
Heterointerface promoted trifunctional electrocatalysts for all temperature high-performance rechargeable Zn–air batteries†
Nayantara K.
Wagh‡
a,
Dong-Hyung
Kim‡
a,
Chi Ho
Lee
b,
Sung-Hae
Kim
a,
Han-Don
Um
c,
Joseph Sang-Il
Kwon
 b,
Sambhaji S.
Shinde
*a,
Sang Uck
Lee
d and
Jung-Ho
Lee
*a
b,
Sambhaji S.
Shinde
*a,
Sang Uck
Lee
d and
Jung-Ho
Lee
*a
aDepartment of Materials Science and Chemical Engineering, Hanyang University, Ansan, Republic of Korea. E-mail: jungho@hanyang.ac.kr; physics.sambhaji2006@gmail.com
bArtie McFerrin Department of Chemical Engineering, Texas A&M Energy Institute, College Station, Texas 77843, USA
cDepartment of Chemical Engineering, Kangwon National University, Chuncheon, Gangwon-do 24341, Republic of Korea
dSchool of Chemical Engineering, Sungkyunkwan University, 2066 Seobu-ro, Jangan-gu, Suwon, Gyeonggi-do 16419, Republic of Korea
First published on 15th May 2023
Abstract
The rational design of wide-temperature operating Zn–air batteries is crucial for their practical applications. However, the fundamental challenges remain; the limitation of the sluggish oxygen redox kinetics, insufficient active sites, and poor efficiency/cycle lifespan. Here we present heterointerface-promoted sulfur-deficient cobalt-tin-sulfur (CoS1−δ/SnS2−δ) trifunctional electrocatalysts by a facile solvothermal solution-phase approach. The CoS1−δ/SnS2−δ displays superb trifunctional activities, precisely a record-level oxygen bifunctional activity of 0.57 V (E1/2 = 0.90 V and Ej=10 = 1.47 V) and a hydrogen evolution overpotential (41 mV), outperforming those of Pt/C and RuO2. Theoretical calculations reveal the modulation of the electronic structures and d-band centers that endorse fast electron/proton transport for the hetero-interface and avoid the strong adsorption of intermediate species. The alkaline Zn–air batteries with CoS1−δ/SnS2−δ manifest record-high power density of 249 mW cm−2 and long-cycle life for >1000 cycles under harsh operations of 20 mA cm−2, surpassing those of Pt/C + RuO2 and previous state-of-the-art catalysts. Furthermore, the solid-state flexible Zn–air battery also displays remarkable performance with an energy density of 1077 Wh kg−1, >690 cycles for 50 mA cm−2, and a wide operating temperature from +80 to −40 °C with 85% capacity retention, which provides insights for practical Zn–air batteries.
New conceptsFlexible solid Zn–air batteries demonstrate important promise as effective energy storage technologies for electric transport and portable electronics owing to their high energy-to-cost ratios, intrinsic safety, and wide-temperature operations. However, the sluggish kinetics of the cathodes, resistance against broad temperatures, high discharge–charge polarizations, and unsatisfactory service lifespan for oxygen electrochemical reactions during the charge–discharge processes limit the commercialization. In this work, we synthesized asymmetric CoS1−δ/SnS2−δ interface-promoted electrocatalysts with superior electronic couplings and numerous reactive sites using two-step solvothermal/pyrolyzation processes, which demonstrate outstanding trifunctional catalytic activities and long-term stability towards the ORR, OER, and HER. The CoS1−δ/SnS2−δ interfaces display the lowest oxygen reversibility overpotential of 0.57 V to obtain realistic Zn–air batteries. Flexible solid-state Zn–air batteries with CoS1−δ/SnS2−δ and CBC interfaces demonstrate a high energy density of 256 Wh kgcell−1 including all active and inactive components of fabricated cells, high operating rates of 50 to 100 mA cm−2, and a wide working temperature range from +80 to −40 °C with harsh operations, outperforming the commercial LIBs and DOE/Battery Consortium, USA 2025 targets. Furthermore, alkaline ZABs demonstrate a record-breaking power density of 249 mW cm−2 and cycle life of above 1000 cycles at 20 mA cm−2. |
1 Introduction
Increased electrical transport requires high-energy rechargeable batteries that safely operate under harsh conditions and a wide range of temperatures.1–4 Lithium-ion batteries (LIBs) suffer from limited capacity/power/energy, poor rate performances, and a short cycle life owing to the temperature-sensitive electrode dynamics and electrolyte freezing points with severe dendritic growth and lithium precipitation.5,6 Zinc-air batteries (ZABs) are considered as the most promising next-generation green power devices featuring high theoretical energy density (1086 Wh kg−1), zero carbon emissions, low cost of electrode materials, earth abundance, and intrinsic safety.1–4,7,8 ZABs can reveal feasibility against wide-temperature environments due to the ultralow and ultrahigh activation energies for the anode and cathode reactions resulting in temperature impervious electrode kinetics according to the Arrhenius equation.9 However, ZABs suffer from poor reversibility, poor charging/discharging rates with high/low charge–discharge voltages, limited electrochemical and thermodynamic stability mainly because of the sluggish electrochemical oxygen reduction/evolution reaction (ORR/OER), and poor ion transport properties of the electrolytes.1–4,7–10Pt-, Ir/Ru-based alloys or oxides are preeminent for the ORR, OER, and hydrogen evolution reaction (HER). However, scarcity, high cost, poor electrochemical stability, and single-functional activity limit widespread industrialization.11,12 Extensive efforts have been devoted to determining the ORR/OER/HER trifunctional catalysts regarding heteroatom-doped carbon, carbon-based composites, metal oxides, sulfides, nitrides, hydroxides, phosphides, and perovskites with unusual/comparable activities with noble-metals.1–4,7–20 However, the actual ZAB performances remain unsatisfactory under practical operating conditions due to the incapability of three-phase boundary electrochemical functions, underperforming catalytic activities/stabilities even for lower current densities, and limited accessibility for fabrication processes. Therefore, the rational construction of robust non-precious trifunctional advanced catalysts with outstanding activities for realizing high-performance solid/liquid ZABs under harsh operating conditions remains challenging.21,22
Typically, the electrochemical reactions (ORR, OER, and HER) determine the multistep intermediates (desorption/adsorption subclasses) by large energy barriers that demand multiscale controlled crystal and electronic structures, and chemical stoichiometries of electrocatalysts to obtain ideal reaction pathways by appropriate binding to key intermediates with the Sabatier principle.23,24 Transition metal catalysts (i.e., multishells, alloys, and heterojunctions) can enlarge the strengths and adsorption sites for different reaction intermediates with promising flexible multifunctional and synergetic interactions. Metal sulfides (layered/non-layered, WS2, MoS2, and MxSy where M= Co, Fe, Cu, Ni, etc.) have emerged as exciting candidates for building effective catalysts. Introducing promoters such as Cu, Ni, or Co can considerably increase the catalytic activities by providing more active sites of two transition metals compared to simple binary metal chalcogenides. However, the self-oxidation brings about structural and morphological collapse, which leads to instability of the catalysts.25–27
First, combining metal species and defect engineering can manifest the underlying intrinsic activities for reasonable adsorptions. Secondly, multiple heterointerface engineering can jointly modify the electronic properties of cations by breaking limitations for selectivity and activities for both core and shell materials. Thus, it is critical to construct interfacial structures to obtain the desirable catalytic functionalities. According to theoretical and experimental reports, heterointerfaces comprise: (1) Gibb's free energy modifications, (2) regulations for density of states at the Fermi level of the components, (3) abundant active site preferences, (4) interfacial coupling effects and charge transfer, (5) interfacial dislocations and structural discontinuities that induce the surface binding energies by intermediate moieties, and 6. appropriate catalytic kinetics.28–30 Thus, heterointerface engineering has been regarded as a promising strategy for the rational design of electrocatalysts.
Furthermore, there are two types of interfacial structures for heterogeneous catalysts: the boundary interface (oxide/oxide, metal/oxide, metal/sulfide, and sulfide/sulfide etc.) and active species anchored with a supporting conductive matrix interface (metal/C, metal-based catalysts/C etc.).24 Since the boundary interface controls the lattice structure, spatial distribution, conductivity, and active sites, it has a favorable impact on the electrochemical performances. In comparison, the conducting C support leads to severe agglomeration and instability of the catalysts with corrosive CO and CO2 formation. Recently, cobalt-based sulfides (CoSx) showed considerable electrochemical properties for the HER or OER. Theoretical calculations also predicted that CoSx offers favorable heterointerfaces for sulfides/oxides that can promote charge transfer through S-bonds due to severe reduction in the kinetic energy barriers for the nano-interfaces.26 Interactions among Sn in SnS2 and S in metal sulfides enhance the active catalytic sites owing to an increment of open metals and S-vacancies on the basal planes and edge sites. Furthermore, Sn-metal has weak hydrogen binding capacity compared to transition metals. The highest occupied d-orbital in Sn-metal deteriorates the bond strength of M*–OH, which can boost oxygen functionality to accelerate the OER/ORR processes.31,32 In this context, the appropriate configuration of well-designed heterointerface materials can be an admirable approach to attain exceptional specific activities by modulating physicochemical properties.33 Furthermore, such interactions promote dispersion and stability during electrolysis. In particular, electrochemical reactions occur at three-phase interfaces relating to the solid-phase electrode, liquid-phase electrolyte, and gas-phase feedstock. Beyond the three-phase interfaces, it cannot perform intrinsic catalytic activity. Therefore, designing advanced electrocatalysts with numerous robust multiphase interfaces is the fundamental challenge for achieving higher utilization efficiency of the catalysts and ZAB performances.
In this work, we report a scalable and facile strategy to fabricate CoS1−δ/SnS2−δ heterointerfaced high-performance trifunctional electrocatalysts in alkaline environments. It reveals advanced electrocatalysts by constructing multi-integrated distinct active species with synergistic influences. Accordingly, it demonstrated outstanding intrinsic ORR (half-wave potential of 0.90 V), OER (overpotential of 1.47 V at 10 mA cm−2), and HER (41 mV at 10 mA cm−2) activities and remarkable long-term durability. The lowest bifunctional overpotential gap (ΔE) of 0.57 V for 0.1 M KOH fulfills prominent oxygen bifunctional activity for high-performance, fast-charging, and low-charge/high-discharge voltages. Adopting CoS1−δ/SnS2−δ as the air cathode for alkaline ZABs shows comprehensive merits, including a high power density of 249 mW cm−2, the smallest discharge–charge overpotential of 0.70 V at 20 mA cm−2, and long cycle life of over 1000 cycles. Moreover, flexible solid-state Zn–air batteries work in a wide temperature range from +80 to −40 °C, and at high power and energy densities of 229 mW cm−2 and 1077 Wh kg−1, showing a cycle life of over 690 cycles at 25 °C and 264 cycles at −40 °C for 50 mA cm−2.
2 Experimental methods
2.1 Fabrication of CoS1−δ/SnS2−δ heterointerface catalysts
Cobalt chloride (60 mg, CoCl2·6H2O) and tin chloride (57 mg) were dissolved in a mixture of N,N-dimethylformamide and deionized water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) under sonication for 30 min. After that, 115 mg of sodium sulfide was added to the above reaction mixture under magnetic stirring for 15 min to obtain homogeneous suspensions. Then, hydrazine hydrate (200 μl) was slowly poured into the reaction mixture with vigorous sonication for 15 min. After that, the reaction suspension was transferred to a Teflon-lined autoclave and heat-treated at 180 °C for 12 h. After cooling to room temperature, the resulting blackish catalyst was collected by filtering, rinsed repetitively with deionized water, and then freeze-dried overnight under a vacuum. The CoS1−δ/SnS2−δ catalysts were achieved after heat treatment at 300 °C for 1 h under a nitrogen atmosphere and utilized for further characterization. For comparison, pristine CoS (120 mg cobalt chloride) and SnS2 (114 mg cobalt chloride) were also fabricated using similar experimental processes except for the insertion of tin chloride and cobalt chloride resources. Additionally, different compositions of Co and Sn with interfaces of Co0.2S1−δ/Sn1.8S2−δ, Co0.6S1−δ/Sn1.4S2−δ, Co1.4S1−δ/Sn0.6S2−δ, and Co1.8S1−δ/Sn0.2S2−δ were also prepared for comparison. Additional details are provided in the supplementary experimental methods (see ESI†).
