
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerfect cubic metallo-borospherenes TM8B6 (TM = Ni, Pd, Pt) as superatoms following the 18-electron rule†
Mei-Zhen
Ao
ab,
Yuan-Yuan
Ma
c,
Yue-Wen
Mu
 *a and
Si-Dian
Li
*a
*a and
Si-Dian
Li
*a
aNanocluster Laboratory, Institute of Molecular Science, Shanxi University, Taiyuan 030006, P. R. China
bFenyang College of Shanxi Medical University, Fenyang 032200, China. E-mail: ywmu@sxu.edu.cn; lisidian@sxu.edu.cn
cShanxi Institute of Energy, Taiyuan 030006, China
First published on 16th October 2023
Abstract
Transition-metal (TM)-doped metallo-borospherenes exhibit unique structures and bonding in chemistry which have received considerable attention in recent years. Based on extensive global minimum searches and first-principles theory calculations, we predict herein the first and smallest perfect cubic metallo-borospherenes Oh TM8B6 (TM = Ni (1), Pd (2), Pt (3)) and Oh Ni8B6− (1−) which contain eight equivalent TM atoms at the vertexes of a cube and six quasi-planar tetra-coordinate face-capping boron atoms on the surface. Detailed canonical molecular orbital and adaptive natural density partitioning bonding analyses indicate that Oh TM8B6 (1/2/3) as superatoms possess nine completely delocalized 14c–2e bonds following the 18-electron principle (1S21P61D10), rendering spherical aromaticity and extra stability to the complex systems. Furthermore, Ni8B6 (1) can be used as building blocks to form the three-dimensional metallic binary crystal NiB (4) (Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) in a bottom-up approach which possesses a typical CsCl-type structure with an octa-coordinate B atom located exactly at the center of the cubic unit cell. The IR, Raman, UV-vis and photoelectron spectra of the concerned clusters are computationally simulated to facilitate their experimental characterization.
m) in a bottom-up approach which possesses a typical CsCl-type structure with an octa-coordinate B atom located exactly at the center of the cubic unit cell. The IR, Raman, UV-vis and photoelectron spectra of the concerned clusters are computationally simulated to facilitate their experimental characterization.
Introduction
As a typical electron-deficient element, boron exhibits unique structures and bonding in both polyhedral molecules and bulk allotropes.1 Researchers have spared no efforts to explore the possibility of all-boron fullerenes since the discovery of buckminsterfullerene (C60) in 1985.2 The first theoretically proposed perfect cage-like Ih B80 fullerene (which is, in fact, not a true minimum of the system) iso-valent with C60 was later proved to favour core–shell structures in thermodynamics at the density functional theory (DFT) level.3,4 The first all-boron fullerenes or borospherenes D2d B40−/0 were discovered in 2014 by joint experimental and theoretical investigations, followed by the experimental observations of the axially chiral borospherenes C3 B39− and C2 B39−, seashell-like C2 B28−/0, and bilayer D2h B48−/0.5–8 Cage-like C1 B41+, C2 B422+, Cs B382−, Cs B373− and Th B364− were also later predicted at the first-principles theory level, presenting a Bnq (n = 36–42, q = n − 40) borospherene family in boron nanoclusters.9–12 Transition-metal-doping has proven to induce dramatic structural and bonding pattern changes in boron clusters.13,14 Exohedral metallo-borospherenes Cs M ∈ B40 (M = Be, Mg) with a quasi-planar hepta-coordinate metal center in an η7-B7 heptagon on the cage surface were the first metallo-borospherenes predicted in theory.15 The first experimentally observed spherical trihedral metallo-borospherene La3B18− with three equivalent deca-coordinate La atoms as integral parts of the cage surface was later extended to the smallest core–shell-like metallo-borospherene D3h La3B20− (La3 & [B2@B18]−) which contains a B2 core at the center at the first principles theory level.16,17 The smallest metallo-borospherene D3h Ta3B12− with three equivalent octa-coordinate Ta centers in three η8-B8 rings was also reported in theory.18 The recently reported Archimedean beryllo-borospherene Be4B12+ was a truncated tetrahedron with four B6 rings capped with four beryllium atoms.19 Exohedral metallo-borospherene Td Ta4B18 and endohedral metallo-borospherene D2d U@B40 were predicted at the DFT level to be superatoms following the 18-electron rule and 32-electron rule, respectively.20,21 Bottom-up approaches were recently proposed at the DFT level from medium-sized bilayer boron nanoclusters to bilayer borophene nanomaterials22 and from heptacoordinate transition-metal-decorated metallo-borospherenes to quasi-multiple-helix metallo-boronanotubes.23Cubic ZnI8-bearing compounds [ZnI8(HL)4(L)8]12− (L = tetrazole dianion) have been experimentally observed, where the aromatic cubic [ZnI8] motif can be used as building blocks to form self-assembled materials.24 Transition metal (TM) clusters based on a TM8 cube with twelve organic ligands on twelve edges have been extensively studied.25–27 A three-dimensional aromatic Ci Li8B6 was proposed which contains eight Li-atoms on the eight triangular faces of a distorted B6 octahedron.28 Recently, a perfect cubic Oh CBe8H12 with an octa-coordinate carbon at the center was predicted at the DFT level as a superatom following a two-fold superatomic electron configuration (1S21P62S22P62D102F6).29 However, transition-metal–non-metal binary clusters with face-capping non-metal atoms on the six tetragonal faces of a perfect TM8 cube still remain unknown to date in both theory and experiments. It is interesting and important to ask at the current stage whether perfect cubic TM8B6 clusters with six face-capping B atoms on the tetragonal faces of a TM8 cube can be formed and whether such cubic complexes can be used as building blocks to form boron-based low-dimensional nanomaterials in bottom-up approaches.
Based on extensive GM searches and first-principles theory calculations, we predicted in this work a series of perfect cubic metallo-borospherenes Oh TM8B6 (TM = Ni (1), Pd (2), Pt (3)) and monoanion Oh Ni8B6− (1−) which contain eight TM atoms at the vertexes of a cube and six quasi-planar tetra-coordinate face-capping boron atoms on the surface. These high-symmetry metallo-borospherenes TM8B6 (1/2/3) satisfy the 18-electron principle (1S21P61D10) in electron configurations and are spherically aromatic in nature. In addition, the structural motif of aromatic cubic Ni8B6 (1) can be extended to construct the metallic three-dimensional (3D) crystal NiB (4) (Pmm) in a bottom-up approach which possesses a characteristic CsCl-type structure with an octa-coordinate B atom located exactly at the center of the cubic unit cell.
Computational details
Extensive GM searches are performed on TM8B6 (TM = Ni, Pd, Pt) using the TGmin2 program,30 in conjunction with manual structural constructions. More than 3000 trial structures were explored for each species on their respective potential energy surfaces in both singlet and triplet states at the PBE/TZVP level. The resulting low-lying isomers for TM8B6 (TM = Ni, Pd, Pt) were then fully optimized using both PBE0 (ref. 31) and TPSSh32 methods with the 6-311+G(d)33 basis set for B and Ni and Stuttgart relativistic small-core pseudopotential for Pd and Pt.34 Vibrational frequencies of low-lying isomers were checked at the same level to ensure that the reported isomers were true minima on the potential surfaces. The UV-vis and photoelectron (PE) spectra were simulated using the time-dependent TD-DFT-PBE0 approach.35 All the DFT calculations in this work were implemented using the Gaussian 09 program.36 Wiberg bond indices (WBIs) and natural atomic charges were obtained by natural bond orbital (NBO) analyses using the NBO 6.0 program.37 Born–Oppenheimer molecular dynamics (BOMD) simulations with a time step of 1 fs in 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps were carried out with the GTH-PBE pseudopotential and the TZVP-MOLOPTSR-GTH basis set employing the CP2K code.38 Chemical bonding patterns were elucidated by canonical molecular orbital (CMO) and adaptive natural density partitioning (AdNDP) analyses.39 Nucleus independent chemical shifts (NICSs)40,41 and iso-chemical shielding surfaces (ICSSs) were computed to reveal the aromaticity of the concerned systems. Orbital compositions and ICSSs were examined using Multiwfn 3.7 code.42 The visualization for the isosurfaces of various functions was realized with VMD software.43
000 steps were carried out with the GTH-PBE pseudopotential and the TZVP-MOLOPTSR-GTH basis set employing the CP2K code.38 Chemical bonding patterns were elucidated by canonical molecular orbital (CMO) and adaptive natural density partitioning (AdNDP) analyses.39 Nucleus independent chemical shifts (NICSs)40,41 and iso-chemical shielding surfaces (ICSSs) were computed to reveal the aromaticity of the concerned systems. Orbital compositions and ICSSs were examined using Multiwfn 3.7 code.42 The visualization for the isosurfaces of various functions was realized with VMD software.43
First-principles calculations on 3D crystal NiB (4) were performed using the Vienna ab initio simulation package (VASP),44,45 within the framework of the projector augmented wave (PAW)46,47 pseudopotential method and PBE generalized gradient approximation (GGA).48,49 The energy cutoff of plane waves was set to 450 eV. Atomic structures were fully relaxed using the conjugate gradient method until the maximum force on each atom was less than 0.01 eV Å−1 and the energy precision was set to 10−5 eV per atom. The Brillouin zone was sampled by 25 × 25 × 25 and 32 × 32 × 32 k-point meshes with the Monkhorst–Pack scheme for geometry optimizations and further calculations on electronic properties, respectively. The Coulomb-corrected local spin density approximation (LSDA + U) was utilized for both structural optimizations and static calculation with the Hubbard value of U = 6 eV for Ni.50–53 The phonon spectrum was calculated using the finite displacement method with the Phonopy code combined with VASP at the PBE level.54
Results and discussion
Structures and stabilities
The obtained singlet GM structures of Oh Ni8B6 (1, 1A1g), Oh Pd8B6 (2, 1A1g) and Oh Pt8B6 (3, 1A1g) are collectively depicted in Fig. 1, with more alternative low-lying isomers tabulated in Fig. S1–S3.† Metallo-borospherenes 1/2/3 possess perfect cubic structures with eight TM atoms (TM = Ni, Pd, Pt) on the vertexes of a cube and six quasi-planar tetra-coordinate face-capping B atoms on the surface, presenting the first and smallest perfect cubic metallo-borospherenes reported to date. 1/2/3 have the smallest vibrational frequencies of 124.4/90.3/54.6 cm−1 and large calculated HOMO–LUMO energy gaps of ΔEgap = 2.31/2.50/2.96 eV at the PBE0/6-311+G(d) level, respectively, well supporting their high chemical stabilities. 1/2/3 possess the optimized TM–B bond lengths of rTM–B = 1.91/2.07/2.08 Å and calculated Wiberg bond orders of WBI = 0.53/0.45/0.54, respectively. These distances are very close to the standard TM–B single bond lengths (1.95, 2.05, and 2.08 Å for TM = Ni, Pd, and Pt, respectively).55 Interestingly, with the calculated total Wiberg bond orders of WBI = 3.71/3.38/3.52, the face-capping B atoms in 1/2/3 can be viewed as quasi-planar tetra-coordinate B atoms interconnected by the triply coordinated TM atoms at the corners, while with the TM–TM distances of rTM–TM = 2.61/2.81/2.82 Å and Wiberg bond orders of WBI = 0.13, 0.06, and 0.08 in 1/2/3, the weak TM–TM covalent interactions in these complex clusters can be practically neglected. Extensive structural searches indicate that Oh Ni8B6 (1, 1A1g) is the well-defined GM of the system, being at least 0.66 eV more stable than other low-lying isomers. Its slightly distorted C2h triplet counterpart (1b) and C2v quintet counterpart (1l) due to the Jahn–Teller effect are 0.66 and 1.38 eV less stable than 1 at the PBE0 level, respectively (Fig. S1†). Similarly, both singlet Oh Pd8B6 (2, 1A1g) and Oh Pt8B6 (3, 1A1g) are true GMs of the systems and are at least 0.21 and 1.11 eV more stable than their nearest counterparts at the PBE0 level, respectively (Fig. S2 and S3†). Interestingly, as collectively shown in Fig. S1–S3,† the top fourteen low-lying isomers of TM8B6 (TM = Ni/Pd/Pt) within 1.45/1.75/1.61 eV all possess cage-like structures. Attachment of one extra electron to Oh Ni8B6 (1, 1A1g) generates its doublet monoanion Oh Ni8B6− (1−, 2A1g) which maintains the same symmetry as Oh Ni8B6 (1) with a non-degenerate singly occupied SOMO (a1g) originating from the LUMO (a1g) of the neutral Oh Ni8B6 (Fig. 3(a)). As clearly shown in Fig. S4,†Oh Ni8B6− (1−) is also the well-defined GM of the monoanion which lies 0.40 eV lower than its second lowest-lying counterpart. | ||
| Fig. 1 Optimized structures of Oh Ni8B6 (1), Oh Ni8B6− (1−), Oh Pd8B6 (2), and Oh Pt8B6 (3) clusters at the PBE0 level and 3D NiB (4) crystal assembled from the structural motif of Ni8B6 (1) at the GGA-PBE level, with the necessary bond lengths and lattice parameters indicated in Å. | ||
Extensive BOMD simulations were performed to check the dynamic stabilities of these cubic metallo-borospherenes. As shown in Fig. S5,†1, 2, and 3 are dynamically stable at 600, 600, and 500 K, respectively, with the small calculated root-mean-square-deviations of RMSD = 0.11, 0.10, and 0.10 Å and maximum bond length deviations of MAXD = 0.29, 0.30, and 0.28 Å. The structural integrities of these metallo-borospherenes are well maintained during the BOMD simulations, with no high energy isomers observed for the 30 ps in the simulating processes.
As demonstrated in Fig. 1, the structural motif of Ni8B6 (1) can be extended in three (x, y, z) dimensions periodically to form the 3D NiB (4) (Pmm) in a bottom-up approach which possesses a typical CsCl-type crystal structure with the optimized lattice parameters of a = b = c = 2.448 Å at the GGA-PBE level. To evaluate the relative stabilities of 3D NimBn crystals, we computed their average cohesive energy per atom (Ecoh) using the formula Ecoh = (mENi + nEB − ENimBn)/(m + n), where ENimBn, EB, and ENi are the total energies of NimBn, a doublet B atom, and a triplet Ni atom, and m and n are the numbers of Ni and B atoms in the unit cell, respectively. 3D NiB (4) has the calculated cohesive energies of Ecoh = 4.57 eV per atom which is obviously higher than the corresponding value of Eb = 3.41 eV per atom obtained for the Oh Ni8B6 (1) cluster, though it is slightly lower than the corresponding value of Ecoh = 4.86 eV per atom calculated for the experimentally reported Ni4B4 crystal (Cmmm).56 As a true minimum of 3D Ni–B binary crystals, NiB (4) contains an octa-coordinate B atom located exactly at the center of the cubic unit cell with the Ni–B coordination bond length of rNi–B = 2.120 Å. The calculated band structures and projected densities of states (PDOS) of 3D NiB (4) depicted in Fig. 2(a) clearly indicate that NiB (4) is metallic in nature, with Ni-3d orbitals contributing mainly to the calculated densities of states near the Fermi level. The calculated phonon dispersion curves of NiB (4) with a 4 × 4 × 4 supercell in Fig. 2(b) exhibit clearly no imaginary frequencies, demonstrating its high dynamical stability. Detailed Bader charge analyses indicate that the central B atom and vertex Ni atom possess the average atomic charge of qB = −0.22|e| and qNi = +0.22|e|, respectively, similar overall to the charge distribution in Ni8B6 (1).
 | ||
| Fig. 2 (a) Calculated band structures and PDOS of 3D NiB (4) and (b) calculated phonon dispersion curves of a 4 × 4 × 4 supercell of 3D crystal NiB (4) at the GGA-PBE level. | ||
Bonding analyses of metallo-borospherenes
Detailed NBO analyses were performed on these cubic complexes to better comprehend their high stabilities. B atoms in 1/2/3 all possess negative calculated net atomic charges of qB = −0.809–−0.313|e| and electronic configurations of [He]2s1.30–1.412p1.88–2.32, while the TM atoms have the corresponding positive net atomic charges of qTM = +0.232–+0.607|e| and electronic configurations of Ni[Ar]3d9.064s0.33, Pd[Kr]4d9.435s0.35, and Pt[Xe]5d9.296s0.49, respectively. These NBO results indicate that TM atoms on the vertexes donate approximately one and a half electrons from their 4s/5s/6s atomic orbitals to the face-capping B atoms, while, in return, they accept roughly one electron in their partially filled 3d/4d/5d orbitals from the surrounding B atoms via effective B(2p) → Ni(3d) backdonations, suggesting the formation of effective electron delocalization in these complexes. To help unveil the bonding nature of these cubic metallo-borospherenes, we further analysed the frontier molecular orbitals (MOs) and densities of states (DOS) of Oh Ni8B6 (1). As clearly shown in Fig. 3(a), the fully filled non-degenerate HOMO−14 (a1g) of Ni8B6 (1) (1S2) exhibits no nodal plane on the cube surface, the triply degenerate HOMO−9 (t1u) (Px2, Py2, and Pz2) has one nodal plane, and the doubly degenerate HOMO−1 (eg) (Dx2−y22 and Dz22) and triply degenerate HOMO (t2g) (Dxz2, Dyz2, and Dxy2) possess two nodal planes. Such an orbital occupation of the cubic complex well corresponds to the electronic configuration of 1S21P61D10 of a superatom in a cubic coordination field. It is noticed that Ni-3d orbitals contribute 67.5–83.5% to these totally delocalized molecular orbitals. As shown in Fig. 3(a), Ni-3d orbitals also make the major contribution to the overall density of states of the complex.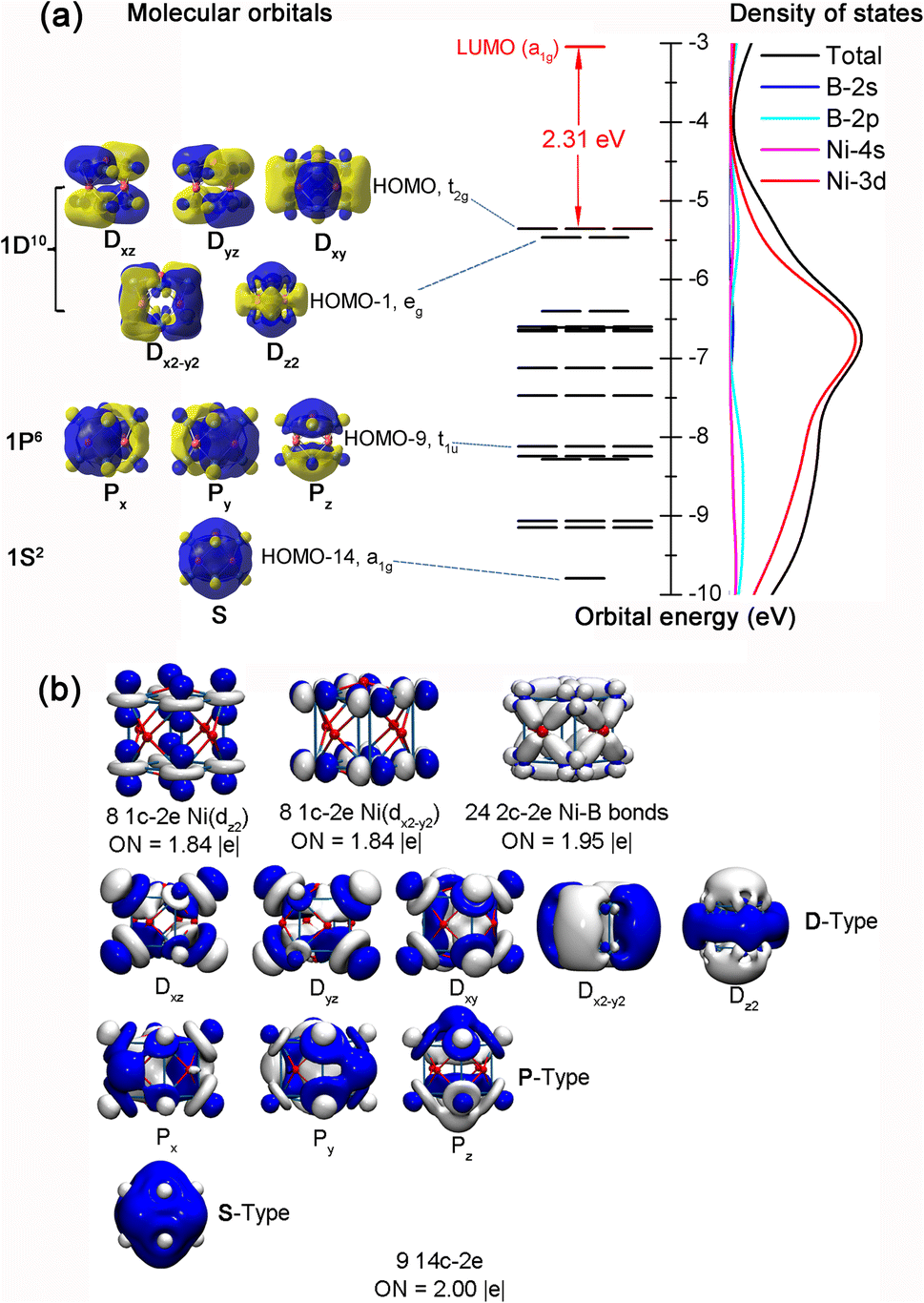 | ||
| Fig. 3 (a) The 1S21P61D10 18-electron configuration and calculated densities of states of Oh Ni8B6 (1). The black and red solid lines refer to occupied and unoccupied orbitals, respectively. (b) AdNDP bonding patterns of Oh Ni8B6 (1) with the occupation numbers (ONs) indicated. | ||
Detailed AdNDP bonding analyses of Oh Ni8B6 (1) shown in Fig. 3(b) unveil clearly the one-center–two-electron (1c–2e) lone pairs, two-center–two-electron (2c–2e) localized bonds, and fourteen-center–two-electron (14c–2e) delocalized bonds in the complex. Ni8B6 (1) possesses 49 pairs of valence electrons in 49 bonds in total. As shown in the first row of Fig. 3(b), each Ni atom at the vertex in Ni8B6 (1) carries two 1c–2e lone pairs in vertical and horizontal directions (3dz2 and 3dx2−y2) with the occupation numbers of ON ≈ 1.84|e|. There exist four equivalent localized 2c–2e Ni–B σ bonds around each quasi-planar tetra-coordinate face-capping B atom with ON = 1.95|e|. The remaining 18 electrons are totally delocalized in nine delocalized 14c–2e bonds in Oh Ni8B6 (1) with ON = 2.00|e|, including 1 14c–2e S-type bond (1S), 3 14c–2e P-type bonds (1Px, 1Py, and 1Pz), and 5 14c–2e D-type bonds (1Dxz, 1Dyz, 1Dxy, 1Dx2−y2, and 1Dz2). Similar to the nine totally delocalized MOs depicted in Fig. 3(a), such an AdNDP bonding pattern with nine totally delocalized 14c–2e bonds further indicates that Ni8B6 (1) as a superatom follows the 18-electron principle (1S21P61D10). As clearly shown in Fig. S6,† both Pd8B6 (2) and Pt8B6 (3) possess similar superatomic molecular orbital occupations and AdNDP bonding patterns to Ni8B6 (1).
Such bonding patterns render spherical aromaticity to cubic metallo-borospherenes 1/1−/2/3, as evidenced by the negative calculated NICS values of NICS = −65.53/−60.80/−67.08/−60.53 ppm at their geometrical centers, respectively. The spherical aromatic nature of 1/1−/2/3 is further evidenced by their iso-chemical shielding surfaces (ICSSs) based on the calculated NICS-ZZ components. As collectively presented in Fig. 4, the areas highlighted in yellow inside the cubes and within about 1.0 Å above the cube surfaces in the vertical direction belong to chemical shielding regions with negative NICS-ZZ values, while the chemical de-shielding regions with positive NICS-ZZ values highlighted in green are located in belt-like areas around the cage waists in the horizontal direction. The ICSSs of metallo-borospherenes 1/2/3 appear to be similar to that observed in the spherically aromatic closo borane dianion Oh B6H62− (Fig. 4(e)), further evidencing that these metallo-borospherenes are spherically aromatic in nature,57,58 well supporting their high thermodynamic stabilities.
 | ||
| Fig. 4 Calculated iso-chemical shielding surfaces (ICSSs) of (a) Oh Ni8B6 (1), (b) Oh Ni8B6− (1−), (c) Oh Pd8B6 (2), and (d) Oh Pt8B6 (3), compared with that of (e) Oh B6H62−, with the main molecular axes designated as the z axis in the vertical direction. Yellow and green regions stand for chemical shielding and chemical de-shielding areas, respectively. | ||
Simulated IR, Raman, UV-vis, and PE spectra
The IR, Raman, and UV-vis spectra of Ni8B6 (1) are computationally simulated at the PBE0/6-311+G(d) level in Fig. 5(a–c) to assist their future experimental characterization. Oh Ni8B6 (1) possesses highly simplified IR and Raman spectra due to its high symmetry, including three sharp IR peaks at 202 (t1u), 381 (t1u), and 683 (t1u) cm−1 and five Raman active vibrations at 124 (t2g), 193 (t2g), 261 (a1g), 390 (eg), and 452(a1g) cm−1, respectively. Detailed vibrational analyses indicate that the symmetrical vibrations at 452 cm−1 (a1g) represent typical radial breathing modes (RBMs) of the metallospherene which can be used to characterize single-walled hollow boron nanostructures. The simulated UV-vis spectrum of Ni8B6 (1) exhibits strong absorption peaks at 289, 331, 417, 469, and 525 nm. The strong UV bands around 331, 417, 469, and 525 nm mainly originate from electron transitions from the deep inner shells to the lowest unoccupied molecular orbitals (LUMO, a1g) of Ni8B6 (1), while UV bands around 289 nm mainly originate from electron transitions from the deep inner shells to the high-lying unoccupied molecular orbitals. As shown in Fig. S7,†Oh Pd8B6 (2) and Oh Pt8B6 (3) exhibit similar spectral features to Oh Ni8B6 (1). The simulated PE spectrum of doublet monoanion Ni8B6− (1−) is depicted in Fig. 4(d) at the TD-DFT-PBE0 level. The first vertical detachment energy (VDE) at 2.01 eV for Ni8B6− (1−) was calculated as the energy difference between the anionic ground state and the neutral ground state at the optimized anion geometry. Ni8B6− (1−) exhibits six well-separated vertical detachment energies centered at VDE = 2.01, 2.60, 3.52, 3.84, 4.40 and 4.78 eV, respectively. It is noticed that Ni8B6− (1−) has an extremely low calculated first VDE at 2.01 eV which is even lower than the measured first VDE (2.62 eV) of the prototypical borospherene anion B40− due to the superb stability of neutral Ni8B6 (1) which has a superatomic electron configuration as discussed above. | ||
| Fig. 5 Simulated (a) IR, (b) Raman, and (c) UV-vis spectra of neutral Oh Ni8B6 (1), and (d) PE spectroscopy of monoanion Ni8B6− (1−) at the PBE0/6-311+G(d) level. | ||
Conclusions
Based on extensive GM searches and first-principles theoretical calculations, we have predicted in this work the GM structures of metallo-borospherenes Oh Ni8B6 (1), Oh Pd8B6 (2), Oh Pt8B6 (3), and Oh Ni8B6− (1−). These perfect cubic metallo-borospherenes possess typical spherical aromaticity, with 18 valence electrons occupying nine totally delocalized 14c–2e bonds following the superatomic electron configuration of 1S21P61D10. The 3D crystal NiB (4) as an assembly of Oh Ni8B6 (1) with an octa-coordinate B atom located exactly at the center of the cubic unit cell exhibits metallic behavior. These high-symmetry metallo-borospherenes and their assembled low-dimensional nanomaterials with interesting geometrical and electronic structures may form novel transition-metal-boron binary nanomaterials as potential chemical catalysts and electronic devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the National Natural Science Foundation of China (22373061, 21720102006 and 21973057 to S.-D. Li).References
- B. Albert and H. Hillebrecht, Angew. Chem., Int. Ed., 2009, 48, 8640 CrossRef CAS PubMed.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162 CrossRef CAS.
- N. G. Szwacki, A. Sadrzadeh and B. I. Yakobson, Phys. Rev. Lett., 2007, 98, 166804 CrossRef PubMed.
- A. Sadrzadeh, O. V. Pupysheva, A. K. Singh and B. I. Yakobson, J. Phys. Chem. A, 2008, 112, 13679 CrossRef CAS PubMed.
- H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu, Y. B. Wu, Y. W. Mu, G. F. Wei, Z. P. Liu, J. Li, S. D. Li and L. S. Wang, Nat. Chem., 2014, 6, 727 CrossRef CAS PubMed.
- Q. Chen, W. L. Li, Y. F. Zhao, S. Y. Zhang, H. S. Hu, H. Bai, H. R. Li, W. J. Tian, H. G. Lu, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, ACS Nano, 2015, 9, 754 CrossRef CAS PubMed.
- Y. J. Wang, Y. F. Zhao, W. L. Li, T. Jian, Q. Chen, X. R. You, T. Ou, X. Y. Zhao, H. J. Zhai, S. D. Li, J. Li and L. S. Wang, J. Chem. Phys., 2016, 144, 064307 CrossRef PubMed.
- W. J. Chen, Y. Y. Ma, T. T. Chen, M. Z. Ao, D. F. Yuan, Q. Chen, X. X. Tian, Y. W. Mu, S. D. Li and L. S. Wang, Nanoscale, 2021, 13, 3868 RSC.
- Q. Chen, S. Y. Zhang, H. Bai, W. J. Tian, T. Gao, H. R. Li, C. Q. Miao, Y. W. Mu, H. G. Lu, H. J. Zhai and S. D. Li, Angew. Chem., Int. Ed., 2015, 54, 1 CrossRef.
- Q. Chen, H. R. Li, C. Q. Miao, Y. J. Wang, H. G. Lu, Y. W. Mu, G. M. Ren, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2016, 18, 11610 RSC.
- Q. Chen, H. R. Li, W. J. Tian, H. G. Lu, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2016, 18, 14186 RSC.
- W. J. Tian, Q. Chen, H. R. Li, M. Yan, Y. W. Mu, H. G. Lu, H. J. Zhai and S. D. Li, Phys. Chem. Chem. Phys., 2016, 18, 9922 RSC.
- J. Barroso, S. Pan and G. Merino, Chem. Soc. Rev., 2022, 51, 1098 RSC.
- W. Liang, J. Barroso, S. Jalife, M. Orozco-Ic, X. Zarate, X. Dong, Z. Cui and G. Merino, Chem. Commun., 2019, 55, 7490 RSC.
- H. Bai, Q. Chen, H. J. Zhai and S. D. Li, Angew. Chem., Int. Ed., 2014, 53, 1 CrossRef.
- T. T. Chen, W. L. Li, W. J. Chen, X. H. Yu, X. R. Dong, J. Li and L. S. Wang, Nat. Commun., 2020, 11, 2766 CrossRef CAS PubMed.
- X. Y. Zhao, M. Yan, Z. H. Wei and S. D. Li, RSC Adv., 2020, 10, 34225 RSC.
- Y. Zhang, X. Y. Zhao, M. Yan and S. D. Li, RSC Adv., 2020, 10, 29320 RSC.
- X. Dong, Yu. Liu, X. Liu, S. Pan, Z. Cui and G. Merino, Angew. Chem., Int. Ed., 2022, 61, e202208152 CrossRef CAS PubMed.
- Y. Zhang, X. Q. Lu, M. Yan and S. D. Li, ACS Omega, 2021, 6, 10991 CrossRef CAS PubMed.
- T. Yu, Y. Gao, D. Xu and Z. Wang, Nano Res., 2018, 11, 354 CrossRef CAS.
- Q. Q. Yan, T. Zhang, Y. Y. Ma, Q. Chen, Y. W. Mu and S. D. Li, Nanoscale, 2022, 14, 11443 RSC.
- M. Z. Ao, F. Zhang, Y. Y. Ma, Y. W. Mu and S. D. Li, Nanoscale, 2023, 15, 2377 RSC.
- P. Cui, H. S. Hu, B. Zhao, J. T. Miller, P. Cheng and J. Li, Nat. Commun., 2015, 6, 6331 CrossRef CAS PubMed.
- N. Masciocchi, S. Galli, V. Colombo, A. Maspero, G. Palmisano, B. Seyyedi, C. Lamberti and S. Bordiga, J. Am. Chem. Soc., 2010, 132, 7902 CrossRef CAS PubMed.
- H. C. Hu, H. S. Hu, B. Zhao, P. Cui, P. Cheng and J. Li, Angew. Chem., Int. Ed., 2015, 54, 11681 CrossRef CAS PubMed.
- C. A. Barboza, A. Gambetta, R. Arratia-Pérez, P. L. Rodríguez-Kessler, A. Muñoz-Castro and D. MacLeod-Carey, Polyhedron, 2021, 195, 114878 CrossRef CAS.
- T. B. Tai and M. T. Nguyen, Chem. Comm., 2013, 49, 913 RSC.
- J. C. Guo, L. Y. Feng, C. Dong and H. J. Zhai, New J. Chem., 2020, 44, 7286 RSC.
- Y. Zhao, X. Chen and J. Li, Nano Res., 2017, 10, 3407 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. M. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef CAS.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- P. E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, 2013 Search PubMed.
- J. V. Vondele, M. Krack, F. Mohamed, M. Parrinello, T. Chassaing and J. Hutter, Comput. Phys. Commun., 2005, 167, 103 CrossRef.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207 RSC.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- I. V. Solovyev and P. H. Dederichs, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 16861 CrossRef CAS PubMed.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943 CrossRef CAS PubMed.
- Y. Gao, X. Wang, J. Ma, Z. Wang and L. Chen, Chem. Mater., 2015, 27, 3456 CrossRef CAS.
- F. Zhou, M. Cococcioni, C. Marianetti, D. Morgan and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235121 CrossRef.
- A. Togo, F. Oba and I. Tanaka, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 134106 CrossRef.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326 CrossRef PubMed.
- E. Lugscheider, O. Knotek and H. Reimann, Monatsh. Chem., 1974, 105, 80 CrossRef CAS.
- G. Merino, T. Heine and G. Seifert, Chem. Eur. J., 2004, 10, 4367 CrossRef CAS PubMed.
- G. Merino, M. Solà, I. Fernández, C. Foroutan-Nejad, P. Lazzeretti, G. Frenking, H. L. Anderson, D. Sundholm, F. P. Cossío, M. A. Petrukhina, J. Wu, J. I. Wu and A. Restrepo, Chem. Sci., 2023, 14, 5569 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00551h |
| This journal is © The Royal Society of Chemistry 2023 |