
Thermally stable carbazole tagged Au(I)–mesoionic N-heterocyclic carbene complexes with diverse gold–hydrogen bonds†
Subramaniyam
Kalaivanan
 a,
Vaddamanu
Moulali
a,
Kumar
Siddhant
b,
Kavitha
Velappan
c,
Kyohei
Hisano
b,
Osamu
Tsutsumi
*b and
Ganesan
Prabusankar
*a
a,
Vaddamanu
Moulali
a,
Kumar
Siddhant
b,
Kavitha
Velappan
c,
Kyohei
Hisano
b,
Osamu
Tsutsumi
*b and
Ganesan
Prabusankar
*a
aDepartment of Chemistry, Indian Institute of Technology Hyderabad, Hyderabad, Kandi, Telangana 502285, India. E-mail: prabu@chy.iith.ac.in
bDepartment of Applied Chemistry, Ritsumeikan University, Kusatsu 525-8577, Japan. E-mail: tsutsumi@sk.ritsumei.ac.jp
cDAV-IITH, Indian Institute of Technology Hyderabad, Kandi, Telangana 502 284, India
First published on 22nd November 2022
Abstract
Carbazole-substituted N-heterocyclic carbene (NHC)–gold complexes have exhibited diverse structural features and interesting thermal properties. The role of the remotely linked carbazole group to mesoionic carbene in gold(I)–NHC complexes has been addressed in this paper. Thus, we have synthesized and characterized neutral gold–mesoionic carbene monomers tagged with carbazole groups. The mononuclear gold(I)–carbene complexes [(L1)AuCl] (1) and [(L2)AuCl] (2), where L1.HI = 1-(naphth-1-yl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazolium iodide and L2.HI = 1-(mesityl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazolium iodide, were synthesized and characterized. This new class of complexes exhibited interesting gold–hydrogen bonding. In addition, the thermal properties of 1 and 2 were investigated. Density functional theory (DFT) calculations and natural bond orbital (NBO) analysis were carried out on a model system, [(L′)AuCl] (1A), where L′ = 1-phenyl-4-methyl-carbazole-1,2,3-triazol-ylidene, to realize the bonding situations. The calculated metrics agreed reasonably well with the experimental observations.
Introduction
Carbazole tagged gold(I)–NHC complexes (NHC = N-heterocyclic carbene), known as carbene–gold-amides (CGAs), have been proven to have potential application in spectacular high-performance organic light-emitting diodes (HP-OLEDs).1–5 The strong chromophoric nature of carbazole, along with good hole-transporting properties, imparts enhanced optical properties.6 In addition, the carbazole moiety can add rich structural diversities such as π–π interactions,7,8 aurophillic interactions,4 and gold–hydrogen bonding6 to the gold–NHC molecules.Understanding the thermal stability of gold-hydrogen bond is a critical aspect due to the highest electronegativity and electron affinity of gold along with the lowest possible coordination number.9–14 The known carbazole substituted Au–NHC complex with gold–hydrogen bonds is shown in Scheme 1. The first gold–hydrogen bond in carbazole substituted [(Me2-NHC)Au(cbz)], (Me2-NHC = N,N′-methyl-imidazol-2-ylidene) (cbz = carbazolate), was reported in 2005, in which the Au⋯HC(sp2) bond distance is 2.93 Å.7 In addition, the weak gold hydrogen bonding has capability to even bend the angle around the gold center at C(carbene)–Au–Cl in [(EF-NHC)AuCl] (EF-NHC = N,N′-9-ethyl-9-fluorenyl-benzimidazol-2-ylidene) and [(BF-NHC)AuCl] (BF-NHC = N,N′-9-butyl-9-fluorenyl-benzimidazol-2-ylidene) substituted NHC–gold complexes.15 Notably, the gold–hydrogen bond plays a significant role in the thermal stability of gold–NHC complexes.14–18
 | ||
| Scheme 1 Authentic report on the carbazole substituted gold–NHC complexes with gold–hydrogen bonds. | ||
However, carbazole-substituted gold–MIC (MIC = mesoionic NHC) complexes with gold–hydrogen bonds are not known.7,19–24 MIC ligands are more basic than normal NHCs, which enhances the donating ability of MIC.25,26 The Tolman electronic parameter explains that MICs are stronger σ donors than NHCs.27–29 Besides, the following questions still need to be answered and shall be addressed in this work. What is the function of the remotely linked carbazole group to carbene in gold(I)–MIC complexes? How does the 4-methyl-carbazole substituent contribute to the aurophillic interactions or gold–hydrogen bonding interactions or thermal stability of the gold(I)–MIC complexes? To answer these questions, we synthesized and characterized the new carbazole derivatives of mononuclear gold(I)–MIC molecules [(L1)AuCl] (1) and [(L2)AuCl] (2), (L1.HI = 1-(naphth-1-yl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazolium iodide and L2.HI = 1-(mesityl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazolium iodide) with gold–hydrogen bonds. The thermal properties of 1 and 2 were investigated. The experimental results of 1 and 2 were compared with theoretical values of the related model compound [(L′)AuCl] (1A), L′ = 1-phenyl-4-methyl-carbazole-1,2,3-triazol-ylidene.
Experimental section
Materials and methods
The reactions were carried out using oven-dried glassware employing standard Schlenk line techniques. The reaction with silver oxide was carried out in the absence of light. The solvents were dried and purified as reported.30 All the starting materials were used as received from commercial sources. [Au(tht)Cl] was synthesized as reported.31L1.HI = 1-(naphth-1-yl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazolium iodide and L2.HI = 1-(mesityl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazolium iodide were synthesized as reported.32 FT-IR spectroscopy (neat) was carried out using a Bruker Alpha-P Fourier transform spectrometer. Elemental analysis was performed using a Euro E.A.-CHNSO elemental analyser. NMR spectra were obtained at 27 °C using Bruker Ultrashield-400 spectrometers. Chemical shifts are expressed relative to TMS, with solvent resonances being provided as internal standards. The thermal stability of 1 and 2 was analysed by thermogravimetric/differential thermal analysis (TG/DTA) using a DTG-60AH analyser (Shimadzu, Japan) at a heating rate of 5 °C min−1. An Oxford SuperNova, Dual, Cu at home/near, Eos diffractometer was used for determining the crystal structure of L1.HI at 273 K. A Rigaku Rigaku Saturn724+ (2 × 2 bin mode) diffractometer was used for determining the crystal structure of 1 at 293 K, while a Rigaku XtaLAB AFC12 (RINC): Kappa single diffractometer was used for determining the crystal structure of 2 at 119.99 K. The structures were solved using olex2.solve structure solution software and refined using olex2.refine in a refining package that uses Gauss-Newton minimization with Olex2.33 Non-hydrogen atoms were refined using an anisotropic technique. Hydrogen atoms riding on their carrier atoms were refined according to the calculated position.Caution! Azides are shock and heat sensitive. Azides are highly explosive substances. Never treat them with strong acids that can yield hydrazoic acid, which is highly toxic, explosive and volatile. The azides should be carefully handled in a sophisticated fume hood along with protective equipment and a respiratory system.
All calculations were carried out using the Gaussian 16 suit of program.34 The input files were produced using GaussView, version 6.35 The structural metrics of model compound [(L′)AuCl] (1A), L′ = 1-phenyl-4-methyl-carbazole-1,2,3-triazol-ylidene were generated from the single crystal X-ray crystal data of 1. Complex 1A was optimized at the B3LYP/Def2TZvp level of theory using the 6-31++G** basis set. The energy minima of the structure on the potential energy surface were optimized by vibrational frequency calculations. Natural bond orbital (NBO) analysis was carried out with Gaussian09.