:1 v/v) under sonication for 30 min. After that, 115 mg of sodium sulfide was added to the above reaction mixture under magnetic stirring for 15 min to obtain homogeneous suspensions. Then, hydrazine hydrate (200 μl) was slowly poured into the reaction mixture with vigorous sonication for 15 min. After that, the reaction suspension was transferred to a Teflon-lined autoclave and heat-treated at 180 °C for 12 h. After cooling to room temperature, the resulting blackish catalyst was collected by filtering, rinsed repetitively with deionized water, and then freeze-dried overnight under a vacuum. The CoS1−δ/SnS2−δ catalysts were achieved after heat treatment at 300 °C for 1 h under a nitrogen atmosphere and utilized for further characterization. For comparison, pristine CoS (120 mg cobalt chloride) and SnS2 (114 mg cobalt chloride) were also fabricated using similar experimental processes except for the insertion of tin chloride and cobalt chloride resources. Additionally, different compositions of Co and Sn with interfaces of Co0.2S1−δ/Sn1.8S2−δ, Co0.6S1−δ/Sn1.4S2−δ, Co1.4S1−δ/Sn0.6S2−δ, and Co1.8S1−δ/Sn0.2S2−δ were also prepared for comparison. Additional details are provided in the supplementary experimental methods (see ESI†).
3 Results and discussion
3.1 Electrocatalyst fabrication and structural analysis
The in situ CoS1−δ/SnS2−δ heterointerface catalysts were fabricated by a facile solvothermal approach in the reaction suspensions of CoCl2·6H2O, tin chloride, sodium sulfide, and hydrazine hydrate (see experimental methods for details). Finally, the as-prepared heterointerface catalysts were annealed at 300 °C in a N2 environment. Furthermore, the heterointerface stoichiometric compositions, time, loading levels, and temperature were also optimized according to their overall bifunctional performances (Fig. S1, S2 and Tables S1–S3, ESI†). The Co:Sn:S proportion for synthesized optimal CoS1−δ/SnS2−δ is 1:1:2.85 (Table S3, ESI†). The surface morphologies and fine nanostructures for the CoS1−δ/SnS2−δ heterointerface-based electrocatalysts were determined using scanning electron microscopy (SEM) and transmission electron microscopy (TEM). It displays three-dimensional (3D) well-distributed cumulation of microspheres with a large rough surface, in which microspheres are coalesced from intersected and ultrathin nanosheets/nanoflakes of 3–16 nm thickness (Fig. 1a, b and Fig. S1, ESI†). These distinctive morphological structures of CoS1−δ/SnS2−δ heterointerface present abundant active site preferences by facilitating superior electrolyte penetrations/ion diffusions that also contribute to the profound degree of construction of nanointerface-based active sites to enhance the oxygen reactions. Pristine CoS and SnS2 catalysts show nanospherical particles and microspheres with numerous sheets. The HRTEM image displays the interlayered lattice fringes of 0.1689 nm and 0.6181 nm corresponding to the (110) and (001) crystal reflections, respectively, featuring the heterointerface of both CoS1−δ and SnS2−δ phases (Fig. 1b and c). The selected area electron diffraction (SAED) pattern manifests the well-defined crystalline structures in good agreement with the HRTEM image (Fig. 1c). High-angle annular dark-field scanning TEM (HAADF-STEM) and elemental maps present the altered distribution of Co, Sn, and S elements across the heterointerface (Fig. 1d). The high-resolution HAADF-STEM maps at the edge-sites (i.e., the specified area of interface) validate the interlayered structures of CoS1−δ and SnS2−δ phases (Fig. 1e).
 | ||
| Fig. 1 Microscopic and structural visualizations of CoS1−δ/SnS2−δ hetero-interface catalysts. (a) STEM image. (b) High-resolution STEM (HRTEM) image hetero-interface of CoS1−δ and SnS2−δ. Scale 5 nm. (c) SAED pattern. (d) HAADF-STEM image and the corresponding maps for Co, Sn, and S. Scale 200 nm. (e) HAADF-STEM image and the corresponding maps at a specified edge site region for Co, Sn, and S with CoS1−δ/SnS2−δ phase. Scale 5 nm. | ||
Powder X-ray diffraction (XRD) patterns display two characteristic phases of SnS2 (Berndtite-2T) and CoS for the interfaced CoS1−δ/SnS2−δ catalysts along with hexagonal crystal structures (JCPDS: 065-3418 for CoS and JCPDS: 023-0677 for SnS2, Fig. 2a and Fig. S2, ESI†). The CoS1−δ/SnS2−δ displays the lattice parameters a = b = 3.368 Å and c = 5.17 Å for CoS1−δ and a = b = 3.649 Å and c = 5.899 Å for SnS2−δ, respectively. Furthermore, the CoS1−δ and SnS2−δ consist of P63/mmc and P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1(164) space groups, respectively. The Raman spectra (Fig. S3, ESI†) display the characteristic peaks related to metallic hexagonal CoS for 469, 512, and 664 cm−1 corresponding to the E2g, F2g, and A1g vibrational modes, respectively.34,35 But, the intense peaks for 310 cm−1 and 224 cm−1 represent the A1g and E2g vibrational modes of SnS2.36,37 The reverse shifting of the diagnostic phase transitions implies the opposite charge transfers, which vindicates the formation of a heterointerface of CoS1−δ and SnS2−δ.34 X-ray photoelectron spectroscopy (XPS) was adopted to evaluate the surface chemical states, valence states, and chemical compositions (Fig. 2 and Tables S1, S2, ESI†), which indicates the prevalent oxidation states of Co, Sn, and S in CoS1−δ/SnS2−δ heterointerface-based catalysts. The elemental composition of CoS1−δ/SnS2−δ shows Co, Sn, and S of 21.41 at%, 21.68 at%, and 56.91 at%, in which lower S content confirms the S-vacancies by ∼5.1% (Tables S1 and S2, ESI†). Co 2p XPS spectra (Fig. 2b) display the characteristic spin–orbit 2p3/2 and 2p1/2 states of the Co–S bond (781.6 and 796.8 eV), Co2+ (785.6 and 800.7 eV) and satellites, respectively.3,38,39 The Sn 3d spectra (Fig. 2c) show significant peaks at 486 and 494.4 eV ascribed to the 3d5/2 and 3d3/2 states of Sn–S bonds with Sn4+ valence.40,41 The blue shifts for Sn 3d and Co 2p spin–orbitals in the CoS1−δ/SnS2−δ heterointerface compared to those of pristine CoS and SnS2 indicate the formation of strong interactions between CoS1−δ and SnS2−δ phases. S 2p XPS spectra (Fig. 2d) demonstrate the presence of metal sulfide (Co–S or Sn–S)-related characteristics peaks for 162.5 and 163.7 eV corresponding to the 2p3/2 and 2p1/2 core states.3,42 Structural and chemical characterizations suggest the favorable electron delocalization in the sub-lattices of sulfides, which illustrates the enhanced charge transfer mechanism for electrochemical reactions.10,43 The BET surface areas of CoS/SnS2, CoS, and SnS2 are 135, 112, and 65 m2 g−1 (Fig. S4, ESI†), respectively.
m1(164) space groups, respectively. The Raman spectra (Fig. S3, ESI†) display the characteristic peaks related to metallic hexagonal CoS for 469, 512, and 664 cm−1 corresponding to the E2g, F2g, and A1g vibrational modes, respectively.34,35 But, the intense peaks for 310 cm−1 and 224 cm−1 represent the A1g and E2g vibrational modes of SnS2.36,37 The reverse shifting of the diagnostic phase transitions implies the opposite charge transfers, which vindicates the formation of a heterointerface of CoS1−δ and SnS2−δ.34 X-ray photoelectron spectroscopy (XPS) was adopted to evaluate the surface chemical states, valence states, and chemical compositions (Fig. 2 and Tables S1, S2, ESI†), which indicates the prevalent oxidation states of Co, Sn, and S in CoS1−δ/SnS2−δ heterointerface-based catalysts. The elemental composition of CoS1−δ/SnS2−δ shows Co, Sn, and S of 21.41 at%, 21.68 at%, and 56.91 at%, in which lower S content confirms the S-vacancies by ∼5.1% (Tables S1 and S2, ESI†). Co 2p XPS spectra (Fig. 2b) display the characteristic spin–orbit 2p3/2 and 2p1/2 states of the Co–S bond (781.6 and 796.8 eV), Co2+ (785.6 and 800.7 eV) and satellites, respectively.3,38,39 The Sn 3d spectra (Fig. 2c) show significant peaks at 486 and 494.4 eV ascribed to the 3d5/2 and 3d3/2 states of Sn–S bonds with Sn4+ valence.40,41 The blue shifts for Sn 3d and Co 2p spin–orbitals in the CoS1−δ/SnS2−δ heterointerface compared to those of pristine CoS and SnS2 indicate the formation of strong interactions between CoS1−δ and SnS2−δ phases. S 2p XPS spectra (Fig. 2d) demonstrate the presence of metal sulfide (Co–S or Sn–S)-related characteristics peaks for 162.5 and 163.7 eV corresponding to the 2p3/2 and 2p1/2 core states.3,42 Structural and chemical characterizations suggest the favorable electron delocalization in the sub-lattices of sulfides, which illustrates the enhanced charge transfer mechanism for electrochemical reactions.10,43 The BET surface areas of CoS/SnS2, CoS, and SnS2 are 135, 112, and 65 m2 g−1 (Fig. S4, ESI†), respectively.
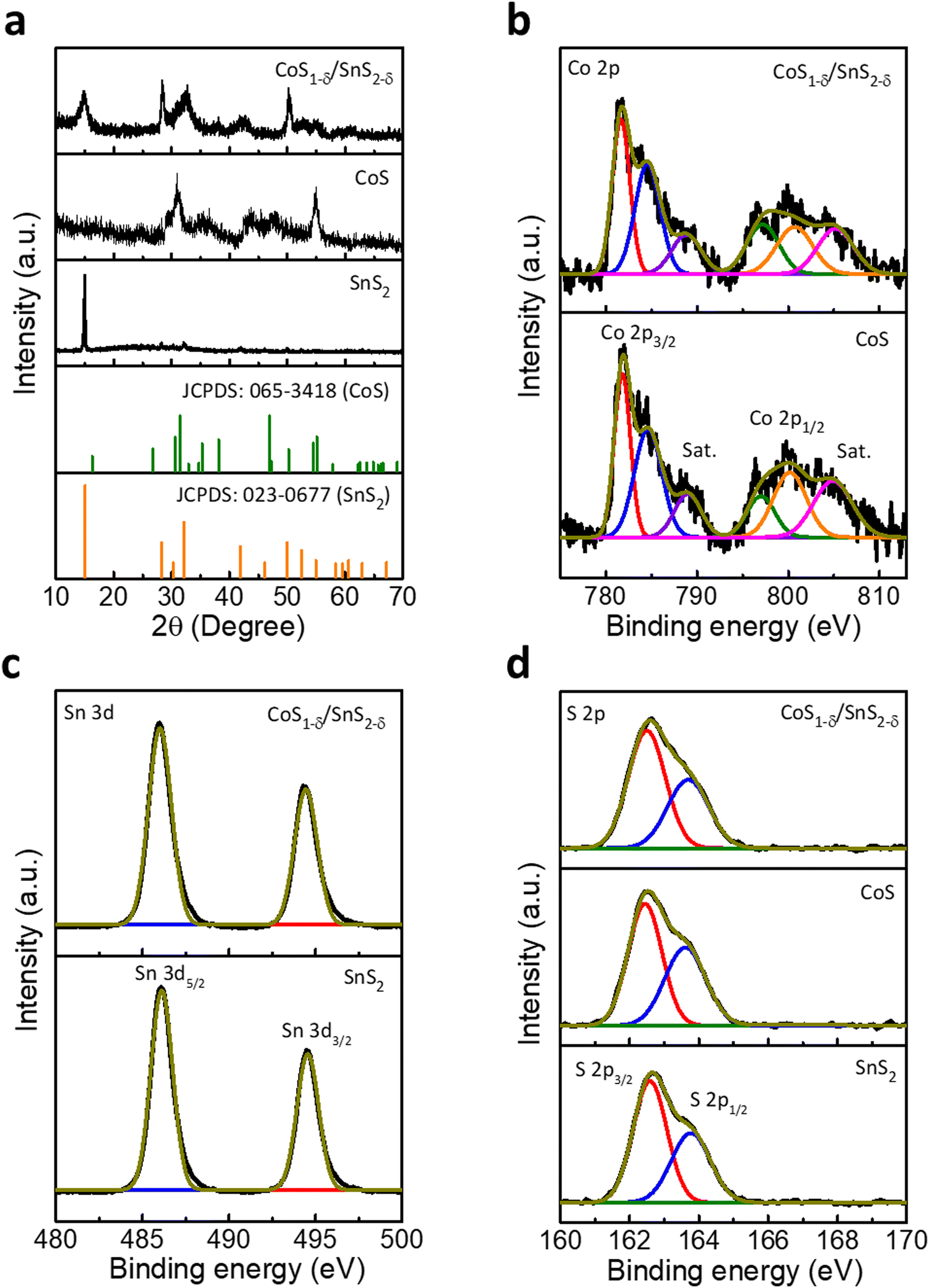 | ||
| Fig. 2 Structural and chemical characterizations of the fabricated catalysts. (a) Powder X-ray diffractions for SnS2, CoS, and CoS1−δ/SnS2−δ catalysts. (b) Co 2p XPS spectra for the CoS1−δ/SnS2−δ heterostructure related to those of pristine CoS materials. (c) Sn 3d XPS spectra for CoS1−δ/SnS2−δ heterostructures related to those of SnS2 catalysts. (d) S 2p XPS spectra for CoS1−δ/SnS2−δ compared to those of SnS2 and CoS catalysts. | ||
The presence of S vacancies is characterized by experimental techniques, as displayed in Tables S1 and S2 (ESI†). The HRTEM image displays the crystal lattice spacing of 0.1689 and 0.6181 nm corresponding to the (110) and (001) planes of the CoS1−δ and SnS2−δ phases. However, bulk CoS and SnS2 show 0.167 and 0.589 nm for the respective (110) and (001) planes. This enlarged lattice interlayer distance implies that the S vacancies cause the lattice distortions. Furthermore, the XRD patterns display the shifting of the (110) and (001) crystal planes towards lower diffraction angles compared to those of the pristine phases, which indicates the expansion of the interplanar distances between (Co–S or S–Sn–S) well consistent with the HRTEM results. The enlarged interlayer spacing is attributed to the weak van der Waals interactions due to the increased number of S-vacancies on the basal planes. The Raman results also clarify the XRD and HRTEM results. The Raman spectra display characteristic peaks of 469, 664, and 310, 224 cm−1 corresponding to the out-of-plane A1g and in-plane E2g vibration modes of CoS1−δ and SnS2−δ phases, whereas the pristine CoS and SnS2 spectra have peaks at 473, 669, and 315, 235 cm−1, illustrating the redshifts in the Co–S and Sn–S phonon modes in the basal planes. This is ascribed to the gradual decrease in the Co–S and Sn–S chemical bonds along with in-plane S vacancies.44,45
3.2 Trifunctional electrochemical performance
The trifunctional electrochemical performances for the ORR, OER, and HER for the CoS1−δ/SnS2−δ heterointerface catalysts were assessed in alkaline environments (0.1 M KOH) using a three-electrode system. The linear sweep voltammetry (LSV) profiles for the CoS1−δ/SnS2−δ heterointerface manifest the superior half-wave potential (E1/2) of 0.90 V (vs. RHE) compared to those of Pt/C (E1/2 = 0.84 V) and state-of-the-art of previous champion catalysts (Fig. 3a and Table S4, ESI†). The reaction kinetics, electron transfer number, peroxide (H2O2) yield, and selectivity were determined by RRDE and the Koutecky–Levich (K–L) equation. A linear increase for the limiting current densities (or fitted straight lines for K–L plots, Fig. 3b) relating to rotating speed from 400 to 2000 rpm was detected for rotating disk electrode (RDE) measurements revealing the first-order reaction kinetics for the ORR. the electron transfer number (n) for CoS1−δ/SnS2−δ heterointerface catalysts is 3.97 with a H2O2 yield of ∼2% (3.9 with a Pt/C yield of 5%), suggesting the 4e− pathway and more excellent selectivity for the ORR (Fig. S5, ESI†). This indicates that the heterointerface between CoS1−δ and SnS2−δ plays a vital role in the catalysts. Chronoamperometry tests (Fig. 3c) for CoS1−δ/SnS2−δ manifest the retention of 97% of the initial current for 240 h, whereas the Pt/C deteriorates severely over 32% within 15 h, which illustrates the excellent ORR stability of the CoS1−δ/SnS2−δ catalysts. Furthermore, the CoS1−δ/SnS2−δ electrode displays no noticeable change in the half-wave potential after 8000 accelerated durability test (ADT) cycles, while that of Pt/C drastically drops, limiting the current density and half-wave potential, validating the outstanding tolerance against electrochemical reactions (Fig. 3d). Electrochemical impedance spectroscopy (EIS) verifies the smallest charge transfer resistance for CoS1−δ/SnS2−δ related to those of CoS and SnS2 materials, which reveals the faster reaction kinetics and electron transfer rates (Fig. S6, ESI†). Furthermore, the electrochemical active surface area (ECSA) was evaluated using double-layer capacitance (Cdl) analysis for the prepared catalysts (Fig. S7, ESI†). The ECSA values are 28.2, 41.8, and 57.1 mF cm−2 for SnS2, CoS, and CoS1−δ/SnS2−δ, respectively, indicating the large active electrochemical surface area for the heterointerface-based catalysts.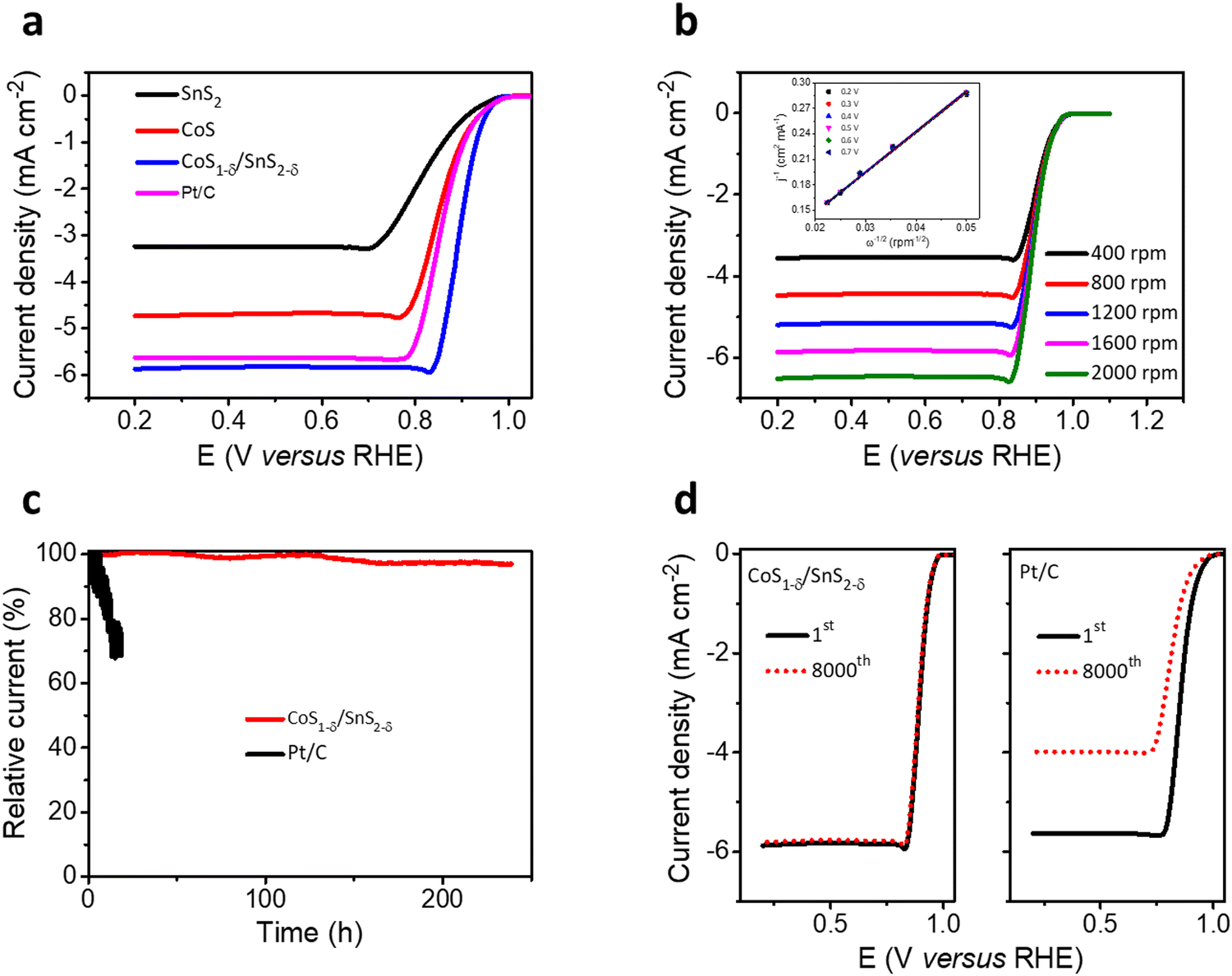 | ||
| Fig. 3 ORR electrochemical performances. (a) LSV polarizations for the CoS1−δ/SnS2−δ hetero-interface related to those of SnS2, CoS, and reference Pt/C materials. (b) RDE profiles for CoS1−δ/SnS2−δ materials. Sweeping rates of 400–2000 rpm. K–L polarizations for electrochemical kinetics with CoS1−δ/SnS2−δ materials from 0.2 to 0.7 V (inset). (c) Chronoamperometric (i–t) tests for CoS1−δ/SnS2−δ referenced to that of commercial Pt/C at their relevant half-wave potentials. (d) ORR-accelerated operations for CoS1−δ/SnS2−δ and reference Pt/C materials for the initial cycle and after 8000 cycles. (Conditions: 0.1 M KOH and geometrical surface area utilized for current density calibration.) | ||
From the perspective of Zn–air batteries’ potential applications, the OER activities and robustness for the fabricated materials have been compared to those of commercial RuO2. The CoS1−δ/SnS2−δ delivers the lowest overpotential of 240 mV at 10 mA cm−2 related to those of RuO2 (330 mV), CoS (360 mV), SnS2 (430 mV), and previously reported OER catalysts (Fig. 4a and Table S4, ESI†). The Tafel slopes of CoS1−δ/SnS2−δ for the ORR and OER are 46 mV dec−1 and 55 mV dec−1, respectively, which are the lowest related to those of the Pt/C, RuO2, CoS, and SnS2 catalysts, demonstrating promising intrinsic ORR and OER kinetics processes (Fig. 4b). Furthermore, the overall bifunctional oxygen performance (ΔE = Ej=10 – E1/2, Fig. 4c) manifests the smallest overpotential of 0.57 V for CoS1−δ/SnS2−δ outperforming those of Pt/C (1.19 V), RuO2 (0.99 V), CoS, and SnS2, demonstrating the lowest polarization for oxygen electrochemistry. Fig. S8 (ESI†) displays paramount retention of the current response (95.5%) for CoS1−δ/SnS2−δ after 230 h compared to RuO2 (56%) after 19 h. LSV tests manifest negligible overpotential degradation (12 mV) for CoS1−δ/SnS2−δ after 8000 cycles, whereas RuO2 displayed drastic degradation in overpotential by 340 mV. This exceptional stability for CoS1−δ/SnS2−δ is because distinctive microstructures forming a strong heterointerface between CoS1−δ and SnS2−δ protect the active sites from dissolution or corrosion. The record-breaking reversible oxygen (ORR–OER) performance permits the CoS1−δ/SnS2−δ heterointerface, compared to those of champion electrocatalysts previously reported, standing for the best applicability for high-performance Zn–air batteries (Fig. 4d and Table S4, ESI†).1,2,5–27,46–50
