DOI:
10.1039/D3NA00281K
(Paper)
Nanoscale Adv., 2023,
5, 4447-4463
Preparation of cellulose nanocrystal (CNCs) reinforced polylactic acid (PLA) bionanocomposites filaments using biobased additives for 3D printing applications†
Received
28th April 2023
, Accepted 27th July 2023
First published on 4th August 2023
Abstract
This work presents the experimental steps taken towards the preparation of 3D printable bionanocomposites using polylactic acid (PLA) biopolymer containing 0.1, 0.5 and 1 wt% CNCs. Optimized amounts of bio-based additives were added to improve the processability and flexibility of the bionanocomposites. The 3D printable bionanocomposite filaments were drawn using a single screw extruder. The bionanocomposites filament was used to 3D print prototypes and test specimens for dynamic mechanical analysis (DMA). Characterization of the CNCs and bionanocomposites was performed using Fourier Transform Infrared Spectroscopy (FTIR) analysis, differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA). The nucleating effect of CNCs enhanced the crystallization behaviour of bionanocomposites by 5%, 15% and 11%, for the different CNCs loadings. The TGA analysis revealed a ∼20 °C improvement in the thermal stability of the bionanocomposites. Meanwhile, the tensile analysis showed a ≥48% increase in the tensile strength of the bionanocomposites filaments which was attributed to the reinforcing effects of CNC. The addition of CNCs significantly increased the melt viscosity, storage and loss modulus of PLA. In summary, the bionanocomposite filaments produced in this study exhibited excellent processibility and superior mechanical and thermal properties.
1. Introduction
The fused deposition modelling (FDM) 3D printing technique is a material extrusion (MEX)-based technique that operates via a layer-by-layer melt deposition of thermoplastics following a pre-determined digitally generated build-path.1 Presently, polylactic acid (PLA) remains the most widely used biopolymer in 3D printing due to its low melting point, minimal thermal shrinkage and warping.2 However, the overall performance of pristine PLA is limited by its brittle nature, slow crystallization, and poor melt strength.2,3 Strategies adopted towards improving the properties of PLA includes the incorporation of nano-reinforcements,4,5 the use of biobased plasticizers5,6 and blending with other biopolymers.7–9 The development of eco-friendly 3D printable biopolymer-based composites has gained much attention in recent times. This is due to the drive for widespread adoption of eco-friendly, and biobased alternative material. Bionanocomposites of polylactic acid (PLA) biopolymer and nano cellulose possess promising physical and mechanical qualities desirable for FDM 3D printing applications. Attributes such as biocompatibility, potential biodegradability and wide-ranging applications make them an ideal material for a circular bioeconomy.10
Cellulose nanocrystals (CNCs) are rigid rod-shaped crystalline nanomaterials with diameters ranging from 1–100 nm and lengths between 50 to 1160 nm.11–13 The interest in CNCs is due to their high aspect ratio, low density, renewability, biocompatibility, and biodegradability. CNCs are typically isolated from the crystalline domains of naturally occurring cellulosic sources like plant lignocellulosic biomass,1–6 sea animal (tunicate)14 and bacterial cellulose.15 Presently sulphuric acid hydrolysis of cellulosic fibres is the most widely used CNC extraction method.16,17 The introduction of unmodified CNCs into the PLA matrix has been shown to result in the lowering of tensile strength.18 This is largely due to the tendency of CNCs to agglomerate inside the PLA matrix, which leads to poor interfacial interactions and low-stress transfer.19,20 However, some studies have successfully demonstrated the use of CNCs to improve the mechanical properties and crystallization behaviour of PLA filaments.21–23 A recent study reported improved crystallinity and an 18% increase in the tensile strength of PLA after incorporating 1% CNCs powder.24 The flexibility and processibility of bionanocomposites have been improved by the addition of bio-based plasticizers.25,26
The major challenges faced during the production of bionanocomposites are poor dispersion of nanomaterials in the polymer matrix, potential thermal degradation, poor compatibility and weak interfacial interactions.25 Modification strategies such as surface functionalization of CNCs (via esterification, silylation and acetylation reactions), grafting, and the addition of coupling agents, compatibilizers and surfactants have been adopted to mitigate this problem.18,27,28 Presently, direct liquid feeding and dry mixing process have been explored as viable strategies for producing CNC/PLA bionanocomposites. Typically, CNCs are dispersed in water, making them suitable for direct liquid feeding methods. However, this is undesirable as the water can initiate thermo-hydrolytic degradation of PLA during melt-mixing. To mitigate this, the moisture in CNCs can be expelled by sublimation via the freeze-drying process. However, free-drying results in the aggregation and agglomeration of CNC fibres, making them difficult to disperse in the polymer matrix.
Bagheriasl et al. (2018)3 conducted a comparative study between the direct melt-mixing process and the solution-casting method of preparing CNC-reinforced PLA bionanocomposites. The results from this study indicated that the solution casting method achieved better CNC dispersion, whereas there was significant CNC agglomeration in the bionanocomposites prepared via the direct melt-mixing method. Oksman et al. (2006)19 prepared PLA/CNC bionanocomposites by direct liquid-feeding of CNC dispersed in a solution of N,N-dimethylacetamide (DMAc) containing lithium chloride (LiCl) into PLA melt. Similarly, Herrera et al. (2015)5 dispersed nano cellulose using a mixture of water, acetone, and glycerol triacetate. Afterwards, they prepared well-dispersed PLA bionanocomposites using melt-extrusion and liquid feeding of nano cellulose suspension.5 However, these methods' drawbacks include toxicity concerns, high cost, limited scalability, and increased hydrolytic degradation of PLA.
This work explores an environmentally friendly and scalable method of producing FDM 3D printable PLA bionanocomposite filaments containing different CNCs loading. Freeze-dried CNCs obtained from forestry waste residues were re-dispersed using a minimal volume of green solvents and without performing any additional chemical modifications. Afterwards, the redispersed CNCs were directly incorporated into PLA during melt-mixing to obtain masterbatches. The melt-mixing was done at a low temperature to minimize the thermal degradation of CNCs and PLA. Optimised amounts of bio-based additives (triacetin and glycerol triacetate) and a food-grade multifunctional chain extender were used as a processing aid, compatibilizer, and plasticizer. Afterwards, a single-screw extruder was used to draw the PLA/CNC bionanocomposites into filaments suitable for 3D printing. The chemical, thermal, rheological, and mechanical properties of the bionanocomposite filaments were investigated. Finally, the 3D printability of the PLA/CNC bionanocomposite filaments was elucidated on a fused deposition modelling (FDM) printer.
2. Materials and methods
2.1. Materials
Polylactic acid (PLA), Ingeo™ PLA6202D, (Mw = 140 kDa, ≤2% D-lactide, fibre grade) was procured from NatureWorks USA. Polyethene glycol, triacetin and a food-safe styrene–acrylic multifunctional chain extender (Joncryl ADR-4368-C) were added to improve flexibility, processability and melt strength. Other solvent-grade chemicals used include ethanol, acetone and ethyl acetate which were procured from Sigma Aldrich.
The freeze-dried cellulose nanocrystals (CNCs) used in this current study were extracted on a pilot scale from sawdust of Eucalyptus grandis, using a propriety technology developed by the Biorefinery Industry Development Facility (BIDF), Natural Resources and the Environment Research Area, Council for Scientific and Industrial Research (CSIR), Durban, South Africa. The freeze-dried CNC were oven dried at 50 °C overnight in an air-circulation oven before use. The transmission electron microscopy (TEM) image of the CNCs has been provided by the supplier and is presented in the ESI.†
2.2. Re-dispersion of cellulose nanocrystals (CNC)
The sublimation of water during the freeze-drying process of CNCs leads to the aggregation and agglomeration of individual fibres due to the high amount of hydrogen bonding.20 A combination of solvents and mechanical agitation was used to partly destabilize the strong interactions between individual CNC fibres. The fluffy CNCs fibres were carefully transferred into a beaker with the protic and aprotic polar solvents and the mixture was stirred on a magnetic stirrer for 48 hours. The protic and aprotic polar solvent mixture contains acetone, ethanol, and ethyl acetate in the following ratios 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1. This was followed by sonication for another 24 hours to achieve a fine dispersion. Before use, the re-dispersed CNC was constantly stirred using an overhead stirrer to avoid sedimentation.
:3:1. This was followed by sonication for another 24 hours to achieve a fine dispersion. Before use, the re-dispersed CNC was constantly stirred using an overhead stirrer to avoid sedimentation.
2.3. Composition and sample description
The sample containing 100% PLA6202D is denoted as PLA100. Whereas the sample labelled as PLASCE contains PLA6202D with bio-based additives and a food-grade chain extender (CE). Furthermore, the sample description PLACNC0.1%, PLACNC0.5%, and PLACNC1% represents the bionanocomposite samples containing 0.1%, 0.5% and 1% cellulose nanocrystals (CNCs). Each bionanocomposite contains equal amounts of additives and chain extender. Table 1 shows the final composition of each sample per mixing cycle.
Table 1 Formulation for PLA and bionanocomposites
| Sample description |
Composition (%) |
Mixing time (minutes) |
| PLA |
Biobased additives |
CNCs |
|
Virgin PLA.
PLA containing plasticizer, chain extender.
PLA containing plasticizer, chain extender and cellulose nanocrystals.
|
| PLA100a |
100 |
0 |
0 |
6 |
| PLASCEb |
89 |
11 |
0 |
6 |
| PLACNC0.1%c |
88.9 |
11 |
0.1 |
8 |
| PLACNC0.5%c |
88.5 |
11 |
0.5 |
10 |
| PLACNC1%c |
88 |
11 |
1.0 |
14 |
2.4. Preparation of PLA/CNC bionanocomposites
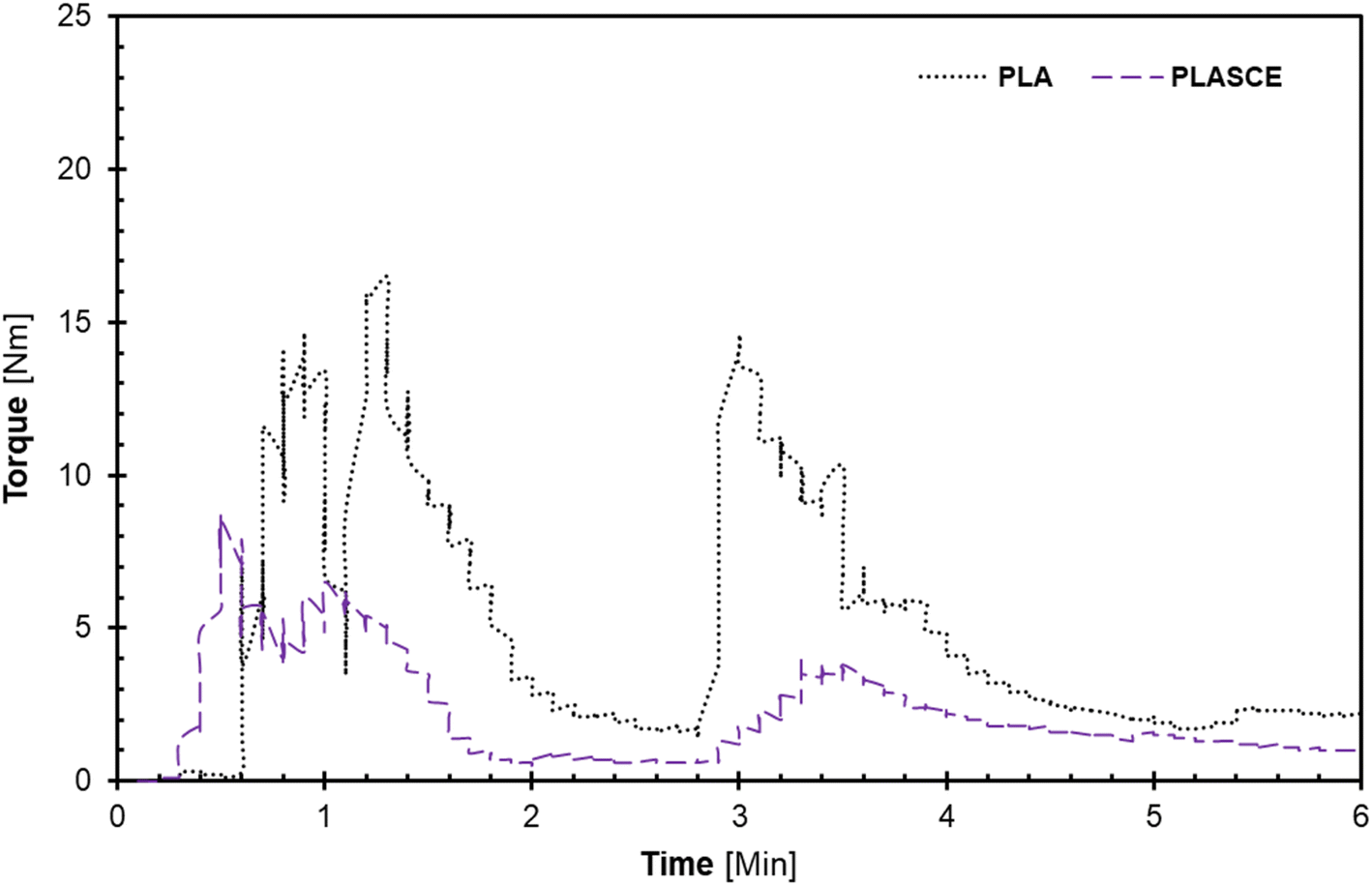
The melt-mixing of bionanocomposites was conducted using a Haake™ Rheomix QC internal mixer (Thermo Fisher Scientific, Waltham, MA USA) set at 165 °C at 75% fill factor. The PLA was divided into two parts and mixed in 2-steps: firstly, PLA alone was fed into the mixing chamber and mixed for 2 min at 30 rpm. Afterwards, the remaining PLA and additives (TA/PEG/CE) were carefully added into the mixing chamber and mixed for additional 3 min at 60 rpm to complete the 2nd mixing step. The evolution of torque on the mixer's rotors and mixing temperature as a function of time was plotted by Rheomix software as seen in Fig. 1(a and b). The additives improved processibility hence the significant reduction in torque [Nm] as seen in Fig. 1. To make the PLA/CNC bionanocomposites, the re-dispersed nano cellulose was introduced after the first mixing step. A pneumatically controlled laboratory hot press set at 165 °C was used to compress each specimen into approximately 0.3 mm sheets. The melt-mixed specimen was placed between Teflon sheets and compressed for 1 minute. Afterwards, the compressed sheets were air-cooled and shredded into extrudable pellets. The shredded samples were then dried at 50 °C for 24 h and vacuumed and sealed in zip-lock bags for further processing.
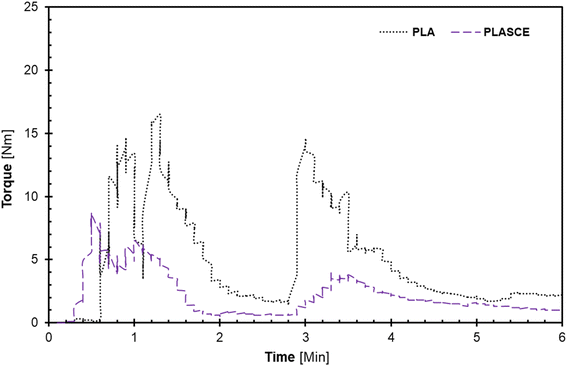 |
| | Fig. 1 A plot of torque vs. time plots during melt-mixing of PLA and plasticized PLA; (a) first, and (b) second mixing regimen. | |
2.5. Preparation of 3D printable filaments
The monofilaments of the various samples were drawn using a compact 3D filament extruder system (Felfil evo, Felfil, Torino). The extruder features a 2 mm nozzle and a K100 steel extrusion screw with a conic profile for an improved compression rate (12.7 L/D ratio). The extrusion temperature was set at 175 °C, and the extrusion speed was varied between 5–7 rpm depending on the sample. The filaments were extruded and spooled using a Felfil spooler system (Felfil, Torino). The spooler system also features a 4 radial high-speed fan array, that cools down the filaments before winding. Fig. 2 below shows the filament extrusion setup. Finally, the filaments were air dried in a convection oven set at 50 °C for 24 h and stored in vacuumed sealed bags for further processing.
 |
| | Fig. 2 Filament extrusion set-up, (A) single screw extruder, (B) spooling fan, (C) filament spooler. | |
2.6. 3D printing application
3D printing was performed using a desktop Fused Deposition Modelling (FDM) 3D printer (Wanhao Duplicator i3 plus) fitted with a 0.4 mm nozzle. Printing conditions were as follows, bed temperature of 50 °C, print speed of 60 mm s−1, 2 perimeter walls, 0 top and bottom layers, and a primary layer height of 0.26 mm. The printing temperature was optimized using a temperature tower calibration step and would be discussed later. A circular disk of 25 mm diameter and 1.5 mm thickness was 3D printed and used for melt-rheological analysis. All specimens were 3D printed lying flat with 90° raster angles, infill directions running parallel to the length in the XY direction. Simply3D® slicing software was used to slice the various STL files and program the 3D printing parameters.
3. Characterization techniques
3.1. Fourier Transform Infrared Spectroscopy (FTIR)
The cellulose nanocrystals (CNCss) were analysed using a TENSOR II FTIR spectrophotometer (BRUKER, Billerica, MA, USA) fitted with a pure diamond crystal and Platinum Attenuated Total Reflectance (ATR) accessory. The FTIR spectrum was recorded by scanning the sample 30 times at 1 cm−1 resolution between 4000 and 400 cm−1. CNCs were collected from the top, middle and bottom areas and tested to check for reproducibility. FTIR spectroscopy was also used to analyse the chemical structure of all the PLA and PLA/CNC bionanocomposite filaments.
3.2. Crystallinity analysis using X-ray powder diffraction (XRD)
A D2 PHASER benchtop powder X-ray diffractometer (BRUKER Billerica, MA, USA) was used to elucidate the crystallinity of the freeze-dried CNC. The samples were dried at 50 °C for 12 hours, afterwards the X-ray diffraction (XRD) pattern was measured over the angular 2θ = 5–40° range with a scanning step of 0.02°/step and 4 s per step with sample rotations using a Cu k-α1 radiation source (λ = 0.15406 nm). A silicon (Si) zero background sample holder was used, and the sample rotated in the horizontal plane at 10 rpm. In addition, a 3 mm air scatter slit was in place to reduce the low-angle erroneous air scattering that could be misinterpreted. After subtracting the air scattering, the diffraction patterns acquired were resolved into crystalline reflections and non-crystalline scattering using DIFFRAC.EVA software. The crystallinity index (Ic) was estimated using the semi-empirical method proposed by Segal et al.29eqn (1);| | 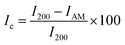 | (1) |
The I200 represents the height maximum diffraction peak of the (200) lattice (at 2θ = 21°) and the IAM was deduced from the minimum between the 200 and 110 lattice peaks (IAM, 2θ = 18°). The I200 represents both the crystalline and amorphous cellulose components, while IAM represents only the amorphous component. The crystallite size was calculated using the Scherrer equation as represented in eqn (2):30
| | 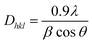 | (2) |
where
Dhkl = crystallite size in the direction normal to the
hkl lattice planes,
λ = X-ray radiation wavelength (1.542 Å),
β = full width at half-maximum (FWHM) of the diffraction peak in radians, and
θ = corresponding Bragg angle.
3.3. Polarised optical microscopy (POM)
Samples of the re-dispersed CNCs were examined under a Leica S6D light microscope (Leica Microsystems, Germany). A few drops of the CNCs sample were placed on a glass slide and allowed to dry in a desiccator. Afterwards, the glass slide containing the CNCs specimen was viewed under polarised and unpolarised light. Subsequently, images were captured using a 3.1-megapixel Leica EC3 colour camera (Leica Microsystems, Germany) at 64× magnification.
3.4. Scanning electron microscopy
High-resolution images of filament cross-sections were acquired using a JSM-IT100 InTouchScope™ scanning electron microscope (SEM) (JEOL Ltd. Japan). Filaments were cryo-fractured in liquid nitrogen and then attached to a metal substrate using double-sided carbon tape. Gold coating of the filament specimen was performed using a DESK V sputter coater (DENTONVACUUM, NJ, USA).
3.5. Thermal analysis
3.5.1. Thermal degradation of cellulose nanocrystals (CNCs).
Thermal degradation analysis of the cellulose nanocrystal (CNCs) specimen was conducted on a thermogravimetric analyser (Discovery Series TG 5500, TA Instrument). Platinum samples and reference pans were used for this experiment. About ∼4 mg of CNCs sample was heated at a rate of 10 °C min−1 from ambient temperature (∼20 °C) to 600 °C. This experiment was conducted under a nitrogen atmosphere at a gas flow rate of 100 mL min−1.
3.5.2. Melt crystallization analysis of bionanocomposites.
The melt-crystallization behaviour of the bionanocomposites was studied using a differential scanning calorimeter (Discovery series, TA Instruments). About ∼1.5 mg sample was weighed and sealed in an aluminium pan whereas an empty aluminium pan was used as reference. The sample was heated from 30 °C to 200 °C at a rate of 20 °C min−1 to erase the previous heat history. Afterwards, the melt crystallization behaviour was measured by cooling the samples at 5 °C min−1 from 200 °C to 0 °C. This was followed by a heating scan from 0 °C to 200 °C at a heating rate of 20 °C min−1. The heating and cooling cycles were conducted under nitrogen as the purge gas, with a flow rate of 25 mL min−1 for all samples. The first heating cycle was performed on all the samples to erase the previous heat history.
3.5.3. Thermal stability analysis of bionanocomposite filaments.
The thermal behaviour of all filament samples was studied on a simultaneous differential scanning calorimeter and thermogravimetric analyser (DSC-TGA) (SDT Q600, TA instrument, New Castle, USA). Samples weighing about ∼6.5 mg were carefully placed in ceramic pans and heated from ambient temperature (∼25 °C) until 600 °C at a 10 °C min−1 heating rate under a nitrogen purge atmosphere at a flow rate of 100 mL min−1.
3.6. Rheological analysis of bionanocomposites
The melt rheological analysis was conducted using an Anton-Paar modular compact rheometer (MCR-502) fitted with a 25 mm (diameter) parallel plate measuring system and a Peltier temperature control system. Circular-shaped (25 mm diameter and 1.5 mm thickness) test specimens were 3D printed for this experiment. Strain sweep measurements were conducted in the 0.1–100% deformation range at 10 rad s−1 to determine the linear viscoelastic region (LVR). Based on the LVR, dynamic frequency sweep measurements (0.1–100 rad s−1) were performed on all the samples at a constant temperature (190 °C) fixed strain (1%) and 1 mm gap size. Low-temperature drying and conditioning of the test specimen were performed at 45 °C for 5 hours before rheological analysis to avoid moisture absorption and thermal ageing. Each sample was analysed in triplicates and the data presented is the averaged result obtained from each sample.
3.7. Tensile analysis of filaments
Tensile analysis was performed using a Tinius Olsen tensile tester fitted with a loadcell of 10 kN loadcell. A gauge length of 25 mm was set throughout the analysis. Filaments were tested at a 50 mm min−1 speed according to ASTM D638 standard.
4. Results and discussion
4.1. Morphological analysis of re-dispersed cellulose nanocrystals
Fig. 3 shows the pictures of freeze-dried cellulose nanocrystals (CNCs) before and after re-dispersion in a mixture of polar protic (ethanol) and aprotic (acetone and ethyl acetate) solvents. The re-dispersion was achieved using a mixture of protic (ethanol) and aprotic (acetone and ethyl acetate) polar solvents. The mechanical stirring and ultra-sonication performed during this step aided the diffusion of solvents onto the surfaces of the CNCs bundles. Furthermore, the strong hydrogen bonding between the cellulose fibres became weakened because of the interactions with the polar solvents. The dissolution of cellulose in a mixture of aprotic and protic solvents involves solvent diffusion and chain disentanglement processes, made possible because of the weakening of the strong hydrogen bonds between agglomerated CNCs.31 Fortunati et al. (2012)20 employed the use of a surfactant to mitigate the agglomeration of CNC during the freeze-drying process, however, the process required additional use of chemicals.
 |
| | Fig. 3 Freeze-dried cellulose nanocrystals (CNCs) before and after re-dispersion in a mixture of polar protic and aprotic solvents. | |
The optical microscopy image presented in Fig. 4A revealed the size anisotropy of CNCs fibres after redispersion, whereas the image captured under polarised light in Fig. 4B reveals the birefringence of aggregated cellulose nanocrystals. The observed birefringence is characteristic of CNCs; this behaviour emanates from the refraction of polarised light passing through highly ordered cellulose crystals.32
 |
| | Fig. 4 Optical microscopy image of re-dispersed cellulose nanocrystals (CNCs); (A), high contrast image under normal light, (B) CNCs showing birefringence under polarised light (64× magnification). | |
4.2. Chemical composition analysis of cellulose nanocrystals
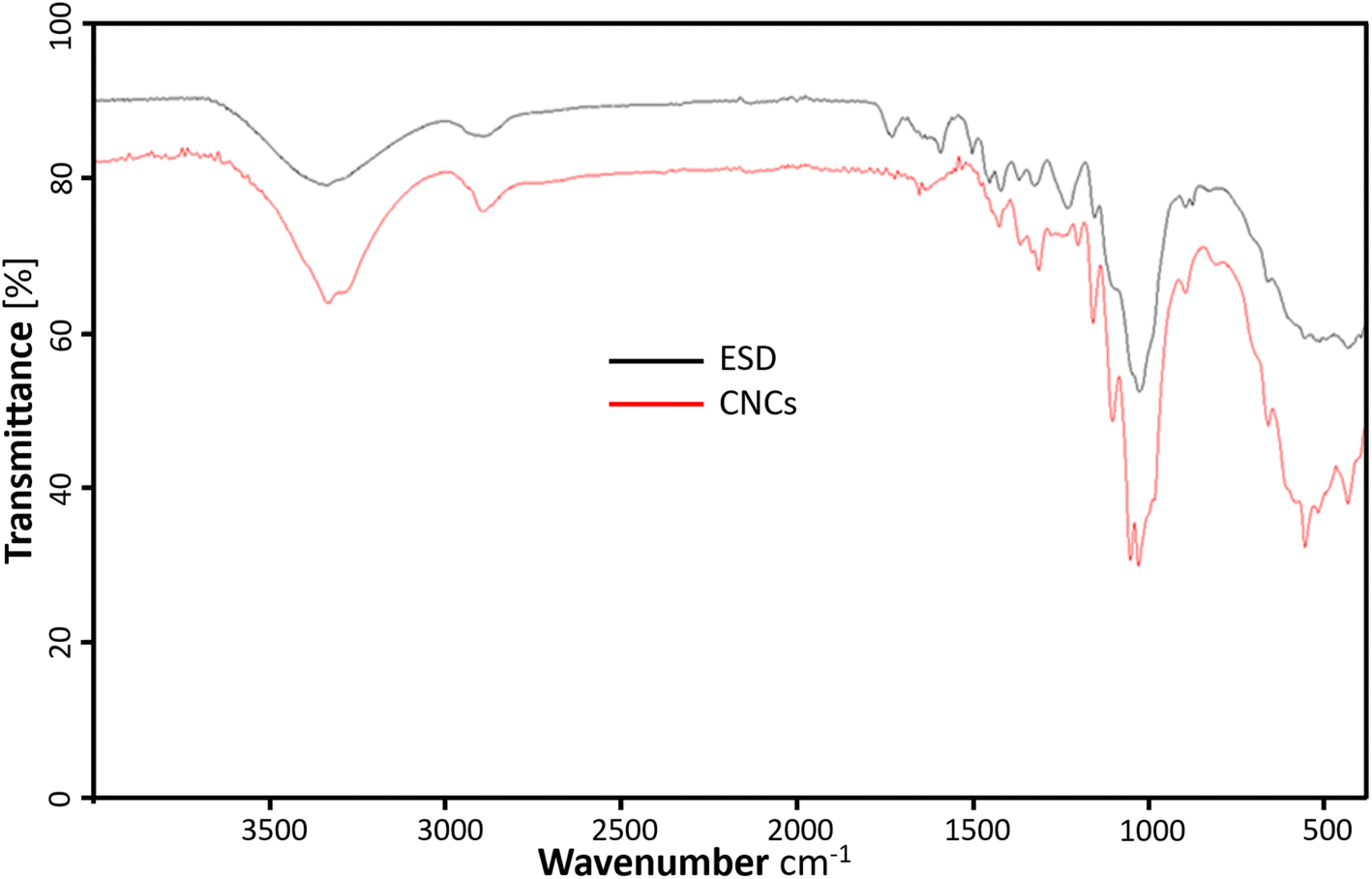
The FTIR spectrum of the freeze-dried cellulose nanocrystals (CNCs) and Eucalyptus saw-dust (ESD) are represented in Fig. 5. The CNCs show characteristic adsorption of chemical functional groups present in cellulosic materials. The prominent peaks include –O–H stretching vibrations responsible for the broad bands ranging from 3000–3600 cm−1. C–H stretching of methylene groups in cellulose around 2898.66 cm−1. Also, the peak around 1620 cm−1 is consistent with the –O–H bending vibrations of absorbed water and hydrogen-bonded hydroxyl groups of cellulose.33 The peak around 1450 cm−1 is assigned to –CH2 bending vibrations and the peak at 1339 cm−1 is assigned to –C–H asymmetric deformation of the crystalline domains.34 In the fingerprint region, the stretching vibrations from the –C–O–C– glucose and pyranose ring skeleton of cellulose are assigned to the 1160 and 1047 cm−1 peaks.35 Elanthikkal et al.36 suggested that the increase in the vibration observed at these peaks corresponds with increasing cellulose content. Furthermore, the C–O stretching of the β-glycosidic linkages between glucose rings in cellulose is attributed to the small peak at 897 cm−1.37 The characteristic aromatic ring vibration of lignin usually seen around 1230.54 cm−1 and 1503.84 cm−1 are absent in CNCs but present in ESD.16 Also, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of hemicellulose and lignin (1600–1750 cm−1) present in ESD is almost negligible in the spectrum of CNCs. This indicates the lignin and hemicellulose components were removed during the extraction of CNCs.16,38 There is no evidence of surface-absorbed sulfate groups39 as the characteristic 800 cm−1 peak from S–O bond stretching vibrations is not noticeable.
O stretching of hemicellulose and lignin (1600–1750 cm−1) present in ESD is almost negligible in the spectrum of CNCs. This indicates the lignin and hemicellulose components were removed during the extraction of CNCs.16,38 There is no evidence of surface-absorbed sulfate groups39 as the characteristic 800 cm−1 peak from S–O bond stretching vibrations is not noticeable.
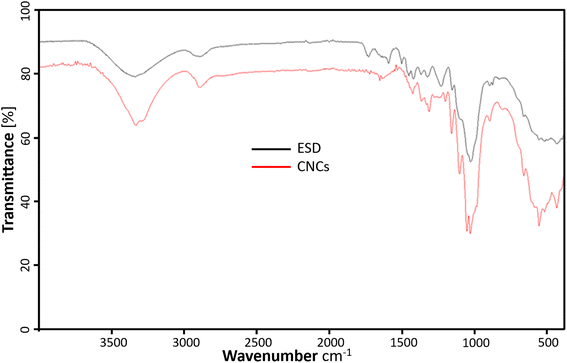 |
| | Fig. 5 ATR-FTIR profiles showing the chemical composition of cellulose nanocrystals (red), and Eucalyptus grandis saw dust (black). | |
4.3. XRD crystallinity analysis of cellulose nanocrystals (CNCs)
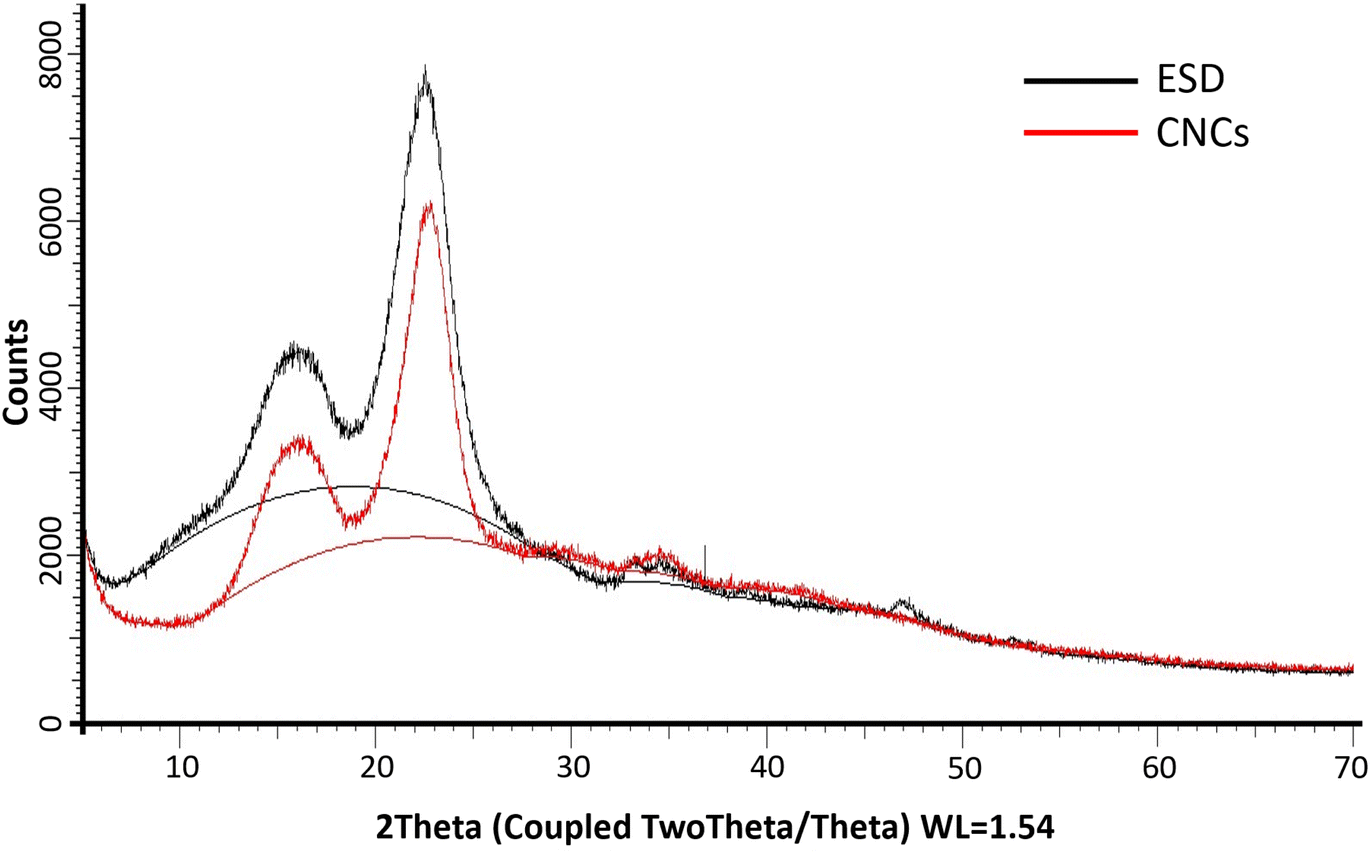
The XRD diffractograms of the freeze-dried CNCs and eucalyptus saw dust (ESD) exhibited patterns consistent with the crystalline structure of cellulose type Iβ (ref. 39) (Fig. 6). The peaks observed at 2θ = 14–17°, 23° and 35° are assigned to the (1−10) and (110), (200) and (004) lattice planes of monoclinic cellulose type Iβ lattice.40,41 The empirically determined crystallinity index (Ic%) was calculated to be 70% for CNCs, and 71.92% for ESD. The slight reduction in Ic% indicates minimal loss of crystalline domains during the extraction of cellulose nanocrystals. Noteworthily, Ic% only describes the relative among of crystalline and amorphous domains present, so it can only be used as a basis of comparison as it provides no information about the crystallinity of the crystalline region or the size of crystallites.29,39 The crystallite size (Dhkl) was deduced using the Scherrer equation and was found to be 36.6 Å for CNCs compared to 29.55 Å for ESD. The larger crystallite size indicates the successful removal of amorphous cellulose.
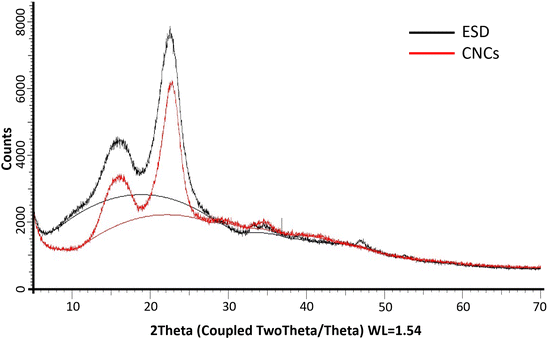 |
| | Fig. 6 PXRD patterns of Eucalyptus grandis saw dust (black) and extracted cellulose nanocrystals (red); (solid lines between 2θ = 10° and 30° show amorphous hump). | |
4.4. Thermal stability analysis of cellulose nanocrystals (CNCs)
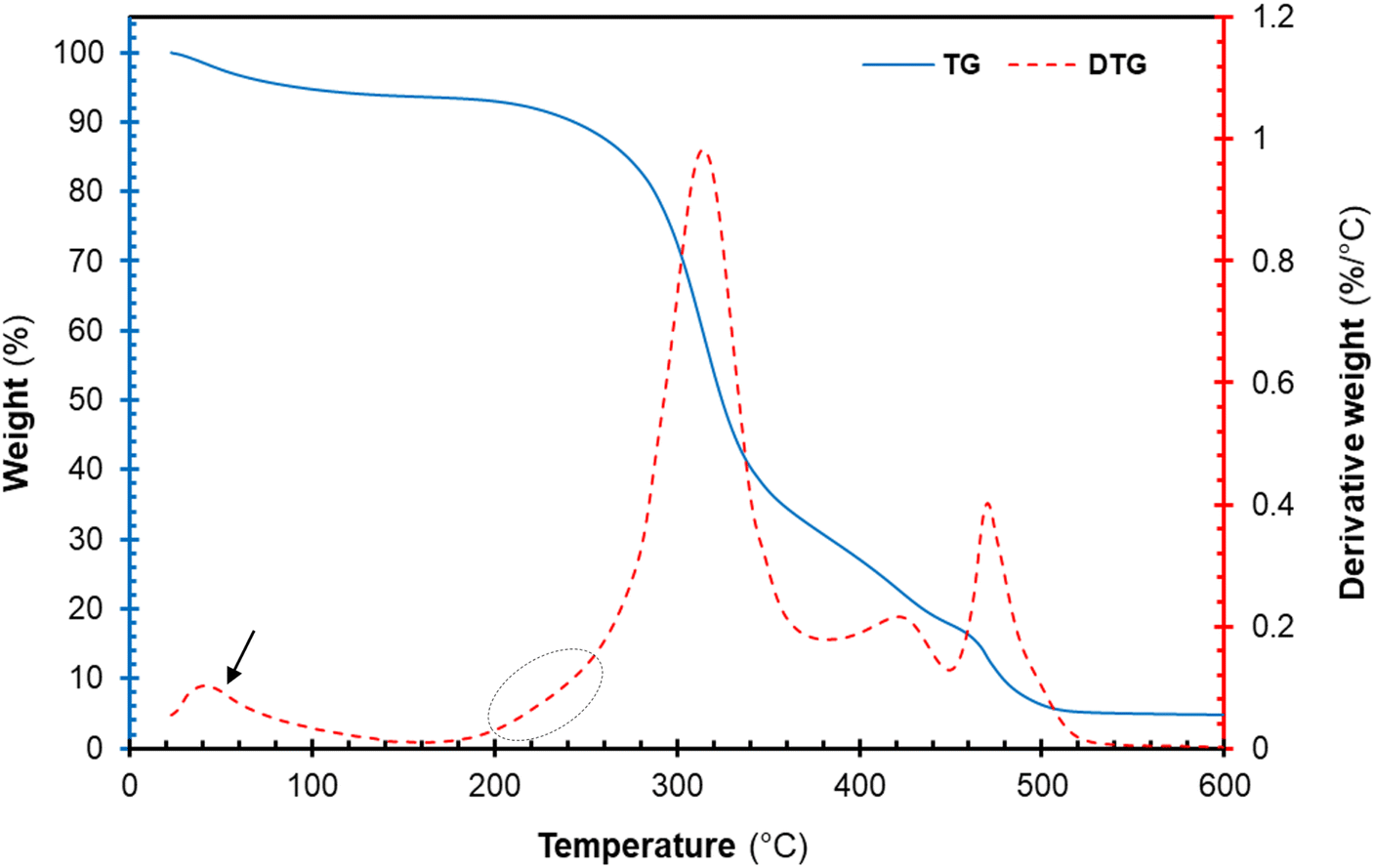
The thermal degradation profile of the freeze-dried cellulose nanocrystal (CNCs) is represented in Fig. 7. The thermogravimetric (TG) and derivative thermogravimetric (DTG) plots of CNCs displayed an early weight loss around 40–100 °C. This early weight loss is attributed to the evaporation of volatiles and moistures present in each sample.16,42 Furthermore, the TG and DTG plots of the cellulose component displayed multi-step degradation as seen in Fig. 7. The early decomposition (circled in Fig. 7) is attributed to the thermal degradation of highly accessible sulphated cellulose regions resulting from excessive acid hydrolysis.16,43 The sulphate groups present on the surfaces of CNCs are known to catalyse the thermal degradation process.20 Roman and Winter (2004)44 reported that the introduction of sulphate groups on the surface of CNCs during sulphuric acid hydrolysis significantly lowers the activation energies of thermal degradation. Nonetheless, the thermal degradation of cellulose glycosyl regions was not affected by sulphuric acid occurring at 315.54 °C. Furthermore, the peaks around 400–480 °C are attributed to the decomposition of thermal stable pyrolysis products formed during the degradation of the cellulose region charred residues occurring around. Overall, the degradation of the CNCs proceeded via dehydration, depolymerization and decomposition of glycosyl regions, similar to observations reported in the literature.16,42
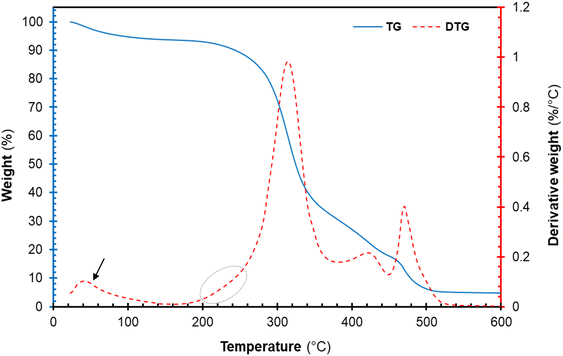 |
| | Fig. 7 Thermal decomposition profile of cellulose nanocrystals (CNCs): thermogravimetric curves (solid blue line), derivative TG curve (red broken lines). | |
4.5. Analysis of the chemical structure of bionanocomposite filaments
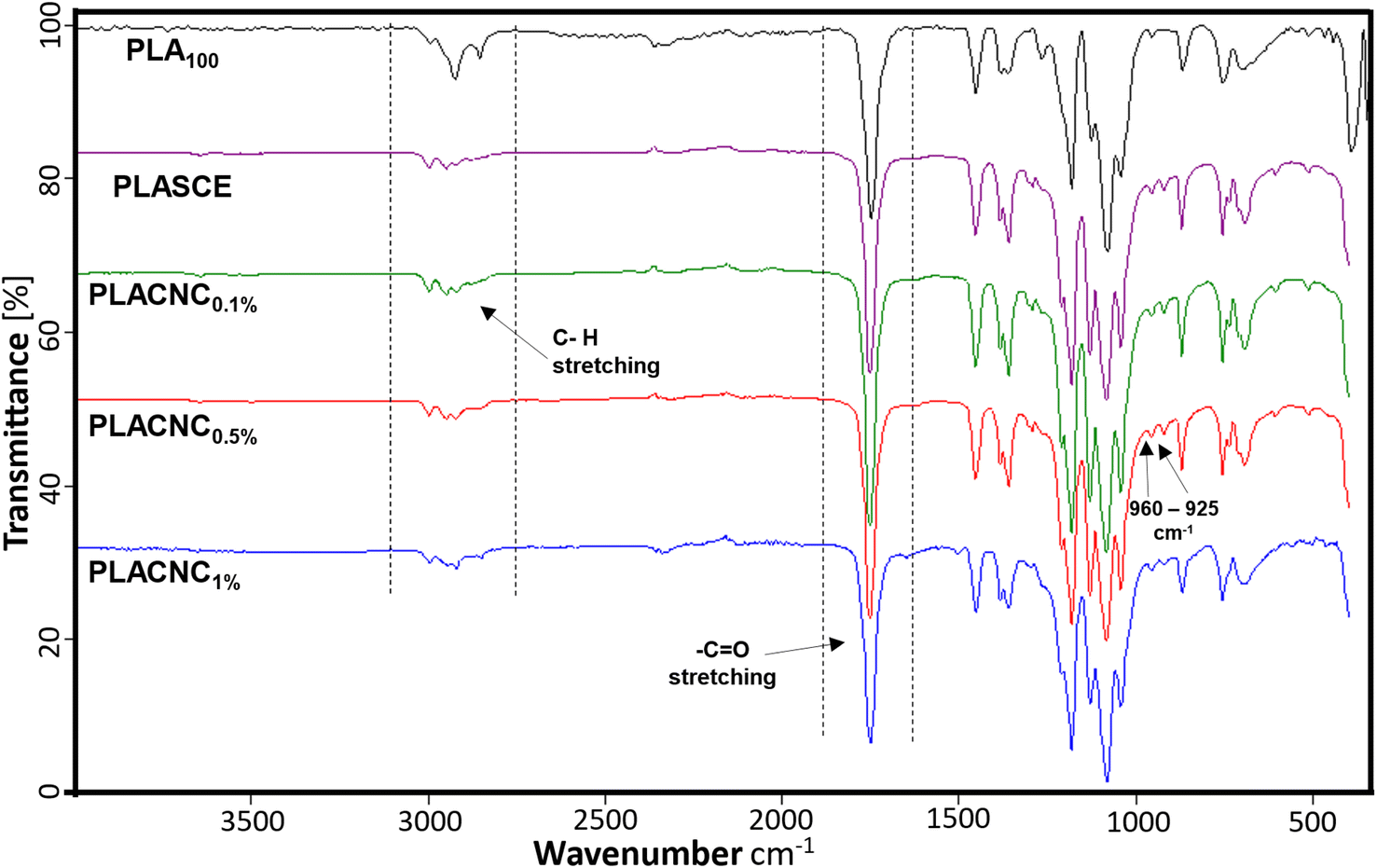
Fig. 8 shows the FTIR spectra of the neat PLA, plasticized PLA (PLASCE) and the PLA/CNCs bionanocomposite filaments. All the specimens exhibited transmittance peaks consistent with the chemical functional groups present in PLA. The peaks in the 3050–2900 cm−1 range are assigned to the C–H stretching. The CO stretching and –CH3 asymmetric bending vibrations are assigned to the peaks at 1750 cm−1 and 1450 cm−1 respectively. The 1260 cm−1 peak is assigned to carbonyl (CO) bending vibrations, whilst the skeletal stretching vibrations of the PLA-ester groups (C–O–C) are assigned to 1200–700 cm−1 peaks. Additionally, the peaks at 960 cm−1 and 925 cm−1 seen in the FTIR spectra of bionanocomposite filaments are assigned to amorphous and crystalline vibrations of PLA.45 The filaments containing CNCs, and additives all exhibited higher carbonyl signals around 1750 cm−1, whilst their methyl group signals (3050–2900 cm−1) were reduced. This observation is attributed to the reaction between PLA and the multifunctional chain extender, which formed new carbonyl bonds. Grigora et al. (2021)46 also reported a similar observation after studying the influence of multifunctional chain extenders on the chemical structure of PLA using FTIR spectroscopy.
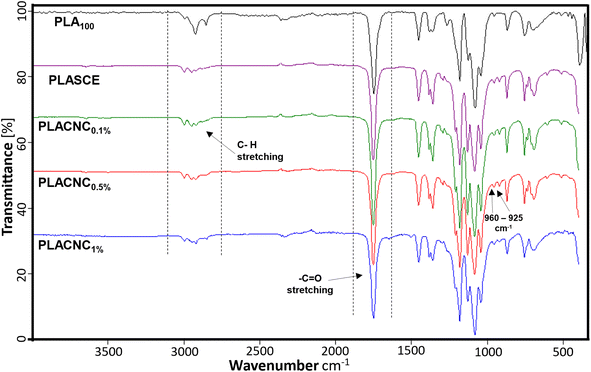 |
| | Fig. 8 Chemical composition of PLA and PLA/CNC bionanocomposites filaments. | |
4.6. Thermal analysis of PLA/CNC bionanocomposite filaments
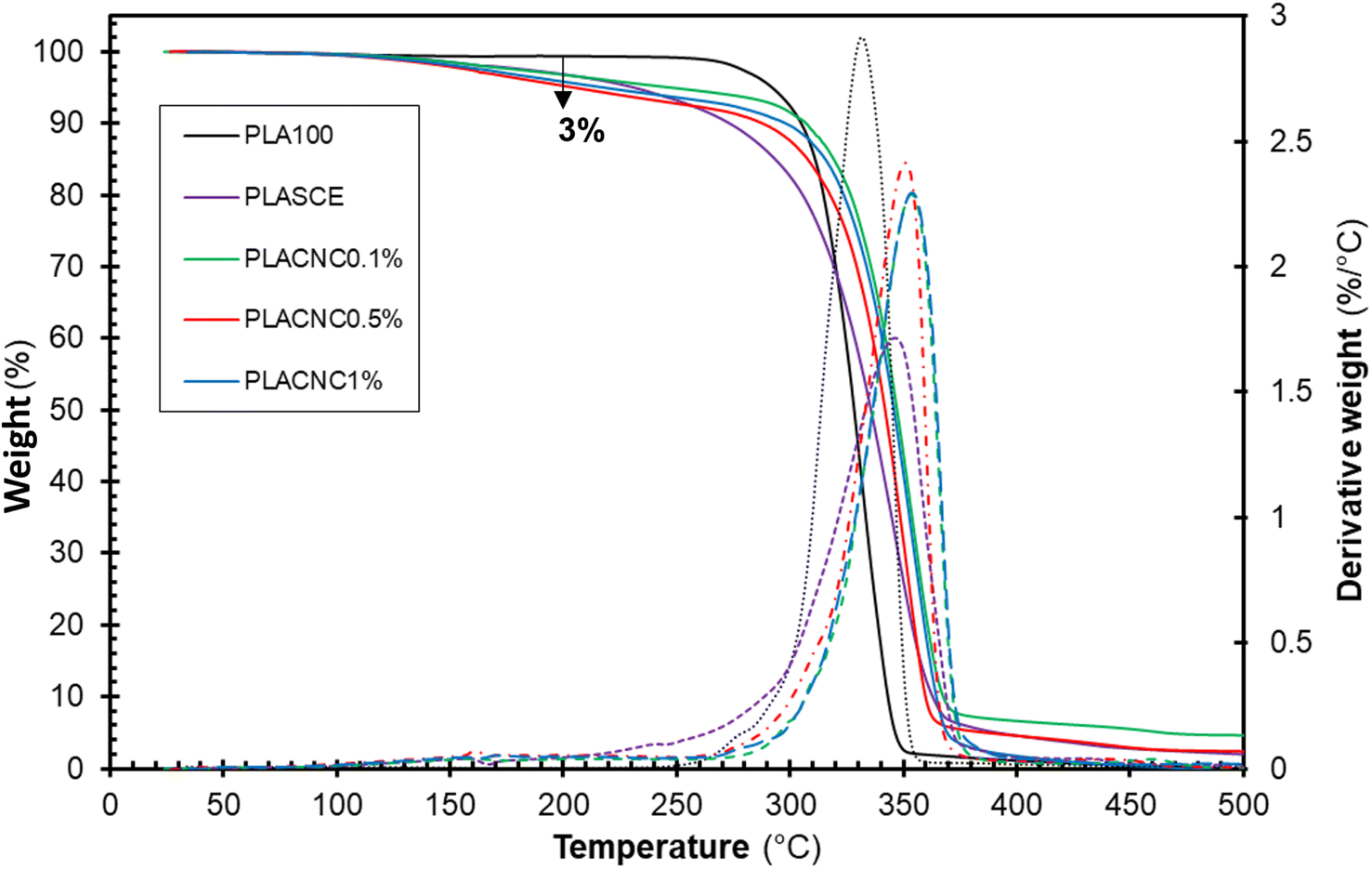
Fig. 9 shows the thermogravimetric weight loss (TG) and derivative (DTG) curves of the filaments of each specimen. The plasticized PLA (PLASCE) and the bionanocomposite filaments all exhibited a weight loss event around 150 °C. This weight loss is attributed to the evaporation of the plasticizer and is absent in the TG/DTG profile of the neat PLA (PLA100). However, only 3 wt% of the plasticizer was lost at 200 °C, which is above the maximum 3D printing temperature used for bionanocomposite. The main weight loss started around 270 °C and represents the onset of thermal degradation of cellulose nanocrystals and PLA matrix. The main degradation peak occurred at 331.8 °C for neat PLA (PLA100), 346.79 °C for the plasticized sample (PLASCE) and around 350 °C for the samples containing CNCs. A comparison of the peak degradation temperature reveals a ∼20 °C improvement upon the addition of CNCs. The increase in thermal stability of PLA upon incorporating CNCs has been reported elsewhere.23,47 Furthermore, the diminished height of the DTG curve for the bio-based plasticized sample (PLASCE) is attributed to decreased rate of thermal decomposition. However, the height of the DTG curves of the bionanocomposite specimens infers that the addition of CNCs improved the rate of thermal degradation.
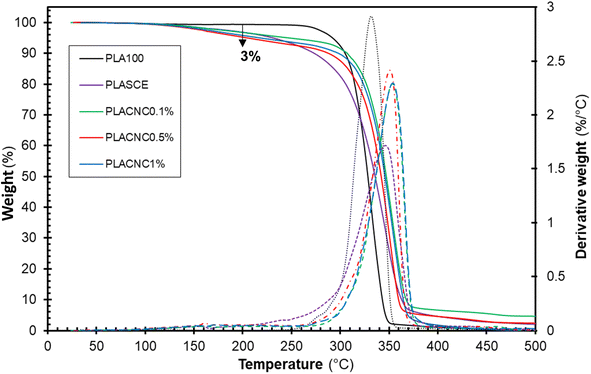 |
| | Fig. 9 Thermal decomposition profiles showing thermogravimetric (TG) and derivative (DTG) curves of the PLA and bionanocomposites filaments. | |
4.7. Melt-crystallization analysis of PLA/CNC biocomposites
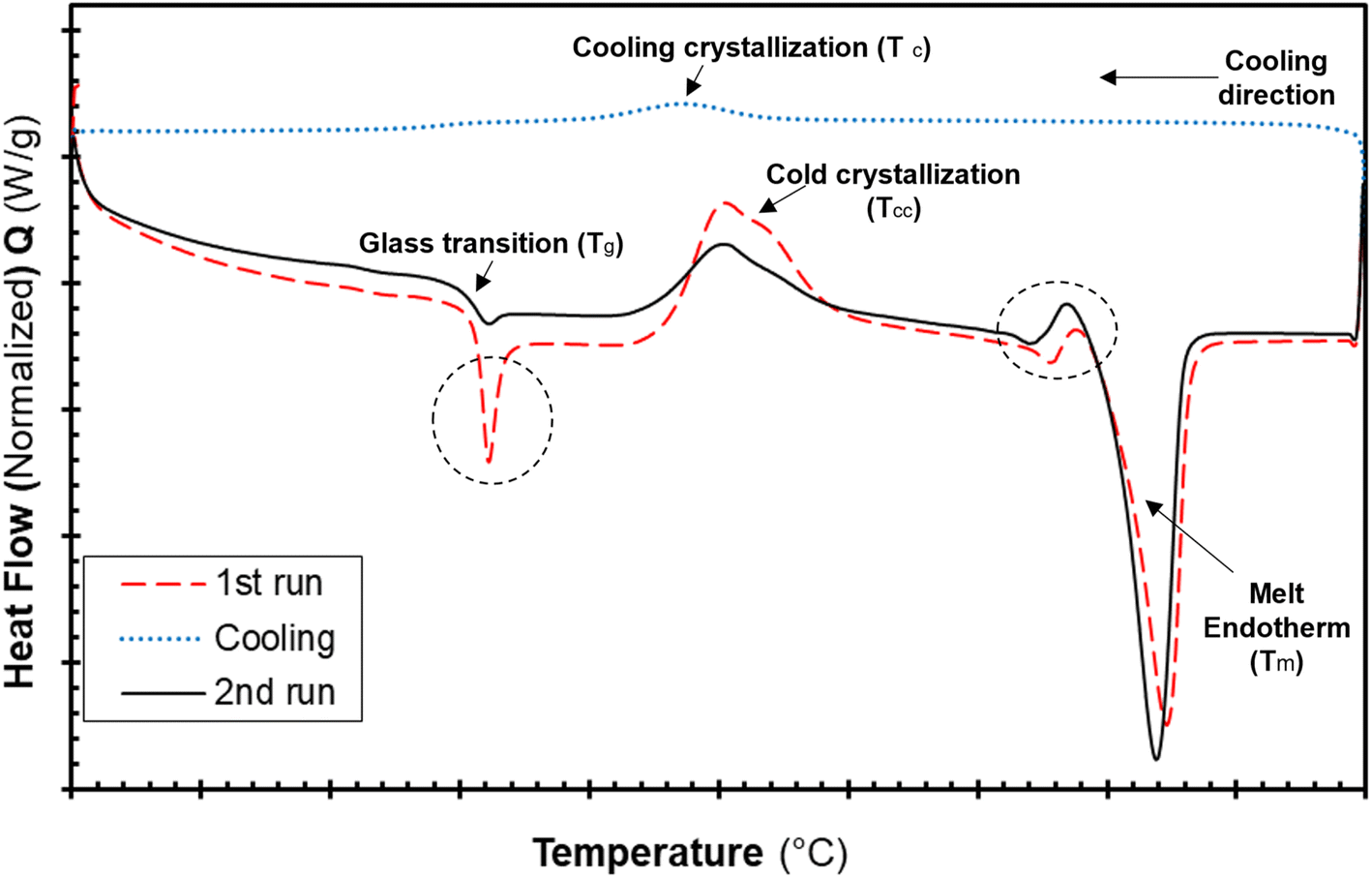
The differential calorimetric profile in Fig. 10 shows the first heating run, cooling and the 2nd heating run of PLA100. Thermal events consistent with PLA such as glass transition (Tg ∼ 62.72 °C), cold crystallization (Tcc ∼ 100.19 °C), crystalline melt temperature (Tm ∼ 167.47 °C) and cooling crystallization (Tc ∼ 94.18 °C) were recorded. The endotherm is visible just after Tg has been linked with a complex thermal ageing phenomenon.27 However, this endotherm is largely invisible in the second heating run as the previous history was erased. The additional exothermic peak which occurred just before the main melting point, is attributed to the additional melting/recrystallization mechanism. The percentage degree of crystallinity from the cooling and second heating run was mathematically deduced using the equations below.| | 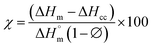 | (3) |
| | 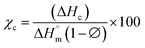 | (4) |
where χc is the crystallinity of the cooling cycle (eqn (3)) and χ refers to the overall degree of crystallinity after the 2nd heating cycle (eqn (4)), ΔHm is the enthalpy of melting for the 2nd heating run, ΔHc is the enthalpy of crystallization for the cooling run and ΔHcc is the enthalpy of cold crystallization. The values of ΔHm, ΔHc and ΔHcc were deduced by integrating the area of the thermal transition.  is the melting enthalpy of a 100% crystalline PLA reported as 93 J g−1 in literature,4 whereas (1 − Ø) represents the mass fraction of PLA. Table 2 shows all the melt-crystallization information obtained from the DSC analysis.
is the melting enthalpy of a 100% crystalline PLA reported as 93 J g−1 in literature,4 whereas (1 − Ø) represents the mass fraction of PLA. Table 2 shows all the melt-crystallization information obtained from the DSC analysis.
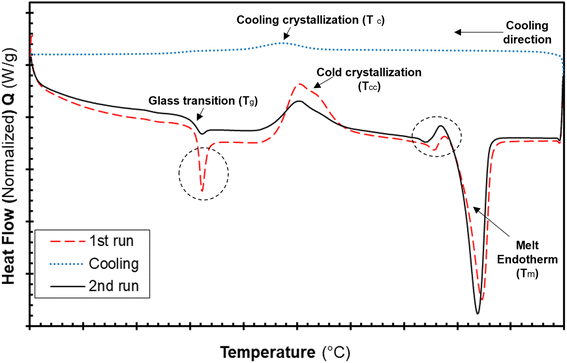 |
| | Fig. 10 DSC thermograms of the neat PLA filaments showing the first, cooling and second heating runs. | |
Table 2 DSC data for the cooling and second heating run PLA and PLA/CNC bionanocomposites
| Sample description |
Cooling run |
2nd heating run |
Crystallinity |
Glass transition |
|
T
c (°C) |
ΔHc (J g−1) |
χ
c (%) |
T
cc (°C) |
ΔHcc (J g−1) |
T
m (°C) |
ΔHm (J g−1) |
Χ (%) |
T
g (°C) |
| PLA100 |
94.18 |
11.40 |
12.26 |
100.19 |
12.09 |
167.47 |
40.34 |
30.38 |
62.72 |
| PLASCE |
97.11 |
18.62 |
20.02 |
102.41 |
4.27 |
166.89 |
27.96 |
22.67 |
56.72 |
| PLACNC0.1% |
90.62 |
16.28 |
17.51 |
103.53 |
12.88 |
167 |
29.79 |
16.17 |
56.29 |
| PLACNC0.5% |
93.33 |
25.89 |
27.84 |
97.05 |
6.068 |
165.88 |
31.17 |
23.89 |
54.48 |
| PLACNC1% |
89.15 |
21.7 |
23.33 |
95.47 |
7.457 |
166.63 |
35.3 |
26.35 |
49.22 |
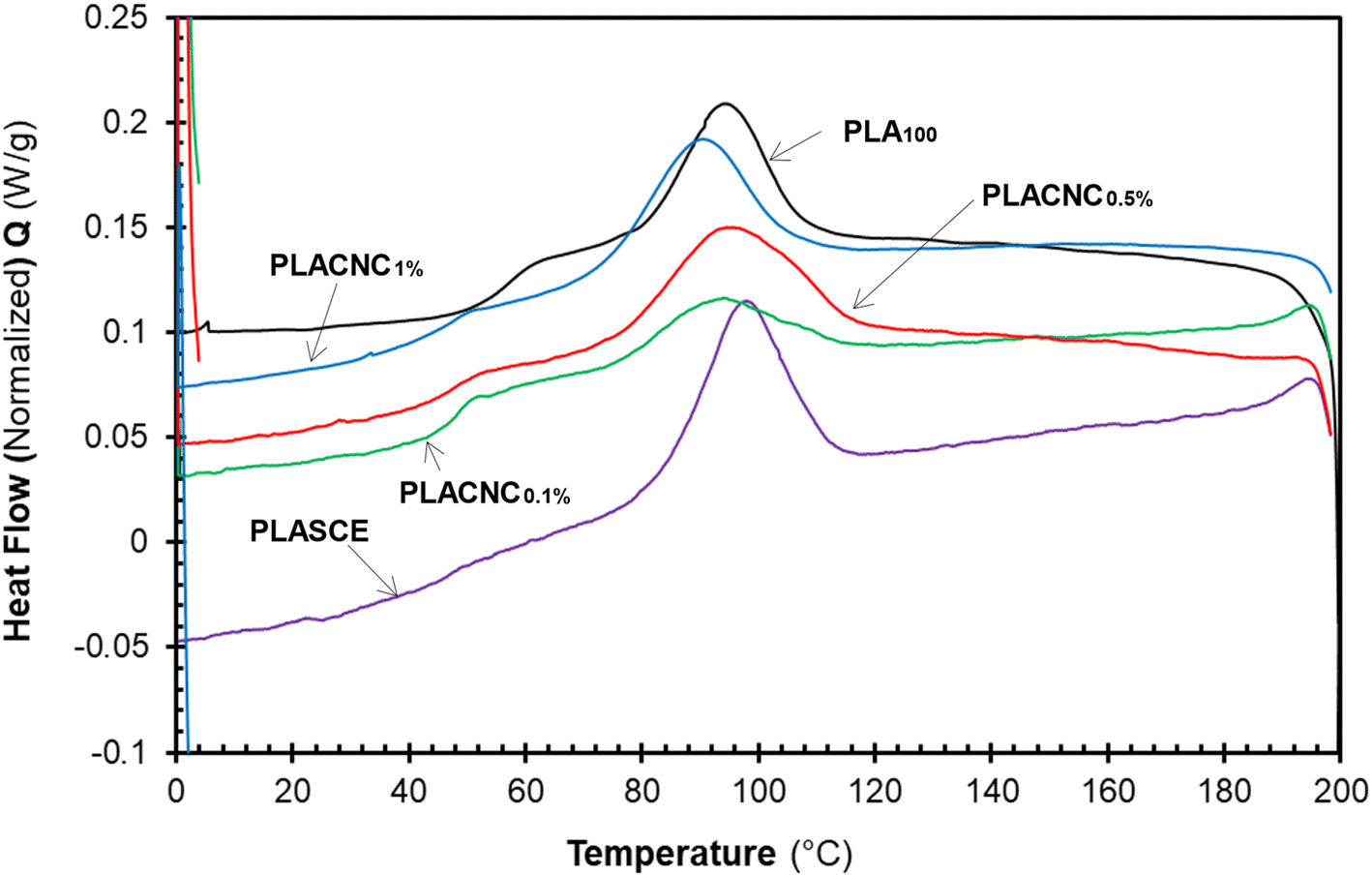
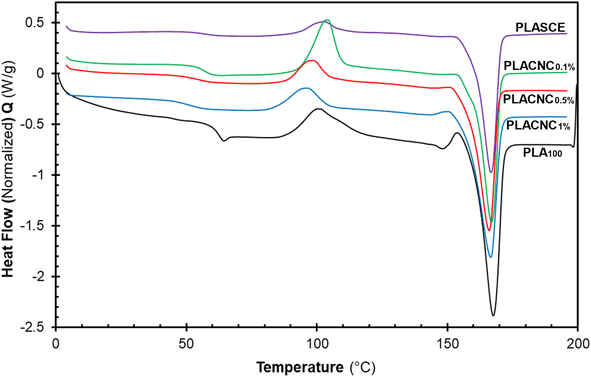
The results in Table 2 reveal a slight delay in the onset of melt crystallization temperature (Tc) after the addition of CNCs. The delay in Tc is more prominent in the specimen containing 1% CNCs (Tc ∼ 89.15 °C). The crystallization behaviour of polymers in the presence of nanoparticles is controlled by two competing phenomena. The first phenomenon occurs when the nanoparticles act as nucleation sites for crystal growth, thus resulting in increased crystallinity.27 The second phenomenon leads to lower crystallinity as the nanoparticles physically hinder crystal growth.48 Although the CNCs present in the bionanocomposites hindered the migration and diffusion of polymer chains to the surface of the growing crystal. The slight decrease in crystallization temperature and the broad Tc exotherms in Fig. 11 is attributed to the second phenomenon. The χc of the neat PLA was determined to be 12.26%; however, the specimens containing CNCs exhibited higher crystallization during the cooling run. Compared to PLA100, their χc increased by 5%, 15% and 11% for PLACNC0.1%, PLACNC0.5%, PLACNC1% respectively. This observation agrees with the results presented by Bagheriasl et al. (2018).49
 |
| | Fig. 11 DSC cooling curves showing the melt crystallization behaviour of the PLA and bionanocomposites filaments. | |
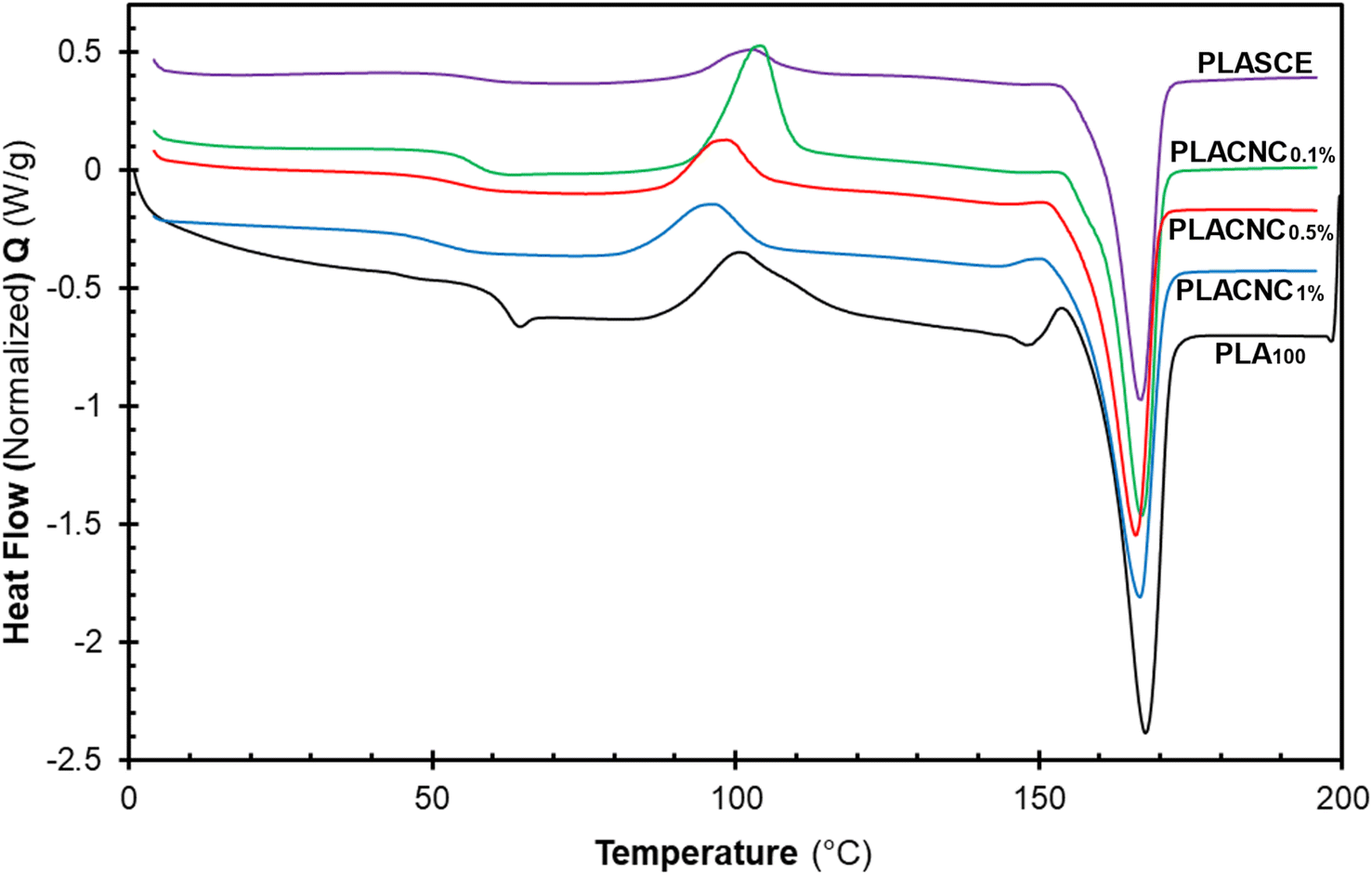
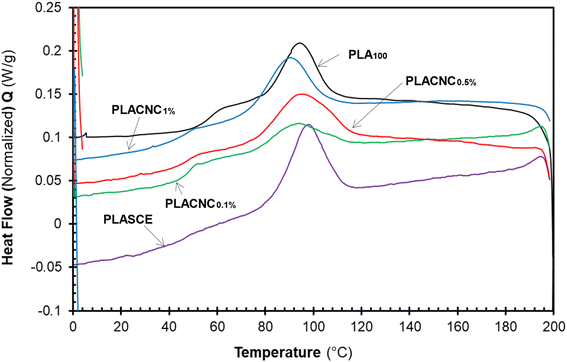
Fig. 12 shows the evolution of thermal events during the second heating run for all the specimens. The glass transition temperature (Tg) of the plasticized specimen (PLASCE) and the bionanocomposites were barely noticeable. This is attributed to the increased mobility of the PLA chains due to the lubricating effect of the plasticizer. The lubricating effect also resulted in the lowering of the Tg temperatures in all the samples containing plasticizer as shown in Table 2. Also, Fig. 12 shows that the PLA crystallites' melting endotherms (Tm) occurred at around 166 °C in all the samples. However, PLA100 exhibited the highest melting enthalpy (ΔHm ∼ 40.34 J g−1) due to the higher cold crystallization (Tcc) behaviour and the residual crystallization that occurred just before the onset of melting. The high ΔHm corresponded with the increased crystallinity of the PLA100 specimen (χ ∼ 30.38%). Meanwhile, the plasticized specimen (PLASCE) exhibited the lowest melting enthalpy (ΔHm ∼ 27.96 J g−1). The incorporation of CNCs into plasticized PLA resulted in slightly higher melting enthalpy compared to plasticized PLA where ΔHm was calculated as ∼29.79, 31.17 and 35.30 J g−1 for samples containing 0.1, 0.5 and 1% CNC respectively. Furthermore, the plasticized PLASCE and bionanocomposites exhibited lower overall percentage crystallinity (χ) which was calculated from the second melting endotherm. The low overall crystallinity (χ) is attributed to the slow cold crystallization (Tcc) process caused by the lubricating effect of the plasticizer.
 |
| | Fig. 12 The evolution of thermal events in the second heating cycle of the PLA and bionanocomposites filaments. | |
4.8. Tensile analysis of PLA/CNC biocomposite filaments
The tensile properties of the bionanocomposite filaments were compared to the neat (PLA100) and plasticized (PLASCE) filaments. Table 3 lists the tensile strength, elongation at the break and maximum breaking force of the analysed filament materials. The neat PLA filament (PLA100) was very brittle and repeatedly broke during tensile analysis. However, the CNCs-reinforced PLA bionanocomposites filaments exhibited improved flexibility and tensile strength as shown in Table 3. As expected, the maximum force at break (N) was higher for samples with improved tensile strength. The improvements in tensile strength and flexibility are attributed to the homogenous dispersion of CNCs, good interfacial interactions and successful plasticization. Additionally, CNCs could have also improved the tensile properties of PLA by influencing the formation of structurally stable crystallites during the cooling process.50 The neat PLA filament (PLA100) exhibited a tensile strength of 22.5 ± 3.8 MPa and 0.3% elongation at break. Meanwhile, the bionanocomposite filaments exhibited significant improvement in tensile strength of 35.3 ± 1.79 (PLACNC0.1%), 33.4 ± 2.07 (PLACNC0.5%), and 35.1 ± 4.07 (PLACNC1%) MPa respectively. The observed increase in tensile strength is attributed to the effective stress transfer from the PLA matrix to the CNCs, which is in agreement with similar findings reported in the literature.24,28
Table 3 Summary of the tensile results of the PLA and bionanocomposites filaments
| Sample description |
CNCsa content (wt%) |
Tensile strength (MPa) |
Maximum force (N) |
Elongation at break (%) |
|
Cellulose nanocrystal.
|
| PLA100 |
0 |
22.5 (±3.80) |
53.5 (±9.00) |
0.3 (±0.05) |
| PLASCE |
0 |
28.9 (±1.52) |
62.7 (±6.03) |
15.8 (±4.34) |
| PLACNC0.1% |
0.1 |
35.3 (±1.79) |
78.4 (±18.3) |
15.4 (±4.31) |
| PLACNC0.5% |
0.5 |
33.4 (±2.07) |
71.3 (±2.38) |
13.1 (±4.61) |
| PLACNC1% |
1 |
35.1 (±4.07) |
86.9 (±8.42) |
25.5 (±5.38) |
4.9. Analysis of PLA and PLA/CNC bionanocomposite filaments
The poor melt strength of virgin PLA affects its draw ability during filament production. This directly affects the final diameter and ovality (i.e., the deviation from circularity) of PLA filaments. Also, the filaments produced using PLA alone were very brittle and unsuitable for FDM 3D printing. These issues are undesirable as they can lead to inadequate extrusion and cause catastrophic breakage of filaments during FDM 3D printing. The biobased additives (triacetin/polyethylene glycol) and food-safe-multifunctional chain extender (Joncryl CE) were added to mitigate these issues. Fig. 13 shows that the modification resulted in a noticeable improvement in the melt strength of PLA during filament extrusion. The minimal ovality seen in the scanning electron microscopy images presented in Fig. 13 also revealed improved filament roundness for the modified specimen. The biobased additives served as a processing aid, plasticizer and compatibilizer, whereas the multifunctional chain extender (Joncryl CE) improved the melt strength of the PLA during filament production. The improvement in melt strength has been attributed to the formation of a covalent bond between the hydroxyl of PLA and the epoxide group of the Joncryl CE.46 Also, the addition of Joncryl CE results in increased PLA molecular weight and mitigates the occurrence of thermos-hydrolytic degradation.46,51,52 Other major contributors to the diameter inconsistencies during filament production include feeding rate, compression, extrusion speed, temperature, rate of cooling, and filament winding speed.
 |
| | Fig. 13 Enhanced melt strength during filament extrusion: minimal drooping of filament during extrusion. | |
Fig. 14 shows the higher SEM magnification images of the cryo-fractured bionanocomposite filament. The surface topography shows distinct brittle failure patterns which were expected since the filaments were cryo-fractured. Also, the SEM image reveals bundles of CNCs embedded into the PLA matrix. A few locations where CNCs got pulled out of the PLA matrix are also visible in figure. The average flat width of the CNCs embedded in the PLA matrix was 1.26 micrometres. The small oval-shaped voids indicate the clear boundary between the phased separated plasticizer and PLA matrix. The plasticizer only permeates and interacts with the amorphous domains in PLA; whereas its phase separates from the PLA crystalline domains.53
 |
| | Fig. 14 Higher magnification SEM image of PLA/CNC bionanocomposite filament. | |
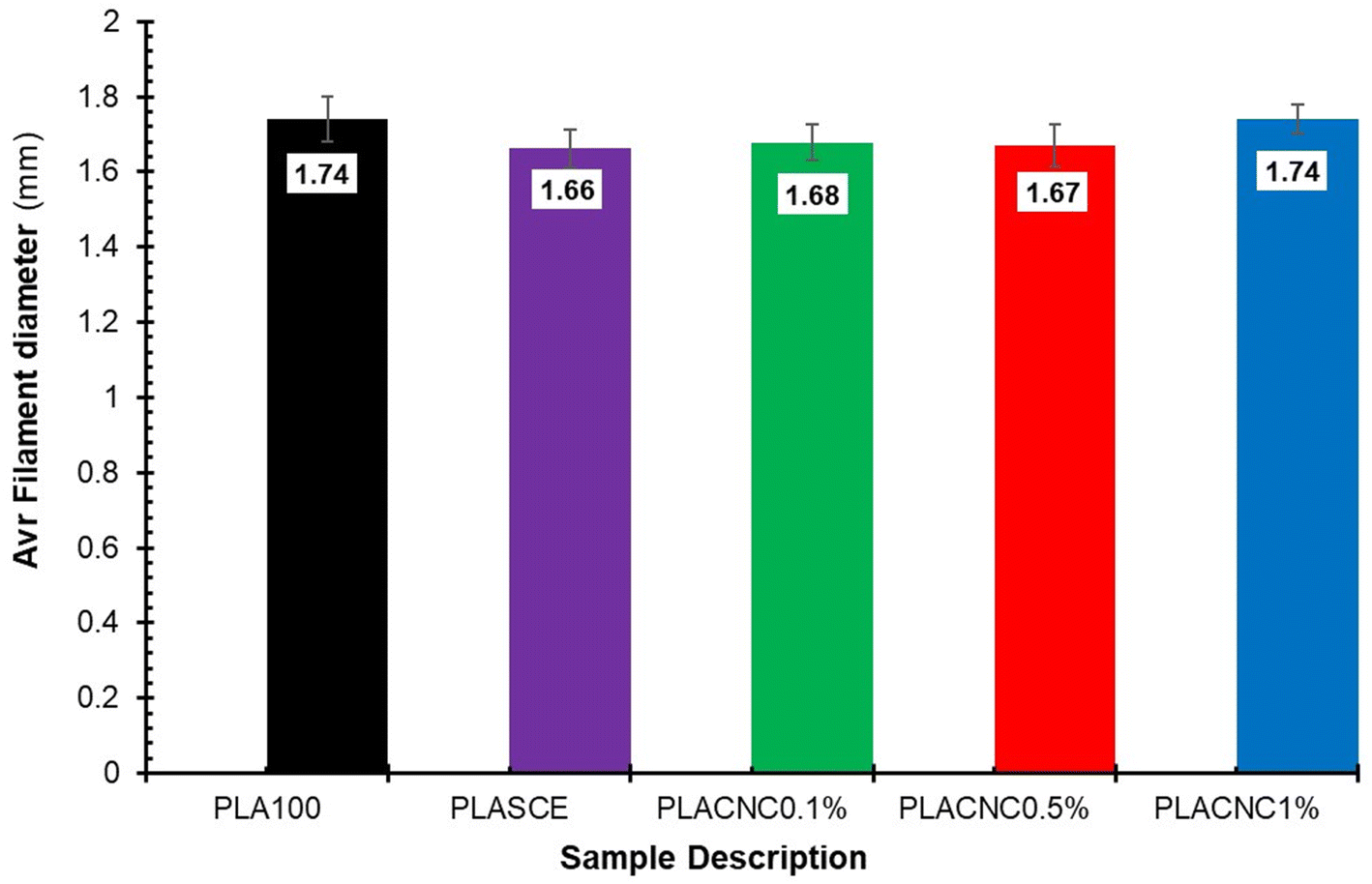
Although the real-time filament diameter was displayed by the Felfil™ filament spooler, the system is not equipped to store the measured data. So, a Vernier calliper was used to measure filament diameters at 10 locations across a 1 meter-long filament sample. Fig. 15 shows the comparison between the average diameters of neat PLA, plasticised PLA, and bionanocomposite filament. There are no major differences between the averaged filament diameter of PLASCE (1.66 mm ± 0.05), PLACNC0.1% (1.68 mm ± 0.05), and PLACNC0.5% (1.67 mm ± 0.06). The average filament diameter of PLA100 and PLACNC1% was 1.74 mm ± 0.06 and 1.74 mm ± 0.04 respectively. Most FDM 3D printers are factory calibrated to operate using a 1.75 mm filament. Using filaments with smaller or larger diameters increases the risks of inconsistent extrusion as fewer or more material gets deposited. The resulting over- or under-extrusion can severely affect the final overall dimensional and structural integrity of 3D-printed objects. In this study, the applicability of the filaments was adapted by increasing the extrusion rate of the FDM printer from 100% to 105%. This adjustment implemented on the slicing software interface of Simplify3D™ was done to compensate for the variation in the bionanocomposite filaments.
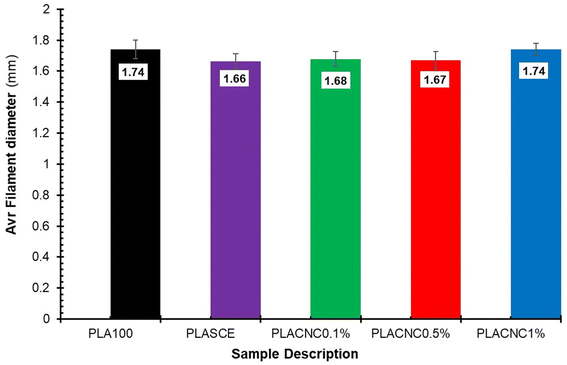 |
| | Fig. 15 The average filament diameters of the PLA and PLA/CNC bionanocomposites filaments. | |
4.10. Analysis of 3D printability of PLA/CNC biocomposite filaments
The 3D printability of all the filament materials was tested on a fused deposition modelling (FDM) 3D printer. The brittle nature of the neat PLA filament (PLA100) caused it to repeatedly shatter during 3D printing. However, the plasticized filament (PLASCE) and the bionanocomposite filaments did not break during 3D printing. The ability of these filaments to withstand the dynamic stresses exerted on them during the 3D printing process is attributed to successful plasticization. Furthermore, a temperature calibration step involving the 3D printing of a vertical (temperature) tower with multiple horizontal slabs (bridge) was conducted. Fig. 16 shows the temperature tower printed using PLACNC0.5% (left) and PLASCE (right) filaments. The printing temperature was varied between 190–170 °C by lowering the temperature by 5 °C at the last layer of the bridge. It was observed that the quality of bridging deteriorated at lower printing temperatures. The deterioration of bridging at lower temperatures indicates that the 3D printer struggled to extrude/flow. Therefore, 190 and 185 °C appear to be the optimal 3D printing temperature for the materials. Afterwards, the PLACNC0.5% filament was employed in the 3D printing of a spiral vase with Voronoi tessellation. The successful 3D printing of these parts highlights the potential use of the bionanocomposite filaments in the production of complex items.
 |
| | Fig. 16 FDM 3D printed temperature test tower and Voronoi tessellation vase using PLA/CNC bionanocomposite filament. | |
4.11. Rheological analysis of 3D printed PLA/CNC bionanocomposites
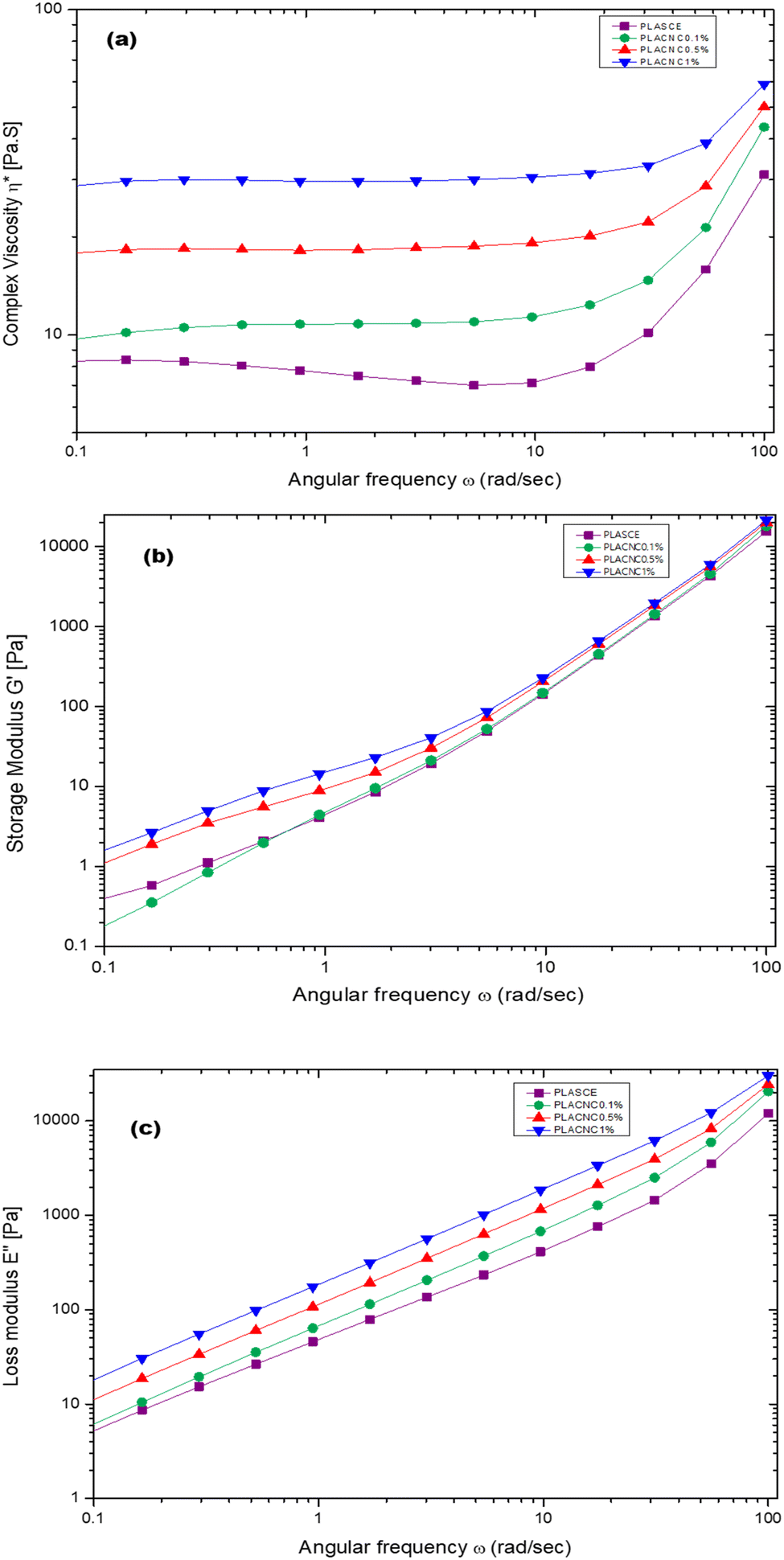
Fig. 17a shows the plots of complex viscosity (η*) as a function of angular frequency (ω) for plasticised PLA (PLASCE) and bionanocomposites. The incorporation of cellulose nanocrystals (CNCs) resulted in higher melt viscosity, which is attributed to reduced free volume and restricted mobility of PLA chains. PLACNC1% showed the highest viscosity indicative of higher mobility inhibition of the PLA chains. However, a gradual decrease in η* between ω = 0.1–10 rad s−1 for the PLASCE specimen indicates pseudoplastic (shear thinning) behaviour because of polymer chain disentanglement. Contrarily, the samples containing CNCs all exhibited constant complex viscosity (η*) (Newtonian behaviour) within the same shear range region (ω = 0.1–10 rad s−1). Nevertheless, all the specimens exhibited a sudden increase in complex viscosity (η*) at higher angular frequencies (ω = >10 rad s−1). This sudden shear thickening (dilatant) behaviour is attributed to increased PLA chain entanglement which is more prominent in PLASCE.
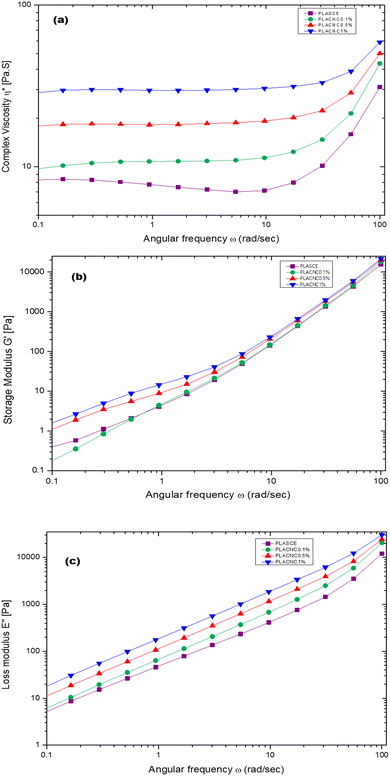 |
| | Fig. 17 Melt rheological behaviour of PLA/CNC bionanocomposites showing the evolution of; (a) complex viscosity (η*), (b) storage (G′) modulus, (c) loss (G′′) modulus. | |
Meanwhile, the storage (G′) and loss (G′′) modulus increased with higher loading of CNCs as shown in Fig. 17b and c. The increase in G′ and G′′ is also attributed to the restricted mobility of PLA chains from the interactions with CNCs. The observed increase of the storage modulus (G′) and low angular frequency (ω) indicates good CNCs dispersion and increased interaction with the PLA chains.54Fig. 17b clearly shows the effect of this interaction on storage modulus G′ was prominent in PLACNC0.5% and PLACNC1% specimens between ω = 0.1–5 rad s−1. However, all the samples exhibited similar G′ values at higher angular frequencies as the interaction between CNCs and PLA chains decayed.
5. Summary
This work demonstrated the process of fabricating PLA/CNC bionanocomposites using freeze-dried cellulose nanocrystals re-dispersed in a mixture of green solvents. The study followed the method of direct liquid feeding of re-dispersed CNCs into the PLA melt which is a scalable process. The melt strength, viscosity and processibility of the bionanocomposites were improved by incorporating bio-based additives and food grade multifunctional chain extender. Thermal analysis of the bionanocomposites revealed improved crystallization behaviour during cooling, which resulted in the formation of mechanically and structurally stable PLA crystallites. This improvement together with the reinforcing effect of CNCs resulted in better tensile strength, tensile modulus, and flexibility of the PLA/CNC bionanocomposites filaments. The morphological analysis of the bionanocomposite filaments revealed improved filament ovality and adequate CNCs dispersion in the PLA matrix. Furthermore, the printability of the PLA/CNC bionanocomposite filaments was demonstrated using a fused deposition modelling (FDM) 3D printer. The 3D printing temperature optimization test showed that the filaments can be processed well below the thermal decomposition temperature.
Author contributions
“Conceptualization, V. C. A. and M. J. J.; methodology, V. C. A.; 3D printing, V. C. A.; formal analysis, V. C. A. and S. P. H.; investigation, V. C. A.; resources, M. J. J., S. P. H. and J. A.; data curation, V. C. A.; writing – original draft preparation, V. C. A., S. P. H. and M. J. J.; writing – review and editing, V. C. A., S. P. H. and M. J. J.; visualization, V. C. A.; supervision, M. J. J. and S. P. H.; project administration, M. J. J. and S. P. H.; funding acquisition, M. J. J., all authors have read and agreed to the published version of the manuscript.
Conflicts of interest
All the study participants provided informed consent, so there are no conflicts of interest to declare.
Acknowledgements
This research was funded by the Department of Science and Innovation (DSI), South Africa – Biorefinery Consortium Project – under the DSI Bioeconomy program, Grant number DST/CON0188/2017.
References
- S. Kumar, V. N. Kannan and G. Sankaranarayanan, Parameter optimization of ABS-M30i parts produced by fused deposition modelling for minimum surface roughness, Int. J. Current Eng. Technol., 2014, 3, 93–97 Search PubMed.
- B. Coppola, N. Cappetti, L. Di Maio, P. Scarfato and L. Incarnato, 3D printing of PLA/clay nanocomposites: Influence of printing temperature on printed samples properties, Materials, 2018, 11(10), 1947 CrossRef PubMed.
- D. Bagheriasl, F. Safdari, P. J. Carreau, C. Dubois and B. Riedl, Development of cellulose nanocrystal-reinforced polylactide: A comparative study on different preparation methods, Polym. Compos., 2019, 40(S1), E342–E349 CrossRef CAS.
- H. Li,
et al., Crystallisation, mechanical properties and rheological behaviour of PLA composites reinforced by surface modified microcrystalline cellulose, Plast., Rubber Compos., 2016, 45(4), 181–187 CrossRef CAS.
- N. Herrera, A. P. Mathew and K. Oksman, Plasticized polylactic acid/cellulose nanocomposites prepared using melt-extrusion and liquid feeding: Mechanical, thermal and optical properties, Compos. Sci. Technol., 2015, 106, 149–155 CrossRef CAS.
- F. Hassouna, J.-M. Raquez, F. Addiego, V. Toniazzo, P. Dubois and D. Ruch, New development on plasticized poly (lactide): Chemical grafting of citrate on PLA by reactive extrusion, Eur. Polym. J., 2012, 48(2), 404–415 CrossRef CAS.
- D. C. França,
et al., Tailoring PBAT/PLA/Babassu films for suitability of agriculture mulch application, J. Nat. Fibers, 2019, 16(7), 933–943 CrossRef.
- Y. Lyu, Y. Chen, Z. Lin, J. Zhang and X. Shi, Manipulating phase structure of biodegradable PLA/PBAT system: Effects on dynamic rheological responses and 3D printing, Compos. Sci. Technol., 2020, 200, 108399 CrossRef.
- M. J. John, N. Dyanti, T. Mokhena, V. Agbakoba and B. Sithole, Design and development of cellulosic bionanocomposites from forestry waste residues for 3d printing applications, Materials, 2021, 14(13), 3462 CrossRef CAS PubMed.
- N. M. Holden, A. M. Neill, J. C. Stout, D. O'Brien and M. A. Morris, Biocircularity: a framework to define sustainable, circular bioeconomy, Circ. Econ. Sustainability, 2023, 3(1), 77–91 CrossRef PubMed.
- A. Hachaichi, B. Kouini, L. K. Kian, M. Asim and M. Jawaid, Extraction and characterization of microcrystalline cellulose from date palm fibres using successive chemical treatments, J. Polym. Environ., 2021, 29(6), 1990–1999 CrossRef CAS.
- L. K. Kian, M. Jawaid, H. Ariffin and Z. Karim, Isolation and characterization of nanocrystalline cellulose from roselle-derived microcrystalline cellulose, Int. J. Biol. Macromol., 2018, 114, 54–63 CrossRef CAS PubMed.
- M. Pan, X. Zhou and M. Chen, Cellulose nanowhiskers isolation and properties from acid hydrolysis combined with high-pressure homogenization, BioResources, 2013, 8(1), 933–943 CrossRef.
- M. N. Angles and A. Dufresne, Plasticized starch/tunicin whiskers nanocomposites. 1. Structural analysis, Macromolecules, 2000, 33(22), 8344–8353 CrossRef CAS.
- M. Grunert and W. T. Winter, Nanocomposites of cellulose acetate butyrate reinforced with cellulose nanocrystals, J. Polym. Environ., 2002, 10(1), 27–30 CrossRef CAS.
- A. Mtibe, L. Z. Linganiso, A. P. Mathew, K. Oksman, M. J. John and R. D. Anandjiwala, A comparative study on properties of micro and nanopapers produced from cellulose and cellulose nanofibres, Carbohydr. Polym., 2015, 118, 1–8 CrossRef CAS PubMed.
- F. Fahma, S. Iwamoto, N. Hori, T. Iwata and A. Takemura, Effect of pre-acid-hydrolysis treatment on morphology and properties of cellulose nanowhiskers from coconut husk, Cellulose, 2011, 18(2), 443–450 CrossRef CAS.
- Y. Zhang,
et al., Poly (lactic acid)/cellulose nanocrystal composites via the Pickering emulsion approach: Rheological, thermal and mechanical properties, Int. J. Biol. Macromol., 2019, 137, 197–204 CrossRef CAS PubMed.
- K. Oksman, A. P. Mathew, D. Bondeson and I. Kvien, Manufacturing process of cellulose whiskers/polylactic acid nanocomposites, Compos. Sci. Technol., 2006, 66(15), 2776–2784 CrossRef CAS.
- E. Fortunati,
et al., Multifunctional bionanocomposite films of poly (lactic acid), cellulose nanocrystals and silver nanoparticles, Carbohydr. Polym., 2012, 87(2), 1596–1605 CrossRef CAS.
- S. D. Kumar, K. Venkadeshwaran and M. Aravindan, Fused deposition modelling of PLA reinforced with cellulose nano-crystals, Mater. Today: Proc., 2020, 33, 868–875 CrossRef.
-
D. Rigotti, A. Dorigato, A. Cataldi, L. Fambri, and A. Pegoretti, Nanocellulose as Reinforc-Ing Agent for Biodegradable Polymers in 3D Printing Fused Deposition Modelling, ECCM, 2018 Search PubMed.
- Q. Wang,
et al., Kinetic thermal behaviour of nanocellulose filled polylactic acid filament for fused filament fabrication 3D printing, J. Appl. Polym. Sci., 2020, 137(7), 48374 CrossRef CAS.
- N. D. Ahmad and M. W. Wildan, Preparation and properties of cellulose nanocrystals-reinforced Poly (lactic acid) composite filaments for 3D printing applications, Results Eng., 2023, 17, 100842 CrossRef CAS.
- M. Eichers, D. Bajwa, J. Shojaeiarani and S. Bajwa, Biobased plasticizer and cellulose nanocrystals improve mechanical properties of polylactic acid composites, Ind. Crops Prod., 2022, 183, 114981 CrossRef.
- V. C. Agbakoba, P. Hlangothi, J. Andrew and M. J. John, Mechanical and Shape Memory Properties of 3D-Printed Cellulose Nanocrystal (CNC)-Reinforced Polylactic Acid Bionanocomposites for Potential 4D Applications, Sustainability, 2022, 14(19), 12759 CrossRef CAS.
- A. N. Frone, S. Berlioz, J.-F. Chailan and D. M. Panaitescu, Morphology and thermal properties of PLA–cellulose nanofibers composites, Carbohydr. Polym., 2013, 91(1), 377–384 CrossRef CAS PubMed.
- J. Shojaeiarani, D. S. Bajwa and K. Hartman, Esterified cellulose nanocrystals as reinforcement in poly (lactic acid) nanocomposites, Cellulose, 2019, 26(4), 2349–2362 CrossRef CAS.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, An Empirical Method for Estimating the Degree of Crystallinity of Native Cellulose Using the X-Ray Diffractometer, Text. Res. J., 1959, 29(10), 786–794, DOI:10.1177/004051755902901003.
- J. I. Langford and A. Wilson, Scherrer after sixty years: a survey and some new results in the determination of crystallite size, J. Appl. Crystallogr., 1978, 11(2), 102–113 CrossRef CAS.
- L. Lin and K. Tsuchii, Dissolution behaviour of cellulose in a novel cellulose solvent, Carbohydr. Res., 2022, 511, 108490 CrossRef CAS PubMed.
- Y. Habibi, H. Chanzy and M. R. Vignon, TEMPO-mediated surface oxidation of cellulose whiskers, Cellulose, 2006, 13(6), 679–687 CrossRef CAS.
- W. He, S. Jiang, Q. Zhang and M. Pan, Isolation and characterization of cellulose nanofibers from Bambusa rigida, BioResources, 2013, 8(4), 5678–5689 CrossRef.
- G. Tonoli,
et al., Properties of cellulose micro/nanofibers obtained from eucalyptus pulp fibre treated with anaerobic digestate and high shear mixing, Cellulose, 2016, 23(2), 1239–1256 CrossRef CAS.
- Q. Lu, Z. Cai, F. Lin, L. Tang, S. Wang and B. Huang, Extraction of cellulose nanocrystals with a high yield of 88% by simultaneous mechanochemical activation and phosphotungstic acid hydrolysis, ACS Sustainable Chem. Eng., 2016, 4(4), 2165–2172 CrossRef CAS.
- S. Elanthikkal, U. Gopalakrishnapanicker, S. Varghese and J. T. Guthrie, Cellulose microfibres produced from banana plant wastes: Isolation and characterization, Carbohydr. Polym., 2010, 80(3), 852–859 CrossRef CAS.
- W. Chen, H. Yu, Y. Liu, P. Chen, M. Zhang and Y. Hai, Individualization of cellulose nanofibers from wood using high-intensity ultrasonication combined with chemical pretreatments, Carbohydr. Polym., 2011, 83(4), 1804–1811 CrossRef CAS.
- J.-M. Raquez,
et al., Surface-modification of cellulose nanowhiskers and their use as nanoreinforcers into polylactide: a sustainably-integrated approach, Compos. Sci. Technol., 2012, 72(5), 544–549 CrossRef CAS.
- D. Gaspar,
et al., Nanocrystalline cellulose applied simultaneously as the gate dielectric and the substrate in flexible field effect transistors, Nanotechnology, 2014, 25(9), 094008 CrossRef CAS PubMed.
- I. Besbes, S. Alila and S. Boufi, Nanofibrillated cellulose from TEMPO-oxidized eucalyptus fibres: effect of the carboxyl content, Carbohydr. Polym., 2011, 84(3), 975–983 CrossRef CAS.
- K. Heritage, J. Mann and L. Roldan-Gonzalez, Crystallinity and the structure of celluloses, J. Polym. Sci., Part A: Gen. Pap., 1963, 1(2), 671–685 CrossRef CAS.
- H. Yousefi, M. Faezipour, S. Hedjazi, M. M. Mousavi, Y. Azusa and A. H. Heidari, Comparative study of paper and nanopaper properties prepared from bacterial cellulose nanofibers and fibres/ground cellulose nanofibers of canola straw, Ind. Crops Prod., 2013, 43, 732–737 CrossRef CAS.
- H. A. Silvério, W. P. F. Neto, N. O. Dantas and D. Pasquini, Extraction and characterization of cellulose nanocrystals from corncob for application as reinforcing agent in nanocomposites, Ind. Crops Prod., 2013, 44, 427–436 CrossRef.
- M. Roman and W. T. Winter, Effect of sulfate groups from sulfuric acid hydrolysis on the thermal degradation behaviour of bacterial cellulose, Biomacromolecules, 2004, 5(5), 1671–1677 CrossRef CAS PubMed.
- F. Beltrán, M. De La Orden, V. Lorenzo, E. Pérez, M. Cerrada and J. M. Urreaga, Water-induced structural changes in poly (lactic acid) and PLLA-clay nanocomposites, Polymer, 2016, 107, 211–222 CrossRef.
- M.-E. Grigora,
et al., Influence of reactive chain extension on the properties of 3d printed poly (Lactic acid) constructs, Polymers, 2021, 13(9), 1381 CrossRef CAS PubMed.
- M. R. Ketabchi, M. Khalid, C. T. Ratnam and R. Walvekar, Mechanical and thermal properties of polylactic acid composites reinforced with cellulose nanoparticles extracted from kenaf fibre, Mater. Res. Express, 2016, 3(12), 125301 CrossRef.
- J.-G. Gwon,
et al., Mechanical and thermal properties of toluene diisocyanate-modified cellulose nanocrystal nanocomposites using semi-crystalline poly (lactic acid) as a base matrix, RSC Adv., 2016, 6(77), 73879–73886 RSC.
- D. Bagheriasl, P. J. Carreau, B. Riedl and C. Dubois, Enhanced properties of polylactide by incorporating cellulose nanocrystals, Polym. Compos., 2018, 39(8), 2685–2694 CrossRef CAS.
- J. Dong, C. Mei, J. Han, S. Lee and Q. Wu, 3D printed poly (lactic acid) composites with grafted cellulose nanofibers: Effect of nanofiber and post-fabrication annealing treatment on composite flexural properties, Addit. Manuf., 2019, 28, 621–628 CAS.
- N. Najafi, M. Heuzey, P. Carreau and P. M. Wood-Adams, Control of thermal degradation of polylactide (PLA)-clay nanocomposites using chain extenders, Polym. Degrad. Stab., 2012, 97(4), 554–565 CrossRef CAS.
- Q. Meng, M.-C. Heuzey and P. J. Carreau, Control of thermal degradation of polylactide/clay nanocomposites during melt processing by chain extension reaction, Polym. Degrad. Stab., 2012, 97(10), 2010–2020 CrossRef CAS.
- N. Ljungberg and B. Wesslen, The effects of plasticizers on the dynamic mechanical and thermal properties of poly (lactic acid), J. Appl. Polym. Sci., 2002, 86(5), 1227–1234 CrossRef CAS.
- F. Wu,
et al., Interfacial relaxation mechanisms in polymer nanocomposites through the rheological study on polymer/grafted nanoparticles, Polymer, 2016, 90, 264–275 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Percy
Hlangothi
b,
Jerome
Andrew
c and
Maya Jacob
John
ab
*ab,
Percy
Hlangothi
b,
Jerome
Andrew
c and
Maya Jacob
John
ab

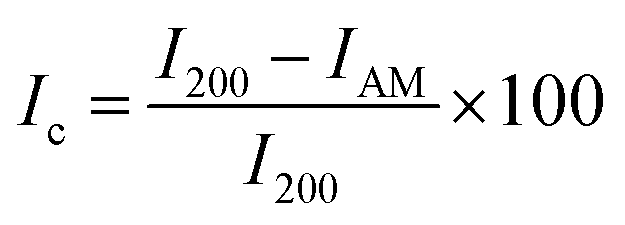
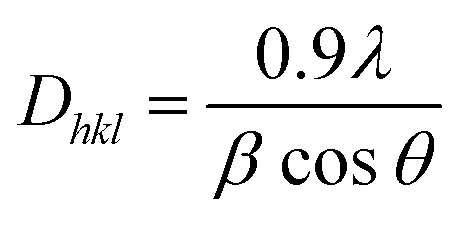


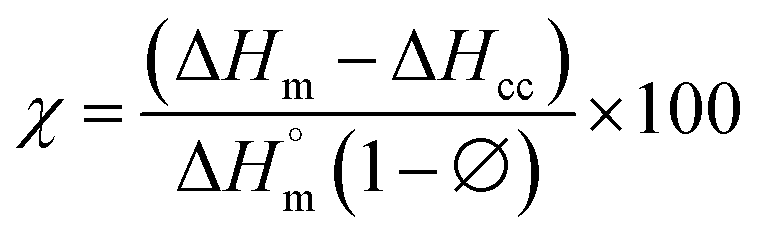
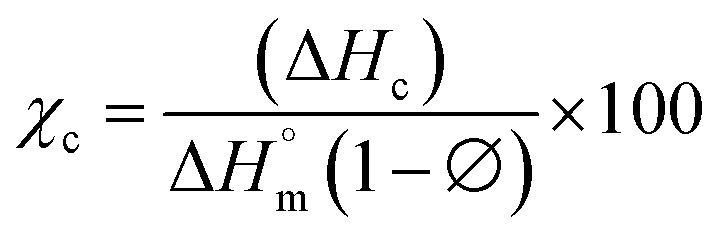
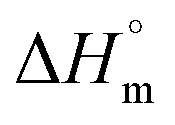 is the melting enthalpy of a 100% crystalline PLA reported as 93 J g−1 in literature,4 whereas (1 − Ø) represents the mass fraction of PLA. Table 2 shows all the melt-crystallization information obtained from the DSC analysis.
is the melting enthalpy of a 100% crystalline PLA reported as 93 J g−1 in literature,4 whereas (1 − Ø) represents the mass fraction of PLA. Table 2 shows all the melt-crystallization information obtained from the DSC analysis.