
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeneration of sub-5 nm AuNPs in the special space of the loop-cluster corona of a polymer vesicle: preparation and its unique catalytic performance in the reduction of 4-nitrophenol†
Wen-Li
Wang‡
,
Ayaka
Kanno‡
,
Amika
Ishiguri
and
Ren-Hua
Jin
 *
*
Department of Material and Life Chemistry, Kanagawa University, 3-27-1 Rokkakubashi, Yokohama 221-8686, Japan. E-mail: rhjin@kanagawa-u.ac.jp
First published on 22nd February 2023
Abstract
The hybrid vesicle AuNP@LCCV, in which a large number of AuNPs with an average size of about 2.8 nm were densely and uniformly distributed in an isolated state throughout the corona of the unusual polymer vesicle, was prepared via in situ reduction of Au3+ ions, which were encapsulated in advance in the unique polymer vesicle (LCCV) consisting of a hydrophobic membrane of poly(2-phenyl-2-oxazoline) and a hydrophilic loop-cluster corona of polyethyleneimine. The vesicle was formed via self-assembly from a comb-like block copolymer in which a polystyrenic main chain was grafted densely with diblock polyethyleneimine-b-poly(2-phenyl-2-oxazoline) and acted as a reactor for the reduction of Au3+. The hybrid vesicle AuNP@LCCV showed powerful catalytic ability in the reduction of nitrophenols (NPs). Interestingly, the reduction reactions of NPs showed a remarkably long induction time, which could be shortened dramatically from 60 min to 1–2 min by greatly increasing the concentration of NaBH4. It is revealed that the oxygen adsorbed on the AuNPs significantly inhibited the reduction, causing the induction time. Once the oxygen is chemically cleaned from the surface of the AuNPs, the reduction of 4-NP proceeds gradually for a while and then completes suddenly. The reduction mechanism accompanying the oxygen-dependent induction time is proposed from the view of the strong oxygen affinity of the catalyst AuNP@LCCV.
1. Introduction
Gold nanoparticles (AuNPs) are very important materials that can play versatile roles in many fields based on their unique physical and chemical properties.1–4 In particular, AuNPs have attracted extensive attention in the field of catalysis due to their powerful catalytic activity and their ability to promote various reactions under mild conditions, such as hydrogenation of unsaturated hydrocarbon groups, reduction of nitro groups, C–C coupling reactions, etc.2,5 Generally, smaller AuNPs with higher specific surface area and more negative redox potential exhibit higher catalytic activity.6 However, the preparation and immobilization of smaller AuNPs (especially those with a particle size below 10 nm in liquid media) has always been a challenge due to their high surface energy.6,7 As a typical method of preventing the aggregation of AuNP particles, stabilizers such as organic small molecules, organic ligands, inorganic ligands and polymers are often added during the preparation of AuNPs.6–10 Different to stabilizer systems in which nanoparticles are capped by stabilizers, AuNPs can also be efficiently prepared using amphiphilic copolymeric assemblies such as vesicles and micelles, by which an organized hybrid in which a lot of AuNPs are distributed in one vesicular or micellar entity is produced.11 This preparation of AuNP involves the in situ reduction of Au ions in polymeric assemblies. Since the polymeric assemblies have dispersed and solidified features in solution, they can effectively hinder the enlargement and aggregation of the AuNPs formed inside the assemblies.11 More notably, the relatively loose framework of the amphiphilic polymeric assemblies facilitates the penetration of the reaction substrate into assembly to collide with the AuNPs. This can endow the AuNPs in the assemblies with catalytic activity.12Generally, in the preparation of AuNPs using polymeric assemblies, the particle size, dispersion, and catalytic activity of the AuNPs are also affected by the structure and density of the polymers. The coronal layer on vesicles or micelles with a loose structure is unfavorable in terms of hindering the growth and aggregation of AuNPs, and usually results in large AuNPs.13 In contrast, AuNPs produced in the membrane of a vesicle or the core of a micelle usually show smaller particle size and good dispersion because the membrane or the core consists of dense polymeric chains and cured structures. However, the dense hydrophobic polymeric chain will hinder the penetration of the substrate, resulting in a poor catalytic effect.7 Therefore, the preparation of AuNPs on the interface of the coronal layer and the core/membrane has been one of the major strategies to improve the catalytic activity of AuNPs.7,14,15
Over the long term, AuNPs have received extensive attention in the field of water purification. For example, nitroaromatics are one of the major classes of water pollutants, and originate from the industries of herbicides, pesticides, and synthetic dyes. They have been listed as “priority pollutants” by the US Environmental Protection Agency (EPA).16 Using AuNP as a catalyst, nitroaromatics can be hydrogenated to their amino analogues, which can be used for producing antipyretics and analgesics. This is a green strategy for water purification. Other materials, such as noble metal nanoparticles (Ag, Pd),17–19 nanocomposites,20,21 carbonaceous materials22 and enzymes,23 can also be used as catalysts for the hydrogenation of nitroaromatics. Among the many catalyst candidates, AuNPs are one of the most promising catalytic materials due to their simple preparation procedure and powerful catalytic activity.
In our previous reports,24,25 we synthesized a comb-like copolymer, c-iEP, possessing diblock side chains of polyethyleneimine-b-poly(2-phenyl-2-oxazoline) [PEI-b-PPOZ] (the PEI block is near the main chain, while PPOZ is far from the main chain). We found that this comb-like copolymer could self-assemble into a unique vesicle covered by a loop-cluster PEI corona (LCCV). This loop-cluster PEI corona may be a good independent space for controlling the growth and hindering the aggregation of AuNPs due to the strong coordination ability of PEI and the unique loop structure of the corona.26 The hydrophilic corona will facilitate the penetration of substrates dissolved in the aqueous phase. Therefore, this may be a new strategy for preparing AuNPs with powerful catalytic functions. In this work, we prepared the hybrid vesicle AuNP@LCCV via an in situ reduction of Au3+ in the corona of LCCV (the preparation process is shown in Scheme 1). The AuNPs formed on the LCCV showed a small size of about 3 nm and were distributed uniformly in the outer coronal layer of LCCV. AuNP@LCCV showed powerful catalytic ability for the reduction of 4-nitrophenol (4-NP), but with an accompanying unusually long induction time. It was found that the catalyst AuNP@LCCV is very sensitive to oxygen, which could occupy the gold surface to cause the induction time.