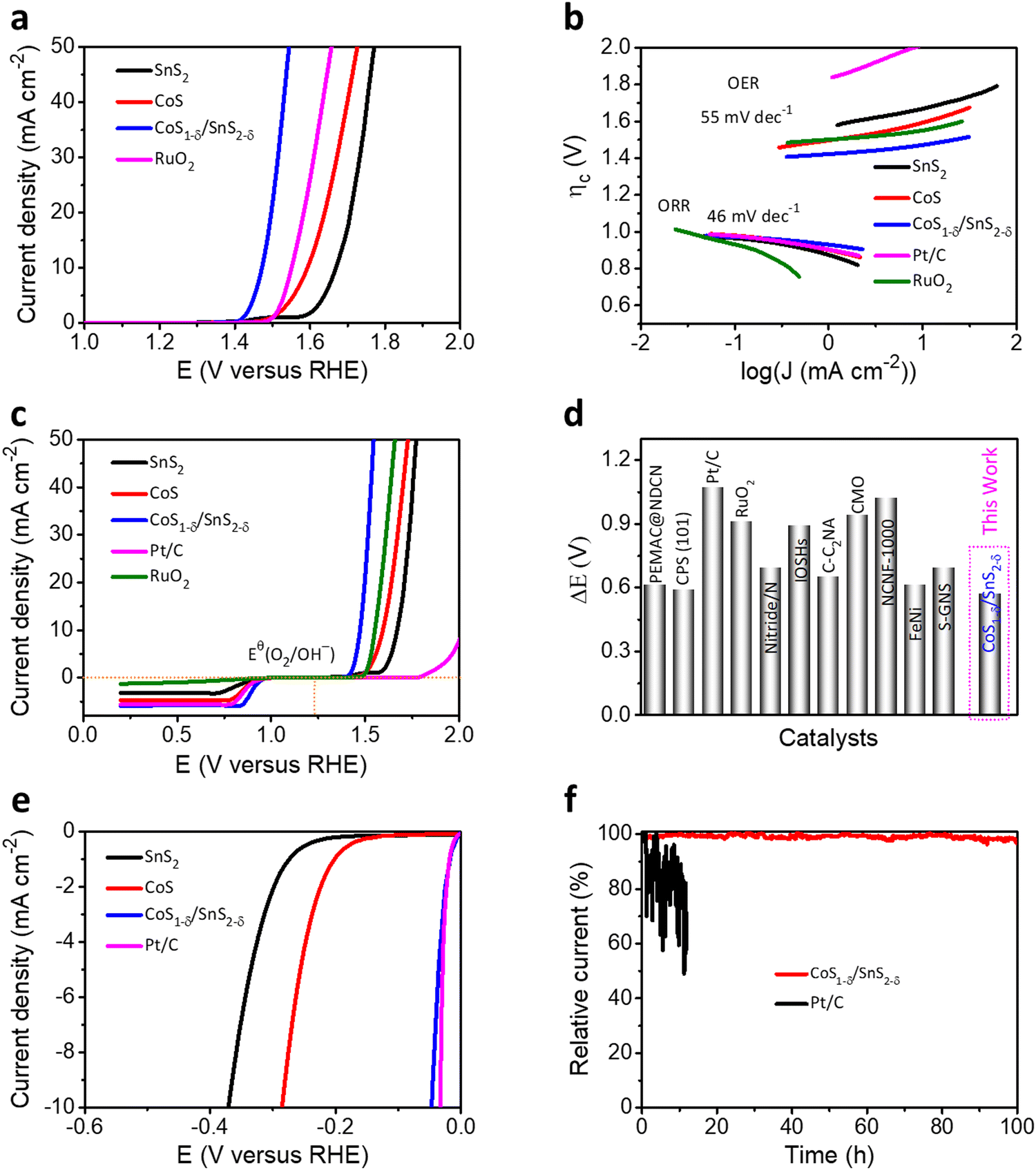 | ||
| Fig. 4 Trifunctional electrochemical performances. (a) OER LSV polarizations for the CoS1−δ/SnS2−δ hetero-interface related to those of SnS2, CoS, and reference RuO2 materials. (b) Tafel plots for both ORR and OER electrochemical kinetics for CoS1−δ/SnS2−δ, SnS2, CoS, Pt/C, and RuO2 materials. (c) Oxygen bifunctional polarizations of hetero-interface-based catalysts related to those of Pt/C, RuO2 and pristine SnS2 and CoS materials. (d) State-of-the-art of oxygen bifunctional reversibility (ΔE) of CoS1−δ/SnS2−δ hetero-interfaced catalysts compared with those of the previous catalytic materials (for details see ESI† Table S4). (Conditions: 0.1 M KOH and geometrical surface area utilized for current density calibration.) (e) HER LSV profiles of the CoS1−δ/SnS2−δ hetero-interface compared to those of SnS2, CoS, and Pt/C materials. (f) i–t stability tests for CoS1−δ/SnS2−δ and Pt/C catalysts for their overpotentials. | ||
In addition, CoS1−δ/SnS2−δ displays superb catalytic performances for the HER and obtains the overpotential of 41 mV at 10 mA cm−2 comparable to Pt/C (33 mV) with negligible decay after 100 h of chronoamperometry response tests (Fig. 4e, f and Table S5, ESI†). Furthermore, it possesses a Tafel slope of 36 mV dec−1 related to that of Pt/C (35 mV dec−1, Fig. S9, ESI†), implying the Heyrovsky mechanism, in which hydrogen desorption from the catalyst surface is the rate-determining step.12,16,32,42 The fabricated catalysts follow the performances in the sequence of CoS1−δ/SnS2−δ > Pt/C > CoS > SnS2 for ORR, CoS1−δ/SnS2−δ > RuO2 > CoS > SnS2 for the OER, and Pt/C ≥ CoS1−δ/SnS2−δ > CoS > SnS2 for the HER, which illustrates significantly enhanced trifunctional activities.
To determine the structural and morphological insights of the catalysts during the ORR and OER catalytic processes, XRD, Raman, TEM, and EDS analyses were performed. Fig. S10 (ESI†) displays slight surface oxidation of CoS1−δ/SnS2−δ after the OER. In contrast, during the ORR process, XRD, Raman, TEM, and EDS (Fig. S11, ESI†) results did not show obvious variation in the structures, morphologies, and oxygen contents of CoS1−δ/SnS2−δ, demonstrating that no surface oxidation was observed on the surface of the catalysts during the ORR.
3.3 Thermodynamic and electronic analysis
Based on the experimental observation, we conducted density functional theory (DFT) calculations to gain insights into the origin of the HER and OER/ORR trifunctional activities on the CoS/SnS2 heterojunction structures with S defects. To investigate their improved performance, we preferentially designed optimal CoS/SnS2 heterojunction structures that minimize lattice mismatch by considering the T phase geometry between defect-free (CoS and SnS2) and S defective structures (CoS1−δ and SnS2−δ). However, combining two different structures always causes different structural stress with specific strains, where tensile and compressive strains are applied to the CoS and SnS2 structures, respectively (Fig. 5a). Also, it is well known that an adequate defect and strain are essential to increase the catalytic activity by enhancing the adsorbent binding.51–55 Therefore, we systematically considered a total of four different structures (CoS, SnS2, CoS1−δ/SnS2, and CoS/SnS2−δ) to understand the effects of heterojunctions with S defects on the enhanced trifunctional activities.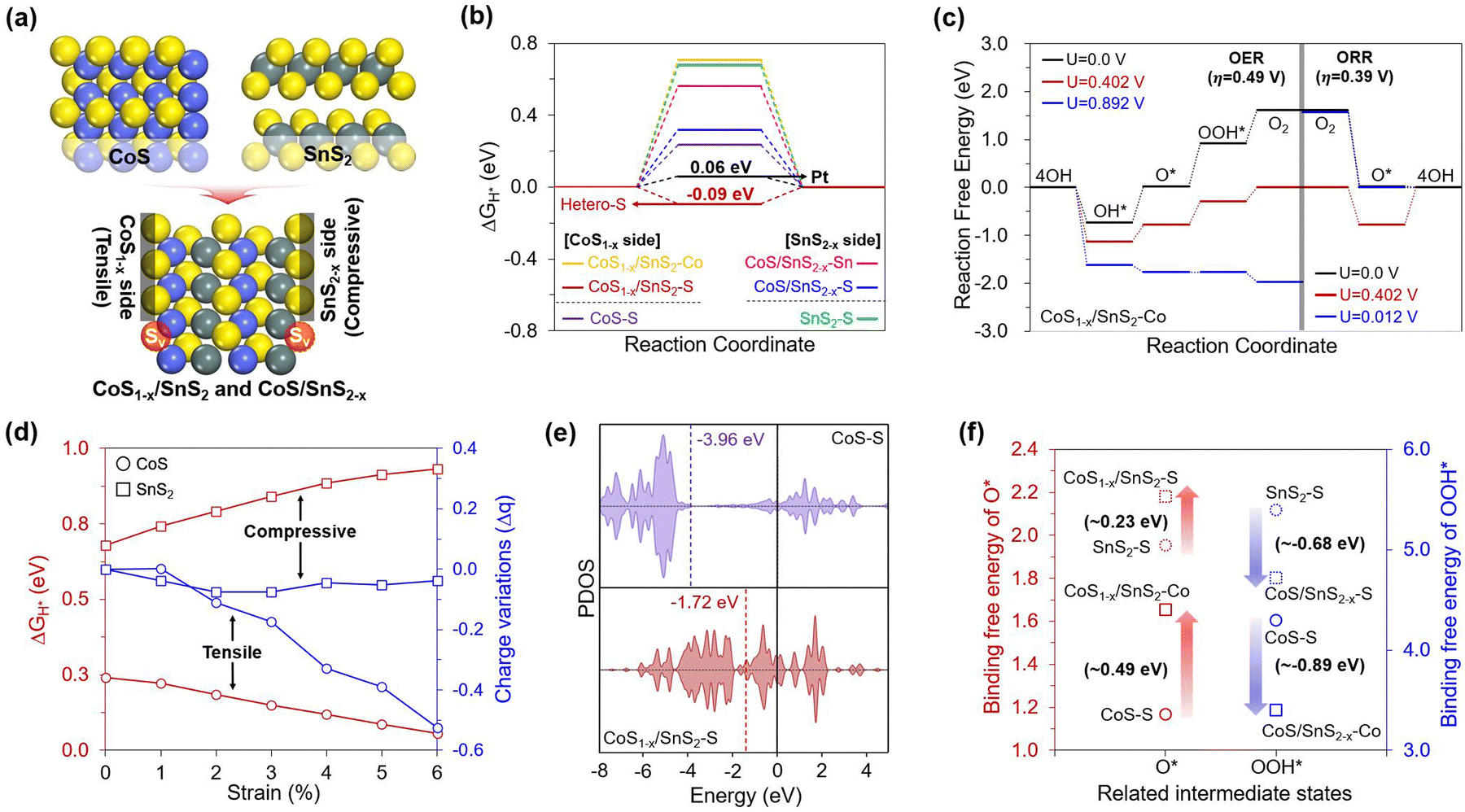 | ||
Fig. 5 Theoretical kinetics. (a) Designed CoS, SnS2, CoS1−δ/SnS2 and CoS/SnS2−δ structures. (b) HER free energy diagram (FED) for the S sites of CoS and SnS2, S and Co sites of CoS1−δ/SnS2, and S and Sn sites of CoS/SnS2−δ. H* binding free energy (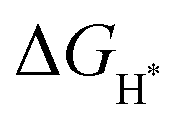 ) of Pt was obtained at optimal coverage (1.08 ML). (c) OER and ORR free energy diagrams on the most active Co site of the CoS1−δ/SnS2 structure. (d) H* binding free energy and charge variations according to the compressive and tensile strains applied to the SnS2 and CoS structures, respectively. (e) Partial density of states (PDOS) for Sp-orbitals of CoS and CoS1−δ/SnS2. (f) Binding free energy variations of intermediate states (O* and OOH*) on the most active sites of CoS, SnS2, and heterojunction structures. ) of Pt was obtained at optimal coverage (1.08 ML). (c) OER and ORR free energy diagrams on the most active Co site of the CoS1−δ/SnS2 structure. (d) H* binding free energy and charge variations according to the compressive and tensile strains applied to the SnS2 and CoS structures, respectively. (e) Partial density of states (PDOS) for Sp-orbitals of CoS and CoS1−δ/SnS2. (f) Binding free energy variations of intermediate states (O* and OOH*) on the most active sites of CoS, SnS2, and heterojunction structures. | ||
Based on these well-established structures, we theoretically evaluated the HER and OER/ORR overpotential values (ηHER and OER/ORR) using a free energy diagram (FED) approach for all possible active sites (Fig. S12, ESI†).1,56 The calculated results revealed that the heterojunction structure significantly lowered the HER and OER/ORR overpotentials (0.09 and 0.49/0.39 V), outperforming those of defect-free structures (Fig. 5b and c), and these values also represent competitive performances compared to conventional Pt and RuO2 for the HER/ORR and OER, respectively. Interestingly, the heterojunction structure could uniquely activate the HER and OER/ORR at the S and Co sites on the CoS1−δ side, respectively. Looking at the overall HER activity trend, firstly, the FED results in Fig. 5b showed that the S defects of the heterojunction structure significantly enhanced the HER performance compared to the defect-free structures. Such an activity increase was prominently featured in the CoS1−δ side compared to the SnS2−δ side, based on the following order of the overpotentials: Pt (0.06 V) < CoS1−δ/SnS2–S (−0.09 V) < CoS–S (0.24 V) < CoS/SnS2−δS (0.32 V). This result suggests that the additional strain to the defective heterojunction is essential to promote H* binding strength.
