
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfrared spectroscopic study of solvation and size effects on reactions between water molecules and neutral rare-earth metals†
Tiantong
Wang
ab,
Shangdong
Li
ab,
Wenhui
Yan
ab,
Shuai
Jiang
ab,
Hua
Xie
 a,
Gang
Li
*a and
Ling
Jiang
*ac
a,
Gang
Li
*a and
Ling
Jiang
*ac
aState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: gli@dicp.ac.cn; ljiang@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
cHefei National Laboratory, Hefei 230088, China
First published on 26th October 2023
Abstract
Elucidating the solvation and size effects on the reactions between water and neutral metals is crucial for understanding the microscopic mechanism of the catalytic processes but has been proven to be a challenging experimental target due to the difficulty in size selection. Here, MO4H6 and M2O6H7 (M = Sc, Y, La) complexes were synthesized using a laser–vaporization cluster source and characterized by size-specific infrared-vacuum ultraviolet spectroscopy combined with quantum chemical calculations. The MO4H6 and M2O6H7 complexes were found to have H˙M(OH)3(H2O) and M2(μ2-OH)2(η1-OH)3(η1-OH2) structures, respectively. A combination of experiments and theory revealed that the formation of H˙M(OH)3(H2O) and M2(μ2-OH)2(η1-OH)3(η1-OH2) is both thermodynamically exothermic and kinetically facile in the gas phase. The results indicated that upon the addition of water to H˙M(OH)3, the feature of the hydrogen radical is retained. In the processes from mononuclear H˙M(OH)3 to binuclear M2(μ2-OH)2(η1-OH)3(η1-OH2), the active hydrogen atom undergoes the evolution from hydrogen radical → bridging hydrogen → metal hydride → hydrogen bond, which is indicative of a reduced reactivity. The present system serves as a model for clarifying the solvation and size effects on the reactions between water and neutral rare-earth metals and offers a general paradigm for systematic studies on a broad class of the reactions between small molecules and metals at the nanoscale.
1 Introduction
Rare-earth metals (Sc, Y, Ln) and their complexes have been widely used in various catalytic processes.1–3 For instance, Ni@CeO2-doped MgH2 hydrogen storage material reduces the dehydrogenation activation energy by 20.3 kcal mol−1, which significantly improves the hydrogen release property of MgH2 material.4–6 Rare-earth single-atom catalysts (SAC) are extensively exploited in the photocatalytic/electrocatalytic processes of O2/CO2/N2, etc.7–12 Theoretical studies have verified that in the electrocatalytic reduction of O2, the rare-earth SAC undergoes the transformation from the poly-hydroxyl structure to the solvated water structure.9,13 CeO2 is utilized as the catalyst in the hydrogenation reduction of alkenes/alkynes,14 in which the key intermediates contain important metal hydride structures and poly-hydroxyl structures. Ultrathin 2D rare-earth nanomaterials of the layered rare-earth hydroxides were synthesized in solution15,16 and were confirmed to exhibit great potential in various applications, such as optoelectronics,17 magnetic devices,18 and high-efficiency catalysts.19 Therefore, the studies on the reactions between small molecules and rare-earth metals at the cluster scale are expected to provide microscopic information to unravel the structure–reactivity relationship of catalysts.Various efforts have been devoted to the spectroscopic studies of the reactions of water molecules with rare-earth metals20–27 owing to their important roles in renewable sources and transport/storage of hydrogen energy.28,29 The investigation of the solvation and size effects on the reactions between water with neutral metals is crucial to understanding the microscopic mechanism of catalytic processes but has been proven to be a challenging experimental target due to the difficulty in size selection. Matrix-isolation infrared spectroscopy has provided valuable insights into the reactions of neutral single rare-earth metal atoms with water, suggesting the formation of the inserted HMOH structures and the solvated M(H2O)1–2 structures.20,24,26,27 Recently, we have characterized two different types of rare-earth hydroxides (MO3H4 and M2O4H4 (M = Sc, Y, La))30,31 using size-specific infrared-vacuum ultraviolet (IR-VUV) spectroscopy, in which MO3H4 has a hydrogen radical structure and M2O4H4 has a water-split structure. In this work, we increased the number of water molecules to study the solvation and size effects on the reactions between water with neutral rare-earth metals. Two new rare-earth metal hydroxides with the chemical formulas MO4H6 and M2O6H7 (M = Sc, Y, La) were prepared and characterized by IR-VUV spectroscopy and quantum chemical calculation. MO4H6 and M2O6H7 complexes were found to have the H˙M(OH)3(H2O) and M2(μ2-OH)2(η1-OH)3(η1-OH2) structures, respectively, in which the hydrogen radical features were retained with the solvation of water in the mononuclear complex MO4H6 and the reactivity of the active hydrogen was reduced in the formation processes of binuclear complex M2O6H7.
2 Experimental and theoretical methods
The experiment was performed using the IR-VUV apparatus, which has been described in detail in previous reports.32–34 Neutral MO4H6 and M2O6H7 (M = Sc, Y, La) complexes were prepared using the laser–vaporization cluster source in a pulsed supersonic expansion of 0.2% H2O/helium. The second harmonic of a Nd: YAG laser (532 nm) was employed to ablate the metal target, with a pulse energy of 1.0 mJ. The reaction gas was introduced using a Parker pulse valve (Series 009), with a pulse width of 180 μs. The molecular beams that passed through a 4 mm-diameter skimmer were ionized by a VUV laser and then detected by a reflectron time-of-flight mass spectrometer (TOF-MS).VUV light with a wavelength of 193 nm was produced by an ArF excimer laser (COMPex 50 F, Coherent, Germany), with an energy of 14 mJ per pulse. The infrared laser was delivered by a potassium titanyl phosphate/potassium titanyl arsenate optical parametric oscillator/amplifier system (OPO/OPA, LaserVision) pumped by another seeded Nd: YAG laser (Continuum, Surelite EX). The OPO/OPA system was tunable from 700 to 7000 cm−1, with a line width of 1 cm−1.
The pulse valve, the vaporization laser, and VUV light were operated at 20 Hz. The tunable IR light pulse was introduced at approximately 150 ns prior to the VUV pulse in a crossed manner and operated at 10 Hz. When the resonant vibrational transition was irradiated by the IR laser light and caused vibrational predissociation, a depletion of a selected neutral cluster mass signal was detected. The infrared spectra were recorded from the difference between the mass spectral signals with and without the infrared laser (IR laser ON minus IR laser OFF). The IR spectra of the size-selected neutral MO4H6 and M2O6H7 complexes were obtained from the depletion spectrum of the ion signal intensity as a function of the IR wavelength. Typical spectra were recorded by scanning the IR wavelength in the steps of 2 cm−1 and averaging over 400 laser shots at each wavelength. The infrared wavelength was calibrated using a commercial wavelength meter (HighFinesse GmbH, WS6-200 VIS IR).
Quantum chemical calculations were performed using the Gaussian 16 package35 at the TPSSh/def2-TZVP level of theory. Our test calculations indicated that the TPSSh functional has a lower computational cost than B2PLYPD3 and a better computational performance in the spectra and energetics than B3LYP for MO4H6 and M2O6H7 (M = Sc, Y, La). Accordingly, we chose the TPSSh functional instead of the B2PLYPD3 or B3LYP functionals that we used in previous works.30,31 For the possible isomerization paths, the initial structures of the transition states were constructed manually and optimized in the Berny algorithm. Intrinsic reaction coordinates (IRC) of all the transition states were carried out to confirm that the transition states were connected to the initial and final states. The relative energies and energy barriers were calculated for 0 K structures, including the zero-point vibrational corrections. The harmonic vibrational frequencies were scaled by a factor of 0.968 in order to account for the method-dependent systematic errors.36 The resulting stick spectra were convoluted by a Gaussian line shape function with a 15 cm−1 width at half-maximum (FWHM).
3 Results and discussion
As exemplified by the Sc + H2O reaction, the mass spectrum of the species is shown in Fig. S1.† The experimental IR spectra of neutral MO4H6 and M2O6H7 (M = Sc, Y, La) complexes in the OH stretching region are shown in Fig. 1a–c and 2a–c, respectively. The experimental band positions of MO4H6 and M2O6H7 (M = Sc, Y, La) are listed in Tables S1 and S2,† respectively. As shown in Fig. 1a–c, the experimental IR spectrum of each MO4H6 complex comprises three groups of bands (labeled bands A, B, and C). B and A in the IR spectra of ScO4H6, YO4H6, and LaO4H6 are centered at 3398, 3104, and 2952 cm−1, respectively, indicating redshift down through the group 3 metal of the periodic table. The B bands in the IR spectrum of ScO4H6, YO4H6, and LaO4H6 are observed at 3694, 3704, and 3722 cm−1, respectively, showing a slight blueshift down through the group 3 metal of the periodic table. B and C in the IR spectra of ScO4H6, YO4H6, and LaO4H6 are located at 3786, 3784, and 3748 cm−1, respectively, showing a slight redshift down through the group 3 metals of the periodic table. Similar spectral features were observed for M2O6H7 (labeled bands a, b, and c), except for the absence of band a in Sc2O6H7.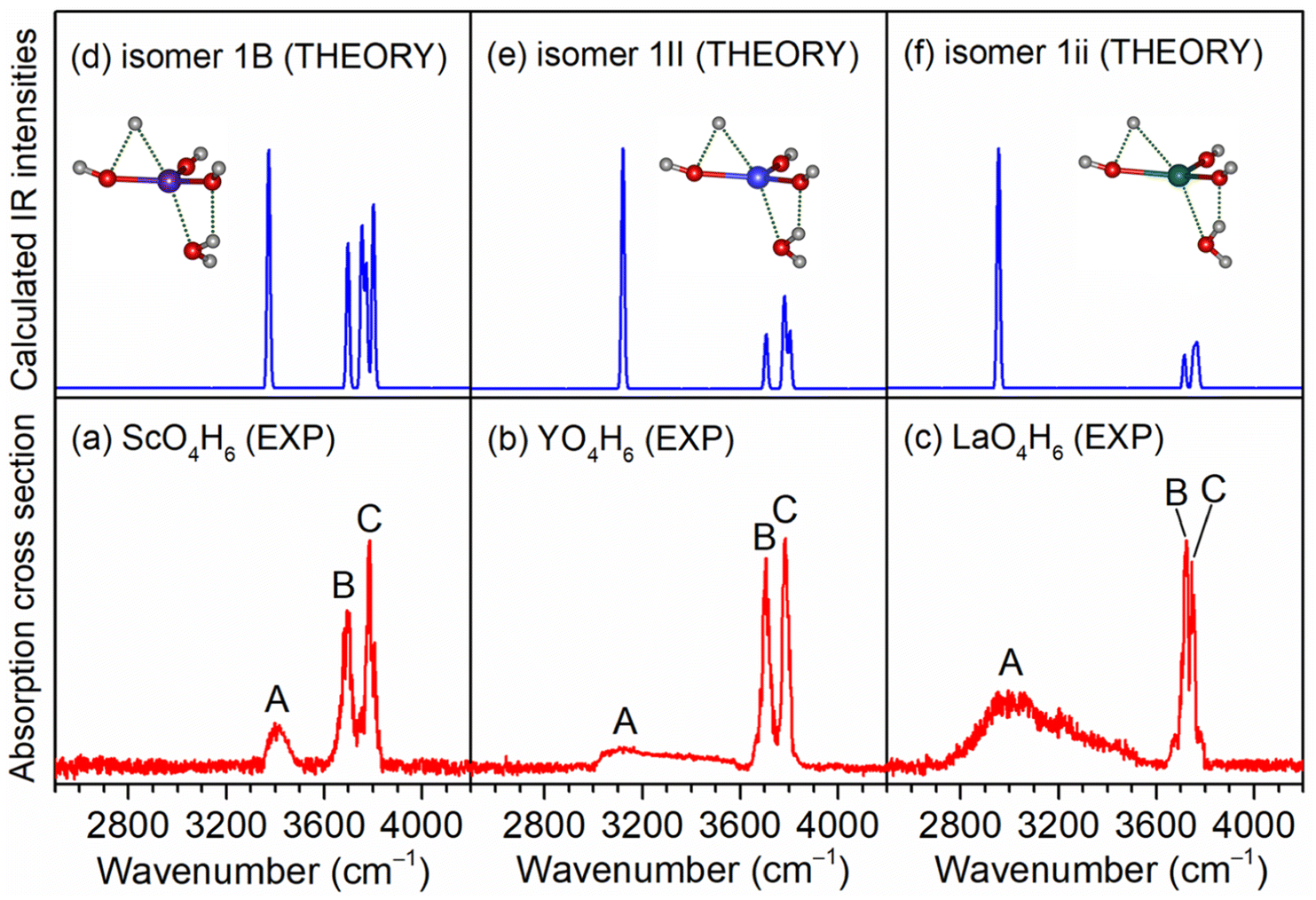 | ||
| Fig. 1 Comparison of the experimental IR spectra of neutral MO4H6 complexes (M = Sc, Y, La) (a–c) and calculated IR spectra of isomers 1B, 1II, and 1ii (d–f). Calculations were performed at the TPSSh/def2-TZVP level of theory, with the harmonic vibrational frequencies scaled by a factor of 0.968. The structures are embedded in the inset (O, red; H, light gray; Sc, purple; Y, blue; La, olive). | ||
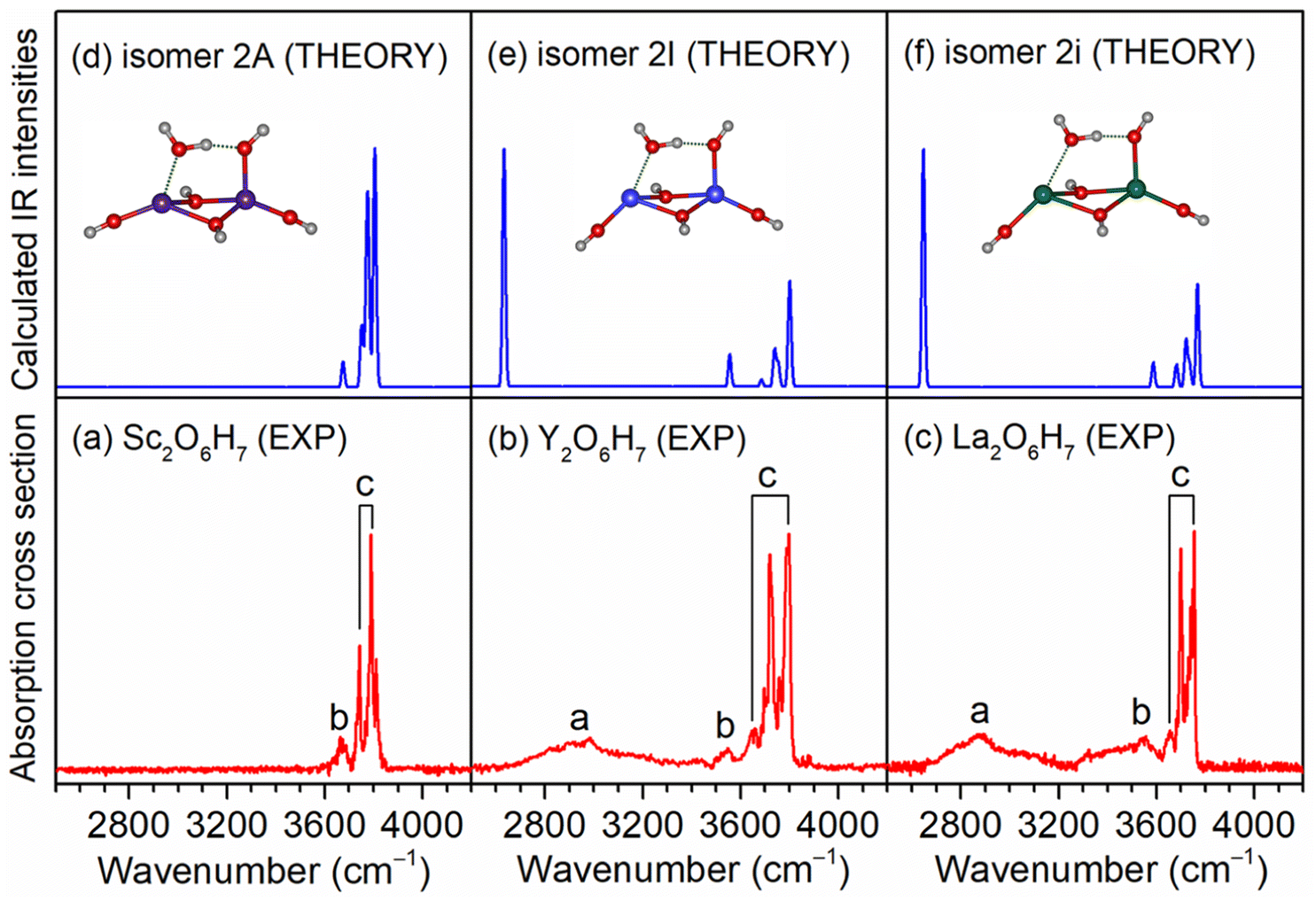 | ||
| Fig. 2 Comparison of the experimental IR spectra of neutral M2O6H7 complexes (M = Sc, Y, La) (a–c) and the calculated IR spectra of isomers 2A, 2I, and 2i (d–f). Calculations were performed at the TPSSh/def2-TZVP level of theory, with the harmonic vibrational frequencies scaled by a factor of 0.968. The structures are embedded in the inset (O, red; H, light gray; Sc, purple; Y, blue; La, olive). | ||
To understand the experimental IR spectra and identify the structures of MO4H6 and M2O6H7 (M = Sc, Y, La) complexes, quantum chemical calculations were carried out at the TPSSh/def2-TZVP level of theory. The structures of the identified isomers and the comparison of their calculated IR spectra with the experimental ones are shown in Fig. 1 and 2, respectively. The structures of other low-lying isomers and the comparison of their calculated IR spectra with the experimental ones are illustrated in Fig. S2 and S3,† respectively.
As shown in Fig. S2,† the most stable isomer of ScO4H6 (labeled 1A) has a Sc(OH)2(H2O)2 structure with a doublet electronic ground state, in which the scandium hydroxide Sc(OH)2 are solvated with two water molecules. The 1B isomer has a H˙Sc(OH)3(H2O) structure with the involvement of a hydrogen radical, as evidenced by the calculated Mulliken spin densities [H(0), 0.72; Sc(0), 0.10] (for the labeling of the atoms, see Table S1†). The 1B isomer lies slightly higher in energy than the 1A isomer by 2.6 kcal mol−1. The 1C isomer lies 36.0 kcal mol−1 above the 1A isomer and has a ScO(H2O)3 structure, in which three water molecules are weakly bound to ScO. It can be seen from Fig. S2† that the calculated IR spectrum of the isomer 1B agrees best with the experimental one. As listed in Table S1,† the calculated band at 3374 cm−1 is attributed to the symmetric stretching mode of the O(4)H(4) and O(4)H(5) groups, which is consistent with the experimental band A (3398 cm−1); the antisymmetric stretching mode of the O(4)H(4) and O(4)H(5) groups was calculated to be 3698 cm−1, which is in accordance with the experimental band B (3694 cm−1); the stretching modes of the O(1)H(1), O(2)H(2), and O(3)H(3) groups were calculated to 3802, 3772, and 3758 cm−1, respectively, which were observed as one band in the experimental spectrum (band C, 3786 cm−1). However, the calculated IR spectrum of the isomer 1A (Fig. S1b†) consists of the bands at 2693 and 2955 cm−1, corresponding to the hydrogen-bonded OH stretching modes of the solvated water molecules, which are not observed experimentally. Analogously, the presence of isomer 1C can be ruled out based on the calculated band at 2640 cm−1. Similar results are also obtained for the YO4H6 and LaO4H6 complexes (Fig. S2 and Table S1†). As summarized in Fig. 1, the agreement of the simulated IR spectra of the 1B, 1II, and 1ii isomers with the experimental spectra of ScO4H6, YO4H6, and LaO4H6 is reasonable to identify the presence of H˙M(OH)3(H2O) (M = Sc, Y, La) structures.
For Sc2O6H7, the most stable isomer (labeled 2A) has a Sc2(μ2-OH)2(η1-OH)3(η1-OH2) structure with a doublet electronic ground state (Fig. S3d†), in which one water molecule forms one weakly bond with one scandium hydroxide and one hydrogen-bond with the hydroxyl group of another scandium hydroxide. The 2B isomer has a similar structure and lies only 0.1 kcal mol−1 above 2A, in which one water molecule forms one weak bond with one scandium hydroxide and one hydrogen bond with the hydroxyl group of the same scandium hydroxide. In the calculated IR spectrum of isomer 2A (Fig. S3 and Table S2†), the band at 3675 cm−1 is attributed to the stretching mode of the O(6)H(6) group, which is consistent with the experimental band b (3662 cm−1); the stretching modes of the O(n)H(n) (n = 1–5) groups is calculated to 3806, 3804, 3778, 3770, and 3751 cm−1, respectively, which are observed in the experimental spectrum at 3810, 3788, and 3742 cm−1 (band c). The absence of stretching mode of the O(6)H(7) group (2474 cm−1) (Table S2†) could be rationalized that the photon energy of the IR laser in this wavelength region is not sufficient to cause the vibrational predissociation of Sc2O6H7. In the calculated IR spectrum of the isomer 2B (Fig. S3g†), the hydrogen-bonded OH stretching mode of the solvated water molecule is predicted at 3275 cm−1, which is not observed in the experimental spectrum. As summarized in Fig. 2, the agreement of the simulated IR spectra of the 2A, 2I, and 2i isomers with the experimental spectra is reasonable to confirm the M2(μ2-OH)2(η1-OH)3(η1-OH2) (M = Sc, Y, La) structures responsible for Sc2O6H7, Y2O6H7, and La2O6H7.
Considering that the reactions in the laver–vaporization processes are quite intricate and their intermediates are very difficult to characterize experimentally, quantum chemical calculations were performed to explore the possible reaction mechanisms. Due to the similarities in the IR spectra and structures of MO4H6 and M2O6H7 (M = Sc, Y, La) complexes, we used ScO4H6 and Sc2O6H7 as examples to investigate the possible formation pathways. Previous studies have demonstrated that the Sc + H2O → Sc(H2O) reaction is more favorable than the dimerization of Sc atoms.31 As shown in Fig. S1,† the mononuclear complexes (i.e., Sc, ScO, HScO, Sc(OH)2, and H˙Sc(OH)3) were observed in the mass spectrum, for which the formation mechanisms have been reported previously.20,23,25,30 Consequently, we will mainly discuss the dominant pathways that begin from the mononuclear complexes.
The potential energy profiles for the formation of ScO4H6 (isomer 1B) initiated by the reaction of ScO with H2O are shown in Fig. 3. ScO reacts with the first water molecule to form a solvation structure ScO(H2O), which is predicted to be exothermic by 18.5 kcal mol−1 at the TPSSh/def2-TZVP level of theory. The ScO(H2O) → Sc(OH)2 isomerization is highly exothermic by 41.3 kcal mol−1via a transition state (TS1) with a very small barrier of 1.1 kcal mol−1. The addition of H2O to Sc(OH)2 forms Sc(OH)2(H2O), which could undergo isomerization to H˙Sc(OH)3. TS2 lies below the energy of Sc(OH)2 + H2O reactants by 14.4 kcal mol−1. Sc(OH)2 + H2O → H˙Sc(OH)3 overall reaction is exothermic by 16.1 kcal mol−1 and is thus thermodynamically favorable. The mechanism for kinetically trapping H˙Sc(OH)3 by the soft helium expansion has been recently discussed in detail.30 The H˙Sc(OH)3 complex combines with the water molecule to form the target product ScO4H6 (H˙Sc(OH)3(H2O), isomer 1B), which is exothermic by 16.3 kcal mol−1.
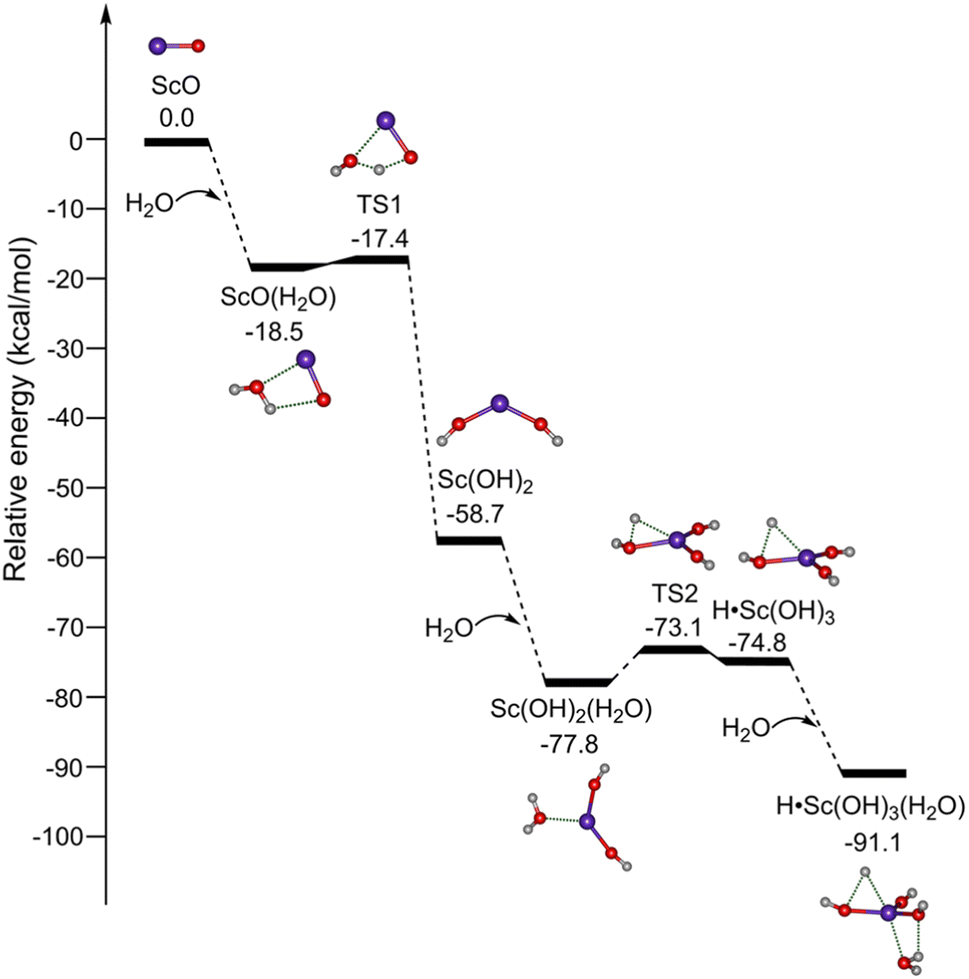 | ||
| Fig. 3 Potential energy profiles for the formation of ScO4H6 (H˙Sc(OH)3(H2O), isomer 1B). The abbreviation “TS” denotes the transition state. Calculations were performed at the TPSSh/def2-TZVP level of theory. Corresponding structures are embedded in the inset (O, red; H, light gray; Sc, purple). | ||
Fig. 4 shows the potential energy profiles for the formation of Sc2O6H7 (isomer 2A) initiated from the mononuclear non-radical complexes. The ScO + HScO → HSc2O2 reaction is predicted to be extremely exothermic at 106.1 kcal mol−1. The most stable isomer of HSc2O2 has a Sc2(μ2-O)2(η1-H) structure, which combines with one water molecule to form IM1 and IM2. IM1 can be isomerized to IM5 via TS4. The calculated energy of TS4 is lower than that of IM1, which is due to the zero-point energy correction.37–39 This indicates that the energy barrier for the IM1 → IM5 isomerization is very low. IM5 undergoes dehydrogenation to form IM6 with a slightly endothermic value of 1.1 kcal mol−1, which could be compensated by the highly exothermic overall reaction of HSc2O2 + H2O → IM6 + H2. Similarly, the reaction path of IM2 → TS3 → IM3 → TS5 → IM4 → IM6 is also both thermodynamically and kinetically facile in the gas phase. IM3 could also be generated by the combination of Sc(OH)2 and HScO, which is predicted to be highly exothermic by 83.8 kcal mol−1. The addition of H2O to IM6 forms IM7, which is exothermic by 16.8 kcal mol−1. IM7 undergoes isomerization to the most stable hydroxyl structure IM8 via TS6 with a tiny barrier of 0.9 kcal mol−1. IM8 reacts with the second water molecule to form IM9, which is exothermic by 20.7 kcal mol−1. IM9 can be isomerized to an all-hydroxyl structure IM10 via TS7 with a barrier of 8.3 kcal mol−1. The addition of the third water molecule to IM10 generates the target product Sc2O6H7 (isomer 2A), which is exothermic by 14.6 kcal mol−1.
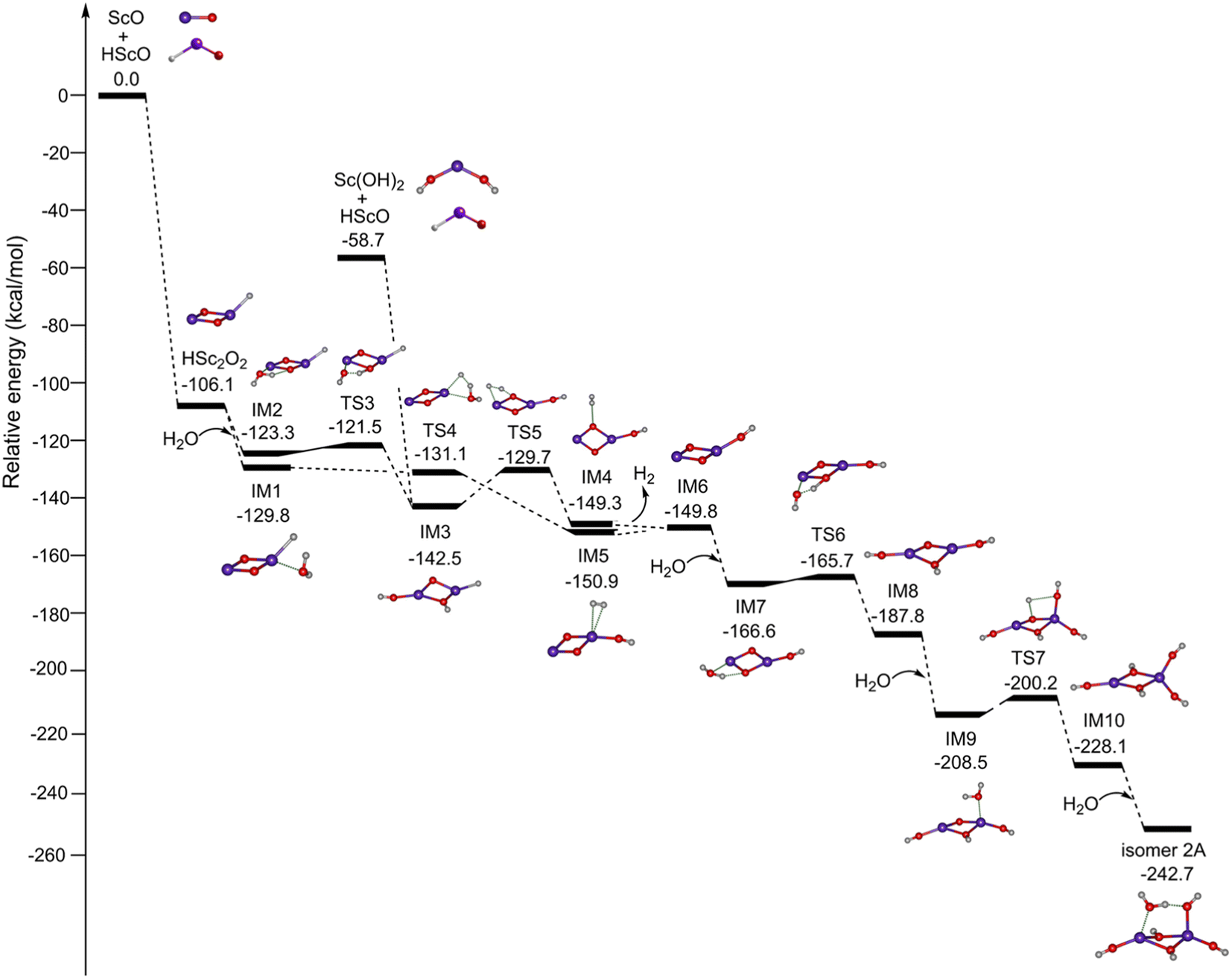 | ||
| Fig. 4 Potential energy profiles for the formation of Sc2O6H7 (isomer 2A) initiated from the mononuclear non-radical complexes. The abbreviation “IM” stands for intermediate and “TS” for the transition state. Calculations were performed at the TPSSh/def2-TZVP level of theory. Corresponding structures are embedded in the inset (O, red; H, light gray; Sc, purple). | ||
Considering that H˙Sc(OH)3 and OH radicals are also generated in the laser–vaporization process,30,34 Sc2O6H7 might also be formed via reactions (1) and (2).
| H˙Sc(OH)3 + Sc → Sc2O3H4 | (1) |
| Sc2O3H4 + 3OH → Sc2O6H7 | (2) |
Accordingly, the potential energy profiles for the formation of Sc2O6H7 initiated from the mononuclear radical complexes are shown in Fig. 5. H˙Sc(OH)3 reacts with the Sc atom to produce the binuclear complex IM11; the reaction is predicted to be highly exothermic by 97.3 kcal mol−1. IM11 can be isomerized to IM12 via TS8 with a small barrier of 1.6 kcal mol−1. The reaction of IM12 with the first, second, and third OH radicals is predicted to be extremely exothermic by 128.4, 122.2, and 51.3 kcal mol−1, respectively. The subsequent isomerization from IM15 to isomer 2B is exothermic by 1.2 kcal mol−1via TS9 with a barrier of 9.3 kcal mol−1. Isomer 2B can be further isomerized to form the most stable isomer 2A via TS10 with a small barrier of 3.8 kcal mol−1. Consequently, the formation of isomer 2A is both thermodynamically exothermic and kinetically facile in the gas phase, which is consistent with the experimental observation.
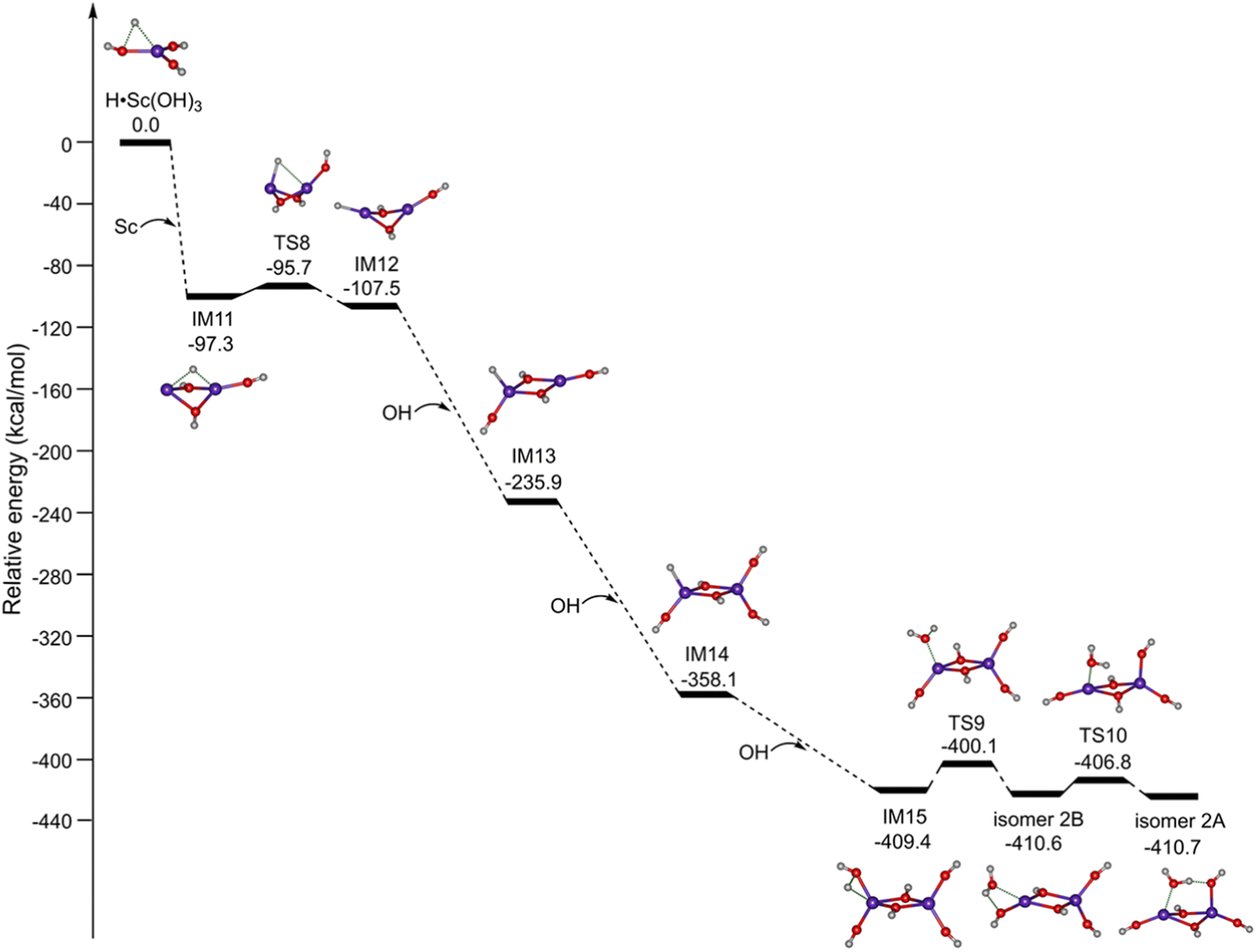 | ||
| Fig. 5 Potential energy profiles for the formation of Sc2O6H7 (isomer 2A) initiated from the mononuclear radical complexes. The abbreviation “IM” stands for intermediate and “TS” for the transition state. Calculations were performed at the TPSSh/def2-TZVP level of theory. Corresponding structures are embedded in the inset (O, red; H, light gray; Sc, purple). | ||
The reaction mechanisms of rare-earth metals with water molecules can be inferred from the aforementioned formation pathways of ScO4H6 and Sc2O6H7. Mononuclear rare-earth metal oxide (MO (M = Sc, Y, La)) could induce the decomposition of water molecules until the hydrogen-radical adduct H˙M(OH)3 was generated. The addition of the subsequent water molecule forms the solvated complex H˙M(OH)3(H2O). The binuclear complex M2O3H (labeled IM6 in Fig. 4) might also induce the successive decomposition of water molecules to produce the poly-hydroxyl complex M2O5H5 (labeled IM10 in Fig. 4). The additional water molecule forms a coordination bond with metals and a delocalized hydrogen-bond with the hydroxyl group.
The evolution mechanism from mononuclear to binuclear rare-earth metal complexes might be summarized as follows. The addition of water to H˙M(OH)3 forms H˙M(OH)3(H2O), in which the radical feature is retained. However, upon the addition of the Sc atom to MO3H4 to produce binuclear complex M2O3H4 (labeled IM11 in Fig. 5), the hydrogen radical first binds to the two metals and then undergoes isomerization to a hydride structure (labeled IM12 in Fig. 5). During the reactions of IM12 with the first two OH radicals, the hydride structures remain. The subsequent reaction with the third OH radical results in the formation of M2O6H7, in which the hydrogen atom in the metal hydride unit is transformed into one hydrogen bond. Therefore, during the MO3H4 → M2O6H7 processes, the active hydrogen atom undergoes the evolution of the hydrogen radical → bridging hydrogen → metal hydride → hydrogen bond, indicative of reduced reactivity.
The mononuclear complexes MO4H6 (M = Sc, Y, La) with an M(OH)3 plane as its basic skeleton, are highly relevant to the ultrathin 2D rare-earth nanomaterials of layered rare-earth hydroxides M(OH)3·nH2O,15,17 and the coordination bonding fashion of water with M(OH)3 is similar to that of the reported rare-earth single-atom catalysis system.9,10 The hydrogen radical is the key intermediate in a variety of processes (i.e., catalysis, molecular hydrogen production, and new particle formation), but it is difficult to detect because of its high reactivity and a very short lifetime. In this work, the feature of hydrogen radicals in the rare-earth single-atom complexes H˙M(OH)3(H2O) is fortunately observed under the presence of additional water, which would have important implications for understanding the solvation effects of the catalysts and would suggest that the ultrathin 2D rare-earth nanomaterials of layered rare-earth hydroxides have the potential to trap hydrogen radicals and participate in more chemical processes.
The binuclear complexes M2O6H7 (M = Sc, Y, La) contain an M2(μ2-O)2 unit, which is reminiscent of the actual rare-earth metal oxide catalysts. In the formation of M2O6H7 from MO3H4, three bonding modes of hydrogen atoms are involved with the reactivity order of η1-H > μ2-OH > η1-OH. Such reduced reactivity of hydrogen atoms might contribute to reducing the activation energy of the dehydrogenation reaction of the hydrogen storage materials.4–6 These three types of hydrogen atoms were also observed in the process of ethylene/acetylene hydrogenation catalyzed by CeO2,14 and the reactivity sequence was consistent with our results. The deactivation of the CeO2 catalyst has been rationalized by the overproduction with the formation of strongly bound M–OH groups that are difficult to remove from the surface.14 Our findings imply that the formation of M2(η1-OH)(η1-OH2) unit or hydrogen-bonding networks on the surfaces may also deactivate the catalysts, which provides insights into the strategy for tuning the active sites.
4 Conclusions
In this work, MO4H6 and M2O6H7 (M = Sc, Y, La) complexes were prepared by a laser–vaporization supersonic expansion cluster source. Infrared-vacuum ultraviolet (IR-VUV) spectroscopy combined with quantum chemical calculations indicates that the MO4H6 and M2O6H7 complexes have the H˙M(OH)3(H2O) and M2(μ2-OH)2(η1-OH)3(η1-OH2) structures, respectively. The joint experimental and theoretical results reveal that the formation of H˙M(OH)3(H2O) and M2(μ2-OH)2(η1-OH)3(η1-OH2) is both thermodynamically exothermic and kinetically facile in the gas phase. The feature of hydrogen radicals in the mononuclear complexes H˙M(OH)3(H2O) is found to be retained under the presence of additional water. The addition of a metal atom to H˙M(OH)3 and subsequent combination with water produces the binuclear complex M2(μ2-OH)2(η1-OH)3(η1-OH2), by which the reactivity of active hydrogen is reduced during the evolution of hydrogen radical → bridging hydrogen → metal hydride → hydrogen bond. The present experimental technique has the potential for size-selected infrared spectroscopic studies on a large variety of reactions between small molecules and neutral clusters, which will help to understand the structure–reactivity relationship of catalysts with isolated metal atoms/clusters dispersed on supports.Author contributions
Ling Jiang and Gang Li conceived the idea, supervised the project and presented the project outline, guided the entire experiment, and provided funding support. Tiantong Wang conducted the experiments, calculations, and data analysis and wrote the original draft. Shangdong Li, Wenhui Yan, and Shuai Jiang conducted the experiments and data analysis. Hua Xie provided some advice for the characterization and analysis. All authors discussed the results, commented on the manuscript, and contributed to the final polishing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Dalian Coherent Light Source (DCLS) and Specreation Co., Ltd., for their support and assistance. This work was supported by the National Natural Science Foundation of China (grant no. 22125303, 92061203, 22373102, 22103082, 22273101, 22288201, and 21327901), the National Key Research and Development Program of China (2021YFA1400501), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (CAS) (2020187), Innovation Program for Quantum Science and Technology (2021ZD0303304), CAS (GJJSTD20220001), Dalian Institute of Chemical Physics (DICP DCLS201702), and International Partnership Program of CAS (121421KYSB20170012).Notes and references
- M. Zimmermann and R. Anwander, Homoleptic Rare-Earth Metal Complexes Containing Ln-C σ-Bonds, Chem. Rev., 2010, 110, 6194–6259 CrossRef CAS PubMed.
- F. Saraci, V. Quezada-Novoa, P. R. Donnarumma and A. J. Howarth, Rare-Earth Metal–Organic Frameworks: From Structure to Applications, Chem. Soc. Rev., 2020, 49, 7949–7977 RSC.
- A. Mehtab, J. Ahmed, S. M. Alshehri, Y. Mao and T. Ahmad, Rare Earth Doped Metal Oxide Nanoparticles for Photocatalysis: A Perspective, Nanotechnology, 2022, 33, 142001 CrossRef PubMed.
- K. Iwase, K. Sakaki, Y. Nakamura and E. Akiba, In Situ XRD Study of La2Ni7H(X) during Hydrogen Absorption–Desorption, Inorg. Chem., 2013, 52, 10105–10111 CrossRef CAS PubMed.
- Z. Yu, W. Zhang, Y. Zhang, Y. Fu, Y. Cheng, S. Guo, Y. Li and S. Han, Remarkable Kinetics of Novel Ni@CeO2–MgH2 Hydrogen Storage Composite, Int. J. Hydrogen Energy, 2022, 47, 35352–35364 CrossRef CAS.
- H. Zhang, L. Bao, Y. Pan and J.-Y. Ge, Enhanced Hydrogen Storage Performances of Binary Mg-Y Nanoscale Particles, Chem. Phys. Lett., 2022, 796, 139573 CrossRef CAS.
- X. Wang, Y. Zhu, H. Li, J. M. Lee, Y. Tang and G. Fu, Rare-Earth Single-Atom Catalysts: A New Frontier in Photo/Electrocatalysis, Small Methods, 2022, 6, e2200413 CrossRef PubMed.
- S. Nundy, D. Tatar, J. Kojčinović, H. Ullah, A. Ghosh, T. K. Mallick, R. Meinusch, B. M. Smarsly, A. A. Tahir and I. Djerdj, Bandgap Engineering in Novel Fluorite-Type Rare Earth High-Entropy Oxides (RE-HEOs) with Computational and Experimental Validation for Photocatalytic Water Splitting Applications, Adv. Sustain. Syst., 2022, 6, 2200067 CrossRef CAS.
- F. He, H. Li, Y. Ding, K. Li, Y. Wang and Z. Wu, The Oxygen Reduction Reaction on Graphitic Carbon Nitride Supported Single Ce Atom and CexPt6−X Cluster Catalysts from First-Principles, Carbon, 2018, 130, 636–644 CrossRef CAS.
- J. Liu, X. Kong, L. Zheng, X. Guo, X. Liu and J. Shui, Rare Earth Single-Atom Catalysts for Nitrogen and Carbon Dioxide Reduction, ACS Nano, 2020, 14, 1093–1101 CrossRef CAS PubMed.
- J. Feng, X. Zhang, J. Wang, X. Ju, L. Liu and P. Chen, Applications of Rare Earth Oxides in Catalytic Ammonia Synthesis and Decomposition, Catal. Sci. Technol., 2021, 11, 6330–6343 RSC.
- X. Liu, Y. Luo, C. Ling, Y. Shi, G. Zhan, H. Li, H. Gu, K. Wei, F. Guo, Z. Ai and L. Zhang, Rare Earth La Single Atoms Supported MoO3−X for Efficient Photocatalytic Nitrogen Fixation, Appl. Catal., B, 2022, 301, 120766 CrossRef CAS.
- N. Liu, D. Cao, W. Liu, H. Zhang, Y. Zhu, L. Chang, D. Wu and D. Cheng, Constructing La-Doped Ultrathin Co-Based Nanostructured Electrocatalysts for High-Performance Water Oxidation Process, Int. J. Hydrogen Energy, 2022, 47, 14504–14514 CrossRef CAS.
- J. Moon, Y. Cheng, L. L. Daemen, M. Li, F. Polo-Garzon, A. J. Ramirez-Cuesta and Z. Wu, Discriminating the Role of Surface Hydride and Hydroxyl for Acetylene Semihydrogenation over Ceria through In Situ Neutron and Infrared Spectroscopy, ACS Catal., 2020, 10, 5278–5287 CrossRef CAS.
- J. Xu, X. Chen, Y. Xu, Y. Du and C. Yan, Ultrathin 2D Rare-Earth Nanomaterials: Compositions, Syntheses, and Applications, Adv. Mater., 2020, 32, 1806461 CrossRef CAS PubMed.
- B.-I. Lee, H. Jeong, J.-S. Bae and S.-H. Byeon, A Layered Polymorph of Rare Earth Hydroxides, Chem. Commun., 2013, 49, 6051–6053 RSC.
- Y.-S. Yoon, S.-H. Byeon and I. S. Lee, Unexplored Thermal Transformation Behavior of Two-Dimensionally Bound Gadolinium Hydroxide Layers: Fabrication of Oriented Crystalline Films of Gadolinium Oxychloride Nanosheets Suitable for the Multicolor Luminescence with Color Tunability, Adv. Mater., 2010, 22, 3272–3276 CrossRef CAS PubMed.
- G. Abellan, G. Minguez Espallargas, G. Lorusso, M. Evangelisti and E. Coronado, Layered Gadolinium Hydroxides for Low-temperature Magnetic Cooling, Chem. Commun., 2015, 51, 14207–14210 RSC.
- F. Gandara, J. Perles, N. Snejko, M. Iglesias, B. Gomez-Lor, E. Gutierrez-Puebla and M. A. Monge, Layered Rare-Earth Hydroxides: A Class of Pillared Crystalline Compounds for Intercalation Chemistry, Angew. Chem., Int. Ed., 2006, 45, 7998–8001 CrossRef CAS PubMed.
- L. Zhang, J. Dong and M. Zhou, Matrix-Isolation Fourier Transform Infrared and Theoretical Studies of Laser-Ablated Sc Atom Reactions with Water Molecules, J. Phys. Chem. A, 2000, 104, 8882–8886 CrossRef CAS.
- J. Guo and J. M. Goodings, A Density Functional Study of The Structures and Ionization Energies of Some Scandium Compounds with Hydrogen and Oxygen, J. Mol. Struct.: THEOCHEM, 2001, 549, 261–273 CrossRef CAS.
- J. Guo and J. M. Goodings, A Density-Functional Study of the Reaction of Neutral Scandium with Water, Chem. Phys. Lett., 2001, 342, 169–176 CrossRef CAS.
- D.-Y. Hwang and A. M. Mebel, Theoretical Study of the Reaction Mechanism of ScO with Molecular Hydrogen, Chem. Phys. Lett., 2001, 341, 393–399 CrossRef CAS.
- L. Zhang, L. Shao and M. Zhou, Reactions of Laser-Ablated Y and La Atoms With H2O Infrared Spectra and Density Functional Calculations of The HMO, HMOH and M(OH)2 Molecules in Solid Argon, Chem. Phys., 2001, 272, 27–36 CrossRef CAS.
- X. Wang and L. Andrews, Infrared Spectra and Density Functional Calculations for The Sc(OH)2,3 and HOScO Molecules and the Sc(OH)2+ Cation in Solid Argon, J. Phys. Chem. A, 2006, 110, 1850–1858 CrossRef CAS PubMed.
- J. Xu and M. Zhou, Reactions of Early Lanthanide Metal Atoms (Nd, Sm, Eu) with Water Molecules. A Matrix Isolation Infrared Spectroscopic and Theoretical Study, J. Phys. Chem. A, 2006, 110, 10575–10582 CrossRef CAS PubMed.
- J. Xu, X. Jin and M. Zhou, Reactions of Late Lanthanide Metal Atoms with Water Molecules: A Matrix Isolation Infrared Spectroscopic and Theoretical Study, J. Phys. Chem. A, 2007, 111, 7105–7111 Search PubMed.
- A. Fujishima and K. Honda, Electrochemical Photolysis of Water at a Semiconductor Electrode, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- A. Ali, F. Long and P. K. Shen, Innovative Strategies for Overall Water Splitting Using Nanostructured Transition Metal Electrocatalysts, Electrochem. Energy Rev., 2023, 6, 1 Search PubMed.
- S. Jiang, H. Zheng, W. Yan, T. Wang, C. Wang, S. Li, H. Xie, G. Li, X. Zheng, H. Fan, X. Yang and L. Jiang, Capturing Hydrogen Radicals by Neutral Metal Hydroxides, J. Phys. Chem. Lett., 2023, 14, 2481–2486 CrossRef CAS PubMed.
- S. Jiang, H. Zheng, W. Yan, T. Wang, C. Wang, Y. Zhao, H. Xie, G. Li, X. Zheng, H. Fan and L. Jiang, Spectroscopic Identification of Water Splitting by Neutral Group 3 Metals, Chin. Chem. Lett., 2023, 34, 108244 CrossRef CAS.
- B. Zhang, Y. Yu, Z. Zhang, Y. Y. Zhang, S. Jiang, Q. Li, S. Yang, H. S. Hu, W. Zhang, D. Dai, G. Wu, J. Li, D. H. Zhang, X. Yang and L. Jiang, Infrared Spectroscopy of Neutral Water Dimer Based on a Tunable Vacuum Ultraviolet Free Electron Laser, J. Phys. Chem. Lett., 2020, 11, 851–855 CrossRef CAS PubMed.
- G. Li, C. Wang, H.-J. Zheng, T.-T. Wang, H. Xie, X.-M. Yang and L. Jiang, Infrared Spectroscopy of Neutral Clusters Based on a Vacuum Ultraviolet Free Electron Laser, Chin. J. Chem. Phys., 2021, 34, 51–60 CrossRef CAS.
- H. Zheng, S. Jiang, W. Yan, T. Wang, S. Li, H. Xie, G. Li, X. Yang and L. Jiang, Size-Specific Infrared Spectroscopic Study of the Reactions between Water Molecules and Neutral Vanadium Dimer: Evidence for Water Splitting, J. Phys. Chem. Lett., 2023, 14, 3878–3883 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. A.03, Wallingford, CT, 2016 Search PubMed.
- D. O. Kashinski, G. M. Chase, R. G. Nelson, O. E. Di Nallo, A. N. Scales, D. L. VanderLey and E. F. Byrd, Harmonic Vibrational Frequencies: Approximate Global Scaling Factors for TPSS, M06, and M11 Functional Families Using Several Common Basis Sets, J. Phys. Chem. A, 2017, 121, 2265–2273 CrossRef CAS PubMed.
- S. Feyel, J. Dobler, R. Hockendorf, M. K. Beyer, J. Sauer and H. Schwarz, Activation of Methane By Oligomeric (Al2O3)x+ (x = 3,4,5): The Role of Oxygen-Centered Radicals in Thermal Hydrogen-Atom Abstraction, Angew. Chem., Int. Ed., 2008, 47, 1946–1950 CrossRef CAS PubMed.
- X. N. Wu, Y. X. Zhao, W. Xue, Z. C. Wang, S. G. He and X. L. Ding, Active Sites of Stoichiometric Cerium Oxide Cations (CemO2m+) Probed by Reactions with Carbon Monoxide and Small Hydrocarbon Molecules, Phys. Chem. Chem. Phys., 2010, 12, 3984–3997 RSC.
- Y.-X. Zhao, X.-N. Wu, J.-B. Ma, S.-G. He and X.-L. Ding, Experimental and Theoretical Study of the Reactions between Vanadium–Silicon Heteronuclear Oxide Cluster Anions with n-Butane, J. Phys. Chem. C, 2010, 114, 12271–12279 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00873h |
| This journal is © The Royal Society of Chemistry 2023 |