Synthesis of L1.HI
1-(Naphth-1-yl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazol (748 mg, 2 mmol) was added to a solution of iodomethane (0.30 mL, 5.0 mmol) in CH3CN (0.30 mL) in a ACE pressure tube. The temperature of the reaction mixture was maintained at 85 °C for 24 hours. The desired product was isolated as a off-white solid. The solid substance was purified and dried under reduced pressure after washing with ethyl acetate. Yield: 60% (based on 1-(naphth-1-yl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazol). M.p.: 163–166 °C (decomp.). Elemental analysis calcd (%) C26H21N4I (516.38): C, 60.48; H, 4.10; N, 10.85; found: C, 59.0; H,4.0; N, 10.9. FT-IR (neat,![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) , cm−1): 3848(w), 3742(w), 3618(m), 3027(vs), 2886(s), 2672(m), 2362(s), 2063(s), 1697(m), 1519(m), 1449(s), 1332(m), 1280(m), 1216(m), 1160(m), 1070(m), 1003(m), 941(vs), 863(vs), 749(vs), 640(m), 557(m). 1H NMR (DMSO-d6, 400.1 MHz): δ 8.98 (s, 1H, Tri-CH), 8.30–8.14 (m, 4H, Ar-CH), 7.88–7.83 (t, 3H, Ar-CH), 7.68 (s, 4H, Ar-CH), 7.53–7.50 (t, 2H, Ar-CH), 7.31–7.27 (t, 2H, Ar-CH), 6.20 (s, 2H, N-CH2), 4.63 (s, 3H, N-CH3). 13C NMR (DMSO-d6, 100.6 MHz): δ 141.5 (Tri-C), 139.8 (C-Ar), 133.4 (C-Ar), 132.4 (C-Ar), 131.1 (C-Ar), 130.8 (C-Ar), 128.6 (C-Ar), 128.5 (C-Ar), 127.7 (C-Ar), 126.9 (C-Ar), 126.1 (C-Ar), 125.2 (C-Ar), 125.1 (C-Ar), 122.9 (C-Ar), 121.2 (C-Ar), 120.5 (C-Ar), 119.9 (C-Ar), 109.7 (C-Ar), 37.4 (N-CH2), 37.3 (N-CH3).
, cm−1): 3848(w), 3742(w), 3618(m), 3027(vs), 2886(s), 2672(m), 2362(s), 2063(s), 1697(m), 1519(m), 1449(s), 1332(m), 1280(m), 1216(m), 1160(m), 1070(m), 1003(m), 941(vs), 863(vs), 749(vs), 640(m), 557(m). 1H NMR (DMSO-d6, 400.1 MHz): δ 8.98 (s, 1H, Tri-CH), 8.30–8.14 (m, 4H, Ar-CH), 7.88–7.83 (t, 3H, Ar-CH), 7.68 (s, 4H, Ar-CH), 7.53–7.50 (t, 2H, Ar-CH), 7.31–7.27 (t, 2H, Ar-CH), 6.20 (s, 2H, N-CH2), 4.63 (s, 3H, N-CH3). 13C NMR (DMSO-d6, 100.6 MHz): δ 141.5 (Tri-C), 139.8 (C-Ar), 133.4 (C-Ar), 132.4 (C-Ar), 131.1 (C-Ar), 130.8 (C-Ar), 128.6 (C-Ar), 128.5 (C-Ar), 127.7 (C-Ar), 126.9 (C-Ar), 126.1 (C-Ar), 125.2 (C-Ar), 125.1 (C-Ar), 122.9 (C-Ar), 121.2 (C-Ar), 120.5 (C-Ar), 119.9 (C-Ar), 109.7 (C-Ar), 37.4 (N-CH2), 37.3 (N-CH3).
Synthesis of L2.HI
The synthesis procedure of L2.HI was the same as reported for L1.HI. The product L2.HI was obtained as a white solid. Yield: 58% (based on 1-(mesityl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazol). M.p.: 151–153 °C (decomp.). Elemental analysis calcd (%) C25H25N4I (508.11): C, 59.06; H, 4.96; N, 11.02; found: C, 59.0; H, 4.9; N, 11.0. FT-IR (neat,, cm−1): 2988(w), 2923(m), 2253(s), 1735(s), 1595(vs), 1449(vs), 1327(vs), 1275(vs), 1212(m), 1150(m), 1076(m), 1028(m), 849(vs), 805(m), 746(vs), 629(vs), 562(m). 1H NMR (DMSO-d6, 400.1 MHz): δ 8.73 (s, 1H, Tri-CH), 8.24–8.22 (m, 2H, Ar-CH), 7.79–7.77 (t, 2H, Ar-CH), 7.52–7.49 (t, 2H, Ar-CH), 7.31–7.28 (t, 2H, Ar-CH), 7.11 (t, 2H, Ar-CH), 6.12 (s, 2H, N-CH2), 4.52 (s, 3H, N-CH3), 2.30 (s, 3H, para-Mes-CH3), 1.93 (s, 3H, ortho-Mes-CH3); 13C NMR (100.6 MHz, DMSO-d6): δ 141.6 (Tri-C), 139.7 (Ar-C), 134.1 (Ar-C), 131.1 (Ar-C), 130.8 (Ar-C), 129.2 (Ar-C), 126.0 (Ar-C), 122.9 (Ar-C), 120.6 (Ar-C), 119.9 (Ar-C), 109.7 (Ar-C), 59.7 (N-CH2), 37.5 (N-CH3), 20.6 (para-Mes-CH3), 16.6 (ortho-Mes-CH3).
Synthesis of 1
The compound L1.HI (0.200 g, 0.4 mmol) was treated with Ag2O (0.268 g, 1.2 mmol) in DCM at ambient temperature. The reaction mixture was stirred for 5 hours in the absence of light. The Ag2O completely reacted with L1.HI, which was confirmed by TLC. Then Au(tht)Cl (0.124 g, 0.4 mmol) was added and stirred for 4 hours. The reaction mixture was filtered using Celite and the filtrate was dried under reduced pressure. The off-white crude product was purified using silica-gel column chromatography (chloroform:methanol). The product was further purified by recrystallisation technique using CHCl3 through slow evaporation. Yield: 60% (based on Au(tht)Cl). M.p.: 243–245 °C (decomp.). Elemental analysis calcd (%) C26H20AuClN4 (620.89): C, 50.30; H, 3.25; N, 9.02; found C, 50.3; H,3.3; N,9.0. FT-IR (neat,, cm−1): 3053(w), 2996(w), 1670(m), 1598(vs), 1448(vs), 1396(m), 1326(vs), 1260(s), 1214(vs), 1164(m), 1075(m), 1010(m), 960(m), 847(m), 742(vs), 663(s), 615(m), 561(m). 1H NMR (400.1 MHz, CDCl3): δ 8.15–8.13 (d, 2H, Ar-CH), 7.84–7.82 (d, 1H, Ar-CH), 7.68–7.66 (d, 1H, Ar-CH), 7.52–7.41 (m, 7H, Ar-CH), 7.35–7.31 (m, 3H, Ar-CH), 7.19–7.15 (t, 1H, Ar-CH), 5.41 (s, 2H, N-CH2), 3.59 (s, 3H, N-CH3). 13C NMR (100.6 MHz, CDCl3): δ 163.2 (Carbene-Au), 142.9 (Ar-C), 140.0 (Ar-C), 135.4 (Ar-C), 133.8 (Ar-C), 131.4 (Ar-C), 128.4 (Ar-C), 128.3 (Ar-C), 127.9 (Ar-C), 127.1 (Ar-C), 126.4 (Ar-C), 125.1 (Ar-C), 124.7 (Ar-C), 123.3 (Ar-C), 122.2 (Ar-C), 120.6 (Ar-C), 120.2 (Ar-C), 109.7 (Ar-C), 38.7 (N-CH2), 38.1 (N-CH3).
