
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into the kinetics and self-assembly order of small-molecule organic semiconductor/quantum dot blends during blade coating†
Daniel T. W.
Toolan
 *a,
Michael P.
Weir
bc,
Shuangqing
Wang
c,
Simon A.
Dowland
d,
Zhilong
Zhang
e,
James
Xiao
e,
Jonathan
Rawle
f,
Neil
Greenham
d,
Richard H.
Friend
e,
Akshay
Rao
e,
Richard A. L.
Jones
g and
Anthony J.
Ryan
a
*a,
Michael P.
Weir
bc,
Shuangqing
Wang
c,
Simon A.
Dowland
d,
Zhilong
Zhang
e,
James
Xiao
e,
Jonathan
Rawle
f,
Neil
Greenham
d,
Richard H.
Friend
e,
Akshay
Rao
e,
Richard A. L.
Jones
g and
Anthony J.
Ryan
a
aDepartment of Chemistry, The University of Sheffield, Dainton Building, Brook Hill, Sheffield S3 7HF, UK. E-mail: d.toolan@sheffield.ac.uk
bSchool of Physics and Astronomy, University of Nottingham, University Park, Nottingham, NG7 2RD, UK
cDepartment of Physics and Astronomy, The University of Sheffield, Hicks Building, Hounsfield Road, Sheffield, S3 7RH, UK
dCambridge Photon Technology, J. J. Thomson Avenue, Cambridge, CB3 0HE, UK
eCavendish Laboratory, Cambridge University, J. J. Thomson Avenue, Cambridge, CB3 0HE, UK
fDiamond Light Source Ltd, Harwell Science & Innovation Campus, Didcot, Oxfordshire OX11 0DE, UK
gJohn Owens Building, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 25th May 2023
Abstract
Organic–inorganic nanocomposite films formed from blends of small-molecule organic semiconductors and colloidal quantum dots are attractive candidates for high efficiency, low-cost solar energy harvesting devices. Understanding and controlling the self-assembly of the resulting organic–inorganic nanocomposite films is crucial in optimising device performance, not only at a lab-scale but for large-scale, high-throughput printing and coating methods. Here, in situ grazing incidence X-ray scattering (GIXS) gives direct insights into how small-molecule organic semiconductors and colloidal quantum dots self-assemble during blade coating. Results show that for two blends separated only by a small difference in the structure of the small molecule forming the organic phase, crystallisation may proceed down two distinct routes. It either occurs spontaneously or is mediated by the formation of quantum dot aggregates. Irrespective of the initial crystallisation route, the small-molecule crystallisation acts to exclude the quantum dot inclusions from the growing crystalline matrix phase. These results provide important fundamental understanding of structure formation in nanocomposite films of organic small molecules and colloidal quantum dots prepared via solution processing routes. It highlights the fundamental difference to structural evolution which can be made by seemingly small changes in system composition. It provides routes for the structural design and optimisation of solution-processed nanocomposites that are compatible with the large-scale deposition manufacturing techniques that are crucial in driving their wider adoption in energy harvesting applications.
New conceptsA new generation of solution-processable films and coatings combining organic semiconductors with inorganic nanoparticles hold great promise as optoelectronic materials where the overall function arises from a rationally-designed nanoscale structure. Potential applications include high efficiency, low-cost solar energy harvesting devices, photon detectors, and novel LEDs for displays, communications, and chemical diagnostics. As in many optoelectronic materials, there is a key relationship between a material's nanomorphology and the resultant device performance. The present study provides new conceptual insights into the mechanisms of self-assembly in hybrid organic–inorganic nanoparticle blends, with respect to interactions between the crystallisation of the organic matrix, formed of small-molecule organic semiconductors (OSC), and the ordering of the nanoparticle inclusions, in the form of colloidal nanocrystal quantum dots (QD). Two cases are observed: (i) QD aggregation acts to nucleate the crystallisation of the OSC and the subsequent crystallisation of the OSC acts to expel the QD nanoparticle inclusions; or (ii) crystallisation of the OSC occurs spontaneously and subsequently expels the QD nanoparticle inclusions. |
Main
Colloidal nanocrystal quantum dot (QD) nanocomposite films have optoelectronic applications including solar cells,1–4 light-emitting diodes,5,6 photon detectors and recently, photon multipliers.7,8 In all such nanocomposites, control of the nanoscale morphologies that form via self-assembly is critical for maximizing device performance.This work focuses on understanding the self-assembly that occurs during the processing of QD nanocomposite films that comprise small molecule organic semiconductors (OSC) blended with QDs. These nanocomposite films are of direct relevance to the development of photon multipliers, a technology that holds significant promise for enhancing the performance of silicon photovoltaics (Si-PV). The current dominant commercial PV technology is Si-PV which holds the potential for providing low-cost renewable energy. However, single-junction Si-PV efficiencies are approaching a fundamental limit [known as the Shockley–Queisser (SQ) limit],9 presenting a significant obstacle for continued gains in performance. The SQ limit arises from energy losses (via heat generation) that are incurred when high-energy photons within the solar spectrum are absorbed above the bandgap of the PV material. One route to circumvent this limit is via the process of singlet exciton fission (SF), where a singlet exciton from a single high energy photon in an organic semiconductor decays into two independent triplet excitons, that are transferred to a QD and subsequently re-emitted at an energy much closer by design to the PV bandgap.4 Thus, energy that would without intervention be lost through thermalisation is harnessed, revising the absolute Shockley–Queisser efficiency limit upwards from 33% to 44% – a substantial gain.10–13 A route to achieve this has been demonstrated through the harvesting of dark triplet excitons generated by SF by a quantum dot emitter material, followed by the emission of light and its optical coupling into the PV module.11,13–15
This approach converts the exciton multiplication process into a photon multiplication process and has recently been demonstrated in both liquid-16–18 and solid-phase,7 for an archetypal OSC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :QD blend, comprising 5,12-bis((triisopropylsilyl)ethynyl)tetracene (TIPS-Tc), a widely studied SF small molecule, with lead sulfide (PbS) QDs functionalised with a carboxylic acid derivative of TIPS-Tc, bis((triisopropylsilyl)ethynyl)tetracene-2-carboxylic acid (TET-CA). In the solid state, the TET-CA ligand not only facilitates efficient triplet transfer between TIPS-Tc and PbS QDs, but also plays a critical role in facilitating QD dispersibility within the crystalline TIPS-Tc host material. While this initial proof-of-principle work is highly promising, understanding relationships between the highly complex morphologies that OSC:QD blends form and correlating these with their consequent photon multiplication efficiency will be critical for realising the potential of photon multiplier devices to increase Si-PV efficiencies. It has been demonstrated that aggregated QD domains within the OSC matrix lead to relatively poor photon multiplication performance and so the optimal photon multiplier film would possess a morphology where QDs are fully dispersed within a crystalline OSC matrix.19
:QD blend, comprising 5,12-bis((triisopropylsilyl)ethynyl)tetracene (TIPS-Tc), a widely studied SF small molecule, with lead sulfide (PbS) QDs functionalised with a carboxylic acid derivative of TIPS-Tc, bis((triisopropylsilyl)ethynyl)tetracene-2-carboxylic acid (TET-CA). In the solid state, the TET-CA ligand not only facilitates efficient triplet transfer between TIPS-Tc and PbS QDs, but also plays a critical role in facilitating QD dispersibility within the crystalline TIPS-Tc host material. While this initial proof-of-principle work is highly promising, understanding relationships between the highly complex morphologies that OSC:QD blends form and correlating these with their consequent photon multiplication efficiency will be critical for realising the potential of photon multiplier devices to increase Si-PV efficiencies. It has been demonstrated that aggregated QD domains within the OSC matrix lead to relatively poor photon multiplication performance and so the optimal photon multiplier film would possess a morphology where QDs are fully dispersed within a crystalline OSC matrix.19
A successful route for achieving improved QD dispersibility within a host matrix employs compatibilising QD ligands with a tendency to mix with the host matrix phase. This was demonstrated for a QD:polymer nanocomposite, where grafting block copolymers to QDs enabled effective dispersion in the bulk polymer matrix (albeit at relatively low QD loadings of 2.5 wt%).20 Recently, well-dispersed QDs have been achieved in a phenylethylammonium (PEA) perovoskite:QD system by performing a QD ligand exchange with a PEA hydrobromide salt.21 Thus the functionalised QDs have an increased solubility in the perovskite precursor solution and trigger fast nucleation of perovskite to achieve homogeneous incorporation of QDs into the perovskite matrix without detrimental QD aggregation. In an OSC:QD nanocomposite, we have recently demonstrated significantly improved QD dispersibility through modifying the PbS QD surface with a carboxylic acid functionalised analogue of the host OSC matrix, enabling good QD dispersibility.8 A further optimization route has been demonstrated that is based on blending two different OSCs to form a fully mixed OSC matrix phase, which was shown to improve QD dispersibilities.22
Current understanding of small-molecule OSC:QD nanocomposites largely stems from evaluation of final film morphologies. Accordingly, understanding of how self-assembly must have proceeded, and the nature of the interplay between small-molecule OSC crystallisation and QD aggregation/dispersibility, are highly qualitative. In a drying OSC:QD:solvent thin film, a number of different self-assembly processes occur as solvent evaporation proceeds: crystallisation of the OSC, aggregation or colloidal crystallisation of the QDs, and interplays between these two. The interaction between OSC crystallisation and QD aggregation processes is crucial. Aggregates solely comprising QDs have the potential to nucleate OSC crystallisation. OSC crystallisation can itself lead to QD aggregation, if the solubilities of the QD ligand and the OSC are not suitably well matched.
In order to establish universal design rules that will enable QDs to be well-dispersed within different small-molecule hosts, it is imperative to understand the relationships between OSC crystallisation and QD ordering that control the generation of the self-assembled nanomorphology generated during film formation. Such understanding will pave the way for the rational design of processing strategies that enable optimal nanocomposite morphologies to be obtained.
In situ grazing incidence X-ray scattering (GIXS) has been widely employed to provide fundamental insights into the self-assembly of binary QD:solvent mixtures.23–25 However, insights into the self-assembly of ternary OSC:QD:solvent mixtures are still in their infancy. A recent study of drop-cast OSC:QD blends demonstrated that the QD ligand envelope plays a critical role in controlling the relative dispersibility or aggregation of the QD species and that when QDs ligated with molecules with high chemical similarity to the host small-molecule species, crystallisation of the host small-molecule is significantly enhanced.8 However, due to the intensity of laboratory based X-ray sources, which significantly limits temporal resolution, this work was not able to uncover the self-assembly order of the small-molecule and QD component effects.
Here, in situ GIXS is performed during the blade coating of two model OSCs, TIPS-Tc and 9,10-Bis[(triisopropylsilyl)ethynyl]anthracene (TIPS-Ac), with PbS QDs functionalised with the carboxylic acid ligand bearing analogue of TIPS-Tc, TET-CA. The study aims to provide new insight into the relationships between OSC crystallisation and QD ordering that control the generation of the self-assembled nanomorphology generated during film formation. Whilst the TET-CA ligand is not a direct match to the TIPS-Ac, being four- and three-ringed polyacenes respectively, we expect that there is sufficient similarity between the TET-CA ligand and the TIPS-Ac matrix to promote greater miscibility between the QD ligand and the organic host, when compared to the native as-synthesised oleic acid ligated PbS quantum dots. However, despite this key strucutral similarity, a key difference between TIPS-Tc and TIPS-Ac should also be highlighted. It is well established that small differences between similar chemical species often lead to different crystal packing motifs,26–28 as is the case for the two polyacene host materials investigated here. TIPS-Ac has the TIPS groups centrally located at carbons 9 & 10 and is thus highly symmetrical. This symetry is broken in TIPS-Tc, with the TIPS groups located off-centre, at carbons 5 & 12, resulting in an additional acene ring on one side of the molecule. This impacts the crystal structures typcially formed by the these two molecules. TIPS-Ac orients in a herringbone pattern, with either face-to-edge or slipped face-to-face stacking, depending upon the polymorph,29 whilst, crystalline packing in TIPS-Tc is an intermediate between herringbone and brickwall packing, with the molecules offset with little overlap of the tetracene backbones.30 It has recently been demonstrated how small changes to the OSC host molecule from TIPS-Tc to TIPS-Ac can have a detrimental effect on QD dispersibility within the host organic semiconductor matrix.22
The different materials studied herein and the experimental setup are shown in Fig. 1. Deposition via blade coating was performed using a custom-built blade coater installed on the beamline (I07, Diamond Light Source) that enabled deposition and spreading of the OSC:QD solutions under a nitrogen atmosphere whilst GIXS was performed at a fixed point on the substrate (for further details see ESI†). PbS quantum dots were synthesised via previously reported methods,31 with the as-synthesised oleic acid ligands exchanged with TET-CA, to obtain TET-CA-ligated PbS quantum dots (PbS-TET-CA).16–18 To enable direct comparison between the self-assembly order of the TIPS-Tc:PbS-TET-CA and TIPS-Ac:PbS-TET-CA blends cast from toluene and to normalise against different film drying rates, data is displayed as a function of effective solvent fraction, normalised using the area under the scattering intensity curve in the solvent halo region (full details on how the time-resolved data was re-sampled as a function of solvent fraction are available in the ESI†). To aid visualisation of the evolution of the 2D scattering patterns, movies showing the in situ scattering data are also available in the ESI.†
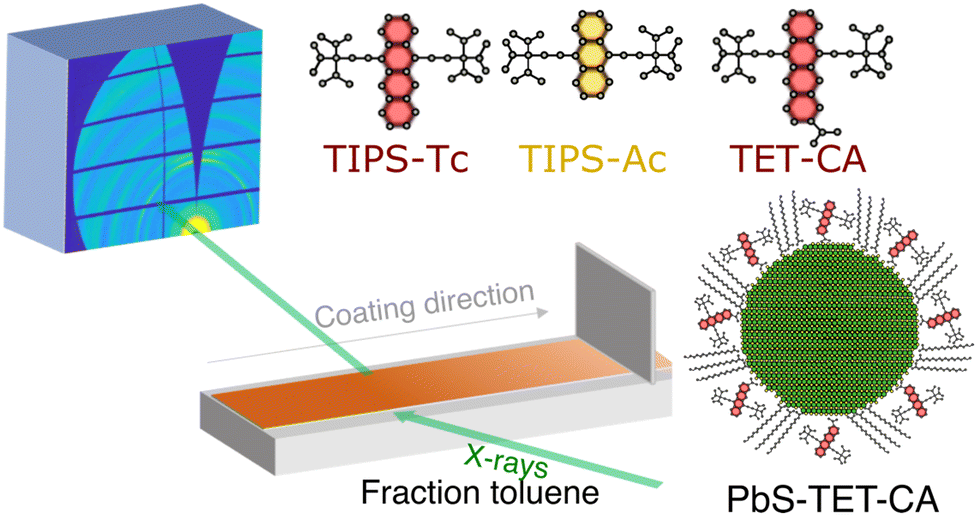 | ||
| Fig. 1 Schematic of in situ grazing incidence X-ray scattering blade coating setup and the structures of the OSCs and QDs explored in this study. | ||
For the purposes of the forthcoming discussion it is important to distinguish QD order from QD orientation. QDs form colloidal crystals when interaction energies between QDs and QD-matrix interaction energies are favourable for a collection of QD to reduce their free energy by forming a regular array. Order here refers to the degree of spatial ordering between the scattering centres within a QD crystallite, with highly ordered meaning corresponding precisely to a defined packing pattern, e.g. FCC. Orientation refers to the direction in which the crystallographic axes of a given crystallite are pointing. Two neighbouring crystallites with identical order could have completely different orientations. In that case two distinct sets of scattering features from the two crystallites would be discernible as sets of spots; in principle, two single-crystal diffraction patterns. When the number of randomly oriented crystallites becomes very large, scattering occurs in all orientations and this results in scattering rings identical to a powder diffraction pattern.
In situ scattering data for TIPS-Tc:PbS-TET-CA and TIPS-Ac:PbS-TET-CA is presented in Fig. 2 and 3, respectively [with the radially integrated data (c) also presented as waterfall plots for convenience in ESI,† Fig. S4 and S5]. When the TIPS-Tc/TIPS-Ac:PbS-TET-CA solutions are initially spread to form wet films on the substrate surface the low q scattering data ∼0.05–0.3 Å−1 is consistent with completely dispersed quantum dots, described as quasi-spherical nanoparticles with an average PbS core radius of 22.8 Å with a 10% size polydispersity (fits and associated parameters available in ESI,† Fig. S6). At higher q, 0.3–1.8 Å−1 a substantial scattering peak is observered and is attributed to the solvent, toluene. As the film dries and the volume fraction of solvent decreases, the amplitude of the solvent scattering feature decreases accordingly.
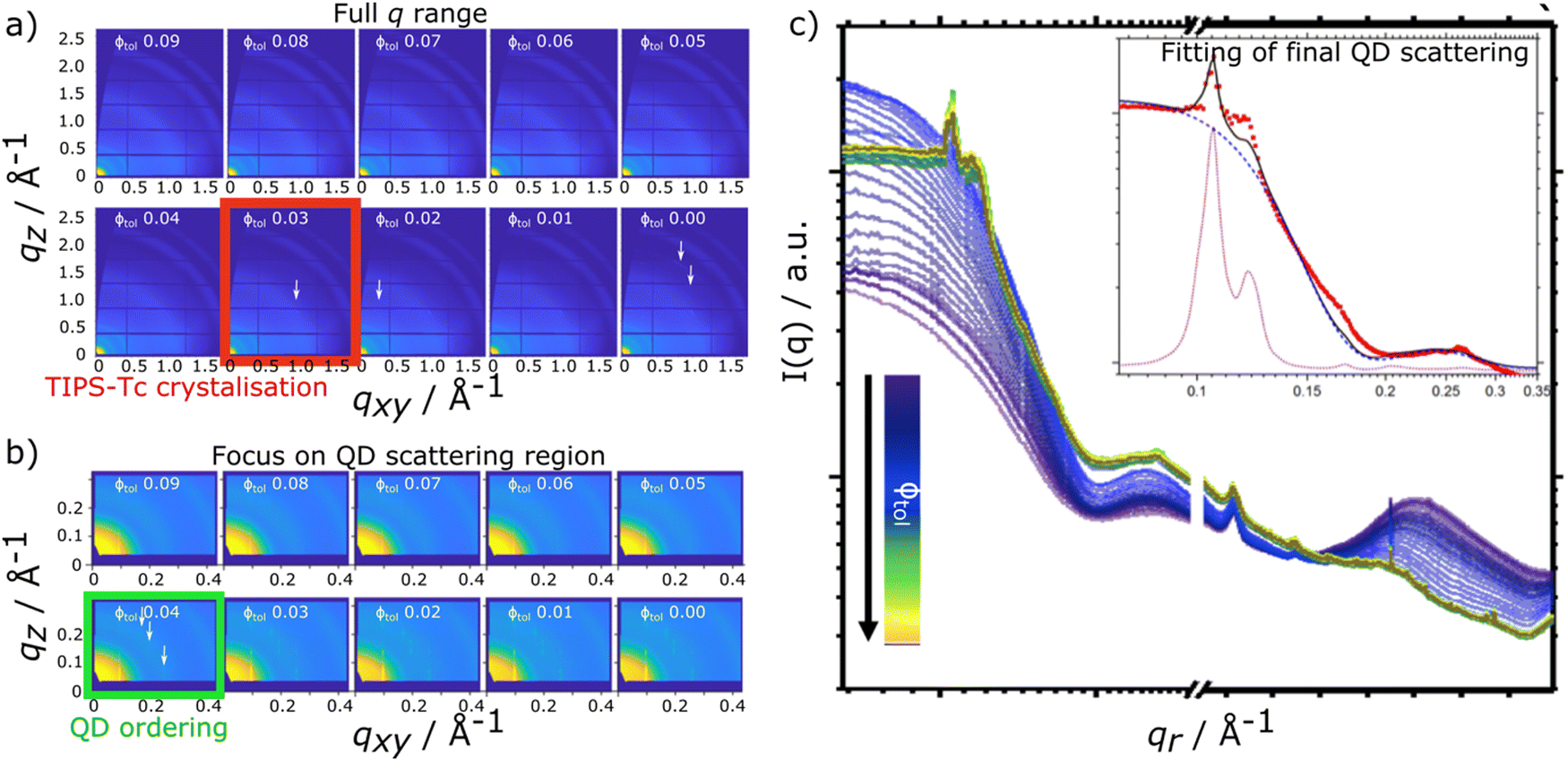 | ||
| Fig. 2 In situ GIXS data for TIPS-Tc:PbS-TET-CA showing the latter stages of film-formation (0.09 > 0.00 volume fraction toluene), with (a) showing the full q range (QD + OSC lengthscales) and (b) focusing on the small angle q range (QD lengthscales). The TIPS-Tc and QD ordering is highlighted with red & green boxes and white arrows, respectively. Radially integrated in situ scattering data for (c) as a function of solvent fraction, where the insert shows the fit (black line) of the final frame of the kinetic data, for a sphere-hard sphere + FCC paracrystal crystal model [showing the constituent sphere-hardsphere (blue dashed line) and FCC paracrystal (purple dotted line) components of the model fits]. The data show that at volume fraction of solvent Φtol = 0.09 the QDs are predominantly well-dispersed. As the solvent fraction decreases, QD ordering scattering features become visible at Φtol = 0.04. Weak scattering peaks from the crystallisation of the TIPS-Tc become visible at Φtol = 0.03, with further TIPS-Tc crystal peaks emerging as the solvent fraction further decreases. | ||
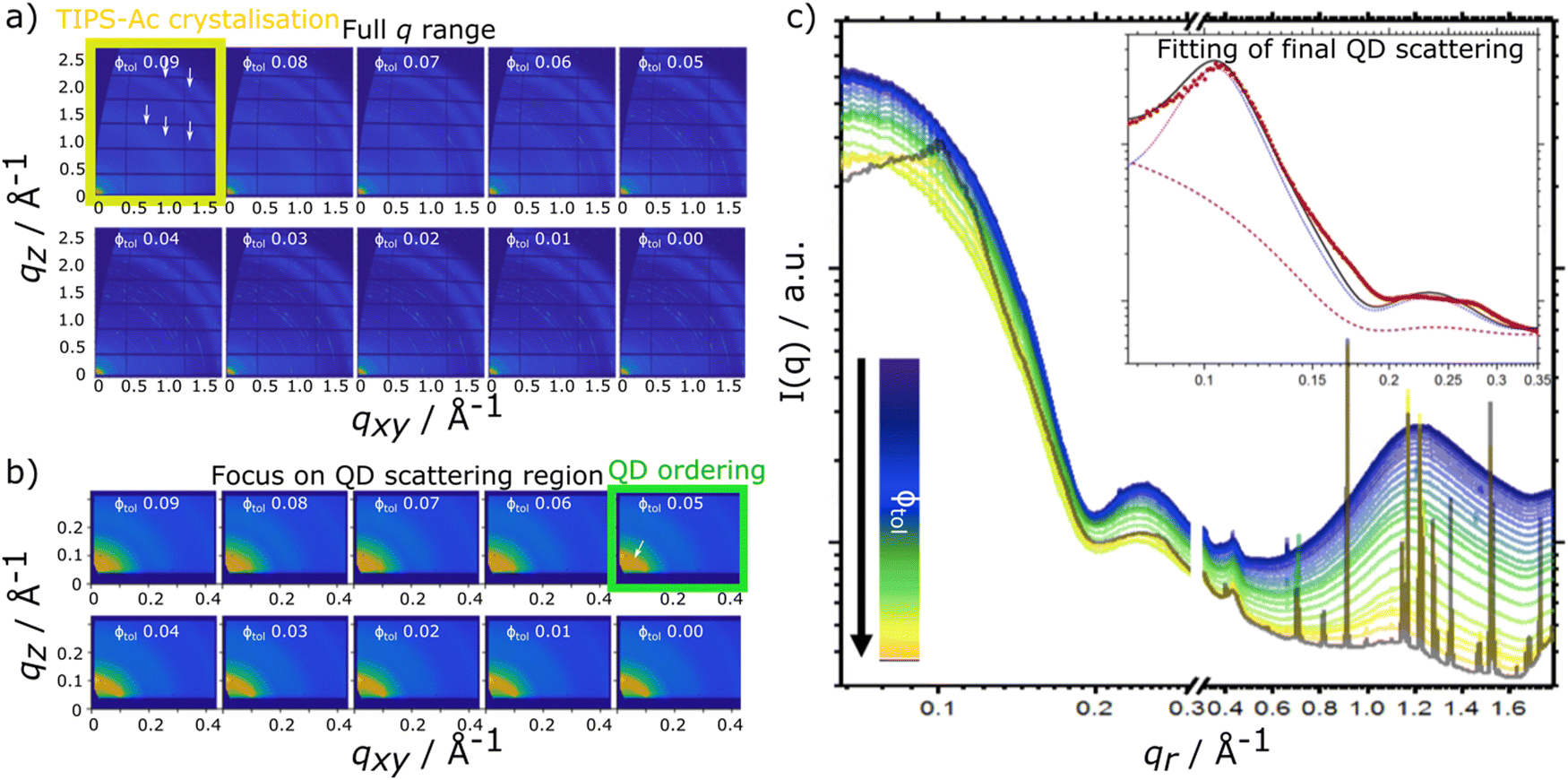 | ||
| Fig. 3 In situ GIXS data for TIPS-Ac:PbS-TET-CA showing the latter stages of film-formation (0.09 > 0.00 volume fraction toluene), with (a) showing the full q range (QD + OSC lengthscales) and (b) focusing on the small angle q range (QD lengthscales). The TIPS-Ac and QD ordering is highlighted with yellow & green boxes and white arrows, respectively. Radially integrated in situ scattering data for (c) as a function of solvent fraction, where the insert shows the fit (black line) of the final frame of the kinetic data, for a sphere-hard sphere + FCC paracrystal crystal model [showing the constituent sphere-hardsphere (blue dashed line) and FCC paracrystal (purple dotted line) components of the model fits]. The data show that at a volume fraction of solvent Φtol = 0.09 the onset of crystallisation of the TIPS-Ac commences. As the solvent fraction decreases, QD ordering scattering features become visible at Φtol = 0.05. As the solvent fraction further decreases the amplitude of the aggregated QD scattering and the TIPS-Ac crystal peaks increasingly dominate the scattering. | ||
For the drying TIPS-Tc:PbS-TET-CA film, no significant changes in the scattering patterns are observed until the toluene fraction reaches 0.04, at which point bright diffraction spots along qxy = 0.1, 0.25 and 0.3 Å−1 become visible. These distinct diffraction features lie within the quantum dot q range and represent the formation of quantum dot crystallites in identical orientations (i.e. whose crystallographic axes are all pointing in the same direction), as opposed to a collection of crystallites whose crystallographic axes had a random set of orientations with respect to one another that would generate paracrystalline scattering rings at qr ∼0.25 Å−1. The observed diffraction features are commensurate with a highly ordered face-centred cubic (FCC) quantum dot phase with a lattice constant of ∼102 Å, as illustrated by simulated scattering data (Fig. 4a and b). Simulated grazing incidence scattering patterns were obtained using SimDiffraction32 using an FCC quantum dot crystal lattice generated in CrystalMaker X. The emergence of the first order diffraction peak at qr 0.105 Å−1 is particularly prominent in the radially integrated scattering data (with integrations performed between 0–90°). Whilst this scattering feature is indeed prominent, there are also significant scattering contributions from well-dispersed quantum dots. To demonstrate this, radial integrations were taken at a number of smaller sectors with χ ranges of, 0 to 22.5°, 22.5 to 45°, 45 to 77.5° and 77.5 to 86° (Fig. 4c and d). Data for χ in the ranges of 0 to 22.5° and 22.5 to 45° do not include contributions from the FCC colloidal crystal and as such, the scattering is commensurate with earlier observations of well-dispersed quantum dot morphologies. As the fraction of solvent further decreases to 0.03, a weak wide-angle scattering feature at qr ∼1.1 Å−1 becomes visible, with a large number of additional isotropic scattering rings commensurate with TIPS-Tc crystallisation becoming visible as the film further dries. The data clearly shows that for the TIPS-Tc:PbS-TET-CA blend, quantum dot ordering occurs prior to the crystallisation of the OSC. We hypothesise that the formation of the observed quantum dot superlattice type structure likely acts as a nucleating agent for the OSC crystallisation. The quantum dot superlattice structure formed at early periods of the drying process in the TIPS-Tc:QD blend films is highly similar to those observed for drop-cast quantum dot films,33 indicating that for this system, prior to the crystallisation of the OSC matrix, film formation proceeds highly similarly to a bulk quantum dot film.
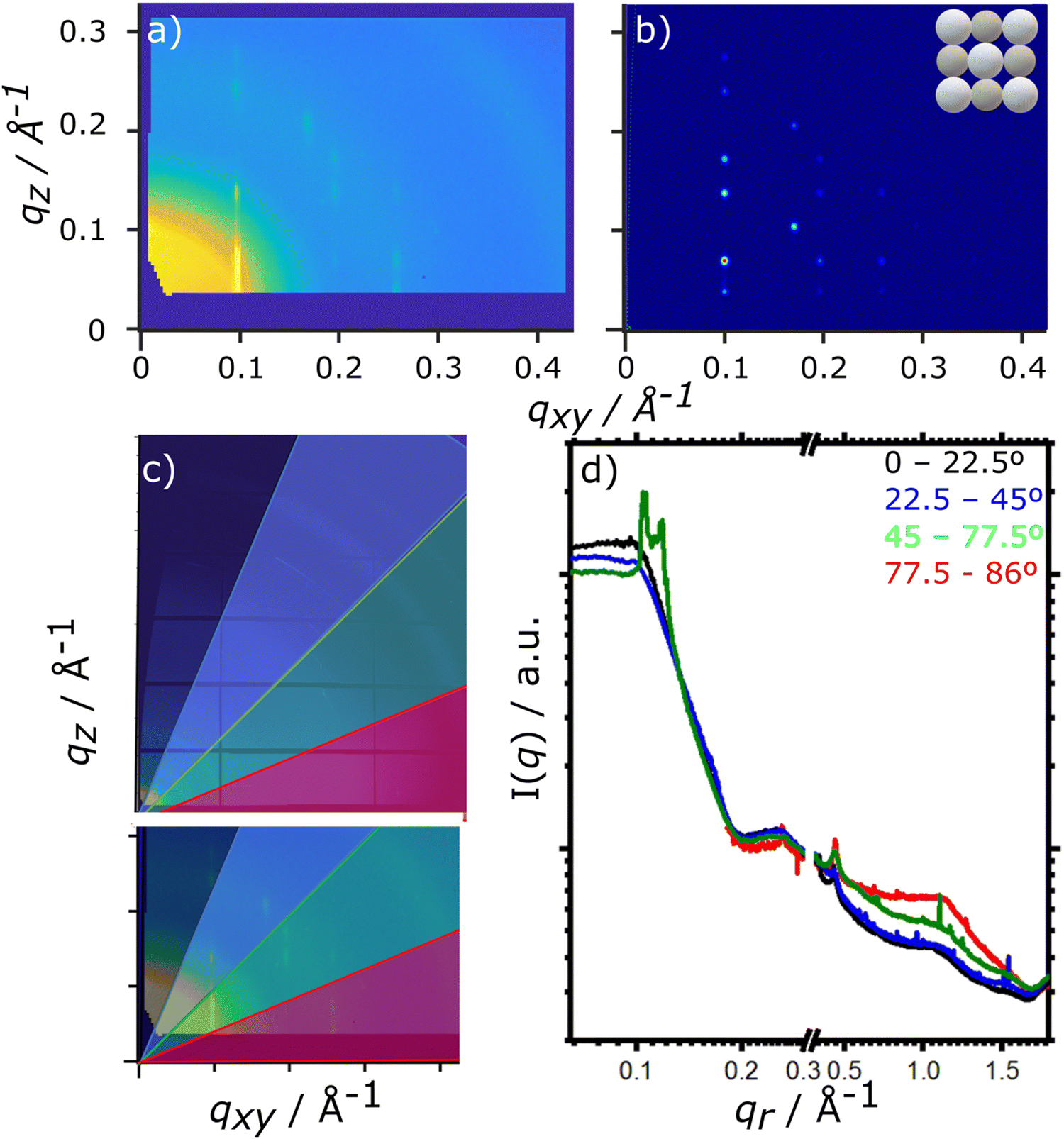 | ||
| Fig. 4 (a) Grazing incidence X-ray scattering patterns of the final TIPS-Tc:PbS-TET-CA film and (b) a simulated scattering pattern of a bulk FCC QDs with a lattice constant of 102 Å, with insert showing unit cell. There is a high degree of similarity between the experimental and simulated scattering patterns, albeit with diffraction spots in simulated scattering data being more well-defined as disorder of the FCC QD lattice is not accounted for. (c and d) Analysis of specific sectors between 0–22.5 (black), 22.5–45 (blue), 45–77.5 (green) and 77.5–86° (red) with overlays of these regions shown on (c) the 2D data and (d) the 1D data, simply illustrating the presence of scattering from well-dispersed QD dots (0–22.5, 22.5–45), that is underlying the FCC scattering pattern. | ||
For the TIPS-Ac:PbS-TET-CA blend film, data show that as film drying proceeds, clear diffraction spots in the high q scattering region become visible at a solvent volume fraction of 0.09, indicating the formation of TIPS-Ac crystallites within the drying film. At this point, scattering in the low q region remains largely unchanged and is consistent with well dispersed isolated quantum dots in solution.
As the film dries and the solvent fraction further reduces, the amplitude of the high q scattering features increases and distinct isotropic rings become visible, indicative of a large number of randomly oriented OSC crystallites within the film. When the solvent fraction reaches 0.05, scattering in the low q region distinctly changes from dispersed quantum dots in solution with the emergence of a distinct structural peak at qr = 0.101 Å−1 and represents quantum dots becoming packed together as opposed to being well-dispersed within the film. As the solvent fraction approaches 0, both TIPS-Ac crystalline features and those associated with aggregated/ordered quantum dots become increasingly strong. Similarly to the TIPS-Tc:PbS-TET-CA blend, the TIPS-Ac:PbS-TET-CA blend exhibits scattering features that likely arise from two distinct quantum dot populations, namely dispersed, non-interacting quantum dots and aggregated, superlattice-like quantum dots.
To confirm the existence of two QD populations, data for the final frame of the kinetic data were fitted to a composite model scattering model with two different components. To aid the discussion we first define the components individually. The first describes scattering from QDs distributed randomly throughout the volume of the film, which comprises a spherical form factor with a hard sphere structure factor (named hereafter as “sphere-hardsphere”). The second describes QDs that have (colloidal) paracrystalline order. This means the QD have a recognisable degree of colloidal crystallinity but with a level of disorder in the form of deviations in the position of the colloids from their ideal lattice positions. A packing disorder parameter describes the distribution of the QD from their ideal lattice positions; at zero disorder the perfect colloidal crystal superlattice is recovered, whilst as disorder values approach unity, QDs are essentially randomly distributed as in the aforementioned sphere-hardsphere scattering model. This second scattering model takes into account regions of the film where the QD are close-packed, albeit without perfect ordering. The model employed is a face-centred-cubic paracrystal (named hereafter as “FCC paracrystal”). Additional details of the models are included in the ESI.† The composite model is a simple arithmetic sum of the two models, representing scattering contributions from populations of QD in both physical arrangements, and is named hereafter as “sphere-hardsphere + FCC paracrystal”.
This sphere-hardsphere + FCC paracrystal model is separated into its component parts (Fig. 1d/e insert) and shows the contributions to the model for quantum dot morphologies where there is a significant proportion of scattering material in both sphere-hard sphere and FCC paracrystal arrangements, with associated fit parameters presented in ESI,† Table S3. The quantum dots in the TIPS-Tc blend are significantly more ordered and therefore poorly dispersed within the OSC matrix, compared to the TIPS-Ac blend, as shown by lattice distortion factors of the FCC paracrystal model of 0.06 and 0.16, respectively. This fits with observations of the distinct quantum dot diffraction spots observed early in the drying process for the TIPS-Tc:PbS-TET-CA blend, commensurate with both a high degree of order but also with colloidal crystallites that are only at one, specific orientation. On the other hand, for the TIPS-Ac:PbS-TET-CA blend the quantum dot aggregates are randomly oriented and dispersed throughout the drying film as evidenced by the isotropic nature of the scattering ring.
The TIPS-Ac:PbS-TET-CA in situ GIXS data not only shows that in this case, OSC crystallisation occurs earlier than for the TIPS-Tc system, but also that OSC crystallisation occurs prior to the onset of quantum dot ordering. We therefore hypothesise that in the case of the TIPS-Ac:PbS-TET-CA blend, a large number of OSC crystal nuclei spontaneously form and subsequently grow. As crystal growth proceeds, the quantum dot inclusions are excluded along the growing TIPS-Ac crystal fronts (as impurities tend to be expelled from a crystallising medium). Quantum dots become closely packed together as the growth front progresses. Illustrations of the two different OSC:QD self-assembly mechanisms are presented in Fig. 5.
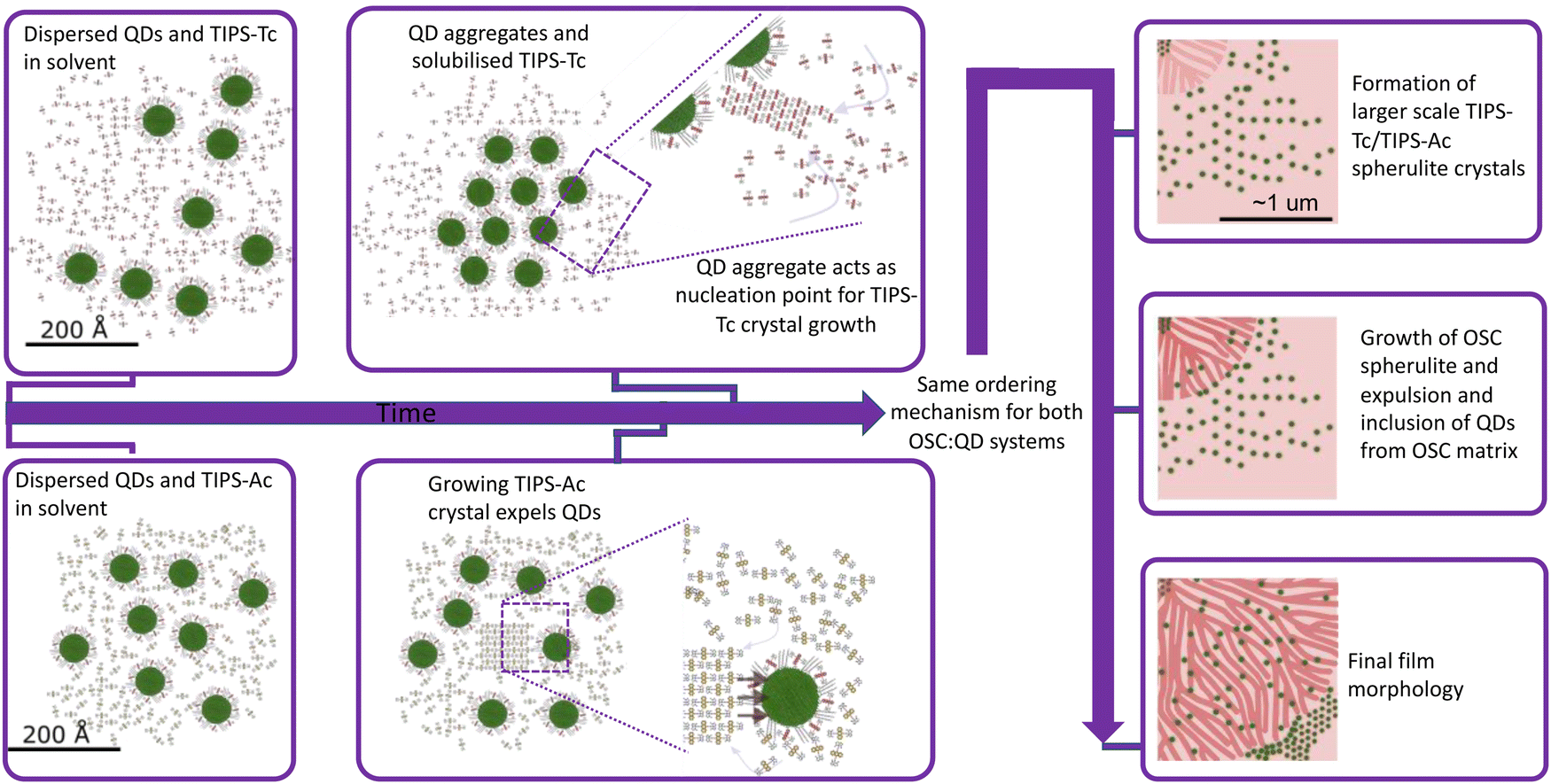 | ||
| Fig. 5 Illustrations of the self-assembly mechanisms of the blade coated blends of TIPS-Tc:PbS-TET-CA and TIPS-Ac:PbS-TET-CA. [The small molecules and quantum dots are drawn to scale.] Initially both TIPS-Tc:PbS-TET-CA and TIPS-Ac:PbS-TET-CA are dispersed in solution. At later points in time, as solvent evaporation proceeds and the film dries, self-assembly of the small molecule and QD components occurs. For TIPS-Tc:PbS-TET-CA, small molecule crystallisation is nucleated by QD ordering, and for TIPS-Ac:PbS-TET-CA, small molecule crystallisation occurs independent of QD ordering. For both TIPS-Tc:PbS-TET-CA and TIPS-Ac:PbS-TET-CA blends, as the OSC crystallisation proceeds QD inclusions are expelled from lamellar crystals in growing spherulites and are concentrated in the remaining amorphous regions. | ||
