
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of bridge structure manipulation on the electrochemical performance of π-conjugated molecule-bridged silicon quantum dot nanocomposite anode materials for lithium-ion batteries†
Young-Hwa
Choi
a,
Jiyoung
Bang
a,
Sunyoung
Lee
b and
Hyun-Dam
Jeong
 *ab
*ab
aDepartment of Chemistry, Chonnam National University, Gwangju 61186, Republic of Korea. E-mail: hdjeong@chonnam.ac.kr
bQURES Co., Ltd., Gwangju 61186, Republic of Korea
First published on 13th June 2023
Abstract
To assess the influence of bridge structure manipulation on the electrochemical performance of π-conjugated molecule-bridged silicon quantum dot (Si QD) nanocomposite (SQNC) anode materials, we prepared two types of SQNCs by Sonogashira cross-coupling and hydrosilylation reactions; one is SQNC-VPEPV, wherein the Si QDs are covalently bonded by vinylene (V)–phenylene (P)–ethynylene (E)–phenylene–vinylene, and the other is SQNC-VPV. By comparing the electrochemical performances of the SQNCs, including that of the previously reported SQNC-VPEPEPV, we found that the SQNC with the highest specific capacity varied depending on the applied current density; SQNC-VPEPV (1420 mA h g−1) > SQNC-VPV (779 mA h g−1) > SQNC-VPEPEPV(465 mA h g−1) at 800 mA g−1, and SQNC-VPV (529 mA h g−1) > SQNC-VPEPEPV (53 mA h g−1) > SQNC-VPEPV (7 mA h g−1) at 2000 mA g−1. To understand this result, we performed EIS and GITT measurements of the SQNCs. In the course of investigating the lithium-ion diffusion coefficient, charge/discharge kinetics, and electrochemical performance of the SQNC anode materials, we found that electronic conductivity is a key parameter for determining the electrochemical performance of the SQNC. Two probable causes for the unique behavior of the electrochemical performances of the SQNCs are anticipated: (i) the SQNC with predominant electronic conductivity is varied depending on the current density applied during the cell operation, and (ii) the degree of surface oxidation of the Si QDs in the SQNCs varies depending on the structures of the surface organic molecules of the Si QDs and the bridging molecules of the SQNCs. Therefore, differences in the amount of oxides (SiO2)/suboxides (SiOx) on the surface of Si QDs lead to significant differences in conductivity and electrochemical performance between the SQNCs.
Introduction
Silicon (Si)-based electrodes are considered as one of the most promising candidates for next-generation lithium-ion batteries (LIBs) because of their environmental compatibility, natural abundance, nontoxicity, low costs, low discharge potential plateau (∼0.37 V vs. Li/Li+), and high theoretical specific and volumetric capacities.1,2 The high theoretical capacity (3590 mA h g−1) at room temperature, corresponding to the maximum lithiation state of Li15Si4, is about one order of magnitude higher than that of commercial graphite.3 In addition, their volumetric capacity (9786 mA h cm−3, calculated based on the initial volume of Si) is higher than that of lithium metal.4 However, the practical use of Si anodes is hampered by the significant volume changes in Si (up to ∼300%) during the lithiation/delithiation process.5 The mechanical stress that occurs over an extended cycle inevitably leads to degradation, electrode pulverization, a subsequent loss of electrical contact between the active materials and the current collector, and the formation of an unstable solid electrolyte interface (SEI), resulting in a rapid capacity fading and low coulombic efficiency.6Considerable efforts have been devoted to overcome the aforementioned challenges of Si-based electrodes, including controlling the cycling voltage window,7 developing novel binder systems,8 designing electrolyte additives,9 performing prelithiation,10 depositing artificial surface coatings,11 as well as nanoscale engineering of Si with various structures such as nanoparticles (NPs),12,13 nanowires,14 nanotubes,15 and nanoporous.16,17 Among the many nanostructured materials developed to date, porous materials have garnered considerable attention, because they can provide free space to accommodate the volume expansion and thus help in maintaining the structural integrity during the cycling.18 In addition, porous structures have a large surface area, which is accessible to the electrolyte and provides a short diffusion path for the lithium-ions to migrate from the electrolyte to the electrode material, consequently facilitating the charging/discharging at high current rates.18,19 Porous Si anode materials can be prepared by various methods, including electroless or electrochemical etching of bulk Si,20,21 deposition into a porous template,22 and magnesiothermic reduction of porous silica.18,23
Assembly into a hierarchical architecture, with NPs as the basic structural unit, is an efficient strategy for fabricating high-performance porous materials for LIBs.18,19,24–29 In recent years, NP assemblies have come under the spotlight as high-performance anode materials for LIBs owing to their lower interfacial area compared to that of NPs, which can significantly reduce the interparticle resistance and mitigate the risk of side reactions.25,26 In addition, their high tap density results in thinner electrodes at the same mass loading, which allows shorter electron pathways and increases the volumetric specific capacity of the whole cell.25,26 Recently, it has been reported that the electrochemical performance of the NP-assembly electrodes can be improved by a robust interconnection of the NPs.26,27 The robust connections between adjacent NPs can significantly reduce the interphase resistance and thus provide a continuous electron pathway, which facilitates electronic connections within the electrode.25–27 However, the influence of the structure, connecting the NPs, on the electrochemical performance has been rarely investigated. Moreover, designing such a structure for the development of high-performance NP-assembly electrodes remains a considerable challenge.
This study was conducted to examine the effect of structural manipulation of the bridge on the electrochemical performance of anode materials containing π-conjugated molecule-bridged Si quantum dot nanocomposites (SQNCs). We synthesized two types of SQNCs by Sonogashira cross-coupling and hydrosilylation reactions: SQNC-VPEPV, wherein the Si QDs are covalently bonded by vinylene (V)–phenylene (P)–ethynylene (E)–phenylene–vinylene, and SQNC-VPV. The Si QD, which is a Si nanocrystal with a diameter smaller than the bulk exciton's Bohr radius,30 was adopted as the basic structural unit for the nanocomposite-based anode material to minimize the fracturing of the anode active material during the lithiation/delithiation process.31 The pore structure characteristic and inter-QD electron transfer rate of the SQNC depend on the structure of the bridging molecule introduced between the Si QDs in the SQNC. To investigate the influence of bridge structure manipulation of the SQNCs on the electrochemical performance, the VPEPV and VPV molecules, whose lengths are shorter than that of VPEPEPV (previously reported by our group), were adopted as the bridges.24 A comparison between the electrochemical performances of the SQNCs, including that of the previously reported SQNC-VPEPEPV, indicated that at different applied current densities, different SQNCs showed the highest specific capacity: SQNC-VPEPV (1420 mA h g−1) > SQNC-VPV (779 mA h g−1) > SQNC-VPEPEPV(465 mA h g−1) at 800 mA g−1, and SQNC-VPV (529 mA h g−1) > SQNC-VPEPEPV (53 mA h g−1) > SQNC-VPEPV (7 mA h g−1) at 2000 mA g−1. To analyze this result in detail, we performed EIS and GITT measurements of the SQNCs. During the course of investigating the lithium-ion diffusion coefficient and charge/discharge kinetics of the SQNC anode materials, it could be speculated that electronic conductivity is a key parameter for determining the electrochemical performance of the SQNCs. Two probable causes for the unique behavior of electrochemical performances of the SQNCs are anticipated: (i) the SQNC with predominant electronic conductivity is varied depending on the current density applied during the cell operation, and (ii) the degree of surface oxidation of the Si QDs in the SQNCs varies depending on the structures of the surface organic molecules of the Si QDs and the bridging molecules of the SQNCs. Therefore, differences in the amount of oxides (SiO2)/suboxides (SiOx) on the surface of Si QDs lead to significant differences in conductivity and electrochemical performance between the SQNCs.
Experimental
Materials
All the chemical reagents were used as received, without further purification. Toluene (anhydrous, 99.8%), borane–tetrahydrofuran (THF) complex solution (BH3OC4H8, 1.0 M in THF), 1,4-diethynylbenzene (C10H6, 96%), 1-bromo-4-ethynylbenzene (C8H5Br, >98.0%), 1-octene (C10H16, 98%), vinylene carbonate (VC) (99%), and poly(acrylic acid) (PAA) (average Mv ∼ 450![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were purchased from Sigma-Aldrich. Triethylamine (TEA) (99%) was purchased from Alfa Aesar. Hydrofluoric acid (HF, 48–51%) was purchased from J.T. Baker. Super P carbon black, n-methyl-2-pyrrolidone (NMP), Cu foil, and lithium metal were obtained from Wellcos Corporation (South Korea). Ethanol (EtOH) (C2H5OH, 99.5%), methanol (MeOH) (CH3OH, 99.5%), toluene (99.5%), and acetone were purchased from Dae-Jung (South Korea). One molar lithium hexafluorophosphate (LiPF6) in ethylene carbonate (EC)/ethyl methyl carbonate (EMC) (1:1 v/v) was obtained from Soul-Brain (South Korea).
000) were purchased from Sigma-Aldrich. Triethylamine (TEA) (99%) was purchased from Alfa Aesar. Hydrofluoric acid (HF, 48–51%) was purchased from J.T. Baker. Super P carbon black, n-methyl-2-pyrrolidone (NMP), Cu foil, and lithium metal were obtained from Wellcos Corporation (South Korea). Ethanol (EtOH) (C2H5OH, 99.5%), methanol (MeOH) (CH3OH, 99.5%), toluene (99.5%), and acetone were purchased from Dae-Jung (South Korea). One molar lithium hexafluorophosphate (LiPF6) in ethylene carbonate (EC)/ethyl methyl carbonate (EMC) (1:1 v/v) was obtained from Soul-Brain (South Korea).
Synthesis of 4-Bs/Oct Si QDs and 4-Es/Oct Si QDs
Hydride-terminated Si QDs (H–Si QDs) were prepared by HF etching of oxide-coated Si nanocrystals (Si NCs@SiOx), which were synthesized by magnesiothermic reduction of silica NPs (SiO2 NPs). The synthetic procedures of SiO2 NPs and Si NCs@SiOx are described in the ESI.† Freestanding 4-bromostyryl/octyl co-capped Si QDs (4-Bs/Oct Si QDs) were prepared by borane-catalyzed hydrosilylation reactions between 1-bromo-4-ethynylbenzene/1-octene and H–Si QDs, as shown in Fig. 1(a).24,32 The H–Si QDs were synthesized by HF etching of Si NCs@SiOx (0.5 g) for 2 h, using a mixture of HF (20 mL), EtOH (20 mL), and distilled water (20 mL). After the completion of the etching process, the H–Si QDs were extracted from the etching solution using toluene (3 × 100 mL). Then, they were separated from the toluene solvent by centrifugation at 12000 rpm for 10 min, and redispersed in anhydrous toluene. After transferring the H–Si QDs to a 250 mL two-neck flask equipped with a condenser connected to an argon-filled Schlenk line, we added the borane–THF complex solution (0.6 mL), 1-octene (6 mL), and 1-bromo-4-ethynylbenzene (0.2 g), and the reaction mixture was stirred for 48 h at room temperature. After the reaction was complete, the aggregated or unreacted materials were removed by centrifugation and filtration. The product was purified five times by solvent precipitation using methanol and toluene as the antisolvent and solvent, respectively. Finally, the 4-Bs/Oct Si QDs were obtained as bright-yellow solids after drying using a rotary evaporator.
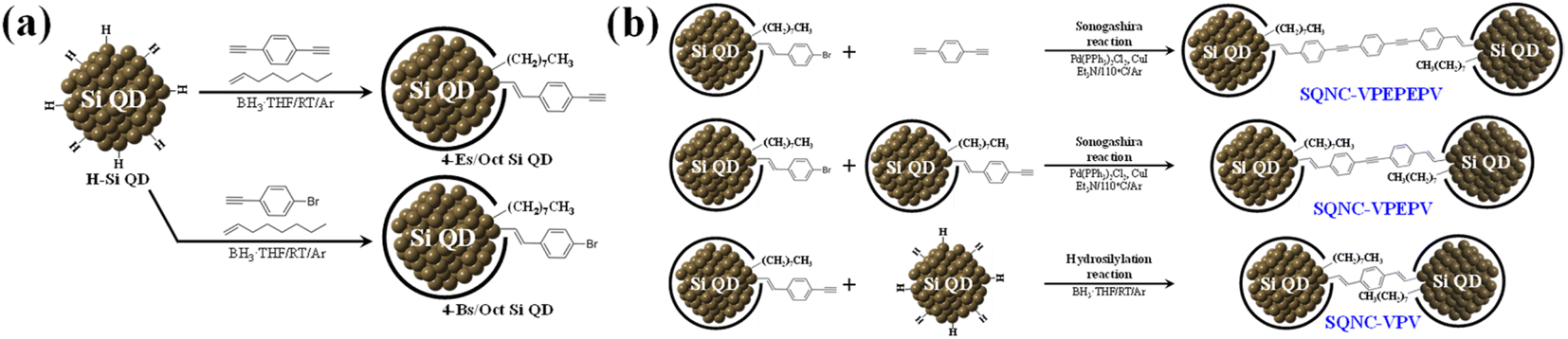 | ||
| Fig. 1 (a) Synthesis of the 4-Es/Oct and 4-Bs/Oct Si QDs by the borane-catalyzed hydrosilylation reaction. (b) Synthesis of SQNC-VPEPEPV, SQNC-VPEPV, and SQNC-VPV by the Sonogashira C–C cross-coupling and borane-catalyzed hydrosilylation reactions. | ||
The 4-ethynylstyryl/octyl co-capped Si QDs (4-Es/Oct Si QDs) were synthesized by a borane-catalyzed hydrosilylation reaction between 1,4-diethynylbenzene/1-octene and H–Si QDs, as shown in Fig. 1(a).32 Except for the use of 1-bromo-4-ethynylbenzene (0.2 g) instead of 1,4-diethynylbenzene (0.14 g), the synthesis and purification conditions of the 4-Es/Oct Si QDs were the same as those employed for the 4-Bs/Oct Si QDs. After drying, the 4-Es/Oct Si QDs were obtained as deep-yellow solids.
Synthesis of SQNC-VPEPEPV, -VPEPV, and -VPV
SQNC-VPEPEPV was prepared via a Sonogashira cross-coupling reaction following the procedure described in our previous report.24 The C–C cross-coupling reaction between the 4-Bs/Oct Si QDs and 1,4-diethynylbenzene was carried out in an argon atmosphere using the standard Schlenk line technique. The 4-Bs/Oct Si QDs (50 mg), 1,4-diethynylbenzene (63 mg), Pd(PPh3)2Cl2 (20 mg), and CuI (8 mg) were dissolved/dispersed into anhydrous toluene (30 mL); then, the reaction mixture was transferred to a 100 mL two-neck flask equipped with a condenser connected to an argon-filled Schlenk line. After the addition of TEA (3 mL) to the flask, the reaction mixture was stirred at 110 °C for 2 h to obtain a dark-brown solution. After cooling the reaction mixture, MeOH was added to precipitate SQNC-VPEPEPV, which was subsequently centrifuged at 12000 rpm for 10 min and washed three times with MeOH (100 mL) to remove the catalyst. Finally, SQNC-VPEPEPV was obtained as a dark-brown powder after drying using a rotary evaporator.
The SQNC-VPEPV material was prepared via a Sonogashira cross-coupling reaction between the 4-Bs/Oct Si QDs (50 mg) and the 4-Es/Oct Si QDs (25 mg). Except for the use of the 4-Es/Oct Si QDs instead of 1,4-diethynylbenzene, the synthesis and purification conditions of SQNC-VPEPV were the same as those employed for SQNC-VPEPEPV.
SQNC-VPV was prepared via a hydrosilylation reaction between the 4-Es/Oct Si QDs and the H–Si QDs. The H–Si QDs (20 mg) were transferred to a 100 mL two-neck flask equipped with a condenser connected to an argon-filled Schlenk line. Then, after adding the 4-Es/Oct Si QDs (100 mg) and borane–THF complex solution (0.6 mL), the reaction mixture was stirred for 48 h at room temperature. Furthermore, to stabilize the surface of the SQNCs, 1-octene (8 mL) was added after stirring for 48 h, and the final mixture was stirred for 12 h. Subsequently, the mixture was filtered through a 0.25 μm polytetrafluoroethylene filter. Finally, SQNC-VPV was obtained as a brown powder after drying using a rotary evaporator.
Material characterization
Fourier-transform infrared (FT-IR) spectroscopy measurements were performed using a Nicolet 380 spectrometer (Waltham, MA) operated in the mid-IR range (4000–400 cm−1); the spectra were obtained at a resolution of 2 cm−1 in the transmittance mode. Proton nuclear magnetic resonance (1H-NMR) data were collected with a superconducting FT-NMR spectrometer at 300 MHz (Varian Inc., Palo Alto, California). The chemical shifts were observed in parts per million (ppm) in a chloroform-d solvent (99.8 at% deuterium). Field-emission transmission electron microscopy (FE-TEM) measurements were performed using a JEM-2100F electron microscope (JEOL, Japan) with an accelerating voltage of 400 kV. For the TEM sampling, 0.1 wt% solutions of SQNC-VPEPV and SQNC-VPV in toluene were drop-cast onto a lacey carbon-coated copper grid, and the solvent was evaporated in vacuum. The carbon, hydrogen, nitrogen, sulfur, and oxygen contents were measured using a Thermo Scientific Flash 2000 organic elemental analyzer and Vario MICRO Cube elemental analyzer. Nitrogen adsorption–desorption isotherms were recorded using a Belsorp mini II surface area and porosimetry analyzer to evaluate the Brunauer–Emmett–Teller (BET) specific surface areas, pore volumes, and pore size distributions. Before the measurements, the SQNC-VPEPV and SQNC-VPV samples were degassed under vacuum at 70 °C for 6 h. The specific surface areas were obtained using the BET method below 0.99 P/P0. The total pore volumes and pore size distributions were obtained by the Barrett–Joyner–Halenda (BJH) method, using the adsorption branch of the nitrogen isotherms.Electrochemical characterization
The electrochemical performances of the SQNC-VPEPV and SQNC-VPV samples were evaluated using CR 2032 coin-type half cells with a lithium foil as the counter/reference electrode. The working electrodes were prepared by casting a mixture of the test material, conductive agent (Super P carbon black), and binder material (PAA), at a weight ratio of 70:15:15, onto a copper foil. The copper foils were dried in vacuum at 60 °C for 5 h to remove the solvent (NMP) used for the slurry preparation. The mass loading of all the electrodes was ∼1.0 mg cm−2. The cells were assembled in an argon-filled glovebox with concentrations of moisture and oxygen below 0.5 ppm. Glass fiber (GF/F, Whatman) and 1 M LiPF6 in EC/EMC (1:1 v/v), containing a 5 wt% VC solution, were used as the separator and electrolyte, respectively. The cycling performance and rate capability of the cells were tested in the potential range of 0.01–2.5 V vs. Li/Li+ at room temperature using a battery cycling system (WonATech-WBCS 3000L). Electrochemical impedance spectroscopy (EIS) analyses were performed using a CompactStat (Ivium Technologies) potentiostat, at a frequency range of 100 kHz to 0.01 Hz and a voltage amplitude of 10 mV. Galvanostatic intermittent titration technique (GITT) measurements of the SQNCs were performed at a current density of 200 mA g−1 in the potential range of 0.01 to 2.5 V.
