
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevisiting the roles of dopants in g-C3N4 nanostructures for piezo-photocatalytic production of H2O2: a case study of selenium and sulfur†
Dat Do
Tran‡
abc,
Hoai-Thanh
Vuong‡
abcd,
Duc-Viet
Nguyen
abce,
Pho Phuong
Ly
abc,
Pham Duc
Minh Phan
abc,
Vu Hoang
Khoi
abce,
Phong Thanh
Mai
abc and
Nguyen Huu
Hieu
 *abc
*abc
aVNU-HCM, Key Laboratory of Chemical Engineering and Petroleum Processing (Key CEPP Lab), Ho Chi Minh City University of Technology (HCMUT), 268 Ly Thuong Kiet Street, District 10, Ho Chi Minh City, Vietnam. E-mail: nhhieubk@hcmut.edu.vn
bFaculty of Chemical Engineering, Ho Chi Minh City University of Technology (HCMUT), 268 Ly Thuong Kiet Street, District 10, Ho Chi Minh City, Vietnam
cVietnam National University Ho Chi Minh City (VNU-HCM), Linh Trung Ward, Thu Duc City, Ho Chi Minh City, Vietnam
dDepartment of Chemistry and Biochemistry, University of California Santa Barbara (UCSB), Santa Barbara, California 93106, USA
eSchool of Chemical Engineering, University of Ulsan, Ulsan, South Korea
First published on 3rd April 2023
Abstract
The sustainable production of hydrogen peroxide (H2O2) from oxygen and water has become an exciting research hotspot in the scientific community due to the importance of this fine chemical in various fields. Besides, piezo-photocatalysis is an emerging star for generating H2O2 from these green reagents. For developing catalysts for this specific application, doping heteroatoms into carbon-based materials such as graphitic carbon nitrides (g-C3N4) is a growing fascination among worldwide scientists. However, systematic study on the effects of doping precursors on the catalytic results is still rare. Herein, we fabricated sulfur (S) and selenium (Se) doped g-C3N4 with various doping precursors to evaluate the effects of these agents on the production of H2O2 under light and ultrasound irradiation. Based on the results, Se-doped g-C3N4 gave an outstanding catalytic performance compared to S-doped g-C3N4, even in a significantly low quantity of Se. In order to fully understand the chemical, physical, optical, and electronic properties of pristine g-C3N4 and its derivatives, the as-prepared materials were thoroughly analyzed with various tools. Thus, this study would give more profound insights into doping techniques for carbon-based materials and encourage further research on the design and development of piezo-photocatalysts for practical applications.
1. Introduction
Hydrogen peroxide (H2O2) is a considerably indispensable chemical due to its diverse practical applications from laboratory to industry. Since the 1940s, when the anthraquinone oxidation (AO) process was developed, over 90% of current commercial H2O2 has come initially from the process. On account of using organic compounds and metal catalysts, the process could pose some demerits. Firstly, although the concentration of produced H2O2 during the process could be up to 70%, it instantly requires further extraction and purification to remove impurities from side reactions of organic precursors. Besides that, the environmental issues could be critical to evaluate in this scenario. Secondly, due to the exothermic nature of the reaction, extra energy for maintaining the operating temperature is compulsory. Finally, H2O2 is greatly dangerous at high concentrations, posing a risk during the transportation process, in which further dilution into low concentration is another mandatory requirement. As a result, these drawbacks could burden production costs and energy, leading to an urgent need for other manufacturing methods.1–3In the recent century, there has been a strong interest in discovering novel, green, and sustainable technologies for fine chemical production, especially H2O2. Many methods for manufacturing H2O2 have been proposed in the literature for decades, such as bio-catalysis or electrochemical synthesis.4,5 Besides that, solar H2O2 unprecedentedly emerges as a research hotspot in recent years with the rapid rise in the significant number of published papers in this research direction. For example, He and co-workers reported the generation of H2O2 coupling with organic synthesis by applying TiO2 composited with Bi2O3 immobilized floatable polystyrene spheres to form step scheme (S-scheme) photocatalysts in 2022 with a drastic enhancement in production yields.6 Apart from traditional inorganic semiconductor photocatalysts, organic polymeric materials based on carbon-based materials have been widely scrutinized due to their unique properties. As a rising star, graphitic carbon nitrides (g-C3N4) were first unraveled with photocatalytic abilities in 2009 by Wang and his co-authors.7 Afterward, Shiraishi and colleagues observed the direct formation of H2O2 under light irradiation in the presence of g-C3N4.8 This research has opened the era of g-C3N4-based materials for directly generating H2O2.
Although solar production could reinforce the sustainability of fine chemical synthesis, the materials' rapid charge recombination and other intrinsic properties could prevent practical applications due to lower process efficiencies.9 Many methods were used to modify the materials to overcome these challenges, such as doping, heterojunction structures, and functionalization.10 Since doping is the simplest way to regulate the electronic structures of g-C3N4, accelerating catalytic performances, this technique has been globally studied with various dopants ranging from metallic to nonmetallic elements.11–13 In the periodic table, sulfur (S) and selenium (Se) are placed in the same column because they exhibit similar physicochemical properties. Several papers were published in the past decades to understand the impact of these types of dopants on H2O2 photogeneration. However, an intensive effort was put into S instead of Se. For example, in 2021, Feng et al. reported S-doped g-C3N4 by using sublimed sulfur with g-C3N4 nanosheets by a three-step calcination process for H2O2 photoproduction with a rate of 566.69 μmol g−1 h−1.14 The other group used thiourea calcinated with melamine–cyanuric complex to synthesize S-doped g-C3N4 for H2O2 evolution, presenting a performance of 24.22 μM min−1.15 Unfortunately, no existing publications, based on our knowledge, scrutinize Se as a dopant for g-C3N4 to produce H2O2. Thus, it would be helpful to investigate the impact of Se-doped g-C3N4 on H2O2 evolution under light irradiation.
Furthermore, doping can contribute to the formation of defects on g-C3N4 structures in which the centrosymmetric properties of the materials were altered into the non-centrosymmetric types. When a mechanical strain, such as ultrasound, was applied in this case, the materials would become polarized, leading to interfacial redox reactions.16 Consequently, coupling light and ultrasonic waves could enhance the catalytic performance of doping materials like g-C3N4. Lei and co-workers published their study on applying piezo-catalytic effects for eliminating aqueous dichlorophenols by employing g-C3N4.17 Further validation of g-C3N4 for piezo-photocatalytic H2O2 production was recently described by Tang et al. using isotype g-C3N4 in 2022.18 Until now, not many reports illustrate the exploitation of modified g-C3N4 for H2O2 production by the simultaneous application of piezo-photocatalytic effects. These things could strongly motivate researchers to explore the promising potential of g-C3N4, given piezo-photocatalytic abilities to maximize the process efficiency.
Herein, we developed the one-step facile fabrication of modified g-C3N4 for piezo-photocatalytic H2O2 production. Se and S from various precursors were employed as a dopant for modulating g-C3N4 structures. The highest amount of produced H2O2 reached up to 903.01 μM for 1 hour of irradiation, with SeO2 acting as a precursor during the calcination process, illustrating the potential for practical applications of the fabricated catalysts compared with other published results in Table S1.† Based on diverse evidence from modern analyses, the findings discovered that the catalytic outperformance was mainly related to the germination of charge recombination and intrinsic properties in the electronic structures of the materials paralleled with the changes in chemical or morphological structures. Intriguingly, using SeO2 to introduce Se into the g-C3N4 lattices could significantly extend the light absorption abilities to 600 nm compared to other precursors, leading to superior catalytic results. Therefore, this research revealed the dopants' role and precursors' impacts on g-C3N4. It would be critical to motivate researchers to perform further studies to get complete insights into H2O2 production via metal-free catalytic materials for the catalysts' design and development.
2. Experimental section
2.1 Synthesis of piezo-photocatalysts
In a typical synthesis, the calculated amount of thiourea (0.8 g), sulfur powder (0.8 g), selenium powder (0.8 g), or selenium dioxide (0.8 g) was separately added into a mixture containing 3 g of melamine, 2 g of urea, and 5 g of NH4Cl. Subsequently, the mixture was ground by ball milling for 10 minutes to obtain homogeneous powders. Then it was transferred to a crucible, carefully covered with aluminum foil, and heated at 550 °C at a heating rate of 5 °C min−1 for 4 h in air. After cooling to room temperature, the yellow product was collected, ground, washed, and stored for further use. The products were named SCN, p-SCN, p-SeCN, and SeCN for using thiourea, sulfur, selenium, and SeO2 as doping precursors. g-C3N4 was prepared by the same method without the presence of other agents and denoted as GCN.2.2 Piezo-photocatalytic evaluations
The piezo-photocatalytic activity was evaluated by applying H2O2 production. Firstly, 1 mg of the catalyst was added into a 150 mL beaker containing 50 mL of distilled water and 10% isopropanol (IPA) as a sacrificial agent. Then, the mixture was put in a homemade piezo-photocatalytic system equipped with an overhead stirrer and an ultrasonic cleaner (60 W, 40 kHz). The reaction was started by exposing the mixture to visible light irradiation using two Philips halogen lamps (50 W). The system was continuously stirred, sonicated, and bubbled with oxygen with simultaneously circulating water. At specific time intervals of the 1 h catalytic reaction, 1.5 mL of the solution was withdrawn, filtrated through a 0.22 μm membrane, and diluted ten times before the measurements. H2O2 concentration was monitored by an iodometric method with the absorbance at 350 nm.193. Results and discussion
3.1 Crystallization and morphological analyses
The crystallinity of the as-prepared catalysts was analyzed with X-ray diffraction (XRD) measurements as shown in Fig. 1a. It is clear that there are two prominent peaks belonging to the crystal structures of g-C3N4 and its modified derivatives. Whereas the peak around 13° corresponding to the (100) plane represents the in-plane structural stacking of the heptazine units, the second peak at 27.3° could demonstrate the interlayer stacking and be indexed to the (002) plane.20 In Fig. S1a,† the peak at 13° presented a left shift to the low angle degree from GCN to SeCN, suggesting an increase in the interlayer distance of aromatic units. It could hint at the insertion of S or Se into the heptazine units, which could be following published results.21 In contrast, the right shift to a more considerable angle degree was observed in Fig. S1b† for the doped materials compared to GCN, indicating smaller d-spacing values between layers of modified catalysts as calculated and represented in Table S3.† Previous studies postulated that doping some elements with a larger atomic radius, such as B, S, and K, compared to C and N, could cause the left shift at 27.3° in the XRD peak for C3N4.22–24 Thus, the left shift should be feasibly observed for the synthesized materials in this study. Nevertheless, the contrast phenomenon appeared in the research due to other doping elements in the systems, such as oxygen with a dominant content compared to S or Se in the materials, producing a right shift. Since oxygen possessed relatively higher electronegativity than substituted nitrogen atoms, the interplanar distances in g-C3N4 should be shortened, owing to strong and attractive forces between layers.25 Additionally, the right shift in the patterns would indicate the loss of carbon atoms in the materials to produce a smaller distance between layers in the systems when introducing other elements into the lattice.26 Based on the evidence, it could validate the success of introducing heteroatoms, including O, Cl, and S or Se, into g-C3N4 structures. | ||
| Fig. 1 (a) XRD patterns of GCN, SCN, p-SCN, p-SeCN, and SeCN, and (b) XRD patterns of SeCN and SeCNused. | ||
Moreover, to examine the stability of the materials, the XRD measurement of SeCN samples after irradiation (SeCNused) was performed. The XRD patterns of SeCN and SeCNused are depicted in Fig. 1b with the drop in the intensity of SeCNused compared to that of SeCN. This scenario would possibly originate from the structural alterations of the materials after exposure to light and ultrasound, in which the stacking layers would break into small fragments during the reactions.27 Generally, the pattern for SeCNused exhibits similarities in peak locations to the pristine sample, recommending the stability of the materials.
Further investigations on morphological structures of as-synthesized samples are shown in Fig. 2. GCN exhibited large aggregations in Fig. 2a, where the layer structures were stacked together to form a block. This type of morphology would give an insufficient surface-active area and limited pore sizes, hampering catalytic results. The morphology of SCN in Fig. 2b also showed agglomerations with more pores than GCN, while p-SCN in Fig. 2c demonstrated significant damage on the stacking structures with a bunch of pores and fragments. Meanwhile, Fig. 2d and e illustrate the porous structures of p-SeCN and SeCN. Thus, it would validate better catalytic abilities of these samples due to increased surface areas and pore sizes, providing more active sites for the reactions. The FE-SEM results would be consistent with the data from BET measurements in Table S3.† From Fig. S2a,† N2 adsorption–desorption plots of the prepared samples exhibit the type IV isotherm with an H3 hysteresis loop based on the Brunauer–Deming–Deming–Teller classification.28,29 The porosities could be created due to the evaporation and decomposition of NH4Cl into HCl and NH3, evidenced by the results in Table S3 and Fig. S2b†.30 From Fig. 2f and g, the layer structures of SeCN were validated by the exploitation of HRTEM measurements. In addition, SeCN after irradiation (SeCNused) in Fig. 2h mostly shares similarities in morphological features with SeCN before catalytic tests. Nevertheless, the size of SeCNused from the HRTEM image became fragmental compared to pristine samples, proving the effects of ultrasonic waves to break the stacking layers during the catalytic process. This is in great consistency with the XRD results for the samples before and after irradiation.
 | ||
| Fig. 2 FE-SEM images of (a) GCN, (b) SCN, (c) p-SCN, (d) p-SeCN, and (e) SeCN, and HRTEM of (f) and (g) SeCN, and (h) SeCNused. | ||
3.2 Physical properties
To recognize the other physical properties of the as-prepared materials, zeta potential measurements were undertaken in water to determine the water-dispersive abilities of the as-prepared catalysts. It has been stated that the attachment of –NH2 and other oxygen-containing groups on the edges of g-C3N4 would increase the dispersion capacities in water. From the results, the significant negative values of zeta potentials are recorded in Table S3,† plausibly explaining the water-stable dispersive properties of the materials.31 From the other point of view, due to the rearrangement of electrons on the surface of catalysts due to the doping effects of heteroatoms, the zeta potential values would change depending on the dopants' properties.32 Moreover, the thermal stability of the as-prepared catalysts was characterized using thermogravimetric analysis (TGA) and is presented in Fig. S3.† It would be comprehensive to see that all samples exhibit distinct stability up to 700 °C, in which the results were in perfect accordance with initial publications.33,343.3 Chemical structures
Chemical structures of as-fabricated materials were confirmed by using FTIR and 13C solid-state NMR spectra to get insight into doping effects on the materials. In terms of FTIR characterization, the spectra of all as-prepared samples are presented in Fig. S4a,† showing similar features to GCN and SeCN in Fig. 3a. As can be seen in Fig. 3a, the sharp peak at near 810 cm−1 would be attributed to the structures of heptazine units in the network. In contrast, the peaks from 1200 to 1600 cm−1 belong to the stretching mode of C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N heterocycles in g-C3N4. The other peak at nearly 884 cm−1 would be assigned to the presence of N–H bonds. A broad region from 3000 to 3500 cm−1 was related to the vibrations of N–H or N–H2 and O–H on the surface of g-C3N4 on account of incomplete polymerizations of precursors, the nature of the materials, and the absorbed water molecules.30,35Fig. 3b presents the solid-state 13C NMR spectra of GCN and SeCN. There are two typical signals of g-C3N4. While the former, located at 166.181 and 164.194 ppm, was assigned to –NC(N)–NH2, the latter, at 157.929 ppm, presented C–N3 groups in the materials.36 The spectra of other samples share similarities in analytical features, as demonstrated in Fig. S4b and c.† It is inferred that the local environments in the network were altered with the addition of heteroatoms, which is proved by the drop in the intensity of doped samples compared to pristine g-C3N4, as presented in Fig. 3b, S4b and c,† indicating the successful incorporation of hetero-elements to damage the typical structures in the materials.
N heterocycles in g-C3N4. The other peak at nearly 884 cm−1 would be assigned to the presence of N–H bonds. A broad region from 3000 to 3500 cm−1 was related to the vibrations of N–H or N–H2 and O–H on the surface of g-C3N4 on account of incomplete polymerizations of precursors, the nature of the materials, and the absorbed water molecules.30,35Fig. 3b presents the solid-state 13C NMR spectra of GCN and SeCN. There are two typical signals of g-C3N4. While the former, located at 166.181 and 164.194 ppm, was assigned to –NC(N)–NH2, the latter, at 157.929 ppm, presented C–N3 groups in the materials.36 The spectra of other samples share similarities in analytical features, as demonstrated in Fig. S4b and c.† It is inferred that the local environments in the network were altered with the addition of heteroatoms, which is proved by the drop in the intensity of doped samples compared to pristine g-C3N4, as presented in Fig. 3b, S4b and c,† indicating the successful incorporation of hetero-elements to damage the typical structures in the materials.
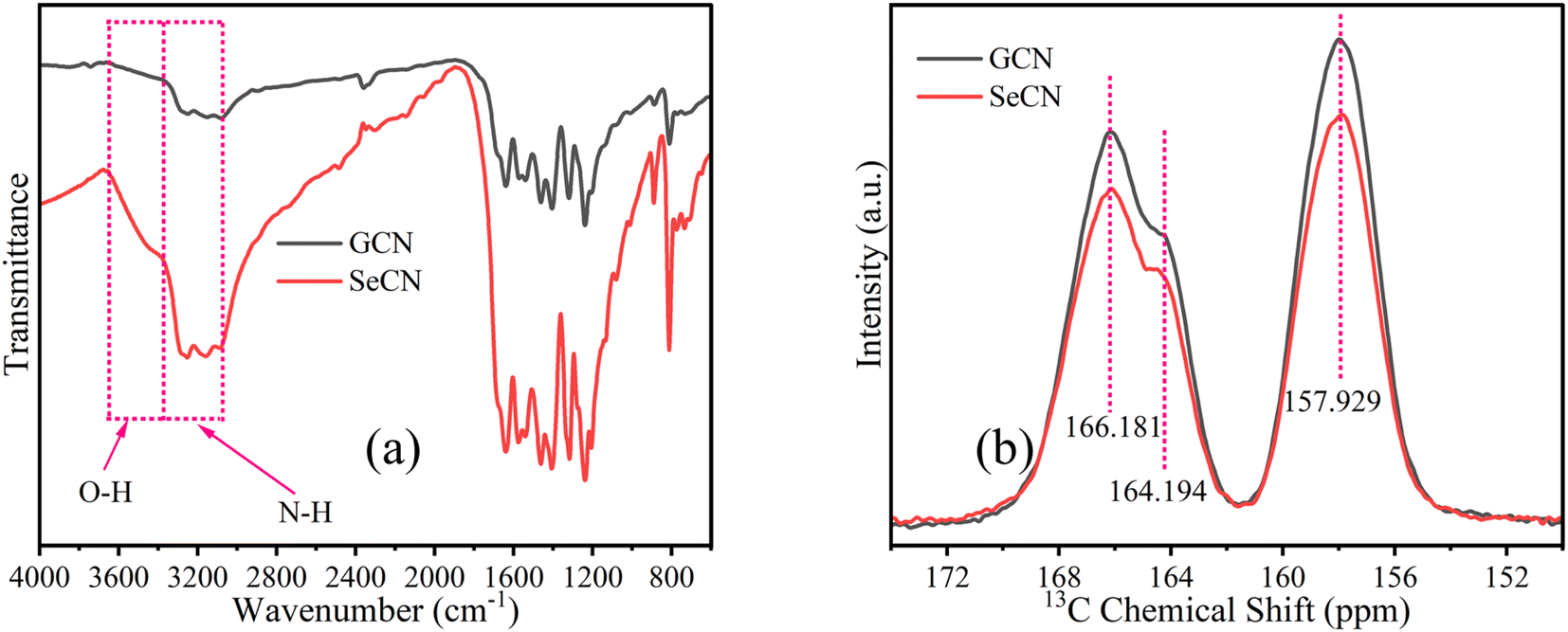 | ||
| Fig. 3 (a) FTIR and (b) 13C solid-state NMR spectra of GCN and SeCN. | ||
Moreover, X-ray photoelectron spectroscopy (XPS) is a powerful tool to confirm the chemical states of elements further. Thereby, XPS analyses were applied in this study to gain deeper insights into the chemical states and bonding in the g-C3N4 network. In Fig. S5a,† the survey spectra of all samples showed that the synthetic materials consist of C, N, and O. Due to the small atomic percentage in the materials, it may be impossible to observe Cl, Se, and S in the spectra. However, the surface atomic percentage of these elements evidenced their existence and is presented in Table S2.† The doping content of Se was also validated with the use of ICP-MS and is shown in Table S2.†
Fig. 4a illustrates the C 1s spectra of all materials with four deconvolution peaks. In particular, the deconvoluted peak located at 293.8, 293.6, 293.9, 294, and 293.9 eV for GCN, SCN, p-SCN, p-SeCN, and SeCN individually could be described as π–π* electron transitions in g-C3N4 and its modified samples.37 While the peak situated at the exact position of 284.8 eV for GCN, SCN, p-SCN, p-SeCN, and SeCN would be attributed to graphitic carbon structures, the remaining peaks at 288 and 288.5 eV for GCN, 287.8 and 288.5 eV for SCN, 288.1 and 288.4 eV for p-SCN, 288 and 288.4 eV for p-SeCN, and 288 and 288.2 eV for SeCN could be associated with C–N and NC–N bonds in the systems.38–40
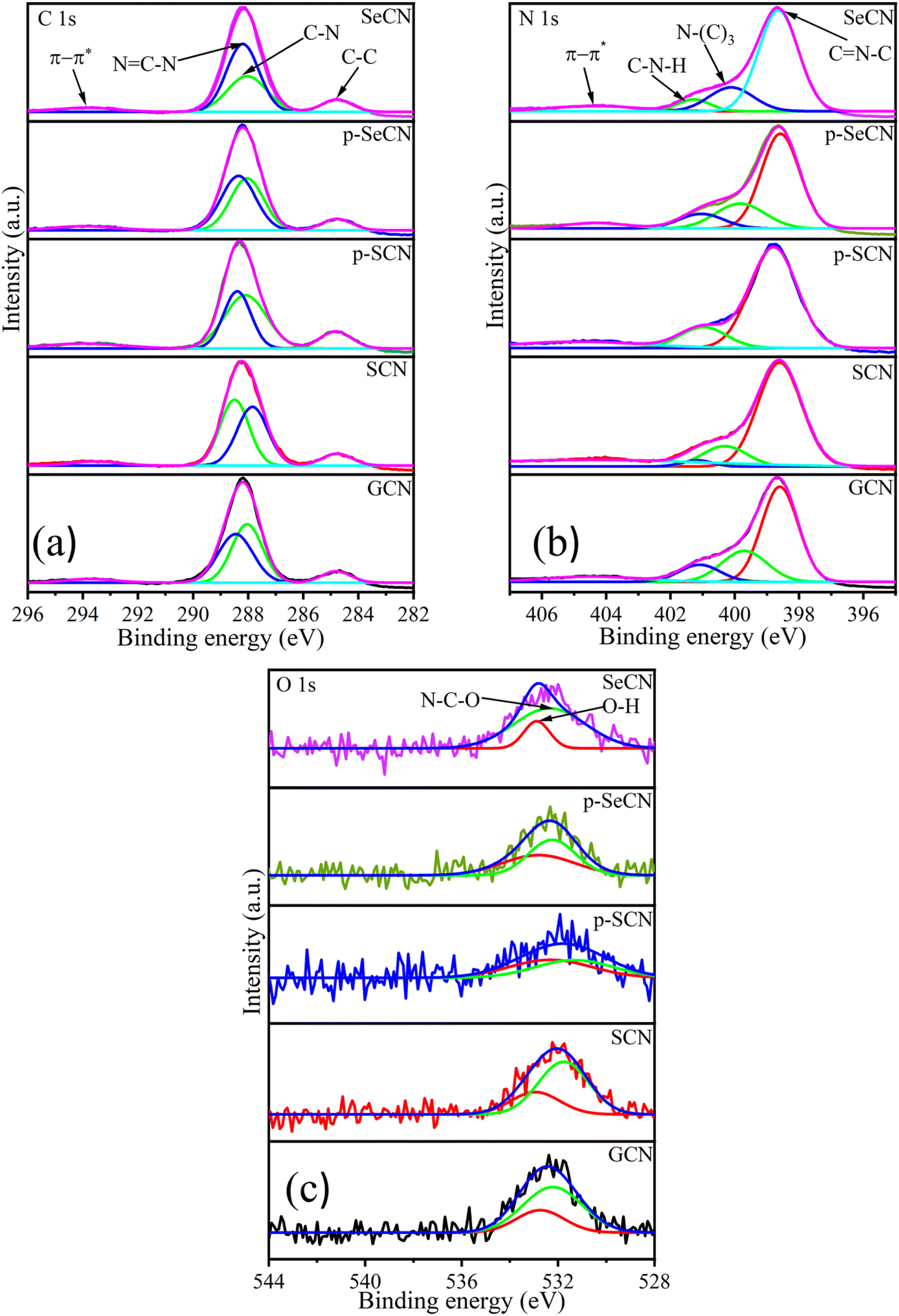 | ||
| Fig. 4 XPS spectra of C 1s (a), N 1s (b), and O 1s (c) of the materials. | ||
The core species of N 1s is illustrated in Fig. 4b with four distinct peaks, comprising π–π* electron transitions in the networks at around 404.5 eV for all samples, graphitic N, pyrrolic N, and pyridinic N.37,41 Notably, the peak was at 399.7, 400.3, 400.2, 399.8, and 400.1 eV for GCN, SCN, p-SCN, p-SeCN, and SeCN, respectively, indicating the presence of graphitic N. In contrast, the peaks centered at 398.6, 398.6, 398.7, 398.6, and 398.6 eV for these samples were associated with pyridinic N in the systems. The remaining peak located at 401.1, 401.2, 401.4, 401, and 401.3 eV belonging individually to GCN, SCN, p-SCN, p-SeCN, and SeCN was ascribed to pyrrolic N. It has been stated that pyridinic N would favor the indirect two-electron oxygen reduction reaction (2e− ORR), in contrast with a direct pathway of graphitic N.42,43 Thus, estimating the dominative reaction mechanism in the research is feasible via computing the ratio between these features. The calculated area ratios between graphitic N and pyridinic N are presented in Table S4,† which are 0.199, 0.199, 0.346, 0.341, and 0.265 for GCN, SCN, p-SCN, p-SeCN, and SeCN. Fundamentally, the values were lower than 1, suggesting that the reaction mechanisms mainly relied on indirect 2e− ORR instead of the direct pathway. The formation of superoxide radicals strongly impacted the yield of H2O2, which could be discussed in the next section.
Further analyses of O 1s, Cl 2p, and S 2p are shown in Fig. 4c and S5.† While the two spin–orbit peaks for O 1s of all samples were centered at around 532.9 eV in Fig. 4c, correlating to O–H species and absorbed moisture in the network, the remaining peak would be attributed to N–C–O bonds in the system. Additionally, there was only one observed peak at approximately 201 eV of Cl 2p in Fig. S5c† due to the small number of chlorine atoms in the system.44,45 The presence of NH4Cl could explain the unintentional grafting of chlorine in the system during thermal polymerization. This leads to substituting nitrogen with chlorine in the network, enhancing the catalytic performances. In addition, S 2p spectra of p-SCN were also analyzed in the research and are exhibited in Fig. S5b† with two deconvolution peaks at 164.4 and 169.3 eV, relating to C–S and SO42− bonds, respectively.46 Due to the low amount of Se detected in the prepared samples, Se could be hard to deconvolute accurately. Based on published results, Se would cooperate with the system via Se–C and SeC(NH2)2 bonds.47
In addition, electron spin resonance (ESR) or electron paramagnetic resonance (EPR) experiments were performed to evaluate the defective states in GCN and SeCN, as shown in Fig. S6,† with the Lorentzian absorption line at g = 2.006. It is well-known that this line would trace the unpaired electrons of the sp2 hybridized carbon atoms in the heterocycles.48 From the data, GCN illustrates a powerful EPR signal compared to SeCN, illustrating that some loss of carbon atoms happened in the modified samples.49,50 Based on the analysis, it would recommend that there were some carbon defects during the thermal polymerization process when introducing S or Se into the catalysts' structures, thus enhancing catalytic outcomes thanks to strengthening the piezo effects.
3.4 Light absorption properties
It is critical to understand that light absorption ability is vital to improving photo-active materials, especially in photocatalysts owing to its positive effects on the charge separation process. Fig. 5a demonstrates the UV-Vis absorption spectra of the prepared catalysts with the distinct characteristics of g-C3N4 and modified samples.51 While the optical absorption bands from 200 to 400 nm would be associated with π–π* electron transitions of the conjugated systems, the tail bands over 400 nm could be frequently stated as n–π* transitions.52 In addition, SeCN exhibited broader absorption spectra than the other samples, suggesting that SeO2 would be suitable for a Se source to dope into g-C3N4 networks to enhance photocatalytic activities. Further calculations of the bandgap energy using a Kubelka–Munk model showed a decrease in the bandgap energy of the synthesized materials, indicating better catalytic activities. Notably, the computed bandgap energy of pristine g-C3N4 at 2.68 eV in Fig. 5b was higher than that of SeCN at 2.61 eV in Fig. 5c. The bandgap energy values of other samples can be found in Fig. S7.† Moreover, SeCN samples also exhibit the mid-gap states in the nanostructures, as can be seen in Fig. 5c, which could enormously enhance the catalytic performances.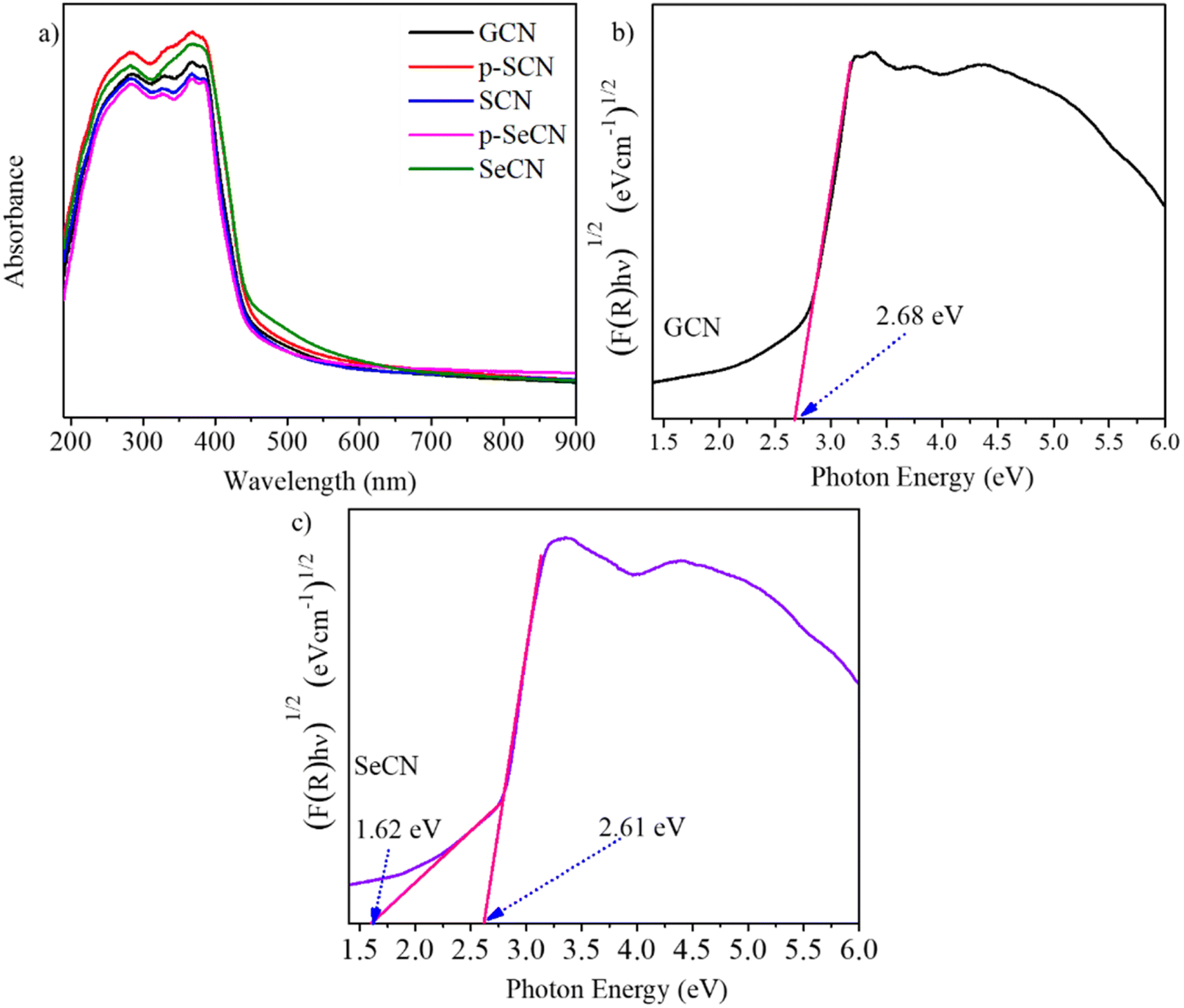 | ||
| Fig. 5 (a) UV-DRS absorption of samples, and bandgap energy of (b) GCN and (c) SeCN. | ||
3.5 Charge separation and migration properties
Although the results from the UV-Vis measurements could validate the prominent light absorption ability of SeCN for showing better catalytic outcomes, it is not adequate to conclude the charge behaviors in the system. Consequently, photoluminescence (PL) spectroscopy was utilized to study charge separation and migrations properties. Specifically, PL results could be a powerful tool for determining the charge recombination rate in photocatalysts in which a higher intensity would indicate higher electron–hole pair recombination.53Fig. 6a shows that GCN presents the fastest charge recombination compared to the modified samples. When introducing other heteroatoms into the network, the recombination phenomenon was hindered due to the possibility of providing exciton dissociation traps, leading to longer charge carrier lifetimes where higher catalytic performances could be observed. In addition, the spectra suggest that Se would be more efficient in suppressing photoinduced charge recombination in the system than introducing S into g-C3N4 structures. Furthermore, when carefully comparing the intensity between SeCN and p-SeCN, SeCN shows a better charge carrier lifetime than p-SeCN, owing to the doping efficiency of Se with different Se sources.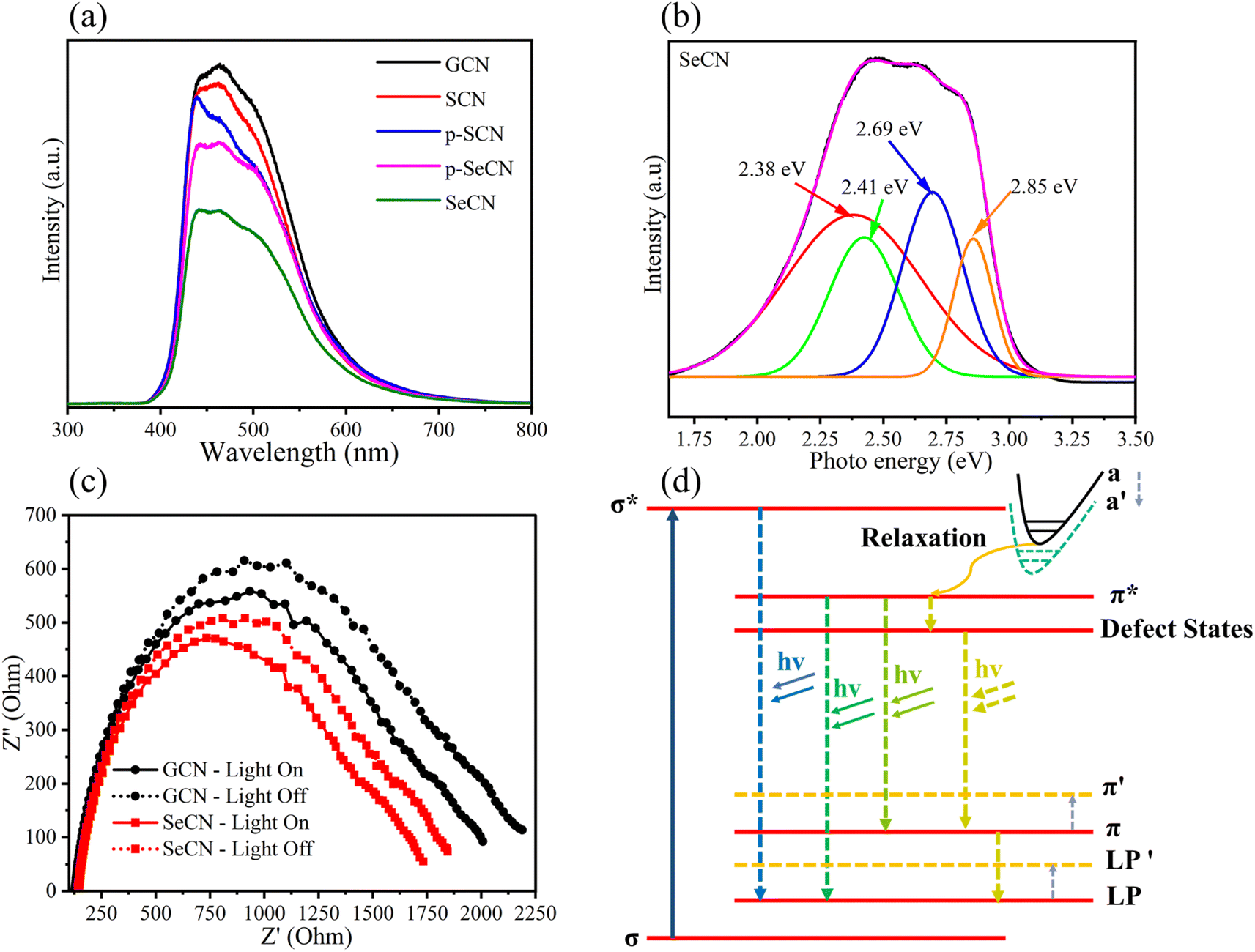 | ||
| Fig. 6 (a) PL spectra of GCN, SCN, p-SCN, p-SeCN, and SeCN, (b) PL-energy converted spectra of SeCN, (c) EIS Nyquist spectra of GCN and SeCN under darkness and illumination, and (d) energy diagram from PL analysis. | ||
Additionally, photocatalyst defects could enhance photocatalytic results by critically creating trapping states to prevent charge recombination. To determine the defect levels in the materials, the typical PL spectra were transformed into energy spectra based on the formation reported by Mooney and Kambhampati in 2013.54 The transformation would include two main steps. Firstly, eqn (1) would convert the wavelength into photon energy. Secondly, the intensity would be recalculated by applying eqn (2) to preserve the emission area spectra. Fig. 6b exhibits the analysis of SeCN samples with four deconvolution peaks by employing Gaussian fitting models. The remaining characterization of other samples can be found in Fig. S8.† Previous studies showed that the bandgap states of g-C3N4 would correspond to sp3 C–N σ bands, sp2 C–N π bands, and the lone pair (LP) states, rooting from the bridge nitride atom.55 Based on published records, three peaks in Fig. 6b at 2.41, 2.69, and 2.85 eV could be related to π–π*, π*–LP, and σ*–LP, respectively.56 The other peak in Fig. 6b at 2.38 eV might correlate with defective states in the conjugated system. In this case, it is understandable to determine the percentage of defects in the fabricated photocatalysts by using PL energy spectra. The results showed that the most significant proportion of defects in SeCN would occupy 45.5% compared to the 6.4, 5.2, 41.4, and 41% from GCN, SCN, p-SCN, and p-SeCN, respectively. These results suggest that SeCN could give better catalytic results due to the largest trapping agents in the system. Besides, the role of doping precursors could be observed by introducing defects in the synthetic process. Based on the results, using thiourea as a doping agent for a sulfur source could not be efficient in enhancing catalytic activities. In other words, using sulfur powder, selenium powder, and SeO2 would be more effective in introducing the other heteroatoms for g-C3N4 networks, posing defective states to ameliorate catalytic outcomes.
 | (1) |
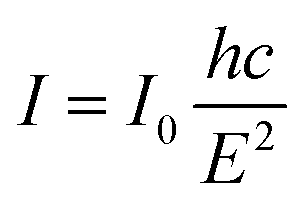 | (2) |
In addition, electrochemical impedance spectroscopy (EIS) was applied to validate system charge behaviors. Theoretically, the minor arc of the Nyquist diagrams could reinforce the lower charge recombination, whereas a larger arc could exhibit the contrasting scenario.57Fig. 6c demonstrates the EIS results of GCN and SeCN under light irradiation and darkness. The spectra for all samples can be seen in Fig. S9a and b.† As shown in Fig. 6c and S8,† GCN depicts the most significant radius arc even under darkness or illumination, suggesting the difficulty in overpassing charge recombination. Furthermore, the modified samples exhibit a lower radius arc in EIS outcomes compared to that of GCN, illustrating the improvement in charge separation and migration behaviors. These results follow PL characterizations, proving the efficiency of heteroatom doping into g-C3N4 frameworks with prolonging charge carrier lifetime.
Further investigations of PL spectra would lead to a plausible explanation for the reduction in the bandgap energy of the materials and a prolonged charge carrier lifetime. Fig. 6d presents a good insight into the tunable PL mechanism of g-C3N4 and its modified samples. The band a stands for the overlap of the σ* and the π* bands. Throughout the relaxation process, the formed channel paves the way for photoexcited electrons to move forward to the π* conduction band. When introducing heteroatoms into the network, the band a′ would appear when the band a would move to overlap with the π* conduction band. Furthermore, a narrow bandgap energy value could be recorded when LP and π overlap with LP′ and π′, respectively. Other observations in Fig. 6d could demonstrate the trapping mechanism of defects in the catalytic system. Photoexcited electrons relaxing to π* need a longer time to recombine with holes in the valence band since they tend to jump into various defect states, validating the efficiency of creating defects in catalytic performances.58,59
3.6 Electronic structural and electrochemical analyses
From an electrochemical perspective, the valence band (VB) and the conduction band (CB) positions in the photocatalysts should be urgently determined in addition to the bandgap energy. To calculate the VB of the materials, XPS-VB spectra would give information on the materials' VB maximum (VBM). As shown in Fig. 7a and S9,† the VBM of GCN, SCN, p-SCN, p-SeCN, and SeCN was at 2.03, 2.05, 1.95, 2.08, and 2.06 eV, individually. Thus, compared to pristine g-C3N4, the modified materials exhibit a small downshift in VBM. Abnormally, the observed electron states below the VBM were also recorded from the measurements, with the data being at −0.75, 0.17, 0.14, −0.46, and −0.34 eV. It is explainable that some positions in the network lack nitrogen atoms due to the replacement of dopants. Hence, some extra electrons would be redistributed in the conjugated system to the nearest carbon atoms, which could reduce C4+ to C3+.60Viaeqn (3), the VB position could be exported.61 In Table S3,† the VB of the materials was computed at 1.47, 1.49, 1.39, 1.52, and 1.50 eV for GCN, SCN, p-SCN, p-SeCN, and SeCN. The lower VB position could lead to a higher CB position in which the reduction ability of the materials increases. Through eqn (4), the theoretical CB position was exported and is shown in Table S5,† with the data at −1.21, −1.18, −1.23, −1.19, and −1.11 eV for GCN, SCN, p-SCN, p-SeCN, and SeCN, respectively. These strongly negative CB positions would help drive the oxygen reduction reaction (ORR) in the water medium.| EVB = Φ + EXPS-VB − 4.44 | (3) |
| ECB = Eg + EVB | (4) |
| ENHE = EAg/AgCl + 0.197 | (5) |
| ECB = ENHE − 0.1 | (6) |
| W = 4.5 − ENHE | (7) |
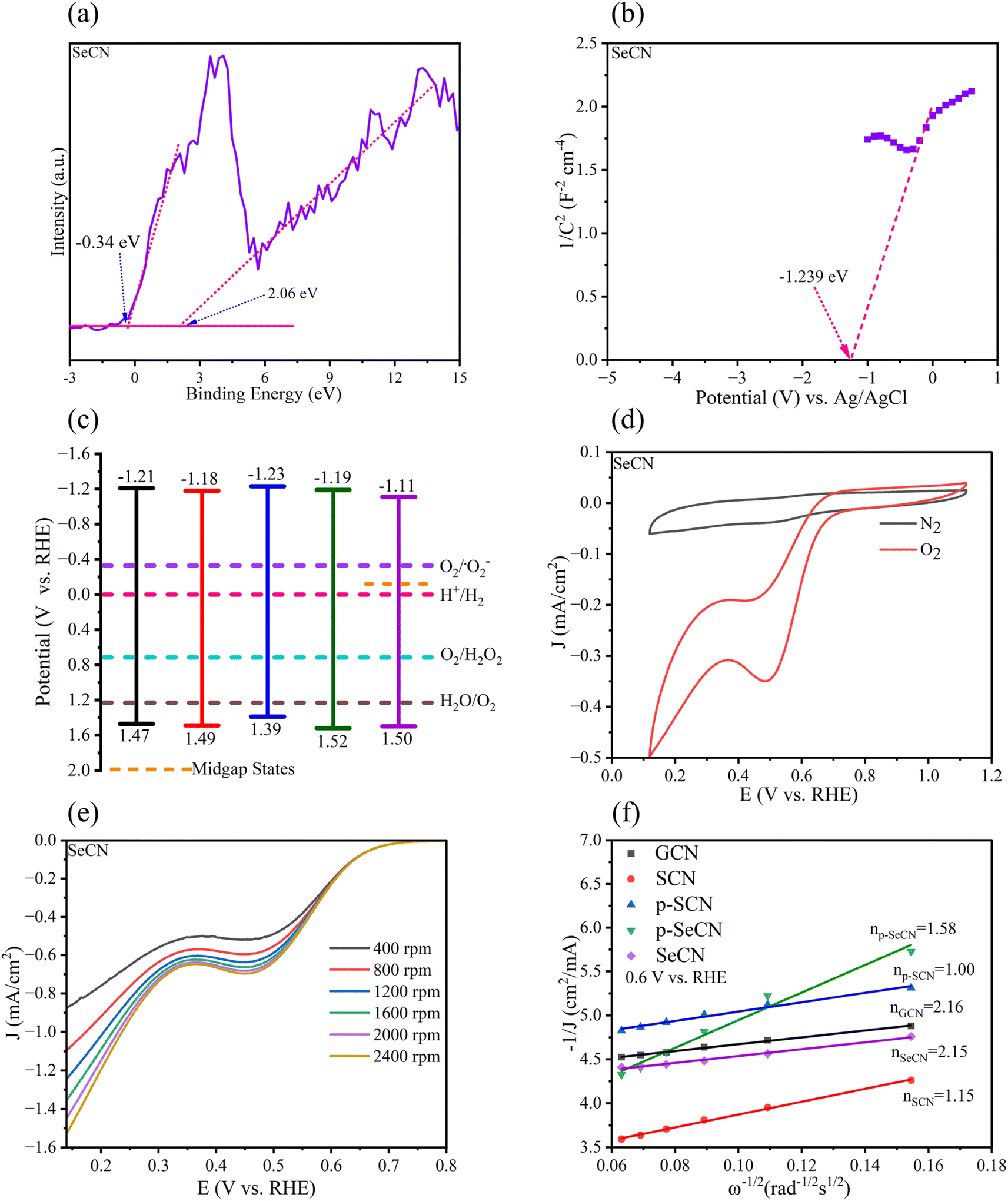 | ||
| Fig. 7 (a) XPS-VB spectra of SeCN, (b) Mott–Schottky plot of SeCN, (c) band structure diagram of fabricated samples, (d) CV curves of SeCN under N2 and O2 environments, (e) LSV curves of SeCN with different speeds, and (f) Koutecky–Levich plots of as-prepared samples. | ||
Additional confirmations of Mott–Schottky plots could give strong evidence for the research. From the analytic results in Fig. 7b and S9,† the positive slope of all samples was determined, indicating that all samples would possess n-type semiconducting properties.57 It has been reported that for n-type semiconductors, the flat band potentials could be equal to their Fermi level.62 This energy level (ENHE) can be computed viaeqn (5). Therefore, the calculated flat band potential value versus the Ag/AgCl electrode for GCN, SCN, p-SCN, p-SeCN, and SeCN was at −1.13, −1.117, −1.122, −1.105, and −1.042 eV, respectively. The positive shift in the flat band potentials could hint at a downshift in the Fermi level of the materials. Furthermore, for n-type semiconductors, previous studies explored the more negative value at 0.1 eV between the flat-band potential and the CB potential, leading to the converted formula in eqn (6).64 Based on these analyses, the calculated values of the CB of GCN, SCN, p-SCN, p-SeCN, and SeCN were at −1.230, −1.217, −1.222, −1.205, and −1.142 eV, respectively. These values could be close to the calculated CB positions from the conversion between the optical bandgap and VB energy.
In addition, through eqn (7), the work function of the materials could be calculated with the data in Table S6† being at 5.630, 5.617, 5.622, 5.605, and 5.542 eV for GCN, SCN, p-SCN, p-SeCN, and SeCN. The results critically show that the work function value would reduce when introducing S or Se atoms into g-C3N4. It has been stated that a high surface work function fundamentally represents strong electron binding restriction abilities and pronounced surface band bending, which can cause difficulties in generating photo-charge separations and migrations.63 Generally, S and Se doping could effectively enhance the separation of photo-excited electrons and holes and their transportations in the system due to – lowering the work function values in modified samples compared to pristine g-C3N4. Besides, the work function of Se-doped g-C3N4 was lower than that of S-doped g-C3N4, meaningfully suggesting better photoinduced charge separation and transfer in the system, thus accelerating catalytic outcomes. Moreover, the work function of p-SeCN was higher than that of SeCN, hinting that SeO2 would be more effective to dope into g-C3N4 in place of Se powders.
From the combined results of optical and electrochemical measurements, the band structure diagram was established and is shown in Fig. 7c. This illustrates the possibility of the reaction in forming H2O2 since fabricated materials tend to have sufficient potential to reduce oxygen into H2O2. It would be assumed that in addition to the supplementary oxygen from the system, water also participated in the process via water splitting reaction to produce oxygen, which could self-aid the reaction. Further controlled experiments were undertaken in the research to confirm the hypotheses, and the mechanistic studies will be discussed in the next section.
CV and LSV measurements were conducted in KOH (0.1 M) as an electrolyte to get more evidence for the reduction mechanism of oxygen in the reaction. Fig. 7d and S11† depict the CV curves of the as-prepared samples, recording a reduction peak at 0.5 V in the saturated O2 environment. There was no reaction when N2 was injected into the medium instead of O2. The average electron transfer number (n) determines the reaction mechanism. From the results of LSV measurements in Fig. 7e and S12† of all samples, the Koutecky–Levich plots in Fig. 7f were exported with the calculated values of n being 2.16, 1.15, 1.00, 1.58, and 2.15 for GCN, SCN, p-SCN, p-SeCN, and SeCN. As a result, the production of H2O2 would concomitantly follow dual-electron oxygen reduction and single-electron oxygen reduction.65 It is easy to see that the largest value of GCN would indicate the reaction towards the two-electron route, giving better catalytic performances.66 However, the rapid charge recombination in this system hindered its merit, resulting in the lowest efficiency in the generation of H2O2. Besides, an increase in electron transfer in the modified samples would reinforce the positive impact of doping sources. In particular, the introduction of Se would improve the selectivity of the generation of H2O2via a two-electron pathway in comparison with S doping. Moreover, the n value of SeCN was larger than that of p-SeCN, presenting better oxygen reducibility to produce H2O2. The results are consistent with other analyses to validate the roles of doping agents in g-C3N4 structures.
3.7 Piezo-photocatalytic results
To understand the practical piezo-photocatalytic abilities, the production of H2O2 was conducted in this research. As pointed out in Fig. S13a,† five samples would exhibit catalytic performances in the whole process. Without light irradiation, the role of piezo-electronic effects was critically shown with the formation of H2O2 during 30 minutes in darkness. GCN demonstrated the lowest piezo catalytic results. However, when introducing heteroatoms into the systems, the non-centrosymmetric structures could induce the piezo effects on accelerating the outcomes. It is familiar that ultrasonic waves could first prompt the formation of bubbles, followed by collision and explosion in the next steps. These events continuously happen on the catalysts' surface to produce the surface's piezoelectric potentials. As a result, thermally induced charges piled up on the surface of catalysts to help the piezo catalytic reactions occur. In reality, a few induced charges could participate in the reaction due to the rapid recombination phenomenon. Thus, the catalytic performance of the synthesized samples was relatively low.67–69 In addition, when the light was shined into the systems, a strong enhancement in catalytic performances was observed. It would be clarified by the synergistic effects between light and ultrasonic vibrations in which the continuous bombardment of these sources separates effectively electrons and holes, hence improving charge behaviors and suppressing charge recombination to yield H2O2.70 From the data, SeCN illustrated the better kinetic formation of H2O2. Thus, SeCN was selected to conduct the stability test and other comparison evaluations, as shown in Fig. S13b† and 8. Empirically, the data showed no differences during the four cycles, generally suggesting the stability of SeCN for long-run uses.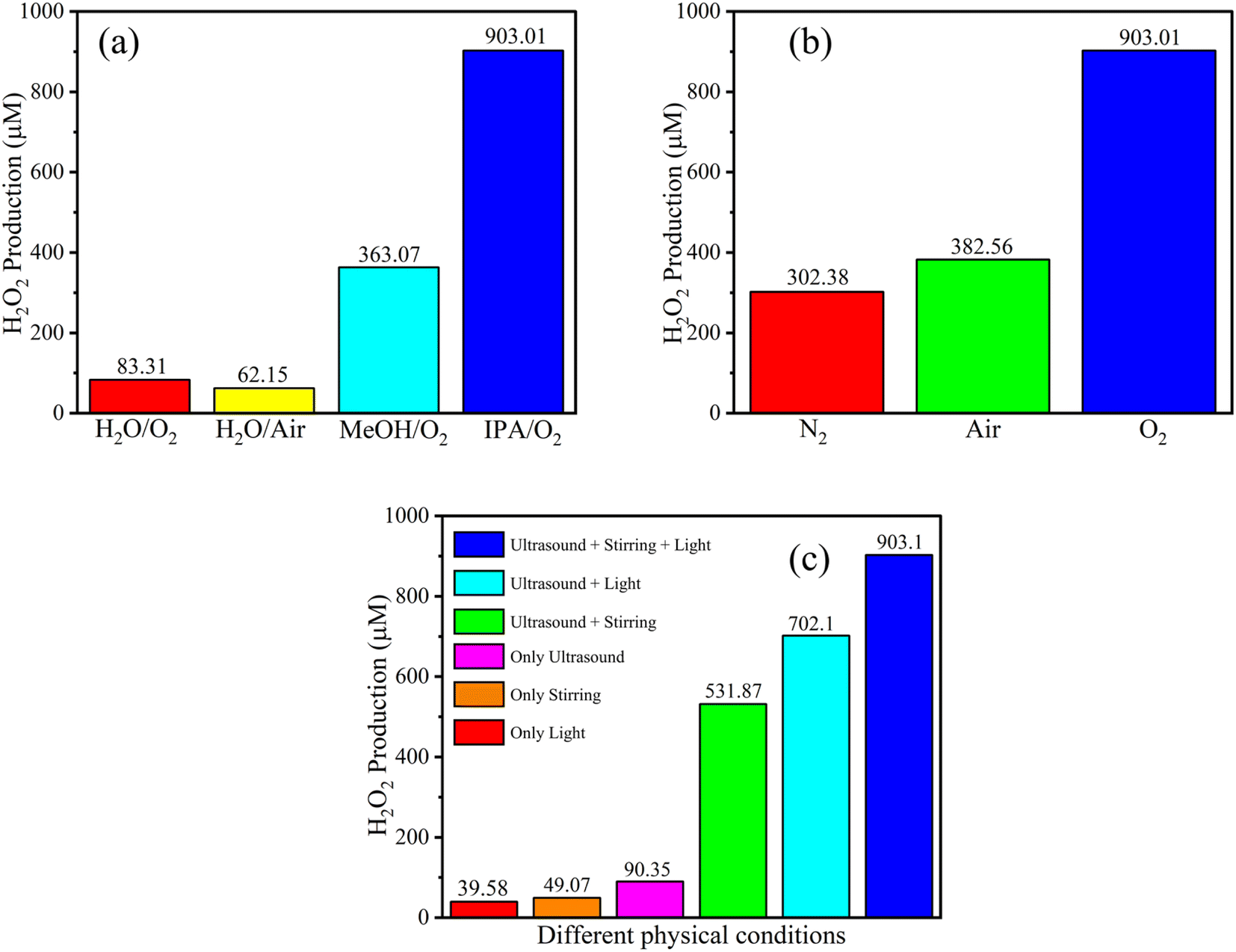 | ||
| Fig. 8 Comparison of piezo-photocatalytic results of SeCN under different sacrificial agents (a), atmospheres (b), and physical-controlled conditions (c). | ||
In order to fully comprehend the catalytic mechanisms in the reaction, various controlled experiments were implemented in the study and are demonstrated in Fig. 8. As can be seen in Fig. 8a, pure water with or without the supplement of oxygen was tested in the system with the formation of a poor amount of H2O2 at 83.31 and 62.15 μM, respectively. The catalytic performance was enormously improved with the addition of methanol (MeOH) or isopropanol (IPA). The highest concentration of H2O2 was recorded using IPA at 903.01 μM. Based on the empirical data, IPA would be more suitable for supplying reaction H+ than MeOH. The results indicate the significant role of sacrificial agents in the reaction, where they tend to provide H+ and trap photoexcited holes to produce other byproducts and prevent the recombination process.
Fig. 8b demonstrates the role of reaction atmospheres in yielding H2O2. Specifically, the saturated nitrogen atmosphere would inhibit the production of H2O2 in the reaction medium. The low performance at 302.38 μM on continuously injecting nitrogen into the reaction was recorded, significantly lower than that of the oxygen injection environment. As mentioned above, the materials possess adequate potential to produce oxygen from water to self-supply for the system, leading to H2O2 even under a nitrogen atmosphere. The research also tested air conditions with 382.56 μM of detected H2O2 in the study, signifying better prospects for practical applications.
The role of physical conditions was evaluated by investigating various physical-controlled experiments. It is easy to see in Fig. 8c that the sample which was solely shined with light or only stirred was sequentially recorded with the highlighted poor result at 39.58 or 49.07 μM. Excitingly, if the ultrasound was applied to the system, a three-time increase was observed in the performance. When the combination of ultrasonic waves with stirring or ultrasonic waves with light was employed in the study, a significant improvement was detected at 531.87 or 702.1 μM, suggesting that the synergistic effects could play a pivotal role in the system. Comprehensively, when light, ultrasound, and stirring were simultaneously integrated, the catalytic outcomes would be highest at 903.01 μM.
To understand the effect of light and ultrasound, the study measured and calculated the intensity of these factors. As a result, the light intensity was at 89 mW cm−2, which was 6.1 times lower than that of ultrasonic intensity at 543 mW cm−2. These analyses could adequately interpret the catalytic performances in the research. Furthermore, it is believed that photocatalysis takes place with two distinct stages comprising the adsorption of reactants on the catalysts' surfaces and the occurrence of photo-redox reactions. Hence, a suitable quantity of reactants should be adsorbed to maximize photocatalytic performances. In this scenario, ultrasonic waves could prompt reversible influences on the system. Ultrasonic waves could help to release the over-adsorption of reactants on the catalysts' surfaces in which the outcomes could be maximized. On the contrary, the excess sonication in terms of power and time would decrease the photocatalytic effects due to the over-desorption of oxygen molecules from modified g-C3N4 surfaces, resulting in low photocatalytic outcomes but enhancing piezo-catalytic performances. Thus, the results could be understandable, and the application of piezo-photocatalytic systems could be a compromise between the power of light and ultrasound.
3.8 Possible catalytic mechanisms of the catalytic production of H2O2
Based on the above-mentioned characterization results, the production of H2O2 would be associated with the two-electron reduction pathway, including either a direct two-electron reduction or a two-step single-electron reduction. The latter was stated as a facile and thermodynamically favorable pathway to accelerate the evolution of H2O2.71 Therefore, to comprehend the possible mechanisms for generating H2O2 in the system, scavenging experiments were carried out in this work to trap active species. In particular, L-tryptophan (L-trp), para-benzoquinone (BQ), EDTA, and AgNO3 were separately added into the reaction to trap singlet oxygen (1O2), superoxides (·O2−), excited holes, and excited electrons. Based on the empirical results in Fig. S14,† with a considerable reduction in catalytic outcomes, evaluating the significant role of intermediate products in the medium would be helpful. Incorporating other evidence from electrochemical measurements, such as the average number of electron transfers from Koutecky–Levich plots, the mechanism to generate H2O2 in this work would be mainly based on a sequential two-step single-electron reduction route. These mechanistic reactions depicted in eqn (8) and (9) would naturally happen thanks to the materials' suitable band positions, helping generate H2O2. Specifically, excited electrons reduced oxygen molecules under irradiation to generate ·O2−, followed by the continuous reduction to yield H2O2. In addition, a part of produced ·O2− might interact with excited holes to give 1O2, which could be further converted into H2O2.27,72 The data from hole-trapping experiments revealed that the conversion from ·O2− to 1O2 is superiorly meaningful to obtain better performance. Thereby, ·O2− and 1O2 would critically contribute as central intermediates in the reaction to generate the final products.| O2 + e− → ·O2− | (8) |
| ·O2− + 2H+ + e− → H2O2 | (9) |
| O2 → ·O2− → 1O2 → H2O2 | (10) |
4. Conclusion
Briefly, the work presented the effects of doping precursors on the production rate of H2O2. Based on the results, the use of SeO2 as a doping agent could be prominent to give a better catalytic performance than Se powders owing to the doping efficiency. Furthermore, the empirical data from spectroscopic and electrochemical studies, such as PL spectra and the average electron transfer number, would prove the positive impacts of Se doping over S doping. Additionally, the characterization of chemical structures showed the successful incorporation of new functional groups into the g-C3N4 networks. These functional groups could behave as a trapping agent to prolong the charge carrier life in the system, causing higher catalytic outcomes. From optical and electrochemical analyses, the effects of doping precursors on the defective states of the materials were also disclosed, with SeO2 prompting the highest percentage of defects in the structures. Besides that, the controlled experiments from chemical and physical conditions revealed the mechanism of the reaction in the medium relying on the dual-electron oxygen reduction for H2O2 evolution. This research would be helpful for further explorations of doping mechanisms in the design and development of g-C3N4 catalysts in the application of piezo and photo effects. Eventually, the study would validate the efficiency of Se surpassing S in the doping techniques and certify the potential of Se-doped g-C3N4 for the production of H2O2 and other fine chemicals.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support of time and facilities from Ho Chi Minh City University of Technology (HCMUT), VNU-HCM for this study.References
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- M. Seo, H. J. Kim, S. S. Han and K.-Y. Lee, Catal. Surv. Asia, 2017, 21, 1–12 CrossRef CAS.
- L. Pi, J. Cai, L. Xiong, J. Cui, H. Hua, D. Tang and X. Mao, J. Chem. Eng., 2020, 389, 123420 CrossRef CAS.
- H. L. Wapshott-Stehli and A. M. Grunden, Enzyme Microb. Technol., 2021, 145, 109744 CrossRef CAS PubMed.
- S. C. Perry, D. Pangotra, L. Vieira, L.-I. Csepei, V. Sieber, L. Wang, C. P. d. León and F. C. Walsh, Nat. Rev. Chem., 2019, 3, 442–458 CrossRef CAS.
- B. He, Z. Wang, P. Xiao, T. Chen, J. Yu and L. Zhang, Adv. Mater., 2022, 34, 2203225 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS.
- Y. Qu and X. Duan, Chem. Soc. Rev., 2013, 42, 2568 RSC.
- H.-T. Vuong, D.-P. Bui, D.-V. Nguyen, L. P. Phuong, P. P. D. Minh, T. D. Dat and N. H. Hieu, ChemPhotoChem, 2023, e202200299 Search PubMed.
- F. He, Z. Wang, Y. Li, S. Peng and B. Liu, Appl. Catal., B, 2020, 269, 118828 CrossRef CAS.
- J. Wang and S. Wang, Coord. Chem. Rev., 2022, 453, 214338 CrossRef CAS.
- L. Acharya, G. Swain, B. P. Mishra, R. Acharya and K. Parida, ACS Appl. Energy Mater., 2022, 5, 2838–2852 CrossRef CAS.
- C. Feng, L. Tang, Y. Deng, J. Wang, Y. Liu, X. Ouyang, H. Yang, J. Yu and J. Wang, Appl. Catal., B, 2021, 281, 119539 CrossRef CAS.
- Y. Liu, Y. Zheng, W. Zhang, Z. Peng, H. Xie, Y. Wang, X. Guo, M. Zhang, R. Li and Y. Huang, J. Mater. Sci. Technol., 2021, 95, 127–135 CrossRef CAS.
- X. Zhou, B. Shen, A. Lyubartsev, J. Zhai and N. Hedin, Nano Energy, 2022, 96, 107141 CrossRef CAS.
- H. Lei, M. Wu, F. Mo, S. Ji, X. Dong, Y. Jia, F. Wang and Z. Wu, Environ. Sci.: Nano, 2021, 8, 1398 RSC.
- R. Tang, D. Gong, Y. Zhou, Y. Deng, C. Feng, S. Xiong, Y. Huang, G. Peng and L. Li, Appl. Catal., B, 2022, 303, 120929 CrossRef CAS.
- O. Tomita, T. Otsubo, M. Higashi, B. Ohtani and R. Abe, ACS Catal., 2016, 6, 1134–1144 CrossRef CAS.
- L. Chen, C. Chen, Z. Yang, S. Li, C. Chu and B. Chen, Adv. Funct. Mater., 2021, 31, 2105731 CrossRef CAS.
- Y. Yang, H. Jin, C. Zhang, H. Gan, F. Yi and H. Wang, J. Alloys Compd., 2020, 821, 153439 CrossRef CAS.
- P. Chen, P. Xing, Z. Chen, H. Lin and Y. He, Int. J. Hydrogen Energy, 2018, 43, 19984–19989 CrossRef CAS.
- J. Zhang, W. Zhang, L. Yue, X. Hu, H. Lin, L. Zhao and Y. He, Appl. Surf. Sci., 2022, 592, 153337 CrossRef CAS.
- Q. Zhang, P. Chen, L. Chen, M. Wu, X. Dai, P. Xing, H. Lin, L. Zhao and Y. He, J. Colloid Interface Sci., 2020, 568, 117–129 CrossRef CAS PubMed.
- J. Li, B. Shen, Z. Hong, B. Lin, B. Gao and Y. Chen, Chem. Commun., 2012, 48, 12017–12019 RSC.
- Y. Li, W. Ho, K. Lv, B. Zhu and S. C. Lee, Appl. Surf. Sci., 2018, 430, 380–389 CrossRef CAS.
- H.-T. Vuong, D.-V. Nguyen, P. P. Ly, P. D. M. Phan, T. D. Nguyen, D. D. Tran, P. T. Mai and N. H. Hieu, ACS Appl. Nano Mater., 2023, 6, 664–676 CrossRef CAS.
- S. Cao, B. Fan, Y. Feng, H. Chen, F. Jiang and X. Wang, Chem. Eng. J., 2018, 353, 147–156 CrossRef CAS.
- C.-Q. Cheng, Y. Feng, Z.-Z. Shi, Y.-L. Zhou, W.-J. Kang, Z. Li, J. Mao, G.-R. Shen, C.-K. Dong, H. Liu and X.-W. Du, Langmuir, 2022, 38, 1471–1478 CrossRef CAS PubMed.
- M. K. Hussien, A. Sabbah, M. Qorbani, M. H. Elsayed, P. Raghunath, T.-Y. Lin, S. Quadir, H.-Y. Wang, H.-L. Wu, D.-L. M. Tzou, M.-C. Lin, P.-W. Chung, H.-H. Chou, L.-C. Chen and K.-H. Chen, Chem. Eng. J., 2022, 430, 132853 CrossRef.
- I. Mohammed, D. Al Shehri, M. Mahmoud, M. S. Kamal and O. S. Alade, ACS Omega, 2021, 6, 4022–4033 CrossRef CAS PubMed.
- X. Wang, M. He and Z. Nan, Sep. Purif. Technol., 2021, 256, 117765 CrossRef CAS.
- D. Ayodhya, Mater. Chem. Front., 2022, 6, 2610 RSC.
- J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiró, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS PubMed.
- S. Samanta, R. Yadav, A. Kumar, A. K. Sinha and R. Srivastava, Appl. Catal., B, 2019, 259, 118054 CrossRef CAS.
- M. Majdoub, A. Amedlous, Z. Anfar and A. Jada, Adv. Mater. Interfaces, 2022, 9, 2200170 CrossRef CAS.
- T. T. Dang, T. K. A. Nguyen, K. C. Bhamu, T. Mahvelati-Shamsabadi, V. K. H. Van, E. W. Shin, K.-H. Chung, S. H. Hur, W. M. Choi, S. G. Kang and J. S. Chung, ACS Catal., 2022, 12, 13763–13780 CrossRef CAS.
- I. Krivtsov, D. Mitoraj, C. Adler, M. Ilkaeva, M. Sardo, L. Mafra, C. Neumann, A. Turchanin, C. Li, B. Dietzek, R. Leiter, J. Biskupek, U. Kaiser, C. Im, B. Kirchhoff, T. Jacob and R. Beranek, Angew. Chem., Int. Ed., 2020, 59, 487–495 CrossRef CAS PubMed.
- S. Yu, J. Li, Y. Zhang, M. Li, F. Dong, T. Zhang and H. Huang, Nano Energy, 2018, 50, 383–392 CrossRef CAS.
- Z. Gu, Z. Cui, Z. Wang, K. S. Qin, Y. Asakura, T. Hasegawa, S. Tsukuda, K. Hongo, R. Maezono and S. Yin, Appl. Catal., B, 2020, 279, 119376 CrossRef CAS.
- S. Chen, J. Duan, J. Ran and S.-Z. Qiao, Adv. Sci., 2015, 2, 1400015 CrossRef.
- Z. Yang, C. Zhang, Y. Mei, Y. Zhang, Y. Feng, M. Shao and J. Hu, Adv. Mater. Interfaces, 2022, 9, 2201325 CrossRef CAS.
- Y. Zheng, S. Chen, X. Yu, K. Li, X. Ni and L. Ye, Appl. Surf. Sci., 2022, 598, 153786 CrossRef CAS.
- J. Li, X. Liu, H. Che, C. Liu and C. Li, Carbon, 2021, 172, 602–612 CrossRef CAS.
- X. Bai, X. Wang, X. Lu, Y. Liang, J. Li, L. Wu, H. Li, Q. Hao, B.-J. Ni and C. Wang, J. Hazard. Mater., 2020, 398, 122897 CrossRef CAS PubMed.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176, 44–52 Search PubMed.
- F. Qiao, J. Wang, S. Ai and L. Li, Sens. Actuators, B, 2015, 216, 418–427 CrossRef CAS.
- R. Du, K. Xiao, B. Li, X. Han, C. Zhang, X. Wang, Y. Zuo, P. Guardia, J. Li, J. Chen, J. Arbiol and A. Cabot, Chem. Eng. J., 2022, 441, 135999 CrossRef CAS.
- S. Li, G. Dong, R. Hailili, L. Yang, Y. Li, F. Wang, Y. Zeng and C. Wang, Appl. Catal., B, 2016, 190, 26–35 CrossRef CAS.
- B. Yang, J. Han, Q. Zhang, G. Liao, W. Cheng, G. Ge, J. g. Liu, X. Yang, R. Wang and X. Jia, Carbon, 2023, 202, 348–357 CrossRef CAS.
- E. M. Dias, K. C. Christoforidis, L. Francas and C. Petit, ACS Appl. Energy Mater., 2018, 1, 6524–6534 CrossRef CAS.
- V. Hasija, P. Singh, S. Thakur, K. Stando, V.-H. Nguyen, Q. V. Le, S. M. Alshehri, T. Ahamad, K. C.-W. Wu and P. Raizada, Appl. Mater. Today, 2022, 29, 101676 CrossRef.
- H.-T. Vuong, T. Mahvelati-Shamsabadi, T. T. Dang, Q. D. Dao, E. W. Shin and J. S. Chung, Int. J. Hydrogen Energy, 2022, 47, 23249–23263 CrossRef CAS.
- J. Mooney and P. Kambhampati, J. Phys. Chem. Lett., 2013, 4, 3316–3318 CrossRef CAS.
- Y. Yuan, L. Zhang, J. Xing, M. I. B. Utama, X. Lu, K. Du, Y. Li, X. Hu, S. Wang, A. Genç, R. Dunin-Borkowski, J. Arbiol and Q. Xiong, Nanoscale, 2015, 7, 12343 RSC.
- D. Das, S. L. Shinde and K. K. Nanda, ACS Appl. Mater. Interfaces, 2016, 8, 2181–2186 CrossRef CAS PubMed.
- J. Cao, J. Cai, R. Li, J. H. Han, J. Liu and M. Huang, J. Phys. Chem. C, 2022, 126, 4939–4947 CrossRef CAS.
- B. Choudhury, K. K. Paul, D. Sanyal, A. Hazarika and P. K. Giri, J. Phys. Chem. C, 2018, 122, 9209–9219 CrossRef CAS.
- Y. Zhang, Q. Pan, G. Chai, M. Liang, G. Dong, Q. Zhang and J. Qiu, Sci. Rep., 2013, 3, 1943 CrossRef PubMed.
- P. Niu, G. Liu and H.-M. Cheng, J. Phys. Chem. C, 2012, 116, 11013–11018 CrossRef CAS.
- C. Liang, H.-Y. Niu, H. Guo, C.-G. Niu, D.-W. Huang, Y.-Y. Yang, H.-Y. Liu, B.-B. Shao and H.-P. Feng, Chem. Eng. J., 2020, 396, 125395 CrossRef CAS.
- X. Yue, L. Cheng, J. Fan and Q. Xiang, Appl. Catal., B, 2022, 304, 120979 CrossRef CAS.
- C. Wan, L. Zhou, L. Sun, L. Xu, D.-g. Cheng, F. Chen, X. Zhan and Y. Yang, Chem. Eng. J., 2020, 396, 125229 CrossRef CAS.
- J. Zhang, L. Yue, Z. Zeng, C. Zhao, L. Fang, X. Hu, H. Lin, L. Zhao and Y. He, J. Colloid Interface Sci., 2023, 636, 480–491 CrossRef CAS PubMed.
- Y. Pan, X. Liu, W. Zhang, B. Shao, Z. Liu, Q. Liang, T. Wu, Q. He, J. Huang, Z. Peng, Y. Liu and C. Zhao, Chem. Eng. J., 2022, 427, 132032 CrossRef CAS.
- X. Zeng, Y. Liu, Y. Kang, Q. Li, Y. Xia, Y. Zhu, H. Hou, M. H. Uddin, T. R. Gengenbach, D. Xia, C. Sun, D. T. Mccarthy, A. Deletic, J. Yu and X. Zhang, ACS Catal., 2020, 10, 3697–3706 CrossRef CAS.
- K. Wang, B. Li, C. Zhao, S. Yuan, C. Zhang, X. Liang, J. Wang, Y. Wu and Y. He, Ultrason. Sonochem., 2023, 92, 106285 CrossRef CAS PubMed.
- S. Zheng, X. Li, J. Zhang, J. Wang, C. Zhao, X. Hu, Y. Wu and Y. He, J. Environ. Sci., 2023, 125, 1–13 CrossRef PubMed.
- C. Zhao, L. Cai, K. Wang, B. Li, S. Yuan, Z. Zeng, L. Zhao, Y. Wu and Y. He, Environ. Pollut., 2023, 319, 120982 CrossRef CAS PubMed.
- L. Chen, J. Wang, X. Li, J. Zhang, C. Zhao, X. Hu, H. Lin, L. Zhao, Y. Wu and Y. He, Green Energy Environ., 2023 Search PubMed , in press.
- W. Hou, Y. Li, S. Ouyang, H. Chen, J. Ye, X. Han and Y. Deng, Chem. Commun., 2019, 55, 13279 RSC.
- Z. Yang, L. Li, S. Zeng, J. Cui, K. Wang, C. Hu and Y. Zhao, ACS Appl. Mater. Interfaces, 2023, 15(6), 8232–8240 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Catalytic summarized table, physical properties, bandgap energy of SCN, p-SCN, p-SeCN, N2 adsorption–desorption isotherms, pore size distribution curves, TGA results, 13C NMR spectra, XPS spectra, PL energy spectra, EIS results, CV curves, LSV curves, catalytic performances, scavenging studies. See DOI: https://doi.org/10.1039/d2na00909a |
| ‡ These authors equally contribute to the research. |
| This journal is © The Royal Society of Chemistry 2023 |