To understand the HER activity-dependence on the strain effect in detail, we explored their geometric and electronic features that could be changed when two structures were combined. From a geometric point of view, the heterojunction induced different structural stresses of the respective structures with specific strains of up to 6%. So, we artificially applied the tensile and compressive strains to the CoS and SnS2 structures to identify the systematic improvements in their HER activities. Then, we found that the greater the tensile strains to the CoS, the greater the H* binding strength improvement. In contrast, the compressive strains to the SnS2 weakened the H* binding strength (Fig. 5d). Additionally, the Bader charge analysis showed that the number of electrons accumulated in the S active sites became higher by increasing the tensile strains to the CoS, while the compressive strains to the SnS2 did not change significantly. Therefore, we note that the increased electron quantities can help in enhancing the HER activity by transferring more electrons from the S active sites to H* under more reductive environments. Our results demonstrated the critical role of the S active sites in promoting HER activity, and thus, we turned to investigate the p-orbital-dependence on the S defects and heterojunction to determine the filling of the anti-bonding state that can be generated when the H* binds on the active sites. Fig. 5e reveals that the p-band center of the S active sites in the CoS1−δ/SnS2 is shifted to a higher position than that of CoS, with the following order: CoS (−3.96 eV) < CoS1−δSnS2 (−1.72 eV). This suggests that the CoS1−δ/SnS2 has enhanced H* binding strength due to a less-filled anti-bonding state, whereas the CoS has weak H* binding strength due to a more filled antibonding state.
We also investigated the basis for differences in the OER and ORR performances between defect-free and heterojunction structures. Through the constructed free energy diagrams (FEDs) in Fig. 5c and Fig. S13–S15 (ESI†), we found that the heterojunction has the best OER/ORR activities, based on the following order of ηOER and ORR values: CoS1−δ/SnS2 (0.49 and 0.39 V) < CoS (1.90 and 0.65 V) < CoS/SnS2−δ (1.84 and 1.11 V). This activity trend suggested that the OER and ORR activities improved with the synergistic effect between the S defects and heterojunction, but this phenomenon was featured in Co sites of the CoS1−δ side, unlike the HER, which prefers S sites. This implies that the selective role of S and Co active sites can separate HER and OER/ORR catalysis.
To first uncover the reason for the enhanced OER performance on the Co sites, we investigated the thermodynamic FED results (Fig. 5c and Fig. S13, ESI†). We found that the potential-determining step (PDS) of the catalysts is the third step (ΔG3). This result is derived from the combination of strong O* and weak OOH* intermediates. The high OER activity should be accompanied by a decrease in the difference between their binding strengths. Hence, we focused on the PDS differences between defect-free and heterojunction structures. The results in Fig. S13 (ESI†) showed that the CoS1−δ/SnS2 structure has the lowest ΔG3 value (0.893 V), which leads to good OER activity competitive with the conventional RuO2 catalyst (ηOER = 0.43 V). However, from the theoretically emphasized ΔG3 value, one might wonder which of the O* and OOH* intermediates plays a more vital role in effectively decreasing the OER overpotential. Hence, we deeply explored their binding free energy changes between defect-free and heterojunction structures considering all possible active sites (Fig. 5e) and found that the OOH* binding strength increases much more significantly than the extent to which the O* binding strength is weakened on the Co sites of the heterojunction. Based on this theoretical evaluation, one can envision that the enhanced OOH* binding strength can highly influence the OER activity by reducing the ΔG3 value. Furthermore, we explored the electronic properties based on the charge density difference and partial density of states (PDOS) for the heterojunction because the binding strength of the intermediates is closely related to the valence orbital trend of active sites.56,57 As shown in Fig. S16 (ESI†), the charge difference revealed that the Co sites have positive charges, leading to efficient binding with O-based intermediates with negative charges compared to S sites with negative charges. The PDOS results showed that the Co sites of CoS1−δ/SnS2 have a significantly high d-band center compared to that of CoS, leading to enhanced OOH* binding strength via a less-filled antibonding state.
In contrast to the four-step pathway for the OER process, the ORR can proceed via either a four-step or two-step pathway, depending on the relative stability between O2* → OOH* and O2* → O*.54 To determine which way is more favorable, we compared their reaction-free energies (Table S6, ESI†) and found that the two-step pathway is more efficient, resulting in a significantly lower overpotential value on the Co sites (0.39 V, Fig. S14 and S15, ESI†), which is competitive with the conventional Pt catalyst (0.45 V). Using this pathway, we investigated the reason for the improved ORR activity on the Co sites of CoS1−δ/SnS2, especially rather than the Sn site of CoS/SnS2−δ.
Considering the essential O2 binding for the ORR process, we could correlate the activity difference between Co and Sn sites with their electronic configurations that play a critical role in the electron back-donation from occupied d-orbital to unoccupied π*-orbital, ultimately leading to the weakening of the double bond of O2. To enable strong back-donation of electrons, the metal site should have empty d-orbitals first to accept electrons from the O2*. However, the Sn4+ site (i.e., 4d10) of CoS/SnS2−δ has a fully-filled d-orbital compared to the Co2+ site (i.e., 3d7) of CoS1−δ/SnS2. Therefore, one can envision that the ORR process on Co2+ sites occurs more favorably by weakening the double bond of O2 through efficient electron back-donation between Co and O2. On the other hand, Sn4+ sites are inefficient even for the O2 binding step due to inefficient electron transfer, even though the O* binding is strong. Based on these theoretical investigations, we concluded that the CoS1−δ/SnS2 heterojunction exhibited superior trifunctional HER and OER/ORR activities on the separated S and Co active sites.
3.4 Alkaline Zn–air battery performances
To validate the practical applications of high-performance Zn–air batteries, home-made aqueous ZABs with CoS1−δ/SnS2−δ catalysts as air-cathode, zinc sheet as a negative electrode, and 6 M KOH + 2M Zn acetate as electrolyte have been preliminarily evaluated (Fig. S17, ESI†). For contrast, Pt/C + RuO2-based ZABs were constructed as a reference. In Fig. 6a, the CoS1−δ/SnS2−δ based ZABs display an open-circuit potential (OCP) of 1.56 V, superior to those of Pt/C + RuO2 (1.44 V), CoS (1.4 V), and SnS2 (1.37 V). The CoS1−δ/SnS2−δ cathode offers high discharge and low charge voltages and the highest power density of 249 mW cm−2 at 300 mA cm−2 related to those of reported air cathodes (Fig. 6a and Table S7, ESI†), validating the superior ORR/OER performance. The Pt/C + RuO2-based ZAB has 230 mW cm−2. As we know, the lower the overpotential in electrochemical catalysis, the more prominent the catalytic activities. Likewise, the lower voltage gap in the charge–discharge profiles leads to superior ZAB overall performance. The CoS1−δ/SnS2−δ ZAB manifests the smallest charge–discharge potential gap from 1–300 mA cm−2 compared to those of Pt/C + RuO2, CoS, and SnS2-based ZABs (Fig. 6b). Notably, the CoS1−δ/SnS2−δ ZAB offers high working rates from 5 to 50 mA cm−2 with average discharge potentials of 1.33, 1.30, 1.25, 1.22, and 1.18 V for current densities of 5, 10, 20, 30 and 50 mA cm−2, respectively (Fig. 6c). For comparison, Pt/C + RuO2 displays lower potentials of 1.24, 1.20, 1.13, 1.07, and 1 V for respective current rates, illustrating superior reversibility and outstanding rate performances. Furthermore, the discharge voltage recovered ∼100% for CoS1−δ/SnS2−δ and ∼98% for Pt/C + RuO2. Moreover, the CoS1−δ/SnS2−δ-based ZABs manifest the specific capacity and energy density of 814 mAh gZn−1 and 1066 Wh kgZn−1, outperforming those of ZABs with Pt/C + RuO2 (776 mAh gZn−1 and 869 Wh kgZn−1, Fig. 6d), which recognizes the enhanced Zn utilization, fast mass transfer, and construction of excess nano-interfaces. Long-term cycling measurements were performed to realize the applicability of CoS1−δ/SnS2−δ for rechargeable alkaline ZABs under harsh operating conditions (Fig. 6e). For 20 mA cm−2, the ZAB with CoS1−δ/SnS2−δ displays outstanding cycling durability over 1008 cycles (168 h, 10 min/cycle) with minimal change in the charge–discharge voltaic efficiency from 62 to 59%, suggesting superior cycling stability. In contrast, Pt/C + RuO2 shows just 180 cycles (30 h, Fig. S18, ESI†) with rapid degradation in voltaic efficiency from 55% to 37%. The aqueous ZAB performance with CoS1−δ/SnS2−δ cathodes reveals the superior energy parameters (i.e., power density, capacity, and energy density) under harsh operations compared to those of previous reports so far (Fig. 6f and Tables S7, S8, ESI†). The improved ZAB performances are ascribed to the full utilization of the coated CoS1−δ/SnS2−δ catalysts for the feedstocks. The heterointerface realizes the high utilization efficiency and superior electron transfer rates for the catalysts. Furthermore, the structural and chemical properties of the CoS1−δ/SnS2−δ air cathodes are also well maintained even after long-term cycling tests (Fig. S19 and S20, ESI†).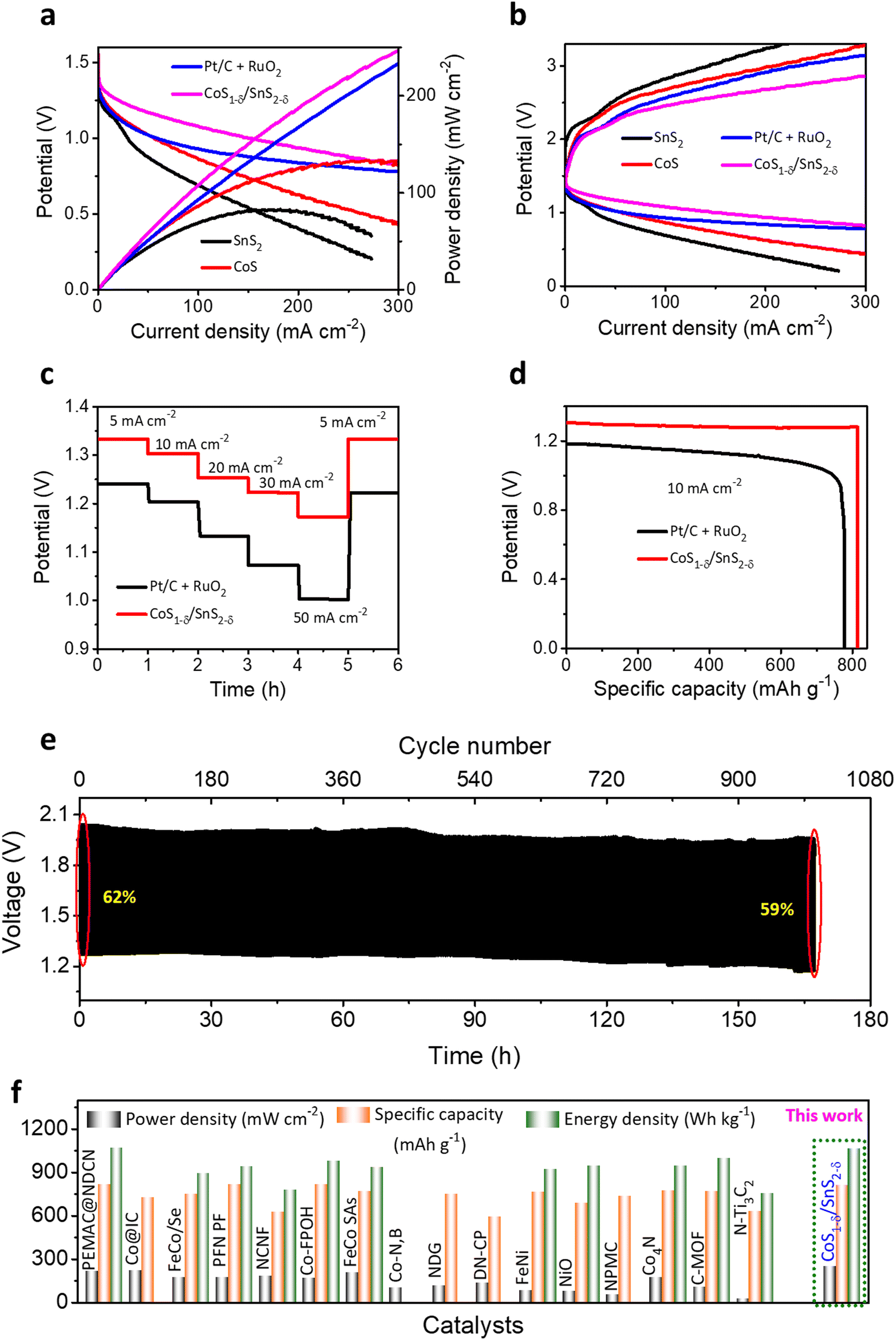 | ||
| Fig. 6 Alkaline Zn–air battery performance with CoS1−δ/SnS2−δ hetero-interface-based cathodes. (a) Galvanostatic polarizations and power densities for ZABs with the CoS1−δ/SnS2−δ hetero-interface, SnS2, CoS, and Pt/C + RuO2 cathode materials. (b) Discharge and charge profiles for the CoS1−δ/SnS2−δ hetero-interface, SnS, CoS, and Pt/C + RuO2 cathode materials. (c) Rate capacity performances for the CoS1−δ/SnS2−δ hetero-interface related to those of Pt/C + RuO2 cathodes for a current density of 5 to 50 mA cm−2. (d) Specific capacity and energy density for CoS1−δ/SnS2−δ and Pt/C +RuO2 cathodes with KOH electrolyte. (e) Cycle life performances for CoS1−δ/SnS2−δ//KOH//Zn cells for 20 mA cm−2 current rate (time: 10 min per cycle). (f) Ragone plots for power/energy densities and capacities of the CoS1−δ/SnS2−δ//KOH//Zn cells related to those of previous champion reports. (Conditions: cathode – CoS1−δ/SnS2−δ hetero-interface-based air cathode, electrolyte – 6 M KOH + 0.2 M Zn acetate, and anode – a Zn plate.) | ||
3.5 Flexible solid-state Zn–air battery performances
We constructed flexible ZABs to validate the commercial applications for electronic and portable devices using CoS1−δ/SnS2−δ heterointerface-based air cathodes, chitosan biocellulosics (CBCs)1 as solid-state electrolytes, and Zn foil as a negative electrode covered with aluminum-based insulation (Fig. S21, ESI†). Flexible ZABs with a CoS1−δ/SnS2−δ air cathode reveal a stable OCP of 1.51, 2.98, and 4.43 V for serial connections of one, two, and three cells (Fig. S22, ESI†). Meanwhile, the CoS1−δ/SnS2−δ-derived ZAB delivers the highest power density of 229 mW cm−2 with high discharge voltage polarizations and fast kinetic responses related to those of Pt/C + RuO2-derived ZABs (Fig. 7a). The charge–discharge polarizations in Fig. 7b display that the CoS1−δ/SnS2−δ cathodes offer smaller voltage gaps compared to those of Pt/C + RuO2 even for higher current rates. Galvanostatic discharge polarizations for flexible ZABs with CoS1−δ/SnS2−δ cathodes manifest the smallest voltage drops related to those of Pt/C + RuO2 from 10 to 100 mA cm−2 current densities (Fig. 7c). In the meantime, the discharge voltage recovers reversibly as the current density returns to the initial value of 10 mA cm−2, demonstrating that the CoS1−δ/SnS2−δ cathode has the smallest degree of polarization and superior rate capability. Furthermore, flexible ZABs with CoS1−δ/SnS2−δ display a specific capacity of 810 mAh gZn−1 and energy density of 1077 Wh kgZn−1 for the current density of 20 mA cm−2 (Fig. 7d), higher than those of Pt/C + RuO2 (759 mAh gZn−1 and 850 Wh kgZn−1). Flexible CoS1−δ/SnS2−δ-based ZABs exhibit 256 Wh kgcell−1 including all active and inactive components of fabricated cells comparable to or exceeding the commercial LIBs. In the galvanostatic charge–discharge cycles in Fig. 7e, flexible ZABs (CoS1−δ/SnS2−δ) deliver a stable cycling lifespan after 690 cycles (∼115 h) at a harsh current density of 50 mA cm−2 with retention of voltage efficiency of 61%. The Pt/C + RuO2 has a limited lifespan of ∼140 cycles (23.3 h) and voltage efficiency of 18% (Fig. S23, ESI†). Notably, the CoS1−δ/SnS2−δ cathode shows high feasibility for harsh operations such as a large current density and area capacity. Comprehensive performances for flexible solid-state ZABs with CoS1−δ/SnS2−δ surpass those of reference catalysts, previous reports, and other storage devices, including LIBs, LiS, ZIBs, etc. (Fig. 7f and Table S9, ESI†). Furthermore, the solid-ZABs manifest stable discharging–charging cycle performances for different folding environments from 0 to 180° and vice versa (Fig. 7g), vindicating the excellent mechanical flexibility as well. These results clearly indicate the superiority of heterointerface structures of cathodes to guarantee total coverage of catalytic reactive sites with electrolytes and enhanced diffusion of ions or reactive molecules through the interfaces.3,11 Furthermore, post-treatment physical, chemical, structural, and elemental characterizations demonstrate the preservation of the air cathodes after long-term continuous cycling tests (Fig. S24–S26, ESI†).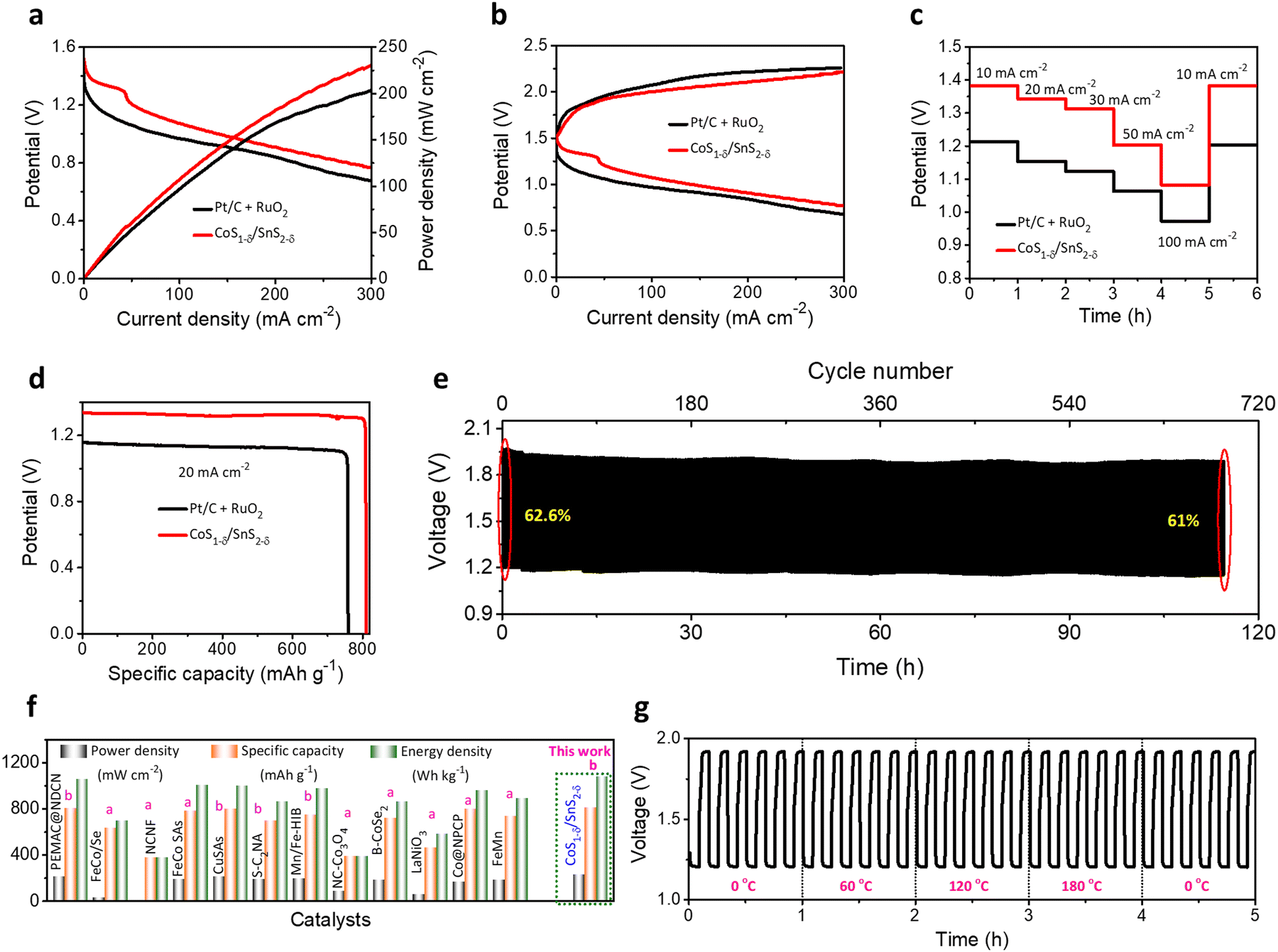 | ||
| Fig. 7 Electrochemical performances for flexible ZABs. (a) Power densities, (b) galvanostatic discharge–charge polarizations for CoS1−δ/SnS2−δ and Pt/C +RuO2 cathodes with CBCs and Zn foil as solid electrolytes and anodes, respectively. (c) Discharge rate capacity performances for CoS1−δ/SnS2−δ and Pt/C +RuO2 cathodes from 10 to 100 mA cm−2. (d) Discharge capacity profiles. (e) Cycle life operations for CoS1−δ/SnS2−δ//CBCs//Zn cells for 50 mA cm−2 current density (time: 10 min per cycle). (f) Ragone plots projecting the state-of-the-art cell capacities and power/energy densities for the assembled CoS1−δ/SnS2−δ based cathodes related to those of reported superior flexible ZABs. Note that “a” and “b” denote the current density ranges applied for measurements for 1–5 mA cm−2 and 25–50 mA cm−2, respectively. (g) Cycle operations for different mechanical bent tests (0, 60, 120, and 180°) for 50 mA cm−2. (Conditions: cathode – CoS1−δ/SnS2−δ, electrolyte – CBCs,1 and anode – a Zn plate). | ||
3.6 Wide temperature performances of Zn–air batteries
Obtaining ZAB performances for broader working temperature ranges is significant for practical applications under harsh operating conditions.1,58 We determined the flexible ZAB performances for +80 to −40 °C. The flexible solid-state ZABs with a CoS1−δ/SnS2−δ cathodes retain the power density of 194 mW cm−2 (∼84.7%) and lower charge–discharge polarizations compared to room temperature (RT, 229 mW cm−2) for the extreme subzero temperatures of −40 °C (Fig. 8a), illustrating the capability for better charge transfer and reversibility. The reliable increment of the discharge voltages of 1.26, 1.29, 1.33, 1.35, and 1.49 V with temperatures of −40, −20, 0, 25, and 80 °C at 10 mA cm−2, respectively, and decrease in charge voltages, demonstrate the smaller voltage gaps and reaction dynamics for elevated temperatures.1 For high temperatures, the electrolyte viscosity decreases with increasing ion conductivity, accelerating the mass transfer and diminishing the ohmic resistance. However, it restricts the cycle lifespan to a poor value due to the loss of hydroxide ions and self-corrosion of the electrodes.1,42 Furthermore, the flexible CoS1−δ/SnS2−δ ZABs exhibit a specific capacity of 685 mAh gZn−1 and energy density of 860 Wh kgZn−1 at 20 mA cm−2 at −40 °C, which is ∼85% retention of the RT capacity (Fig. 8c). The flexible ZAB in Fig. 8d also displays superior cycling durability for more than 264 cycles at 50 mA cm−2 with a voltage efficiency of 58% at −40 °C outperforming those of reported solid-state ZABs.1,10,42,59 Significantly, flexible ZABs with CoS1−δ/SnS2−δ cathodes vindicate the outstanding electrochemical performances in the energy evaluation parameters such as power density, capacity, energy density, and cycle lifespan for wide operating temperatures, relating to those of previous reports (Fig. 8e and Table S10, ESI†).1,10,42,59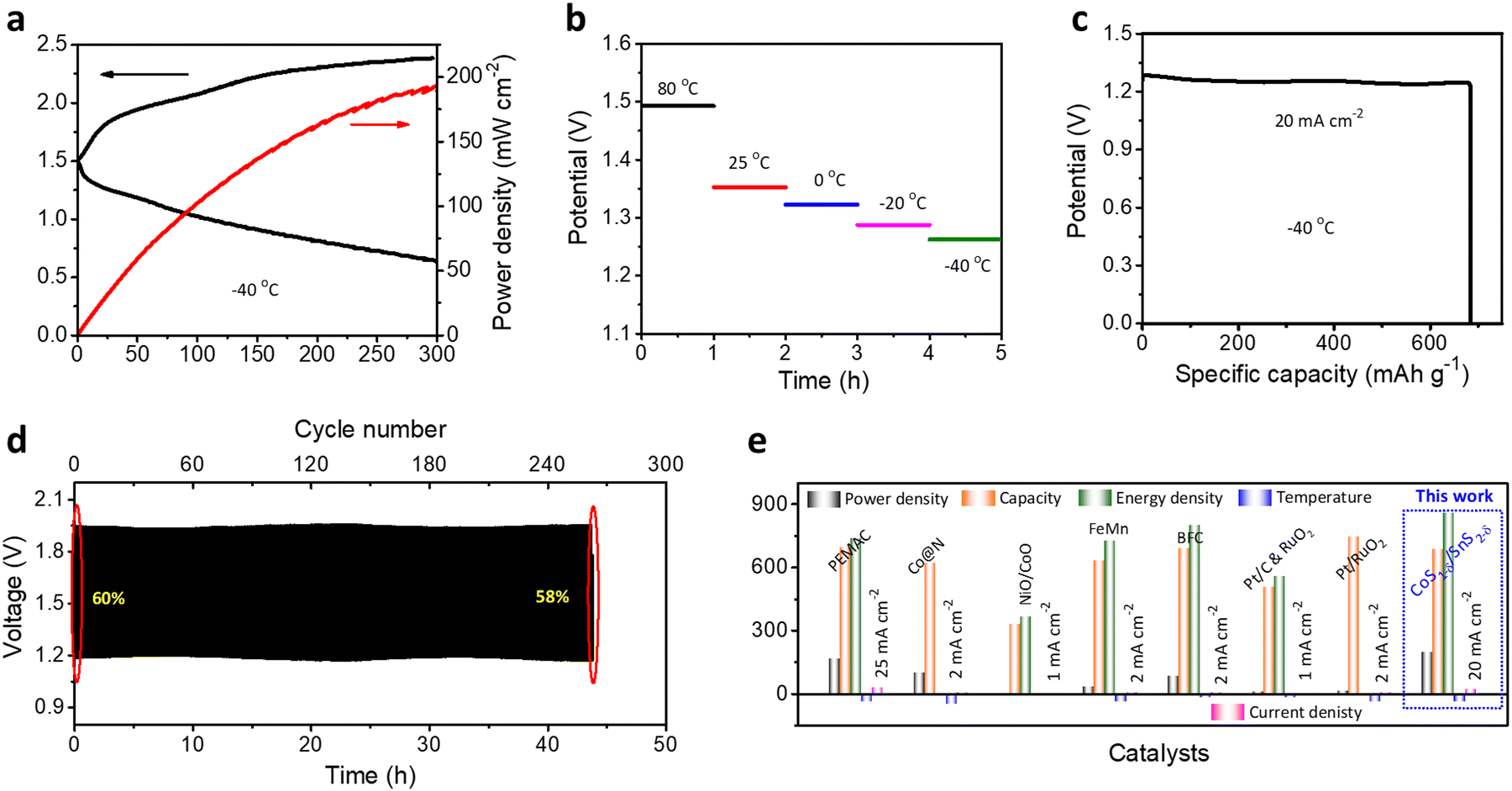 | ||
| Fig. 8 All temperature performance of flexible ZABs. (a) LSV and power density measurements of CoS1−δ/SnS2−δ at −40 °C. (b) Discharge rate voltages for wide operating temperatures from 80 to −40 °C at 10 mA cm−2. (c) Capacity and energy density for CoS1−δ/SnS2−δ at −40 °C for 20 mA cm−2. (d) Cycle life performance for CoS1−δ/SnS2−δ-based Zn–air cells for −40 °C at 50 mA cm−2. (Time: 10 min per cycle.) (e) Ragone plots projecting the state-of-the-art cell capacities and power/energy densities (i.e., capacity (mAh g−1), power (mW cm−2), energy (Wh kg−1), temperature (°C), and current densities (mA cm−2)) related to those of previous reports for sub-zero temperatures. | ||
4 Conclusions
In summary, high-performance rechargeable ZABs for practical harsh operating conditions such as current density and wide temperature range were proposed by employing CoS1−δ/SnS2−δ heterointerface air cathodes. Experimental and theoretical results reveal that the electronic interactions of the asymmetric CoS1−δ/SnS2−δ interface facilitate the numerous reactive sites with the formation of hydrophobic–aerophilic surfaces to improve the utilization efficiency of the catalysts. The CoS1−δ/SnS2−δ manifests an outstanding trifunctional ORR, OER, and HER activity and durability with optimal reaction pathways. Alkaline ZABs demonstrate a record-breaking power density of 249 mW cm−2 and superior cycle life for 1000 cycles at 20 mA cm−2. Moreover, flexible solid-state ZABs exhibit the high energy density of 1077 Wh kgZn−1, the fast-charge capability of 50 to 100 mA cm−2 for lower charge–discharge voltage polarizations, the outstanding cycle lifespans of 690 and 264 cycles at RT (25 °C) and subzero (−40 °C), respectively at 50 mA cm−2 and wide operating temperatures from +80 to −40 °C. This work offers fundamental insights for designing advanced trifunctional catalysts for practical Zn–air batteries.Author contributions
N. K. W., D. H. K., S. S. S., and J. H. L. conceptualized the idea. N. K.W., D. H. K., and S. S. S. designed the experiments and analyzed the results. C. H. L., S. I. K., and S. U. L. performed the DFT calculations. Structural and microscopic analyses were performed by S. H. K. and H. D. U. S. S. S. and J. H. L. drafted the manuscript. S. S. S. and J. H. L. supervised the project. All the authors contributed to the writing – review and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Creative Materials Discovery Program (grant no. 2018M3D1A1057844) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT. J. H. L. acknowledges the Basic Science Research Program of the NRF (NRF-2023R1A2C3003788). S. U. L. thanks the Basic Science Research Program of the NRF (NRF-2021R1A2B5B01002879).References
- S. Shinde, J. Jung, N. Wagh, C. Lee, D. Kim, S. Kim, S. Lee and J. Lee, Nat. Energy, 2021, 6, 592–604 CrossRef CAS.
- S. Shinde, C. Lee, J. Jung, N. Wagh, S. Kim, D. Kim, C. Lin, S. Lee and J. Lee, Energy Environ. Sci., 2019, 12, 727–738 RSC.
- L. Yan, B. Xie, C. Yang, Y. Wang, J. Ning, Y. Zhong and Y. Hu, Adv. Energy Mater., 2023, 2204245 CrossRef CAS.
- M. Zhao, H. Liu, H. Zhang, W. Chen, H. Sun, Z. Wang, B. Zhang, L. Song, Y. Yang, C. Ma, Y. Han and W. Huang, Energy Environ. Sci., 2021, 14, 6455–6463 RSC.
- X. Fan, X. Ji, L. Chen, J. Chen, T. Deng, F. Han, J. Yue, N. Piao, R. Wang, X. Zhou, X. Xiao, L. Chen and C. Wang, Nat. Energy, 2019, 4, 882–890 CrossRef CAS.
- G. Assat and J. Tarascon, Nat. Energy, 2018, 3, 373–386 CrossRef CAS.
- H. Zhang, M. Zhao, H. Liu, S. Shi, Z. Wang, B. Zhang, L. Song, J. Shang, Y. Yang, C. O. Ma, L. Zheng, Y. Han and W. Huang, Nano Lett., 2021, 21, 2255–2264 CrossRef CAS PubMed.
- J. Li, P. Liu, J. Yan, H. Huang and W. Song, Adv. Sci., 2023, 2206165 CrossRef CAS PubMed.
- C. Zhao, J. Liu, N. Yao, J. Wang, D. Ren, X. Chen, B. Li and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 15281–15285 CrossRef CAS PubMed.
- N. Wagh, S. Shinde, C. Lee, S. Kim, D. Kim, H. Um, S. Lee and J. Lee, Nano-Micro Lett., 2022, 14, 190 CrossRef CAS PubMed.
- J. Liu, C. Zhao, D. Ren, J. Wang, R. Zhang, S. Wang, C. Zhao, B. Li and Q. Zhang, Adv. Mater., 2022, 34, 2109407 CrossRef CAS PubMed.
- A. Yang, K. Su, W. Lei, Y. Tang and X. Qiu, Adv. Energy Mater., 2023, 13, 2203150 CrossRef CAS.
- D. Li, C. Li, L. Zhang, H. Li, L. Zhu, D. Yang, Q. Fang, S. Qiu and X. Yao, J. Am. Chem. Soc., 2020, 142, 8104–8108 CrossRef CAS PubMed.
- K. Yoon, C. Hwang, S. Kim, J. Jung, J. Chae, J. Kim, K. Lee, A. Lim, S. Cho, J. Singh, J. Kim, K. Shin, B. Moon, H. Park, H. Kim, K. Chae, H. Ham, I. Kim and J. Kim, ACS Nano, 2021, 15, 11218–11230 CrossRef CAS PubMed.
- Q. Lu, H. Wu, X. Zheng, Y. Cao, J. Li, Y. Wang, H. Wang, C. Zhi, Y. Deng, X. Han and W. Hu, Adv. Energy Mater., 2022, 12, 2202215 CrossRef CAS.
- L. An, Z. Zhang, J. Feng, F. Lv, Y. Li, R. Wang, M. Lu, R. Gupta, P. Xi and S. Zhang, J. Am. Chem. Soc., 2018, 140, 17624–17631 CrossRef CAS PubMed.
- X. Zheng, X. Han, Y. Cao, Y. Zhang, D. Nordlund, J. Wang, S. Chou, H. Liu, L. Li, C. Zhong, Y. Deng and W. Hu, Adv. Mater., 2020, 32, 2000607 CrossRef CAS PubMed.
- T. Shen, X. Huang, S. Xi, W. Li, S. Sun and Y. Hou, J. Energy Chem., 2022, 68, 184–194 CrossRef CAS.
- S. Shinde, A. Sami and J. Lee, J. Mater. Chem. A, 2015, 3, 12810–12819 RSC.
- X. Huang, T. Shen, T. Zhang, H. Qiu, X. Gu, Z. Ali and Y. Hou, Adv. Energy Mater., 2020, 10, 1900375 CrossRef CAS.
- H. Li, Q. Li, P. Wen, T. Williams, S. Adhikari, C. Dun, C. Lu, D. Itanze, L. Jiang, D. Carroll, G. Donati, P. Lundin, Y. Qiu and S. Geyer, Adv. Mater., 2018, 30, 1705796 CrossRef PubMed.
- Z. Yang, C. Zhao, Y. Qu, H. Zhou, F. Zhou, J. Wang, Y. Wu and Y. Li, Adv. Mater., 2019, 31, 1808043 CrossRef PubMed.
- T. Sun, S. Mitchell, J. Li, P. Lyu, X. Wu, J. Pérez-Ramírez and J. Lu, Adv. Mater., 2020, 33, 2003075 CrossRef PubMed.
- Y. Tian, L. Xu, M. Li, D. Yuan, X. Liu, J. Qian, Y. Dou, J. Qiu and S. Zhang, Nano-Micro Lett., 2020, 13, 3 CrossRef PubMed.
- M. Cabán-Acevedo, M. Stone, J. Schmidt, J. Thomas, Q. Ding, H. Chang, M. Tsai, J. He and S. Jin, Nat. Mater., 2015, 14, 1245 CrossRef PubMed.
- D. Nguyen, D. Tran, T. Luyen Doan, D. Kim, N. Kim and J. Lee, Adv. Energy Mater., 2020, 10, 1903289 CrossRef CAS.
- K. Min, S. Kim, E. Lee, G. Yoo, H. Ham, S. Shim, D. Lim and S. Baeck, J. Mater. Chem. A, 2021, 9, 17344–17352 RSC.
- W. Li, L. Wu, X. Wu, C. Shi, Y. Li, L. Zhang, H. Mi, Q. Zhang, C. He and X. Ren, Appl. Catal., B, 2022, 303, 120849 CrossRef CAS.
- L. An, B. Huang, Y. Zhang, R. Wang, N. Zhang, T. Dai, P. Xi and C. Yan, Angew. Chem., Int. Ed., 2019, 58, 9459–9463 CrossRef CAS PubMed.
- P. Rao, Y. Deng, W. Fan, J. Luo, P. Deng, J. Li, Y. Shen and X. Tian, Nat. Commun., 2022, 13, 5071 CrossRef CAS PubMed.
- R. Jeyagopal, Y. Chen, M. Ramadoss, K. Marimuthu, B. Wang, W. Li and X. Zhang, Nanoscale, 2020, 12, 3879–3887 RSC.
- Q. Geng, Y. Li and H. Xie, Vacuum, 2022, 203, 111308 CrossRef CAS.
- J. He, G. Hartmann, M. Lee, G. Hwang, Y. Chen and A. Manthiram, Energy Environ. Sci., 2019, 12, 344 RSC.
- D. Ma, B. Hu, W. Wu, X. Liu, J. Zai, C. Shu, T. Tsega, L. Chen, X. Qian and T. Liu, Nat. Commun., 2019, 10, 3367 CrossRef PubMed.
- C. Chen, Z. Shih, Z. Yang and H. Chang, J. Power Sources, 2012, 215, 43–47 CrossRef CAS.
- A. Smith, P. Meek and W. Liang, J. Phys. Chem. C, 1977, 1321, 1321 Search PubMed.
- C. Gurnani, S. Hawken, A. Hector, R. Huang, M. Jura, W. Levason, J. Perkins, G. Reid and G. Stenning, Dalton Trans., 2018, 47, 2628–2637 RSC.
- Y. Lian, K. Shi, H. Yang, H. Sun, P. Qi, J. Ye, W. Wu, Z. Deng and Y. Peng, Small, 2020, 16, 1907368 CrossRef CAS PubMed.
- D. Lyu, S. Yao, A. Ali, Z. Q. Tian, P. Tsiakaras and P. K. Shen, Adv. Energy Mater., 2021, 11, 2101249 CrossRef CAS.
- J. Li, Y. Liu, X. Tang, L. Xu, L. Min, Y. Xue, X. Hu and Z. Yang, Microchim. Acta, 2020, 187, 80 CrossRef CAS PubMed.
- M. Jiang, Y. Huang, W. Sun and X. Zhang, J. Mater. Sci., 2019, 54, 13715–13723 CrossRef CAS.
- A. Wang, X. Zhang, S. Gao, C. Zhao, S. Kuang, S. Lu, J. Niu, G. Wang, W. Li, D. Chen, H. Zhang, X. Zhou, S. Zhang, B. Zhang and W. Wang, Adv. Mater., 2022, 34, 2204247 CrossRef CAS PubMed.
- C. Zhao, L. Tian, Z. Zou, Z. Chen, H. Tang, Q. Liu, Z. Lin and X. Yang, Appl. Catal., B, 2020, 268, 118445 CrossRef CAS.
- C. Tsai, H. Li, S. Park, J. Park, H. han, J. Norskov, X. Zheng and F. Abid-pedersen, Nat. Commun., 2017, 8, 15113 CrossRef PubMed.
- S. Wang, Y. Zhang, H. Si, Q. Zhang, J. Wu, L. Gao, X. Wei, Y. Sun, Q. Liao, Z. Zhang, K. Ammarah, L. Gu, Z. Kang and Y. Zhang, J. Am. Chem. Soc., 2020, 142, 4298 CrossRef PubMed.
- S. Shinde, C. Lee, J. Yu, D. Kim, S. Lee and J. Lee, ACS Nano, 2018, 12, 596–608 CrossRef CAS PubMed.
- S. Shinde, C. Lee, A. Sami, D. Kim, S. Lee and J. Lee, ACS Nano, 2017, 11, 347–357 CrossRef CAS PubMed.
- N. Wagh, S. Shinde, C. Lee, J. Jung, D. Kim, S. Kim, C. Lin, S. Lee and J. Lee, Appl. Catal., B, 2020, 268, 118746 CrossRef CAS.
- N. Wagh, D. Kim, S. Kim, S. Shinde and J. Lee, ACS Nano, 2021, 15, 14683–14696 CrossRef CAS PubMed.
- S. Shinde, J. Yu, J. Song, Y. Nam and D. Kim, Nanoscale Horiz., 2017, 2, 333–341 RSC.
- A. Ahmed, C. Lee, A. Ansari, S. Pawar, J. Han, S. Park, G. Shin, S. Yeon, S. Cho, J. Seol, S. Lee, H. Kim and H. Im, Appl. Surf. Sci., 2022, 592, 153196 CrossRef.
- J. Lee, D. Yim, J. Park, C. Lee, J. Ju, S. Lee and J. Kim, J. Mater. Chem. A, 2020, 8, 13490–13500 RSC.
- L. Lin, N. Miao, Y. Wen, S. Zhang, P. Ghosez, Z. Sun and D. Allwood, ACS Nano, 2016, 10, 8929–8937 CrossRef CAS PubMed.
- D. Escalera-Lopez, Y. Niu, J. Yin, K. Cooke, N. Rees and R. Palmer, ACS Catal., 2016, 6, 6008–6017 CrossRef CAS PubMed.
- Y. Ouyang, C. Ling, Q. Chen, Z. Wang, L. Shi and J. Wang, Chem. Mater., 2016, 28, 4390–4396 CrossRef CAS.
- C. Lee, B. Jun and S. Lee, ACS Sustainable Chem. Eng., 2018, 6, 4973–4980 CrossRef CAS.
- C. Lee, B. Jun and S. Lee, RSC Adv., 2017, 7, 27033–27039 RSC.
- A. Thenuwara, P. Shetty, N. Kondekar, S. Sandoval, K. Cavallaro, R. May, C. Yang, L. Marbella, Y. Qi and M. McDowell, ACS Energy Lett., 2020, 5, 2411 CrossRef CAS.
- X. Shu, Q. Chen, M. Yang, M. Liu, J. Ma and J. Zhang, Adv. Energy Mater., 2023, 13, 2202871 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00108c |
| ‡ NKW and DHK have made equal contributions. |
| This journal is © The Royal Society of Chemistry 2023 |