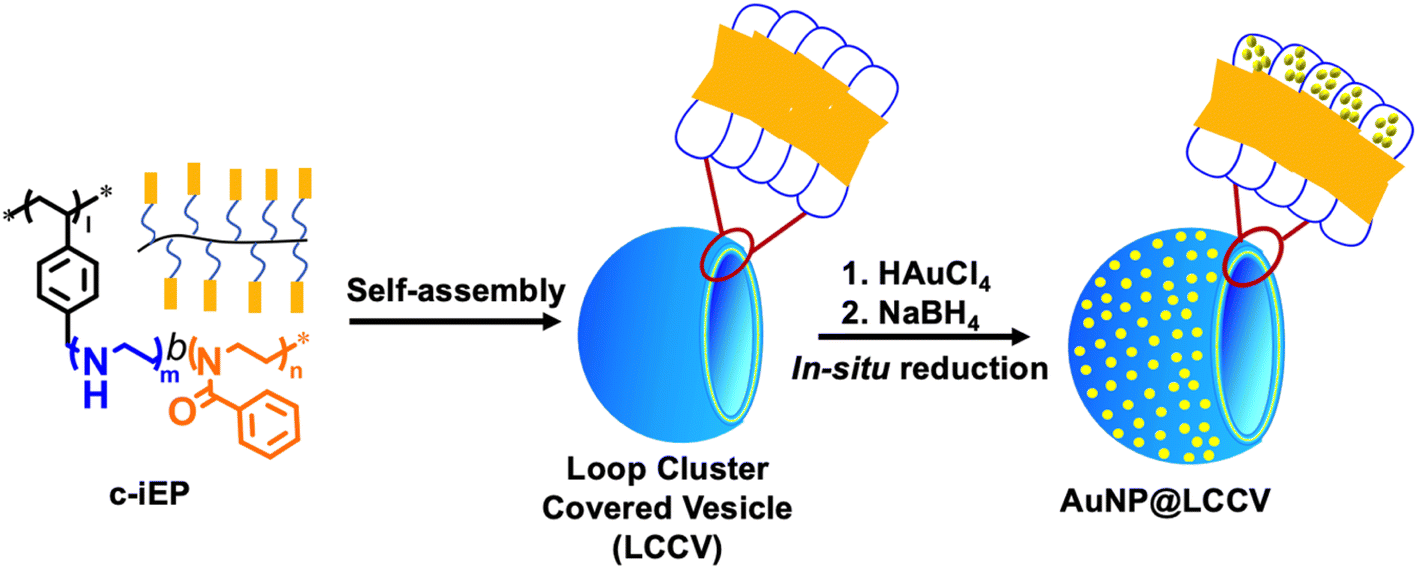 | ||
| Scheme 1 Preparation process of the composite vesicle (AuNP@LCCV) containing AuNPs with a particle size of less 3 nm by self-assembly of comb-like block copolymer c-iEP (I = 104, m = 50, n = 48) and in situ reduction of Au ions in the loop corona of the LCCV vesicle. | ||
2. Experimental
2.1 Materials
A comb-like block copolymer (c-iEP) possessing polyethyleneimine-b-poly(2-phenyl-2-oxazoline) diblock side chains (the degree of polymerization [DP] of the polystyrenic main chain is 104, the DP of polyethyleneimine in the side chain is 50, and the DP of poly(2-phenyl-2-oxazoline) in the side chain is 48) was synthesized according to our previous method.13 The LCCV vesicle was prepared via self-assembly of c-iEP using solvent-switching of methanol and water.14 Hydrogen tetrachloroaurate (III) tetrahydrate (HAuCl4·4H2O, >99.0%, Fujifilm wako), 4-nitrophenol (4-NP, >99.0%, TCI), sodium borohydride (NaBH4, 95.0%, TCI), distilled water (Fujifilm wako) and other chemicals were used as received.2.2 Characterizations
The vesicle (LCCV) and composite vesicle (LCCV@AuNP) were visualized using a HITACHI SU8010 scanning electron microscope (SEM) after samples were sputtered with Pt particles. The TEM observation was performed using a JEOL JEM-2010 instrument with an acceleration voltage of 200 kV. The average particle size and particle size distribution of the AuNPs were determined using the statistical software of J-image. DLS measurement was performed using a fiber-optics particle analyzer with an auto-sampler (FPAR-1000, Otsuka Electronic Co., Ltd). The reduction of 4-nitrophenol was monitored via UV-vis analysis using a Shimadzu UV-2500 spectrophotometer.2.3 Preparation of AuNP@LCCV
A mixture containing 100 mL of an LCCV aqueous solution (0.1 mg mL−1, ethyleneimine units: 75 × 10−6 mol) and 3.2 mL of an aqueous solution of HAuCl4·4H2O (1.64 mg mL−1, 12.7 × 10−6 mol, 1 equiv.) was prepared and stirred for 1 h. To the mixture, 0.95 mL of a fresh aqueous solution of NaBH4 (1.5 mg mL−1, 38 × 10−6 mol, 3 equiv. relative to Au) was added to perform the in situ reduction of Au3+ to AuNP in the corona of LCCV. After stirring for 1 h at room temperature, the reaction mixture was dialyzed against water for 3 days to remove byproducts and excess NaBH4. The recovered aqueous solution of AuNP@LCCV was diluted in a 150 mL measuring flask to prepare a definite concentration (0.084 μmol mL−1) of AuNP.2.4 Preparation of AuNP@C
In a two-necked 300 mL round-bottom flask equipped with a condenser, 85.08 mg (0.33 mmol) of trisodium citrate was dissolved in 150 mL of deionized water and stirred for 30 min at 110 °C. Then, 1.0 mL of an aqueous solution of HAuCl4·4H2O (5.91 mg mL−1, 0.014 mmol) was added and stirred at the same temperature for 20 min. After that, the reaction solution ([Au] = 0.0956 μmol mL−1) was kept at room temperature and used as a sample for SEM observation, DLS, and UV-vis measurement and as a catalyst in the reduction of 4-nitrophenol.2.5 Process of the reduction reaction of 4-nitrophenol
3. Results and discussion
3.1 Preparation of AuNP@LCCV
As mentioned above, we have established the unique polymer vesicle LCCV using the specially designed comb polymer c-iEP. Different to typical polymer vesicles, LCCV has amazing stability, being able to maintain its spherical morphology even in the dried state and/or high vacuum conditions without collapse and even after being kept in acidic and/or basic water for a period of years. This stability should be attributed to the PEI loop cluster corona, which does not have the free ends seen in usual brush corona.24,25 This is a very desirable property from the viewpoint of trapping metallic components on the LCCV, because the PEI in the loop clusters is a good polymeric ligand for transition metal ions.27–29First, using the comb-like copolymer c-iEP, we prepared LCCV vesicle at a c-iEP concentration of 0.1 mg mL−1via the solution switching method with methanol and water according to our previous report.25 The vesicle solution was a milk-like opaque dispersion with no absorption in the wavelength range of 400–600 nm (see Fig. 1). Afterwards, as shown in Scheme 1, a mixture containing LCCV aqueous solution and an aqueous solution of HAuCl4 was mixed with a fresh aqueous solution of NaBH4 to perform in situ reduction of Au+ to AuNP in the corona of LCCV. After stirring for 1 h at room temperature, the reaction mixture was dialyzed against water for 3 days to remove byproducts and excess NaBH4. The recovered aqueous solution of AuNP@LCCV was diluted in a measuring flask to prepare a definite concentration of AuNP. Here, using NaBH4 as a reductant is important because NaBH4 benefits the generation of small AuNPs due to its higher reducibility and faster reduction process compared to other reductants such as ascorbic acid, citric acid, etc.5,30–32 Generally, solutions of AuNPs show a wine-red color and often give sharp and strong absorptions at 500–600 nm due to the surface plasmon resonance of AuNPs.1,11 In our case, the plasmon absorption of AuNP@LCCV appeared very weak as a broadened band in the wavelength region of 500–600 nm (Fig. 1), although the prepared AuNP@LCCV solution with a concentration of 0.222 mg mL−1 (which is sufficiently high) showed a wine-red color (see inset in Fig. 1).
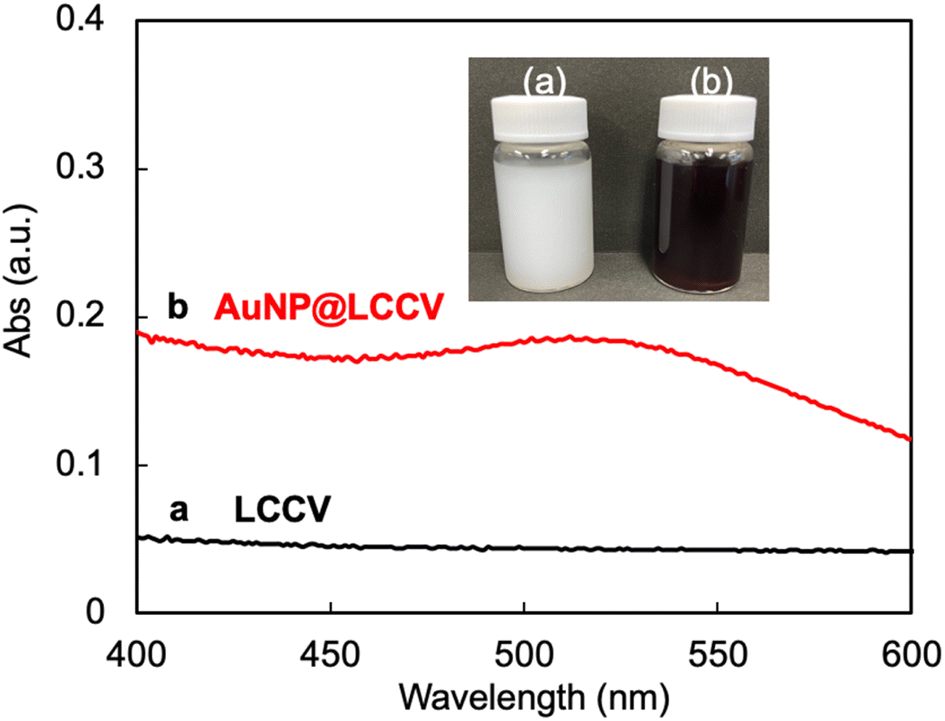 | ||
| Fig. 1 UV-vis spectra of (a) LCCV and (b) AuNP@LCCV. The inset is a photograph of the dispersion solutions before and after the loading of gold nanoparticles on LCCV. | ||
To clarify the state of the generated AuNPs and the morphology of AuNP@LCCV, we compared the vesicles before and after loading with AuNPs using SEM and TEM. As shown in the SEM and TEM images (Fig. 2), the vesicles showed a spherical morphology before and after loading with gold. We found that there were not any collapsed circular images, which are often seen for typical polymer vesicles. After loading Au, different to the case of LCCV, many spots can be seen on AuNP@LCCV (Fig. 2e). From the magnified edge area, we can clearly see that gold nanoparticles smaller than 5 nm were uniformly distributed in an isolated state on the vesicle surface. Each of the particles is almost free of contact and fusion. This is very rare in the preparation of AuNPs on vesicles. The average particle size of the AuNPs was calculated to be 2.8 nm via statistical analysis through counting 70 AuNPs on a vesicle (Fig. 2f). In addition, after the hybridization with AuNP, the size of the vesicles was significantly reduced from 456 to 338 nm in DLS (Fig. 2c). Probably, the coordinative interaction of the PEI loops of the corona with the AuNPs causes the PEI chains to arrange more tightly, which leads to the contraction of the polymer vesicles.
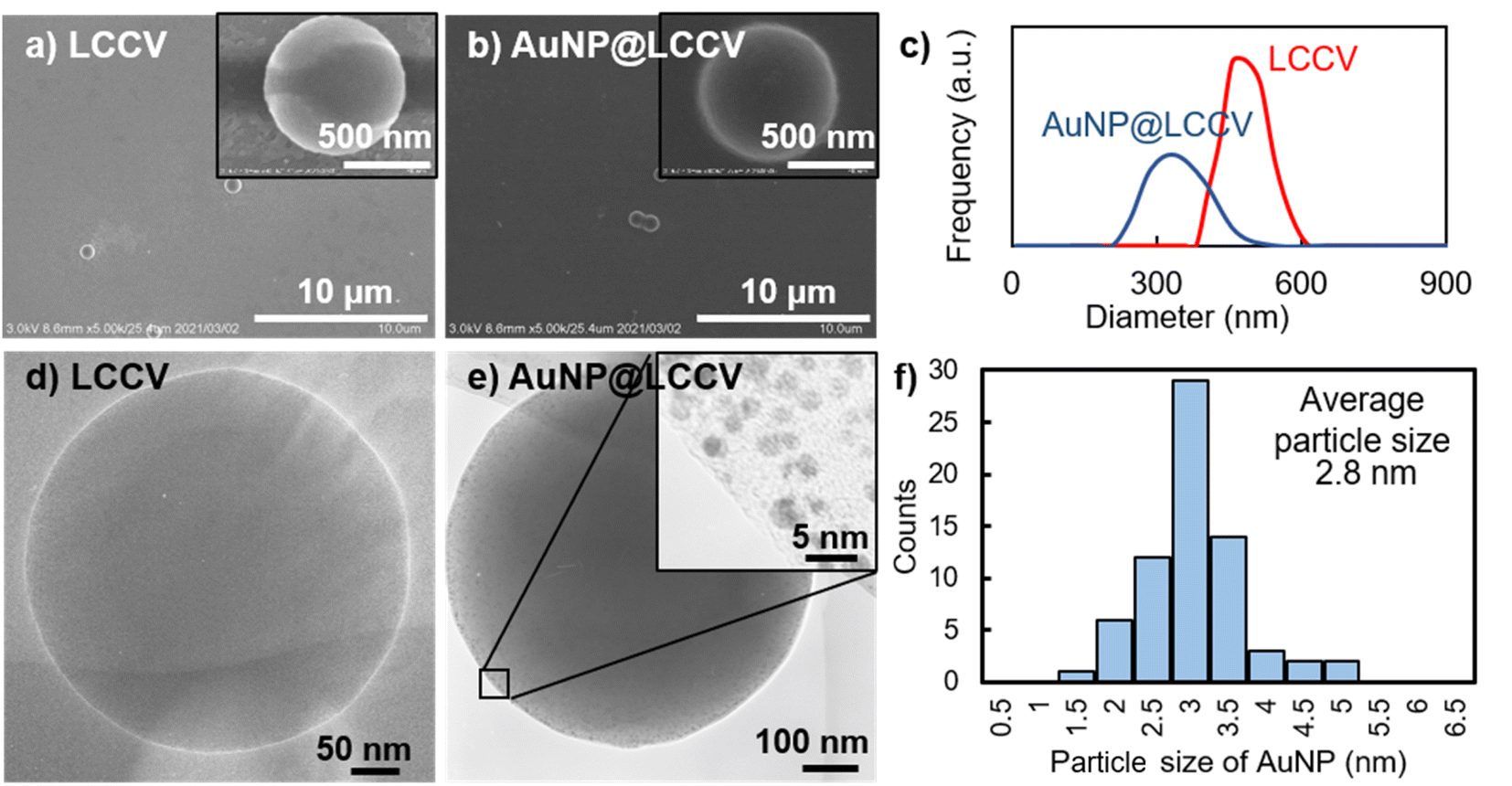 | ||
| Fig. 2 Electron microscope studies and dynamic light scattering of the vesicles (LCCV, without Au NP loading) and (AuNP@LCCV, with Au NP loading): (a) SEM and (d) TEM image of LCCV. (b) SEM and (e) TEM image of AuNP@LCCV; the inset image in (e) clearly shows that the size of the AuNPs on the vesicle is about 3 nm. (c) Dynamic light scattering (DLS) of LCCV and AuNP@LCCV. (f) Particle size distribution of AuNPs measured by counting 70 AuNPs on the vesicle in the TEM image in (e). The average particle size of the AuNPs was calculated to be 2.8 nm. SEM images visualized from multiple AuNP@LCCVs are shown in Fig. S4,† demonstrating the homogeneity of the AuNP@LCCVs. | ||
3.2 Catalysis of AuNP@LCCV in reduction of nitrophenol
It is well known that gold nanoparticles with a smaller particle size have higher catalytic activity in many organic reactions.5 Among them, the reduction reaction of 4-nitrophenol (4-NP) is the most-used model reaction to evaluate the catalytic activity of AuNPs.5,33 The gold nanoparticles in AuNP@LCCV are very small, with an average size of 2.8 nm, and are encapsulated in the unusual vesicular corona with loop-clusters. To understand the performance and/or features of these unique gold nanoparticles, we attempted to use AuNP@LCCV as a catalyst in the reduction of 4-nitrophenol (4-NP).Here, we evaluated the catalytic function of AuNP@LCCV according to the following procedure: 1.0 mL of an aqueous solution of AuNP@LCCV (Au = 0.084 μmol, 1 equiv.) was mixed with an aqueous solution with a definite concentration of 4-NP and a fresh aqueous solution of NaBH4 with different concentrations. The reaction solution was quickly transferred to a quartz cell, capped without stirring and monitored using UV-vis spectroscopy at fixed time intervals (Fig. S1†). The time-course of the absorbance of 4-NP monitored at 400 nm is shown in Fig. 3. From Fig. S1† and 3, it can be seen that the reduction depended strongly on the concentration of NaBH4. When the molar ratio of NaBH4/Au was 251/1, no reaction took place even after standing for 90 min. After changing the molar ratio to 314/1, the reaction took place completely after a 57 min blank time. When the molar ratio was increased greatly to over 2000, the blank time was significantly reduced to about 1 min. The interesting point here is that regardless of the molar ratio of NaBH4 to Au, the reaction proceeds slowly for a period (ca. 10 min) after the blank time, but after that completes suddenly with a falling-cascade-like trend within 1–5 min. The blank time before the reaction proceeds is often referred to as the induction time.5 Such an induction period is often observed in the reduction of 4-NP using metallic nanoparticles as catalysts, and there are several arguments to explain the reasons for the induction time. One of the typical arguments is that the induction time is caused by the tight molecular coverage of the AuNP surface, which hinders the restructuration of molecules on the AuNP surface.5,27,29,34,35 The other is the oxygen effect, which claims that the induction time is caused by dissolved oxygen in the reaction mixture.36–38 It has been proven that oxygen dissolved in an aqueous solution not only consumes the hydride (H−) from NaBH4, but also oxidizes the intermediate 4-nitrosophenol reduced from 4-NP. In many reported cases, however, the induction time is short (less than 5 min).11,29,36–38 In contrast, in our case, using AuNP@LCCV, the induction time is remarkably longer, ranging from 60 minutes to several minutes depending on the molar ratio of the reductant to the catalyst. This is a rare observation in the reduction of 4-NP catalyzed by metallic nanoparticles.
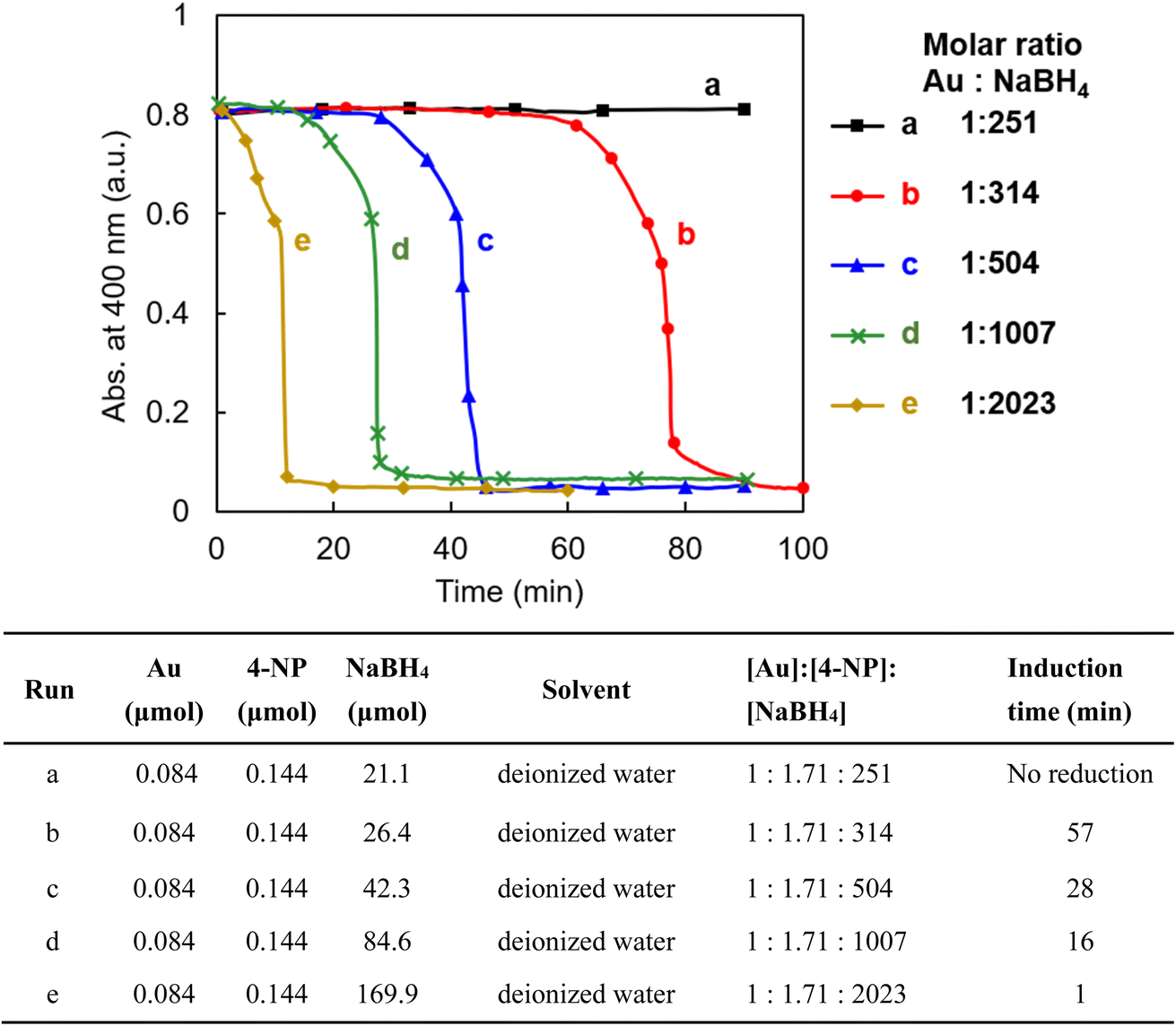 | ||
| Fig. 3 Time-course of the reduction of 4-NP at different concentrations of NaBH4, as monitored at 400 nm using UV-vis spectroscopy. The reduction was performed under the conditions indicated in the table by setting the concentration of [Au] at 0.021 μmol mL−1. | ||
To elucidate further the reason for the induction time, we performed the reduction under three different conditions, namely using O2-bubbled water, N2-bubbled water and non-bubbled water (control) as the solvent for the solutions of 4-NP and NaBH4. The results are displayed in Fig. 4. In the cases of the non-bubbled and N2-bubbled conditions, the reaction showed an induction time of 7–8 min, after which the reaction suddenly completed within about 4 min. There was no difference between the control and N2-bubbled conditions. In contrast, significantly differently, under the O2-bubbled conditions, the induction time was extended to a much-longer period of 33 min (prolonged 4 times), after which the reduction proceeded rapidly. These results revealed that the induction time is caused by the dissolved oxygen in the reaction solution; a high oxygen concentration in the solution easily inhibits the starting of the reduction. It seems that the reduction could proceed only after the consumption of the oxygen in the mixture solution. It must be noted here that although N2-bubbling could make it possible to physically remove the dioxygen dissolved in solution, this treatment could not get rid of the induction time (this will be discussed later).
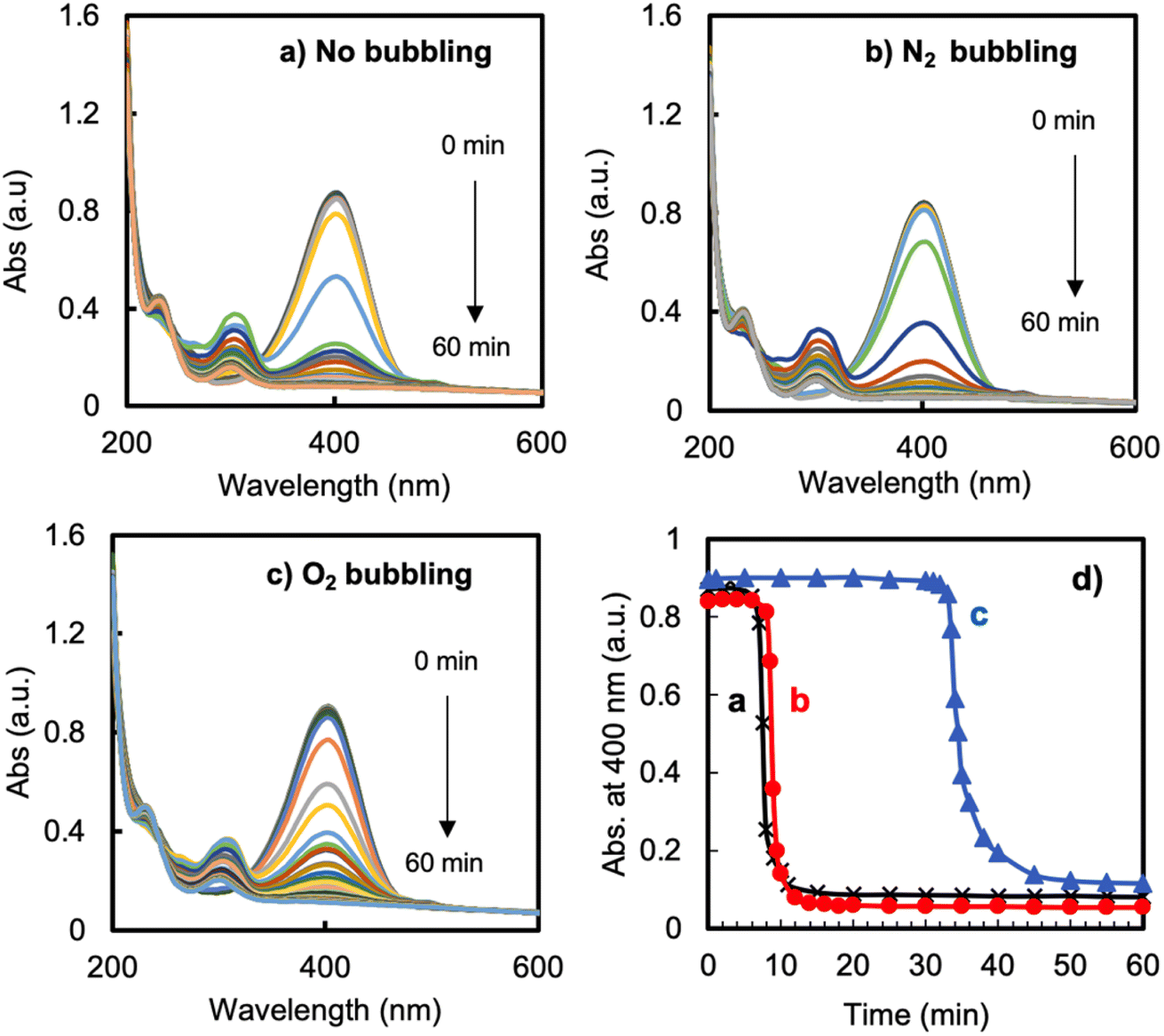 | ||
| Fig. 4 Reduction of 4-nitrophenol catalyzed by LCCV@AuNP in aqueous solution with a feed ratio of [Au]/[4-NP]/[NaBH4] = 1/1.71/1693 ([Au] = 0.021 μmol mL−1). (a–c) UV-vis spectra of the reaction recorded at fixed time intervals. (d) Plots of the absorbance at 400 nm at different reaction times from the spectra in (a–c). | ||
For comparison of the effect of the gold nanoparticle component on the catalytic performance in 4-NP reduction, we also prepared gold nanoparticles via the reaction of HAuCl4 with trisodium citrate without using any capping reagents. These gold nanoparticles (denoted as AuNP@C) had an averaged size of ca. 27 nm in DLS, strong plasmon absorbance around 540 nm in the UV-vis spectrum and a spherical shape in the SEM image (see Fig. S2†). Using the AuNP@C as a catalyst, we performed the same reduction of 4-NP under similar conditions to those in Fig. 3 while changing the molar ratio of NaBH4/Au. As can be seen clearly from Fig. 5, remarkably different to the case of AuNP@LCCV, the reaction proceeded slowly even using lower concentrations of NaBH4 (molar ratio of NaBH4/Au = 122/1) without an apparent induction time and was completed in approximately 60 min (Fig. 5a). This is accordance with the results reported by Cerimedo et al., who reported that the induction time is always negligible when using Au NP reduced by citrate as a catalyst.10 When the molar ratio of NaBH4/Au was increased to 492 and 979, the reaction was accelerated and finished in 17 and 8 min, respectively. However, there was no sudden completion of the reduction with a falling-cascade-like trend like that appearing when the reaction was catalyzed by AuNP@LCCV. From this result, we can conclude that the catalytic activity of AuNP@C is not affected by the dissolved oxygen, indicating at least that the relatively larger gold nanoparticles without a polymeric capping environment in AuNP@C have no considerable affinity toward oxygen. Therefore, once the reactants and catalyst are mixed, both hydride (H−) and 4-phenolate (4-NP−) can be adsorbed smoothly on the gold surface, and thus, the reaction between hydride (H−) and 4-phenolate (4-NP−) takes place without inhibition. This could be considered as a reason why there is no oxygen-induced induction time. Additionally, in Fig. 5, in addition to the absorption band at 400 nm, which decreased with time, an apparent absorption band appeared at 530 nm, which originated from the plasmon absorption of AuNP@C. This plasmon absorption signal did not appear when AuNP@LCCV was used as the catalyst (see Fig. 4) because of the very weak plasmon absorption of AuNP@LCCV (Fig. 1).
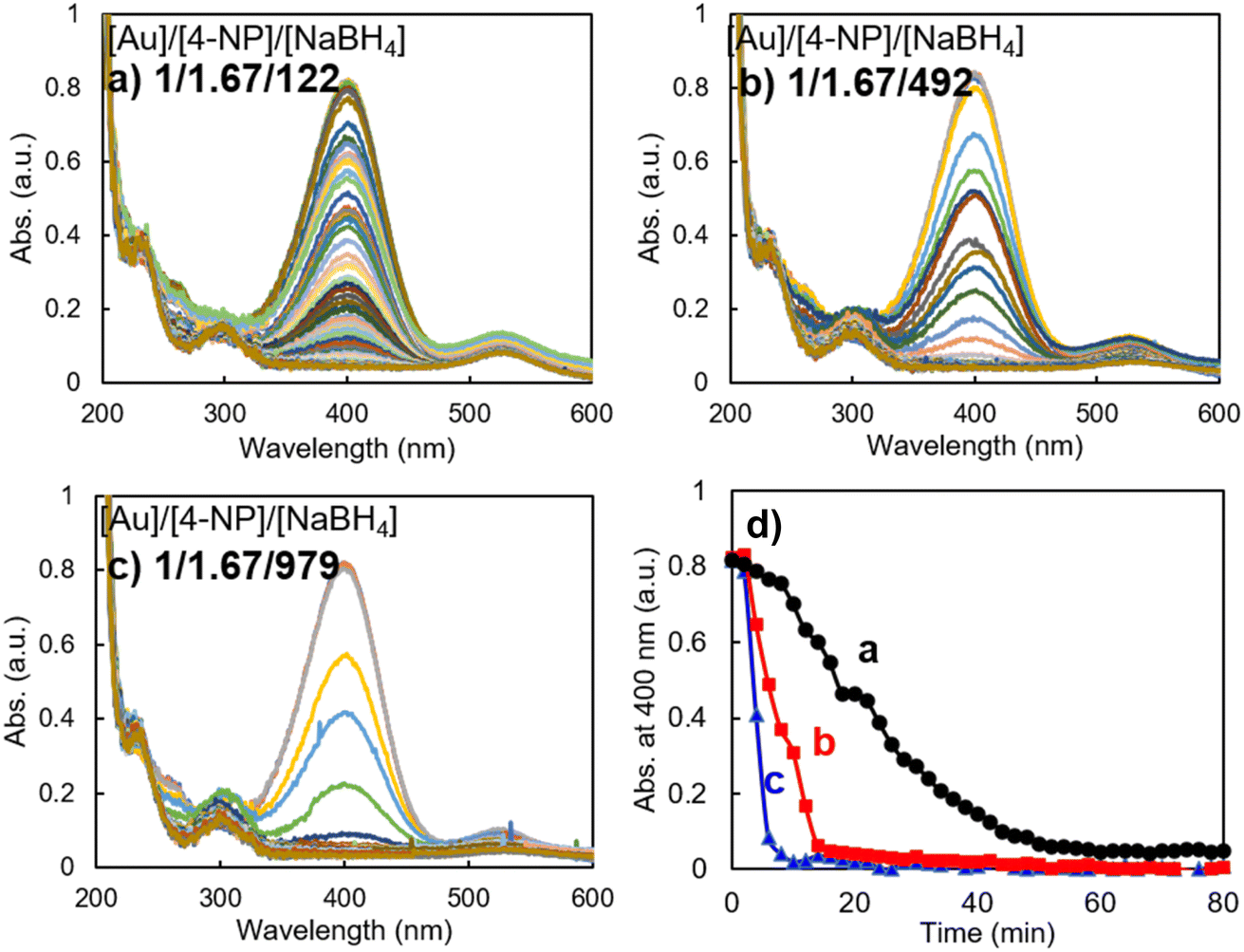 | ||
| Fig. 5 Reduction of 4-NP monitored by UV-vis absorption spectra (left), and time course (right) of the absorbance changes of 4-NP at 400 nm. Reaction conditions: the feed ratios of [Au]/[4-NP]/[NaBH4] were as follows: (a) 1/1.67/122, (b) 1/1.67/492, and (c) 1/1.67/979, with [Au NPs] fixed at 0.021 μmol mL−1. (d) Plot of the absorbance at 400 nm vs. reaction time from the spectra of (a–c). | ||
We should note here that one of the characteristic features of small gold nanoparticles is in their oxygen adsorption and oxygen activation on their surface, and thus, gold nanoparticles can be used as very useful oxidation catalysts.39–45 To understand the reason for the oxygen-induced long induction time in our AuNP@LCCV system, the interactions between gold nanoparticles and oxygen should be considered. It is well known that when 4-NP is mixed with NaBH4, the solution becomes yellow due to the formation of 4-nitrophenolate anions, whose reduction causes the yellow color to disappear. To prove the effect of oxygen on the reaction, we further followed the naked-eye-observable changes in the reaction using camera visual tracking (Fig. 6 and Movie S1†). That is, the change in the yellow state of the 4-nitrophenolate anions in the reaction solution was visualized under stirring and without stirring. We set up two systems for the visualization of the reaction solution. In one, 4.0 mL of the reaction solution was placed in a 5 mL screw vial with a cap and observed under stirring. In the other, the same reaction solution, but with different volumes of 4.0, 3.0, 2.0, 1.0 and 0.5 mL, was placed in a 5 mL screw vial with a cap, and the solutions were observed without stirring. In the latter, the decrease in the solution volume corresponds to an increase of the volume of air in contact with the surface of the reaction solution. It can be seen clearly that under stirring, the reaction solution stirred for 14 min was still yellow and remained pale yellow even after stirring for 60 min (yellow dotted box in Fig. 6). In comparison, very interestingly, under the conditions without stirring, the reaction with the same solution volume of 4.0 mL showed apparent disappearance of the yellow colour by 14 min and remained colourless thereafter, indicating that the reaction was close to complete. At the same time, however, the 2.0 mL solution showed apparent two-phase-like layers with a yellow upper layer and a colourless lower layer, while the 0.5 mL solution was still yellow. In the case of the 2.0 mL reaction solution, the appearance of the yellow-tinged upper layer indicates that, at least in the upper layer, AuNP@LCCV was in contact with more oxygen, because the air volume in this vial was twice that compared to the case of the 4.0 mL reaction solution. This causes a delay in the reduction of the fraction of the 4-nitrophenolate anions that are near the air-solution interface. In the case of the 0.5 mL reaction solution, the interface of the solution was surrounded by sufficient oxygen because the air volume (ca. 4.5 mL) was the largest in this vial, and thus reduction was heavily inhibited. The above phenomena revealed that the incorporation of air into the reaction solution via the interface easily inhibits the reduction of 4-NP. Both reaction under a large air volume without stirring and reaction with vigorous stirring would enhance the probability of the adsorption of oxygen on the small gold nanoparticles located on the stable LCCV, and the adsorbed oxygen blocks the reduction reaction. The significant air sensitivity in our AuNP@LCCV-catalyzed reduction system is probably related to the unique structure of the catalyst AuNP@LCCV. First, the hybrid vesicles of AuNP@LCCV can be easily and uniformly dispersed in aqueous media, even for over 1 year, without fusion and precipitation. Thus, it is conceivable that the hybrid vesicles of AuNP@LCCV would frequently emerge at the interface between the reaction solution and air. Secondly, the gold nanoparticles in AuNP@LCCV are very small, with an average size of 2.8 nm (i.e., larger surface area) and are uniformly distributed in the PEI loop-cluster box. These two structural characteristics endow the small gold nanoparticles floating at the aqueous solution–air interface with strong ability to adsorb dioxygen molecules. It is certain that the vigorous stirring would enhance the adsorption of oxygen onto AuNP@LCCV because of the incorporation of air under stirring, and would thus delay the disappearance of the yellow color of the reaction solution. Despite this drawback as a reduction catalyst, in some sense, the reaction solution mixed with 4-NP, NaBH4 and AuNP@LCCV could be used as an oxygen sensor.
 | ||
| Fig. 6 Time-course photographs of the reduction of 4-NP using AuNP@LCCV with or without stirring. The reduction with stirring is shown in the yellow dotted box and the reduction without stirring is shown in the blue dotted box. By reducing the volume of the reaction solution (keeping the same concentration with a defined feeding ratio of [Au]/[4-NP]/[NaBH4] = 1/5.33/1470 ([Au] = 0.21 μmol mL−1) in deionized water), the yellow color of unreduced 4-NP at the air/water interface was more clearly visible. | ||
We also investigated the catalytic activity of AuNP@LCCV on other substrates, namely, 2-nitrophenol, 2-methyl-4-nitrophenol, 2-amino-4-nitrophenol and 2-chloro-4-nitrophenol. As shown in Fig. S3,† AuNP@LCCV exhibited excellent catalytic activity in each case, reducing all these nitrophenols to aminophenols after an induction time of about 20 min.
3.3 Mechanism of the reduction of 4-NP accompanying induction time
The reduction reaction of 4-NP by NaNH4 in the presence of metallic nanoparticle catalysts is determined by the reaction between the two species hydride (H−) and 4-nitrophenolate (4-NP−) adsorbed on the surface of the metallic nanoparticles. However, there are several factors that are not well understood that practically affect the reduction process before the initiation of the surface reaction of the two species. The retardation by oxygen is the most apparent riddle in this reaction. There are several proposed mechanisms to explain the oxygen-induced induction time observed in the reduction of 4-NP in water.8–10,36–38,46,47 Compared to the early mechanistic model of the reconstruction of reactants on gold nanoparticles, later work is based on the reduction model involving the intermediate 4-nitrosophenol, because the intermediate 4-nitrosophenol can revert to 4-NP due to its easy oxidation by dissolved oxygen. Other factors such as catalyst reduction/reoxidation are also considered as causing the induction time; that is, hydride (H−) on the catalytic surface (reduced state) is oxidized by dissolved oxygen. We think that each mechanism proposed based on each experimental case has its own reliable rationale. In our catalytic system, the induction time phenomenon may also be related to oxygen consumption by the intermediate 4-nitrosophenol. However, we gather that our case would not fall perfectly into this explanation, but relates to an unexpected special event.Based on our experimental results, the oxygen affinity of AuNP@LCCV must be considered. As mentioned above, gold nanoparticles smaller than 5 nm could adsorb/activate dioxygen and thus be effectively used as oxidation catalysts. We think that the dissolved oxygen in our system prefers to interact with the Au nanoparticles on the vesicles and to occupy the surface of the Au nanoparticles to form a stable oxygen complex. As shown in Fig. 1 and S2,† the plasmon absorbance of AuNP@LCCV is significantly weaker than that of AuNP@C, although the solution color of AuNP@LCCV seems sufficiently wine-red. This weak plasmon absorption is similar to cases of gold nanoparticles treated by O2/Ar-plasma or oxygen adsorption, in which oxygen complexing weakens the plasmon absorbance of the gold nanoparticles.48,49 We predict that the small gold nanoparticles encapsulated by the PEI loop-clusters are in a state of oxygen trapping. Therefore, we propose the reaction mechanism shown in Scheme 2. In the reduction reaction of 4-NP, the hydride produced early in the process from the reductant NaBH4 is not able to complex with the Au surface, but first reacts with dioxygen adsorbed on the Au. This is the first reaction before the reduction of 4-NP starts. Once the oxygen adsorbed on the AuNPs is removed completely by H−, the gold surface could be sufficiently occupied by the reactants of hydride (H−) and nitrophenolate (NP−), and the reaction between the two species proceeds rapidly to give aminophenol (AP). In other words, the gold surface of the as-prepared hybrid of AuNP@LCCV has already been saturated by the adsorbed dioxygen. Once adsorbed on AuNP@LCCV, the dioxygen is too stable to remove using N2 bubbling, although the dissolved dioxygen molecules in solution could be replaced by nitrogen gas. Due to this, the reaction requires the use of a large excess of NaBH4, which acts as a cleaner to remove the adsorbed oxygen via the formation of water through reaction between the adsorbed oxygen and hydride. This surface cleaning process depends strongly on the concentration of NaBH4; a higher concentration rapidly removes oxygen and thus significantly shortens the induction time. In this sense, we conclude that in the reduction of NP, the catalyst AuNP@LCCV must undergo a reconstruction of the gold surface via removal of the adsorbed dioxygen. This process should be a main cause of the long induction time in our AuNP@LCCV system. Although the driving force strengthening the complexing interactions between AuNPs and dioxygen in aqueous dispersion is not clear at present, the idea that oxygen adsorbed on the gold surface causes the induction time should shed light to understand more clearly the reduction of nitrophenol catalyzed by gold nanoparticles. We expect that differently structured gold nanoparticles would adsorb oxygen more strongly or weakly on their surface, and this state would cause shorter or longer induction periods.
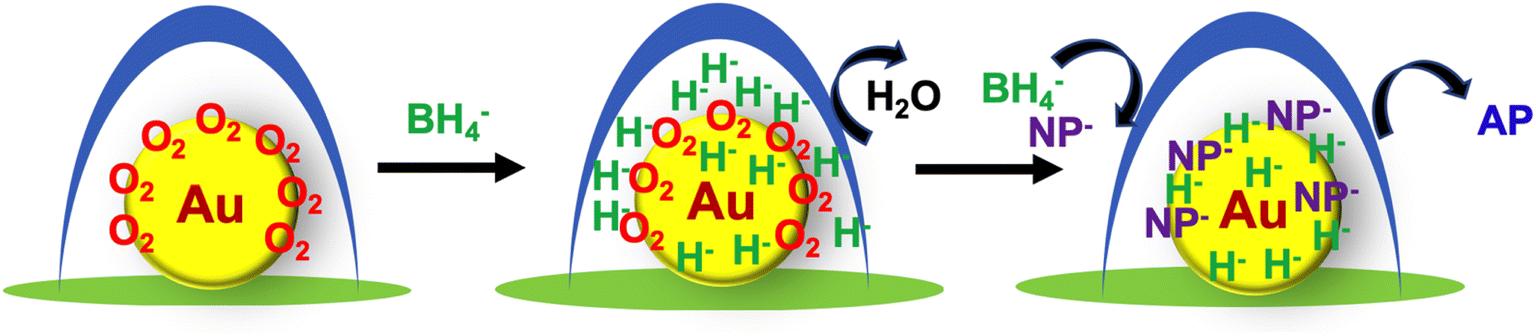 | ||
| Scheme 2 Representation of the mechanism of the oxygen-induced induction time in the reduction of nitrophenol (NP) into aminophenol (AP) catalyzed by AuNP@LCCV. | ||
3.4 Recycling the catalyst in continuous reduction
To explore the catalytic potential of AuNP@LCCV, we also performed recycling of the catalyst of AuNP@LCCV in the continuous reduction reaction of 4-NP. The initial catalytic reduction reaction of 4-NP was carried out in deionized water with a ratio of [Au]/[4-NP]/[NaBH4] = 1/1.71/500 (stage-a in Fig. 7). After the complete consumption of 4-NP in stage-a, another 1.71 eq. of 4-NP was added to the reaction mixture (stage-b in Fig. 7). Interestingly, the newly added 4-NP was rapidly reduced without an induction time. Thus, 4-NP (1.71 eq.) was again added to the mixture solution, but no reduction took place in this stage, even after a long time had passed (stage-c in Fig. 7). Afterwards, a small amount of NaBH4 (30.5 eq. to Au) was added to the mixture and monitored continuously for 1 hour. However, the reduction of 4-NP did not occur (stage-d in Fig. 7). When NaBH4 was added again (122 eq. relative to Au), the absorption at 400 nm began to decrease and disappeared after about 20 min (stage-e in Fig. 7). This demonstrates not only that the catalyst AuNP@LCCV is recyclable in the reduction of 4-NP, but also that the induction time reappears during the continuous reduction reaction procedure due to the incorporation of oxygen.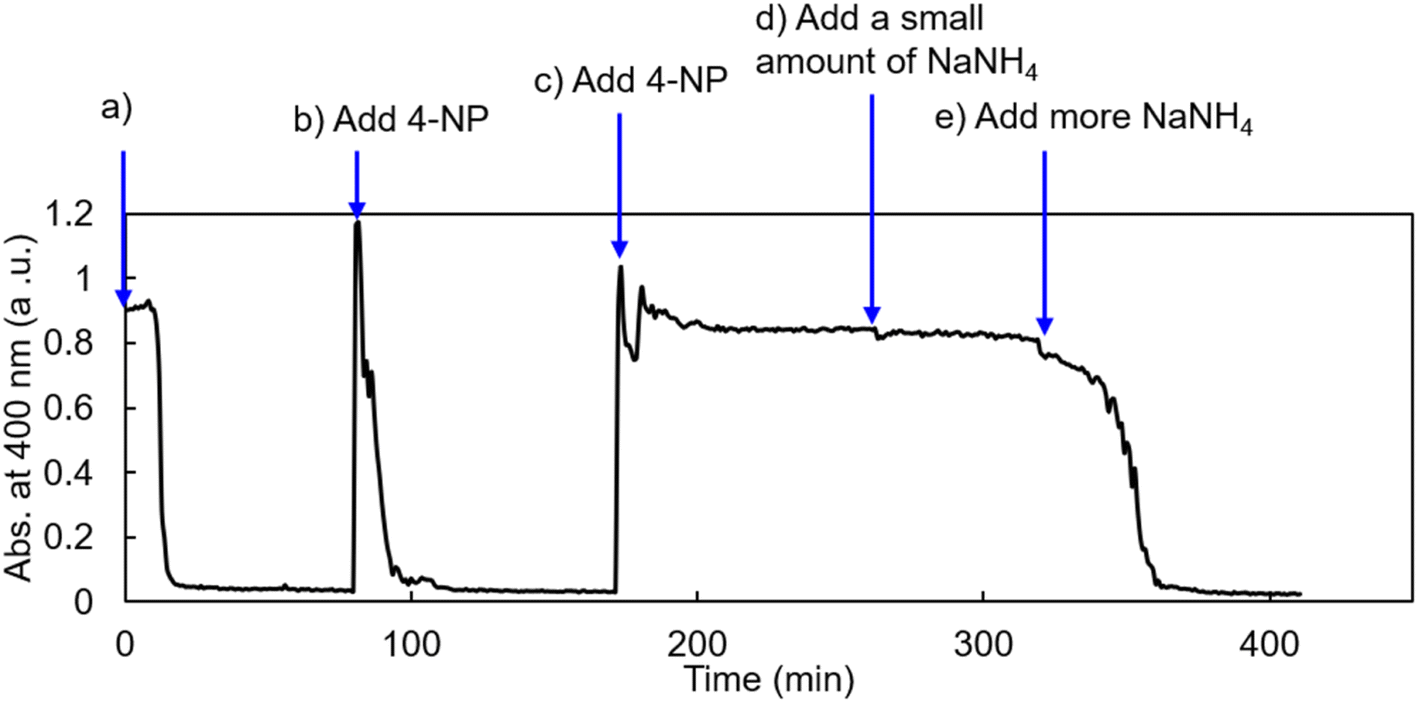 | ||
| Fig. 7 Recycling of the catalyst AuNP@LCCV in the continuous reduction reaction of 4-NP monitored using UV-vis at 400 nm. (stage-a): Reduction reaction of 4-NP using AuNP as the catalyst carried out in an aqueous mixture containing AuNP@LCCV, 4-NP and NaBH4 in a ratio of [Au]/[4-NP]/[NaBH4] = 1/1.71/500; (stage-b): an additional 1.71 eq. of 4-NP was added; (stage-c): another 1.71 eq. of 4-NP was added; (stage-d): 30.5 eq. of NaBH4 was added to the mixture; (stage-e): an additional 122 eq. of NaBH4 was added to the mixture. | ||
To investigate the state of AuNP@LCCV after recycling, the above recycling reaction solution at stage-e was dialyzed using deionized water to remove the reactants, and the remaining solution containing the catalyst AuNP@LCCV was subjected to SEM and TEM measurements. It can be seen from the SEM image (Fig. 8b) that the spherical vesicles remained without corruption despite the appearance of some protrusions in the vesicular surface. In the lower-magnification TEM image (Fig. 8c), we can see some larger dark spots that did not appear in the intact vesicle (see Fig. 2e). However, in the higher-magnification TEM image (Fig. 8d), it can still be observed that a large amount of small AuNPs with a size of about 3 nm were evenly distributed on the vesicle, although there were some fused gold nanoparticles. These results strongly indicate the excellent encapsulating performance of LCCV for AuNP.
 | ||
| Fig. 8 SEM images of AuNP@LCCV as intact (a) and recovered (b) from the recycling reaction solution at stage-e. TEM images (c and d) of AuNP@LCCV recovered from the recycling reaction solution at stage-e. | ||
4. Conclusion
In this work, a unique hybrid polymeric vesicle (AuNP@LCCV) composed of AuNPs and a polyethyleneimine (PEI) loop-cluster covered vesicle (LCCV) was successfully prepared via simple in situ reduction of AuCl3− trapped in LCCV. The loop-cluster coronal structure of PEI effectively controlled the growth of the AuNPs to generate AuNPs with an average radius of 2.8 nm in the corona of the vesicles. This hybrid vesicle AuNP@LCCV is extraordinarily stable under dried and vacuum conditions, retaining its spherical vesicular shape whose outer coronal surface was occupied by a large number of small gold nanoparticles. These extremely small AuNPs exhibited excellent catalytic activity in the reduction of NP using AuNP@LCCV as a catalyst, although accompanied by an unusually long induction period before the reduction of NP started. The induction time depended strongly upon the concentration of NaBH4, with a large excess of NaBH4 remarkably shortening the induction time. Based on the air-dependence results, we expected that the long induction time originated from the dioxygen adsorbed on the gold nanoparticles. Once the adsorbed dioxygen was removed by the hydride provided from NaBH4, the reduction of NP completed suddenly. In addition, AuNP@LCCV is reusable as catalyst in the procedure with the continuous addition of NP and NaBH4.This work presents a new strategy for the preparation of metal–polymer hybrid vesicles with an extremely small AuNP particle size and new inspiration for the coronal engineering of polymeric vesicles. We expect that, potentially, AuNP@LCCV could act as an oxygen detector/sensor by formulating the reactants nitrophenol and sodium borohydride.
Conflicts of interest
There are no conflicts to declare.References
- Y. Huang, P. Huang and J. Lin, Small Methods, 2019, 3, 1800394 CrossRef.
- L. Q. Zheng, X. D. Yu, J. J. Xu and H. Y. Chen, Chem. Commun., 2015, 51, 1050–1053 RSC.
- Z. D. Pozun, S. E. Rodenbusch, E. Keller, K. Tran, W. Tang, K. J. Stevenson and G. Henkelman, J. Phys. Chem. C, 2013, 117, 7598–7604 CrossRef CAS PubMed.
- Y. Tani and T. Kaneta, R. Soc. Open Sci., 2019, 6, 190293 CrossRef CAS PubMed.
- Y. S. Seo, E. Y. Ahn, J. Park, T. Y. Kim, J. E. Hong, K. Kim, Y. Park and Y. Park, Nanoscale Res. Lett., 2017, 12, 7 CrossRef PubMed.
- X. Fang, J. Li, B. Ren, Y. Huang, D. Wang, Z. Liao, Q. Li, L. Wang and D. D. Dionysiou, J. Membr. Sci., 2019, 579, 190–198 CrossRef CAS.
- Y. Dai, T. Ren, Y. Wang and X. Zhang, Gold Bull., 2017, 51, 21–26 CrossRef.
- R. Ongaratto, N. Conte, C. R. Montes D'Oca, R. C. Brinkerhoff, C. P. Ruas, M. A. Gelesky and M. G. Montes D'Oca, New J. Chem., 2019, 43, 295–303 RSC.
- S. Xu, Y. Nie, L. Jiang, J. Wang, G. Xu, W. Wang and X. Luo, Anal. Chem., 2018, 90, 4039–4045 CrossRef CAS PubMed.
- M. S. Álvarez Cerimedo, L. G. Baronio, C. E. Hoppe and M. A. Ayude, ChemistrySelect, 2019, 4, 608–616 CrossRef.
- Y. Zhu, L. Fan, B. Yang and J. Du, ACS Nano, 2014, 8, 5022–5031 CrossRef CAS PubMed.
- Y. Dai and X. Zhang, Macromol. Mater. Eng., 2018, 303, 1800105 CrossRef.
- X. Chen, D. Zhao, Y. An, Y. Zhang, J. Cheng, B. Wang and L. Shi, J. Colloid Interface Sci., 2008, 322, 414–420 CrossRef CAS PubMed.
- Y. Dai, T. Ren, Y. Wang and X. Zhang, Gold Bull., 2017, 50, 123–129 CrossRef CAS.
- Y. Dai, P. Yu, X. Zhang and R. Zhuo, J. Catal., 2016, 337, 65–71 CrossRef CAS.
- F. Shukla, M. Das and S. Thakore, J. Mol. Liq., 2021, 336, 116217 CrossRef CAS.
- M. Chenouf, C. Megías-Sayago, F. Ammari, S. Ivanova, M. Á. Centeno and J. A. Odriozola, C. R. Chim., 2019, 22, 621–627 CrossRef CAS; S. Varshney, R. Bar-Ziv and T. Zidki, ChemCatChem, 2020, 12, 4680–4688 CrossRef.
- Y.-C. Chang and D.-H. Chen, J. Hazard. Mater., 2009, 165, 664–669 CrossRef CAS PubMed.
- A. Iben Ayad, D. Luart, A. Ould Dris and E. Guenin, Nanomaterials, 2020, 10, 1169 CrossRef PubMed.
- F.-H. Lin and R.-A. Doong, Appl. Catal., A, 2014, 486, 32–41 CrossRef CAS.
- D. W. Cho, S. Kim, Y. F. Tsang and H. Song, Environ. Geochem. Health, 2019, 41, 1729–1737 CrossRef CAS PubMed.
- J. Qian, A. Yuan, C. Yao, J. Liu, B. Li, F. Xi and X. Dong, ChemCatChem, 2018, 10, 4747–4754 CrossRef CAS.
- J. Chen, R. J. Dai, B. Tong, S. Y. Xiao and W. Meng, Chin. Chem. Lett., 2007, 18, 10–12 CrossRef CAS.
- W.-L. Wang and R.-H. Jin, RSC Adv., 2020, 10, 13260–13266 RSC.
- W.-L. Wang and R.-H. Jin, Polymer, 2021, 212, 132289 Search PubMed.
- A. K. Sharma, S. Shankhwar and M. S. Gaur, J. Exp. Nanosci., 2013, 9, 892–905 CrossRef.
- B. Gao, F. An and K. Liu, Appl. Surf. Sci., 2006, 253, 1946–1952 CrossRef CAS.
- R. Qu, J. Liu, C. Sun, Y. Zhang, C. Ji and P. Yin, J. Chem. Eng. Data, 2010, 55, 4650–4659 CrossRef CAS.
- W. Zhao, W. Jia, M. Sun, X. Liu, Q. Zhang, C. Zong, J. Qu and H. Gai, Sens. Actuators, B, 2016, 223, 411–416 CrossRef CAS.
- J. Chen, G. Qin, Q. Chen, J. Yu, S. Li, F. Cao, B. Yang and Y. Ren, J. Mater. Chem. C, 2015, 3, 4933–4944 RSC.
- N. Li, M. Echeverria, S. Moya, J. Ruiz and D. Astruc, Inorg. Chem., 2014, 53, 6954–6961 CrossRef CAS PubMed.
- M. Li and G. Chen, Nanoscale, 2013, 5, 11919–11927 RSC.
- J. Han, M. Wang, R. Chen, N. Han and R. Guo, Chem. Commun., 2014, 50, 8295–8298 RSC.
- R. Ciganda, N. Li, C. Deraedt, S. Gatard, P. Zhao, L. Salmon, R. Hernandez, J. Ruiz and D. Astruc, Chem. Commun., 2014, 50, 10126–10129 RSC.
- S. Wunder, Y. Lu, M. Albrecht and M. Ballauff, ACS Catal., 2011, 1, 908–916 CrossRef CAS.
- J. Strachan, C. Barnett, A. F. Masters and T. Maschmeyer, ACS Catal., 2020, 10, 5516–5521 CrossRef CAS.
- E. Menumerov, R. A. Hughes and S. Neretina, Nano Lett., 2016, 16, 7791–7797 CrossRef CAS PubMed.
- J. G. You, D. Y. Jin, W. B. Tseng, W. L. Tseng and P. C. Lin, ChemCatChem, 2020, 12, 4558–4567 CrossRef CAS.
- B. E. Salisbury, W. T. Wallace and R. L. Whetten, Chem. Phys., 2000, 262, 131–141 CrossRef CAS.
- A. Franceschetti, S. J. Pennycook and S. T. Pantelides, Chem. Phys. Lett., 2003, 374, 471–475 CrossRef CAS.
- A. Staykov, T. Nishimi, K. Yoshizawa and T. Ishihara, J. Phys. Chem. C, 2012, 116, 15992–16000 CrossRef CAS.
- B.-T. Teng, J.-J. Lang, X.-D. Wen, C. Zhang, M. Fan and H. G. Harris, J. Phys. Chem. C, 2013, 117, 18986–18993 CrossRef CAS.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- A. P. Woodham, G. Meijer and A. Fielicke, Angew. Chem., Int. Ed., 2012, 51, 4444–4447 CrossRef CAS PubMed.
- S. Mitiche, J.-F. Audibert, S. Marguet, B. Palpant and R. B. Pansu, J. Photochem. Photobiol., A, 2021, 410, 113170 CrossRef CAS.
- P. Zhao, X. Feng, D. Huang, G. Yang and D. Astruc, Coord. Chem. Rev., 2015, 287, 114–136 CrossRef CAS.
- A. Buonerba and A. Grassi, Catalysts, 2021, 11, 714 CrossRef CAS.
- X. Deng, Y. Ding, X. Wang, X. Jia, S. Zhang and X. Li, Nanomaterials, 2021, 12, 106 CrossRef PubMed.
- Y. Borensztein, L. Delannoy, R. G. Barrera and C. Louis, Eur. Phys. J. D, 2011, 63, 235–240 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00893a |
| ‡ These two authors W. W. and A. K. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |