
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe smallest superatom Au4(PPh3)4I2 with two free electrons: synthesis, structure analysis, and electrocatalytic conversion of CO2 to CO†
Cheng
Zhang
,
Mei
Ding
,
Yonggang
Ren
,
Along
Ma
,
Zhengmao
Yin
*,
Xiaoshuang
Ma
 * and
Shuxin
Wang
*
* and
Shuxin
Wang
*
College of Materials Science and Engineering, Qingdao University of Science and Technology, Qingdao 266042, Shandong, P. R. China. E-mail: yzm198752@163.com; xiaoshuang_Ma@qust.edu.cn; shuxin_wang@qust.edu.cn
First published on 15th May 2023
Abstract
Atomically precise metal nanoclusters (NCs) have emerged as a new class of ultrasmall nanoparticles with both free valence electrons and precise structures (from the metal core to the organic ligand shell) and provide great opportunities to understand the relationship between their structures and properties, such as electrocatalytic CO2 reduction reaction (eCO2RR) performance, at the atomic level. Herein, we report the synthesis and the overall structure of the phosphine and iodine co-protected Au4(PPh3)4I2 (Au4) NC, which is the smallest multinuclear Au superatom with two free e− reported so far. Single-crystal X-ray diffraction reveals a tetrahedral Au4 core stabilized by four phosphines and two iodides. Interestingly, the Au4 NC exhibits much higher catalytic selectivity for CO (FECO: > 60%) at more positive potentials (from −0.6 to −0.7 V vs. RHE) than Au11(PPh3)7I3 (FECO: < 60%), a larger 8 e− superatom, and Au(I)PPh3Cl complex; whereas the hydrogen evolution reaction (HER) dominates the electrocatalysis when the potential becomes more negative (FEH2 of Au4 = 85.8% at −1.2 V vs. RHE). Structural and electronic analyses reveal that the Au4 tetrahedron becomes unstable at more negative reduction potentials, resulting in decomposition and aggregation, and consequently the decay in catalytic performance of Au based catalysts towards the eCO2RR.
Introduction
The electrochemical CO2 reduction reaction (eCO2RR) can be regarded as an efficient method to convert CO2 into a variety of high-value chemicals and fuels using electricity generated from renewable energy sources such as solar, wind, and tidal energy.1–5 Due to the chemical inertness of CO2, reducing CO2 to high value-added chemicals requires sufficient activation energy to break C–O bonds. At present, the development of efficient catalysts to reduce activation energy and improve CO2 conversion efficiency has become a research hotspot in this field, and remarkable results have been achieved.6–10 However, the unclear structure–property relationship limits our deep understanding of eCO2RR catalytic mechanisms at the atomic level.Atomically precise metal nanoclusters (NCs) protected by ligands have attracted lots of interest for their unique properties and precise structures.11,12 And NCs can be considered as an ideal model catalyst widely employed in the study of the catalytic mechanism of the eCO2RR. For instance, the Jin group systematically studied the influence of the Au NCs on their eCO2RR properties such as the shape effect,13 ligand effect,14 and doping effect.15,16 Subsequently, many studies have been devoted to the accurate identification of catalytically active sites of gold nanoclusters.17,18 In 2021, the Lee group finally determined that the catalytic active site was the demercaptan gold atom at the interface through different sizes of Au25, Au38, and Au144 NCs, and found a positive correlation between catalytic activity and cluster size.19 The Wang group reported Au28 and Au55 NCs, showing high selectivity for CO (FECO > 90%).20,21 The Wu group introduced Cd to capture CO2 at the interface of gold nanoclusters, forming a Cd–O–C(OH)–Au structure, which can effectively inhibit the competitive hydrogen evolution reaction (HER).22 Zhu and co-workers found that C–S bond breaking on partial partitions makes it easier for open S sites to bind CO2 to form catalytically active sites.23 Recently, Zhu found that monomer Au24Au20 with a superatomic electronic configuration has better eCO2RR catalytic reaction activity than dimer Au43Ag38 nanoclusters.24 However, the relationship between the eCO2RR performance and the size of Au NCs is still unclear. We are trying to push the limit and produce the smallest Au NC to answer the question. Moreover, few studies have been reported to explain the decay of eCO2RR catalytic performance for Au NC-based catalysts at negative reduction potentials.
In this work, we report the synthesis, structure analysis and electroreduction performance of a novel Au4(PPh3)4I2 (Au4 hereafter) cluster with two free electrons, which is the smallest cluster with free electrons reported so far. This structure is determined by single-crystal X-ray diffraction (SC-XRD). Furthermore, for comparison, Au11(PPh3)7I3 (Au11 hereafter) and the Au(I) complex (Au(I)P hereafter) were prepared to explore the structure and electron effects of clusters on the eCO2RR. The three catalysts exhibited drastically different catalytic performances toward the eCO2RR, which is not simply related to the size of the clusters. This work will provide guidance for the rational design of the Au-based catalysts for the eCO2RR.
Experimental
Chemicals
All chemicals are of commercial grade and were used without further purification. Hydrogen tetrachloroaurate tetrahydrate (HAuCl4·4H2O, 99.95%), triphenylphosphine (PPh3, 98%), ethanol (HPLC grade, 99.9%), sodium hexafluoroantimonate (NaSbF6,99%), tetra-n-octylammonium bromide (TOAB, 98%), methylene chloride (CH2Cl2, HPLC grade, 99.9%), hexane (HPLC grade, 99.9%), sodium borohydride (NaBH4, 99.99%), methanol (HPLC grade, 99.9%), ethyl ether (HPLC grade, 99.9%), n-pentane (HPLC grade, 99.9%), silver nitrate (AgNO3, 99.9%), and sodium iodide (NaI, 99.9%). We purchased ultrapure water from Wahaha Co. Ltd. We cleaned all glassware with aqua regia (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mix of hydrochloric acid and nitric acid), rinsed it with ultrapure water, and dried it before use.
:1 mix of hydrochloric acid and nitric acid), rinsed it with ultrapure water, and dried it before use.
Synthesis of the Au(I)(PPh3)Cl complex (Au(I)P)
The Au complex was prepared according to a reported protocol with some minor modifications.25 In a typical synthesis, HAuCl4·3H2O (500 mg, 1.21 mmol) was first dissolved in 35 mL of deaerated EtOH in a 100 mL round-bottom flask. Subsequently, PPh3 (350 mg, 1.34 mmol) dissolved in 40 mL of deaerated EtOH was injected into the above solution under vigorous stirring, and white precipitates formed immediately. After 30 min, the white precipitates were filtered, washed with diethyl ether three times, and finally dissolved in CH2Cl2. Excess n-pentane was added dropwise to recrystallize the product to obtain white needle crystals of Au(PPh3)Cl.Synthesis of the Au(I)(PPh3)NO3 complex
Au(PPh3)NO3 was obtained by the counterion exchange reaction. Briefly, AgNO3 (169.8 mg, 1 mmol) was dissolved in 10 mL MeOH and added to Au(PPh3)Cl (446 mg, 0.9 mmol) dissolved in 50 mL CH2Cl2, and white precipitates AgCl formed immediately. After vigorous stirring for about 10 min, the mixture was centrifuged to remove AgCl, and the solvent organic phase was removed through rotary evaporation. The residual solid was washed with methanol several times to obtain white solids of Au(PPh3)NO3.Synthesis of Au4(PPh3)4I2 nanoclusters (Au4)
Au(PPh3)NO3 (25 mg, 0.05 mmol), and NaI (37.5 mg, 0.25 mmol) was added to 6 mL mixed solution of MeOH and CH2Cl2 (v:v = 1:2) with vigorous stirring. After about 20 min, NaSbF6 (17.6 mg, 0.068 mmol) was added and stirred for 15 min. A freshly prepared NaBH4 solution (2 mg in 4 mL of ice methanol) was added directly with vigorous stirring. The color changed from colorless to pale brown and finally to dark brown. After aging for 48 h, the solvent in the organic phase was removed through rotary evaporation, and then the product was washed with n-hexane three times and dissolved in CH2Cl2. After diffusion of n-hexane into CH2Cl2 at room temperature for about one week, orange-red crystals of Au4(PPh3)4I2 were obtained (yield: ca. 15% based on the Au atom).
Synthesis of Au11(PPh3)7I3 nanoclusters (Au11)
Au11(PPh3)7I3 was synthesized according to the method described in the literature with slight modifications.26 To a clean round-bottomed flask was added HAuCl4·4H2O (78 mg, 0.19 mmol), toluene (10 mL), TOAB (200 mg, 0.36 mmol), and stirred at room temperature for 1 hour until the color of the solution gradually deepened. After the gold salt was transferred from the aqueous phase to the organic layer, the water layer was removed by suction or centrifugation. The red organic layer was collected in a new round-bottomed flask, and NaI (5 mg, 0.033 mmol) was added, and stirred for 20 min. With intense agitation, PPh3 (210 mg, 0.8 mmol) was added. The liquid changed from bright red to colorless, indicating the formation of the Au complex. After 20 min, NaBH4 (90 mg, 2.38 mmol) ice water solution (5 mL) was added, stirred and aged for 24 h at room temperature. The color of the solution changed from colorless to brownish red. The reaction mixture was centrifuged to remove the water layer, and the crude product obtained after evaporation and drying was washed several times with n-hexane (30 mL) to remove impurities and obtain black precipitate. A single crystal Au11 NC was obtained by a stratified method. 2 mg of the product was dissolved with CH2Cl2 (2 mL), and was centrifuged to remove insoluble impurities, and then the black solution was layered with n-hexane (5 mL). After three days red crystals were obtained. The yield of Au11 is ca. 50% (based on the Au atom).X-ray crystallography
The data collections for single crystal X-ray diffraction were carried out on a STOE Stradivari diffractometer at 150 K, using Mo Kα radiation (λ = 0.71073 Å). Data reductions and absorption corrections were performed using the X-Area. The structure was solved by intrinsic phasing and refined with full-matrix least squares on F2 using the SHELXTL software package. All non-hydrogen atoms were refined anisotropically, and all the hydrogen atoms were set in geometrically calculated positions and refined isotopically using a riding model. Detailed crystal data and structure refinements for the cluster are given in Table S1.† CCDC 2251680 for Au4(PPh3)I2 contains the ESI crystallographic data for this paper. These data are provided free of charge by the Cambridge Crystallographic Data Centre.Electrochemical measurements
The catalyst sample was prepared by loading three as-prepared NCs on acidic multi-walled carbon nanotubes (CNTs) with a mass ratio of 1 (5 mg NCs and 5 mg CNTs). The catalyst ink was obtained by dispersing the sample in isopropyl alcohol and sonicating for 5 min. Then, 1 mL of the catalyst suspension and 10 μL of Nafion (5 wt%) were mixed uniformly to form the final catalyst ink. Next, 40 μL of the catalyst ink was drop-cast onto a carbon cloth (1.0 × 1.0 cm2) and dried at room temperature to make the working electrode. All electrochemical measurements were performed in a custom gas-tight H-cell with two compartments separated by a Nafion 117 membrane. Each compartment contained 25 mL of electrolyte (0.5 M KHCO3:pH = 7.2 when saturated with CO2 and pH = 8.8 when saturated with N2) with 15 mL headspace. The electrochemical measurements were conducted on an electrochemical workstation (CHI 760E) with a Pt sheet as the counter electrode and an Ag/AgCl electrode (KCl saturated) as the reference electrode. All the potentials were calibrated to a reversible hydrogen electrode (RHE) according to the Nernst equation:| R(RHE) = E(Ag/AgCl) + 0.059 × pH + 0.21 | (1) |
The gas flow from the cathode chamber was directed into a gas chromatograph (GC3900Plus, RUI NENG) to identify and quantify the gaseous products. The GC was purged for 30 min with an average rate of 10 mL min−1 (at room temperature and ambient pressure) before the test. The GC was equipped with a thermal conductivity detector (TCD) for H2 detection and a flame ionization detector (FID) for hydrocarbon detection. A methanizer (Agilent) was connected to the FID for CO detection. High-purity argon (99.9999%) was used as the carrier gas for all compartments of the GC. The faradaic efficiency (FEX) and the corresponding partial current density (jX) of X (X = CO or H2) were calculated as follows:
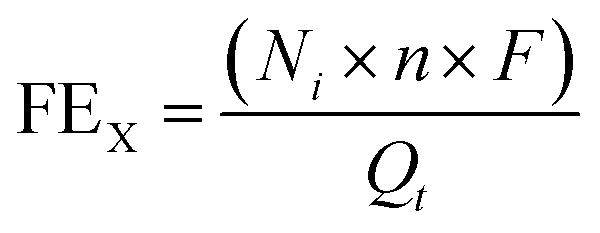 | (2) |
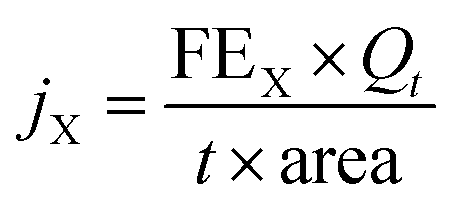 | (3) |
485 C mol−1), t = reaction time (s), and area = the geometric area of the electrode (1 cm2).
The electrochemically active surface area (ECSA) of the catalyst was obtained via the linear fit of the corresponding current density with respect to the scan rate (Fig. S5†). The results indicated that the double layer capacitance of the electrode (CdI) for Au(I)P, Au4, and Au11 was 0.648, 1.154, and 0.805 mF, respectively. The ECSA was calculated using the following formula:
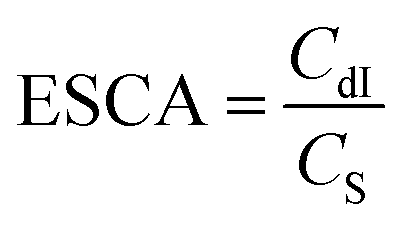 | (4) |
To evaluate catalyst stability, after the eCO2RR, the samples were rinsed with CH2Cl2 from working electrodes and collected for characterization with UV-vis absorbance.
Results and discussion
Preparation and structure analysis of Au4 and Au11 NCs
Au4(PPh3)I2 was first prepared by following a modified one-pot method (see experimental details). Briefly, in the presence of NaI and NaSbF6, Au(I)(PPh3)NO3 were reduced by NaBH4 in the mixed solvent of dichloromethane and methanol. The reaction was aged for two days during which the solution gradually changed from colorless to pale brown, and finally to dark brown. An orange-red block crystal was obtained by diffusing n-hexane into the dichloromethane solution containing the crude product. Subsequently, Au11(PPh3)7I3 with eight electrons was prepared by following the previously reported work to compare eCO2RR catalytic performance with Au4.First, we compared the optical absorbance properties of Au4 and Au11. For Au4, as shown in Fig. 1a and c, there is only one prominent peak at 422 nm (2.94 eV). The energy bandgap derived from the absorbance spectrum is 2.51 eV. For Au11, as presented in Fig. 1b and d, there are also one prominent peak at 419 nm (2.96 eV), and two weak shoulders at 300 nm (4.13 eV) and 315 nm (3.94 eV), while the energy bandgap of 1.88 eV agree well with the previously reported work.26 Furthermore, one week stability of Au4 and Au11 in liquid state at room temperature was tested (Fig. S1†). It exhibits varying degrees of absorbance reduction, indicating that the stability of two clusters decreased due to structural collapse, while the deterioration rate of Au4 is significantly faster than that of Au11. It is concluded that the stability of Au4 is relatively poor compared to that of Au11 at room temperature.
 | ||
| Fig. 1 The optical absorbance properties of two clusters. (a and b) UV-vis absorbance spectra and (c and d) the spectra plotted in the energy axis of Au4 and Au11 in CH2Cl2, respectively. Insets are the photos of Au4 and Au11 in CH2Cl2. | ||
Subsequently, the atomic packing structure of Au4(PPh3)4I2 was examined using single crystal X-ray diffraction (SC-XRD). As illustrated in Fig. S2,†Au4 crystallizes in the space group C2/c in the monoclinic crystal system, and each unit cell has no counterion, indicating that Au4 is electrically neutral. Therefore, Au4(PPh3)4I2 is a superatom containing two free electrons (N = 4 − 2 − 0 = 2). More detailed structural parameters are summarized in Table S1.† The comparison of overall structures of Au4 and Au11 is shown in Fig. 2. Au4 consists of a tetrahedral structure composed of four gold atoms, each of which is coordinated with one PPh3 ligand (Fig. 2a). In addition, there are two iodine atoms, each of which forms a triangle with two gold atoms via σ bonding. This triangle, together with the PPh3 ligands connected to the gold atoms, that is, two staple motifs and two iodine atoms together form Au4. Along the line connecting the two iodine atoms as the axis, half of the cluster structure, that is, one staple motif, rotates 90° clockwise along the axis and overlaps with the projection of the other half of the staple motif. The bond lengths of Au–P are 2.282 and 2.292 Å, the bond lengths of Au–I are 2.898 and 2.972 Å, and the bond lengths of the adjacent Au–Au are 2.65, 2.74, and 2.83 Å (Fig. S3 and Table S2†). Furthermore, the structure of Au11(PPh3)7I3 consists of an Au11 core and seven PPh3 ligands in the ligand shell (Fig. 2b). The remaining three gold atoms are bonded to iodine atoms, and the three iodine atoms form an approximate equilateral triangle with the center of mass located at the core gold atom. Ten of the eleven gold atoms form an incomplete icosahedral structure (missing one vertex and one edge gold atom), with only ten triangular faces and three non-planar quadrilateral faces. The remaining gold atom is located at the center of this incomplete icosahedral structure. The gold core can be viewed as consisting of a ring with four gold atoms and a ring with five gold atoms, as well as a gold atom located at the center of the pentagon and one at the top. The crystal belongs to the P21/n space group (Fig. S3†). The Au–P bond length is between 2.271 and 2.297 Å, the Au–I bond length is between 2.595 and 2.609 Å, and the length of adjacent Au–Au bonds is between 2.608 and 3.109 Å (Fig. S3 and Table S2†). Moreover, the tetrahedron composed of four gold atoms is the smallest subunit for the structural evolution of Au nanoclusters.28
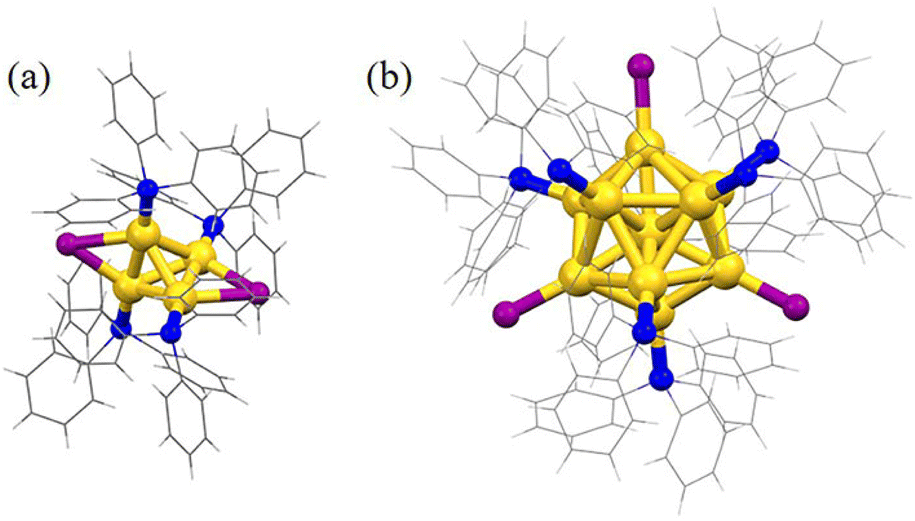 | ||
| Fig. 2 Structural analysis of Au4(PPh3)4I2 and Au11(PPh3)7I3. Overall structure of monocationic (a) Au4 and (b) Au11, respectively (color labels: yellow = Au, blue = P, grey = C, purple = I and white = H). | ||
Electrocatalytic CO2 reduction performance of the three catalysts
To evaluate the catalytic performance of cluster catalysts, the electrochemical CO2 reduction reaction (eCO2RR) was conducted. Given the abundant surface defects and functional groups of acidic multi-walled carbon nanotubes (CNTs), the two clusters and the Au(I)PPh3I complex (Au(I)P) were deposited onto CNTs with 50% wt. loading to form the Au(I)P/CNT, Au4/CNT and Au11/CNT electrocatalysts. By using gas chromatography (GC), it was discovered that CO and H2 were the only two gaseous products compared to the reversible hydrogen electrode (RHE) at all applied potentials (without IR correction). Moreover, there were no liquid products as confirmed by 1H-NMR spectra. We first performed linear scanning voltammetry (LSV) for the three catalysts (Fig. 3a). Au11/CNTs show much higher current density and a more positive onset potential in CO2 saturated 0.5 M KHCO3 aqueous solutions than in N2 purged aqueous solutions, indicating higher CO2 reduction selectivity. Au4/CNTs have similar onset potential and current density in both solutions, and did not exhibit high CO2 reduction selectivity. This implies that the HER rather than the eCO2RR is the main reduction process of the three catalysts. Interestingly, Au(I)P/CNTs exhibit more positive onset potential and higher current density in CO2 saturated 0.5 M KHCO3 aqueous solution than in N2 purged aqueous solution, suggesting some CO2 reduction selectivity. Thus, the eCO2RR catalytic activity can be arranged in descending order as Au11 > Au(I)P > Au4, which are not simply related to the size of the clusters. Furthermore, electrochemical impedance spectroscopy (EIS) was carried out to investigate the electron transport properties of these catalysts at the electrode/electrolyte interface (Fig. 3b). These three samples had significantly different electrochemical impedances, indicating different electron transport capabilities. The interface charge transfer rate was highest for Au4, followed by Au11 and Au(I)P. This suggests that Au4 would donate electrons to the intermediates more efficiently than Au11 and Au(I)P during the electroreduction. As shown in Fig. 3c, Au(I)P/CNTs and Au11/CNTs show excellent selectivity for CO at all tested potentials, with CO faradaic efficiency (FECO) more than 50% at all test potentials. Specifically, Au11/CNTs exhibit higher selectivity for CO with the highest FECO (75.8% at −0.9 V vs. RHE) than Au(I)P/CNTs (FECO: 72.6% at −0.9 V vs. RHE). Furthermore, the selectivity (HER vs. eCO2RR) is shown in Fig. 3d. Au11/CNTs show higher CO selectivity than Au(I)P/CNTs from −0.9 to −1.2 V vs. RHE. This is due to the electron effect, indicating that clusters with free electrons is more favourable than the Au(I) complex for selective reduction of CO2 to CO. In contrast, Au4/CNTs exhibit selectivity for CO at more positive potentials, while the hydrogen evolution reaction dominates over the eCO2RR as the potential becomes more negative. The FECO of Au4/CNTs is 62.8% at −0.6 V vs. RHE with the highest FECO of 74.3% at −0.7 V vs. RHE, suggesting that Au4/CNTs have a smaller eCO2RR overpotential than the other two catalysts. Note that, although Au4 has two free electrons, the cluster surface does not expose as much active site metal as Au11. Moreover, H2 is a competitive reaction product, and at a more negative applied potential, Au4/CNTs exhibit a higher selectivity for H2 (FEH2: 85.8% at −1.2 V vs. RHE) than Au(I)P/CNTs (FEH2: 48.9%) and Au11/CNTs (FEH2: 26.7%).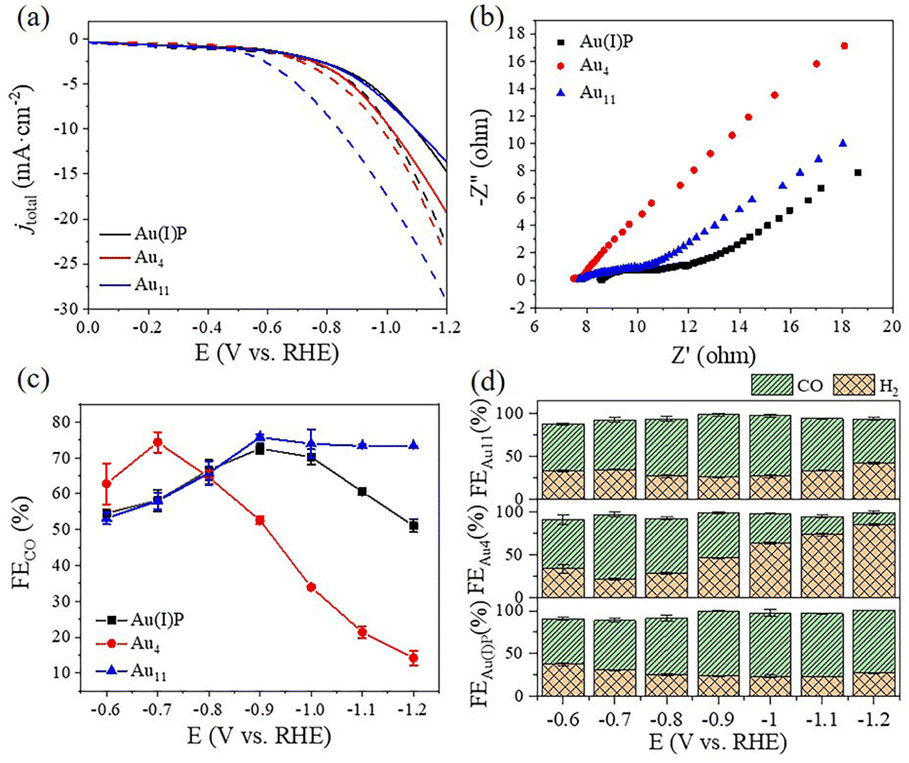 | ||
| Fig. 3 The electrocatalytic performance of Au(I)P, Au4 and Au11 in the eCO2RR. (a) LSV curves of the three catalysts in an N2-saturated (dotted line) and a CO2-saturated (full line) 0.5 M KHCO3 solution. (b) EIS and (c) FECO of the three catalysts. (d) Selectivity for various eCO2RR products (CO vs. H2) obtained on Au11, Au4, and Au(I)P. The error bars represent the standard deviation of three tests at the same test potential. | ||
The CO partial current density (jCO) increased with increasing applied potential for the three electrocatalysts (Fig. 4a). However, Au4/CNTs show a slightly higher jCO (1.8 mA cm−2) than Au11/CNTs (0.7 mA cm−2) and Au(I)P/CNTs (1.0 mA cm−2) at −0.6 V vs. RHE. Furthermore, the jH2 value of Au4/CNTs increased more sharply than that of Au11/CNTs (6.0 mA cm−2) and Au(I)P/CNTs (9.8 mA cm−2) as the potential became more negative (Fig. 4b). The electrochemically active surface area (ECSA) was measured to further reveal the reason for the difference in the catalytic performance of the three catalysts in the eCO2RR. We recorded the current density of Au(I)P/CNTs, Au4/CNTs and Au11/CNTs at different scan rates ranging from 0.02 to 0.1 V s−1 (Fig. S4†). We evaluated the double-layer capacitance (Cdl) of the three catalysts to be 0.648, 1.154 and 0.99 mF, respectively (Fig. 4c). And the ECSA of Au(I)P/CNTs, Au4/CNTs and Au11/CNTs was calculated to be 16.20, 30.25 and 24.75 cm2, respectively. Therefore, active surface area of the three catalysts can be arranged in descending order as Au4> Au11> Au(I)P. Electrokinetic studies of CO2 electroreduction were performed on these clusters to identify the origin of the eCO2RR activities of the three Au-based catalysts. The Tafel plots were then recorded to analyse the reaction kinetics, where the Tafel slopes are calculated to be 448 mV dec−1, 480 mV dec−1 and 422 mV dec−1 for Au(I)P/CNTs, Au4/CNTs and Au11/CNTs, respectively (Fig. 4d). The High Tafel slopes of ca. 400 mV dec−1 of the three catalysts suggest that the first electron transfer step is the eCO2RR rate-determining step. Furthermore, using the fingerprint absorbance peak (422 nm for Au4; 419 nm for Au11) as the metric, the absorbance change can be used as an indicator of the catalyst stability (Fig. S6†). Notably, the entire absorbance feature of Au4 and Au11 remains constant, though the intensity of the characteristic peaks of Au4 is drastically decreased, indicating that the structure of Au4 is difficult to maintain during the electroreduction.
 | ||
| Fig. 4 (a) CO and (b) H2 partial current density of Au(I)P, Au4 and Au11 at different potentials. (c) The corresponding linear curves of current density and the scan rate (CdI) for the three catalysts. (d) Tafel plots constructed for the eCO2RR on the three catalysts in the 0.5 M KHCO3 solution. | ||
Discussion on structure and electronic effects of the catalysts toward the eCO2RR
Based on the experimental test results of the three catalysts for the eCO2RR, we would like to discuss the effect of free electrons and stability of the three clusters toward the eCO2RR. Note that, Au(I)P and Au11 can convert CO2 to CO with FECO > 50% at all test potentials, suggesting that the +1 valence Au is the catalytically active site. However, cationic gold is quite oxidizing (Au+/0 = 1.83 V), indicating that the Au(I) complex was reduced into small particles by Au–P bond breaking during electroreduction. Therefore, as the reduction potential becomes negative, Au(I)P becomes less selective for CO. Although Au4 contains +1 valence Au atoms, its Au(I)/Au(0) ratio (Au4:Au11 = 2:3) is less than that of Au11, resulting in the highest FECO (74.3%) of Au4 being slightly less than that of Au11 (FECO: 75.8%). However, as the particle size decreases, the adsorption capacity of H increases greatly. That means, for Au4, the HER process became dominant at more negative potentials.
Conclusions
In conclusion, the smallest superatom with free electrons of Au4(PPh3)I2 is synthesized by a one-pot method, and the Au4 tetrahedron could be considered as the smallest unit constituting the Au11 core. Moreover, Au(I)P, Au4 and Au11 exhibited drastically different catalytic performances toward the eCO2RR. Au(I)P and Au11 could convert CO2 into CO at all tested potentials with high FECO (>50%). In contrast, Au4 can exclusively convert CO2 into CO at more positive potentials, and as the potential becomes more negative, H2 is the main product. According to structure analysis, the Au4 tetrahedron could not remain stable at more negative reduction potentials, resulting in decomposition and aggregation, which is the reason for the decay of catalytic performance of Au based catalysts towards the eCO2RR. Moreover, Au(I) sites on the surface of the cluster were the active sites. This work provides an example at the atomic level to construct a structure–activity relationship between the structure and electroreduction of CO2 of Au based catalysts.Author contributions
S. W., Z. Y. and X. M. guided the whole experiment and conceived the idea. C. Z. conducted most of the experiments. M. D. and X. M. gave some advice for the characterization and analysis. S. W., Z. Y. and X. M. provided the funding support, and all the authors contributed to the final polishing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support provided by the National Natural Science Foundation of China (22171156, 52072194 and 2022KJ305), Taishan Scholar Foundation of Shandong Province (China), Shandong Province Excellent Youth Innovation Team and Startup Funds from Qingdao University of Science and Technology.Notes and references
- A. Wagner, C. D. Sahm and E. Reisner, Nat. Catal., 2020, 3, 775–786 CrossRef CAS.
- Y. Y. Birdja, E. Perez-Gallent, M. C. Figueiredo, A. J. Gottle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- X. Y. Wang, Q. D. Zhao, B. Yang, Z. J. Li, Z. Bo, K. H. Lam, N. M. Adli, L. C. Lei, Z. H. Wen, G. Wu and Y. Hou, J. Mater. Chem. A, 2019, 7, 25191–25202 RSC.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- Y. T. Liu, D. H. Deng and X. H. Bao, Chem, 2020, 6, 2497–2514 CAS.
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2020, 367, eabb0992 CrossRef CAS PubMed.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Norskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- Y. Zou and S. Wang, Adv. Sci., 2021, 8, 2003579 CrossRef CAS PubMed.
- M. B. Ross, P. De Luna, Y. F. Li, C. T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- M. Ding, L. Tang, X. Ma, C. Song and S. Wang, Commun. Chem., 2022, 5, 172 CrossRef CAS PubMed.
- Y. Li, H. K. Kim, R. D. McGillicuddy, S. L. Zheng, K. J. Anderton, G. J. Stec, J. Lee, D. Cui and J. A. Mason, J. Am. Chem. Soc., 2023, 145, 9304–9312 CrossRef CAS PubMed.
- Y. Li, M. Zhou, Y. Song, T. Higaki, H. Wang and R. Jin, Nature, 2021, 594, 380–384 CrossRef CAS PubMed.
- S. Zhao, N. Austin, M. Li, Y. B. Song, S. D. House, S. Bernhard, J. C. Yang, G. Mpourmpakis and R. C. Jin, ACS Catal., 2018, 8, 4996–5001 CrossRef CAS.
- S. Li, A. V. Nagarajan, Y. Li, D. R. Kauffman, G. Mpourmpakis and R. Jin, Nanoscale, 2021, 13, 2333–2337 RSC.
- S. Li, D. Alfonso, A. V. Nagarajan, S. D. House, J. C. Yang, D. R. Kauffman, G. Mpourmpakis and R. Jin, ACS Catal., 2020, 10, 12011–12016 CrossRef CAS.
- S. Li, A. V. Nagarajan, D. R. Alfonso, M. Sun, D. R. Kauffman, G. Mpourmpakis and R. Jin, Angew. Chem., Int. Ed., 2021, 60, 6351–6356 CrossRef CAS PubMed.
- D. R. Alfonso, D. Kauffman and C. Matranga, J. Chem. Phys., 2016, 144, 184705 CrossRef PubMed.
- N. Austin, S. Zhao, J. R. McKone, R. Jin and G. Mpourmpakis, Catal. Sci. Technol., 2018, 8, 3795–3805 RSC.
- H. Seong, V. Efremov, G. Park, H. Kim, J. S. Yoo and D. Lee, Angew. Chem., Int. Ed., 2021, 60, 14563–14570 CrossRef CAS PubMed.
- S. F. Yuan, R. L. He, X. S. Han, J. Q. Wang, Z. J. Guan and Q. M. Wang, Angew. Chem., Int. Ed., 2021, 60, 14345–14349 CrossRef CAS PubMed.
- X. K. Wan, J. Q. Wang and Q. M. Wang, Angew. Chem., Int. Ed., 2021, 60, 20748–20753 CrossRef CAS PubMed.
- S. Zhuang, D. Chen, L. Liao, Y. Zhao, N. Xia, W. Zhang, C. Wang, J. Yang and Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 3073–3077 CrossRef CAS PubMed.
- Y. N. Sun, X. Liu, K. Xiao, Y. Zhu and M. Y. Chen, ACS Catal., 2021, 11, 11551–11560 CrossRef CAS.
- J. Y. Xu, L. Xiong, X. Cai, S. S. Tang, A. C. Tang, X. Liu, Y. Pei and Y. Zhu, Chem. Sci., 2022, 13, 2778–2782 RSC.
- L. C. McKenzie, T. O. Zaikova and J. E. Hutchison, J. Am. Chem. Soc., 2014, 136, 13426–13435 CrossRef CAS PubMed.
- A. K. Das, S. Mukherjee, S. S. R, A. S. Nair, S. Bhandary, D. Chopra, D. Sanyal, B. Pathak and S. Mandal, ACS Nano, 2020, 14, 16681–16688 CrossRef CAS PubMed.
- C. C. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977–16987 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, G. Li and R. Jin, Chem. Commun., 2014, 50, 55–57 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting figures and tables. Details and crystal data of Au4(PPh3)I2 (cif). CCDC 2251680. See DOI: https://doi.org/10.1039/d3na00191a |
| This journal is © The Royal Society of Chemistry 2023 |