
Multifunctional LnIII complexes with SMM, luminescence and MCE properties constructed using selected phosphine oxide and diketonate†
Han
Yan
,
Zi-Wei
Che
and
Wen-Bin
Sun
 *
*
Key Laboratory of Functional Inorganic Material Chemistry Ministry of Education, School of Chemistry and Material Science Heilongjiang University, 74 Xuefu Road, Harbin, 150080, P. R. China. E-mail: wenbinsun@126.com
First published on 15th November 2022
Abstract
A series of mononuclear and binuclear lanthanideIII complexes with formulae [Dy(Cy3PO)(tmhd)3] (1), [Dy2(dppeO2)(tmhd)6] (2) and [Ln2(dppeO2)(acac)6(H2O)2] (3, Ln = Dy, 4, Ln = Gd, 5, Ln = Eu, 6, Ln = Tb) (Cy3PO = tricyclohexyl phosphine oxide, tmhd = 2,2,6,6-tetramethyl-3,5-heptane dionate, acac = acetylacetone, dppeO2 = 1,2-bis(diphenylphosphino)ethane dioxide) constructed using β-diketonate combined with mono- and bi-phosphine oxide ligands were structurally, optically and magnetically characterized. The easy to modify backbone of monodentate phosphine oxide and bidentate β-diketonate enables them to modulate local coordination geometries around lanthanide ions by means of an electronic effect and/or steric hindrance of substituents on the terminal of these ligands and consequently optimize the corresponding optical and magnetic properties. In this vein, a series of Dy containing single-molecule magnets (SMMs) were constructed, in which complex 3 possessed a relatively higher local geometry symmetry (D4d) and displayed a significantly slow magnetic relaxation behavior at a hysteresis temperature of 3 K. In addition, the Gd congener complex 4 showed a pronounced magnetocaloric effect (MCE) with a –ΔSmaxm value of 20.4 J kg−1 K−1, and more strikingly, a field induced SMM behavior was observed in 4 despite the isotropic nature of GdIII ions. Also of note is that the suitable triple state energy of ligands matches well with the excited states of EuIII, TbIII and DyIII ions, which led to the corresponding characteristic metal-centered luminescence emission. In this work, the deliberately selected β-diketonate and phosphine oxide combined with different lanthanide ions led to a series of multifunctional materials with SMM, luminescence and MCE properties.
Introduction
Since the first single-molecule magnetic (SMM) materials were discovered,1 these materials have always attracted increasing attention due to their unique slow magnetic relaxation behavior occurring because of magnetic bistabilities and have enormous potential for application in data storage, quantum computing, etc.2–4 In recent years, because of their significant anisotropy arising from the large unquenched orbital angular momentum, lanthanide (Ln) ions have become the most attractive candidates for constructing SMMs with a large energy barrier (Ueff) and a high blocking temperature (TB),5–7 combined with ligand field effects, and lanthanide-containing species have been dominantly used in many high-performance SMMs reported.8,9 To date, Ln-based SMMs have made a major breakthrough, the highest energy barrier has reached 1541 cm−1 and the blocking temperature has surpassed 80 K which is above the liquid nitrogen temperature.10,11 However, the application of SMMs at room temperature remains a great challenge; in particular, the blocking temperature is only observed at low temperatures, which is mainly due to the obstacle derived from the faster quantum tunnelling of magnetization (QTM) through the barriers. In view of this, crystal-field theory reveals that the local coordination environment surrounding the spin center plays an important role in determining the magnetic anisotropy of an individual spin carrier. Therefore, regulating the symmetry of charge distribution to suppress QTM becomes an effective method to improve SMM performance and is proved to be an efficient strategy to improve energy barriers and blocking temperatures, and many Ln–SMMs with high order axial symmetry including Cn (n ≥ 7), S8/D4d, C5h/D5h and S12/D6d have been reported in the past few years.12–21However, due to the inherent high coordination number and flexible coordination patterns of lanthanide ions, it is not easy to synthesize complexes with high axial symmetry. Among the organic coordination ligands, β-diketone and phosphine oxide compounds and their derivatives, as excellent oxygen-containing ligands, have been widely used to build lanthanide-based optical, electric, magnetic and fluorescent materials.22–27 More importantly, they have become ideal candidates for the construction of Ln–SMMs because of the convenient modification of their backbone and they can be modulated by means of an electronic effect and a steric hindrance effect of substituents of these ligands.
In this vein, we presented a series of mononuclear and binuclear lanthanide complexes, in which β-diketonate and phosphine oxide ligands jointly combined DyIII-based SMMs, a GdIII congener with a MCE and the characteristics of EuIII, TbIII and DyIII centered luminescent species, which provided a new idea for the assembly of multi-functional lanthanide containing materials (Scheme 1).
 | ||
| Scheme 1 Syntheses of complexes 1–6. | ||
Experimental
Materials and methods
All chemicals and solvents except dppeO2 were obtained from commercial sources and were used as received, without further purification. The ligand dppeO2 was prepared according to the literature procedure.28 Elemental (C and H) analyses were performed using a PerkinElmer 2400 analyzer. The magnetic susceptibilities of complexes were measured using a Quantum Design VSM superconducting quantum interference device (SQUID) magnetometer. Data were corrected for the diamagnetism of the samples using Pascal constants and the sample holder by measurements. Excitation spectra and emission spectra were recorded using a PerkinElmer luminescence spectrophotometer (FL6500).Synthesis of [Dy(Cy3PO)(tmhd)3] (1)
A methanol solution (10 mL) of Cy3PO (0.12 mmol, 0.036 g) was added in a round flask. Under stirring, Dy(tmhd)3 (0.12 mmol, 0.086 g) was added to the solution. The mixture was stirred and heated under refluxing for 6 hours. The resultant solution was filtered immediately and set aside for slow evaporation, producing colorless block crystals within one week. Crystal yield: 0.056 g (47.4%). Elemental analysis calcd (%) for 1, C51H72DyO7P: C 61.84, H 7.33; found: C 61.73, H 7.40.Synthesis of [Dy2(dppeO2)(tmhd)6] (2)
A methanol solution (5 mL) of dppeO2 (0.12 mmol, 0.052 g) was added in a round flask. After stirring for 5 minutes, into which the methanol (8 mL) solution of Dy(tmhd)3 (0.12 mmol, 0.086 g) was added. The mixture was stirred and heated under reflux for 5 hours. The resultant solution was filtered immediately and set aside for slow evaporation, producing colorless block crystals within one week. Crystal yield: 0.055 g (49.8%). Elemental analysis calcd (%) for 2, C92H138Dy2O14P2: C 59.57, H 7.50; found: C 59.52, H 7.59.Synthesis of [Dy2(dppeO2)(acac)6(H2O)2] (3)
A methanol solution (5 mL) of dppeO2 (0.12 mmol, 0.052 g) was added in a round flask. After stirring for 5 minutes, into which the methanol (7 mL) solution of Dy(acac)3·H2O (0.12 mmol, 0.058 g) was added. The mixture was stirred and heated under reflux for 5 hours. The resultant solution was filtered immediately and set aside for slow evaporation, producing colorless block crystals within one week. Crystal yield: 0.037 g (45%). Elemental analysis calcd (%) for 3, C56H70Dy2O16P2: C 48.53, H 5.09; found: C 48.44, H 5.10.Synthesis of [Gd2(dppeO2)(acac)6(H2O)2] (4)
Complex 4 was synthesized using the same procedure as that used for complex 3, except that Gd(acac)3·H2O (0.12 mmol, 0.057 g) was used instead of Dy(acac)3·H2O. Crystal yield: 0.040 g (48.7%). Elemental analysis calcd (%) for 4, C56H70Gd2O16P2: C 48.90; H 5.13; found: C 48.8; H 5.11.Synthesis of [Eu2(dppeO2)(acac)6(H2O)2] (5)
Complex 5 was synthesized using the same procedure as that used for complex 3, except that Eu(acac)3·H2O (0.12 mmol, 0.056 g) was used instead of Dy(acac)3·H2O. Crystal yield: 0.038 g (46.5%). Elemental analysis calcd (%) for 5, C56H70Eu2O16P2: C 49.27; H 5.17; found: C 49.32; H 5.19.Synthesis of [Tb2(dppeO2)(acac)6(H2O)2] (6)
Complex 6 was synthesized using the same procedure as that used for complex 3, except that Tb(acac)3·H2O (0.12 mmol, 0.057 g) was used instead of Dy(acac)3·H2O. Crystal yield: 0.036 g (44.2%). Elemental analysis calcd (%) for 6, C56H70Tb2O16P2: C, 48.78; H, 5.12; found: C 48.80; H 5.09.X-Ray crystallographic studies of 1–6.
Data were collected using a Nonius Kappa CCD diffractometer with Mo Ka radiation (λ = 0.71073 Å). The structures of complexes were solved by direct methods and refined on F2 by full-matrix least-squares using the SHELXTL program29 and Olex 2.30 All non-hydrogen atoms were refined with isomorphous displacement parameters. All the crystallographic data of complexes are summarized in Tables S1 and S2 (ESI†), and the selected bond lengths and angles of complexes 1–6 are tabulated in Tables S3–S8 (ESI†).Results and discussion
The phase purity of all complexes is confirmed by PXRD (Fig. S30–S33 ESI†). The crystal structural analysis reveals that complex 1 is a mononuclear structure and complexes 2–6 are binuclear structures, which crystallize in a monoclinic crystal system P21/c for complex 1, and a triclinic crystal system P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) for complexes 2–6 (Tables S1 and S2 ESI†). As shown in Fig. S1 (ESI†), the central DyIII ions of complex 1 is seven-coordinated by six oxygen atoms from the three tmhd ligands and one oxygen atom from the Cy3PO molecule, and the local coordination geometry is a capped trigonal prism with C2v symmetry estimated by using the continuous shape measure (CShM) method31 (Table S9, ESI†).
for complexes 2–6 (Tables S1 and S2 ESI†). As shown in Fig. S1 (ESI†), the central DyIII ions of complex 1 is seven-coordinated by six oxygen atoms from the three tmhd ligands and one oxygen atom from the Cy3PO molecule, and the local coordination geometry is a capped trigonal prism with C2v symmetry estimated by using the continuous shape measure (CShM) method31 (Table S9, ESI†).
In order to improve the axial symmetry of the single ion center and assemble them together, as the initial strategy, the biphosphine oxide ligand dppeO2 instead of Cy3PO was used, which led to binuclear complex 2. Unfortunately, individual DyIII ions remain seven-coordinated coordination geometry and the lowest CShM value suggests that the coordination geometry of the DyIII ions is a capped octahedron with C3v symmetry for complex 2 (Fig. 1 and Table S9, ESI†), in which the bridging dppeO2 links two DyIII ions in a symmetric fashion with a Dy1–O1 distance of 2.290(2) Å. This is most likely due to the larger steric hindrance of six methyl groups on the terminal tmhd, which prevents extra coordination atoms ligating to DyIII ions to improve the coordination number.
 | ||
| Fig. 1 Partially labeled molecular structure of complex 2, and the minor part of the disorder is shown as the front ellipse in the blue line. Color code: Dy (teal), P (pink), O (red), and C (grey). | ||
Further to enhance the molecular axiality, a relatively small β-diketonate acac without six methyl was used and resulted in complex 3, in which the coordination number was enhanced to eight with a higher symmetry (D4d) coordination geometry successfully (Fig. 2 and Table S9, ESI†). By means of this way, the corresponding GdIII, EuIII and TbIII analogues 4–6 (Fig. S2–S4, ESI†) were obtained and the X-ray analysis indicated that they possessed almost the same local coordination environments (Tables S2 and S6–S8, ESI†). Taking complex 3 as an example to describe the whole series molecular structures, in which the central DyIII ion is eight-coordinated by six oxygen atoms from the three acac ligands and one oxygen atom from dppeO2 and one oxygen atom from H2O (Fig. 2). In order to quantify the degrees of distortion of the coordination spheres of the real complexes, the coordination geometry of DyIII ions was estimated by the continuous-shape measure (CShM) method using the SHAPE software.31,32 They lie among the ideal square antiprism (SAPR-8, D4d), biaugmented trigonal prism (BTPR-8, C2v) and triangular dodecahedron (TDD-8, D2d) with relatively small deviation values of 0.373, 1.988, and 2.011, respectively (Table S9, ESI†). Obviously, the relatively small CShM value reveals that the coordination geometries surrounding two DyIII ions are close to the ideal D4d square antiprism (Table S9, ESI†) (CShM = 0.373, CShM = 0 corresponds to the ideal polyhedron). The upper planes of the prism consist of one oxygen atom from dppeO2 and one oxygen atom from H2O and two oxygen atoms from the acac ligand, and the bottom planes of the prism consist of four oxygen atoms from two acac (Fig. 2). By comparing the geometric configurations of complexes 1–3, it is clear that the CShM value of Dy1 in complex 3 is closest to the ideal square antiprism (SAPR-8, D4d). Obviously, using small β-diketonate acac successfully allows the coordination of one H2O molecule and completes the eight-coordinated polyhedron with a D4d symmetry. In other words, complex 3 has the higher coordination symmetry. The significant difference of 1–3 above is mainly due to different ligands with distinct steric hindrance, which likely result in different magnetic behaviors. We boldly predicted that complex 3 with a higher symmetry would exhibit more significant SMM behaviors than complexes 1 and 2.33 Due to large distances, the intra- and intermolecular Dy–Dy interactions are negligible.34
 | ||
| Fig. 2 Partially labeled molecular structure of complex 3 and the local coordination geometry of the DyIII ions. Color code: Dy (teal), P (pink), O (red), and C (grey). | ||
Magnetic properties
The direct current (dc) magnetic properties of complexes 1–4 were investigated under a 1000 Oe field in a temperature range of 2–300 K (Fig. 3). At room temperature, the χMT values of complexes 1–3 are 14.73, 27.98 and 27.84 cm3 K mol−1, which are in good agreement with the theoretical values (14.17 cm3 K mol−1 and 28.34 cm3 K mol−1) of one non-interacting DyIII ion (6H15/2, S = 5/2, L = 5, g = 4/3, C = 14.17 cm3 K mol−1) and two non-interacting DyIII ions (Fig. 3). Upon cooling, the χMT values of complexes 1–3 decreased gradually in a wide temperature range from 300 to 50 K, and then decrease sharply to 2 K (Fig. 3), which is most likely due to the depopulation of the Stark sublevels and/or the significant magnetic anisotropy present in DyIII systems. For complex 5, the χMT value at room temperature of 5.44 cm3 K mol−1 owes to the magnetic contribution of EuIII (Fig. 3). The χMT value tends to zero at low temperatures indicating the presence of a MJ = 0 as a ground state in a compound. The χMT value of 23.56 cm3 K mol−1 for 6 recorded at 300 K is consistent with a theoretical value of 23.64 cm3 K mol−1 expected for two TbIII ions (7F6, S = 3, L = 3, and g = 3/2) (Fig. 3). The M versus H plots under a 0–7 T dc field at 2, 3, 5, and 8 K of complexes 1–3 show a relatively rapid increase in the magnetization under a low field to reach almost saturation for magnetic fields of 7 T, and then the maximum value reaches 4.69, 10.90 and 11.45 Nβ, respectively (Fig. S5 and S6, ESI†). | ||
| Fig. 3 Temperature dependence of the magnetic susceptibility χMT at 1000 Oe for complexes 1–4. | ||
The values are seriously lower than the theoretical saturation values of one DyIII ion (10 Nβ, g = 4/3, J = 15/2) in 1 and two free DyIII ions (20 Nβ) in 2 and 3 (Fig. S5 and S6, ESI†), which is due to crystal-field effects leading to significant magnetic anisotropy and/or low-lying excited states.
Polynuclear GdIII complexes normally exhibit a typical magnetocaloric effect (MCE), whose magnetic entropy varies with an applied magnetic field and is expected to be used in cooling applications due to its adiabatic demagnetization.35 In order to explore the potential MCE properties of the binuclear GdIII ions, complex 4 [Gd2(dppeO2)(acac)6(H2O)2], the dc susceptibility measurement was performed. The χMT value of 15.02 cm3 K mol−1 at 300 K (Fig. 3) is consistent with the expected value of 15.76 cm3 K mol−1 for two uncoupled GdIII ions (8S7/2, S = 7/2, and L = 0). Usually, the antiferromagnetic interaction between GdIII ions in complex 4 is very weak, due to the relatively large separated dppeO2 ligand bridging the two GdIII ions, which is suitable for generating a large magnetocaloric effect. In view of the isotropy nature of 4, the MCE properties were further studied. As expected, the –ΔSm value increases with the applied magnetic field at the same temperature (Fig. 4). Under an external field change with a ΔH of 7.0 T at 2 K for 4, the maximum –ΔSm value of 20.4 J kg−1 K−1 (60.9 mJ cm−3 K−1) (Fig. 4) can be calculated by using the Maxwell eqn (1),36–38 which is close to the theoretical value of 25.69 J kg−1 K−1 calculated by nR ln(2S + 1).39 This –ΔSm value is comparable with series of binuclear Gd2 MCE congeners.40
 | (1) |
 | ||
| Fig. 4 Temperature dependence of the out-of-phase (χ′′) ac susceptibility of 4 (left) under a 1500 dc field and the temperature dependence of the magnetic entropy change (–ΔSm) as calculated from the magnetization data of 4 (right). | ||
More strikingly, the temperature dependence of out-of-phase (χ′′) ac susceptibility was detected for 4 under a 1500 Oe dc field (Fig. 4), although the isotropic nature of GdIII ions. While this is not unexpected for the GdIII congeners, there have been similar reports related to this phenomenon.41,42 In this case, the analysis of the temperature dependence of the relaxation time was performed assuming a τ−1 = CTn dependence. The slow relaxation is not attributed to an anisotropy barrier, but to the phonon-bottleneck effect originated by the trapping of resonant phonons that are unable to release the energy of the spin bath to the thermal.43 A similar analysis performed on 4 provides a better agreement with the experimental data and a best fit value n = 2.07 (Fig. S15, ESI†).44 An exponent equal to 2 is theoretically expected for this phonon-bottleneck phenomenon,42 suggesting that this mechanism could be responsible for the slow magnetic relaxation observed in 4.