Synthesis of 2
Molecule 2 was isolated as reported for 1 using L2.HI (0.200 g, 0.4 mmol), Ag2O (0.274 g, 1.2 mmol), and Au(tht)Cl (0.126 g, 0.4 mmol). Yield: 51% (based on Au(tht)Cl). M.p.: 253–255 °C (decomp.). Elemental analysis calcd (%) C25H24AuClN4 (612.91): C, 48.99; H, 3.95; N, 9.14; found: C, 50.0; H, 4.0; N, 9.0. FT-IR (neat,, cm−1): 2919(vs), 2855(vs), 2016(m), 1732(vs), 1597(s), 1455(m), 1363(m), 1275(s), 1183(s), 1078(vs), 1032(m), 962(vs), 891(m), 849(m), 803(s), 745(vs), 646(m). 1H NMR (400.1 MHz, CDCl3) δ 8.14–8.12 (d, 2H, Ar-CH), 7.63–7.61 (d, 2H, Ar-CH), 7.51–7.47 (t, 2H, Ar-CH), 7.33–7.29 (m, 2H, Ar-CH), 6.93 (s, 2H, N-CH2), 5.90 (s, 3H, N-CH2), 3.51 (s, 3H, N-CH3), 2.26 (s, 3H, para-Mes-CH3), 2.05 (s, 3H, ortho-Mes-CH3). 13C NMR (100.6 MHz, CDCl3) δ 162.8 (C-Au), 143.0 (Ar-CH), 140.9 (Ar-CH), 139.9 (Ar-CH), 135.1 (Ar-CH), 134.3 (Ar-CH), 129.4 (Ar-CH), 126.5 (Ar-CH), 123.3 (Ar-CH), 120.7 (Ar-CH), 120.3 (Ar-CH), 109.5 (Ar-CH), 109.3 (Ar-CH), 39.0 (N-CH2), 38.1 (N-CH3), 21.1 (para-Mes-CH3), 18.0 (ortho-Mes-CH3).
Results and discussion
The compounds 1-(naphth-1-yl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazolium iodide (L1.HI) and 1-(mesityl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazolium iodide (L2.HI) were synthesised by treating methyl iodide with corresponding 1-(naphth-1-yl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazol and 1-(mesityl)-3-methyl-4-(carbazolylmethyl)-1,2,3-triazol, respectively, (Scheme 2).32 The compounds L1.HI and L2.HI were soluble in CH3CN and DMSO but not soluble in acetone or MeOH. L1.HI and L2.HI were characterized by elemental analysis, FTIR, 1H NMR, 13C NMR and single-crystal X-ray diffraction techniques. The formation of L1.HI and L2.HI salts was confirmed from the NMR resonance peaks associated with N-substituents, carbazole and triazole moieties. In the 1H NMR spectrum, the more acidic triazole C–H proton in L1.HI and L2.HI resonates at 8.98 and 8.73 ppm, respectively. In the 13C NMR study, the triazole C–H carbon in L1.HI and L2.HI appears at 141.5 and 141.6 ppm, respectively. | ||
| Scheme 2 Synthesis of L1.HI, L2.HI, 1 and 2. | ||
The solid-state structure of L1.HI salt was unambiguously determined using the single-crystal X-ray diffraction technique (ESI,† Fig. S13 and Tables S4, S5). The single crystals of L1.HI salt were obtained from a CH3CN solution by slow evaporation. The salt L1.HI crystallizes in the monoclinic space group, P21/c. The NMR results of L1.HI are in good agreement with the solid-state structure. Both the naphthyl and carbazole rings are oriented perpendicular to the five-membered triazoline ring system. The charge on the cationic moiety of substituted triazoline is balanced by one iodine anion.
The gold–carbene complexes [(L1)AuCl] (1) and [(L2)AuCl] (2) were isolated via the trans-metalation route. The reactions between Ag2O and corresponding salts L1.HI or L2.HI, followed by the reaction with [Au(tht)Cl] (tht = tetrahydrothiophene) gave 1 and 2 in very good yield (Scheme 2). Molecules 1 and 2 were purified using column chromatography and then recrystallized in CHCl3. These new gold–carbene molecules 1 and 2 were characterised by FTIR, NMR, and single-crystal X-ray techniques.
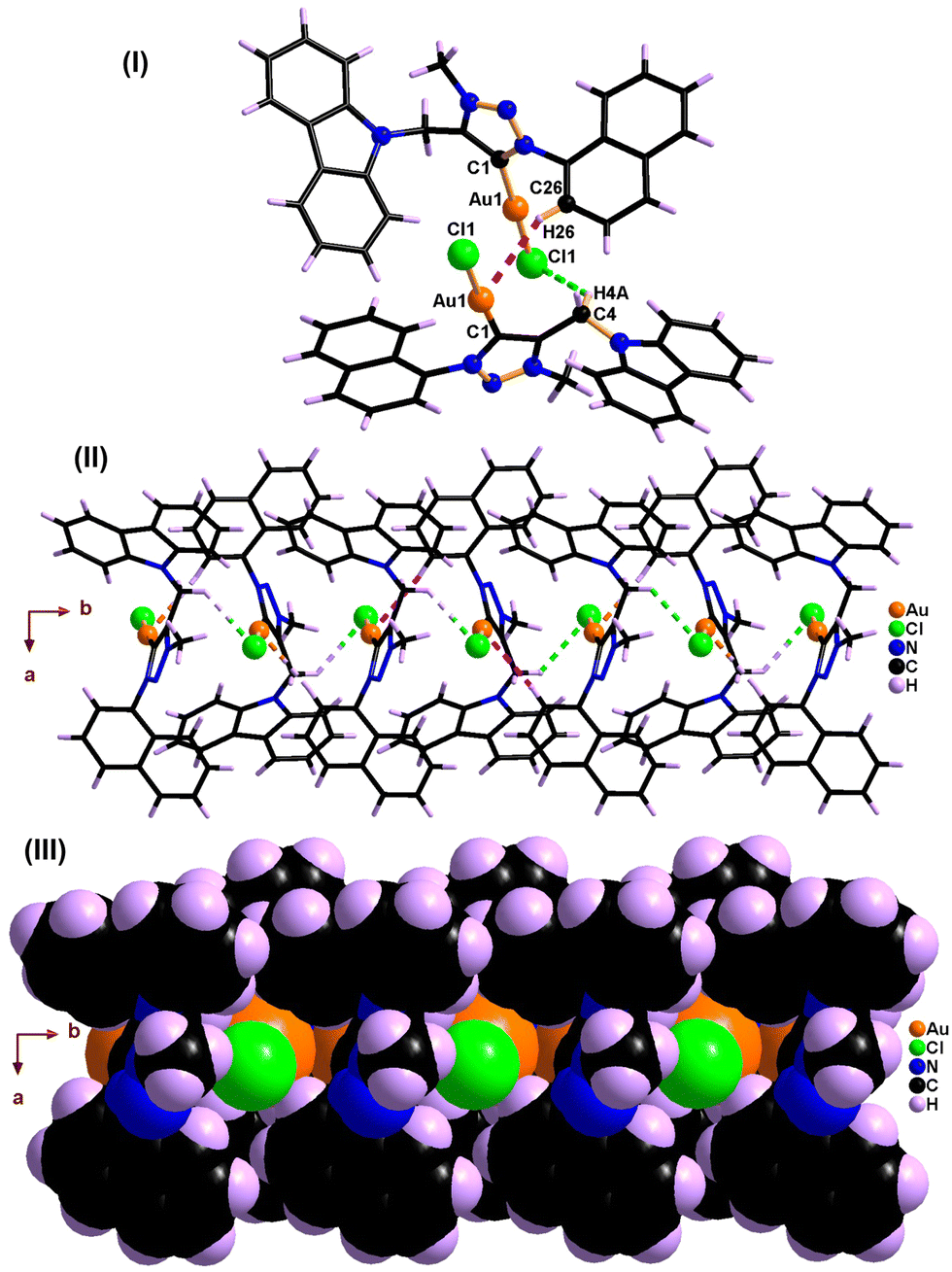
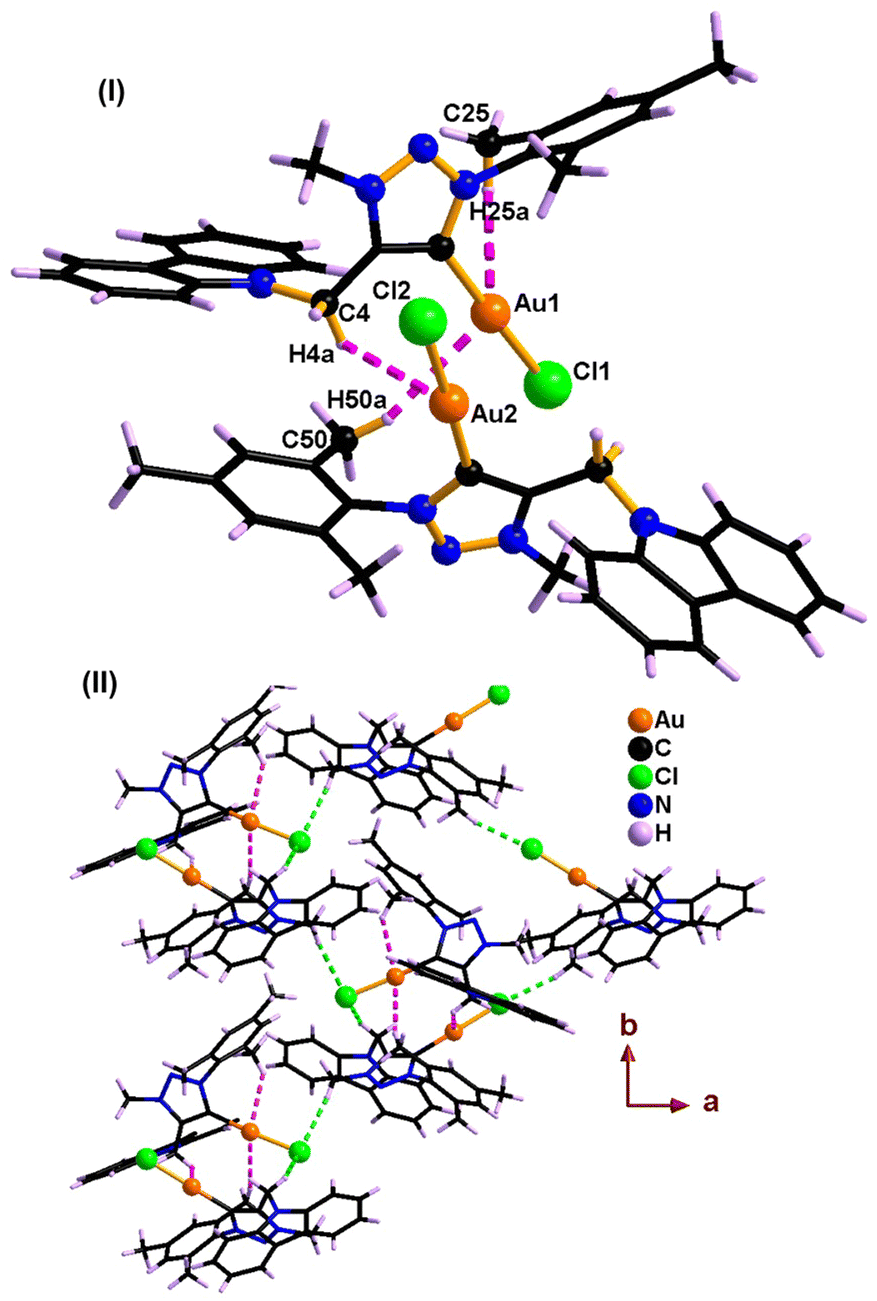
The disappearance of the triazole C–H peak in 1H NMR along with the appearance of a new 13C NMR signal around 163 ppm for 1 and 2 affirms the formation of a carbene metal bond. The 13C NMR chemical shift values of carbene carbon in 1 and 2 are comparable. The solid-state structures of 1 and 2 were confirmed by single-crystal X-ray analysis (Fig. 1–4). The molecules 1 and 2 crystallise in the monoclinic space group, P21/c (for 1) and P21/n (for 2) (Tables 1 and 2).
 | ||
| Fig. 1 Solid-state structure of 1. | ||
 | ||
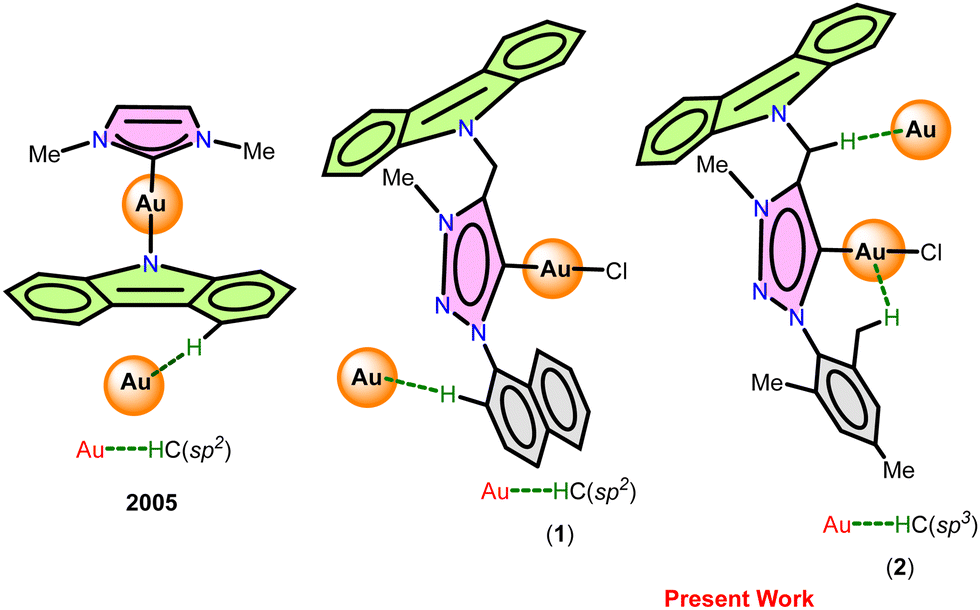
| Fig. 2 (I) Au⋯H–C(sp2) and Cl⋯H–C hydrogen bonds in 1. (II) 1D coordination chain of 1 through intermolecular Au⋯H–C(sp2) and Cl⋯H–C hydrogen bonds. (III) Space-filling model of the 1D coordination chain of 1. | ||
 | ||
| Fig. 3 Solid-state structure of 2. | ||
 | ||
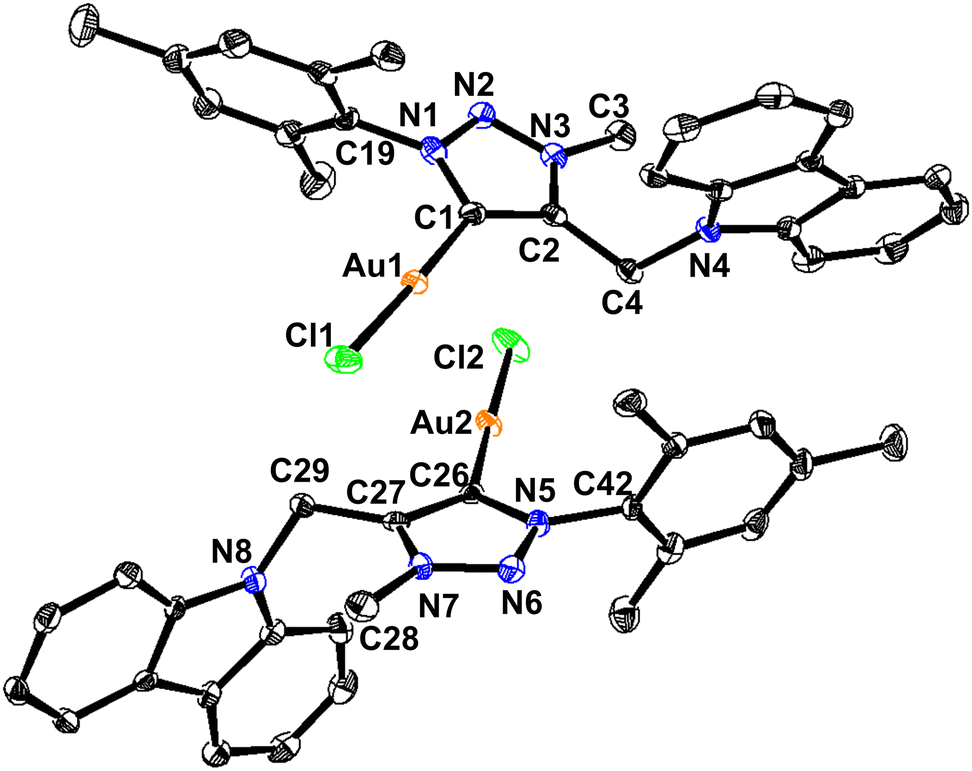
| Fig. 4 (I) Inter- and intra-molecular Au⋯H–C(sp3) hydrogen-bonded dimer of 2. (II) 2D coordination layer of 1 through intermolecular Cl⋯H–C hydrogen bonds. | ||
| Identification code | 1 | 2 |
|---|---|---|
| CCDC | 2182804 | 2182805 |
| Empirical formula | AuC26H20ClN4 | Au2C50H48Cl2N8 |
| Formula weight | 620.895 | 1225.79 |
| Temperature/K | 293.0 | 119.99(10) |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/n |
| a/Å | 13.1307(4) | 16.6671(4) |
| b/Å | 8.5693(2) | 13.2879(3) |
| c/Å | 20.6523(5) | 21.1978(5) |
| α/° | 90 | 90 |
| β/° | 100.607(2) | 97.521(2) |
| γ/° | 90 | 90 |
| Volume/Å3 | 2284.11(10) | 4654.32(19) |
| Z | 4 | 4 |
| ρ calcd/g cm−3 | 1.805 | 1.749 |
| μ/mm | 6.579 | 6.456 |
| F (000) | 1200.0 | 2384.0 |
| Crystal size/mm3 | 0.09 × 0.07 × 0.05 | 0.16 × 0.14 × 0.12 |
| Radiation | Mo Kα (λ = 0.71073) | Mo Kα (λ = 0.71073) |
| 2Θ range for data collection/° | 4.626 to 58.152 | 5.746 to 57.93 |
| Index ranges | −14 ≤ h ≤ 17, −9 ≤ k ≤ 11, −27 ≤ l ≤ 24 | −21 ≤ h ≤ 21, −17 ≤ k ≤ 18, −28 ≤ l ≤ 25 |
| Reflections collected | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 030 030 |
41742 |
| Independent reflections | 5250 [Rint = 0.0274, Rsigma = 0.0462] | 10748 [Rint = 0.0412, Rsigma = 0.0423] |
| Data/restraints/parameters | 5250/0/315 | 10748/0/567 |
| Goodness-of-fit on F2 | 1.0588 | 1.037 |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.0373, wR2 = 0.0575 | R 1 = 0.0268, wR2 = 0.0479 |
| Final R indexes [all data] | R1 = 0.0669, wR2 = 0.688 | R1 = 0.0381, wR2 = 0.0499 |
| Largest diff. peak/hole/e Å−3 | 0.8716/−0.8103 | 0.64/−0.69 |
| 1 | 2 | |
|---|---|---|
| Bond lengths (Å) | ||
| N3–N2 | 1.311(6) | 1.322(3) |
| N3–C2 | 1.361(6) | 1.359(4) |
| N1–N2 | 1.335(6) | 1.336(3) |
| N1–C1 | 1.357(6) | 1.367(4) |
| C1–C2 | 1.379(7) | 1.387(4) |
| Au1–Cl1 | 2.2860(16) | 2.3008(8) |
| Au1–C1 | 1.974(5) | 1.984(3) |
| N5–N6 | — | 1.335(3) |
| N6–N7 | — | 1.333(3) |
| N7–C27 | — | 1.355(4) |
| N5–C26 | — | 1.362(4) |
| C26–C27 | — | 1.370(4) |
| Au2–Cl2 | — | 2.2866(8) |
| Au2–C26 | — | 1.987(3) |
| Bond angles (°) | ||
| N2–N3–C2 | 113.4(4) | 113.1(2) |
| N2–N1–C1 | 114.7(5) | 115.3(2) |
| N1–C1–C2 | 103.2(5) | 102.2(3) |
| N3–C2–C1 | 106.0(5) | 107.0(3) |
| N3–N2–N1 | 102.7(4) | 102.5(2) |
| C1–Au1–Cl1 | 177.06(16) | 176.50(9) |
| N6–N5–C26 | — | 115.0(2) |
| N5–C26–C27 | — | 103.0(3) |
| N6–N7–C27 | — | 112.7(2) |
| N5–N6–N7 | — | 102.2(2) |
| C26–Au2–Cl2 | — | 177.80(9) |
Molecules 1 and 2 are neutral gold–carbene complexes. The carbene C1–Au1–Cl1 bond angle of 1 and 2 corroborates nearly a linear geometry to the gold center. The aryl and carbazole rings are perpendicularly oriented with respect to the triazole ring system. The Au1–C1 bond distance is comparable.
The solid-state structures of molecules 1 and 2 are distinctly different due to the gold–gold and hydrogen-bonding interactions (Fig. 2, 4, Table 3 and Scheme 3). Similar hydrogen-bonding interactions in gold–mesoionic carbene complexes are limited.9–12 The gold–gold interaction and intramolecular hydrogen bonding interactions are absent in 1. The spacefilling model of a 1D chain depicts the possible void around the gold center favoring the gold-hydrogen and chlorine-hydrogen interactions (Fig. 2(III)). Molecule 1 forms a one-dimensional chain through intermolecular Au⋯H–C(sp2) and Cl⋯H–C hydrogen bonds. The Au⋯H–C(sp2) interaction is more favorable than the Cl⋯H–C hydrogen bonding interaction. Similarly, the Au⋯H–C(sp2) interaction is comparable to the known intramolecular hydrogen bonding interactions found in [(Me2-NHC)Au(cbz)] (2.93 Å).7,10a
| Bond lengths (Å) | Bond angles (°) | ||
|---|---|---|---|
| 1 | |||
| Au(1)⋯H(26) | 2.997 | Au(1)⋯H(26)-C(26) | 152.34 |
| Cl(1)⋯H(4A) | 2.855 | Cl(1)⋯H(4A)-C(4) | 130.32 |
| 2 | |||
| Au(1)⋯H(25A) | 2.877 | Au(1)⋯H(25A)-C(25) | 130.29 |
| Au(1)⋯H(50A) | 2.999 | Au(1)⋯H(50A)-C(50) | 160.04 |
| Cl1(1)⋯H(29B) | 2.832 | Cl(1)⋯H(29B)-C(29) | 128.56 |
| Cl(1)⋯H(28C) | 2.815 | Cl(1)⋯H(28C)-C(28) | 166.21 |
| Cl(2)⋯H(44C) | 2.678 | Cl(2)⋯H(44C)-C(44) | 167.57 |
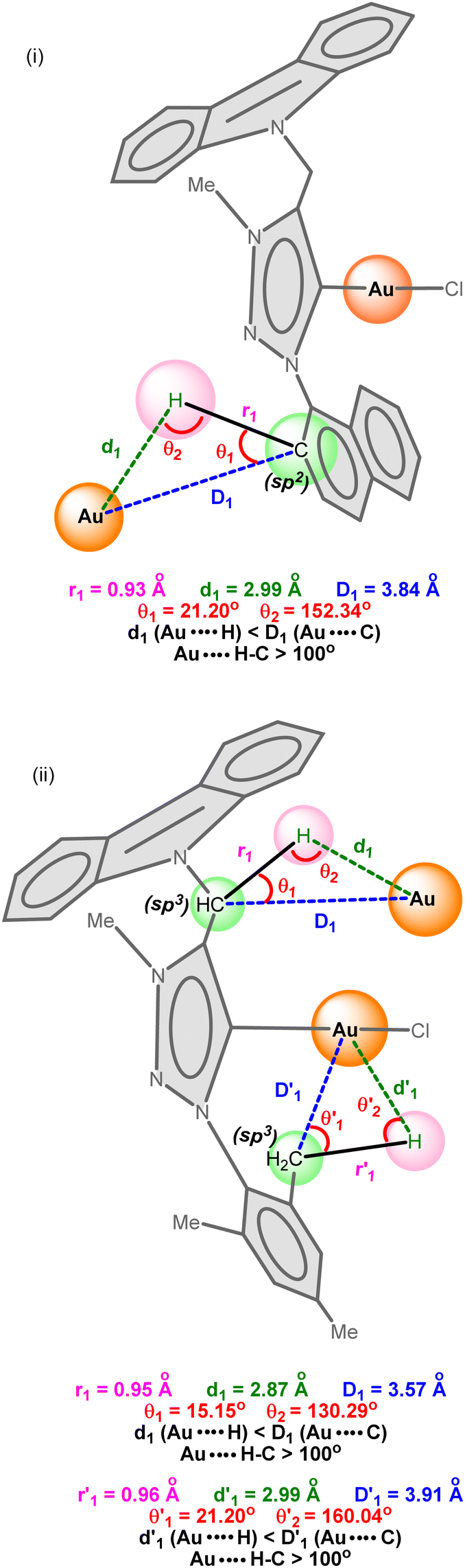 | ||
| Scheme 3 (i) Inter-molecular multicenter hetero-acceptor hydrogen in 1 and (ii) inter-and intra-molecular multicenter hetero-acceptor hydrogen bonding metrics in 2. | ||
As shown in Scheme 3, the C–H bond distances (r1 or r′1) are not elongated and the Au⋯H bond distances (d1 or d′1) are shorter than the Au⋯C bond distances (D1 or D′1) in Au⋯H–C(sp2) and Au⋯H–C(sp3) bonds. Moreover, the Au⋯H–C(sp2) or Au⋯H–C(sp3) bond angle (θ or θ′) is more than 100°. All these metrics support the existence of intermolecular or intramolecular multicenter hetero-acceptor hydrogen bonding in 1 and 2.36 The adjacent molecules in the 1D chain are orientated opposite to each other to minimize the intermolecular steric hindrance and maximize the hydrogen bonding interactions.
The hydrogen bonding patterns of 2 are significantly different from those of 1. Molecule 2 associates as a dimer through both intermolecular and intramolecular hydrogen bonding interactions between gold and methyl hydrogen of mesitylene (Fig. 4). These dimers are further connected through the Cl(1)⋯H(28C)-C(28), Cl(1)⋯H(29B)-C(29) and Cl(2)⋯H(44C)-C(44) hydrogen bonds, resulting in the formation of 2D layers. The intramolecular Au(1)⋯H(25A)-C(25) interaction is shorter than the intermolecular C(50)-H(50A)⋯Au(I) and C(4)–H(4A)⋯Au(I) interactions. Molecule 2 depicts a weak aurophillic interaction between two gold centers (3.728 Å).
The C–Au–Cl bond angle is nearly linear. The Au–C bond distance and C–Au–Cl bond angle are comparable to those of [(Me2-NHC)Au(cbz)].7 The 1,2,3-triazolidene ring system in 1 and 2 exists in the mesoionic carbene form as the N(1)–N(2) and N(2)–N(3) bond distances are not comparable.10a
In addition, the structural and electronic proprieties of 1 and 2 were investigated using a model system [(L′)AuCl] (1A), L′ = 1-phenyl-4-methyl-carbazole-1,2,3-triazol-ylidene. The density functional theory (DFT) calculations were carried out on 1A at the B3LYP level of theory using the Def2TZvp valence basis set for gold and the 6-31++G** basis set for chlorine, carbon, hydrogen and nitrogen. For gold, the core electrons (ncore = 60) were represented using the Stuttgart–Dresden effective core potential (ECP) denoted as def2 or SDD in Gaussian was used (see Fig. 5 and ESI†). The input data for the geometry optimization were furnished from the single-crystal X-ray crystallographic data set of corresponding molecules.
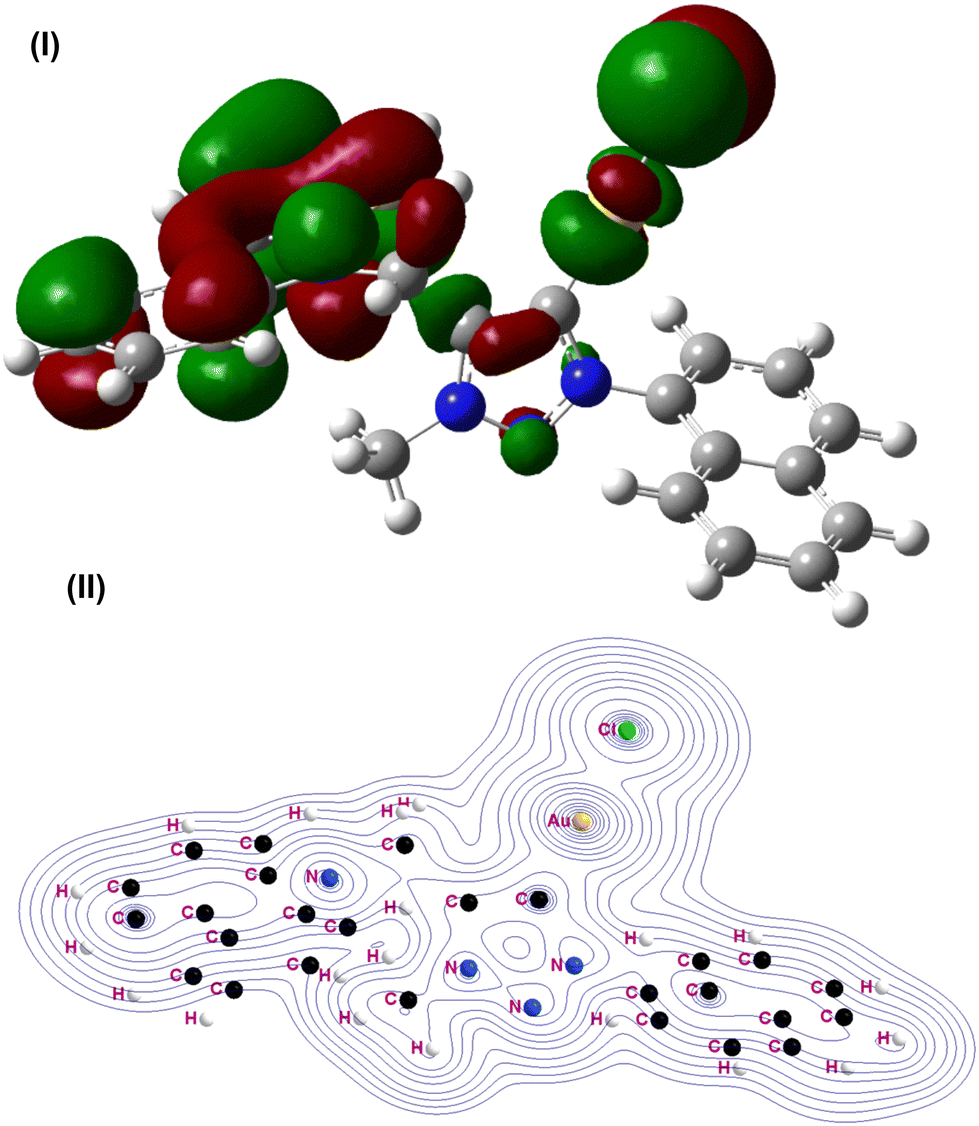 | ||
| Fig. 5 (I) Highest occupied molecular orbital (HOMO) of 1A. (II) Contour line diagrams of the Laplacian distribution ∇2ρ(r) of 1A. | ||
The calculated structural parameters for mononuclear gold(I)-carbene complex 1A agreed reasonably with those of 1 and 2. Note that the hydrogen bonding interactions in 1 and 2 are not accounted for in the present calculations as they are associated with the aryl substituents and molecular packing. Fig. 5b shows the Laplacian distribution of 1A in the molecular plane. The topology of the charge distribution and its associated Laplacian suggest that the gold atom is primarily bonded to carbene and chlorine. The pattern of the charge concentration at the carbene carbon, gold and chlorine center reveals the dominant interactions. There is an area of charge concentration near the carbene carbon atom pointing toward gold. This is in agreement with the charge distribution calculated by the natural bond orbital (NBO) analysis in Gaussian for 1A (see ESI†). The NBO occupancy of Au–C is 1.95744. The s orbital of gold and sp2 orbital of carbene are involved in bonding.
The thermal stability of complexes 1 and 2 was investigated by thermogravimetric analysis (TGA) and differential thermal analysis (DTA) (Fig. 6). In general, the temperature at which the weight is reduced by 3% or 5% is often defined as the thermal decomposition temperature. Both complexes were thermally very stable. The naphthyl group and multiple hydrogen bonds in 2 seem to increase the thermal stability of the mesityl substituent with limited hydrogen bonds in 1. The thermal decomposition of 1 starts at 241 °C, while the thermal decomposition of 2 begins at 257 °C. Complex 2 showed a major weight loss within 341 °C, while 1 depicted a major weight loss until 439 °C due to the loss of organic moieties and chlorine atoms. The residual mass of 1 (found: 30.26%; calcd: 30%) and 2 (found: 40.54%; calcd: 32%) was ascribed to the gold. The 8% deviation in the residual mass of 2 could be due to the nature of sample to undergo gradual decomposition.
 | ||
| Fig. 6 (I) TG/DTA of 1 from 30–600 °C under a nitrogen atmosphere at a heating rate of 10 °C min−1. (II) TG/DTA of 2 from 30–600 °C under a nitrogen atmosphere at a heating rate of 10 °C min−1. | ||
Conclusions
We have synthesized and characterized two new 4-methyl-carbazole tagged mesoionic gold(I) complexes 1 and 2. The present study suggests that even the remotely tagged 4-methyl-carbazole group is important for hydrogen bonds and thermal stability. The molecules were stable until 241 °C. The compound 4-methyl-carbazole shows a substantial contribution to the hydrogen bonds in the solid-state packing. Molecule 1 forms a 1D polymer through hydrogen bonds, while 2 forms a 2D layer structure via hydrogen bonds. From a steric viewpoint, the gold center in 1 prefers to form intermolecular Au⋯H–C(sp2) hydrogen bonds due to the presence of a sterically less crowded naphthalene substituent, while the sterically more crowded mesitylene group allows the formation of both intermolecular and intramolecular Au⋯H–C(sp3) hydrogen bonds in 2. The ability to design and control the gold–hydrogen interaction is critical for the development of gold catalysed C–H activation reactions. Further studies on the isolation of gold–carbene compounds with gold–hydrogen interactions and efforts to understand the bonding nature are underway.Author contributions
Subramaniyam Kalaivanan: data curation, formal analysis, methodology, investigation, and writing – original draft. Moulali Vaddamanu: data curation and formal analysis. Kumar Siddhant: formal analysis. Kavitha Velappan: data curation, investigation, formal analysis, software, and funding acquisition. Kyohei Hisano: project administration and funding acquisition. Osamu Tsutsumi: conceptualization, supervision, project administration, resources, validation, writing – review and editing, and funding acquisition. Ganesan Prabusankar: conceptualization, supervision, project administration, resources, validation, writing – review and editing, and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. P. and O. T. gratefully acknowledge JICA Friendship 2.0 for financial support. This research was also supported by the Japan-India Science Cooperative Program between JSPS and DST (JPJSBP120217716 for O.T., and DST/INT/JSPS/P-332/2021 for G. P.), and the Cooperative Research Program of the Network Joint Research Centre for Materials and Devices (O.T.). S. K. thanks DST-SERB (CRG/2022/000714) and IIT Hyderabad for the fellowship.Notes and references
- D. Di, A. S. Romanov, L. Yang, J. M. Richter, J. P. H. Rivett, S. Jones, T. H. Thomas, M. A. Jalbi, R. H. Friend, M. Linnolahti, M. Bochmann and D. Credgington, Science, 2017, 356, 159–163 CrossRef CAS.
- P. J. Conaghan, C. S. B. Metthews, F. Chotard, S. T. E. Jones, N. C. Greenham, M. Bochmann, D. Credgington and A. S. Romanov, Nat. Chem., 2020, 11:1758, 1–8 Search PubMed.
- A.-P. M. Reponen, F. Chotard, A. Lempelto, V. Shekhovtsev, D. Credgington, M. Bochmann, M. Linnolahti, N. C. Greenham and A. S. Romanov, Adv. Opt. Mater., 2022, 2200312, 1–15 Search PubMed.
- C. C. Beto, C. J. Zeman IV, Y. Yang, J. D. Bullock, E. D. Holt, A. Q. Kane, T. A. Makal, X. Yang, I. Ghiviriga, K. S. Schanze and A. S. Veige, Inorg. Chem., 2020, 59, 1893–1904 CrossRef CAS.
- A. Ruduss, B. Turovska, S. Belyakov, K. A. Stucere, A. Vembris, G. Baryshnikov, H. Ågren, J.-C. Lu, W.-H. Lin, C.-H. Chang and K. Traskovskis, ACS Appl. Mater. Interfaces, 2022, 14, 15478–15493 CrossRef CAS.
- K. R. Justin Thomas, J. T. Lin, Y. T. Tao and C. W. Ko, J. Am. Chem. Soc., 2001, 123, 9404–9411 CrossRef.
- H. M. J. Wang, C. S. Vasam, T. Y. R. Tsai, S.-H. Chen, A. H. H. Chang and I. J. B. Lin, Organometallics, 2005, 24, 486–493 CrossRef CAS.
- L. Cao, S. Huang, W. Liu, H. Zhao, X.-G. Xiong, J.-P. Zhang, L.-M. Fu and X. Yan, Chem. – Eur. J., 2020, 26, 17222–17229 CrossRef CAS PubMed.
- H. Schmidbaur, Angew. Chem., Int. Ed., 2019, 58, 5806–5809 CrossRef CAS.
- (a) L.-A. Schaper, X. Wei, S. J. Hock, A. Pöthig, K. Öfele, M. Cokoja, W. A. Herrmann and F. E. Kühn, Organometallics, 2013, 32, 3376–3384 CrossRef CAS; (b) H. Schmidbaur, H. G. Raubenheimer and L. Dobrzanska, Chem. Soc. Rev., 2014, 43, 345–380 RSC.
- X. Lin, W. Wu and Y. Mo, Coord. Chem. Rev., 2020, 419, 213401 CrossRef CAS.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2011, 50, 52–59 CrossRef CAS.
- M. Rigoulet, S. Massou, E. D. S. Carrizo, S. Mallet-Ladeira, A. Amgoune, K. Miqueu and D. Bourissou, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 46–51 CrossRef CAS.
- M. Teci, E. Brenner, D. Matt, C. Gourlaouen and L. Toupet, Chem. – Eur. J., 2015, 21, 10997–11000 CrossRef CAS PubMed.
- M. Vaddamanu, A. Sathyanarayana, Y. Masaya, S. Sugiyama, O. Kazuhisa, K. Velappan, K. Subramaniyam, K. Hisano, O. Tsutsumi and G. Prabusankar, Organometallics, 2020, 39, 2202–2206 CrossRef CAS.
- M. A. Narayana, M. Vaddamanu, A. Sathyanarayana, K. Siddhant, S. Sugiyama, K. Ozaki, A. K. Rengan, K. Velappan, K. Hisano, O. Tsutsumi and G. Prabusankar, Dalton Trans., 2021, 50, 16514–16518 RSC.
- M. Vaddamanu, A. Sathyanarayana, Y. Masaya, S. Sugiyama, O. Kazuhisa, K. Velappan, M. Nandeshwar, K. Hisano, O. Tsutsumi and G. Prabusankar, Chem. – Asian J., 2021, 16, 521–529 CrossRef CAS.
- M. A. Bakar, M. Sugiuchi, M. Iwasaki, Y. Shichibu and K. Konishi, Nat. Commun., 2017, 8, 1–7 CrossRef CAS.
- (a) M. Straka, E. Andris, J. Vícha, A. Ruzicka, J. Roithova and L. Rulisek, Angew. Chem., Int. Ed., 2019, 58, 2011–2016 CrossRef CAS PubMed; (b) M. A. Bakar, M. Sugiuchi, M. Iwasaki, Y. Shichibu and K. Konishi, Nat. Commun., 2017, 8:576, 1–7 Search PubMed; (c) J. Handelmann, C. N. Babu, H. Steinert, C. Schwarz, T. Scherpf, A. Kroll and V. H. Gessner, Chem. Sci., 2021, 12, 4329–4337 RSC; (d) L. Pallova, L. Abella, M. Jean, N. Vanthuyne, C. Barthes, L. Vendier, J. Autschbach, J. Crassous, S. Bastin and V. César, Chem. – Eur. J., 2022, 28, e202200166 CrossRef CAS.
- (a) L. Estevez, Dalton Trans., 2020, 49, 4797–4804 RSC; (b) G. d P. Gomes, G. Xu, X. Zhu, L.-M. Chamoreau, Y. Zhang, O. Bistri-Aslanoff, S. Roland, I. V. Alabugin and M. Sollogoub, Chem. – Eur. J., 2021, 27, 8127–8142 CrossRef.
- L.-A. Schaper, X. Wei, S. J. Hock, A. Pöthig, K. Öfele, M. Cokoja, W. A. Herrmann and F. E. Kühn, Organometallics, 2013, 32(11), 3376–3384 CrossRef CAS.
- (a) G. Kleinhans, A. K.-W. Chan, M. Y. Leung, D. C. Liles, M. A. Fermandes, V. W.-W. Yam, I. Fernández and D. I. Bezuidenhout, Chem. – Eur. J., 2020, 26, 6993–6998 CrossRef CAS; (b) R. Hamze, M. Idris, D. S. M. Ravinson, M. C. Jung, R. Haiges, P. I. Djurovich and M. E. Thompson, Front. Chem., 2020, 8, 1–9 CrossRef PubMed; (c) N. V. Tzouras, E. A. Martynova, X. Ma, T. Scattolin, B. Hupp, H. Busen, M. Saab, Z. Zhang, L. Falivene, G. Pisanò, K. V. Hecke, L. Cavallo, C. S. J. Cazin, A. Steffen and S. P. Nolan, Chem. – Eur. J., 2021, 27, 11904–11911 CrossRef CAS PubMed; (d) T.-y Li, D. G. Shlian, P. I. Djurovich and M. E. Thompson, Chem. – Eur. J., 2021, 27, 6191–6197 CrossRef CAS PubMed.
- (a) C. Biz, S. Ibáñez, M. Poyatos, D. Gusev and E. Peris, Chem. – Eur. J., 2017, 23, 14439–14444 CrossRef CAS PubMed; (b) J. Dinda, S. D. Adhikary, S. K. Seth and A. Mahapatra, New J. Chem., 2013, 37, 431–438 RSC; (c) A. Ruduss, B. Turovska, S. Belyakov, K. A. Stucere, A. Vembris and K. Traskovskis, Inorg. Chem., 2022, 61, 2174–2185 CrossRef CAS PubMed; (d) S. K. Verma, S. N. Ansari, P. Kumari and S. M. Mobin, Organometallics, 2019, 38, 2591–2596 CrossRef CAS.
- M. Heckenroth, E. Kluser, A. Neels and M. Albrecht, Angew. Chem., Int. Ed., 2007, 46, 6293–6296 CrossRef CAS.
- A. R. Chianese, A. Kovacevic, B. M. Zeglis, J. W. Faller and R. H. Crabtree, Organometallics, 2004, 23, 2461–2468 CrossRef CAS.
- J. W. Ogle and S. A. Miller, Chem. Commun., 2009, 5728–5730 RSC.
- O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed.
- M. Albrecht, Chem. Commun., 2008, 3601–3610 RSC.
- D. D. Perrin and W. L. F. Armarego, Purification of laboratory chemicals, Pergamon Press, London, 3rd edn, 1988 Search PubMed.
- W. J. Ramsay, N. A. W. Bell, Y. Qing and H. Bayley, J. Am. Chem. Soc., 2018, 140, 17538–17546 CrossRef CAS PubMed.
- S. Kalaivanan and G. Prabusankar, Catal. Lett., 2022, 1–11 Search PubMed.
- (a) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS; (b) L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., 2015, A71, 59–75 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- T. S. Thakur and G. R. Desiraju, Chem. Commun., 2006, 552–554 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, FTIR, TG/DTA, DFT, and NBO. CCDC 2182803–2182805. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2nj03215e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |