4.52 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.57, 4.23 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 3.95, 3.91, 3.81 (3 × 1H, 3 pt, J = 9.0, 8.5 Hz in each, H-2′, H-3′, H-4′), 3.76–3.69 (3H, m, H-5′, H-6′a,b); 13C NMR (90 MHz, CDCl3) δ (ppm): 158.9, 158.6 (C-3, C-6), 138.5, 138.2, 138.1, 137.6, 130.0–127.2 (Ar), 126.2, 124.0 (C-4, C-5), 87.0, 81.4, 81.0, 79.6, 78.2 (C-1′–C-5′), 75.7, 75.1, 74.5, 73.5 (PhCH2), 69.1 (C-6′). ESI-HRMS positive mode (m/z): calcd for C44H43N2O5+ [M + H]+ 679.3166; C44H42N2NaO5+ [M + Na]+ 701.2986. Found: [M + H]+ 679.3161; [M + Na]+ 701.2980.
6-Phenyl-3-(2′,3′,4′,6′-tetra-O-benzoyl-β-D-glucopyranosyl)pyridazine (58)
Prepared from tetrazine 38 (30 mg, 0.07 mmol) and 2,5-norbornadiene (15 μL, 0.15 mmol) in toluene according to General procedure IV. Reaction time: 2 h. Purified by column chromatography (hexane–EtOAc 7:2) to give 35 mg (70%) white solid. Rf = 0.33 (hexane–EtOAc 2:1); [α]D = −53 (c 1.0, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.04–7.76, 7.55–7.25 (27H, m, Ar, H-4, H-5), 6.16, 5.87, 5.75 (3 × 1H, 3 pt, J = 9.6, 9.6 Hz and 9.8, 9.7 Hz and 9.8, 9.7 Hz, respectively, H-2′, H-3′, H-4′), 5.33 (1H, d, J = 9.8 Hz, H-1′), 4.69 (1H, dd, J = 12.4, 2.6 Hz, H-6′a), 4.53 (1H, dd, J = 12.4, 5.1 Hz, H-6′b), 4.41 (1H, ddd, J = 9.8, 5.1, 2.6 Hz, H-5′); 13C NMR (90 MHz, CDCl3) δ (ppm): 166.1, 165.7, 165.3, 165.2 (4 × CO), 159.4, 157.1 (C-3, C-6), 135.9, 133.5, 133.3, 133.2, 133.1, 130.2–127.2 (Ar), 125.6, 124.7 (C-4, C-5), 79.9, 76.8, 74.1, 72.2, 69.6 (C-1′–C-5′), 63.2 (C-6′). ESI-HRMS positive mode (m/z): calcd for C44H34N2NaO9+ [M + Na]+ 757.2157. Found: 757.2158.
6-(2-Pyridyl)-3-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)pyridazine (59)
Prepared from tetrazine 26 (20 mg, 0.03 mmol) and 2,5-norbornadiene (6 μL, 0.06 mmol) in toluene according to General procedure IV. Reaction time: 10 min. Purified by column chromatography (hexane–EtOAc 7:2) to give 18 mg (90%) white amorphous solid. Rf = 0.57 (hexane–EtOAc 1:1); [α]D = −16 (c 0.25, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.73–8.71 (2H, m, Py-H-3, Py-H-6), 8.43 (1H, d, J = 8.8 Hz, H-5), 7.89 (1H, dt, J = 7.8, 1.6 Hz, Py-H-4), 7.56 (1H, d, J = 8.8 Hz, H-4), 7.39 (1H, ddd, J = 7.5, 4.7, 1.1 Hz, Py-H-5), 7.36–7.19, 7.09–7.03, 6.92–6.89 (20H, m, Ar), 4.97, 4.93 (2 × 1H, 2 d, J = 11.1 Hz in each, PhCH2), 4.89, 4.64 (2 × 1H, 2 d, J = 10.8 Hz in each, PhCH2), 4.74 (1H, d, J = 9.1 Hz, H-1′), 4.58, 4.52 (2 × 1H, 2 d, J = 12.2 Hz in each, PhCH2), 4.53, 4.17 (2 × 1H, 2 d, J = 11.0 Hz in each, PhCH2), 3.94, 3.92, 3.80 (3 × 1H, 3 pt, J = 9.4, 8.6 Hz in each, H-2′, H-3′, H-4′), 3.76–3.69 (3H, m, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 159.9, 158.3 (C-3, C-6), 153.6 (Py-C-2), 149.4 (Py-C-6), 138.6, 138.2, 138.1, 137.5 (Ar), 137.1 (Py-C-4), 128.4–127.5 (Ar), 126.5, 124.7, 124.6, 121.7 (C-4, C-5, Py-C-3, Py-C-5), 86.9, 81.6, 81.0, 79.7, 78.2 (C-1′–C-5′), 75.7, 75.1, 74.6, 73.5 (4 × PhCH2), 69.1 (C-6′). ESI-HRMS positive mode (m/z): calcd for C43H42N3O5+ [M + H]+ 680.3119; C43H41N3NaO5+ [M + Na]+ 702.2938. Found: [M + H]+ 680.3119; [M + Na]+ 702.2933.
6-(2-Pyridyl)-3-(2′,3′,4′,6′-tetra-O-benzoyl-β-D-glucopyranosyl)pyridazine (60)
Prepared from tetrazine 41 (17 mg, 0.023 mmol) and 2,5-norbornadiene (3.5 μL, 0.035 mmol) in CH2Cl2 according to General procedure IV. Reaction time: 30 min. Purified by column chromatography (hexane–EtOAc 1:1) to give 16 mg (94%) white amorphous solid. Rf = 0.20 (hexane–EtOAc 1:1); [α]D = −60 (c 0.72, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 8.69 (1H, d, J = 4.7 Hz, Py-H-6), 8.61 (1H, d, J = 8.9 Hz, H-5), 8.56 (1H, d, J = 7.9 Hz, Py-H-3), 8.04 (2H, dd, J = 7.1, 1.2 Hz, Ar), 7.96 (2H, dd, J = 7.1, 1.2 Hz, Ar), 7.92 (1H, d, J = 8.9 Hz, H-4), 7.84–7.78 (5H, m, Ar, Py-H-4), 7.57–7.25 (13H, m, Ar, Py-H-5), 6.17, 5.88, 5.79 (3 × 1H, 3 pt, J = 9.6, 9.5 Hz and 9.8, 9.8 Hz and 9.8, 9.7 Hz, respectively, H-2′, H-3′, H-4′), 5.34 (1H, d, J = 9.9 Hz, H-1′), 4.72 (1H, dd, J = 12.2, 2.7 Hz, H-6′a), 4.55 (1H, dd, J = 12.2, 5.0 Hz, H-6′b), 4.43 (1H, ddd, J = 9.6, 5.0, 2.7 Hz, H-5′); 13C NMR (90 MHz, CDCl3) δ (ppm):166.1, 165.8, 165.3, 165.1 (4 × CO), 158.7, 158.4 (C-3, C-6), 153.2 (Py-C-2), 149.4 (Py-C-6), 137.0 (Py-C-4), 133.5, 133.2 (2), 133.1, 129.9–128.3 (Ar), 125.8, 125.4, 124.8 (C-4, C-5, Py-C-5), 121.7 (Py-C-3), 79.9, 76.8, 74.1, 72.2, 69.6 (C-1′–C-5′), 63.2 (C-6′). ESI-HRMS positive mode (m/z): calcd for C43H33N3NaO9+ [M + Na]+ 758.2109. Found: 758.2104.
3,6-Bis-(2′,3′,4′,6′-tetra-O-benzyl-β-D-glucopyranosyl)pyridazine (61)
Prepared from tetrazine 7 (30 mg, 0.03 mmol) and 2,5-norbornadiene (5.4 μL, 0.05 mmol) in toluene according to General procedure IV. Reaction time: 1.5 h. Purified by column chromatography (hexane–EtOAc 7:2) to give 25 mg (83%) white amorphous solid. Rf = 0.38 (hexane–EtOAc 2:1); [α]D = −20 (c 0.60, CHCl3). 1H NMR (360 MHz, CDCl3) δ (ppm): 7.32–6.86 (2 × 21H, m, Ar, H-4, H-5), 4.94, 4.91 (2 × (2 × 1H), 2 d, J = 11.2 Hz in each, PhCH2), 4.89, 4.63 (2 × (2 × 1H), 2 d, J = 10.8 Hz in each, PhCH2), 4.73 (2 × 1H, d, J = 9.6 Hz, H-1′), 4.58, 4.52 (2 × (2 × 1H), 2 d, J = 12.2 Hz in each, PhCH2), 4.43, 4.09 (2 × (2 × 1H), 2 d, J = 10.9 Hz in each, PhCH2), 3.93 (2 × 1H, pt, J = 8.9, 8.8 Hz, H-2′ or H-3′ or H-4′), 3.81–3.67 (2 × 5H, m, H-2′ and/or H-3′ and/or H-4′, H-5′, H-6′a, H-6′b); 13C NMR (90 MHz, CDCl3) δ (ppm): 160.1 (C-3, C-6), 138.5, 138.1 (2), 137.3 (Ar), 128.4–127.6 (Ar), 125.9 (C-4, C-5), 86.9, 81.3, 80.8, 79.5, 78.1 (C-1′–C-5′), 75.7, 75.1, 74.4, 73.5 (4 × PhCH2), 69.2 (C-6′). ESI-HRMS positive mode (m/z): calcd for C72H72N2NaO10+ [M + Na]+ 1147.5079. Found: 1147.5072.
Author contributions
É. B. conceived the research, supervised the synthetic work and wrote the paper. D. T. K., F. G., A. F., E. K., A. D., Zs. V. and E. Sz. synthesized the compounds. L. S. wrote the paper and contributed to the manuscript editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the National Research, Development and Innovation Office of Hungary (Grant OTKA FK 125067).References
- A. Pinner, Über die Einwirkung von Hydrazin auf Imidoäther, Liebigs Ann. Chem., 1897, 297, 221–271 CrossRef CAS.
- N. Saracoglu, Recent advances and applications in 1,2,4,5-tetrazine chemistry, Tetrahedron, 2007, 63, 4199–4236 CrossRef CAS.
- G. Clavier and P. Audebert, s-Tetrazines as Building Blocks for New Functional Molecules and Molecular Materials, Chem. Rev., 2010, 110, 3299–3314 CrossRef CAS PubMed.
- L. Wang, J. W. Zhang, J. Z. Zhao, P. Yu, S. Wang, H. M. Hu and R. Wang, Recent synthesis of functionalized s-tetrazines and their application in ligation reactions under physiological conditions: a concise overview, Catal. Rev.: Sci. Eng., 2020, 62, 524–565 CrossRef CAS.
- S. K. Choi, J. Kim and E. Kim, Overview of Syntheses and Molecular-Design Strategies for Tetrazine-Based Fluorogenic Probes, Molecules, 2021, 26, 1868 CrossRef CAS.
- J. Mondal and A. Sivaramakrishna, Functionalized Triazines and Tetrazines: Synthesis and Applications, Top. Curr. Chem., 2022, 380, 34 CrossRef CAS PubMed.
- D. L. Boger, Diels–Alder Reactions of Azadienes, Tetrahedron, 1983, 39, 2869–2939 CrossRef CAS.
- D. L. Boger, Diels–Alder Reactions of Heterocyclic Azadienes - Scope and Applications, Chem. Rev., 1986, 86, 781–793 CrossRef CAS.
- H. Neunhoeffer, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky and C. V. Rees, Pergamon, Oxford, 1984, vol. 3, ch. 2.21, pp. 531–572 Search PubMed.
- H. Neunhoeffer, in Methoden der Organischen Chemie (Houben-Weyl), Hetarenes III, Part 3, Georg Thieme Verlag, Stuttgart, 4th edn, 1997, vol. E9, pp. 870–916 Search PubMed.
- H. W. Shih, D. N. Kamber and J. A. Prescher, Building better bioorthogonal reactions, Curr. Opin. Chem. Biol., 2014, 21, 103–111 CrossRef CAS PubMed.
- M. King and A. Wagner, Developments in the Field of Bioorthogonal Bond Forming Reactions-Past and Present Trends, Bioconjugate Chem., 2014, 25, 825–839 CrossRef CAS PubMed.
- M. M. Zheng, L. Zheng, P. Y. Zhang, J. B. Li and Y. Zhang, Development of Bioorthogonal Reactions and Their Applications in Bioconjugation, Molecules, 2015, 20, 3190–3205 CrossRef PubMed.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Inverse electron demand Diels–Alder reactions in chemical biology, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- F. Liu, Y. Liang and K. N. Houk, Bioorthogonal Cycloadditions: Computational Analysis with the Distortion/Interaction Model and Predictions of Reactivities, Acc. Chem. Res., 2017, 50, 2297–2308 CrossRef CAS PubMed.
- M. Grammel and H. C. Hang, Chemical reporters for biological discovery, Nat. Chem. Biol., 2013, 9, 475–484 CrossRef CAS.
- R. E. Bird, S. A. Lemmel, X. Yu and Q. A. Zhou, Bioorthogonal Chemistry and Its Applications, Bioconjugate Chem., 2021, 32, 2457–2479 CrossRef CAS.
- K. Porte, M. Riberaud, R. Chatre, D. Audisio, S. Papot and F. Taran, Bioorthogonal Reactions in Animals, ChemBioChem, 2021, 22, 100–113 CrossRef CAS.
- E. Brachet and P. Belmont, Inverse Electron Demand Diels–Alder (IEDDA) Reactions: Synthesis of Heterocycles and Natural Products Along with Bioorthogonal and Material Sciences Applications, Curr. Org. Chem., 2016, 20, 2136–2160 CrossRef CAS.
- E. Kozma, O. Demeter and P. Kele, Bio-orthogonal Fluorescent Labelling of Biopolymers through Inverse-Electron-Demand Diels–Alder Reactions, ChemBioChem, 2017, 18, 486–501 CrossRef CAS.
- S. Mayer and K. Lang, Tetrazines in Inverse-Electron-Demand Diels–Alder Cycloadditions and Their Use in Biology, Synthesis, 2017, 830–848 CAS.
- K. K. Palaniappan and C. R. Bertozzi, Chemical Glycoproteomics, Chem. Rev., 2016, 116, 14277–14306 CrossRef CAS.
- T. J. Sminia, H. Zuilhof and T. Wennekes, Getting a grip on glycans: A current overview of the metabolic oligosaccharide engineering toolbox, Carbohydr. Res., 2016, 435, 121–141 CrossRef CAS PubMed.
- H. Y. Yoon, H. Koo, K. Kim and I. C. Kwon, Molecular imaging based on metabolic glycoengineering and bioorthogonal click chemistry, Biomaterials, 2017, 132, 28–36 CrossRef CAS.
- B. V. Rao, S. Dhokale, P. R. Rajamohanan and S. Hotha, A tetrazine templated method for the synthesis of ternary conjugates, Chem. Commun., 2013, 49, 10808–10810 RSC.
- T. Machida, K. Lang, L. Xue, J. W. Chin and N. Winssinger, Site-Specific Glycoconjugation of Protein via Bioorthogonal Tetrazine Cycloaddition with a Genetically Encoded trans-Cyclooctene or Bicyclononyne, Bioconjugate Chem., 2015, 26, 802–806 CrossRef CAS PubMed.
- T. Cheewawisuttichai and M. Brichacek, Development of a multifunctional neoglycoside auxiliary for applications in glycomics research, Org. Biomol. Chem., 2021, 19, 6613–6617 RSC.
- É. Bokor, S. Kun, D. Goyard, M. Tóth, J.-P. Praly, S. Vidal and L. Somsák, C-Glycopyranosyl arenes and hetarenes: Synthetic methods and bioactivity focused on antidiabetic potential, Chem. Rev., 2017, 117, 1687–1764 CrossRef PubMed.
- L. Somsák, É. Bokor, L. Juhász, S. Kun, L. Lázár, É. Juhász-Tóth and M. Tóth, New Syntheses towards C-Glycosyl Type Glycomimetics, Pure Appl. Chem., 2019, 91, 1159–1175 CrossRef.
- É. Bokor, in Recent Trends in Carbohydrate Chemistry – Synthesis, Structure and Function of Carbohydrates, ed. A. P. Rauter, B. E. Christensen, L. Somsák, P. Kosma and R. Adamo, Elsevier, Amsterdam, 2020, vol. 1, ch. 7, pp. 253–300 Search PubMed.
- E. Szennyes, É. Bokor, P. Langer, G. Gyémánt, T. Docsa, Á. Sipos and L. Somsák, The first general synthesis of 2-C-(β-D-glycopyranosyl)pyrimidines and their evaluation as inhibitors of some glycoenzymes, New J. Chem., 2018, 42, 17439–17446 RSC.
- E. Szennyes, G. Gyémánt, L. Somsák and É. Bokor, Synthesis of new series of 2-C-(β-D-glucopyranosyl)pyrimidines and their evaluation as inhibitors of some glycoenzymes, Molecules, 2020, 25, 701 CrossRef CAS PubMed.
- Á. Sipos, E. Szennyes, N. É. Hajnal, S. Kun, K. E. Szabó, K. Uray, L. Somsák, T. Docsa and É. Bokor, Dual-Target Compounds against Type 2 Diabetes Mellitus: Proof of Concept for Sodium Dependent Glucose Transporter (SGLT) and Glycogen Phosphorylase (GP) Inhibitors, Pharmaceuticals, 2021, 14, 364 CrossRef PubMed.
- E. Szennyes, É. Bokor, G. Batta, T. Docsa, P. Gergely and L. Somsák, Improved preparation of 4(5)-aryl-2-(β-D-glucopyranosyl)-imidazoles, the most efficient glucose analogue inhibitors of glycogen phosphorylase, RSC Adv., 2016, 6, 94787–94794 RSC.
- J. Yang, M. R. Karver, W. L. Li, S. Sahu and N. K. Devaraj, Metal-Catalyzed One-Pot Synthesis of Tetrazines Directly from Aliphatic Nitriles and Hydrazine, Angew. Chem., Int. Ed., 2012, 51, 5222–5225 CrossRef CAS PubMed.
- D. Z. Wang, W. X. Chena, Y. Q. Zheng, C. F. Dai, L. F. Wang and B. H. Wang, A general and efficient entry to asymmetric tetrazines for click chemistry applications, Heterocycl. Commun., 2013, 19, 171–177 CrossRef CAS.
- I. Kacsir, A. Sipos, G. Ujlaki, P. Buglyó, L. Somsák, P. Bai and É. Bokor, Ruthenium half-sandwich type complexes with bidentate monosaccharide ligands show antineoplastic activity in ovarian cancer cell models through reactive oxygen species production, Int. J. Mol. Sci., 2021, 22, 454 CrossRef PubMed.
- Z. Hadady, M. Tóth and L. Somsák, C-(β-D-glucopyranosyl) heterocycles as potential glycogen phosphorylase inhibitors, ARKIVOC, 2004, (vii), 140–149 Search PubMed.
- Protecting Groups: Strategies and Applications in Carbohydrate Chemistry, ed. S. Vidal, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 1st edn, 2019 Search PubMed.
- T. Polat and R. J. Linhardt, Zinc triflate-benzoyl bromide: a versatile reagent for the conversion of ether into benzoate protecting groups and ether glycosides into glycosyl bromides, Carbohydr. Res., 2003, 338, 447–449 CrossRef CAS.
- S. Y. Kang, K. S. Song, J. Lee, S. H. Lee and J. Lee, Synthesis of pyridazine and thiazole analogs as SGLT2 inhibitors, Bioorg. Med. Chem., 2010, 18, 6069–6079 CrossRef CAS PubMed.
- I. Maeba, K. Iwata, F. Usami and H. Furukawa, C-Nucleosides. I. Synthesis of 3-(β-D-ribofuranosyl)pyridazines, J. Org. Chem., 1983, 48, 2998–3002 CrossRef CAS.
- I. Maeba, M. Suzuki, O. Hara, T. Takeuchi, T. Iijima and H. Furukawa, C-Nucleosides. 6. Synthesis of 5-Methoxy-5-(2,3,5-tri-O-benzoyl-β-D-ribofuranosyl)furan-2(5H)-one and Its Ring Transformation, J. Org. Chem., 1987, 52, 4521–4526 CrossRef CAS.
- I. Maeba, T. Iijima, Y. Matsuda and C. Ito, C-Nucleosides. 12. Synthesis of 2’-Deoxy Pyridazinone C-Nucleoside from 2-(2,3,5-Tri-O-benzoyl-β-D-ribofuranosyl)furan, J. Chem. Soc.-Perkin Trans. 1, 1990, 73–76 RSC.
- I. Maeba, K. Osaka, N. Morishita, K. Fujioka and C. Ito, C-Nucleosides. 14. Synthesis of (6S)- and (6R)-6-Methoxy-6-(β-D-ribofuranosyl)pyran-3(2H,6H)-one. Potential Chiral Synthons, and 6-(β-D-Ribofuranosyl)pyridazine-3-carboxamide, a 2-azanicotinamide C-Nucleoside, from 6-Hydroxy-6-(2,3,5-tri-O-benzoyl-β-D-ribofuranosyl)pyran-3(2H,6H)-one, J. Chem. Soc.-Perkin Trans. 1, 1991, 939–944 RSC.
- T. Tomori, K. Nagaoka, L. Takeshita, T. Shiozawa, Y. Miyatake, Y. Masaki, M. Sekine and K. Seio, Deoxynucleoside Triphosphate Containing Pyridazin-3-one Aglycon as a Thymidine Triphosphate Substitute for Primer Extension and Chain Elongation by Klenow Fragments, J. Org. Chem., 2018, 83, 8353–8363 CrossRef CAS PubMed.
- P. G. Baraldi, A. Bigoni, B. Cacciari, C. Caldari, S. Manfredini and G. Spalluto, Nitrile Oxide [3 + 2] Cycloaddition: Application to the Synthesis of 6-Substituted 3(2H)-Pyridazinones and 6-Substituted 4,5-Dihydro-4-hydroxy-3(2H)-pyridazinones, Synthesis, 1994, 1158–1162 CrossRef CAS.
- M. Richter and G. Seitz, 4-(β-D-Ribofuranosyl)pyridazines: Novel C-Nucleosides by [4+2]-Cycloadditions with Inverse Electron Demand, Arch. Pharm., 1994, 327, 365–370 CrossRef CAS.
- H. Wamhoff and H. Warnecke, Synthesis and cycloaddition reactions of [2-deoxy-3,5-bis[O-(p-toluoyl)]-α-D-ribofuranosyl]ethyne, ARKIVOC, 2001, (ii), 1085–1090 Search PubMed.
- D. L. Boger, M. Zhang and N. Haider, Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2001 DOI:10.1002/047084289X.rd389.pub2.
- U. Joshi, S. Josse, M. Pipelier, F. Chevallier, J. P. Pradere, R. Hazard, S. Legoupy, F. Huet and D. Dubreuil, Novel pyrrole C-nucleosides by nitrogen extrusion from pyridazine C-nucleosides, Tetrahedron Lett., 2004, 45, 1031–1033 CrossRef CAS.
- S. Naud, M. Pipelier, G. Viault, A. Adjou, F. Huet, S. Legoupy, A.-M. Aubertin, M. Evain and D. Dubreuil, Synthesis of polyhydroxylated pyrano-pyrrole derivatives from carbohydrate precursors, Eur. J. Org. Chem., 2007, 3296–3310 CrossRef CAS.
- M. X. Zhu and S. Messaoudi, Diastereoselective Decarboxylative Alkynylation of Anomeric Carboxylic Acids Using Cu/Photoredox Dual Catalysis, ACS Catal., 2021, 11, 6334–6342 CrossRef CAS.
- F. Cermola, M. R. Iesce and G. Buonerba, Dye-Sensitized Photooxygenation of Furanosyl Furans: Synthesis of a New Pyridazine C-Nucleoside, J. Org. Chem., 2005, 70, 6503–6505 CrossRef CAS.
- F. Cermola and M. R. Iesce, Dye-sensitized photooxygenation of sugar-furans as synthetic strategy for novel C-nucleosides and functionalized exo-glycals, Tetrahedron, 2006, 62, 10694–10699 CrossRef CAS.
- K. Czifrák, P. Szilágyi and L. Somsák, Anomeric α-azido acid (2-azido-2-deoxy-hept-2-ulopyranosonic acid) derivatives en route to peptides incorporating sugar amino acids, Tetrahedron: Asymm., 2005, 16, 127–141 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nj03920f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |