
Highly efficient electrocatalytic biomass valorization over a perovskite-derived nickel phosphide catalyst†
Wanbing
Gong
a,
Jiayi
Li
a,
Jun
Ma
ab,
Dong
Liu
 *ab,
Ran
Long
a and
Yujie
Xiong
*a
*ab,
Ran
Long
a and
Yujie
Xiong
*a
aHefei National Research Center for Physical Sciences at the Microscale, School of Chemistry and Materials Science, National Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: yjxiong@ustc.edu.cn; dongliu@ustc.edu.cn
bSuzhou Institute for Advanced Research, University of Science and Technology of China, Suzhou, Jiangsu 215123, P. R. China
First published on 8th November 2022
Abstract
In this work, we successfully develop a binder-free phosphorus-engineered perovskite-based catalyst grown on nickel foam via a hydrothermal-phosphorization strategy. For the first time, an as-synthesized perovskite-based nickel phosphide catalyst exhibits excellent electrocatalytic oxidation (ECO) performance for biomass valorization to supersede the competitive oxygen evolution reaction (OER).
New conceptsElectrocatalytic oxidation is a significant but challenging carbon-neutral route for biomass valorization. Nevertheless, it usually requires highly efficient and robust electrocatalysts. As one of the most important biomass-derived platform molecules, 5-hydroxymethylfurfural (HMF) can be oxidized to 2,5-furandicarboxylic acid (FDCA), which can be used to replace petrochemical based terephthalic acid to produce degradable plastic. Herein, we for the first time report a binder-free phosphorus-engineered perovskite-based catalyst grown on nickel foam via a hydrothermal-phosphorization strategy as a cost-effective and highly efficient ECO catalyst to replace the inefficient and low-value oxygen evolution reaction at ambient conditions. The optimized phosphorus-engineered perovskite-derived catalyst exhibits excellent electrocatalytic oxidation activity for the conversion of HMF to FDCA, which affords an almost 100% yield and >90% faradaic efficiency using water as an oxygen source. In addition, in situ Raman tests have shown that the high valence NiOOH species is considered to be the real active site. We expect this strategy to open new prospects for designing highly efficient and low-cost nonprecious metal electrocatalysts for heterogeneous electrocatalytic oxidation reactions. |
Biomass, a naturally abundant and renewable organic resource, has been recognized as a significant and promising feedstock to replace traditional fossil fuel resources for producing value-added fuels and chemicals.1–3 5-Hydroxymethylfurfural (HMF), one of the most important biomass-derived platform molecules, can be generated from C6 monosaccharides via acid hydrolysis and can be subsequently upgraded to serve as a fundamental precursor for the manufacture of fine chemicals, plastics, pharmaceuticals and biofuels.4–6 In terms of functional groups, HMF can be converted into a variety of value-added chemicals via hydrogenation or oxidation routes.6 For example, HMF can be oxidized into some important chemical intermediates such as 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), 2,5-diformylfuran (DFF), 5-formyl-2-furancarboxylic acid (FFCA) and 2,5-furandicarboxylic acid (FDCA).7,8 Among these products, FDCA has emerged as one fascinating substitute for petrochemical based terephthalic acid to produce degradable plastic.9,10 Conventional HMF oxidation to FDCA via a thermo-catalytic approach remains challenging due to the involvement of chemical oxidants, toxic and expensive solvents and/or precious metal catalysts.6,11,12 In recent years, the electrocatalytic oxidation (ECO) of HMF to FDCA has attracted increasing attention owing to the advantages of low energy consumption, mild operating conditions and the use of water as the oxygen source.13–16 In this respect, Choi's group demonstrated a highly efficient ECO of HMF into FDCA using 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO) as a mediator coupled with hydrogen evolution reaction on Au and Pt electrodes.13 However, the use of expensive TEMPO and precious metal electrodes leads to high process costs. Sun's group recently reported the ECO of biomass compounds to value-added products with ∼100% faradaic efficiencies (FEs) over an inexpensive porous Ni3S2/Ni foam (NF) electrode.14 Subsequently, numerous cost-effective electrocatalysts like Ni2P NPA/NF,15 hp-Ni,17 NiFe layered double hydroxide,18 vanadium nitride,19 NiCo2O4,20 CoO-CoSe2,21 Ni3N@C,22 NiBx,23 MoO2-FeP@C,24etc. have been extensively investigated to be used as the electrodes for HMF oxidation with high activities and FEs. Nevertheless, it is still extremely desirable to develop an efficient and robust nonprecious metal-based catalyst for ECO reactions under mild reaction conditions.
Perovskite oxides are promising oxygen electrode catalysts among the versatile developed nonprecious metal-based electrocatalysts because of their low cost, environmental friendliness, highly tunable composition and flexible structure.25,26 The perovskite-structured LaNiO3 shows excellent potential for the oxygen evolution reaction (OER), but its activity and durability are still far from optimal.27–29 Varied approaches to boost the intrinsic electrocatalytic activity of LaNiO3-based perovskite catalysts have been developed to address these issues, mainly including strain engineering, defect engineering, heteroatomic doping, regulation of the structure and the components.27,28,30–32 For instance, Shao's group adopted a top-down strategy to transform bulk crystalline LaNiO3 into a nanostructured amorphous hydroxide by FeCl3 treatment, resulting in an extremely low overpotential of 189 mV at 10 mA cm−2 during the OER process.31 The outstanding OER activities of numerous perovskite oxides have been thoroughly explored.33–36 However, the synthesis of simple, cost-effective, and efficient perovskite-based catalysts for ECO reactions has not yet been reported.
Here, we demonstrate a high-performance phosphorus-engineered perovskite-based electrocatalyst (denoted as LN-400-P-350) grown on nickel foam (NF) by a hydrothermal-phosphorization strategy. Benefiting from improved mass and charge transport abilities, the as-synthesized LN-400-P-350 catalyst shows excellent ECO performance for the conversion of HMF to FDCA with a 100% yield and >90% FE at 1.49 V vs. RHE. In situ Raman tests have confirmed the formation of NiOOH species on the surface, which are considered to be the real active species. Our results provide a simple and highly efficient route to unleash the catalytic potential of perovskite based phosphide electrocatalysts for green synthesis of high value-added chemicals.
The synthesis route of the optimum LN-400-P-350 catalyst is briefly shown in Fig. 1a. To enhance the overall electrical conductivity, NF was employed as a conductive support. Firstly, the LaNiO3 (LN) perovskite based nanosheets (Fig. S1, ESI†) were uniformly grown on the skeleton of the NF through a hydrothermal reaction, and the nanosheets network could promote mass transport and charge transfer. Secondly, the LN based nanosheets were calcinated at 400 °C for 2 h to obtain a perovskite oxide (denoted as LN-400). According to Fig. S2 (ESI†), the regular and smooth nanosheets convert into rough nanosheets and irregular nanorods, which are attributed to NiO and La2O3, respectively. Lastly, the LN-400-P-350 was obtained at 350 °C for 4 h using NaH2PO2·H2O as a phosphorus source in a N2 atmosphere. The scanning electron microscope (SEM) images (Fig. 1b) show that the pristine morphology is well preserved with a rough surface. The transmission electron microscopy (TEM) image (Fig. 1c) reveals that the sample mainly consists of flake-like phosphides. High resolution TEM (HRTEM) images (Fig. 1d) show the lattice fringes with a spacing of 0.22 and 0.19 nm, corresponding to (111) and (210) planes of Ni2P phase, respectively. The high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image and corresponding elemental mapping images show that La, Ni, O and P elements homogeneously distribute throughout the sample (Fig. 1e). The X-ray diffraction (XRD) pattern (Fig. 2a) of LN-400 shows diffraction peaks at ∼26.1°, 30.0°, and 37.3°, 43.3°, 63.0°, 75.6°, which are assigned to La2O3 (JCPDS 96-101-0279) and NiO (JCPDS 96-101-0094), respectively, indicating the formation of the perovskite-derived oxide. Impressively, the diffraction peaks at ∼26.0°, 30.0°, and 40.7°, 44.6°, 47.3° are respectively attributed to LaP (JCPDS 96-900-8660) and the (111), (201) and (210) planes of Ni2P phase (JCPDS 00-003-0953) over the phosphorus-engineered LN-400-P-350 catalyst, which suggests the successful transformation of the perovskite-derived oxide to the perovskite-derived phosphide. The surface elemental composition and chemical states of the samples were determined by X-ray photoelectron spectra (XPS). The survey spectra show that the P element has been successfully introduced into the LN-400-P-350 sample (Fig. S3, ESI†). The La 3d and Ni 2p spectra (Fig. 2b) reveal a negative shift of the Ni 2p1/2 peaks, indicating the successful formation of Ni2P.27 Meanwhile, the disappearance of the characteristic peaks at ∼861 and 880 eV indicates the transformation of oxides to phosphides. Furthermore, the O 1s spectra (Fig. 2c) can be deconvolved into three peaks, where the peaks at ∼529, ∼531 and ∼532 eV correspond to lattice oxygen species (O1), adsorbed oxygen species (O2) and adsorbed hydroxyl groups (O3), respectively.29 The absence of lattice oxygen species after phosphorization indicates the transformation of the perovskite-derived oxide to the perovskite-derived phosphide, which is consistent with the XRD results. Additionally, the P 2p spectrum of the LN-400-P-350 catalyst (Fig. 2d) shows two peaks with binding energies at 129.1 and 133.6 eV, corresponding to the P–Ni and P–O bonds, respectively.37,38 The ICP-MS analysis indicates the atomic ratio of La/Ni is around 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating the formation of stoichiometric perovskite-derived materials.
:1, indicating the formation of stoichiometric perovskite-derived materials.
 | ||
| Fig. 1 (a) The typical synthetic procedure; (b) SEM image (inset: high magnification SEM image); (c) TEM image; (d) HRTEM image (insets: magnified HRTEM images); (e) HAADF-STEM image and elemental mapping images of the LN-400-P-350 catalyst. | ||
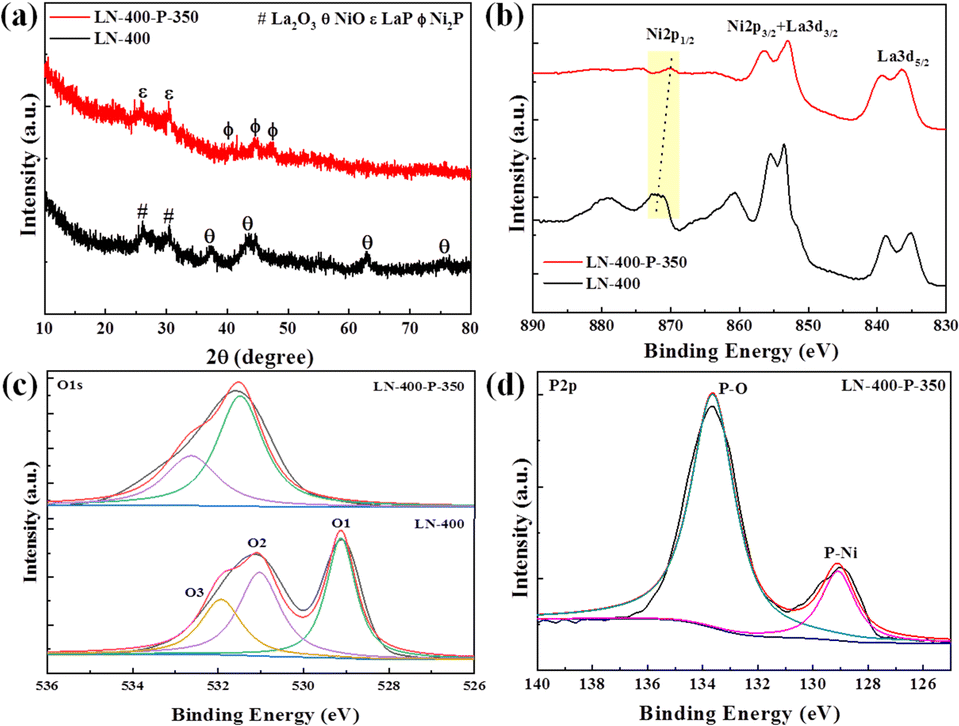 | ||
| Fig. 2 (a) XRD patterns; (b) high resolution La 3d + Ni 2p spectra and (c) high resolution O 1s spectra of the LN-400 and LN-400-P-350 catalysts. (d) High resolution P 2p spectrum of the LN-400-P-350 catalyst. | ||
The electrocatalytic oxidation of HMF to FDCA was chosen as a model reaction to evaluate the catalytic performance of the as-synthesized catalysts. As shown in Fig. 3, the selective oxidation of HMF to FDCA can proceed via two different pathways.39 Path I begins with an initial carbonyl group oxidation to produce the HMFCA intermediate, while path II starts with an initial hydroxy group oxidation to yield the 2,5-diformylfuran (DFF) intermediate. Then both HMFCA and DFF can be further oxidized to produce the FFCA intermediate, which is finally oxidized to generate FDCA. The linear sweep voltammograms (LSV) were first investigated with and without 10 mM HMF in a 1.0 M KOH solution, respectively (Fig. 3a). The LN-400-P-350 catalyst exhibits an excellent OER performance and a current density of 40 mA cm−2 at 1.59 V vs. RHE without 10 mM HMF, which is ∼70 mV lower than the potentials of the LN-400 and NF catalysts at the same current density, respectively. After adding 10 mM HMF, LN-400-P-350 only requires 1.41 V vs. RHE to drive a 40 mA cm−2 current density, which is ∼180 mV smaller than water oxidation at the same current density. Meanwhile, the potentials are slightly lowered to 1.62 and 1.64 V vs. RHE to achieve a current density of 40 mA cm−2 for the referenced LN-400 and NF (Fig. S4, ESI†), respectively. These results indicate that oxidation of HMF is favorable over the OER process, and the phosphorus-engineered LN-400-P-350 catalyst has a superior ECO performance for biomass conversion. The results in Fig. 3b demonstrate that the charge transfer resistance (Rct) of LN-400-P-350 is smaller than that of LN-400, indicating the faster catalytic kinetics of LN-400-P-350. After the addition of 10 mM HMF, the Rct can be effectively decreased, which suggests a faster charge-transfer process for HMF oxidation than water oxidation. The electrochemical surface areas (ECSA) of the samples were estimated through the double-layer capacitance (Cdl) in the non-faradaic potential range. As seen in Fig. 3c, the Cdl value of LN-400-P-350 is 117 mF cm−2, which is significantly more than that of LN-400 (74 mF cm−2). It indicates that there are more active site on LN-400-P-350.40 To identify the HMF oxidation products, the electrocatalytic HMF oxidation reaction was performed at 1.49 V vs. RHE in 1.0 M KOH as the electrolyte containing 10 mM HMF over LN-400-P-350. High-performance liquid chromatography (HPLC) was used to quantify the concentrations of HMF and the yielded oxidation products (Fig. S5, ESI†). First, we investigated the catalytic performance under the different potentials, and found that the optimal potential is 1.49 V vs. RHE (Fig. S6, ESI†). As revealed in Fig. 3d and e, the HMF concentration decreases and the FDCA concentration increases as the reaction time increases, and the complete transformation of HMF to FDCA is achieved after 0.75 h. Moreover, the HMFCA and FFCA can be detected during electrolysis, while DFF is barely detected. These results clearly prove the HMF oxidation over LN-400-P-350 mainly follows path I. After 0.75 h reaction, the FDCA yield and the FE are 100% and >90% at 1.49 V vs. RHE, respectively, which is comparable to previous work (Table S1, ESI†). Under identical reaction conditions, the referenced LN-400 and NF only gave low FDCA yields of 20.3 and 7.2%, respectively. Additionally, the concentration variation of HMF with the increasing time can be fitted to a first-order kinetic model (Fig. 3f). The kinetic constant k of LN-400-P-350 is 3.49 h−1, which is more than ∼4 times that of LN-400 (0.86 h−1). The results further confirm that the phosphorus-engineered perovskite based electrocatalyst exhibits more outstanding electrocatalytic activity for the ECO of HMF.
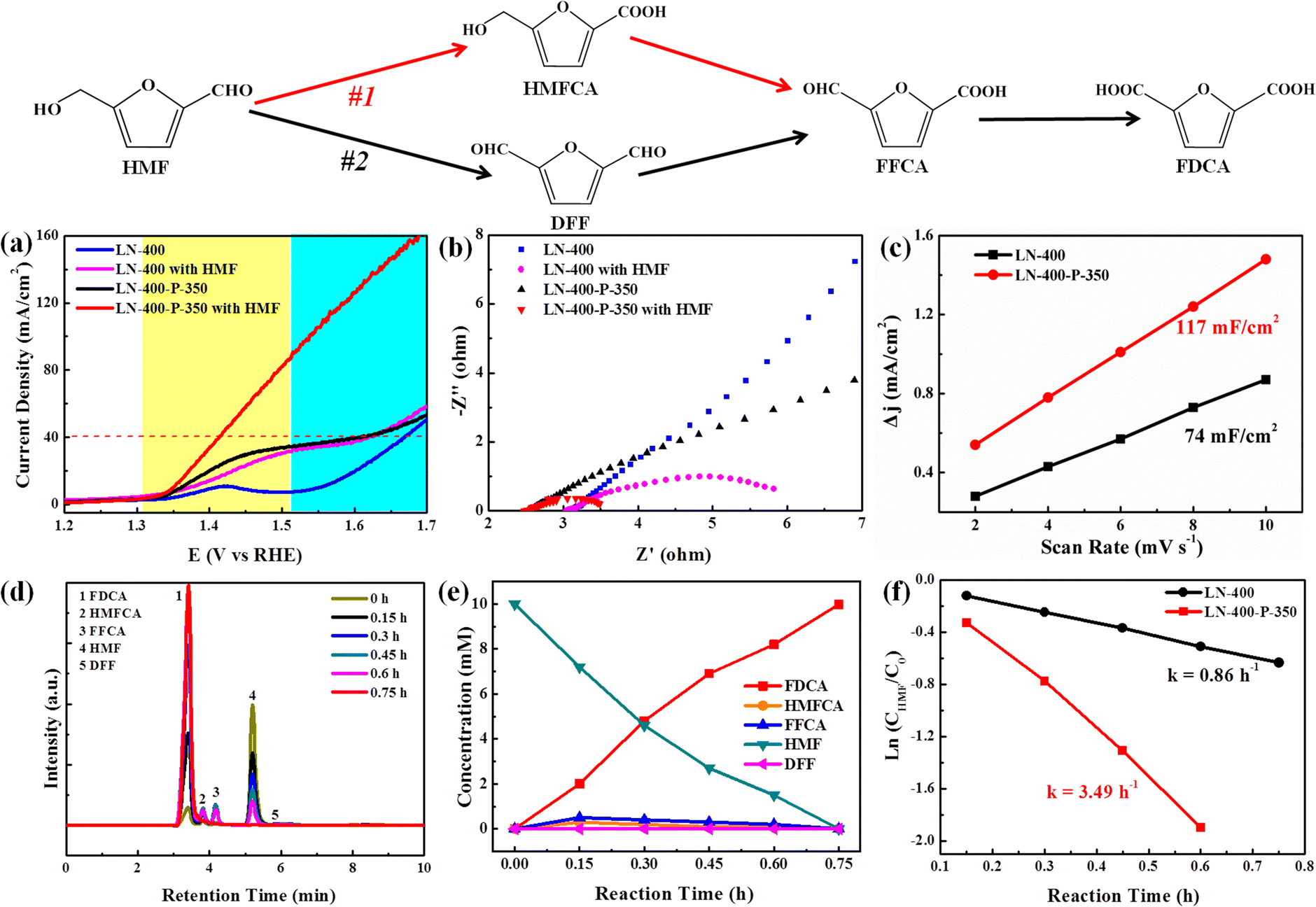 | ||
| Fig. 3 (a) LSV curves of LN-400 and LN-400-P-350 in 1.0 M KOH without and with 10 mM HMF; (b) Nyquist plots of LN-400 and LN-400-P-350 in 1.0 M KOH without and with 10 mM HMF; (c) Cdl of LN-400 and LN-400-P-350 in 1.0 M KOH; (d) HPLC chromatograms of ECO of HMF by LN-400-P-350 at 1.49 V vs. RHE in a 1.0 M KOH solution containing 10 mM HMF at various time; (e) concentrations of HMF and the oxidation products at different reaction times in 1.0 M KOH solution containing 10 mM HMF at 1.49 V vs. RHE and (f) kinetic fitting curves of the ECO of HMF by LN-400 and LN-400-P-350. | ||
Notably, the durability was further explored using the same LN-400-P-350 electrode. As revealed in Fig. 4a, the LSV curves are almost unchanged for nine cycles. After that, the yield of FDCA and the FE remain at >98% and >90% (Fig. 4b), respectively. These results prove the robust stability of LN-400-P-350 for electrocatalytic HMF oxidation. Additionally, the SEM, TEM, HRTEM, HAADF-STEM image and the corresponding elemental mapping images (Fig. S7, ESI†) of reused LN-400-P-350 show that its morphology, structure and constituents remain well after the ECO reactions. The XRD patterns after the reaction (Fig. S8a, ESI†) show the peaks of the Ni2P phase with a decrease in intensity. XPS was then carried out to analyze the change in the surface characteristics of LN-400-P-350. As displayed in Fig. S8b and c (ESI†), the La 3d and Ni 2p spectra exhibit a positive shift of the Ni 2p1/2 peaks, while the O 1s spectra show an obvious negative shift,41 indicating the generation of a high valence Ni species after the reaction. Meanwhile, the intensity of the P 2p peak decreases dramatically, implying that the electrochemically transformed metal (oxy)hydroxides are the real catalytic species, which is consistent with previous reports.42,43
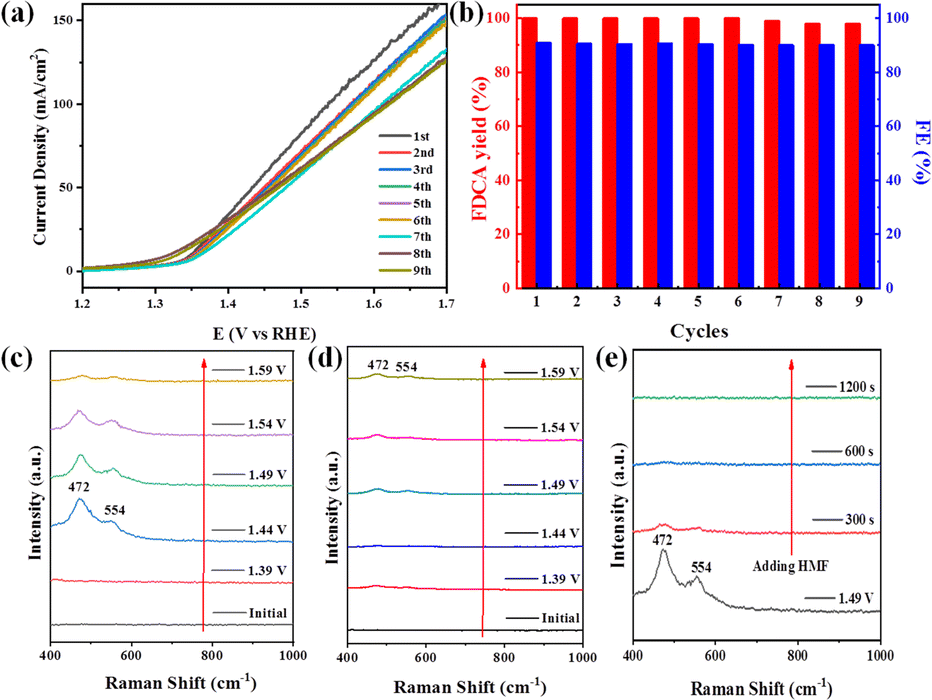 | ||
| Fig. 4 (a) LSV curves of LN-400-P-350 in 1.0 M KOH with 10 mM HMF for nine successive cycles. (b) FDCA yields and FEs of LN-400-P-350 for nine successive cycles. (c) Potential-dependent in situ Raman spectra of LN-400-P-350 without HMF. (d) Potential-dependent in situ Raman spectra of LN-400-P-350 with HMF. (e) In situ Raman spectra at 1.49 V vs. RHE without HMF and after the addition of HMF. | ||
To investigate the real catalytic sites for the ECO of biomass-derived HMF to FDCA, potential-dependent in situ Raman spectroscopy was collected over LN-400-P-350. As seen in Fig. 4c, no characteristic peaks corresponding to Ni-based species are observed at 1.39 V vs. RHE. In comparison, two obvious peaks at 472 and 554 cm−1 associated with the metal (oxy)hydroxides species (NiOOH) appear at 1.44 V vs. RHE,42,44 indicating its partial transformation to a higher valence Ni species. The slight intensity decrease at higher applied potential may be due to the intense competitive OER reaction. However, the NiOOH peaks are effectively inhibited when adding HMF (Fig. 4d). Furthermore, the peaks associated with the NiOOH species are gradually weakened and disappear within 300 s after the addition of HMF during the OER process (Fig. 3e). Therefore, we speculate that metal (oxy)hydroxides species can be formed with the assistance of OH− species from water. When adding HMF, the high valence state NiOOH species is reduced back to a low valence Ni species with the help of the electrons from HMF, and meanwhile, the HMF is therefore oxidized to FDCA, which is similar to prior studies.42,45 Such a simple and effective active site circulation makes the material highly robust under harsh OER and ECO conditions. Consequently, these results demonstrate that the electrochemically transformed metal (oxy)hydroxides are the real catalytic species for the ECO reactions.
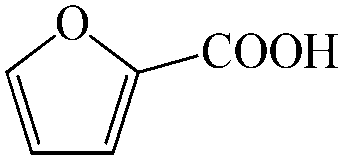
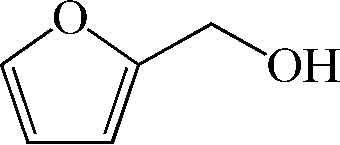
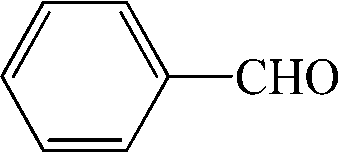
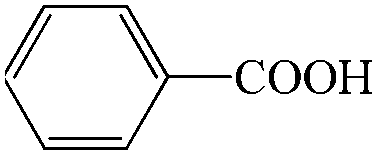
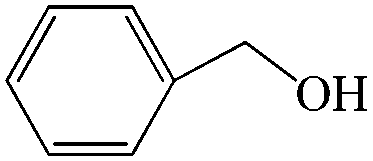
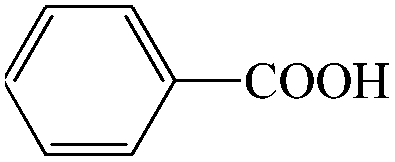
Subsequently, the ECO reactions of various alcohol/aldehyde containing compounds including furfural (FAL), furfuryl alcohol (FOL), benzaldehyde (BAL) and benzyl alcohol (BOL) were further studied using LN-400-P-350 as a catalyst at 1.49 V vs. RHE (Table 1). When using FAL or FOL as the substrate (Fig. S9 and S10, ESI†), furoic acid can be effectively produced with a high yield (>99%) and FE (>90%) within 0.35 or 0.95 h (entries 1 and 2, Table 1). Furthermore, when BAL or BOL were used as substrates (Fig. S11 and S12, ESI†), benzoic acid can be obtained with a high yield of >99% and FE of >90% after 0.25 and 0.55 h (entries 3 and 4, Table 1). These results prove the generic applicability of the LN-400-P-350 catalyst for the selective oxidation of alcohol and aldehyde containing compounds.
| Substrate | Product | Time (h) | Yield (%) | FE (%) |
|---|---|---|---|---|
| a Reaction conditions: 10 mM substrates, 1.0 M KOH, room temperature, 1.49 V vs. RHE. | ||||
|
|
0.35 | >99 | >90 |
|
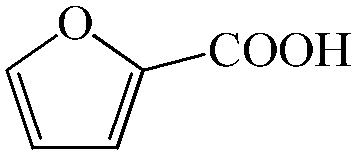
|
0.95 | >99 | >90 |
|
|
0.25 | >99 | >90 |
|
|
0.55 | >99 | >90 |
Conclusions
In summary, we have successfully developed a phosphorus-engineered perovskite oxide-derived electrocatalyst via a hydrothermal and phosphorization method, which can efficiently catalyze the ECO of biomass-derived alcohol/aldehyde containing compounds. For the ECO of HMF in alkaline medium, the as-synthesized phosphorus-engineered LN-400-P-350 catalyst exhibited an almost 100% FDCA yield and >90% FE at 1.49 V vs. RHE within 0.75 h. Furthermore, experimental measurements and in situ Raman results revealed that electrochemically transformed metal (oxy)hydroxides were the real catalytic species for the ECO reactions. The strategy demonstrated in this work opens a new avenue for engineering new perovskite-based phosphide catalysts for electrocatalytic selective oxidation reactions.Author contributions
Y. X. and D. L. conceived the project. W. G. designed and performed the experiments. W. G., D. L. and Y. X. wrote the manuscript. All the authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported in part by the National Key R&D Program of China (2020YFA0406103), NSFC (21725102, 22122506, 22075267, 22279128, 51902311, 91961106, U1832156), the Strategic Priority Research Program of the CAS (XDPB14), the Anhui Provincial Natural Science Foundation (2008085J05), the Youth Innovation Promotion Association of CAS (2019444), Fundamental Research Funds for the Central Universities (WK2060000039, 20720220007), the Gusu Innovation and Entrepreneurship Leading Talents Program (ZXL2022386). The authors thank the USTC Center for Micro- and Nanoscale Research and Fabrication for support.Notes and references
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Chem. Soc. Rev., 2012, 41, 8075–8098 RSC.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gartner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef CAS PubMed.
- J. N. Chheda, G. W. Huber and J. A. Dumesic, Angew. Chem., Int. Ed., 2007, 46, 7164–7183 CrossRef CAS PubMed.
- J. C. Serrano-Ruiz, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- X. Kong, Y. Zhu, Z. Fang, J. A. Kozinski, I. S. Butler, L. Xu, H. Song and X. Wei, Green Chem., 2018, 20, 3657–3682 RSC.
- Y. Wang, B. Liu, K. Huang and Z. Zhang, Ind. Eng. Chem. Res., 2014, 53, 1313–1319 CrossRef CAS.
- E. Hayashi, Y. Yamaguchi, K. Kamata, N. Tsunoda, Y. Kumagai, F. Oba and M. Hara, J. Am. Chem. Soc., 2019, 141, 890–900 CrossRef CAS PubMed.
- M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427–5453 RSC.
- K.-J. Liu, T.-Y. Zeng, J.-L. Zeng, S.-F. Gong, J.-Y. He, Y.-W. Lin, J.-X. Tan, Z. Cao and W.-M. He, Chin. Chem. Lett., 2019, 30, 2304–2308 CrossRef CAS.
- X. Wan, C. Zhou, J. Chen, W. Deng, Q. Zhang, Y. Yang and Y. Wang, ACS Catal., 2014, 4, 2175–2185 CrossRef CAS.
- S. Siankevich, G. Savoglidis, Z. Fei, G. Laurenczy, D. T. L. Alexander, N. Yan and P. J. Dyson, J. Catal., 2014, 315, 67–74 CrossRef CAS.
- H. G. Cha and K. S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef CAS PubMed.
- B. You, X. Liu, N. Jiang and Y. Sun, J. Am. Chem. Soc., 2016, 138, 13639–13646 CrossRef CAS PubMed.
- B. You, N. Jiang, X. Liu and Y. Sun, Angew. Chem., Int. Ed., 2016, 55, 9913–9917 CrossRef CAS PubMed.
- M. Sun, Y. Wang, C. Sun, Y. Qi, J. Cheng, Y. Song and L. Zhang, Chin. Chem. Lett., 2022, 33, 385–389 CrossRef CAS.
- B. You, X. Liu, X. Liu and Y. Sun, ACS Catal., 2017, 7, 4564–4570 CrossRef CAS.
- W.-J. Liu, L. Dang, Z. Xu, H.-Q. Yu, S. Jin and G. W. Huber, ACS Catal., 2018, 8, 5533–5541 CrossRef CAS.
- S. Li, X. Sun, Z. Yao, X. Zhong, Y. Cao, Y. Liang, Z. Wei, S. Deng, G. Zhuang, X. Li and J. Wang, Adv. Funct. Mater., 2019, 29, 1904780 CrossRef CAS.
- L. Gao, Y. Bao, S. Gan, Z. Sun, Z. Song, D. Han, F. Li and L. Niu, ChemSusChem, 2018, 11, 2547–2553 CrossRef CAS PubMed.
- X. Huang, J. Song, M. Hua, Z. Xie, S. Liu, T. Wu, G. Yang and B. Han, Green Chem., 2020, 22, 843–849 RSC.
- N. Zhang, Y. Zou, L. Tao, W. Chen, L. Zhou, Z. Liu, B. Zhou, G. Huang, H. Lin and S. Wang, Angew. Chem., Int. Ed., 2019, 58, 15895–15903 CrossRef CAS PubMed.
- P. Zhang, X. Sheng, X. Chen, Z. Fang, J. Jiang, M. Wang, F. Li, L. Fan, Y. Ren, B. Zhang, B. J. J. Timmer, M. S. G. Ahlquist and L. Sun, Angew. Chem., Int. Ed., 2019, 58, 9155–9159 CrossRef CAS PubMed.
- G. Yang, Y. Jiao, H. Yan, Y. Xie, A. Wu, X. Dong, D. Guo, C. Tian and H. Fu, Adv. Mater., 2020, 32, 2000455 CrossRef CAS PubMed.
- H. J. Song, H. Yoon, B. Ju and D. W. Kim, Adv. Energy Mater., 2021, 11, 2002428 CrossRef CAS.
- C. E. Beall, E. Fabbri and T. J. Schmidt, ACS Catal., 2021, 11, 3094–3114 CrossRef CAS.
- J. Liu, E. Jia, L. Wang, K. A. Stoerzinger, H. Zhou, C. S. Tang, X. Yin, X. He, E. Bousquet, M. E. Bowden, A. T. S. Wee, S. A. Chambers and Y. Du, Adv. Sci., 2019, 6, 1901073 CrossRef CAS PubMed.
- D. U. Lee, H. W. Park, M. G. Park, V. Ismayilov and Z. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 902–910 CrossRef CAS PubMed.
- J. Hu, Q. Liu, Z. Shi, L. Zhang and H. Huang, RSC Adv., 2016, 6, 86386–86394 RSC.
- H. Wang, J. Wang, Y. Pi, Q. Shao, Y. Tan and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 2316–2320 CrossRef CAS PubMed.
- G. Chen, Y. Zhu, H. M. Chen, Z. Hu, S. F. Hung, N. Ma, J. Dai, H. J. Lin, C. T. Chen, W. Zhou and Z. Shao, Adv. Mater., 2019, 31, 1900883 CrossRef PubMed.
- T. V. Pham, H. P. Guo, W. B. Luo, S. L. Chou, J. Z. Wang and H. K. Liu, J. Mater. Chem. A, 2017, 5, 5283–5289 RSC.
- W. Xu, N. Apodaca, H. Wang, L. Yan, G. Chen, M. Zhou, D. Ding, P. Choudhury and H. Luo, ACS Catal., 2019, 9, 5074–5083 CrossRef CAS.
- L. Yan, Y. Lin, X. Yu, W. Xu, T. Salas, H. Smallidge, M. Zhou and H. Luo, ACS Appl. Mater. Interfaces, 2017, 9, 23820–23827 CrossRef CAS PubMed.
- H. Wang, W. Xu, S. Richins, K. Liaw, L. Yan, M. Zhou and H. Luo, Electrochim. Acta, 2019, 296, 945–953 CrossRef CAS.
- W. Xu, L. Yan, L. Teich, S. Liaw, M. Zhou and H. Luo, Electrochim. Acta, 2018, 273, 80–87 CrossRef CAS.
- Q. Wang, Z. Liu, H. Zhao, H. Huang, H. Jiao and Y. Du, J. Mater. Chem. A, 2018, 6, 18720–18727 RSC.
- X. Liu, W. Li, X. Zhao, Y. Liu, C.-W. Nan and L.-Z. Fan, Adv. Funct. Mater., 2019, 29, 1901510 CrossRef.
- D.-H. Nam, B. J. Taitt and K.-S. Choi, ACS Catal., 2018, 8, 1197–1206 CrossRef CAS.
- D. Zhou, Z. Cai, Y. Jia, X. Xiong, Q. Xie, S. Wang, Y. Zhang, W. Liu, H. Duan and X. Sun, Nanoscale Horiz., 2018, 3, 532–537 RSC.
- T. Komanoya, H. Kobayashi, K. Hara, W.-J. Chun and A. Fukuoka, Appl. Catal., A, 2011, 407, 188–194 CrossRef CAS.
- C. Huang, Y. Huang, C. Liu, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2019, 58, 12014–12017 CrossRef CAS PubMed.
- F. Cheng, Z. Li, L. Wang, B. Yang, J. Lu, L. Lei, T. Ma and Y. Hou, Mater. Horiz., 2021, 8, 556–564 RSC.
- X. Deng, G. Y. Xu, Y. J. Zhang, L. Wang, J. Zhang, J. F. Li, X. Z. Fu and J. L. Luo, Angew. Chem., Int. Ed., 2021, 60, 20535–20542 CrossRef CAS PubMed.
- R. Luo, Y. Li, L. Xing, N. Wang, R. Zhong, Z. Qian, C. Du, G. Yin, Y. Wang and L. Du, Appl. Catal., B, 2022, 311, 121357 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization of catalysts and supplementary experimental results. See DOI: https://doi.org/10.1039/d2nh00391k |
| This journal is © The Royal Society of Chemistry 2023 |