The experimental data presented here shows that the TIPS-Tc crystallisation occurred at a lower volume fraction of solvent (Φtol 0.03 & 0.09 for TIPS-Tc:PbS-TET-CA & TIPS-Ac:PbS-TET-CA, respectively). Furthermore, the magnitude of the OSC crystalline peaks for the TIPS-Tc:quantum dot blend films are significantly lower than those observed for the TIPS-Ac:quantum dot blend. This indicates that the TIPS-Tc matrix is likely less crystalline that of the TIPS-Ac matrix.
Conclusions
To conclude, in situ GIXS studies of OSC:quantum dot blends have enabled new insight into two nanoscale structure formation pathways: one where quantum dot aggregate formation seeds the crystallisation of the OSC, and another where OSC crystallisation occurs spontaneously. Irrespective of the OSC crystallisation initiation, quantum dot inclusions are expelled from the growing OSC crystallities. That such two differing pathways are observed despite the high chemical similarity between the TIPS-Tc and TIPS-Ac OSC hosts employed here may initially appear surprising. However, it shows that the small differences between OSCs that lead to the generation different crystal packing motifs also play a critical role in the nanoscale structure formation pathways of OSC:QD blend films. We hypothesise that the asymmetry of TIPS-Tc, combined with the screening of π–π interactions by the TIPS solubilising groups, results in the TIPS-Tc matrix having a relatively low propensity to crystallise during solution processing compared with TIPS-Ac. As such, the TIPS-Tc does not undergo crystallisation until the quantum dots form a distinct supperlattice type structure capable of nucleating TIPS-Tc crystallisation. By comparison, whilst TIPS-Ac still has significantly screened π–π interactions, this symmetric OSC more readily crystallises as the film dries and as such a different nanostructure formation pathway is observed. These results provide important understanding and insight into the structure formation of OSC:quantum dot blends that have important applications as energy materials. It is important to note that the existence of significant proportions of aggregated/packed QDs identified in both the TIPS-Tc:PbS-TET-CA and the TIPS-Ac:PbS-TET-CA blend films are disadvantageous for the intended applications as photon multipliers and would lead to aggregation-induced quenching. As such, the insights into the two self-assembly mechanisms demonstrated here provide the understanding needed for future work to begin to rationally design pathways that improve the dispersabilities of quantum dots in OSC matricies. Future work in on these and related systems will seek to utilise these insights on the interplay between OSC and QD ordering. For systems, such at the TIPS-Tc:PbS-TET-CA blend, approaches to increase the nucleation density of TIPS-Tc via the use of nucleation agents or developing QDs functioning as nucleation agents may provide a pathway for increasing QD dispersibility in such systems. Likewise for the TIPS-Ac:PbS-TET-CA blend, where OSC crystallisation clearly results in QD ordering, processing routes that suppress this crystallisation tendency, such as employing diluents or eutectic OSC mixtures, could be highly promising for improving QD dispersibilities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding through the Engineering and Physical Sciences Research Council (UK) via grants EP/P027814/1 and EP/P027741/1 and the Winton Programme for the Physics of Sustainability. We acknowledge Diamond Light Source for time on I07 under proposal SI23587-1. DTWT and MPW offer particular thanks to Simon Dixon for his expedient work in producing the in situ blade coating set-up. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement no. 654000.References
- O. O. Matvienko, et al., Dispersion and aggregation of quantum dots in polymer–inorganic hybrid films, Thin Solid Films, 2013, 537, 226–230, DOI:10.1016/j.tsf.2013.03.046.
- M. Helgesen, R. Søndergaard and F. C. Krebs, Advanced materials and processes for polymer solar cell devices, J. Mater. Chem., 2010, 20, 36–60, 10.1039/B913168J.
- N. Zhao, et al., Colloidal PbS Quantum Dot Solar Cells with High Fill Factor, ACS Nano, 2010, 4, 3743–3752, DOI:10.1021/nn100129j.
- A. J. Moulé, L. Chang, C. Thambidurai, R. Vidu and P. Stroeve, Hybrid solar cells: basic principles and the role of ligands, J. Mater. Chem., 2012, 22, 2351–2368, 10.1039/C1JM14829J.
- S. Coe, W.-K. Woo, M. Bawendi and V. Bulović, Electroluminescence from single monolayers of nanocrystals in molecular organic devices, Nature, 2002, 420, 800–803, DOI:10.1038/nature01217.
- M. Zorn, et al., Quantum Dot−Block Copolymer Hybrids with Improved Properties and Their Application to Quantum Dot Light-Emitting Devices, ACS Nano, 2009, 3, 1063–1068, DOI:10.1021/nn800790s.
- J. Allardice et al. Ligand Directed Self-Assembly of Bulk Organic-Semiconductor/Quantum-Dot Blend Films Enables Near Quantitative Harvesting of Triplet Excitons. arXiv, 2020, preprint, arXiv:2009.05764.
- D. T. W. Toolan, et al., Insights into the Structure and Self-Assembly of Organic-Semiconductor/Quantum-Dot Blends, Adv. Funct. Mater., 2021, 2109252 Search PubMed.
- W. Shockley and H. J. Queisser, Detailed balance limit of efficiency of p-n junction solar cells, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- J. Lee, et al., Singlet Exciton Fission Photovoltaics, Acc. Chem. Res., 2013, 46, 1300–1311, DOI:10.1021/ar300288e.
- A. Rao and R. H. Friend, Harnessing singlet exciton fission to break the Shockley-Queisser limit, Nat. Rev. Mater., 2017, 2, 17063, DOI:10.1038/natrevmats.2017.63.
- M. J. Y. Tayebjee, A. Rao and T. W. Schmidt, All-optical augmentation of solar cells using a combination of up- and downconversion, J. Photonics Energy, 2018, 8, 022007, DOI:10.1117/1.Jpe.8.022007.
- M. H. Futscher, A. Rao and B. Ehrler, The Potential of Singlet Fission Photon Multipliers as an Alternative to Silicon-Based Tandem Solar Cells, ACS Energy Lett., 2018, 3, 2587–2592, DOI:10.1021/acsenergylett.8b01322.
- M. Tabachnyk, et al., Resonant energy transfer of triplet excitons from pentacene to PbSe nanocrystals, Nat. Mater., 2014, 13, 1033–1038, DOI:10.1038/nmat4093.
- N. J. Thompson, et al., Energy harvesting of non-emissive triplet excitons in tetracene by emissive PbS nanocrystals, Nat. Mater., 2014, 13, 1039–1043, DOI:10.1038/nmat4097.
- J. R. Allardice, et al., Engineering Molecular Ligand Shells on Quantum Dots for Quantitative Harvesting of Triplet Excitons Generated by Singlet Fission, J. Am. Chem. Soc., 2019, 141, 12907–12915, DOI:10.1021/jacs.9b06584.
- N. J. L. K. Davis, et al., Singlet Fission and Triplet Transfer to PbS Quantum Dots in TIPS-Tetracene Carboxylic Acid Ligands, J. Phys. Chem. Lett., 2018, 9, 1454–1460, DOI:10.1021/acs.jpclett.8b00099.
- V. Gray, et al., Direct vs Delayed Triplet Energy Transfer from Organic Semiconductors to Quantum Dots and Implications for Luminescent Harvesting of Triplet Excitons, ACS Nano, 2020, 14, 4224–4234, DOI:10.1021/acsnano.9b09339.
- D. T. W. Toolan, et al., Linking microscale morphologies to localised performance in singlet fission quantum dot photon multiplier thin films, J. Mater. Chem. C, 2022, 10, 11192–11198, 10.1039/D2TC00677D.
- J. Kwak, et al., Characterization of Quantum Dot/Conducting Polymer Hybrid Films and Their Application to Light-Emitting Diodes, Adv. Mater., 2009, 21, 5022–5026, DOI:10.1002/adma.200902072.
- L. Gao, et al., Efficient near-infrared light-emitting diodes based on quantum dots in layered perovskite, Nat. Photonics, 2020, 14, 227–233, DOI:10.1038/s41566-019-0577-1.
- D. T. W. Toolan, et al., Mixed Small-Molecule Matrices Improve Nanoparticle Dispersibility in Organic Semiconductor-Nanoparticle Films, Langmuir, 2023, 39, 4799–4808, DOI:10.1021/acs.Langmuir.3c00152.
- W. Chen, et al., Colloidal PbS quantum dot stacking kinetics during deposition via printing, Nanoscale Horiz., 2020, 5, 880–885, 10.1039/D0NH00008F.
- K. Whitham, D.-M. Smilgies and T. Hanrath, Entropic, Enthalpic, and Kinetic Aspects of Interfacial Nanocrystal Superlattice Assembly and Attachment, Chem. Mat., 2018, 30, 54–63, DOI:10.1021/acs.chemmater.7b04223.
- J. J. Geuchies, et al., In situ study of the formation mechanism of two-dimensional superlattices from PbSe nanocrystals, Nat. Mater., 2016, 15, 1248–1254, DOI:10.1038/nmat4746.
- J.-H. Dou, et al., Fine-Tuning of Crystal Packing and Charge Transport Properties of BDOPV Derivatives through Fluorine Substitution, J. Am. Chem. Soc., 2015, 137, 15947–15956, DOI:10.1021/jacs.5b11114.
- H. Usta, A. Facchetti and T. J. Marks, n-Channel Semiconductor Materials Design for Organic Complementary Circuits, Acc. Chem. Res., 2011, 44, 501–510, DOI:10.1021/ar200006r.
- F. S. Kim, G. Ren and S. A. Jenekhe, One-Dimensional Nanostructures of π-Conjugated Molecular Systems: Assembly, Properties, and Applications from Photovoltaics, Sensors, and Nanophotonics to Nanoelectronics, Chem. Mat., 2011, 23, 682–732, DOI:10.1021/cm102772x.
- K. Bhattacharyya and A. Datta, Polymorphism Controlled Singlet Fission in TIPS-Anthracene: Role of Stacking Orientation, J. Phys. Chem. C, 2017, 121, 1412–1420, DOI:10.1021/acs.jpcc.6b10075.
- X. Xu, et al., Synthesis, solution-processed thin film transistors and solid solutions of silylethynylated diazatetracenes, Chem. Commun., 2014, 50, 12828–12831, 10.1039/C4CC04627G.
- M. A. Hines and G. D. Scholes, Colloidal PbS nanocrystals with size-tunable near-infrared emission: observation of post-synthesis self-narrowing of the particle size distribution, Adv. Mater., 2003, 15, 1844–1849 CrossRef CAS.
- D. W. Breiby, O. Bunk, J. W. Andreasen, H. T. Lemke and M. M. Nielsen, Simulating X-ray diffraction of textured films, J. Appl. Crystallogr., 2008, 41, 262–271 CrossRef CAS.
- T. Hanrath, J. J. Choi and D.-M. Smilgies, Structure/Processing Relationships of Highly Ordered Lead Salt Nanocrystal Superlattices, ACS Nano, 2009, 3, 2975–2988, DOI:10.1021/nn901008r.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00079f |
| This journal is © The Royal Society of Chemistry 2023 |