
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSARS-CoV-2 virus-like-particles via liposomal reconstitution of spike glycoproteins†
Sarah
McColman
 *ab,
Klaidi
Shkalla
ab,
Pavleen
Sidhu
ab,
Jady
Liang
bc,
Selena
Osman
ab,
Norbert
Kovacs
ab,
Zainab
Bokhari
ab,
Ana Carolina
Forjaz Marques
abd,
Yuchong
Li
bce,
Qiwen
Lin
bce,
Haibo
Zhang
bcef and
David T.
Cramb
*abg
*ab,
Klaidi
Shkalla
ab,
Pavleen
Sidhu
ab,
Jady
Liang
bc,
Selena
Osman
ab,
Norbert
Kovacs
ab,
Zainab
Bokhari
ab,
Ana Carolina
Forjaz Marques
abd,
Yuchong
Li
bce,
Qiwen
Lin
bce,
Haibo
Zhang
bcef and
David T.
Cramb
*abg
aDepartment of Chemistry and Biology, Faculty of Science, Toronto Metropolitan University, Toronto, ON, Canada. E-mail: sarah.mccolman@torontomu.ca
bKeenan Research Centre for Biomedical Science, St. Michael's Hospital, Unity Health Toronto, Toronto, ON, Canada
cDepartment of Physiology, University of Toronto, Toronto, ON, Canada
dFaculdade de Ciências Farmacêuticas, Seção Técnica de Graduação, Universidade Estadual Paulista, Araraquara, SP, Brazil
eThe State Key Laboratory of Respiratory Disease, Guangzhou Institute of Respiratory Disease, The First Affiliated Hospital of Guangzhou Medical University, Guangzhou, Guangdong, China
fDepartments of Anaesthesia and Physiology, Interdepartmental Division of Critical Care Medicine, University of Toronto, Toronto, ON, Canada
gDepartment of Chemistry, Faculty of Science, University of Calgary, Calgary, AB, Canada
First published on 14th July 2023
Abstract
The SARS-CoV-2 virus, implicated in the COVID-19 pandemic, recognizes and binds host cells using its spike glycoprotein through an angiotensin converting enzyme 2 (ACE-2) receptor-mediated pathway. Recent research suggests that spatial distributions of the spike protein may influence viral interactions with target cells and immune systems. The goal of this study has been to develop a liposome-based virus-like particle (VLP) by reconstituting the SARS-CoV-2 spike glycoprotein within a synthetic nanoparticle membrane, aiming to eventually establish tunability in spike protein presentation on the nanoparticle surface. Here we report on first steps to this goal, wherein liposomal SARS-CoV-2 VLPs were successfully produced via detergent mediated spike protein reconstitution. The resultant VLPs are shown to successfully co-localize in vitro with the ACE-2 receptor on lung epithelial cell surfaces, followed by internalization into these cells. These VLPs are the first step toward the overall goal of this research which is to form an understanding of the relationship between spike protein surface density and cell-level immune response, eventually toward creating better vaccines and anti-viral therapeutics.
Introduction
SARS-CoV-2, implicated in the COVID-19 pandemic, is a spherical enveloped virus comprised of a lipid bilayer studded with integral membrane proteins that encapsulates viral nucleic acids and associated proteins.1 Upon introduction of its genetic material to a host cell, the virus harnesses the cellular machinery of its target organism to reproduce and proliferate to other cells.2 Three main proteins attached to the viral envelope membrane are known to regulate the ability of this virus to successfully bind and fuse with host cells. Of these, the spike glycoprotein is involved in mediating viral entry into the host cells and as such has attracted substantial attention as a focus of immunological and therapeutic research.3,4 This protein is primarily responsible for recognizing and binding to host cells through an angiotensin converting enzyme 2 (ACE2) receptor mediated pathway.Harnessing nanotechnology to study SARS-CoV-2 has been crucial in the global battle against COVID-19, and this remains essential even as large-scale vaccination efforts have proven highly effective.5 Recent studies have suggested that spatial distributions of the spike protein may influence viral interactions with target cells and with immune systems.6 Therefore, elucidating structure–function relationships of antigen presentation is critical to understanding the virulence of this pathogen. A liposome-based virus-like particle (VLP) could be a useful tool to model this threatening pathogen to help scientists explore fundamental questions about the spike protein and immune responses. In this study, we present a method of synthetic VLP production, namely the development of stable and biomimetic protein-containing liposomes via detergent-mediated reconstitution of viral antigen proteins (Fig. 1).
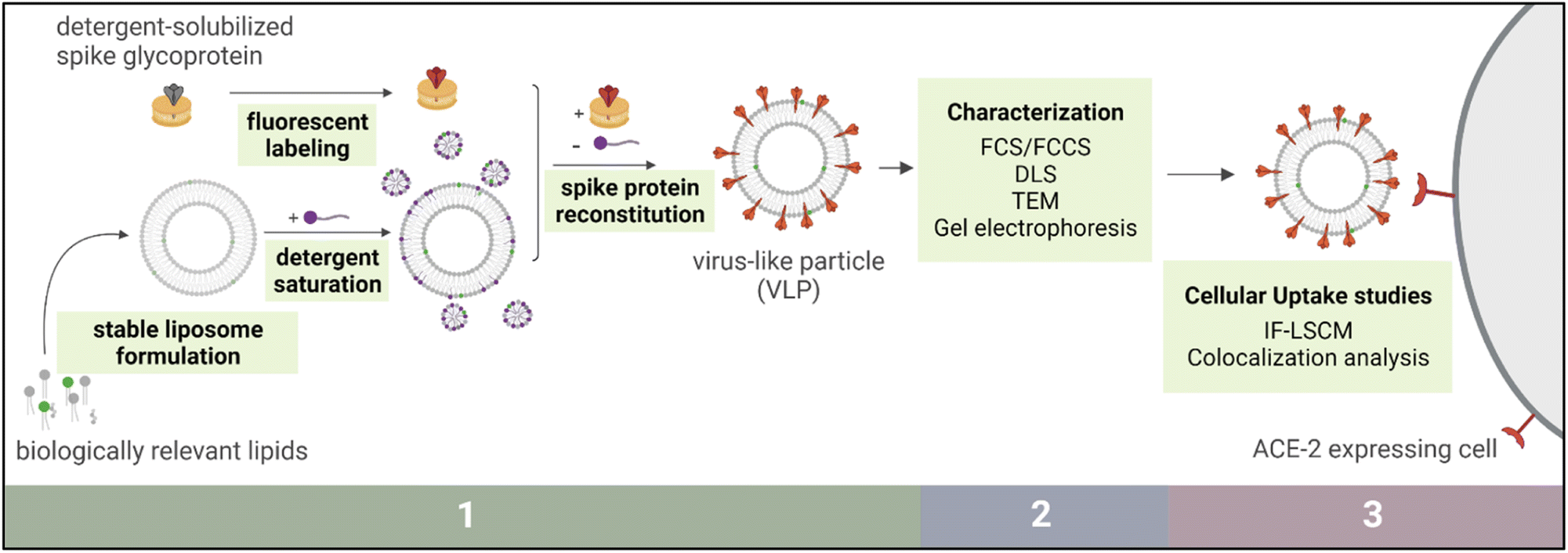 | ||
| Fig. 1 Schematic outline of methodology, grouped into three sections. Section 1 involves virus-like particle (VLP) preparation, Section 2 denotes biophysical characterization of the VLPs, and Section 3 includes VLP cellular uptake studies and image analysis. | ||
Many different groups have developed SARS-CoV-2 VLPs in recent years using other production systems. Protein-based VLPs expressed from transfected cell systems have been reported extensively in literature, typically by transfecting mammalian cells with plasmids encoding antigenic proteins for SARS-CoV-2.4,7–15 In these models, the proteins expressed by these transgenic cells accumulate in the membranes of the endoplasmic reticulum – Golgi intermediate complex (ERGIC). After budding from these ERGIC membranes, protein rich VLPs are excreted from the cell. Importantly, the enrichment of the SARS-CoV-2 membrane glycoprotein is essential for the formation and release of these VLPs.9 Groups have also engineered mammalian expression systems to produce spike glycoprotein receptor-binding domains (RBDs) and encapsulated these protein domains in chitosan-based nanoparticles post-purification.16 Plant cells have been utilized for protein-based VLP production as well, some of which have been developed as vaccine candidates for SARS-CoV-2. These systems are more scalable and economical compared to using mammalian cell systems, and differences in glycosylation patterns for proteins produced in these non-mammalian systems have been shown to be negligible.17 Because the VLPs produced in these cells bud off the plasma membrane, however, lipids are incorporated into these plant-sourced VLPs that may not be present in the authentic virus.17–20
Another method of producing VLPs is through the production of chimeric pseudoviruses. SARS-CoV-2 pseudoviruses have been formed using several viruses including marine leukemia virus,7,21,22 HIV-1 lentivirus,23 cucumber mosaic virus,24 and vesicular stomatitis virus.25 These systems are developed by expressing proteins from SARS-CoV-2 within a scaffold virus framework that has limited replicability.26 Although these systems have the advantage of genetic mutability, their downside is the inevitable expression of cell or viral proteins on the VLP/pseudovirus surface, which can add uncontrolled complexity to the virus-like particle.27
A recent study modeled SARS-CoV-2 by immobilizing spike glycoprotein RBDs on a DNA origami lattice to investigate the relationship between immune activation and surface presentation of antigens.28 This approach has the benefit of precise control over antigen presentation but comes with drawbacks including innate immunogenicity of the DNA origami structures.29
As for liposome-based systems, previous studies have set precedent for the successful incorporation of integral membrane proteins into synthetic membranes,30–34 including studies in which liposomes have modelled other viruses, such as HIV-1.35–37 For SARS-CoV-2, recently natural and synthetic vesicles have been used to create VLPs by chemically conjugating the receptor-binding-domain (RBD) of the spike glycoprotein to membrane components.13,38–41 To our knowledge, however, no liposome-based SARS-CoV-2 VLPs containing full-length membrane anchored spike glycoproteins have been reported to date.35,40 Here we report on the successful synthesis of SARS-CoV-2 VLPs via liposomal reconstitution of spike glycoproteins. We present proof-of-concept preliminary cellular uptake studies and provide evidence of ACE-2 involvement in the intracellular trafficking of these liposomal VLPs.
Results and discussion
Virus-like particle preparation
For this initial characterization, liposome formulations were doped with a fluorescent marker and analysed using fluorescence correlation spectroscopy (FCS).61 Narrowly distributed fluorescent count rate trajectories detected from liposome samples suggested the solution contained uniformly dispersed fluorescent liposomes (Fig. 2A). Formulations were discarded immediately from the pool of candidates if the liposomes appeared highly aggregated upon initial preparation, based on intensity spikes in fluorescence signal leading to wide distributions of recorded count rate trajectories (Fig. 2B). Beyond this count rate analysis, the different trial formulations were evaluated for hydrodynamic diameter immediately post-production by fitting the FCS autocorrelation decays with eqn (1) to obtain average concentrations and diffusion coefficients for the liposomes in solution (Fig. 2C) (characterization data for all 97 formulations not shown). Because the SARS-CoV-2 virion has an approximate diameter between 60 and 140 nm, formulations which appeared to have the desired 60–140 nm diameter upon preparation were preferred.42 The formulations were stored at 4 °C overnight and FCS analysis was performed again the next day to test their overnight storage stability. Again, liposome formulations were rejected if they appeared highly aggregated the next day based on intensity spikes in the count rate trajectories, or if their apparent concentration or hydrodynamic diameter had changed by more than 50% from the measurement on the initial day.
 | ||
| Fig. 2 (A) Example stable fluorescent count rate trajectory with inset count rate histogram of a liposome solution, (B) example unstable fluorescent count rate trajectory and inset count rate histogram of a liposome solution, and (C) example autocorrelation decay curve showing fit line to eqn (1). | ||
All liposome formulations considered stable overnight were then characterized for stability over five days (data not shown). As the duration of a typical VLP preparation spans two days, out of an abundance of caution five days was selected as the preferred time window for stability of liposomes. For these experiments, stability was defined using two key criteria monitored over five days: average hydrodynamic diameter and average number of nanoparticles in the FCS focal volume (N) (a proxy for concentration).
Six formulations with the least degree of change between days 0 and 5 (hereafter: 1, 2, 3, 1PEG, 2PEG, and 3PEG) were prepared in 1× PBS with and without 0.3 M sucrose as a proposed stabilizing agent,62,63 and were characterized to determine their appropriateness for the next stages of VLP development (Fig. 3A and B).64,65 These formulations are all mixed-phase liposomes with 1 and 1PEG being primarily gel-phase and 2, 2PEG, 3, and 3PEG comprised of mostly liquid-crystal phase lipids (Fig. 3B). All six of these formulations were primarily comprised of lipids with phosphatidyl choline (PC) headgroups, with varying relative proportions of phosphatidyl ethanolamine (PE), phosphatidyl inositol (PI), cholesterol, and PE-PEG1000 headgroups making up the remainder of the total membrane components (Fig. 3B).
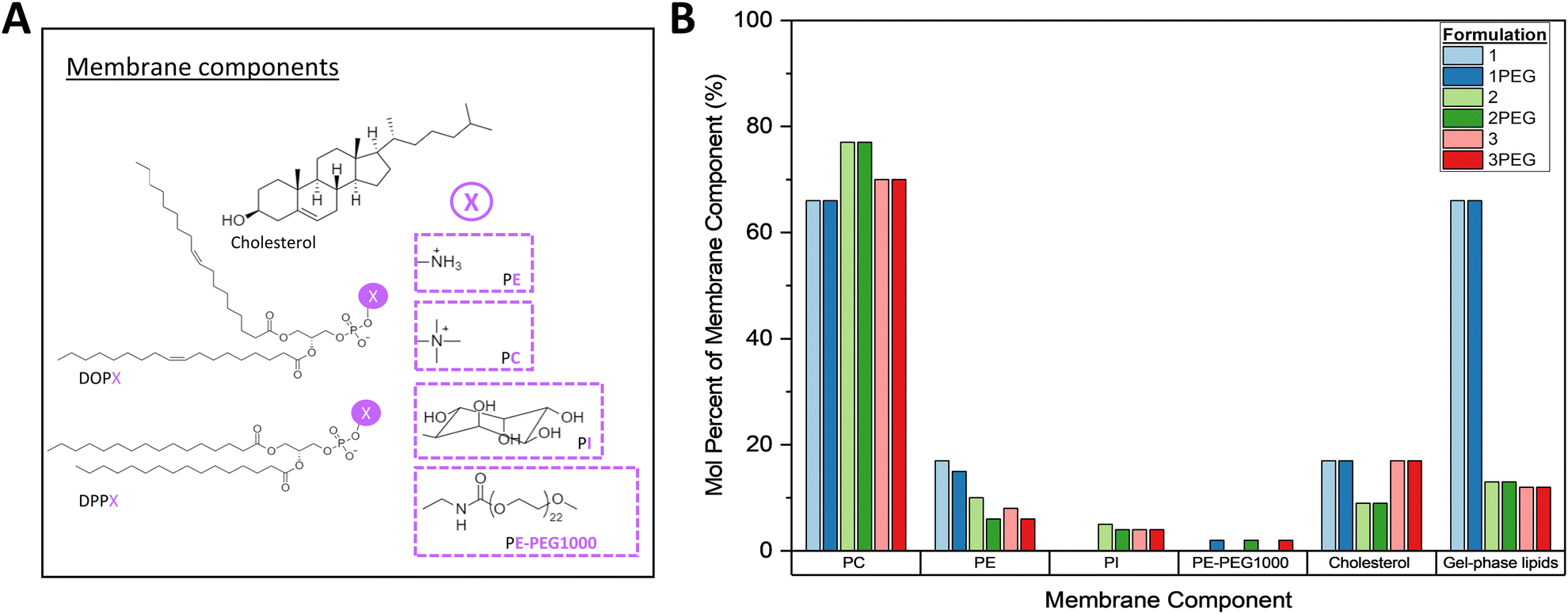 | ||
| Fig. 3 (A) Structures of membrane components comprising formulations 1, 2, 3, 1PEG, 2PEG, and 3PEG. For the lipids, hydrophobic tail groups are shown as DOPX and DPPX where “X” represents the functional moiety of the headgroups, structures of which are within the dashed boxes. (B) Liposome formulations of the top six candidates represented as mole percent of each component. | ||
Examining the average hydrodynamic diameters obtained from FCS measurements of these liposomes between the day of preparation (day 0) and the fifth day (day 4) revealed a significant change in diameter for formulation 1 prepared in 1× PBS (p = 0.0088) (Fig. 4A). Comparing this to the other five formulations which showed no significant difference between hydrodynamic diameters measured on day 0 and day 4 suggests that formulation 1 was the least colloidally stable over time by this metric (Fig. 4A). In 1× PBS with 0.3 M Sucrose, the same comparison showed significant changes in diameter between day 0 and day 4 for formulations 1 (p = 0.0034) and formulation 3PEG (p = 0.016) (Fig. 4C). Thus, these formulations were considered the least colloidally stable over the five days as measured by changes in hydrodynamic diameter.
 | ||
| Fig. 4 (A) Average hydrodynamic diameter (HD) for liposomes in 1× PBS. Data is presented as HD on the day of preparation (day 0) and on the fifth day (day 4), both normalized to the HDday0 value. (B) Liposome concentration in 1× PBS reported by the number of entities diffusing through the FCS interrogation volume (N). Data is presented as N on day 0 and on day 4, both normalized to the Nday0 value. (C) Average hydrodynamic diameter (HD) for liposomes in 1× PBS + 0.3 M sucrose. Data is presented as HD on day 0 and on day 4, both normalized to the HDday0 value. (D) Liposome concentration in 1× PBS + 0.3 M sucrose reported as N on day 0 and on day 4, both normalized to the Nday0 value. Two-tailed t-tests were performed comparing day 4 to day 0 data for all panels, assuming unequal variance (Welch correction) and significance is shown as * (p < 0.05) and ** (p < 0.01). Error bars represent error propagated through averaging three biological replicates, each having five technical replicates, and the error is normalized to the day 0 value for comparison. | ||
Comparing particle concentration derived from FCS data between day 0 and day 4 resulted in significant differences for formulations 2PEG (p = 0.012) and 3 (p = 0.042) (Fig. 4B). This comparison revealed no significant difference in concentration between days for formulations 1, 1PEG, 2, and 3PEG in 1× PBS, but the smaller variance within formulation 2 and 3PEG data suggest these are more reproducible formulations and would be better candidates (Fig. 4B). When conducting similar comparisons for the samples prepared in 1× PBS + 0.3 M sucrose, significant differences were found for concentrations measured between days 0 and 4 for formulations 1 (p = 4.3 × 10−6), 1PEG (p = 1.6 × 10−8), 2 (p = 0.042), and 3 (p = 0.0027) (Fig. 4D). Lack of significant differences between day 0 and day 4 concentration in 1× PBS + 0.3 M sucrose paired with a relatively low degree of variance in the data suggests that formulation 3PEG is the best suited for VLP production from a colloidal stability standpoint (Fig. 4D).
Electrophoretic light scattering (ELS) was performed on formulations 1, 2, 3, 1PEG, 2PEG, and 3PEG to characterize the average surface zeta (ζ) potential of the liposomes in solution (Fig. 5A). This was carried out in 1× PBS as well as in 1× PBS with 0.3 M sucrose. A lack of significance by two-way ANOVA revealed that within error these six liposome formulations had the same ζ potential and therefore the same effective surface charge (p = 0.567 comparing formulations, p = 0.197 comparing buffers). This data also allows us to conclude that the presence or absence of sucrose as a stabilizer did not significantly alter the electrophoretic mobility of the particles. Dynamic light scattering (DLS) was then used to characterize the mean hydrodynamic diameters and polydispersity indices (PDIs) of the liposome particles (Fig. 5B and C). Again, for the selected six formulations named above, no significant difference was found in hydrodynamic diameter between the formulations nor between the buffers by two-way ANOVA (p = 0.104, p = 0.067 for formulations and buffers respectively). The same was shown for the PDIs of the individual formulations (p = 0.536, p = 0.660 for formulations and buffers respectively), but while no significance was found this was likely due to the large variance observed. As such, we interpreted this data to suggest that the formulations displaying smaller PDI ranges were more monodispersed and thus more ideal to work with moving forward. By collectively considering the zeta potential, hydrodynamic diameter, and PDI data, formulations 2, 3, 2PEG, and 3PEG appeared to be equally suitable for VLP production. In combination with the stability data (Fig. 4), however, it became clear that formulations 2 and 3PEG were the most suitable. From this point forward, formulation 2 was selected as a viable option for VLP production, acknowledging that formulation 3PEG would also have been a reasonable choice. We selected formulation 2 hypothesizing that its lack of PEG content might be more favourable for the insertion of proteins into the bilayer due to less steric hindrance, but this hypothesis was not tested in this work.
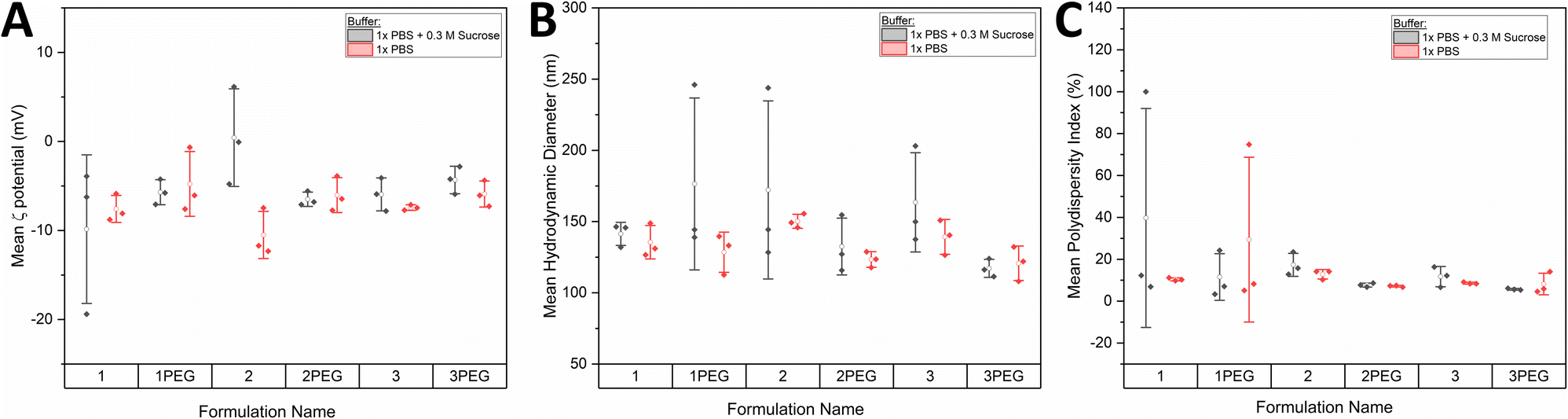 | ||
| Fig. 5 (A) Mean zeta (ζ) potential of the top six candidates using ELS. (B) Mean intensity-weighted hydrodynamic diameters of the top six candidates using DLS. (C) Mean polydispersity indices of the top six candidates using DLS. Data is reported for measurements of formulations in both 1× PBS and 1× PBS + 0.3 M sucrose. Filled points represent mean (n = 3 technical replicates) for each of 3 independent samples. Open points represent the overall mean of the three independent samples and bars represent ± standard deviation for these means. Two-way ANOVA was performed for all panels. | ||
Once the liposomes were treated with 0.2 mM TX100, the LMNG-solubilized fluorescent spike proteins (spike-AF633) were added to the liposome solution and incubated with the liposomes at room temperature for 90 min. To remove the TX100 and promote protein insertion into the liposomal membranes, 3 mg aliquots of polystyrene microbeads (Bio-Beads™ SM-2 resin, Bio-Rad 1523920, Hercules, CA) (henceforth, BioBeads) were added in 120 min intervals, then allowed to incubate with the liposome-protein mixture overnight at 4 °C.32 After the overnight incubation, one final aliquot of BioBeads (in total, 4–5 aliquots) was added at room temperature, and removed after 120 min. The capacity of BioBeads to completely remove TX100 from a liposome solution was confirmed by a set of control experiments showing eradication of TX100 – originating absorbance at 280 nm from a liposome solution after BioBead treatment (ESI Fig. 2†). Other control experiments in which mock reconstitutions were conducted with AF633 resulted in particles with significantly lower fluorescent brightness per particle compared to spike-AF633 reconstituted liposomes (p = 0.0014) (ESI Fig. 3†). This suggests that measured AF633 signal observed in any VLPs produced was due to the presence of spike-AF633 in the membrane rather than non-specific dye adsorption (ESI Fig. 3†).
Biophysical characterization of VLPs
Success of protein reconstitution into VLPs and quantification of such protein loading was first established using FCS.32 To qualitatively probe the success of VLP formation, FCS was used to compare diffusion coefficients between spike-AF633 and the assembled VLP (Fig. 6A). The diffusion coefficient for the proteins in the VLP was an order of magnitude smaller than those of the spike-AF633 (p = 5.7 × 10−20). Since the only fluorescent entity in both samples is the spike-AF633, we suggest that the apparent slower-diffusing entity detected in the VLP channel is the spike-AF633 associating with the liposomes in solution, providing evidence of VLP formation. To assess the degree of reconstitution quantitatively, a brightness analysis was performed wherein the fluorescent brightness per particle of diffusing VLPs was compared to the fluorescent brightness per particle of freely diffusing detergent-solubilized spike proteins (eqn (6)) (Fig. 6C). This produced a value for the average loading ratio of spikes per VLP for the particles in solution, assuming that the brightness of membrane-reconstituted spike proteins is the same as the free spike protein brightness (Table 1). | ||
| Fig. 6 (A) Representative normalized autocorrelation decays for spike-AF633 and VLPs in identical buffer conditions. (B) Mean diffusion coefficients obtained from fitting curves in (A) with eqn (1). Data represents minimum of three technical replicates each of three independent preparations (Spike-AF633) or two independent preparations (VLP-AF633). (C) Brightness per particle for Spike-AF633 compared to VLPs in EBSS with and without 0.3 M sucrose. For these samples, ratio of brightness resulted in an average of 9 ± 4 spikes per VLP. Data represents three technical replicates. For (B) and (C), statistical significance is represented as **p < 0.01 by one-way ANOVA. | ||
| Buffer | Hydrodynamic diameter (nm) | Average spike proteins per VLP |
|---|---|---|
| 1× PBS | 161 ± 19 | 3 ± 1 |
| EBSS | 124 ± 39 | 9 ± 4 |
| EBSS + 0.3 M sucrose | 233 ± 73 | 9 ± 4 |
The produced VLPs were next characterized using DLS to determine their hydrodynamic diameters in solution. For these, VLPs were prepared in 1× PBS, and in addition they were prepared in Earl's balanced salt solution (EBSS) and EBSS + 0.3 M sucrose to maintain appropriate cell growth conditions and to allow for cell adherence to be maintained. Control experiments with liposomes show that using EBSS in place of 1× PBS to prepare liposomes does not significantly alter the measured properties of the particles with and without sucrose added (see ESI Fig. 4†). Significant differences are also not observed between VLP diameters in the different buffer systems (p = 0.054) (Table 1, ESI Fig. 5†).
SDS-PAGE was also employed to characterize the structure of the spike proteins in LMNG micelles as well as in VLPs. For the detergent-solubilized spike proteins, this analysis resulted in a clearly observed band around apparent molecular weight of 227 kDa corresponding to the glycosylated spike monomer which has a molecular weight of approximately 142 kDa according to the manufacturer. Such discrepancy between expected and observed gel migration is a regularly observed phenomenon for SDS-PAGE analysis of membrane proteins.66,67 In addition to these major bands, faint bands around 580 and 81 kDa are observed, likely corresponding to the trimeric spike (formal weight 426 kDa) and S1 or S2 subunits (formal weight 78 and 62 kDa respectively) (Fig. 7, lanes 2–5). VLPs run on the same gel in lanes 6–9 show similar band patterning as was seen for intact spike proteins in lanes 2–5, but also showed more prominent high-molecular weight banding which could indicate multimerization of the spike protein, perhaps due to protein–lipid–cholesterol interactions not completely disrupted by SDS (Fig. 7, lanes 6–9). Performing densitometric analysis using lanes 2–5 to create a standard curve led to an evaluation of the apparent spike concentration in the VLP samples in lanes 6–9 (ESI Fig. 6A†).68 Based on this, a linear relationship appears to exist between spike concentration in the final VLP solution and the original mixing ratio of spikes to liposomes (ESI Fig. 6B†). These data and preliminary FCS brightness data for other VLP preparation conditions (data not shown) support the tunability of these VLPs in terms of antigen concentration, however more experimentation is necessary to resolve this relationship which is the topic of ongoing study in our group.
 | ||
| Fig. 7 Captured image of SDS-PAGE gel analysis of spike glycoproteins (lanes 2–5) and VLPs (lanes 6–9) (see Methods). Gel was stained with Coomassie Brilliant Blue stain. Spike protein concentrations in samples loaded on the gel were 10, 40, 80, and 120 nM for lanes 2, 3, 4, 5 respectively. For the VLP samples, calculated spike concentrations based on initial preparation conditions were 9, 38, 79, and 117 nM for lanes 6, 7, 8, and 9 respectively. Molecular weight ladder in lane 1 is HiMark™ Pre-stained Protein Standard (Thermo Fisher Scientific). | ||
Another method used to characterize VLPs was transmission electron microscopy (TEM), which permitted the visualization of spike proteins embedded within the liposome membrane. Empty liposomes, unlabeled spike proteins, and labeled spike proteins were also characterized using TEM (Fig. 8D–F). Approximately 10–15 nm diameter projections from the liposomal membranes shown in TEM micrographs of VLPs suspended in a cellulose matrix suggest that proteins are inserted in the membrane (Fig. 8A–C, red arrows).42,69,70 In contrast, empty liposomes also suspended in cellulose shown in Fig. 8D do not contain these membrane projections, which suggests that these are not artifacts of the matrix but are more likely related to the presence of spike proteins in the sample. Micrographs of the LMNG-solubilized spike proteins show that the morphology of the proteins within the LMNG micelles appears to be similar with and without the fluorescent tag (Fig. 8E and F).71 While these free proteins in panels E and F appear slightly morphologically different than the proposed membrane-anchored spike proteins in panels A–C, this might be because LMNG solubilization confers a micellar shape. These micrographs also suggest a size agreement to the proposed spike proteins in the VLP images, with the LMNG micelles appearing to have diameters between 10 and 25 nm for both unlabelled and labelled proteins.
 | ||
| Fig. 8 Representative TEM micrographs of (A–C) VLPs in 1× PBS with 0.3 M sucrose, (D) empty formulation 2 liposomes in 1× PBS with 0.3 M sucrose. (E) Spike glycoprotein solubilized in LMNG micelles, suspended in 1× PBS + 5 μM LMNG + 2 mM sodium azide, and (F) spike-AF633 solubilized in LMNG micelles, suspended in 1× PBS + 5 μM LMNG + 2 mM sodium azide. Red arrows denote proposed spike proteins in VLPs, and yellow scale bars represent 100 nm. | ||
VLPs prepared in EBSS with and without 0.3 M sucrose as an additive were also monitored for colloidal stability by measuring hydrodynamic diameter with DLS at 0, 7, and 14 days post-preparation (ESI Fig. 7†). These timepoints were selected as they are typical monitoring times for acute exposure in the case of downstream in vivo applications. Both buffer conditions resulted in VLPS which had no statistically significant change in diameter over 14 days, however notably fewer large aggregates were observed at 7 and 14 days post-production when the VLPs were produced and stored with the 0.3 M sucrose additive. This suggests that the hypothesized stabilizing effect of the sucrose may be a valid consideration when preparing VLPs for future in vivo studies.
Cellular internalization of VLPs
Calu-3 is a cancer-derived bronchiolar epithelial cell line which has been used extensively to model lung epithelia for pharmacological studies.72 Specifically for SARS-CoV-2, this cell line has been used for viral propagation and to model infection.73,74 Based on this precedent for the use of this cell line, Calu-3 cells were used as the cell line for VLP internalization in this study. Cells were treated with VLPs in EBSS or EBSS + 0.3 M sucrose using exposure times of 60 min (Fig. 9A and B) and 150 min (Fig. 9C and D), based on typical in vitro infection times for cells by SARS-CoV-2.74 After treatment, confocal laser scanning microscopy (CLSM) was used to visualize cellular uptake of VLPs. For cell treatment experiments immunofluorescence (IF) was performed on ACE2 receptors and staining of the plasma membranes and nuclei was performed. Four detection channels were used to distinguish fluorescence from the nucleus, plasma membrane, ACE2 secondary antibodies, and fluorescent VLPs. Buffer treatment and IF controls are shown in ESI Fig. 8 and 9.† | ||
| Fig. 9 Confocal fluorescence microscopy images of Calu-3 cells treated for (A) 60 min with VLPs in EBSS, (B) 150 min with VLPs in EBSS, (C) 60 min with formulation 2 liposomes in EBSS, (D) 60 min with VLPs in EBSS + 0.3 M sucrose, (E) 150 min with VLPs in EBSS + 0.3 M sucrose, and (F) 150 min with formulation 2 liposomes in EBSS + 0.3 M sucrose. All VLP samples contained 9 ± 4 spike proteins per particle and a VLP concentration of (2 ± 1) × 1011 VLPs per mL (total spike concentration of approximately 0.4 μg mL−1). Liposomes and VLPs were treated at equimolar concentrations ((2 ± 1) × 1011 particles per mL). White boxes designate the area shown in the 5× zoom panel, and white arrows guide the eye to areas of AF633 fluorescence detected in the images. VLPs were visualized by fluorescence from spike-AF633 (pink) and monitored for colocalization with AF555-labeled ACE-2 (orange). Cell membranes are labelled with WGA-AF488 (green) and nuclear counterstaining via DAPI is shown in blue. Cytosolic AF488 signal is likely due to endosomal recycling pathways internalizing membrane stain, since membrane staining was done pre-fixation. Images were viewed under 63× magnification. Scale bars represent 50 μm. Data presented are representative images from three independent experiments, each imaged in triplicate z-stacks. | ||
Post hoc analysis of CLSM images was performed to determine whether VLP internalization might be an ACE2-dependent process mimicking the internalization pathway of the authentic SARS-CoV-2 virus. Specifically, pixel-by-pixel colocalization of Alexafluor-555 signal from ACE2 and Alexafluor-633 signal from VLPs was studied.75 Control experiments comparing the integrated fluorescence intensity per cell unit area of cells treated with fluorescent spike protein to those treated with excess fluorescent label with no spike protein showed a significantly higher uptake of AF633 signal into the cell cytosol when protein is present (p = 1.39 × 10−36) (ESI Fig. 9 and 11†). This supports the validity of our assumption that fluorescent signal from AF633 visible on CLSM images indeed represents spike proteins and not free AF633. When plotting calculated colocalization coefficients versus the ratio of pixel areas bearing signal from the two interrogated fluorophores, it becomes clear that the amount of ACE-2-AF555 signal is not a limiting factor in the degree of colocalization observed between the two fluorophores (ESI Fig. 12†). Moreover, calculated Manders' colocalization coefficients for this fluorophore pair for all four VLP treatments (shown in Fig. 9A, B, D and E) were significantly higher than colocalization coefficients calculated from control empty liposomes prepared both with and without sucrose present (Fig. 9C and F) (Fig. 10, ESI Fig. 13†) (p = 0.033 with sucrose, p = 0.017 without sucrose).76,77 Two-way ANOVA reveals that the presence of sucrose has no significant effect on the amount of colocalization observed (ESI Fig. 14†).
 | ||
| Fig. 10 Mander's colocalization coefficient (M1) represented as percent of pixels in each image showing signal for AF633 which also show signal for AF555, compared between images of cells treated for 60 min with equivalent concentrations of empty liposomes in EBSS-Sucrose and VLPs in EBSS-Sucrose. (B) M1 compared between images of cells treated for 60 min with equivalent concentrations of empty liposomes in EBSS and VLPs in EBSS. For both (A) and (B), data is represented as mean ± standard deviation of coefficients collected from three images from each of 2 independent biological replicates. Statistical significance is shown as *p < 0.05 as calculated by one-way ANOVA. | ||
This significant difference between VLP and liposome treatments supports the legitimacy of colocalization signal observed in VLP-treated cells and suggests that the presence of the spike proteins in the membrane of the VLP results in a significant amount of colocalization with ACE-2 receptors in vitro.
Experimental methods
Materials
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC),1,2-dioleoyl-sn-glycero-3-phospho-(1′-myo-inositol), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dioleoyl-sn-glucero-3-phospho-(1′-myo-inositol) (DOPI), 1,2-dipalmitoyl-sn-glycero-3-phospho-(1′-myo-inositol) (DPPI), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE), PEG-1000-conjugated 2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE-PEG1K), PEG-1000-conjugated 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-PEG1K), cholesterol, and Oregon Green™ 488 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (OG-DHPE) were purchased from Sigma-Aldrich (DOPC, DPPC, DOPI, DOPE, DPPE, DOPE-PEG1K, DSPE-PEG1K) and Cayman Chemical (DPPI). Stock solutions were prepared in chloroform and stored at −20 °C. Full-sequence SARS-CoV-2 spike glycoprotein trimers solubilized in LMNG detergent micelles were obtained (Cube Biotech, Monheim am Rhein, Germany) and fluorescently labeled with AlexaFluor-633 labeling kits (Thermo Fisher A20170, Waltham, MA).Liposome preparation
Multilamellar liposomes were prepared using a modified lipid film hydration technique.78 The liposome solutions were extruded 29 passes through a polycarbonate membrane with 100 nm diameter pores to produce unilamellar vesicles. For liposome characterization prior to VLP production, OG-DHPE was incorporated at <1 mol% for fluorescence analysis of the nanoparticles via FCS as described below.Fluorescent labeling of spike glycoproteins
Briefly, sodium bicarbonate was added to the spike protein to raise the pH above 7.5. The basic protein solution was added to a vial of reactive Alexa Fluor 633 dye and the reaction tube was stirred at room temperature for 90 min. The reaction mixture was then loaded on a column packed with size exclusion purification resin (Zeba™ spin columns, Thermo Fisher Scientific) and the labeled protein was eluted (elution buffer modified to include 2 mM sodium azide and 5 μM lauryl maltose neopentyl glycol detergent) by centrifuging the columns at 1000×g. Fractions were collected and absorbance at 280 and 632 nm were measured using a NanoDrop™ spectrophotometer to determine the concentration and degree of labeling of the fractions containing labeled protein.Liposome stability tests
Liposome formulations were discarded immediately from the pool of candidates if they appeared highly aggregated upon initial preparation based on FCS data. This process of testing liposome candidates involved FCS measurements of each liposome sample every day for five days, wherein the samples were stored at 4 °C between measurements but brought to room temperature for FCS measurements. Results were reported as percent change compared with that from the preparation date to normalize between samples, and standard deviations obtained from multiple technical replicates of multiple preparations were propagated through relevant calculations.Two-photon excitation fluorescence correlation spectroscopy (TPE-FCS) and two-photon excitation fluorescence cross-correlation spectroscopy (TPE-FCCS)
Two-photon excitation fluorescence correlation and cross-correlation spectroscopy (TPE-FCS and TPE-FCCS) were used to study liposome stability and longevity, and to monitor and verify protein reconstitution. Two-photon excitation was used because its femtolitre-range interrogation volume improves detection of fluctuations in fluorescence and reduces thermal and photo-damage to the sample. FCS allows fluctuations of fluorescence intensity within a given interrogation volume to be monitored over time. FCCS correlates fluorescence intensity fluctuations from two or more distinctly labeled fluorophores moving together in solution.79 Importantly, this is done in a non-invasive manner that does not affect the equilibrium of the system.Fluorescent count rate analysis
Fluorescence emissions from the interrogation volume of the FCS instrument were detected and grouped into 256 separate temporal bins, the length of which depended on the overall collection time of each experimental “run”. Bins were arranged sequentially to form a count rate trajectory, fluctuations in which were autocorrelated as described below. Averaging the count rates across the experimental run allowed for evaluation of brightness per particle (see FCS brightness analysis).Autocorrelation and cross-correlation analysis
By analyzing autocorrelation data from one fluorophore, FCS provides information on changes in relative concentrations of the fluorophore, which can give indirect information about processes such as binding, aggregation, fluorescent quenching, or energy transfer. Diffusion coefficients can also be obtained from the data which can inform about the size of the diffusing entity. During an FCS measurement, molecules diffuse in and out of the excitation volume due to Brownian motion, and their fluorescence is detected when inside the TPE volume. The count rate trajectory describes the average fluorescence intensity per time bin, from the fluorescence emitters within the TPE volume. As this fluorescence intensity fluctuates over time, the signal is autocorrelated temporally to show the self-similarity of the signal as a function of lag time, τ. Physically, this autocorrelation decay represents the likelihood of finding the same emitter particle within the TPE volume at different lag times. The normalized autocorrelation trace can be fitted to the following function: | (1) |
 | (2) |
FCCS can track spectrally distinct fluorophores which are physically linked, by registering simultaneous fluorescence fluctuations resulting from their movement together in and out of the TPE volume. A cross-correlation signal appears only when the movement of these multiple fluorophores is temporally correlated. This cross-correlation decay is modelled using eqn (3),
 | (3) |
 | (4) |
FCS brightness analysis
For solutions where only one type of fluorescent diffuser can be assumed to be present, eqn (2) can be re-written as eqn (5) below, | (5) |
Following this, the brightness per particle can be obtained using eqn (6),
 | (6) |
TPE-FCS/FCCS instrumentation
The TPE-FCS setup has been described elsewhere.61 Briefly, a mode-locked Titanium![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sapphire 100 fs pulsed laser, operating at a 82 MHz repetition rate, with an excitation wavelength of 774 nm was used for all TPE-FCS and FCCS experiments. In the chosen optical arrangement, a sequence of mirrors and lenses direct and expand the excitation laser beam to fill the back aperture of a 40×, 1.2 NA, 0.8 mm working distance water-immersion objective lens. This objective lens collects fluorescence emitted by the diffusing particles in the TPE volume and reflects this emitted light to a dichroic optic which separates it from the excitation light. The fluorescence is then further separated into the two distinct coloured light paths and finally each is passed through a spectral notch filter and directed to a separate avalanche photodiode detector. The fluorescence intensity information from the photodiodes is analyzed by a PC using a correlator card (ALV-6000). All data was plotted and processed using OriginPro software.
:Sapphire 100 fs pulsed laser, operating at a 82 MHz repetition rate, with an excitation wavelength of 774 nm was used for all TPE-FCS and FCCS experiments. In the chosen optical arrangement, a sequence of mirrors and lenses direct and expand the excitation laser beam to fill the back aperture of a 40×, 1.2 NA, 0.8 mm working distance water-immersion objective lens. This objective lens collects fluorescence emitted by the diffusing particles in the TPE volume and reflects this emitted light to a dichroic optic which separates it from the excitation light. The fluorescence is then further separated into the two distinct coloured light paths and finally each is passed through a spectral notch filter and directed to a separate avalanche photodiode detector. The fluorescence intensity information from the photodiodes is analyzed by a PC using a correlator card (ALV-6000). All data was plotted and processed using OriginPro software.
Dynamic and electrophoretic light scattering
The top six liposome candidates, and later the produced VLPs, were characterized using DLS to determine their hydrodynamic diameter and PDI. DLS experiments were performed using a Malvern Panalytical Zetasizer Advance Series – Ultra (Red Label) instrument to measure the mean diameter and PDI of the nanoparticles. Settings included a backscatter measurement angle of 173° and a 633 nm incident laser wavelength for the 4.0 mW He–Ne laser. Fluorescence filters were applied when required. Measurements were performed within 10 mm path length acrylic cuvettes and results were reported as mean ± standard deviation of three technical replicates for each of three independent samples. Zeta potential was measured at room temperature on the same instrument using electrophoretic light scattering (ELS) to approximate the surface charge of the nanoparticles. The measurement settings were consistent with the DLS settings.Transmission electron microscopy
To prevent artifacts from drying or vitrification of liposomes and VLPs for TEM, a methylcellulose matrix-assisted drying process was used as a structural support for the particles before negative staining with uranyl acetate.80,81 Briefly, 3 mm copper grids with carbon film (CF200-CU-50 200 mesh) were glow discharged (PELCO easiGlow), then grids were inverted on drops of sample and incubated at room temperature for 60 min. The grids were washed with 1× PBS for 1 min, fixed for 5 min with 4% paraformaldehyde + 2.5% glutaraldehyde in 0.1 M sodium cacodylate, and then stained with 2% uranyl acetate for 5 min. Next, the grids were transferred to drops of 2% methyl cellulose containing 0.2% uranyl acetate and incubated for 15 min at 4 °C. The grids were blotted and dried on wire loops at room temperature before imaging.Samples were imaged in both high resolution and high contrast modes at 120 kV using a Hitachi HT7800 transmission electron microscope. Images were processed using FIJI by applying a Gaussian blur filter with a blur radius of 2.00 pixels.82
Polyacrylamide gel electrophoresis (PAGE)
A pre-cast polyacrylamide gel (NuPage Bis–Tris 4–12%) was washed twice with 1× MOPS SDS buffer. Protein samples were prepared with 1× LDS sample buffer, 2 mM DTT reducing agent, and AF633-labeled spike protein brought to final concentrations shown in Fig. 7. Similarly, VLP samples were prepared containing calculated spike concentrations ranging from 9 to 117 nM. Samples were centrifuged briefly then heated at 90 °C for 5 min. Samples were loaded into wells and NuPAGE antioxidant was added to the upper gel tank chamber to prevent sample reoxidation. Gels were run for 50 min at 200 V constant voltage. Densitometric analysis was performed using FIJI.82Cell line
We maintained Calu-3 cells from the American Type Culture Collection (ATCC) in Minimum Essential Medium Eagle – alpha modification (α-MEM) (Wisent Biological 310-010-CL) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% antibiotic–antimycotic (Millipore-Sigma a5955). All cells were incubated at 37°C with 5% CO2. Cells were cultured to 90% confluency at which point the supernatant was removed and the cells were rinsed with PBS and later trypsinized with TrypLE (Gibco™ TrypLE™ Express Enzyme (1×), no phenol red, 12604013) for 5 min at 37 °C. The trypsinized cell suspension was then neutralized with an equal volume of culture media and centrifuged at 500×g for 3 min at room temperature to pellet the cells. Cells were resuspended in culture media and seeded on cover slips coated with poly-L-lysine (PLL) (Mandel Scientific) at a density of 250000 cells per well in a Falcon 12-well plate flat bottom tissue culture plate (Corning 353043).
Cell treatment, staining, and immunofluorescence
Calu-3 cells on PLL-coated coverslips (#1.5 thickness) were treated with VLPs at a concentration of (2 ± 1) × 1011 particles per mL, bearing spike proteins at a molar stoichiometry of 9 ± 4 spikes/VLP in EBSS and EBSS + 0.3 M sucrose for 60 and 150 min. Control treatments included liposomes in EBSS, liposomes in EBSS-Sucrose, EBSS, EBSS-sucrose, Spike protein in LMNG-PBS, LMNG-PBS, and Alexa Fluor 633-LMNG-PBS (see ESI†). Cells were stained with Alexa Fluor 488-conjugated WGA (WGA-488) (Invitrogen W11261) diluted 1:500 in HBSS for 5 min at 37 °C before fixation, then fixed with 4% paraformaldehyde in HBSS for 15 min at 37 °C and allowed to cool to room temperature. The cells were next treated with 100 mM glycine for 10 min before immunofluorescence staining. The cells were blocked with goat serum for 60 min and immunostained with human recombinant anti-ACE2 antibody (Abcam ab272500) overnight at 4 °C. Fluorescent secondary antibody (Anti-rab 555) (Invitrogen A32732) was then incubated with the cells for 60 min at room temperature. Appropriate controls were prepared to account for nonspecific binding of primary and secondary antibodies and to provide single-stained optical controls for colocalization analysis. Cell nuclei were counterstained with DAPI. Coverslips were mounted on microscopic slides using Dako mounting medium (Agilent Technologies CS70330-2). Cells were left unpermeabilized to prevent ambiguity about the mechanism of internalized fluorescent signal.
Confocal laser scanning microscopy
To visualize cellular uptake of VLPs, confocal laser scanning microscopy (CLSM) was conducted using a Zeiss LSM700 confocal microscope. Four channels were measured on three tracks, to distinguish spectrally separated fluorescence from (a) DAPI, (b) WGA-488, (c) Anti-rab 555, and (d) VLPs with AF633-labeled spike proteins (VLP-AF633). All images were 16 bit with dimensions of 2048 × 2048 pixels, a resolution of 13.26 pixels per micron, and each z-slice had a thickness of 0.38 μm. Laser excitation and gain parameters were kept consistent between all samples within each biological replicate for image analysis purposes. Images represented in the text body originate from one biological replicate for consistency.CLSM image analysis
Colocalization analysis was performed to quantify the spatial coincidence of fluorescent signal in the ACE2-AF555 and VLP-AF633 detection channels for confocal microscopy images using the BIOP JACoP plugin in FIJI.83,84 Briefly, first, all images were processed by using rolling-ball background subtraction with a rolling-ball radius of 50 pixels. Next, single-stained control sample images were used to set threshold values for non-specific background signal in the two channels of interest. These threshold values were averaged over three images for each control sample, and the average background signal values were used to set thresholds for Manders method of colocalization analysis. Colocalization analysis was then performed on three images each of three independent biological replicates for each treatment. Mean fluorescence intensity measurements were performed using FIJI by first performing a maximum projection z-stack compression of the images, followed by selecting cytosolic regions of interest using the WGA-AF488 cell membrane staining as a guide. Integrated fluorescence intensity was normalized to the integrated cell area for each cell for comparison between treatments. 2 biological replicates, each with three images collected, were used for these analyses.Statistical analyses
For normally distributed data, including data in liposome stability studies, DLS and ELS data, Mander's colocalization data, and FCS brightness analyses, pairwise comparisons were executed via two-tailed t-tests assuming unequal variance with the Welch correction. Tables of p-values are found in ESI Table 2.† Multi-level comparisons or multi-factor comparisons were carried out using one- or two-way ANOVA respectively (ESI Tables 3 and 5† for p values). In some cases, where Smirnov and Shapiro–Wilk normality tests determined that data was not normally distributed, mostly due to cell–cell variation and low population counts, Kruskal–Wallis nonparametric ANOVA and Mann–Whitney U-tests were performed on the data to compare mean fluorescence intensity between treatments (ESI Table 4† for p values). In cases where equal variance could not be assumed as determined by Levene's test, Welch ANOVA was performed (ESI Table 6† for p values).Conclusions
In an ever-evolving COVID-19 pandemic, research into creating better vaccines and anti-viral therapeutics against SARS-CoV-2 remains essential. In this work, we report our successful preparation of a novel liposome-based SARS-CoV-2 VLP containing fluorescent, full-length membrane anchored spike glycoproteins. This work presents evidence that these particles are internalized into relevant lung epithelial cells through a similar ACE-2-mediated pathway to that of the authentic SARS-CoV-2 virus. Because of our interest in understanding relationships between antigen density on virus-like particles and resultant cellular cytokine responses, this study stands as a proof of concept for the further development of these particles into density-tunable systems. One limitation of this work is the inevitable heterogeneity of the prepared VLP solutions and as such the optimization of purification protocols is ongoing and will be featured in a follow-up manuscript. In addition, further developing the tunability of these VLPs will advance research elucidating relationships between spike protein surface density and cell-level immune response.Author contributions
Sarah McColman: conceptualization, funding acquisition, project administration, methodology, investigation, data curation, formal analysis, writing (original draft, review, and editing), visualization. Klaidi Shkalla: investigation, formal analysis, writing (editing). Jady Liang: funding acquisition, investigation, writing (editing). Selena Osman: investigation, formal analysis. Ana Carolina Forjaz Marques: Investigation. Pavleen Sidhu: investigation, formal analysis. Zainab Bokhari: Investigation. Norbert Kovacs: Software. Yuchong Li: investigation. Qiwen Lin: investigation. Haibo Zhang: supervision, resources, funding acquisition, writing (editing). David T. Cramb: supervision, resources, writing (editing), funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding and infrastructure for this study was provided by the National Science and Engineering Research Council of Canada (NSERC Grant #RT691164 to DTC), Toronto Metropolitan University, University of Toronto, Canadian Institute for Health Research (CIHR Grant OV3-170344, SBC-171482 and VS1-175560 to HZ), Unity Health Toronto, and the University of Calgary. We would like to acknowledge the Keenan Research Centre for Biomedical Science Core Facilities at St. Michael's Hospital (Toronto, Canada), specifically Dr Dario Bogojevic and Dr Caterina di Ciano-Oliveira for technical expertise and training. We would also like to acknowledge the Nanoscale Biomedical Imaging Facility at the Hospital for Sick Children (Toronto, Canada) for use of the TEM with special thanks to Dr Ali Darbandi for technical expertise and training. Schematics in Fig. 1 and graphical abstract were created with https://www.biorender.com/.Notes and references
- T. P. Velavan and C. G. Meyer, Trop. Med. Int. Health, 2020, 25, 278–280 CrossRef CAS PubMed.
- P. Hunter, EMBO Rep., 2020, 21, e50334 CrossRef CAS PubMed.
- M. A. Tortorici and D. Veesler, Adv. Virus Res., 2019, 105, 93–116 CAS.
- R. Xu, M. Shi, J. Li, P. Song and N. Li, Front. Bioeng. Biotechnol., 2020, 8, 1026 CrossRef PubMed.
- A. Rauf, T. Abu-Izneid, A. A. Khalil, N. Hafeez, A. Olatunde, M. Rahman, P. Semwal, Y. S. Al-Awthan, O. S. Bahattab, I. N. Khan, M. A. Khan and R. Sharma, Int. J. Surg., 2022, 104, 106818 CrossRef PubMed.
- A. H. Thames, K. L. Wolniak, S. I. Stupp and M. C. Jewett, ACS Cent. Sci., 2020, 6, 1341–1347 CrossRef CAS PubMed.
- L. Zhang, C. B. Jackson, H. Mou, A. Ojha, H. Peng, B. D. Quinlan, E. S. Rangarajan, A. Pan, A. Vanderheiden, M. S. Suthar, W. Li, T. Izard, C. Rader, M. Farzan and H. Choe, Nat. Commun., 2020, 11, 6013 CrossRef CAS PubMed.
- J. Lu, G. Lu, S. Tan, J. Xia, H. Xiong, X. Yu, Q. Qi, X. Yu, L. Li, H. Yu, N. Xia, T. Zhang, Y. Xu and J. Lin, Cell Res., 2020, 30, 936 CrossRef CAS PubMed.
- C. B. Plescia, E. A. David, D. Patra, R. Sengupta, S. Amiar, Y. Su and R. v. Stahelin, J. Biol. Chem., 2021, 296, 100103 CrossRef CAS PubMed.
- H. Swann, A. Sharma, B. Preece, A. Peterson, C. Eldridge, D. M. Belnap, M. Vershinin and S. Saffarian, Sci. Rep., 2020, 10, 21877 CrossRef CAS PubMed.
- B. Boson, V. Legros, B. Zhou, E. Siret, C. Mathieu, F. L. Cosset, D. Lavillette and S. Denolly, J. Biol. Chem., 2021, 296, 100111–100112 CrossRef CAS PubMed.
- A. M. Syed, T. Y. Taha, T. Tabata, I. P. Chen, A. Ciling, M. M. Khalid, B. Sreekumar, P. Y. Chen, J. M. Hayashi, K. M. Soczek, M. Ott and J. A. Doudna, Science (1979), 2021, 374, 1626–1632 CAS.
- D. A. Rothen, P. S. Krenger, A. Nonic, I. Balke, A. C. S. Vogt, X. Chang, A. Manenti, F. Vedovi, G. Resevica, S. M. Walton, A. Zeltins, E. Montomoli, M. Vogel, M. F. Bachmann and M. O. Mohsen, Allergy, 2022, 77, 2446–2458 CrossRef CAS PubMed.
- Q. Geng, W. Tai, V. K. Baxter, J. Shi, Y. Wan, X. Zhang, S. A. Montgomery, S. A. Taft-Benz, E. J. Anderson, A. C. Knight, K. H. Dinnon, S. R. Leist, R. S. Baric, J. Shang, S. W. Hong, A. Drelich, C. T. K. Tseng, M. Jenkins, M. Heise, L. Du and F. Li, PLoS Pathog., 2021, 17, e1009897 CrossRef CAS PubMed.
- M. Bañó-Polo, L. Martínez-Gil, M. M. Sánchez del Pino, A. Massoli, I. Mingarro, R. Léon and M. J. Garcia-Murria, J. Oral Microbiol., 2022, 14, 2030094 CrossRef PubMed.
- I. J. Lee, Y. H. Lan, P. Y. Wu, Y. W. Wu, Y. H. Chen, S. C. Tseng, T. J. Kuo, C. P. Sun, J. T. Jan, H. H. Ma, C. C. Liao, J. J. Liang, H. Y. Ko, C. S. Chang, W. C. Liu, Y. A. Ko, Y. H. Chen, Z. L. Sie, S. I. Tsung, Y. L. Lin, I. H. Wang and M. H. Tao, Emerg. Microb. Infect., 2023, 12(1), 2149353 CrossRef PubMed.
- F. Hemmati, M. Hemmati-Dinarvand, M. Karimzade, D. Rutkowska, M. H. Eskandari, S. Khanizadeh and A. Afsharifar, Biotechnol. Lett., 2022, 44, 45–57 CrossRef CAS PubMed.
- C. Dubé, S. Paris-Robidas, G. Andreani, C. Gutzeit, M. A. D'Aoust, B. J. Ward and S. Trépanier, Vaccine, 2022, 40, 4017–4025 CrossRef PubMed.
- M. A. D'Aoust, M. M. J. Couture, N. Charland, S. Trépanier, N. Landry, F. Ors and L. P. Vézina, Plant Biotechnol. J., 2010, 8, 607–619 CrossRef PubMed.
- K. B. Moon, J. H. Jeon, H. Choi, J. S. Park, S. J. Park, H. J. Lee, J. M. Park, H. S. Cho, J. S. Moon, H. Oh, S. Kang, H. S. Mason, S. Y. Kwon and H. S. Kim, Sci. Rep., 2022, 12, 1005 CrossRef CAS PubMed.
- A. C. Fluckiger, B. Ontsouka, J. Bozic, A. Diress, T. Ahmed, T. Berthoud, A. Tran, D. Duque, M. Liao, M. McCluskie, F. Diaz-Mitoma, D. E. Anderson and C. Soare, Vaccine, 2021, 39, 4988–5001 CrossRef CAS PubMed.
- S. Roy, K. Ghani, P. O. de Campos-Lima and M. Caruso, Virus Res., 2021, 295, 198305 CrossRef CAS PubMed.
- S. Du, W. Xu, Y. Wang, L. Li, P. Hao, M. Tian, M. Wang, T. Li, S. Wu, Q. Liu, J. Bai, X. Qu, N. Jin, B. Zhou, M. Liao and C. Li, J. Virol., 2022, 96, 1897–1918 Search PubMed.
- M. O. Mohsen, I. Balke, S. Zinkhan, V. Zeltina, X. Liu, X. Chang, P. S. Krenger, K. Plattner, Z. Gharailoo, A. C. S. Vogt, G. Augusto, M. Zwicker, S. Roongta, D. A. Rothen, R. Josi, J. J. da Costa, J. M. Sobczak, A. Nonic, L. A. Brand, K. Nuss, B. Martina, D. E. Speiser, T. Kündig, G. T. Jennings, S. M. Walton, M. Vogel, A. Zeltins and M. F. Bachmann, Allergy, 2022, 77, 243–257 CrossRef CAS PubMed.
- A. C. Fluckiger, B. Ontsouka, J. Bozic, A. Diress, T. Ahmed, T. Berthoud, A. Tran, D. Duque, M. Liao, M. McCluskie, F. Diaz-Mitoma, D. E. Anderson and C. Soare, Vaccine, 2021, 39, 4988–5001 CrossRef CAS PubMed.
- M. Chen and X. E. Zhang, Int. J. Biol. Sci., 2021, 17, 1574 CrossRef CAS PubMed.
- P. Pereira Aguilar, T. A. Schneider, V. Wetter, D. Maresch, W. L. Ling, A. Tover, P. Steppert and A. Jungbauer, Vaccine, 2019, 37, 7070–7080 CrossRef CAS PubMed.
- J. Zhang, Y. Xu, M. Chen, Y. Huang, T. Song, C. Yang, Y. Yang and Y. Song, J. Am. Chem. Soc., 2022, 144, 21295–21303 CrossRef CAS PubMed.
- C. R. Lucas, P. D. Halley, A. A. Chowdury, B. K. Harrington, L. Beaver, R. Lapalombella, A. J. Johnson, E. K. Hertlein, M. A. Phelps, J. C. Byrd, C. E. Castro, C. R. Lucas, P. D. Halley, A. A. Chowdury, B. K. Harrington, J. C. Byrd, L. Beaver, R. Lapalombella, A. J. Johnson, E. K. Hertlein, M. A. Phelps and C. E. Castro, Small, 2022, 18, 2108063 CrossRef CAS PubMed.
- W. Chen, Y. Cai, Q. Fu, B. Chen, J. Guo and J. J. Chou, Angew. Chem., Int. Ed., 2019, 58, 9866–9870 CrossRef CAS PubMed.
- I. A. Smirnova, P. Ädelroth and P. Brzezinski, Sci. Rep., 2018, 8, 1–7 CAS.
- P. Simeonov, S. Werner, C. Haupt, M. Tanabe and K. Bacia, Biophys. Chem., 2013, 184, 37–43 CrossRef CAS PubMed.
- J. Kruip, N. v. Karapetyan, I. v. Terekhova and M. Rögner, J. Biol. Chem., 1999, 274, 18181–18188 CrossRef CAS PubMed.
- M. Ando, S. Schikula, Y. Sasaki and K. Akiyoshi, Adv. Sci., 2018, 5, 1870062 CrossRef.
- B. Kratzer, S. Hofer, M. Zabel and W. F. Pickl, Eur. J. Immunol., 2020, 50, 17–32 CrossRef CAS PubMed.
- W. Y. Wholey, J. L. Mueller, C. Tan, J. F. Brooks, J. Zikherman and W. Cheng, Bioconjugate Chem., 2020, 31, 685–697 CrossRef CAS PubMed.
- M. Amidi, M. de Raad, D. J. A. Crommelin, W. E. Hennink and E. Mastrobattista, Synth. Syst. Biol., 2011, 5, 21–31 CrossRef PubMed.
- S. Himbert, I. P. Gastaldo, R. Ahmed, K. M. Pomier, B. Cowbrough, D. Jahagirdar, S. Ros, J. Juhasz, H. D. H. Stöver, J. Ortega, G. Melacini, D. M. E. Bowdish and M. C. Rheinstädter, PLoS One, 2022, 17, e0263671 CrossRef CAS PubMed.
- G. Sulbaran, P. Maisonnasse, A. Amen, G. Effantin, D. Guilligay, N. Dereuddre-Bosquet, J. A. Burger, M. Poniman, M. Grobben, M. Buisson, S. Dergan Dylon, T. Naninck, J. Lemaître, W. Gros, A. S. Gallouët, R. Marlin, C. Bouillier, V. Contreras, F. Relouzat, D. Fenel, M. Thepaut, I. Bally, N. Thielens, F. Fieschi, G. Schoehn, S. van der Werf, M. J. van Gils, R. W. Sanders, P. Poignard, R. Le Grand and W. Weissenhorn, Cell Rep. Med., 2022, 3, 100528 CrossRef CAS PubMed.
- J. Wang, Y. Wen, S. H. Zhou, H. W. Zhang, X. Q. Peng, R. Y. Zhang, X. G. Yin, H. Qiu, R. Gong, G. F. Yang and J. Guo, J. Med. Chem., 2022, 65, 2558–2570 CrossRef CAS PubMed.
- Z. Wang, K. D. Popowski, D. Zhu, B. L. de Juan Abad, X. Wang, M. Liu, H. Lutz, N. De Naeyer, C. T. DeMarco, T. N. Denny, P. U. C. Dinh, Z. Li and K. Cheng, Nat. Biomed. Eng., 2022, 6(7), 791–805 CrossRef CAS PubMed.
- H. Yao, Y. Song, Y. Chen, N. Wu, J. Xu, C. Sun, J. Zhang, T. Weng, Z. Zhang, Z. Wu, L. Cheng, D. Shi, X. Lu, J. Lei, M. Crispin, Y. Shi, L. Li and S. Li, Cell, 2020, 183, 730–738 CrossRef CAS PubMed.
- G. van Meer and A. I. P. M. de Kroon, J. Cell Sci., 2011, 124, 5–8 CrossRef CAS PubMed.
- Q. Wu, L. Zhou, X. Sun, Z. Yan, C. Hu, J. Wu, L. Xu, X. Li, H. Liu, P. Yin, K. Li, J. Zhao, Y. Li, X. Wang, Y. Li, Q. Zhang, G. Xu and H. Chen, Sci. Rep., 2017, 7, 9110 CrossRef PubMed.
- M. Anderson and A. Omri, Drug Delivery, 2004, 11, 33–39 CrossRef CAS PubMed.
- B. Mattei, R. B. Lira, K. R. Perez and K. A. Riske, Chem. Phys. Lipids, 2017, 202, 28–37 CrossRef CAS PubMed.
- B. Mattei, A. D. C. França and K. A. Riske, Langmuir, 2014, 31, 378–386 CrossRef PubMed.
- D. Lichtenberg, H. Ahyayauch, A. Alonso, F. Lix and M. Goñ, Trends Biochem. Sci., 2013, 38, 85–93 CrossRef CAS PubMed.
- S. T. H. Liu, R. Sharon-Friling, P. Ivanova, S. B. Milne, D. S. Myers, J. D. Rabinowitz, H. A. Brown and T. Shenk, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12869–12874 CrossRef CAS PubMed.
- M. Kowalska, M. Broniatowski, M. Mach, Ł. Płachta and P. Wydro, J. Mol. Liq., 2021, 335, 116529 CrossRef CAS.
- B. Brügger, B. Glass, P. Haberkant, I. Leibrecht, F. T. Wieland and H. G. Kräusslich, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 2641–2646 CrossRef PubMed.
- H. D. Klenk, R. Rott and H. Becht, Virology, 1972, 47, 579–591 CrossRef CAS PubMed.
- P. T. Ivanova, D. S. Myers, S. B. Milne, J. L. McClaren, P. G. Thomas and H. A. Brown, ACS Infect. Dis., 2016, 1, 435–442 CrossRef PubMed.
- H.-D. Klenk' and P. W. Choppin, Virology, 1969, 38, 255–268 CrossRef PubMed.
- F. R. Maxfield and D. Wüstner, J. Clin. Invest., 2002, 110, 891 CrossRef CAS PubMed.
- S. Raffy and J. Teissié, Biophys. J., 1999, 76, 2072–2080 CrossRef CAS PubMed.
- A. Pratelli and V. Colao, J. Gen. Virol., 2015, 96, 331–337 CrossRef CAS PubMed.
- F. Monteiro, V. Bernal, M. Chaillet, I. Berger and P. M. Alves, J. Biotechnol., 2016, 233, 34–41 CrossRef CAS PubMed.
- M. L. Briuglia, C. Rotella, A. McFarlane and D. A. Lamprou, Drug Delivery Transl. Res., 2015, 5, 231–242 CrossRef CAS PubMed.
- R. S. Cantor, Biophys. J., 1999, 76, 2625–2639 CrossRef CAS PubMed.
- S. McColman, R. Li, S. Osman, A. Bishop, K. P. Wilkie and D. T. Cramb, Nanoscale, 2021, 13, 20550–20563 RSC.
- T. Nobeyama, M. Mori, K. Shigyou, K. Takata, G. N. Pandian, H. Sugiyama and T. Murakami, ChemistrySelect, 2018, 3, 8325–8331 CrossRef CAS.
- Z. J. Jakubek, S. Chen, J. Zaifman, Y. Y. C. Tam and S. Zou, Langmuir, 2023, 39(7), 2509–2519 CrossRef CAS PubMed.
- T. Yang, F. de Cui, M. K. Choi, J. W. Cho, S. J. Chung, C. K. Shim and D. D. Kim, Int. J. Pharm., 2007, 338, 317–326 CrossRef CAS PubMed.
- D. Guimarães, J. Noro, C. Silva, A. Cavaco-Paulo and E. Nogueira, Front. Bioeng. Biotechnol., 2019, 7, 424 CrossRef PubMed.
- A. Rath, M. Glibowicka, V. G. Nadeau, G. Chen and C. M. Deber, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1760–1765 CrossRef CAS PubMed.
- A. Rath and C. M. Deber, Anal. Biochem., 2013, 434, 67–72 CrossRef CAS PubMed.
- M. Schwamborn, J. Schumacher, J. Sibold, N. K. Teiwes and C. Steinem, Analyst, 2017, 142, 2670–2677 RSC.
- K. Sejwal, M. Chami, P. Baumgartner, J. Kowal, S. A. Müller and H. Stahlberg, Nanotechnol. Rev., 2017, 6, 57–74 CAS.
- X. Yao, X. Fan and N. Yan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 18497–18503 CrossRef CAS PubMed.
- N. G. Aduri, H. A. Ernst, B. K. Prabhala, S. Bhatt, T. Boesen, M. Gajhede and O. Mirza, Biochem. Biophys. Res. Commun., 2018, 495, 1738–1743 CrossRef CAS PubMed.
- H. X. Ong, D. Traini and P. M. Young, Expert Opin. Drug Delivery, 2013, 10, 1287–1302 CrossRef CAS PubMed.
- M. Grossegesse, D. Bourquain, M. Neumann, L. Schaade, J. Schulze, C. MacHe, T. Wolff, A. Nitsche and J. Doellinger, J. Proteome Res., 2022, 21, 459–469 CrossRef CAS PubMed.
- J. J. Baczenas, H. Andersen, S. Rashid, D. Yarmosh, N. Puthuveetil, M. Parker, R. Bradford, C. Florence, K. J. Stemple, M. G. Lewis and S. L. O'Connor, Viruses, 2021, 13, 2434 CrossRef CAS PubMed.
- V. Lalioti, S. González-Sanz, I. Lois-Bermejo, P. González-Jiménez, Á. Viedma-Poyatos, A. Merino, M. A. Pajares and D. Pérez-Sala, Sci. Rep., 2022, 12, 7063 CrossRef CAS PubMed.
- K. W. Dunn, M. M. Kamocka and J. H. McDonald, Am. J. Physiol., 2011, 300, C723–C742 CrossRef CAS PubMed.
- E. M. M. MANDERS, F. J. VERBEEK and J. A. ATEN, J. Microsc., 1993, 169, 375–382 CrossRef CAS PubMed.
- H. Zhang, Thin-Film Hydration Followed by Extrusion Method for Liposome Preparation, Humana Press, New York, NY, 2017, pp. 17–22 Search PubMed.
- J. L. Swift, R. Heuff and D. T. Cramb, Biophys. J., 2006, 90, 1396–1410 CrossRef CAS PubMed.
- Y. Giesecke, S. Soete, K. Mackinnon, T. Tsiaras, M. Ward, M. Althobaiti, T. Suveges, J. E. Lucocq, S. J. McKenna and J. M. Lucocq, Int. J. Mol. Sci., 2020, 21, 1–25 Search PubMed.
- K. Sejwal, M. Chami, P. Baumgartner, J. Kowal, S. A. Müller and H. Stahlberg, Nanotechnol. Rev., 2017, 6, 57–74 CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9(7), 676–682 CrossRef CAS PubMed.
- S. Bolte and F. P. Cordelières, J. Microsc., 2006, 224, 213–232 CrossRef CAS PubMed.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00190c |
| This journal is © The Royal Society of Chemistry 2023 |