In contrast, for the anisotropic DyIII ion, the slow magnetic relaxation behaviour is detected clearly in the corresponding complexes 1–3. To explore their dynamic magnetic properties, alternating-current (ac) measurements were performed on 1–3 in a temperature range of 2–10 K under a zero dc field at frequencies between 1 and 1000 Hz (Fig. S7–S11, ESI†). Under 0 Oe, complex 1 does not exhibit any out-of-phase (χ′′) (Fig. S7, ESI†) peaks. For 2, the temperature and frequency dependences of in-phase (χ′) and out-of-phase (χ′′) ac susceptibilities confirm the zero-field slow magnetization relaxation (Fig. S8 and S10, ESI†). However, the full peak was not observed, only the upturning and tailing of the peak in the low temperature area, which normally indicates the interference of QTM (Fig. S8, ESI†). For 3, the out-of-phase (χ′′) susceptibilities (Fig. S9, ESI†) exhibit a temperature dependent full peak with a maximum of 7.5 K. This indicates that the SMM behaviour of complex 3 is more significant than that of complexes 1 and 2. The results revealed that complexes with a high local axial symmetry display more significant SMM behaviors, which are consistent with the theoretical principle.
Generally, for lanthanide SMM systems, applying an external magnetic field is an effective means to suppress QTM. The frequency dependence of the out-of-phase (χ′′) ac susceptibility component at 2 K under different dc fields for complexes 1–3 is shown in Fig. S20 and S21, ESI.† From the figure, it can be determined that the optimal dc field for complex 1 is 800 Oe, while that for complexes 2 and 3 cannot be determined. In view of this, a moderate dc field of 1500 Oe is applied on samples 2 and 3, respectively. As expected, after applying a dc magnetic field, QTM is significantly compressed in complexes 1–3, and the out-of-phase (χ′′) component of the ac susceptibility clearly exhibits a temperature dependent full peak with maxima of 4.5 K, 8.5 K and 8 K, respectively (Fig. S12–S14, ESI†). Additionally, a frequency dependence of the maximum associated with a single relaxation process appears apparently on the plot of the variation of χ′′ versus the frequency of the oscillating field between 1 and 1000 Hz under a dc field (Fig. S16–S18, ESI†) for 1–3. Meanwhile, the χ′′ frequency dependence of complexes 1–3 shows only a single peak under an external magnetic field (Fig. S16–S18, ESI†), which may indicate a single thermally activated relaxation mechanism.
The graphical representation of χ′′ versus χ′ (Cole–Cole plot44,45) in a certain temperature range further confirms the single relaxation process. The data of complex 1–3 can be fitted using a generalized Debye model46 under a zero dc field and an applied magnetic field by CC-FIT244 (Fig. S22–S25 and Table S14, ESI†). Considering contributions from Raman, Orbach and QTM processes, the plot of ln(τ) versus 1/T of 3 was fitted by CC-FIT244, yielding Ueff/kB = 27 K and τ0 = 5.13 × 10−6 s. In order to suppress QTM, the dc field was applied, and the curvature of the plot of ln(τ) versus 1/T was fitted by CC-FIT244, yielding Ueff/kB = 19 K and τ0 = 3.46 × 10−6 s for 1, Ueff/kB = 45 K and τ0 = 1.03 × 10−6 s for 2, and Ueff/kB = 45 K and τ0 = 6.42 × 10−7 s for 3. The extracted parameters indicating the deviation from the pure Debye model are in Tables S14 and S15 (ESI†).
Further to confirm the magnetic relaxation behaviour, the magnetic hysteresis data were collected with a sweep rate of 200 Oe s−1. Complexes 2 and 3 display butterfly-shaped hysteresis loops at 2.5 K and 3 K (Fig. 5 and Fig. S26, ESI†), respectively, which is indicative that the magnetic relaxation rate is slower under an external dc field and a faster near zero field. When the magnetic field is zero, quantum tunnelling dominates the relaxation, so the remanent magnetization (Mr) and the coercive field (Hc) cannot be observed. This is mainly due to the fast QTM possibly induced by symmetry deviations, dipole–dipole interactions and/or hyperfine interactions.
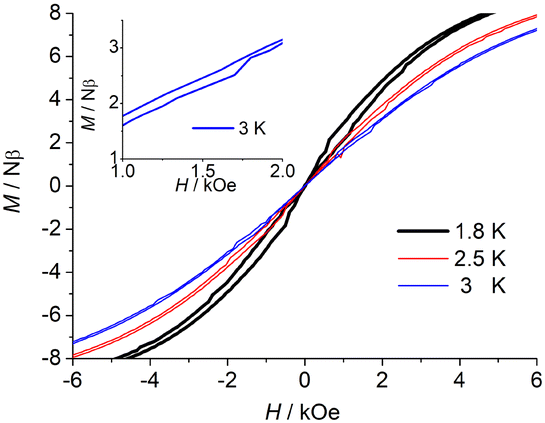 | ||
| Fig. 5 Field dependence of the magnetization of 3 between 1.8 K, 2.5 K, and 3 K at a sweep rate of 200 Oe s−1 (right). | ||
To investigate the anisotropic nature of these systems, MAGELLAN47 is used as an efficient method to estimate the orientation of the anisotropy axes of DyIII ions in complexes 1–3. The main magnetic axes on two DyIII ions are depicted in Fig. 6 and Fig. S27 (ESI†), where the magnetic axes on the two DyIII ions of complexes 2 and 3 have little differences. This is probably the main reason why 2 and 3 show very similar behavior. For 1 and 2, the Dy–O bond length is in the range of 2.184(3)–2.320(3) Å and 2.251(2)–2.320(3) Å, respectively. For 3, compared to the average bond length of Dy1–Oeasy axis (O3, O7, and O8) and the average bond, the Dy1–Operpendicular (O1, O2, O4, O5) bond along the easy axis is longer. As expected, the main magnetic axis of strongly axial Dy compounds or fragments is oriented along the ligand atom that is the shortest Dy–Oaverage bond. This suggests that the strongest bonds between the metal centers and the coordinated atoms may favor an axial nature of the ligand field and thus influences single-ion anisotropy.48 Generally, for the oblate electronic density distribution of DyIII ions, an axially ligand field is favourable to enhance magnetic anisotropies,33 while in 3, the molecular symmetric axis of the DyIII center is not collinear with the anisotropic axis of the spin center which leads to moderately slow magnetic relaxation observed in 3. In addition, the ac susceptibility measurements of Tb-containing complex 6 were carried out to check the potential slow magnetic relaxation behaviour, while there are no any positive signals were detected which are most likely due the unfavourable coordination environment around the TbIII center.
 | ||
| Fig. 6 Orientations of the anisotropy axes for DyIII ions in 1 (left) and orientations of the anisotropy axes for each of the two DyIII ions in 3 (right) as calculated by MAGELLAN. | ||
Luminescence properties
These popular diketonate and phosphine oxide ligands are usually used as ideal chromophores for inducing lanthanide-centered luminescence emission. Therefore, the solid powder luminescence emission spectra of complexes 1–3 and 5 and 6 containing DyIII, EuIII and TbIII ions were recorded (Fig. 7 and Fig. S28, S29, ESI†) at room temperature. With respect to 1, four groups of signals with peak maxima at 480, 573, 662, and 752 nm (λex = 333 nm) can be found in its spectrum, which are assigned to the characteristic 4F9/2 → 6HJ (J = 15/2, 13/2, 11/2, 9/2) transitions of DyIII ions (Fig. S28, ESI†). The emission peaks were clearly observed and implied a more efficient luminescence sensitization via the triple excited states of the ligands in association with the antenna effect. For 2 and 3, upon 342 and 325 nm excitation respectively, both the emission spectra show four emission peaks corresponding to the transitions of DyIII from 4F9/2 to 6HJ transmission (J = 15/2, 13/2, 11/2, and 9/2) (Fig. 7 and Fig. S28, ESI†). The emission spectrum of 5 exhibits five emission peaks at 577, 589, 611, 648, and 698 nm (λex = 393 nm), and they are ascribed to the 5D0 → 7FJ (J = 1, 2, 4) transitions of EuIII (Fig. S29, ESI†). The red emission color is dominated by 5D0 → 7F2 transition (Fig. S29, ESI†). For 6, upon 376 nm excitation, the emission spectrum shows four emission peaks at 487, 543, 581, and 617 nm, corresponding to the transitions of TbIII from 5D4 to 7FJ (J = 6, 5, 4, and 3) (Fig. 7). Complexes 1–3 and 5 and 6 display a strong metal-centered emission, which suggests an efficient energy transfer from the triple energy state of ligands to the excited state of lanthanide ions. More strikingly, the yellow (DyIII), red (EuIII) and green (TbIII) emission colors can be obviously observed by the naked eyes under UV light. This proves that diketonate and phosphine oxide ligands we selected can not only construct high-performance SMMs, but also inspire excellent lanthanide-centered luminescence emission by means of efficient energy transmission between lanthanide ions and the ligand field. | ||
| Fig. 7 Emission spectra of 3 (λex = 325 nm) (left) and 6 (λex = 376 nm) (right). | ||
Conclusions
In conclusion, the selected mono- and bi-phosphine oxide and β-diketonate ligands with different steric hindrance effects jointly coordinated lanthanideIII ions and led to a series of mononuclear and binuclear Dy-based SMMs. Relatively high axial symmetric Dy center complexes with a D4d configuration were successfully constructed by controlling the steric hindrance effect, which displayed significant SMM performance with a hysteresis temperature up to 3 K. More strikingly, the GdIII congener exhibits not only significant MCE properties but also a SMM behaviour, and DyIII, EuIII and TbIII analogues display their characteristic metal centered luminescence properties. The present work reveals that this is an efficient way to design multifunctional materials with SMM, luminescence and MCE properties using selected organic ligands with a tuneable molecular skeleton in association with electronic and steric hindrance effects.Author contributions
H. Yan: synthesis, measurement, luminescence and magnetic data analysis and writing. Z. W. Che: X-ray diffraction analysis of single crystals. W. B. Sun: supervision, discussion, and writing review and editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work was supported by the NSFC (No. 22071047) and the Heilongjiang Provincial Natural Science Foundation of China (LH2019B016).Notes and references
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- M. Yamanouchi, D. Chiba, F. Matsukura and H. Ohno, Nature, 2004, 428, 539–542 CrossRef CAS.
- T. Glaser, Chem. Commun., 2011, 47, 116–130 RSC.
- R. A. Layfield, Organometallics, 2014, 33, 1084–1099 CrossRef CAS.
- R. Sessoli and A. K. Powell, Coord. Chem. Rev., 2009, 253, 2328–2341 CrossRef CAS.
- L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC.
- F. Habib and M. Murugesu, Chem. Soc. Rev., 2013, 42, 3278–3288 RSC.
- M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia, D. Gatteschi and R. Sessoli, Nat. Mater., 2009, 8, 194–197 CrossRef CAS PubMed.
- S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135–1138 CrossRef CAS.
- F.-S. Guo, M. Day Benjamin, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and A. Layfield Richard, Science, 2018, 362, 1400–1403 CrossRef CAS.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño, A. Camón, M. Evangelisti, F. Luis, M. J. Martínez-Pérez and J. Sesé, J. Am. Chem. Soc., 2012, 134, 14982–14990 CrossRef CAS.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- L. Ungur, S.-Y. Lin, J. Tang and L. F. Chibotaru, Chem. Soc. Rev., 2014, 43, 6894–6905 RSC.
- J. Wang, Q.-W. Li, S.-G. Wu, Y.-C. Chen, R.-C. Wan, G.-Z. Huang, Y. Liu, J.-L. Liu, D. Reta, M. J. Giansiracusa, Z.-X. Wang, N. F. Chilton and M.-L. Tong, Angew. Chem., Int. Ed., 2021, 60, 5299–5306 CrossRef CAS.
- K.-X. Yu, J. G. C. Kragskow, Y.-S. Ding, Y.-Q. Zhai, D. Reta, N. F. Chilton and Y.-Z. Zheng, Chemistry, 2020, 6, 1777–1793 CrossRef CAS.
- R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. L. McInnes, L. F. Chibotaru and R. E. P. Winpenny, Nat. Chem., 2013, 5, 673–678 CrossRef CAS PubMed.
- K. R. Meihaus and J. R. Long, J. Am. Chem. Soc., 2013, 135, 17952–17957 CrossRef CAS.
- J.-L. Liu, Y.-C. Chen, Y.-Z. Zheng, W.-Q. Lin, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M.-L. Tong, Chem. Sci., 2013, 4, 3310–3316 RSC.
- J.-L. Liu, Y.-C. Chen and M.-L. Tong, Chem. Soc. Rev., 2018, 47, 2431–2453 RSC.
- J.-P. Sutter, V. Béreau, V. Jubault, K. Bretosh, C. Pichon and C. Duhayon, Chem. Soc. Rev., 2022, 51, 3280–3313 RSC.
- J.-L. Liu, K. Yuan, J.-D. Leng, L. Ungur, W. Wernsdorfer, F.-S. Guo, L. F. Chibotaru and M.-L. Tong, Inorg. Chem., 2012, 51, 8538–8544 CrossRef CAS PubMed.
- E. Colacio, J. Ruiz, E. Ruiz, E. Cremades, J. Krzystek, S. Carretta, J. Cano, T. Guidi, W. Wernsdorfer and E. K. Brechin, Angew. Chem., Int. Ed., 2013, 52, 9130–9134 CrossRef CAS.
- W.-B. Sun, P.-F. Yan, S.-D. Jiang, B.-W. Wang, Y.-Q. Zhang, H.-F. Li, P. Chen, Z.-M. Wang and S. Gao, Chem. Sci., 2016, 7, 684–691 RSC.
- S. Mishra and S. Daniele, Chem. Rev., 2015, 115, 8379–8448 CrossRef CAS.
- W.-B. Sun, B. Yan, Y.-Q. Zhang, B.-W. Wang, Z.-M. Wang, J.-H. Jia and S. Gao, Inorg. Chem. Front., 2014, 1, 503–509 RSC.
- S. Biju, D. B. A. Raj, M. L. P. Reddy and B. M. Kariuki, Inorg. Chem., 2006, 45, 10651–10660 CrossRef CAS.
- M. J. Petersson, W. A. Loughlin and I. D. Jenkins, Chem. Commun., 2008, 4493–4494 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- S. Alvarez, P. Alemany, D. Casanova, J. Cirera, M. Llunell and D. Avnir, Coord. Chem. Rev., 2005, 249, 1693–1708 CrossRef CAS.
- J. Cirera, E. Ruiz and S. Alvarez, Organometallics, 2005, 24, 1556–1562 CrossRef CAS.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- W.-B. Sun, B. Yan, L.-H. Jia, B.-W. Wang, Q. Yang, X. Cheng, H.-F. Li, P. Chen, Z.-M. Wang and S. Gao, Dalton Trans., 2016, 45, 8790–8794 RSC.
- Y.-Z. Zheng, G.-J. Zhou, Z. Zheng and R. E. P. Winpenny, Chem. Soc. Rev., 2014, 43, 1462–1475 RSC.
- Y.-Z. Zheng, M. Evangelisti and R. E. P. Winpenny, Angew. Chem., Int. Ed., 2011, 50, 3692–3695 CrossRef CAS.
- S. Zhang and P. Cheng, Chem. Rec., 2016, 16, 2077–2126 CrossRef CAS PubMed.
- J.-L. Liu, Y.-C. Chen and M.-L. Tong, Chem. Rec., 2016, 16, 825–834 CrossRef CAS.
- Z.-M. Zhang, L.-Y. Pan, W.-Q. Lin, J.-D. Leng, F.-S. Guo, Y.-C. Chen, J.-L. Liu and M.-L. Tong, Chem. Commun., 2013, 49, 8081–8083 RSC.
- A. V. Pavlishchuk and V. V. Pavlishchuk, Theor. Exp. Chem., 2020, 56, 1–25 CrossRef CAS.
- A. Rossin, G. Giambastiani, M. Peruzzini and R. Sessoli, Inorg. Chem., 2012, 51, 6962–6968 CrossRef CAS PubMed.
- E. Ruiz-Bilbao, M. Pardo-Almanza, I. Oyarzabal, B. Artetxe, L. S. Felices, J. A. García, J. M. Seco, E. Colacio, L. Lezama and J. M. Gutiérrez-Zorrilla, Inorg. Chem., 2022, 61, 2428–2443 CrossRef CAS.
- A. C. Anderson and J. E. Robichaux, Phys. Rev. B: Solid State, 1971, 3, 1410–1417 CrossRef.
- D. Reta and N. F. Chilton, Phys. Chem. Chem. Phys., 2019, 21, 23567–23575 RSC.
- S. M. J. Aubin, Z. Sun, L. Pardi, J. Krzystek, K. Folting, L.-C. Brunel, A. L. Rheingold, G. Christou and D. N. Hendrickson, Inorg. Chem., 1999, 38, 5329–5340 CrossRef CAS.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular nanomagnets, Oxford University Press on Demand, 2006 Search PubMed.
- N. F. Chilton, D. Collison, E. J. L. McInnes, R. E. P. Winpenny and A. Soncini, Nat. Commun., 2013, 4, 2551 CrossRef.
- S. Xue, Y.-N. Guo, L. Ungur, J. Tang and L. F. Chibotaru, Chem. – Eur. J., 2015, 21, 14099–14106 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structural, magnetic data for the complexes. CCDC 2181171 (1), 2181172 (2), 2181173 (3), 2181174 (4), 2181175 (5), and 2181176 (6). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2nj03544h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |