
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA greener approach for synthesizing metal-decorated carbogels from alginate for emerging technologies†
Juan I.
del Río
ab,
Laura
Juhász
 c,
József
Kalmár
d,
Zoltán
Erdélyi
c,
María D.
Bermejo
a,
Ángel
Martín
a,
Irina
Smirnova
e,
Pavel
Gurikov
fg and
Baldur
Schroeter
*e
c,
József
Kalmár
d,
Zoltán
Erdélyi
c,
María D.
Bermejo
a,
Ángel
Martín
a,
Irina
Smirnova
e,
Pavel
Gurikov
fg and
Baldur
Schroeter
*e
aBioEcoUva, Bioeconomy Research Institute, PressTech Group, Department of Chemical Engineering and Environmental Technology, Universidad de Valladolid, Prado de La Magdalena S/n, 47011 Valladolid, Spain. E-mail: baldur.schroeter@tuhh.de; Tel: +49 40 42878 3962
bGrupo Procesos Químicos Industriales, Department of Chemical Engineering, Universidad de Antioquia UdeA, Calle 70 No. 52-21, Medellín 050010, Colombia
cDepartment of Solid State Physics, University of Debrecen, Egyetem sqr. 1, H-4032 Debrecen, Hungary
dELKH-DE Mechanisms of Complex Homogeneous and Heterogeneous Chemical Reactions Research Group, Department of Inorganic and Analytical Chemistry, University of Debrecen, Egyetem tér 1., Debrecen, H-4032 Hungary
eInstitute for Thermal Separation Processes, Hamburg University of Technology, Eißendorfer Straße 38, 21073 Hamburg, Germany
fLaboratory for Development and Modelling of Novel Nanoporous Materials, Hamburg University of Technology, Eißendorfer Straße 38, 21073 Hamburg, Germany
gaerogel-it GmbH, Albert-Einstein-Str. 1, 49076 Osnabrück, Germany
First published on 11th October 2023
Abstract
In the present work, a series of metal nanoparticle-decorated carbogels (M-DCs) was synthesized starting from beads of parent metal-crosslinked alginate aerogels (M-CAs). M-CAs contained Ca(II), Ni(II), Cu(II), Pd(II) and Pt(iv) ions and were converted to M-DCs by pyrolysis under a N2 atmosphere up to pyrolysis temperatures of TP = 600 °C. The textural properties of M-CAs are found to depend on the crosslinking ion, yielding fibrous pore networks with a high specific mesoporous volume and specific surface area SV (SV ∼ 480–687 m2 g−1) for M-CAs crosslinked with hard cations, Ca(II), Ni(II) and Cu(II), and comparably loose networks with increased macroporosity and lower specific surface (SV ∼ 240–270 m2 g−1) for Pd(II) and Pt(IV) crosslinked aerogels. The pyrolysis of M-CAs resulted in two simultaneously occurring processes: changes in the solid backbone and the growth of metal/metal oxide nanoparticles (NPs). The thermogravimetric analysis (TGA) showed a significant influence of the crosslinking cation on the decomposition mechanism and associated change in textural properties. Scanning electron microscopy-backscattered electron imaging (SEM-BSE) and X-ray diffraction revealed that metal ions (molecularly dispersed in the parent aerogels) formed nanoparticles composed of elementary metals and metal oxides in varying ratios over the course of pyrolytic treatment. Increasing the TP led to generally larger nanoparticles. The pyrolysis of the nickel-crosslinked aerogel (Ni-CA) preserved, to a large extent, the mesoporous structure and resulted in the evolution of fine (∼14 nm) homogeneously dispersed Ni/NiO nanoparticles. Overall, this work presents a green approach for synthesizing metal-nanoparticle containing carbon materials, useful in emerging technologies related to heterogeneous catalysis and electrocatalysis, among others.
Introduction
Since the discovery of aerogels in the 1930s, special attention has been paid to their development for a number of industrial applications.1,2 Given their nanoporous, thermal and electrical properties, breakthroughs in the past few decades have been made in the sectors of foods, biomedical, wastewater treatment, pollution, construction, electrode materials and catalysis.3,4 Since the late 1980s, carbon aerogels (carbogels) have presented a promising alternative for natural carbonaceous materials, due to the possibility of controlling porosity, purity and mechanical strength by adjustments of the synthesis pathway.5 The tailorable textural and structural features of carbogels allow us to overcome some constraints related to metal-nanoparticle materials, such as formation of metal-species aggregates, pore blocking and leaching. Other features such as large specific area, low density, and low thermal and high electrical conductivity as well as hydrophobicity make them attractive materials as catalyst supports and chemical adsorbents and in energy storage as capacitors.6 The polycondensation of resorcinol with formaldehyde under alkaline conditions was a typical pathway to obtain carbogels in recent decades. Other synthetic polymers, such as polybenzoxazines, polyureas, polyimides, polyamides and polyurethanes, have also been used as porous precursors for carbogels.7 For instance, Raptopoulos et al. produced carbogels from polyurea crosslinked alginate, with an increased carbonization yield of up to 37%, which in turn improves surface functionalities (e.g. electrical conductivity and catalytic activity) of the carbogel by contributing oxygen and nitrogen functional groups.8 Recently, Zhang et al. developed a series of carbon aerogel-based electrocatalysts with transition metal-nitrogen sites for the electroreduction of CO2.9 The starting hydrogel was formed via complexation between 2,2′-bipyridine-4,4′-dicarboxylic acid and selected metal ions (Ni, Fe, Co, Mn and Cu) followed by physical crosslinking by polypyrrole. Baumann et al. incorporated copper in a carbogel matrix by formaldehyde polymerization, achieving a uniform dispersion of spherical metal nanoparticles, ranging from 10 to 50 nm.10 In another study, potassium-doped hydrogels were synthesized via formaldehyde polymerization with the potassium salt of 2,4-dihydroxybenzoic acid: these hydrogels were formed on monodisperse polystyrene particle templates (diameter = 300 or 450 nm). Subsequent ion exchange with Ni, Co or Cu-salts and supercritical drying followed by pyrolysis at 800 °C then led to macroporous ordered carbons.11 The materials obtained presented dense networks of interconnected carbon particles with hierarchical pore structure of meso- and microporosity in the wall that facilitates the accessibility to the metal sites.In the conventional synthesis of metal-containing carbonaceous 3D structures, the incorporation of metals requires subsequent post-functionalization, leading to the need for additional process steps and resource consumption. As an example, a sulphonated starch-based support is first pyrolysed at 400 °C and subsequently impregnated with palladium acetate (10% by weight) in acetone.12 Wet impregnation, along with other elaborate methods, e.g., chemical vapor deposition,13 represent additional challenges such as: (i) post-distribution of metal clusters homogeneously through the adhesive-resistant carbon network, and (ii) aggregation of dispersed metals on the carbon surface resulting in the formation of large metal clusters. Despite the remarkable properties exhibited by materials generated via the aforementioned methods, they rely on expensive chemicals and have adverse environmental impacts. Consequently, there has been growing interest in exploring alternative carbon sources, particularly the non-expensive and abundant polysaccharides. Specifically, polysaccharides derived from species with a large capability to fix carbon dioxide, such as brown algae, have gained much interest as precursors for the synthesis of nanoporous carbon materials.14,15 From a sustainable perspective, this CO2 capture feature can add value to novel algae-derived nanomaterials. On average, brown algae species contains ∼40% alginate, an anionic biopolymer that can be easily crosslinked with cations for the preparation of hydrogels.16,17 For example, Meng et al. crosslinked sodium alginate with CaCl2 to obtain a gel that was dried at 50 °C for 3 h, and pyrolized at temperatures up to 1000 °C, under a nitrogen atmosphere.18 The resulting carbon aerogel was soaked in 2 M of HCl and washed with deionized water to obtain a near neutral sample. In the synthesis of cellulose-based carbogels for the removal of heavy metals from wastewater, Alatalo et al. employed concentrated sulfuric acid to catalyze the degradation of cellulose prior to hydrothermal carbonization.19
The above reports evidence the need for consolidating the production of carbogels based on inexpensive and more abundant biomass-derived carbon sources, while minimizing the use of no-green chemicals during the process. On the basis of the principles of green chemistry and modern aerogel science, the present work aims at developing an easy, flexible, and inexpensive strategy towards green and sustainable metal-decorated carbogels. The selection of alginate as the carbon source allowed the reduction of production costs and the carbon footprint. Therefore, a series of metal nanoparticle-decorated carbogels (M-DCs) were synthesized, starting from the beads of parent metal-crosslinked aerogels (M-CAs), comprising the ions Ni(II), Cu(II), Pd(II), and Pt(iv). Because Ca(II) is the most commonly used ion for alginate crosslinking and in alginate aerogel production, Ca-crosslinked gels were included in the study as a reference material. The in situ generation of monodispersed metal nanoparticles was controlled by the pyrolysis temperature, with no additional post-modification step. The conversion of the biopolymer-aerogel network into the corresponding carbogels was studied with a focus on the changes in textural characteristics during the transition and the evolution of the metallic clusters in the range of low pyrolysis temperatures < 600 °C.
Materials and methods
Chemicals
The fine powder of sodium alginate Hydagen 500, was used as received from BASF (Ludwigshafen, Germany). The metal precursors were calcium chloride dihydrate (CaCl2·2H2O) (>99%, Carl Roth catalog number: T885.3), nickel(II) nitrate hexahydrate (>99%, Bernd Kraft, catalog number: 15306), copper(II) sulfate pentahydrate (>99%, Riedel-de Haën), palladium(II) nitrate dihydrate (∼40% Pd basis; Sigma Aldrich, catalog number: 76070-1G) and platinum(IV) chloride (assay 96%; Sigma Aldrich, catalog number: 206113-1G). All chemicals were used without further purification.Characterization of M-CAs and M-CDs
The metal content was determined via ICP-OES using a PerkinElmer Optima 8300 DV. The samples were previously dissolved in a combination of HNO3, HCl and HF solution. The textural properties of M-CAs and M-DCs were analyzed by N2 adsorption–desorption measurements (Nova 3000e Surface Area Analyzer Quantachrome Instruments, Boynton Beach, FL, USA). An overall sample mass of 20–30 mg was used and all samples were degassed under vacuum at 60 °C for at least 6 h prior to each analysis. The Brunauer–Emmett–Teller (BET) method was used to estimate the specific surface area (SV); the pore volume of the mesopores (Vmeso) and pore diameter (dmeso) were determined by the Barrett–Joyner–Halendia (BJH) method. All gas sorption experiments were carried out as single determinations with an estimated relative measurement error of 5%. The particle size and sphericity, SPH, of M-CA beads were determined using a Camsizer XT system (Retsch Technology) in free fall mode according to our previous studies.21 Skeletal densities ρs were determined via helium pycnometry (Multivolume Micromeritics 1305, 4-fold determination). The bulk densities were calculated from the weight of beads that filled a graduated cylinder (volume = 0.35 cm3). The total porosity ε was estimated based on former work,22 as follows:| ε = (1 − ρb/ρs) × 100% |
The outer skin and inner pore structure morphologies of the M-CAs were characterized by scanning electron microscopy (SEM, Zeiss Supra VP55, Jena, Germany). The measurements were carried out under high vacuum at an accelerating voltage of 3–5 kV.
Reducibility and oxidizability of Pd-CA and M-DCs were determined by means of temperature programmed reduction (H2-TPR). In a pretreatment step, the sample was heated at a rate of 10 °C min−1 to 150 °C, maintained for 0.5 h under a constant flow (50 cm3 min−1) of pure argon and finally returned to room temperature to start the data collection. The H2-TPR experiments were conducted up to temperatures of 1000 °C under a constant flow of H2/Ar (10% v/v; 50 cm3 min−1) at a heating rate of 10 °C min−1, in Autochem II 2920 Micromeritics equipment, coupled to a quadrupole mass spectrometer (Pfeiffer-Vacuum Modelo Omnistar). Hydrogen consumption was monitored by using a thermal conductivity detector. Before the detector, an ice trap was used to retain any water formed during the reduction.
Characterization of M-DCs via backscattered electron imaging and X-ray diffraction
In order to image the metallic nanoparticles (NPs) in M-DCs obtained at different pyrolysis temperatures, low voltage scanning electron microscopy was performed using a ThermoFisher Scientific Scios 2 microscope. Backscattered electrons (BSEs) were detected. BSEs are from a deeper interaction volume of the incident beam. The intensity of BSEs shows a significant dependence on the atomic number, which results in a contrast between the elements in the BSE images. The contrast correlates with the atomic number: brighter regions correspond to higher atomic numbers. The electron beam resolution was 1.6 nm at an optimum working distance < 5 mm at 2 kV accelerating voltage. The samples were fixed with vacuum-resistant carbon tape on the sample holder. Sputter coating was not applied in this case because even a 5 nm thick sputtered gold layer can dramatically alter the morphology and the surface structure of the aerogel.23X-ray diffraction patterns of M-DC samples were collected under Cu-Kα1 radiation (Cu-Kα1 = 0.154059 nm) using a Rigaku SmartLab 9 kW X-ray diffractometer (XRD) with the following measurement conditions: measurements were carried out in Bragg–Brentano (θ/2θ) mode, between 35 and 80° using a scan rate of 5° min−1. The average crystallite size for each component of the NPs was calculated from the XRD data. Quantitative analysis of the XRD pattern was carried out using Rietveld refinement using the SmartStudio II software. In this process, the measured XRD profile is approximated by using a calculated one that includes all instrumental and structural parameters. This method applies a non-linear least squares method based on a wide range of free parameters such as unit cell dimension, crystal structure, and peak shape. Using this fitted graph, quantitative information can be deduced from the XRD pattern.
Results and discussion
Properties of metal ion crosslinked aerogels (M-CAs)
The dripping method using bivalent metal ions resulted in immediate gelation with the formation of hydrogel beads when the solution droplets hit the gelation bath. When converted into aerogels, the beads showed unimodal size distributions with an average particle mean diameter of 1.41 ± 0.06 mm and sphericities in the range of SPH = 0.82–0.91 (Fig. S2 and S3†). In contrast to gelation via bivalent ions, the use of Pt(iv) as a crosslinker led to formation of weak wet gels of an undefined shape during gelation and final dry aerogels of foam-like character (Fig. S3e†)). These observations are in line with previous studies, which showed that the crosslinking of alginate with metals strongly depends on the hard–soft character of the ions.24 The low capability of Pt(iv) to crosslink alginate chains may be attributed to the softer character of the ion as compared to the other crosslinking metal ions in this work, which are well coordinated by hard oxygen atoms in negatively charged carboxylate groups and –OH-groups of β-D-mannurate (M) and of α-L-guluronate (G) building blocks. Additionally, Pt(ii) as well as Pt(IV) tend to form characteristically inert “inner-orbital” type complexes in general;25 therefore, the reaction rates of complex formation change significantly with the type of original ligand, as well as with additional factors, such as pH. In our case, we can conclude that either (1) the coordination of Pt-ions in the alginate matrix was not strong enough or (2) the ligand exchange with hydrated PtCl4 was not fast enough to fix the shape of the alginate solution during gelation before it is dispersed. These effects are also indicated by the results of the ICP-OES analysis, which yielded by far the smallest metal mass with respect to the alginate mass (rmetal) in the Pt-containing aerogel (Table 1). The low rmetal value for Pt(IV) may result from either low affinity of Pt(IV) to the alginate matrix or washing out effects during the washing/solvent exchange steps as a result of weak coordination. Coordination of other metal ions resulted in a significant increase in rmetal. Aerogel skeletal density correlates with the ratio of rmetal and parent metal density (Fig. S4,† left) (with the exception of Ca-CA, since calcium-chloride and alginate show relatively similar parent densities). While no systematic relation between rmetal and textural properties was determined, the type of crosslinking ion has a strong effect on the overall mesopore volumes and specific surface areas. Gelation via comparably hard ions Ca(II), Ni(II) and Cu(II) resulted in higher specific surface areas as well as mesopore volumes as compared to those of Pd(II) and Pt(IV) crosslinked gel, whereas the trend of the specific surface area is in line with the affinity of different bivalent ions to alginate molecules.26 Hereby, values of SV (480–687 m2 g−1) are in the typical range reported for Ca(II), Cu(II) and Zn(II) crosslinked alginate and alginate-polyurea hybrid aerogels.8,20,24,27,28| Material [—] | r metal (wt%) | S V (m2 g−1) | d meso (nm) | V meso (cm3 g−1) | ρ s (g cm−3) | ρ b (mg cm−3) | Total porosity (%) |
|---|---|---|---|---|---|---|---|
| Ca-CA | 8.9 ± 0.5 | 494 ± 25 | 41.6 | 7.0 ± 0.4 | 1.71 ± 0.09 | 90.5 ± 0.3 | 94.7 |
| Ni-CA | 9.5 ± 0.2 | 480 ± 24 | 41.4 | 6.5 ± 0.3 | 1.9 ± 0.2 | 35.8 ± 0.2 | 98.1 |
| Cu-CA | 11.1 ± 0.2 | 687 ± 34 | 17.4 | 6.7 ± 0.3 | 2.3 ± 0.1 | 106.8 ± 0.1 | 95.1 |
| Pd-CA | 12.9 ± 0.6 | 243 ± 12 | 17.0 | 2.3 ± 0.1 | 4.6 ± 0.9 | 85.68 ± 0.04 | 98.1 |
| Pt-CA | 3.40 ± 0.04 | 268 ± 13 | 27.1 | 1.29 ± 0.01 | 1.69 ± 0.07 | — | — |
To the best of our knowledge, this work is the first example of purely Pt(IV) crosslinked alginate aerogels. We also determined a considerably higher specific surface area for Pd-CA (242 m2 g−1) than that reported elsewhere (52 m2 g−1).20 The low surface areas in the compared work could result from the presence of excess free metal salt in the alginate matrix (as also indicated by the high Pd content used of rmetal 17.6 wt%), which would lead to a decrease in SV given the large difference in atomic mass of the metal compounds as compared to that of carbon. In this work, the decrease in SV was also obtained, when increasing the metal salt concentrations of Ca(II), Ni(II) and Cu(II)-salts in the gelation bath above cm = 17 mmol L−1 (Fig. S4,† right) as well as in the work of Miller et al., who obtained a decreased SV with increasing metal loading in the Ru/carbogel nanocomposite.29 In contrast to the trend of the specific surface area, no significant differences were found in the pore size distributions and mean pore diameters if cm increased above 17 mmol L−1 (Fig. S5†). This shows that additional ions in the gelation bath do not additionally contribute to crosslinking and cause no microstructural changes.
Since no metal salt crystals or deposits were detected in SEM analysis in any substrates (Fig. S6–S10†) produced at cm = 17 mmol L−1, we conclude that metal ions were quantitatively integrated into the alginate matrix at the given concentration. Typical macro- to mesoporous (dpore = 5–50 nm) fibrous microstructures are visible in the SEM pictures, with relatively broad mesopore size distributions and mean mesopore diameters dmeso being in the range of 17–42 nm (Fig. 1). Optical evaluation of SEM pictures shows that crosslinking with Pd(II) and Pt(IV) yields comparatively loose networks with a significant fraction of macropores, whereas the Cu(II) crosslinked gel shows the densest network with fewer visible pores in the macroporous range (Fig. 1a–e and S6–S8†). The qualitative results from SEM are in line with the results from gas sorption analysis, which yielded lower mesopore volumes for Pd(II)-CA and Pt(IV)-CA.
 | ||
| Fig. 1 (a)–(e) SEM pictures of inner pore structures of M-CAs crosslinked with different metal ions at cm = 17 mmol L−1 and (f) their pore size distributions. Lines are drawn to guide the eye. | ||
Furthermore, a thin denser but still porous outer layer is identified in SEM pictures of aerogel beads (Fig. S9 and S10†). This result is in line with previous studies, and it is caused by the gelation mechanism. When sol droplets hit the gelation bath, the immediate complexation of alginate by metal ions on the surface of droplets leads to the formation of a comparably dense, porous skin, followed by a slower, diffusion controlled completion of the reaction in the inner droplet part.8 In summary, the microstructure of M-CAs is determined by the metal ion chosen for the crosslinking of alginate, while keeping the total porosity at a constant level. In the next step, the pyrolysis of the produced M-CAs was investigated for gels produced at cm = 17 mmol L−1.
Influence of pyrolytic treatment on mass loss and textural properties
Thermogravimetric analysis (Fig. 2a) and derivative thermogravimetric (DTG, Fig. 2b and c) curves were recorded to investigate the mass loss of M-CAs during pyrolysis in the whole temperature range up to final pyrolysis temperatures of TP = 600 °C at a constant heat rate, and to identify TP values at which the most significant changes occur. Signals in the DTG curves represent major mass loss and are used to determine the thermal behavior steps of the materials,30 whereas the individual peaks and maxima are defined in the order of appearance as peak X and TXmax (with X = 1–3). The decomposition patterns of all samples can generally be divided into three distinct main steps: (1) moisture loss; (2) biopolymer degradation and (3) slow sustained degradation.31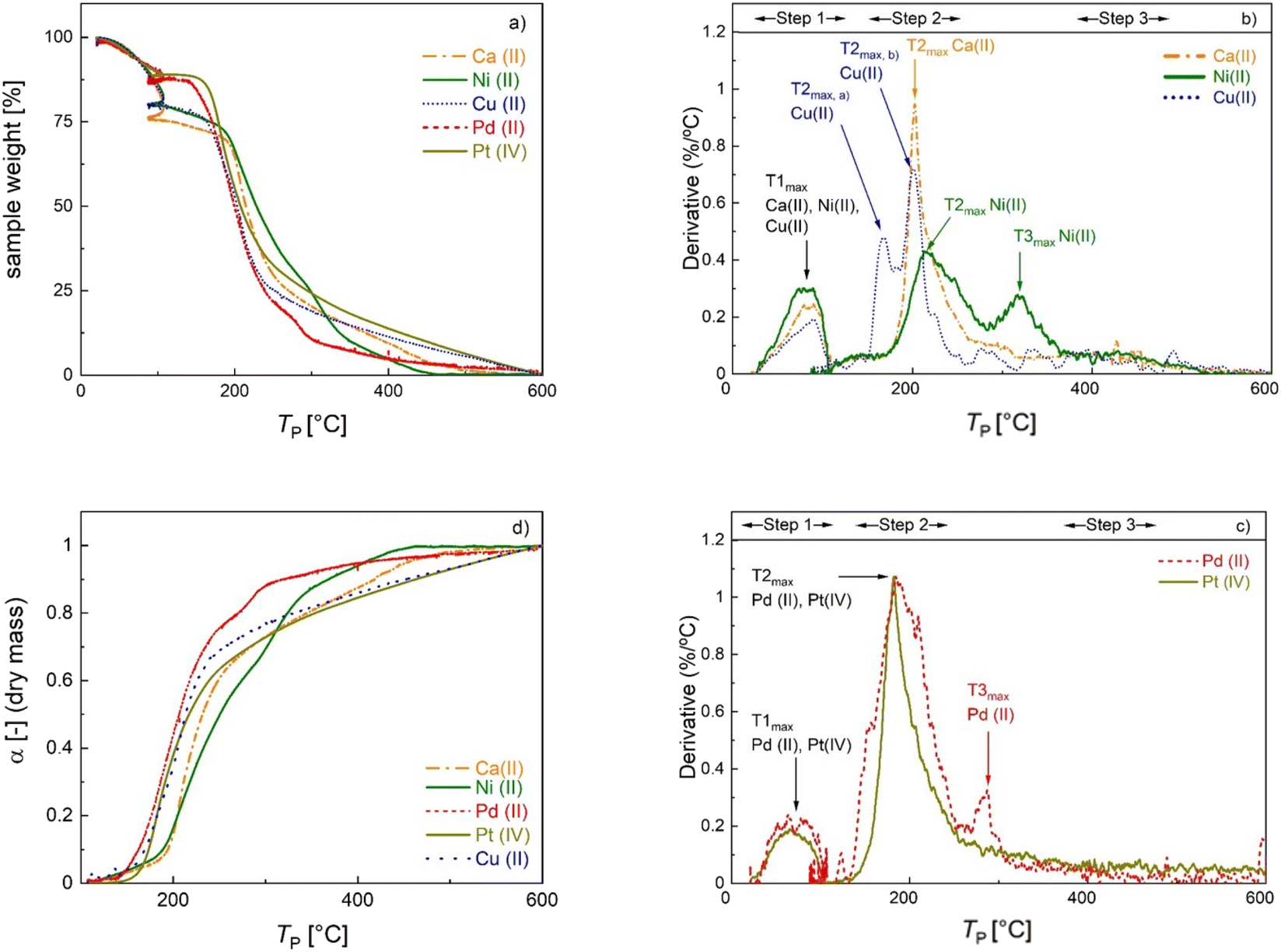 | ||
| Fig. 2 Normalized TGA curves (a), DTG curves (b) and (c) and degree of conversion profiles (d) of M-CAs crosslinked with different metal ions. See the text for details. | ||
Alginate aerogels crosslinked with different metal ions show a metal-specific decomposition profile (Table 2). The first DTG peak (TP = 22–108 °C) is due to moisture loss. A relatively high mass loss (6–18% with respect to the initial mass of the sample, denoted as Δmwater in Table 2) is rather expected at this stage given the hydrophilic and porous nature of the materials. The second DTG peak occurs in the range of 130–255 °C for Ca-, Cu- and Pt-CAs and 130–364 °C for Ni- and Pd-CAs. The second peak corresponds to the major mass loss of 39–61% (denoted as Δmd and calculated with respect to the dry mass, i.e. the mass after the first step). The onset temperature Tonset of peak 2 is assumed to mark the beginning of the thermal scission of the carboxylate/carboxylic acid groups via decarboxylation/decarbonylation and was determined to follow the order: Pd(II) ≈ Pt(IV) < Cu(II) < Ca(II) ≈ Ni(II). In general, the decomposition mechanism of metal-crosslinked alginates is complex and far from being completely understood. For instance, over 20 different degradation products were determined in the decomposition of Ni- and Cu-crosslinked alginate in each case, suggesting metal-specific decomposition mechanisms.32,33
| Material [—] | DTG-peak 1 [°C] | DTG-peak 2 [°C] | DTG-peak 3 [°C] | T 1max [°C] | T 2max [°C] | T 3max [°C] | Δmwater peak1 [%] | Δmd peak2 [%] | Δmd peak3 [%] | Δmd total [%] |
|---|---|---|---|---|---|---|---|---|---|---|
| a See assignment in Fig. 2. | ||||||||||
| Ca-CA | 27–108 | 174–255 | — | 65 | 185 | — | 17.7 | 39.2 | — | 67.9 |
| Ni-CA | 22–108 | 174–283 | 283–365 | 79 | 215 | 318 | 17.1 | 46.0 | 21.8 | 81.3 |
| Cu-CA | 22–108 | 150–255 | — | 87 | 162a, 202a | — | 13.1 | 44.2 | — | 67.4 |
| Pd-CA | 22–107 | 131–255 | 267–309 | 68 | 186 | 285 | 12.2 | 41.6 | 5.7 | 54.5 |
| Pt-CA | 22–107 | 135–255 | — | 65 | 182 | — | 10.2 | 60.9 | — | 95.3 |
The decomposition profiles observed for Ca(II), Cu(II), and Ni(II) are consistent with those in previous studies, whereas the Tonset of Ni(II)-crosslinked alginate is shifted to higher temperatures.8,33 Differences in the values of Tonset and T2,max compared to the cited studies may be attributed to the alginate composition (G/M ratio and molecular weight) as well as to the different degrees of crosslinking.33 For the Cu(II)-crosslinked sample, a DTG duplet peak is recorded at 165 and 203 °C, which was also observed in other studies.8,31 In the decomposition of pure metal salts, the duplet peak at lower temperatures (45–58 °C) corresponds to the formation of trihydrate and monohydrate species of the pentahydrate sulfate.34,35 However, in this work this phenomenon took place at higher temperature, suggesting a strong interaction of the Cu-complex with alginate chains. In contrast to strong coordinating ions, Tonset for alginate crosslinked with soft Pd(II) and Pt(iv) ions is shifted to lower temperatures. Furthermore, a third peak which appeared at TP > 265 °C for Ni(II)- and Pd(II)-crosslinked samples may be ascribed to the decomposition of the remaining metal precursor or to the further decomposition of alginate. In the case of Ni-CA, a relatively high mass loss (Δmd > 20%) in this region points to biopolymer decomposition.36,37
The overall mass loss with respect to the dry mass (“Δmd, total” in Table 2) covers a wide range (55–95%). The literature results for ionic crosslinked alginate aerogels are close to the upper values of this range.8,38 In our case, the variation in the total mass loss is linearly related to rmetal (Fig. S11†), showing that the variations in overall yield can be attributed to the amount of non-pyrolyzed inorganic material. Due to significant differences in the total mass loss caused by variation of inorganic/organic component ratios, additional insight into the effect of individual metal ions on biopolymer decomposition is provided by plotting the degree of conversion α against TP (Fig. 2d), which is defined as:
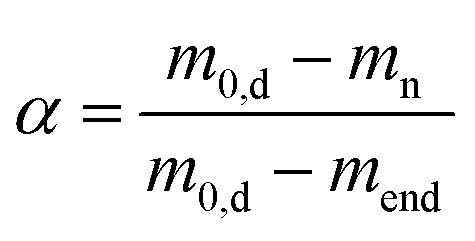
Because the degree of conversion α is normalized with respect to the final mass, samples with significantly different metal/metal oxide contents (such as Pd-CA and Pt-CA) can be directly compared. It is notable that in the temperature range TP < 310 °C the degree of conversion of Ni-CA is the lowest compared to that of other M-CAs, while no further decomposition of Ni-CA took place at TP > 460 °C. Furthermore, the shape of the curves for Ca-, Cu and Pt-CAs is similar for high conversions (α > 63%) which are readily achieved in the low temperature range (TP < 270 °C), followed by a slower decomposition at higher conversions. Pd-CA shows both, a low Tonset as well as rapid decomposition, in the low temperature range.
Since mass loss in aerogel pyrolysis is connected to textural properties, changes in the mesoporous structure during pyrolysis were followed via N2 sorption analysis of samples, which were pyrolyzed at up to four different TP values whereas the pyrolysis process was stopped after the most significant mass loss steps, respectively (TP = 130–150 °C, 265–285 °C, 380 °C, 600 °C). The general trend is that the SV and Vmeso values decrease over the course of the pyrolysis process (Fig. 3a–c), with the most significant changes being directly related to different steps of the mass loss. With the exception of Ni(II)-crosslinked aerogels, pyrolysis up to TP = 285 °C results in a significant loss of mesoporosity. This is also reflected in the applicability of BET/BJH-methods: the M-CAs and materials obtained at low pyrolysis temperatures show typical type IV-isotherms and highly correlated BET-plots (Fig. S12,†R2 0.9991–0.9999), whereas determination of the textural properties via N2-porosimetry is not possible for most M-DCs obtained at values of TP > 300 °C. To date, the exact mechanisms behind the transformation of the intrinsic biopolymer-aerogel pore structure into the corresponding carbon network have not been revealed; due to the complexity of the pyrolysis process, it seems reasonable to assume that different effects might dominate the transition depending on the starting material and its intrinsic pore structure. From a fundamental point of view, the following processes might lead to the diminishing, shrinkage, or formation of porous voids in the system, e.g., (a) partial collapse of the already existing macro- and mesoporous network resulting from mass loss in the form of gases. During this process, effects like pore-opening and pore-enclosure might occur in different temperature ranges of the pyrolytic treatment.38 (b) Creation of new pores by evaporation of material from the bulk phase. In the case of the latter, the existence of a pore network in the starting material is not essential to obtain carbon aerogels, and pores could be formed by starting from a multi-component system followed by pyrolysis or subsequent etching of only one component (template-approach), as has e.g. been shown for polymer crosslinked silica-based xerogel powders.39 In our case (one component system, if metal-ions are neglected), the mass loss during pyrolysis may result in transformation of the already existing pores. This is reflected in the mesoporous range by BJH-pore size distributions of Ni(II)-crosslinked gels: while no changes are observed up to TP = 285 °C, a further increase to TP = 380 °C results in a shift in the mesopore size-distribution to smaller pore sizes, being indicative of the shrinkage of intrinsic mesopores (Fig. 3d). It is interesting to note that this effect coincides with the T3max-signal in the corresponding DTG-curve (Fig. 2b), showing that prior mass-loss of up to 46% (T2max-signal) did not lead to substantial changes in the pore network. The additional increase in the pyrolysis temperature up to TP = 600 °C then leads only to small changes in the textural properties (a slight broadening of the pore size distribution and an increase in the pore volume in the range of larger mesopores), which is consistent with the minor mass loss occurring in the temperature range between TP = 380–600 °C (Fig. 2a and b) In summary, our results suggest that a slower and uniform conversion rate of the organic material, as obtained for Ni-CA, is beneficial for the (partial) preservation of mesoporosity.
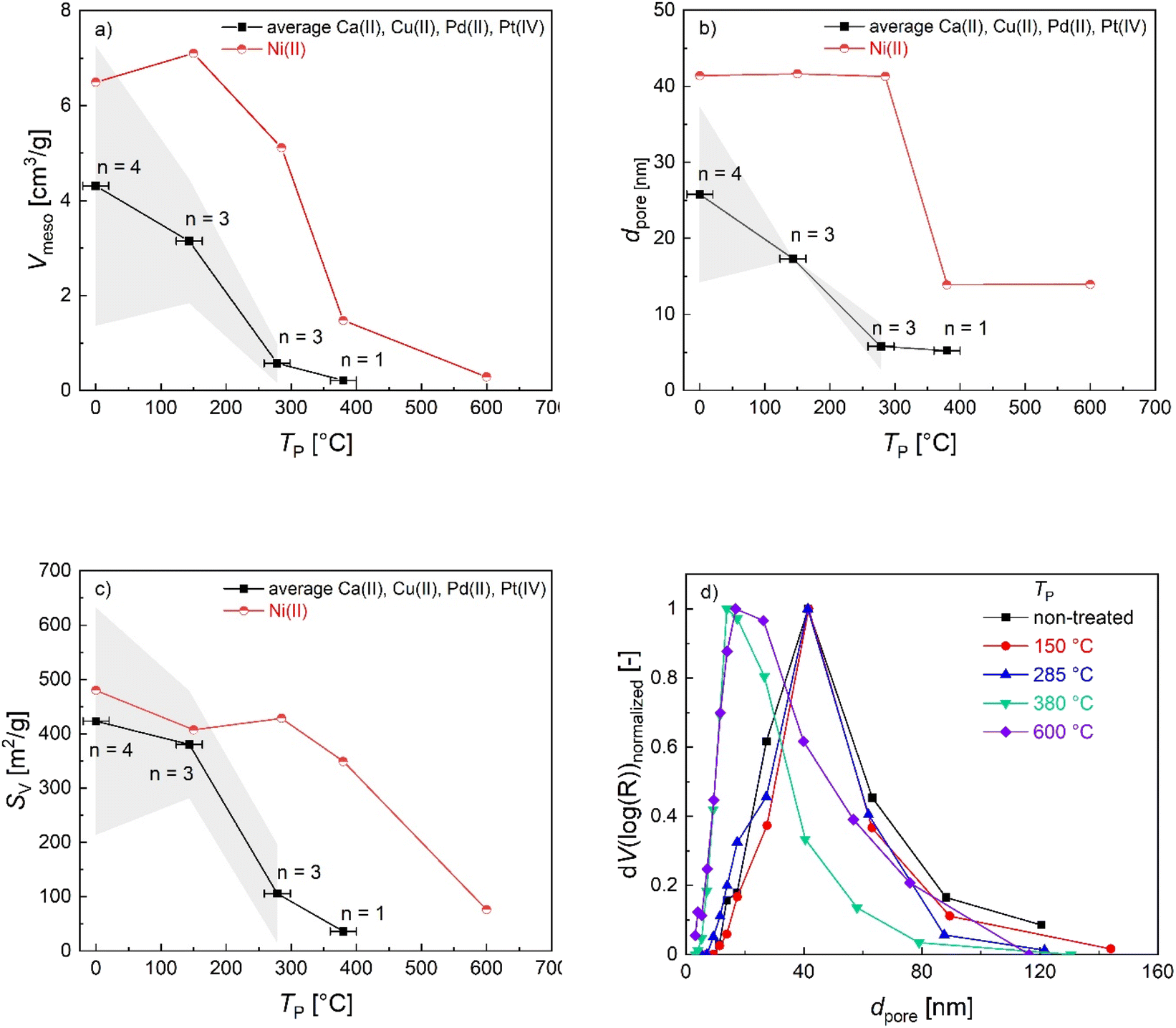 | ||
| Fig. 3 Change in (a) Vmeso, (b) dpore and (c) SV depending on TP. Squares represent the average values from aerogels crosslinked with different metal ions, excluding Ni(II). Lines are drawn to guide the eye, error areas represent the standard deviation from averaged values (y-error), n represents the number of single values taken into account varying at different temperatures, and error bars represent the range of averaged values of TP (x-error). Values at TP = 0 °C represent non-pyrolysed M-CAs. (d) Normalized pore size distributions of Ni-crosslinked gels heated up to different temperatures. Lines are drawn to guide the eye. | ||
Influence of pyrolytic treatment on the backbone morphology and evolution of nanoparticles
Pyrolysis of M-CAs resulted in pronounced volumetric shrinkage of beads (approx. 31–52%), whereas structural integrity was generally maintained (Fig. S13†). However, in the case of Cu-DC and Pd-DC, some morphological changes are visible in SEM pictures, which indicate fast shrinkage or outgassing processes due to the comparably fast conversion in the lower temperature range (see Fig. 2d). In contrast, no defects in Ca-DC and Ni-DC beads were identified. SEM-BSE data provide a qualitative assessment of the morphological changes and especially the evolution of emerging nanoparticles during pyrolysis; as the intensity of BSEs strongly depends on the atomic number, metal-containing structures are visible as brighter zones embedded in the darker biopolymer/carbon backbone (Fig. 4). | ||
| Fig. 4 Exemplary SEM-BSE images of M-DCs obtained after stopping the pyrolysis at three different temperatures TP. | ||
Besides the evolution of the nanoparticles, changes in the backbone can also be observed: all materials obtained at TP = 285 °C still show a fibrous structure that is typical for biopolymer aerogels (Pt-crosslinked aerogel represents an exception). At higher pyrolysis temperatures, the backbone demonstrates further densification and reduction in pore size. While all aerogels remain porous upon pyrolysis, the pore densification process during pyrolysis is highly influenced by the crosslinking metal. Optical evaluation of SEM-BSE pictures allows for a rough categorization: the best resistance against temperature-induced morphological changes is observed in the following order: Ni(II) > Cu(II) > Pd(II) > Pt(IV). This qualitative result highlights the crucial influence of crosslinking metal ions on the materials resulting from the carbonization process.8 The result is also in line with BET/BJH and TGA findings as well as with former work on Ni- and Cu crosslinked alginate.33 In the following section, trends in the evolution of nanoparticles are described for each crosslinking ion. An overview of all results is provided in Table 3.
| M-DC | T P (°C) | Species [—] | d NP estimated via image analysis [nm] | Crystallite size estimated via XRD [nm] |
|---|---|---|---|---|
| Ni-DC | 380 | NiO | — | 8 ± 1 |
| Ni-DC | 600 | NiO | 14 ± 7 | 23 ± 2 |
| Cu-DC | 285 | Cu | 40 ± 19 | 15 ± 5 |
| Cu-DC | Cu2O | 23 ± 15 | ||
| Cu-DC | 600 | Cu | 85 ± 29 | 21 |
| Cu-DC | CuO | 25 ± 6 | ||
| Pt-DC | 525 | Pt | 33 ± 23 | 25 ± 5 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9.6% at 380 °C vs. 95.4% Ni:4.6% at 600 °C. The temperature increase can lead to the sintering of metallic nickel into larger crystallites of metal, driven by the transformation of different crystal forms (e.g., 111, 200, and 220) of nickel.41
:9.6% at 380 °C vs. 95.4% Ni:4.6% at 600 °C. The temperature increase can lead to the sintering of metallic nickel into larger crystallites of metal, driven by the transformation of different crystal forms (e.g., 111, 200, and 220) of nickel.41
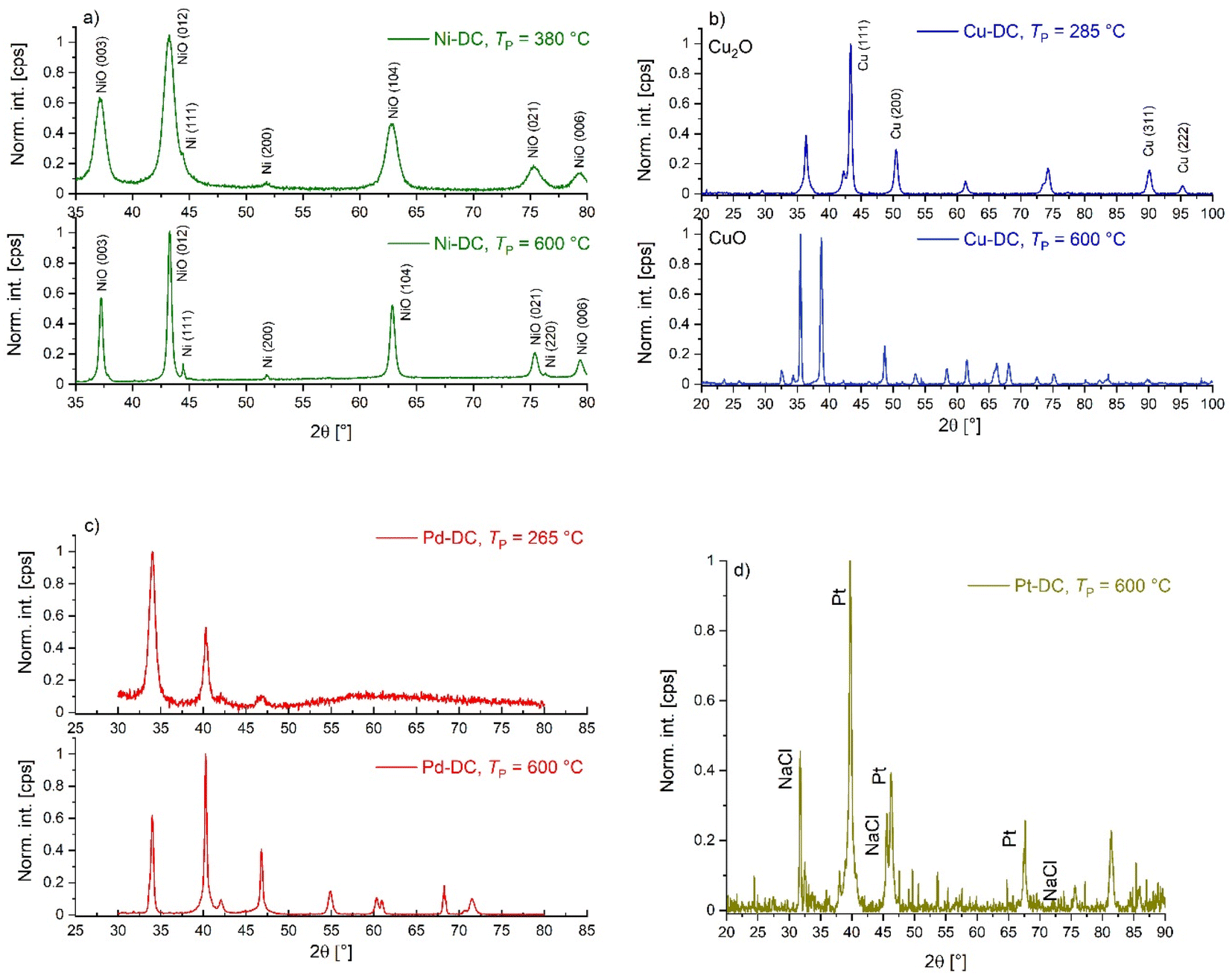 | ||
| Fig. 5 XRD patterns of different metallic metal/metal oxide nanoparticles obtained from different M-Cas and different values of TP: (a) Ni-DC, (b) Cu-DC, (c) Pd-DC, and (d) Pt-DC. | ||
Reducibility of metal oxides
The temperature programmed reduction (TPR) technique allows the reduction temperature of metal oxides to be determined, revealing their reducibility, which is an important parameter to determine the optimal activation conditions for potential applications.42 H2-TPR of Ni-DC (TP = 600 °C) (Fig. 6) showed a main broad peak at 334.3 °C, characteristic of the reduction region of Ni(II) to Ni (0) (378 °C for bulk NiO, and 382 °C for a Ni4.3%/AC catalyst), confirming that there is only one oxidative state in Ni-DC.43,44 The slightly lower reduction temperature (334.3 °C) may be related to the higher metal content in Ni-DC (9.5%), also reflected by the broad shape of the reduction peak, thus providing a weak surface level interaction between Ni and hydrogen. This higher reducibility is comparatively competitive to other Ni based catalysts with similar metal contents and lower reducibility, such as Ni10%/AC (main peak at 430 °C).45 This implies that an easier activation pretreatment of Ni-DC would be needed in further applications. Moreover, Ni-DC did not present significant gasification of the carbon lattice, which usually occurs from 600 °C for carbon supports, hence supporting evidence of its thermal stability.46,47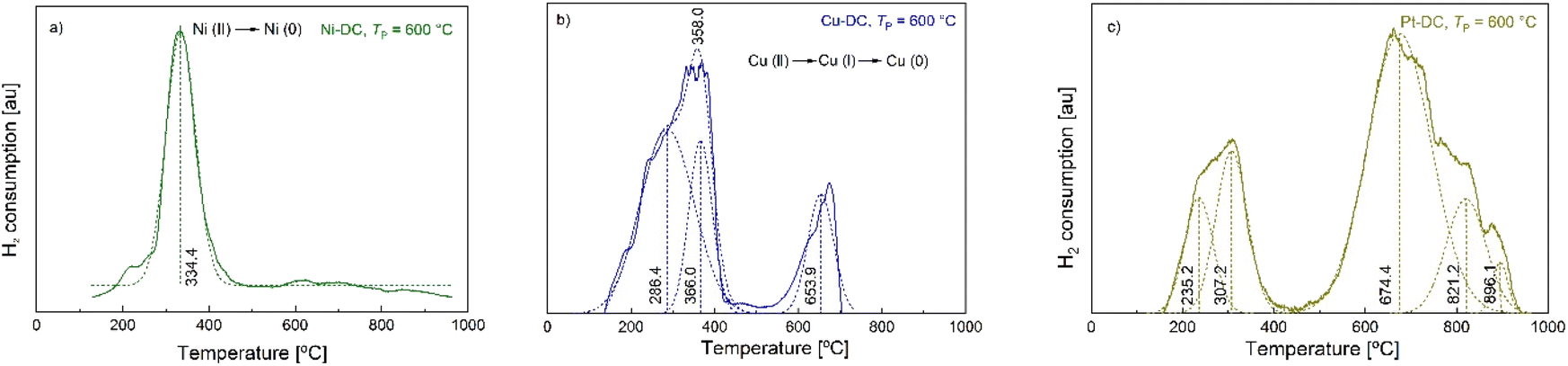 | ||
| Fig. 6 H2-TPR curves of M-DCs produced at TP = 600 °C. Straight lines represent raw data, dashed lines the deconvoluted peaks and the corresponding maxima ((a) Ni-DC, (b) Cu-DC, (c) Pt-DC). | ||
Fig. 6 also illustrates the reduction of copper at different oxidative states, showing two major steps. The first one starts developing from 60 °C and is completely completed at 420 °C, corresponding to the reduction of CuO species (Cu(II)) to Cu2O (Cu(I)) and Cu(0).45 The second one starts at 520 °C, with a maximum at 673 °C. Since copper oxide species completely reduce to metallic Cu(0) at temperatures no further than 420 °C, this second step should be explained by the gasification of the carbon backbone of the Cu-DC sample.48,49
The deconvoluted reduction peaks of Pt-DC at 235.2 and 307.2 °C (Fig. 6c)) can be attributed to the reduction of bulk and inter surficial PtOxCly, as suggested by Hwang et al., originating from traces of the PtCl4 precursor.50 Furthermore, HCl desorption has also been identified within this temperature range.51 It appears that there is an absence of typical PtOx reduction peaks (expected at below 200 °C),52 probably associated with the poor crosslinking that is displayed in Pt-CA. This means that unlike Ni, Cu and Pd, Pt does not catalyze oxide formation during carbonization. Finally, gasification of the Pt-DC sample is observed from 450 to 950 °C. It was not possible to obtain the TPR curves of Pd-DC with the method used, in order to avoid serious damage to the chemisorption equipment, due to the detection of condensable substances. In summary, H2-TPR analysis shows that among the tested M-DCs, Ni-DC has the best performance toward activation by the H2 reduction process, because it presents (i) only one single reduction step, (ii) the lowest reduction temperature, and (iii) the highest thermal stability of the carbon backbone as indicated by minimal gasification.
Conclusions
This work presents a facile method for the production of nanoparticle-decorated porous carbons by pyrolysis of aerogels obtained via crosslinking of sodium alginate with different metal ions and supercritical drying. The broad variety of textural properties obtained for parent aerogels emphasizes that the choice of crosslinking metal ions is a suitable tool for manipulating the pore structure in alginate aerogel synthesis. In the pyrolysis step up to 600 °C, all M-DCs showed a significant mass loss and loss of mesoporosity, whereas the highest stability was identified for the Ni-crosslinked gel with the lowest conversion rate during pyrolysis, which shows the significant influence of the metal ion used on the pyrolysis mechanism. XRD and BSE measurements show the growth of metal(oxide) nanoparticles from the intrinsically molecularly distributed metal ions and the possibility of generating a carbon matrix with homogeneously distributed NPs. While the advantages of this approach lie clearly in its green nature and the avoidance of costly and time consuming post-processing steps (such as, e.g. wet-impregnation our supercritical deposition). in order to distribute NPs in the carbon matrix, it has to be pointed out that limitations are given by the fact that the properties of nanoparticles and the porous matrix cannot be changed independently. In order to achieve the desired nanoparticle sizes with metals of choice, individual strategies have to be developed which could include the use of crosslinking metal ion mixtures, variation of alginate and metal-precursor concentrations and the use of alginates with different M- to G-ratios. Compared to the properties of conventional carbogels based on resorcinol-formaldehyde, the M-DCs developed in this work showed significantly lower specific surface area and higher mass loss during pyrolysis.53 The optimization potential becomes evident when considering that the pyrolysis temperatures employed in this study remain below the threshold at which micropore formation might occur. Hence, the application of a higher TP ≥ 800 °C is a promising next step, which is expected to result in the formation of additional micropores. Enhanced preservation of the mesopore volume/specific surface area may be achieved by employing lower heating rates during pyrolysis (e.g., in the range of 1.0–2.5 °C min−1), as has already been reported54 and is also indicated by our results with the lowest conversion rate yielding comparably best textural features of the carbon matrix. A further possibility to reduce mass loss and to improve textural properties is the introduction of lignins or tannins into the alginate matrix as both biopolymers have reactivity and properties similar to those of resorcinol and phenol and have already successfully been used as non-toxic replacements with natural origin.55 We believe that studies conducted in this work are helpful in identifying suitable conditions and might therefore contribute to the development of catalyst-systems based on green synthesis routes.Abbreviations
| M-CA | Metal-crosslinked aerogel |
| M-DC | Metal nanoparticle-decorated carbogels |
| M | Ca, Ni, Cu, Pd or Pt |
| NPs | Nanoparticles |
Author contributions
Juan I. del Río: investigation, original draft, formal analysis. Laura Juhász: investigation, formal analysis. József Kalmár: conceptualization, formal analysis, editing. Zoltán Erdélyi: investigation. María D. Bermejo: investigation. Ángel Martín: investigation. Irina Smirnova: resources. Pavel Gurikov: project administration, formal analysis, editing. Baldur Schroeter: conceptualization, original draft, formal analysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project was funded by the Ministry of Science and Universities through project RTI2018-097456-B-I00 and by the Junta de Castilla y León through the project by FEDER FUNDS under the BioEcoUVa Strategic Program (CLU-2019-04). Juan Ignacio del Río acknowledges Universidad de Valladolid for the predoctoral and postdoctoral fellowships, as well as the programs (i) MOVILIDAD DOCTORANDOS Y DOCTORANDAS UVa 2021 and (ii) PRÁCTICAS ERASMUS EN EMPRESAS EXTRANJERAS CON SEDE EN EL ESPACIO EUROPEO DE EDUCACIÓN SUPERIOR (EEES) Y PAISES ASOCIADOS DEL PROGRAMA. The XRD and the SEM measurements were financially supported by the TKP2021-NKTA-34 project implemented with the support provided from the National Research, Development and Innovation Fund of Hungary, financed under the TKP2021-NKTA funding scheme. Publishing fees were supported by Funding Programme Open Access Publishing of Hamburg University of Technology (TUHH).References
- S. S. Kistler, Nature, 1931, 127, 741 CrossRef CAS.
- S. S. Kistler, Rubber Chem. Technol., 1932, 5, 600–603 CrossRef.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243–4266 CrossRef CAS PubMed.
- C. Moreno-Castilla and F. J. Maldonado-Hódar, Carbon, 2005, 43, 455–465 CrossRef CAS.
- R. W. Pekala, J. Mater. Sci., 1989, 24, 3221–3227 CrossRef CAS.
- J.-H. Lee and S.-J. Park, Carbon, 2020, 163, 1–18 CrossRef CAS.
- P. Paraskevopoulou, D. Chriti, G. Raptopoulos and G. C. Anyfantis, Materials, 2019, 12, 1543 CrossRef CAS PubMed.
- G. Raptopoulos, et al. , Mater. Adv., 2021, 2, 2684–2699 RSC.
- Y. Zhang, et al. , Adv. Funct. Mater., 2021, 31, 2104377 CrossRef CAS.
- T. F. Baumann, G. A. Fox, J. H. Satcher, N. Yoshizawa, R. Fu and M. S. Dresselhaus, Langmuir, 2002, 18, 7073–7076 CrossRef CAS.
- T. F. Baumann and J. H. Satcher, Chem. Mater., 2003, 15, 3745–3747 CrossRef CAS.
- R. Luque, V. Budarin, J. H. Clark and D. J. Macquarrie, Appl. Catal. B Environ., 2008, 82, 157–162 CrossRef CAS.
- B. A. T. Mehrabadi, S. Eskandari, U. Khan, R. D. White and J. R. Regalbuto, Adv. Catal., 2017, 61, 1–35 CAS.
- O. K. Lee and E. Y. Lee, Biomass Bioenergy, 2016, 92, 70–75 CrossRef CAS.
- R. J. White, N. Brun, V. L. Budarin, J. H. Clark and M. Titirici, ChemSusChem, 2014, 7, 670–689 CrossRef CAS PubMed.
- L. Pereira and J. Cotas, in Alginates: Recent Uses of This Natural Polymer, ed. L. Pereira, Intechopen, London, 1t edn, 2020, vol. 71–150 Search PubMed.
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed.
- X. Meng, X. Tian, Y. Xia and Z. Xiong, J. Dispers. Sci. Technol., 2021, 43, 1–8 Search PubMed.
- S.-M. Alatalo, et al. , ACS Appl. Mater. Interfaces, 2015, b, 25875–25883 CrossRef PubMed.
- Reduction.
- B. Schroeter, et al. , Cellulose, 2021, 28, 223–239 CrossRef CAS.
- L. E. Flint and A. L. Flint, in Methods of Soil Analysis: Part 4 Physical Methods, ed. J. H. Dane and G. C. Topp, Soil Science Society of America, Madison, 2002, pp. 241–254 Search PubMed.
- L. Juhász, et al. , Polymers, 2021, 13, 588 CrossRef PubMed.
- R. P. Narayanan, G. Melman, N. J. Letourneau, N. L. Mendelson and A. Melman, Biomacromolecules, 2012, 13, 2465–2471 CrossRef CAS PubMed.
- C. Richardson-Boedler, Toxicol. Environ. Chem., 2007, 89, 15–70 CrossRef CAS.
- C. Hu, W. Lu, A. Mata, K. Nishinari and Y. Fang, Macromolecules, 2021, 177, 578–588 CrossRef CAS PubMed.
- S. P. Raman, et al. , J. Supercrit. Fluids, 2019, 153, 104545 CrossRef CAS.
- C. Keil, et al. , Polymers, 2020, 12, 1–17 CrossRef PubMed.
- J. M. Miller and B. Dunn, Langmuir, 1999, 15, 799–806 CrossRef CAS.
- S. M. Alay-e-Abbas, K. Mahmood, A. Ali, M. I. Arshad, N. Amin, and M. S. Hasan, in Micro and Nano Technologies, Bionanocomposites, ed. K. M. Zia, F. Jabeen, M. N. Anjum, S. Ikram, Elsevier, 2020, ch. 5, pp. 105–144 Search PubMed.
- J. S. Rowbotham, P. W. Dyer, H. C. Greenwell, D. Selby and M. K. Theodorou, Interface Focus, 2013, 3, 20120046 CrossRef CAS PubMed.
- M. J. Zohuriaan and F. Shokrolahi, Polym. Test., 2004, 23, 575–579 CrossRef CAS.
- Y. Liu, et al. , RSC Adv., 2015, 5, 64125–64137 RSC.
- E. Widjaja, H. H. Chong and M. Tjahjono, J. Raman Spectrosc., 2010, 41, 181–186 CrossRef CAS.
- R. L. White, Thermochim. Acta, 2012, 528, 58–62 CrossRef CAS.
- W. Brockner, C. Ehrhardt and M. Gjikaj, Thermochim. Acta, 2007, 456, 64–68 CrossRef CAS.
- A. Alba, M. A. Aramendia, V. Borau, C. Jimenez and J. M. Marinas, J. Catal., 1986, 98, 288–295 CrossRef CAS.
- S. Seleem, M. Hopkins, J. Olivio and D. A. Schiraldi, Ohio J. Sci., 2017, 117, 50–60 CrossRef CAS.
- R. U. Soni, et al. , Chem. Mater., 2022, 34, 4828–4847 CrossRef CAS.
- C. Teng, et al. , Nanoscale Res. Lett., 2015, 10, 384 CrossRef PubMed.
- E. D. Garbowski, C. Mirodatos, and M. Primet, in Studies in Surface Science and Catalysis, Elsevier, 1982, pp. 235–243 Search PubMed.
- M. A. Reiche, M. Maciejewski and A. Baiker, Catal. Today, 2000, 56, 347–355 CrossRef CAS.
- A. Alihosseinzadeh, B. Nematollahi, M. Rezaei and E. N. Lay, Int. J. Hydrogen Energy, 2015, 40, 1809–1819 CrossRef CAS.
- Q. Zeng, et al. , Environ. Sci. Pollut. Res., 2019, 26, 15420–15435 CrossRef CAS PubMed.
- R. Merabti, K. Bachari, D. Halliche, Z. Rassoul and A. Saadi, React. Kinet. Mech. Catal., 2010, 101, 195–208 CrossRef CAS.
- M. Trépanier, A. Tavasoli, A. K. Dalai and N. Abatzoglou, Appl. Catal. A Gen., 2009, 353, 193–202 CrossRef.
- G. Zhang, Z. Li, H. Zheng, T. Fu, Y. Ju and Y. Wang, Appl. Catal. B Environ., 2015, 179, 95–105 CrossRef CAS.
- G. Fierro, M. Lojacono, M. Inversi, P. Porta, R. Lavecchia and F. Cioci, J. Catal., 1994, 148, 709–721 CrossRef CAS.
- K. Sonobe, M. Tanabe, T. Imaoka, W. Chun and K. Yamamoto, Chem.–Eur. J., 2021, 27, 8452–8456 CrossRef CAS PubMed.
- C.-P. Hwang and C.-T. Yeh, J. Mol. Catal. A Chem., 1996, 112, 295–302 CrossRef CAS.
- M. A. Fraga, E. Jordao, M. J. Mendes, M. M. A. Freitas, J. L. Faria and J. L. Figueiredo, J. Catal., 2002, 209, 355–364 CrossRef CAS.
- T. Huizinga, J. Van Grondelle and R. Prins, Appl. Catal., 1984, 10, 199–213 CrossRef CAS.
- F. Li, et al. , Microporous Mesoporous Mater., 2019, 279, 293–315 CrossRef CAS.
- M. A. T. Jaya, W. M. H. F. W. Harun and M. A. Ahmad, J. Appl. Sci., 2014, 1359–1364 CrossRef CAS.
- D. K. Sam, et al. , Carbohydr. Res., 2020, 491, 107986 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00444a |
| This journal is © The Royal Society of Chemistry 2023 |