Results and discussion
Fig. 1 shows the schematic of the preparation process for the SQNC-VPEPV and SQNC-VPV samples. The 4-Es/Oct Si QDs and 4-Bs/Oct Si QDs were prepared by a borane-catalyzed hydrosilylation reaction between the H–Si QDs and unsaturated molecules (Fig. 1(a)). The successful functionalization of the Si QD surface with 1-octene, 1-bromo-4-ethynylbenzene, and 1,4-diethynylbenzene was demonstrated by 1H NMR spectroscopy (Fig. 2(a)). In the 1H NMR spectra of the 4-Es/Oct Si QDs and 4-Bs/Oct Si QDs, the peaks at 3.6 (vinyl CH2 (a)) and 5.3 ppm (vinyl CH2 (b)) can be assigned to the protons of the vinyl groups bonded to Si and indicate the functionalization of 1-bromo-4-ethynylbenzene or 1,4-diethynylbenzene on the Si atoms.24,32 In addition, the two intense peaks at 1.2 (CH3 (1)) and 0.8 ppm (CH2 (2)) are associated with the octyl group, which was introduced to prevent oxidation of the Si QDs. Based on the 1H-NMR results, we can conclude that the Si QD surface was effectively functionalized by the introduced unsaturated molecules. As shown in Fig. 1(b), SQNC-VPEPV was synthesized by the Sonogashira cross-coupling reaction between the 4-Bs/Oct Si QDs and the 4-Es/Oct Si QDs, wherein the Si QDs were covalently connected by vinylene (V)–phenylene (P)–ethynylene (E)–phenylene–vinylene bonds. SQNC-VPV, in which the Si QDs are bonded by V–P–V, was prepared via a hydrosilylation reaction between the 4-Es/Oct Si QDs and the H–Si QDs. The surface chemical structures of the SQNC-VPEPV and SQNC-VPV samples were characterized by FT-IR spectroscopy. Fig. 2(b) shows the FT-IR spectra of both the SQNCs with the 4-Bs/Oct Si QDs and SQNC-VPEPEPV, as reported in our previous study.24 All the samples show absorption peaks at 2964, 2926, and 2860 cm−1, which can be attributed to sp3-hybridized C–H stretching vibrations and indicate the presence of octyl groups on the surface of the Si QDs. The peaks at 1600–1700 and 2100–2250 cm−1 can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C
C and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C stretching vibrations of the functionalized surface or bridging molecules of the SQNCs, respectively. However, no distinguishable peaks are observed between those of the SQNCs, because all the SQNCs contained CC and CC groups either between the QDs or on the surface. In addition, a broad peak at ∼1000–1150 cm−1 related to Si–O stretching vibration is observed in all samples, which can be attributed to oxidation by incomplete functionalization of the Si QD surface.33
C stretching vibrations of the functionalized surface or bridging molecules of the SQNCs, respectively. However, no distinguishable peaks are observed between those of the SQNCs, because all the SQNCs contained CC and CC groups either between the QDs or on the surface. In addition, a broad peak at ∼1000–1150 cm−1 related to Si–O stretching vibration is observed in all samples, which can be attributed to oxidation by incomplete functionalization of the Si QD surface.33
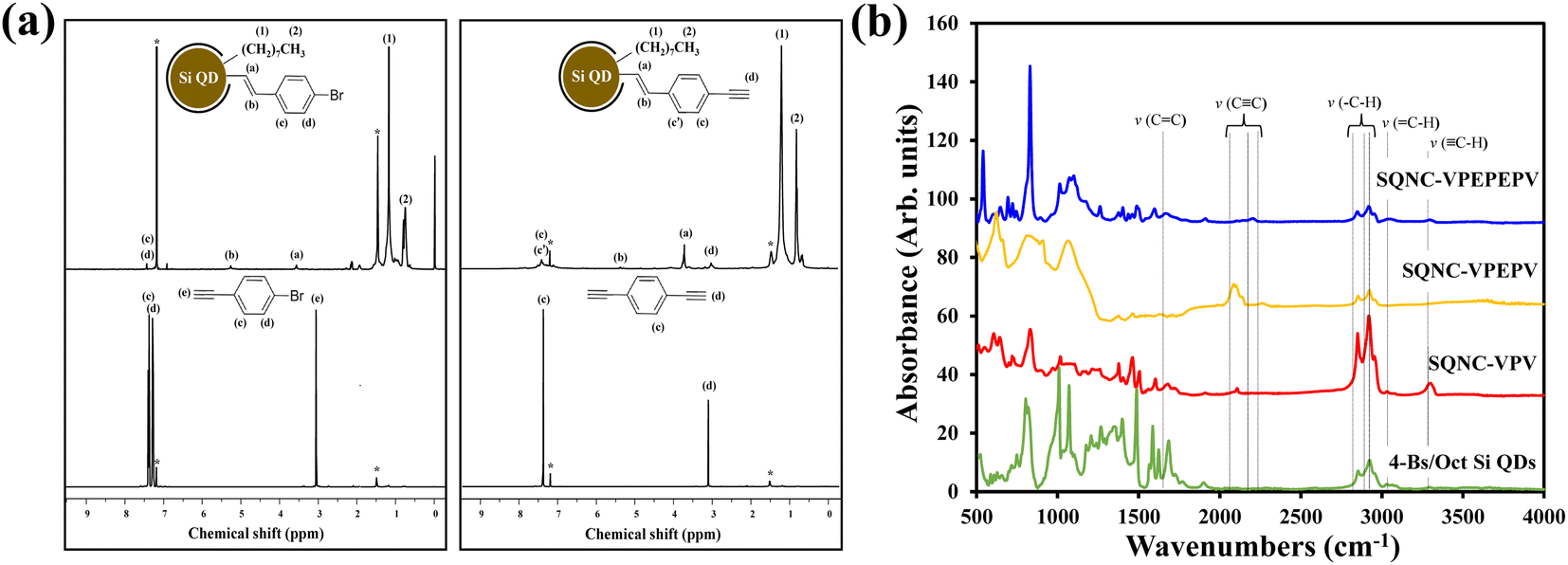 | ||
| Fig. 2 (a) NMR spectra of 4-Bs/Oct Si QDs, 1-bromo-4-ethynylbenzene, 4-Es/Oct Si QDs, and 1,4-diethynylbenzene. (b) FT-IR spectra of SQNC-VPEPEPV, SQNC-VPEPV, SQNC-VPV, and the 4-Bs/Oct Si QDs. | ||
The morphology and microstructure of SQNC-VPEPV and SQNC-VPV were analyzed by FE-TEM. The spherical Si QDs in both the SQNCs are distinguishable from the π-conjugated organic molecular matrix, as shown in Fig. 3(a) and (b). The size distributions of the Si QDs in the SQNC-VPEPV and SQNC-VPV are displayed in Fig. S1(a) and (b),† respectively. The high-resolution TEM images reveal that the Si QDs with a diameter of ∼2.8 nm were highly crystalline with an interplanar distance of 0.32 nm, corresponding to the (111) crystal plane of diamond cubic Si (Fig. S2†). The high dispersibility of the Si QDs in organic matrices could be attributed to the presence of the π-conjugated bridging molecules between them.
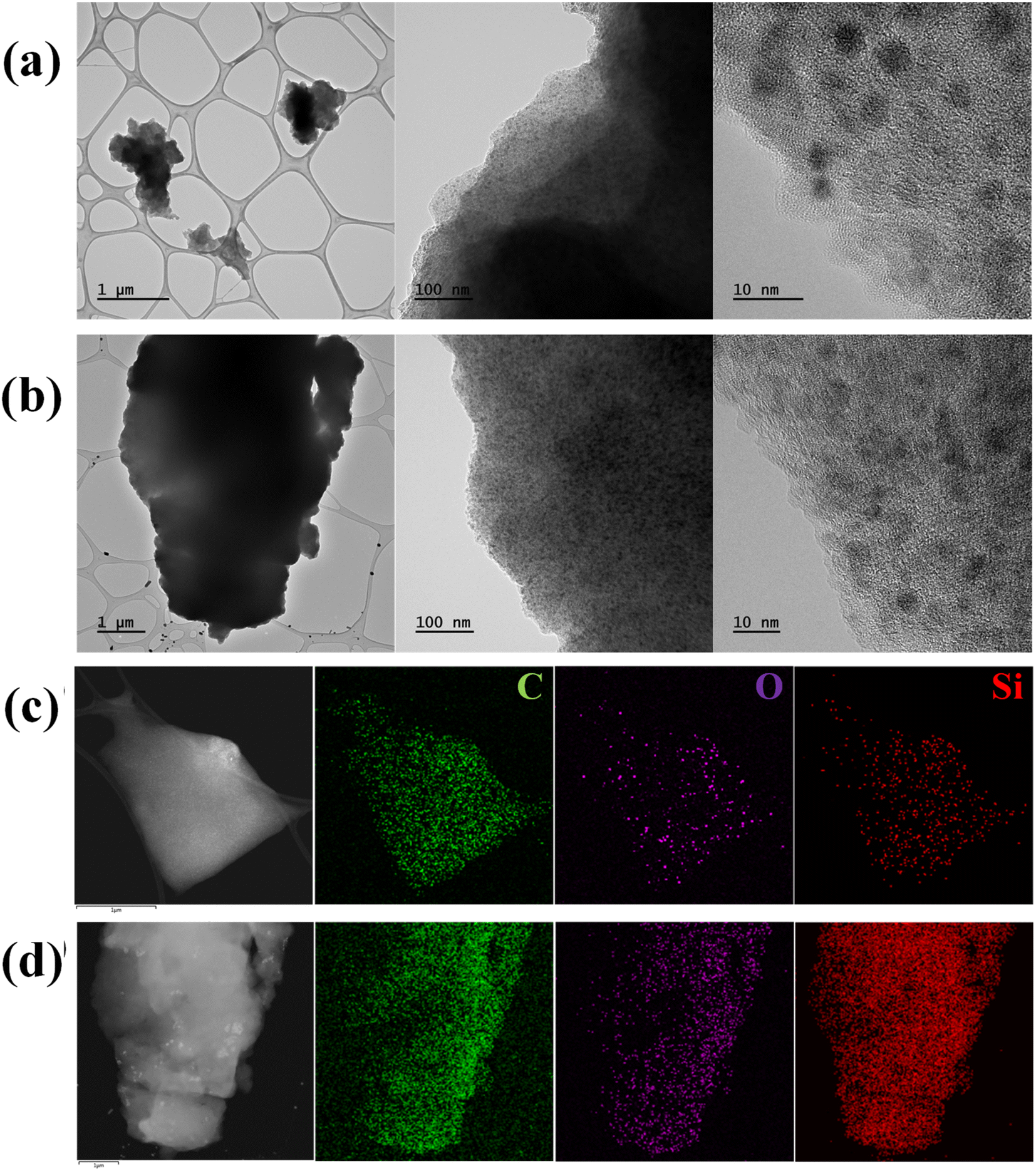 | ||
| Fig. 3 TEM images of (a) SQNC-VPEPV and (b) SQNC-VPV. Secondary electron TEM images and EDS mappings of (c) SQNC-VPEPV and (d) SQNC-VPV. | ||
The elemental distribution and chemical composition of SQNC-VPEPV and SQNC-VPV were determined from energy-dispersive X-ray spectroscopic (EDS) and elemental analyses, respectively. The elemental distributions of SQNC-VPEPV and SQNC-VPV are shown in Fig. 3(c) and (d), respectively. The C, O, and Si elements are uniformly distributed over the SQNCs. Table 1 shows a comparison of the chemical compositions of SQNC-VPEPV and SQNC-VPV with those of the 4-Bs/Oct Si QDs and SQNC-VPEPEPV. In the case of the SQNCs, the Si content increases, while the C content decreased as the length of the π-conjugated molecule, covalently bridging the Si QDs, decreases. The increased O content of the SQNCs compared to that of the 4-Bs/Oct Si QDs can be attributed to incomplete surface functionalization of the Si QDs. The Si QDs appear to be oxidized due to brief air exposure during the further weighing and drying processes for their clustering.
| Sample | % C | % N | % H | % S | % O | % Si (remaining) |
|---|---|---|---|---|---|---|
| 4-Bs/Oct Si QDs | 49.8 | 0.1 | 7.0 | 0.5 | 1.5 | 41.1 |
| SQNC-VPEPEPV | 72.9 | 0.3 | 4.9 | 0.1 | 6.8 | 15.0 |
| SQNC-VPEPV | 63.3 | 0.5 | 8.5 | 0.9 | 2.4 | 24.4 |
| SQNC-VPV | 22.7 | 1.7 | 4.4 | 0.8 | 4.1 | 66.3 |
Nitrogen adsorption–desorption isotherm measurements were carried out to identify the surface areas and pore structures of SQNC-VPEPV and SQNC-VPV. The specific surface areas were estimated using the BET method below 0.99 P/P0, whereas the total pore volumes and pore size distributions were obtained by the BJH method using the adsorption branch of the nitrogen isotherms. Fig. 4 displays the nitrogen adsorption–desorption isotherms and the corresponding BJH pore diameter distributions of SQNC-VPV and SQNC-VPEPV, along with those of the 4-Bs/Oct Si QDs and SQNC-VPEPEPV. The isotherms of the SQNCs show the characteristics of the typical type IV pattern with H3-type hysteresis loops, indicating a mesoporous structure with a wide distribution of pore diameters, whereas the 4-Bs/Oct Si QDs do not show any typical characteristic of a porous structure.34–36 The observed pore characteristics of the SQNCs indicate that clustering of surface-functionalized Si QDs via Sonogashira C–C cross–coupling or hydrosilylation reactions is an effective approach for preparing materials with porous architectures. The specific surface areas, pore volumes, and average pore diameters of the SQNCs are summarized in Table 2. With the decreasing the length of the π-conjugated molecule covalently bridging the Si QDs, the specific surface area and pore volume of the SQNCs decrease. Interestingly, SQNC-VPV exhibits an extremely low specific surface area and pore volume, which can be attributed to the filling of the pores by the octyl functional groups on the surface of the Si QDs. The number of the octyl groups per pore volume can be drastically increased by reducing the inter-QD distance, which might result in pore-filling as schematically shown in Fig. 4(c). However, as the length of the bridging molecule decreases, the average pore diameter increases in the following order: SQNC-VPV > SQNC-VPEPV > SQNC-VPEPEPV. The large pore diameter of SQNC-VPV can be attributed to the intra-cluster pore filling by the octyl groups and the inter-cluster pores (Fig. 4(c) and (d)).
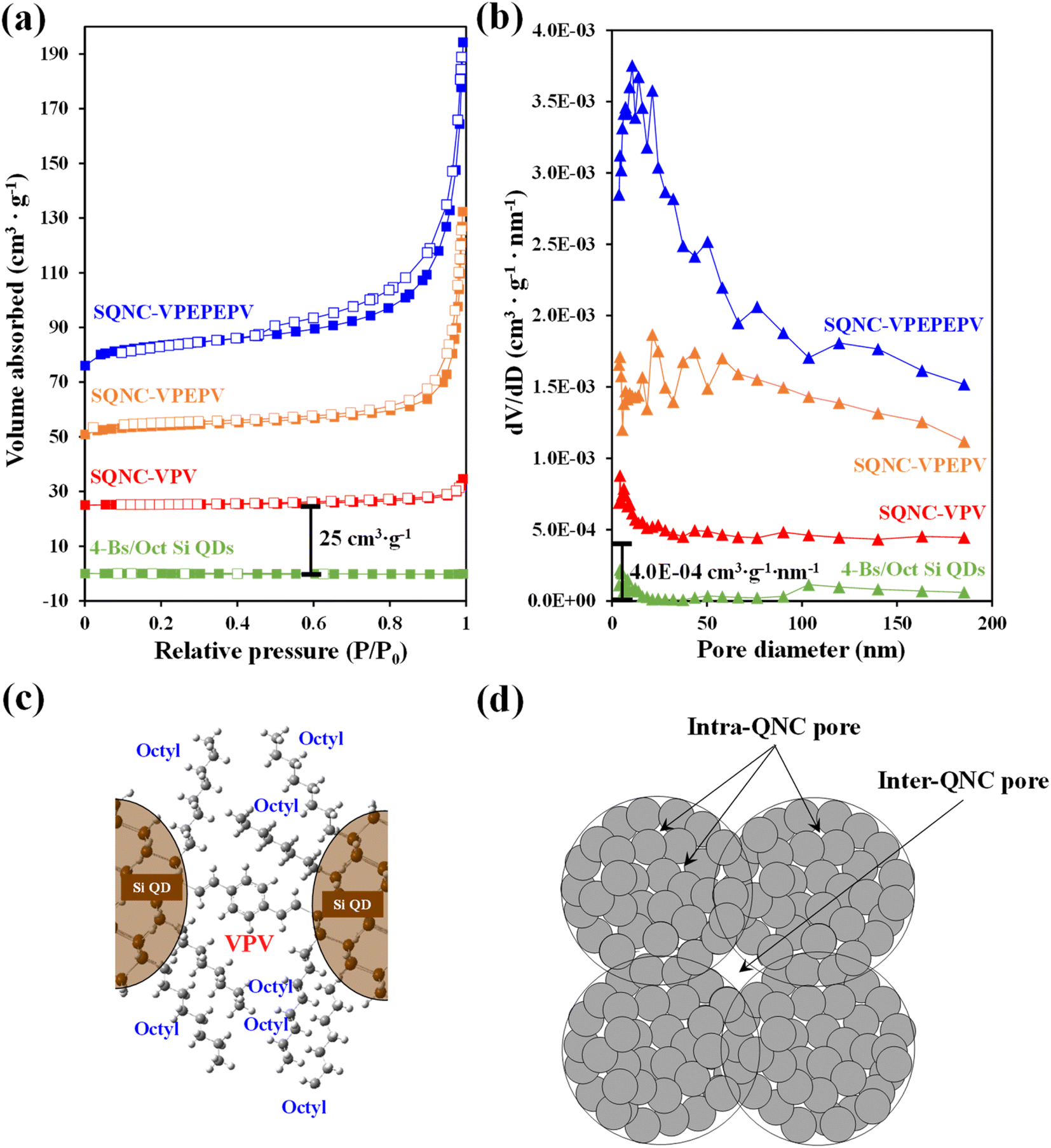 | ||
| Fig. 4 (a) Nitrogen adsorption–desorption isotherms and (b) corresponding BJH pore diameter distributions of the 4-Bs/Oct Si QDs, SQNC-VPV, SQNC-VPEPV, and SQNC-VPEPEPV. Schematic illustrations of (c) pore-filling by the octyl functional groups in SQNC-VPV and (d) intra- and inter-QNC pores. | ||
| Sample | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|
| 4-Bs/Oct Si QDs | — | — | — |
| SQNC-VPEPEPV | 31.53 | 0.174 | 22.0 |
| SQNC-VPEPV | 14.77 | 0.122 | 32.9 |
| SQNC-VPV | 0.77 | 0.013 | 68.5 |
The electrochemical cycling properties of SQNC-VPEPV and SQNC-VPV as anode-active materials were evaluated by fabricating CR 2032-type coin cells in a half-cell configuration with lithium metal as the counter/reference electrode. To examine the effect of structural manipulation of the bridge on the electrochemical performance of the SQNCs, the specific capacity and applied current density of the SQNCs were calculated based on the Si content obtained from the elemental analysis (Table 1). In this study, all the SQNC anode materials were composed of Si QDs with nearly identical diameters, and the SQNC electrodes contained the same electrolyte and conductive agent in equal proportions. Therefore, the difference between the electrochemical performances of the SQNC electrodes was mainly caused by the electronic and lithium-ion conductivities of the SQNCs. Fig. 5(a) shows a schematic illustration of a lithium-ion half coin cell with an SQNC as the anode-active material, along with the corresponding energy diagram. In the charging process, the electrons supplied to the SQNC are consumed by electron transfers to the adjacent Si QD through the bridging molecule or by lithiation reactions of the Si QDs. Notably, the charging process completes when the lithium-rich Si QDs are formed at the outer part of the SQNC, irrespective of the complete participation of the inner Si QDs in the lithiation process. If the electron transfer rate (k2) is sufficiently high compared to the lithiation reaction rate (k1) at a given current density, then the electrons will flow into the SQNC interior. Accordingly, the charging time (T) for attaining electronic equilibrium between the lithium metal (cathode material) and the Si QDs increases, which results in an increased capacity of the SQNC. Contrarily, when the lithiation reaction is significantly faster than the electron transfer, the chemical compositions of the outer Si QDs change to Li3.75Si before the supplied electrons are transferred to the adjacent Si QD. At this time, the capacity of the SQNC is relatively low, because only a very small amount of Si QDs participate in the lithiation reaction. Therefore, the inter-QD electron transfer kinetics in the SQNCs likely influence the electrochemical performances of the SQNCs. Fig. 5(b)–(g) shows the galvanostatic charge–discharge curves and the corresponding differential capacity plots (dQ/dV) at a current density of 200 mA g−1 in the potential range of 0.01–2.5 V for the SQNC-VPEPV and SQNC-VPV electrodes, along with those of the SQNC-VPEPEPV electrode obtained in our previous report.24 The discharge (charge) capacities of the SQNC-VPEPEPV, SQNC-VPEPV, and SQNC-VPV electrodes are 1957 (8151), 2510 (6916), and 1308 (2834) mA h g−1 in the first cycle, corresponding to the initial coulombic efficiency (ICE) values of 24.0%, 36.3%, and 46.2%, respectively. The ICE is associated with the formation of the SEI layer, which is coated on the surface of the anode-active material during the first few lithiation processes. Therefore, a decrease in the surface area of the SQNC may lead to an increase in its ICE. The ICE delivered by SQNC-VPV is higher ICE than those of SQNC-VPEPEPV and SQNC-VPEPV, indicating that the reduction in the surface area of the SQNC by the introduction of a relatively short π-conjugated bridging molecule between the Si QDs limits the space available for the SEI formation. Meanwhile, In the first cycle of the lithiation process, the SQNCs exhibit broad peaks associated with the side reactions on the Si electrode, including the SEI formation and reduction of SiOx on the surface of the Si QDs in the range of 0.5–1.5 V vs. Li/Li+.24 Both the SQNC-VPEPV and SQNC-VPV electrodes show a large peak at around ∼0.10 V vs. Li/Li+, indicating the phase transformation from crystalline Si to amorphous Si–Li alloys (a-LixSi). In the subsequent cycles, distinct peaks of the SQNC-VPEPV and SQNC-VPV electrodes are observed at ∼0.25 and ∼0.10 V, which correspond to the conversion of a-Si to a-Li2.0Si and a-Li2.0Si to a-Li3.5Si, respectively.37 During the delithiation reaction, the SQNC electrodes exhibit peaks at ∼0.37 and ∼0.53 V vs. Li/Li+, indicating that the reversible capacities of the SQNCs mainly originate from the Si QDs.24,37 No significant differences are found in the shapes of the dQ/dV plots of SQNC-VPEPV and SQNC-VPV at the 10th, 50th, and 100th cycle, demonstrating the structural stability of the SQNCs. In contrast, the broad peaks of SQNC-VPEPEPV at ∼0.37 and ∼0.53 V vs. Li/Li+ can be attributed to the participation of the relatively long bridging molecule of the SQNC in the delithiation reaction, as reported earlier.24 The relatively long residence time of the electrons in the bridging molecule, due to the low electron transfer rate, enables insertion/de-insertion of lithium ions into the unsaturated π-conjugated molecules, resulting in these broad peaks.
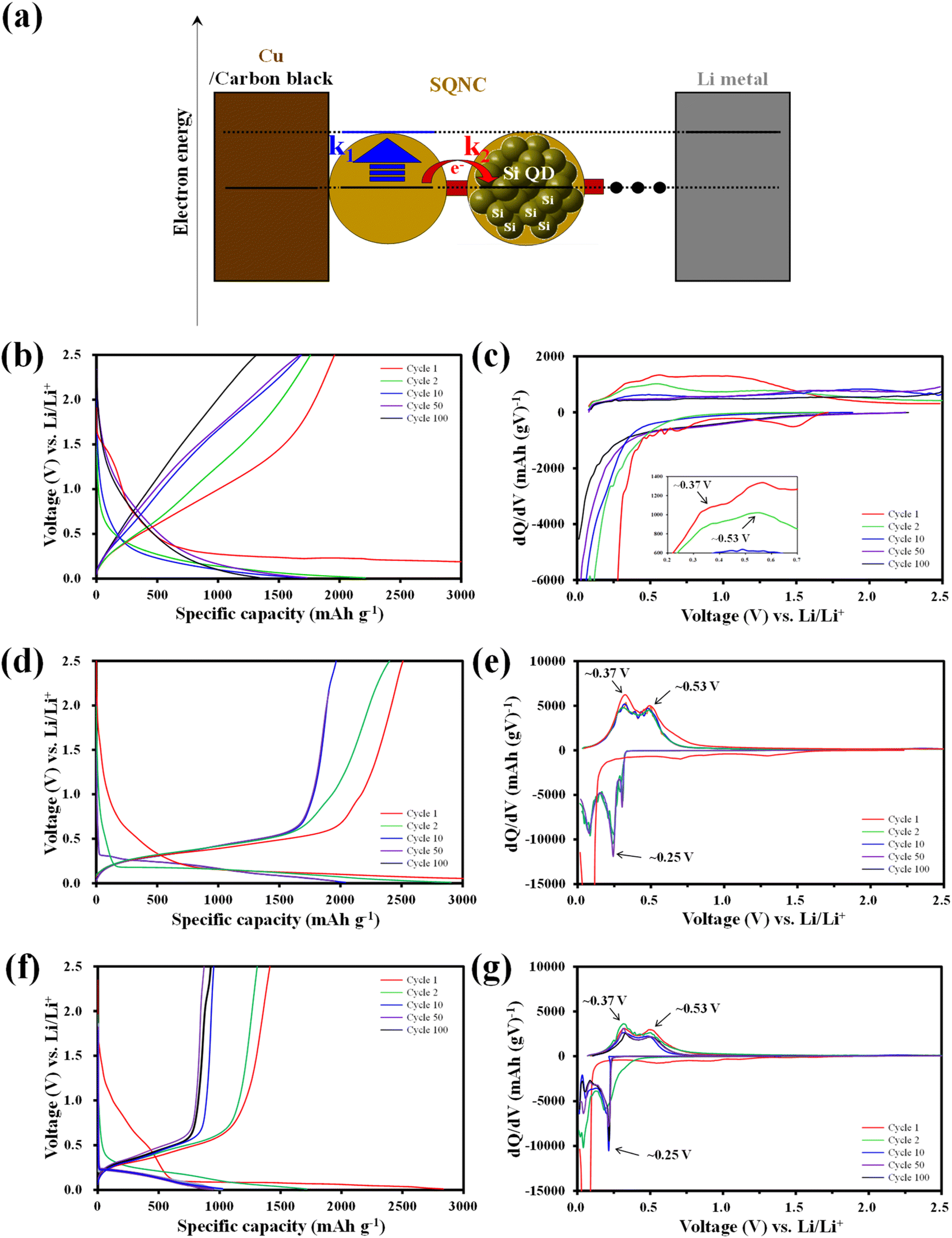 | ||
| Fig. 5 (a) Schematic illustration of a lithium-ion half coin cell with an SQNC as the anode-active material, along with the corresponding energy diagram. Galvanostatic charge–discharge curves and the corresponding differential capacity plots (dQ/dV) of (b and c) SQNC-VPEPEPV, (d and e) SQNC-VPEPV and (f and g) SQNC-VPV. | ||
In Fig. 6(a), the cycling performance of the SQNC electrodes is compared with that of the H–Si QD and 4-Bs/Oct Si QD electrodes obtained in our previous report.24 The reversible capacity of the H–Si QDs electrode sharply decays until the 15th cycle and remains close to zero thereafter. The 4-Bs/Oct Si QDs exhibit a near-zero specific capacity due to the electrical isolation caused by their smaller diameters compared to that of the conducting polymers, as reported earlier.24 However, all the SQNCs show a good cycling stability, possibly due to the presence of the π-conjugated bridging molecules and the porous structure formed between the Si QDs via the clustering of the QDs. We speculate that the porous structure provides a free space that can accommodate the volume expansion, and the π-conjugated molecule serves as a buffer layer, which mitigates the mechanical stresses arising from the alloying reaction of Si with lithium during the extended cycling. Interestingly, among the SQNC electrodes, SQNC-VPEPV shows the highest reversible capacity after 100 cycles; the order is SQNC-VPEPV (1960 mA h g−1) > SQNC-VPEPEPV (1288 mA h g−1) > SQNC-VPV (1027 mA h g−1). Three possible causes of this unique result are anticipated: (i) the trade-off relationship between the electronic and ionic conductivities that depend on the pore structure characteristics of the SQNCs; in this case, each SQNC may have different parameters that determine its electrochemical performance. (ii) The electronic conductivity-dominant SQNC is varied depending on the current density applied during the cell operation. (iii) The degree of surface oxidation of the Si QDs in the SQNCs varies depending on the structures of the surface organic molecules of the Si QDs and the bridging molecules of the SQNCs.
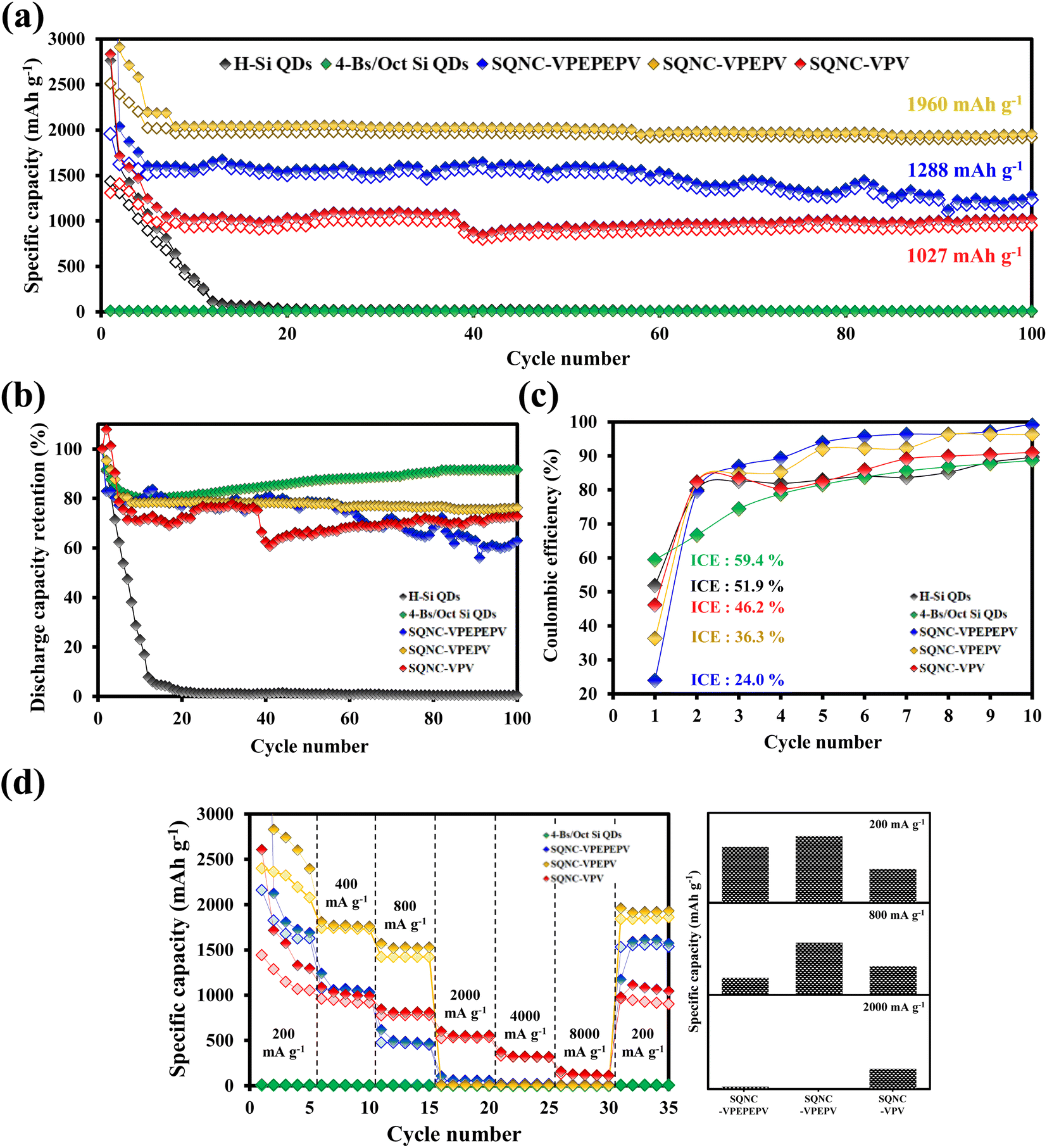 | ||
| Fig. 6 (a) Cycling performances, (b) discharge capacity retentions, and (c) coulombic efficiencies of the H–Si QDs, 4-Bs/Oct Si QDs, SQNC-VPEPEPV, SQNC-VPEPV, and SQNC-VPV electrodes. (d) Rate capabilities of the 4-Bs/Oct Si QDs, SQNC-VPEPEPV, SQNC-VPEPV, and SQNC-VPV, and scheme showing the reversible capacities of SQNC-VPEPEPV, SQNC-VPEPV, and SQNC-VPV at the current densities of 200, 800, and 2000 mA g−1. | ||
The good electrochemical performance of the SQNC-VPEPV was examined by rate performance tests at low current densities (200–800 mA g−1) as shown in Fig. 6(d) and Tables 3 and 4. The reversible capacity of SQNC-VPEPV is higher than those of SQNC-VPEPEPV and SQNC-VPV. Interestingly, as the current density increases from 200 to 800 mA g−1, the specific capacity of SQNC-VPEPEPV rapidly decreases compared to the other SQNCs. Unexpectedly, at high current densities between 2000 and 8000 mA g−1, SQNC-VPEPEPV and SQNC-VPEPV exhibit near-zero reversible capacities, whereas SQNC-VPV delivers a reversible capacity of 529 mA h g−1.
| Sample | Average reversible capacity (mA h g−1) | ||||||
|---|---|---|---|---|---|---|---|
| 200 mA g−1 | 400 mA g−1 | 800 mA g−1 | 2000 mA g−1 | 4000 mA g−1 | 8000 mA g−1 | 200 mA g−1 | |
| 4-Bs/Oct Si QDs | 5.0 | 3.4 | 2.4 | 1.4 | 0.8 | 0.4 | 4.3 |
| SQNC-VPEPEPV | 1782.2 | 1053.7 | 464.7 | 53.4 | 17.2 | 7.1 | 1541.1 |
| SQNC-VPEPV | 2270.9 | 1736.0 | 1420.0 | 7.3 | 4.1 | 2.2 | 1848.0 |
| SQNC-VPV | 1197.8 | 932.6 | 779.1 | 529.4 | 319.3 | 119.9 | 929.7 |
| Sample | T (h) | ||
|---|---|---|---|
| 200 mA g−1 | 800 mA g−1 | 2000 mA g−1 | |
| SQNC-VPEPEPV | 7.57 | 0.64 | 0.0345 |
| SQNC-VPEPV | 9.64 | 1.92 | 0.0038 |
| SQNC-VPV | 5.28 | 1.02 | 0.28 |
To determine the lithium-ion diffusion coefficients of the SQNCs, GITT measurements were performed at a current density of 200 mA g−1 in the range of 0.01–2.5 V vs. Li/Li+. For a more accurate determination of the lithium-ion diffusion coefficients, both the duration and rest times were set to a relatively short 10 min.38,39 The lithium-ion diffusion coefficient can be determined by the following equation, which is based on Fick's second law:38,40
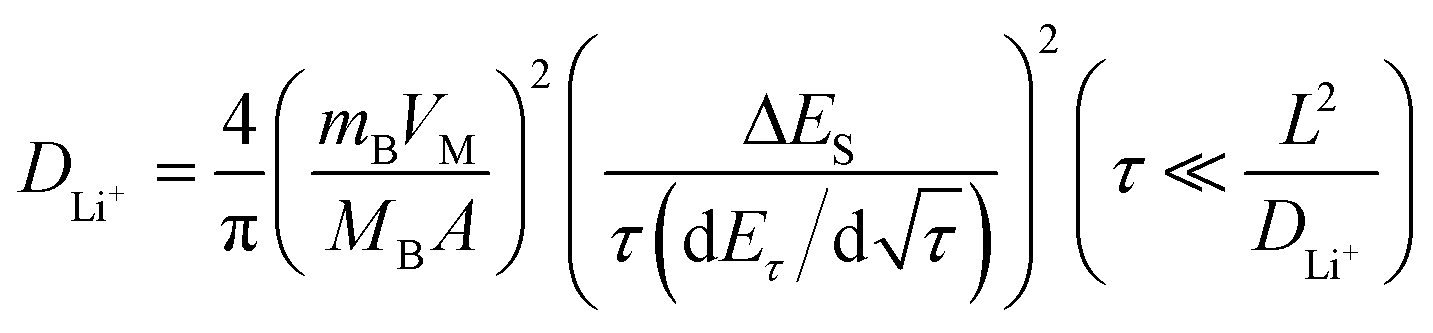 | (1) |
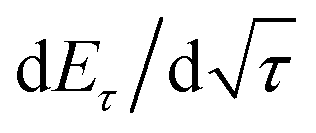 is the slope of the voltage change vs. the square root of τ; L is the lithium diffusion distance (thickness of the electrode); and A is the total interfacial area between the electrolyte and the electrode.41,42 In this study, A was adopted as the apparent geometric area of electrode, and VM was assumed to remain constant with changes in the lithium content in an active material.43 When the change in the cell voltage exhibits a linear relationship with
is the slope of the voltage change vs. the square root of τ; L is the lithium diffusion distance (thickness of the electrode); and A is the total interfacial area between the electrolyte and the electrode.41,42 In this study, A was adopted as the apparent geometric area of electrode, and VM was assumed to remain constant with changes in the lithium content in an active material.43 When the change in the cell voltage exhibits a linear relationship with 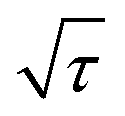 , eqn (1) can be simplified as:42,44
, eqn (1) can be simplified as:42,44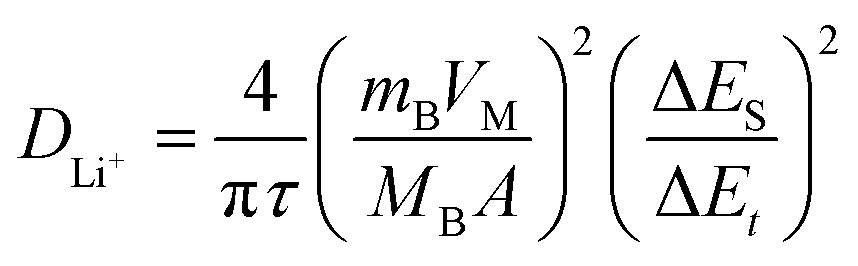 | (2) |
The voltage profiles of the SQNC anodes recorded at a current density of 200 mA g−1 using the GITT method are shown in Fig. S3.† The lithium-ion diffusion coefficients of the SQNCs, at each titration step in the potential ranges of 0.05–0.40 V during the lithiation and 0.30–0.60 V during the delithiation, were determined from the GITT data. Fig. 7(a) and (b) show the lithium-ion diffusion coefficients of the SQNCs as functions of the voltage (vs. Li/Li+). Except for the potential range of 0.45–0.55 V during the delithiation, the diffusion coefficients of SQNC-VPV are smaller than those of SQNC-VPEPV and SQNC-VPEPEPV, might be due to the smaller specific surface area of SQNC-VPV compared to those of the other SQNCs. However, no dependence of the lithium-ion diffusion coefficients on the pore structure properties, such as specific surface area, total pore volume, and average pore diameter, is found. Conversely, the lithium-ion coefficients of all the SQNCs during both the lithiation and delithiation processes range from 10−13 to 10−11 cm2 s−1, which are comparable to that reported for nano-Si electrodes by Ding et al. and larger than that reported for Si thin films and bundle-type Si nanorod electrodes by Nguyen et al.45,46 However, the specific capacities of the SQNCs are lower than those of the Si-based electrode reported by Nguyen et al. The relatively poor capacities of the SQNCs indicate that the ionic conductivity is not a key factor in determining the electrochemical performance of the SQNCs, because it is sufficiently high to deliver the required capacity; hence, the electrochemical performances of the SQNCs were determined by their electronic (rather than ionic) conductivity in this study.
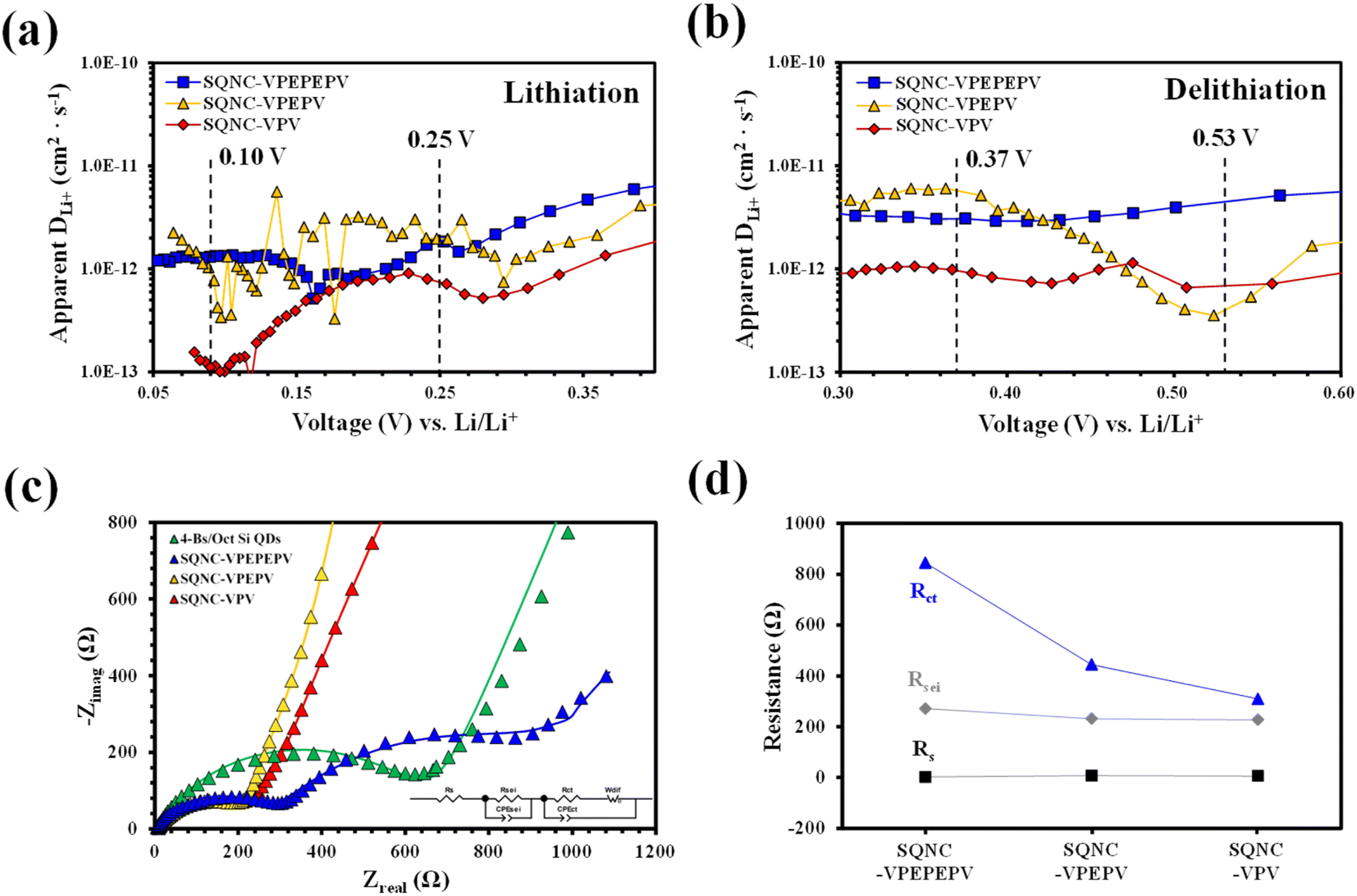 | ||
| Fig. 7 Lithium diffusion coefficients of SQNC-VPEPEPV, SQNC-VPEPV, and SQNC-VPV: (a) lithiation and (b) delithiation processes. (c) Experimental and fitted EIS plots and (d) fitted parameters of SQNC-VPEPV and SQNC-VPV, along with those of the 4-Bs/Oct Si QD and SQNC-VPEPEPV electrodes. | ||
The charge/discharge kinetics of the SQNC electrodes was analyzed by EIS in the fully delithiated state after 100 charge/discharge cycles. Fig. 7(c) displays the Nyquist plots of the SQNC electrodes along with those of the 4-Bs/Oct Si QDs electrode; the curves consist of one or two semicircles in the high- and intermediate-frequency ranges and a sloping line in the low-frequency range. The Nyquist plots are fitted with the equivalent circuit shown in the inset of Fig. 7(c). In the equivalent circuit, Rs is related to the contact resistance associated with the cell components, such as the electrolyte, working electrode, and counter electrode; RSEI and CPESEI are the interface resistance and constant-phase element of the SEI layer, respectively; Rct and CPEct are the charge-transfer resistance and constant-phase element on the electrode/electrolyte surface or within the electrode, respectively, and Wdif is the Warburg contribution related to the diffusion of lithium ions within the active material. The fitted impedance plots, indicated by solid lines, are in good agreement with the experimental EIS curves, and the fitted parameters are summarized in Table 5 and Fig. 7(d). By comparing the values of the SQNC electrodes in Table 5, we observe that the SEI layer resistance decreases slightly as the length of the bridging molecule of the SQNC increases, but is similar (with a value of about 250 Ω) for all the SQNCs. This indicates that all the SQNCs contained an SEI film of a similar thickness and structure. In contrast, the charge transfer resistance of the SQNCs significantly decreases. Consequently, the connection formed using a short bridging molecule between adjacent NPs can greatly reduce the charge transfer resistance and thus provide a continuous electron pathway. This result corresponds to the calculation of the electron transfer rates between Si QDs in simplified molecule-bridged Si QD dimer (SQD) systems (ESI 4†). However, the EIS results could not explain the observed electrochemical performances of the SQNCs at low current densities (200–800 mA g−1).
| Electrode | R e | R SEI | CPESEI–T | CPESEI–P | R ct | CPEct–T | CPEct–P | W–R | W–T | W–P |
|---|---|---|---|---|---|---|---|---|---|---|
| a Resistance (R) and CPE–T parameters have units of Ω and F s(CPE–P)−1, respectively. b Warburg diffusion resistance (W–R) and Warburg diffusion capacitance (W–T) have units of Ω and s, respectively. | ||||||||||
| 4-Bs/Oct Si QDs | 4.00 | 667.2 | 7.33 × 10−6 | 0.69 | 147620 |
3.64 × 10−4 | 0.78 | 815890 |
23.13 | 1.26 |
| SQNC-VPEPEPV | 2.48 | 271.3 | 2.39 × 10−5 | 0.67 | 845 | 1.18 × 10−3 | 0.63 | 1224 | 83.36 | 0.73 |
| SQNC-VPEPV | 7.33 | 231.1 | 4.34 × 10−5 | 0.67 | 444 | 2.74 × 10−4 | 0.97 | 1608 | 0.27 | 0.43 |
| SQNC-VPV | 5.73 | 227.1 | 3.36 × 10−5 | 0.65 | 310 | 4.35 × 10−4 | 0.75 | 14450 |
0.14 | 0.64 |
During the course of investigating the lithium-ion diffusion coefficient, charge/discharge kinetics, and electrochemical performance of the SQNC anode materials, it could be speculated that electronic conductivity is a key parameter for determining the electrochemical performance of the SQNCs. Two probable causes for the unique behavior of the electrochemical performances of the SQNCs are that (i) the SQNC with predominant electronic conductivity is varied depending on the current density applied during the cell operation (this phenomenon is found in ESI 5† for electron transmission calculations in simplified molecule-bridged Si QD dimer model systems in which excess electrons injected into the left Si QD). (ii) The degree of surface oxidation of the Si QDs in the SQNCs varies depending on the structures of the surface organic molecules of the Si QDs and the bridging molecules of the SQNCs. Therefore, differences in the amount of SiO2/SiOx on the surface of Si QDs lead to significant differences in conductivity and electrochemical performance between the SQNCs.47
Conclusions
In this study, we investigated the effect of structural manipulation of the molecule bridge on the electrochemical performance of π-conjugated molecule-bridged Si QD cluster (SQNC)-based anode materials using two types of fabricated SQNCs, viz. SQNC-VPEPV (where the Si QDs are covalently bonded by vinylene (V)–phenylene (P)–ethynylene (E)–phenylene–vinylene) and SQNC-VPV. We compared the electrochemical performances of the SQNCs, including that of the previously reported SQNC-VPEPEPV, and found that the SQNC with the highest specific capacity varied depending on the applied current density. During the course of investigating the lithium-ion diffusion coefficient, charge/discharge kinetics, and electrochemical performance of the SQNC anode materials, it could be speculated that electronic conductivity is a key parameter for determining the electrochemical performance of the SQNCs. Two probable causes for the unique behavior of the electrochemical performances of the SQNCs are that (i) the SQNC with predominant electronic conductivity is varied depending on the current density applied during the cell operation and (ii) the degree of surface oxidation of the Si QDs in the SQNCs varies depending on the structures of the surface organic molecules of the Si QDs and the bridging molecules of the SQNCs. Therefore, differences in the amount of SiO2/SiOx on the surface of Si QDs lead to significant differences in conductivity and electrochemical performance between the SQNCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF 2016R1D1A1B01009523).References
- Y. Wang and J. R. Dahn, J. Electrochem. Soc., 2006, 153, A2188–A2191 CrossRef CAS.
- H. Jung, M. Park, Y.-G. Yoon, G.-B. Kim and S.-K. Joo, J. Power Sources, 2003, 115, 346–351 CrossRef CAS.
- T. D. Hatchard and J. R. Dahn, J. Electrochem. Soc., 2004, 151, A838–A842 CrossRef CAS.
- D. Liu, Z. Liu, X. Li, W. Xie, Q. Wang, Q. Liu, Y. Fu and D. He, Small, 2017, 13, 1702000 CrossRef PubMed.
- U. Kasavajjula, C. Wang and A. J. Appleby, J. Power Sources, 2007, 163, 1003–1039 CrossRef CAS.
- R. Yi, F. Dai, M. L. Gordin, S. Chen and D. Wang, Adv. Energy Mater., 2013, 3, 295–300 CrossRef CAS.
- J. Kim, J. Kwon, M. J. Kim, M. J. O, D. S. Jung, K. C. Roh, J. Jang, P. J. Kim and J. Choi, Nanomaterials, 2021, 11, 3248 CrossRef CAS PubMed.
- D. Wei, J. Mao, Z. Zheng, J. Fang, Y. Luo and X. Gao, J. Mater. Chem. A, 2018, 6, 20982–20991 RSC.
- H. Shobukawa, J. Shin, J. Alvarado, C. S. Rustomji and Y. S. Meng, J. Mater. Chem. A, 2016, 4, 15117–15125 RSC.
- H. J. Kim, S. Choi, S. J. Lee, M. W. Seo, J. G. Lee, E. Deniz, Y. J. Lee, E. K. Kim and J. W. Choi, Nano Lett., 2016, 16, 282–288 CrossRef CAS PubMed.
- B. Wang, W. Li, T. Wu, J. Guo and Z. Wen, Energy Storage Mater., 2018, 15, 139–147 CrossRef.
- H. Kim, M. Seo, M.-H. Park and J. Cho, Angew. Chem., Int. Ed., 2010, 49, 2146–2149 CrossRef CAS PubMed.
- J. Song, S. Chen, M. Zhou, T. Xu, D. Lv, M. L. Gordin, T. Long, M. Melnyk and D. Wang, J. Mater. Chem. A, 2014, 2, 1257–1262 RSC.
- K.-Q. Peng, X. Wang, L. Li, Y. Hu and S.-T. Lee, Nano Today, 2013, 8, 75–97 CrossRef CAS.
- M.-H. Park, M. G. Kim, J. Joo, K. Kim, J. Kim, S. Ahn, Y. Cui and J. Cho, Nano Lett., 2009, 9, 3844–3847 CrossRef CAS PubMed.
- Y. Yu, L. Gu, C. Zhu, S. Tsukimoto, P. A. van Aken and J. Maier, Adv. Mater., 2010, 22, 2247–2250 CrossRef CAS PubMed.
- H. Kim, B. Han, J. Choo and J. Cho, Angew. Chem., Int. Ed., 2008, 47, 10151–10154 CrossRef CAS PubMed.
- M. Ge, X. Fang, J. Rong and C. Zhou, Nanotechnology, 2013, 24, 422001 CrossRef CAS PubMed.
- S. H. Lee, S.-H. Yu, J. E. Lee, A. Jin, D. J. Lee, N. Lee, H. Jo, K. Shin, T.-Y. Ahn, Y.-W. Kim, H. Choe, Y.-E. Sung and T. Hyeon, Nano Lett., 2013, 13, 4249–4256 CrossRef CAS PubMed.
- M. Ge, J. Rong, X. Fang and C. Zhou, Nano Lett., 2012, 12, 2318–2323 CrossRef CAS PubMed.
- D. R. Turner, J. Electrochem. Soc., 1958, 105, C55–C56 CrossRef.
- A. Esmanski and G. A. Ozin, Adv. Funct. Mater., 2009, 19, 1999–2010 CrossRef CAS.
- Z. Bao, M. R. Weatherspoon, S. Shian, Y. Cai, P. D. Graham, S. M. Allan, G. Ahmad, M. B. Dickerson, B. C. Church, Z. Kang, H. W. Abernathy III, C. J. Summers, M. Liu and K. H. Sandhage, Nature, 2007, 446, 172–175 CrossRef CAS PubMed.
- Y.-H. Choi, H. Park, S. Lee and H.-D. Jeong, ACS Omega, 2020, 5, 8629–8637 CrossRef CAS PubMed.
- M. Jiang, J. Chen, Y. Zhang, N. Song, W. Jiang and J. Yang, Adv. Sci., 2022, 9, 2203162 CrossRef CAS PubMed.
- G. Zhu, R. Guo, W. Luo, H. K. Liu, W. Jiang, S. X. Dou and J. Yang, Natl. Sci. Rev., 2021, 8, nwaa152 CrossRef CAS PubMed.
- T. R. Martin, L. Rynearson, M. Kuller, J. Quinn, C. Wang, B. Lucht and N. R. Neale, Adv. Energy Mater., 2023, 2203921 CrossRef CAS.
- C. Kim, J. W. Kim, H. Kim, D. H. Kim, C. Choi, Y. S. Jung and J. Park, Chem. Mater., 2016, 28, 8498–8503 CrossRef CAS.
- T. Pham, D.-S. Lee, T. D. Dao and H.-D. Jeong, J. Ind. Eng. Chem., 2018, 57, 22–27 CrossRef.
- A. Kole and P. Chaudhuri, AIP Adv., 2014, 4, 107106 CrossRef.
- M. T. McDowell, I. Ryu, S. W. Lee, C. Wang, W. D. Nix and Y. Cui, Adv. Mater., 2012, 24, 6034–6041 CrossRef CAS PubMed.
- T.-H. Le, Y.-H. Choi, K.-J. Kim and H.-D. Jeong, ACS Omega, 2019, 4, 3133–3145 CrossRef CAS PubMed.
- M. Aghajamali, H. Xie, M. Javadi, W. P. Kalisvaart, J. M. Buriak and J. G. C. Veinot, Chem. Mater., 2018, 30, 7782–7792 CrossRef CAS.
- S.-S. Chang, B. Clair, J. Ruelle, J. Beauchene, F. D. Renzo, F. Quignard, G.-J. Zhao, H. Yamamoto and J. Gril, J. Exp. Bot., 2009, 60, 3023–3030 CrossRef CAS PubMed.
- F. J. Sotomayor, K. A. Cychosz and M. Thommes, Acc. Mater. Surf. Res., 2018, 3, 34–50 Search PubMed.
- A. Beda, F. Rabuel, M. Morcrette, S. Knopf, P. L. Taberna, P. Simon and C. M. Ghimbeu, J. Mater. Chem. A, 2021, 9, 1743–1758 RSC.
- M. J. Loveridge, M. J. Lain, I. D. Johnson, A. Roberts, S. D. Beattie, R. Dashwood, J. A. Darr and R. Bhagat, Sci. Rep., 2016, 6, 37787 CrossRef CAS PubMed.
- W. Martin, Y. Tian and J. Xiao, J. Electrochem. Soc., 2021, 168, 060513 CrossRef CAS.
- C. Heubner, K. Nikolowski, S. Reuber, M. Schneider, M. Wolter and A. Michaelis, Batteries Supercaps, 2021, 4, 268–285 CrossRef CAS.
- X. H. Rui, N. Ding, J. Liu, C. Li and C. H. Chen, Electrochim. Acta, 2010, 55, 2384–2390 CrossRef CAS.
- J. Kim, S. Park, S. Hwang and W.-S. Yoon, J. Electrochem. Sci. Technol., 2022, 13, 19–31 CrossRef CAS.
- Y.-S. Lee and K.-S. Ryu, Sci. Rep., 2017, 7, 16617 CrossRef PubMed.
- K. Pan, F. Zou, M. Canova, Y. Zhu and J.-H. Kim, J. Power Sources, 2019, 413, 20–28 CrossRef CAS.
- W. Weppner and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 1569–1578 CrossRef CAS.
- N. Ding, J. Xu, Y. X. Yao, G. Wegner, X. Fang, C. H. Chen and I. Lieberwirth, Solid State Ionics, 2009, 180, 222–225 CrossRef CAS.
- S. H. Nguyen, J. C. Lim and J. K. Lee, Electrochim. Acta, 2012, 74, 53–58 CrossRef CAS.
- S. Xun, X. Song, L. Wang, M. E. Grass, Z. Liu, V. S. Battaglia and G. Liu, J. Electrochem. Soc., 2011, 158, A1260–A1266 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00132f |
| This journal is © The Royal Society of Chemistry 2023 |