
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Superior cyclability of high surface area vanadium nitride in salt electrolytes†
James
Kasten
a,
Cheng-Che
Hsiao
a,
Denis
Johnson
 a and
Abdoulaye
Djire
*ab
a and
Abdoulaye
Djire
*ab
aArtie McFerrin Department of Chemical Engineering, Texas A&M University, College Station, TX 77843, USA
bDepartment of Materials Science & Engineering, Texas A&M University, College Station, TX 77843, USA. E-mail: adjire@tamu.edu
First published on 9th June 2023
Abstract
High surface area vanadium nitrides (VNs) have been extensively studied as materials for aqueous supercapacitors due to the high initial capacitance in alkaline media at low scan rates. However, low capacitance retention and safety limit their implementation. The use of neutral aqueous salt solutions has the potential to mitigate both of these concerns, but is limited in analysis. Hence, we report on the synthesis and characterization of high surface area VN as a supercapacitor material in a wide variety of aqueous chlorides and sulfates using Mg2+, Ca2+, Na+, K+, and Li+ ions. We observe the following trend in the salt electrolytes: Mg2+ > Li+ > K+ > Na+ > Ca2+. Mg2+ systems provide the best performance at higher scan rates with areal capacitances of 294 μF cm−2 in 1 M MgSO4 over a 1.35 V operating window at 2000 mV s−1. Furthermore, VN in 1 M MgSO4 maintained a 36% capacitance retention from 2 to 2000 mV s−1 compared to 7% in 1 M KOH. Capacitance in 1 M MgSO4 and 1 M MgCl2 increased to 121% and 110% of their original values after 500 cycles and maintained capacitances of 589 and 508 μF cm−2 at 50 mV s−1 after 1000 cycles, respectively. In contrast, in 1 M KOH the capacitance decreases to 37% of its original value, reaching only 29 F g−1 at 50 mV s−1 after 1000 cycles. The superior performance of the Mg system is attributed to a reversible surface 2 e− transfer pseudocapacitive mechanism between Mg2+ and VNxOy. These findings can be used to further the field of aqueous supercapacitors to build safer and more stable energy storage systems that can charge quicker compared to KOH systems.
Introduction
Supercapacitors are electrochemical energy storage devices that have the potential to replace and/or complement batteries for electronics and large-scale energy applications, due to their high power densities (higher than those of batteries) and moderate energy densities.1–10 In the search to find cost-effective and efficient materials for supercapacitor use, a wide range of composite materials, including polymers,11–15 metal oxides16–22 and sulfides,23–27 and higher surface area transition-metal carbides28–33 and nitrides,34–37 are frequently investigated due to their pseudocapacitive charge storage mechanisms involving fast and reversible faradaic redox reactions which contribute to substantially larger capacitances compared to double layer capacitors. Among this material class, vanadium nitride (VN) has been thoroughly investigated and lauded for achieving high capacitances of up to 1340 F g−1 in alkaline environments.38–40 This high capacitance is attributed to a redox reaction occurring at the oxynitride (VNxOy) surface with hydroxy ions as verified by in situ analysis of the pseudocapacitive mechanism.39,41However, this impressive capacitance attributed to VN in alkaline systems is exclusively reported at low scan rates of 2 mV s−1 due to the significant drop in performance as scan rates increase based on electrolyte concentration. Few literature studies have reported capacitance values beyond 300 mV s−1 with capacitance retention dropping to as low as 20% from 2 to 100 mV s−1.38,42–44 Moreover, at KOH concentrations beyond 1 M, VN exhibits poor voltametric cyclability leading to steep drops in capacitance retention after only a few hundred cycles.38,45 Finally, there is a severe dearth of studies reporting on the performance of high surface area VN in non-alkaline, specifically pH-neutral, electrolytes such as aqueous salts.38,46–48 Given the highly corrosive nature of high concentration (∼6 M) KOH used in standard electrochemical setups for VN supercapacitors, there is an additional safety concern regarding the highly cathodic potentials being reached during the charging process leading to corrosion and additional maintenance.42,49,50 Considering these gaps in KOH systems against a desire to construct supercapacitors with high cyclability and performance at fast charging rates, the practical shortcomings of KOH as a superlative electrolyte for VN supercapacitors become more evident. This serves as an exigence to explore VN in other electrolyte systems which may prove to be more preferential for charging and stability conditions which KOH cannot currently fulfill.
Herein, we report the synthesis and characterization of high surface area VN as a supercapacitor material in a wide variety of aqueous salt electrolytes as well as 1 M and 0.1 M KOH for comparison. The high surface area VN is synthesized via a standardized temperature-programmed reduction and nitridation of a V2O5 precursor in an ammonia (NH3) atmosphere and then passivated at room temperature with a surface oxide layer.51 X-ray diffraction (XRD) analysis is performed on the bulk to confirm the successful reduction and nitridation of the precursor oxide. N2 physisorption is used to analyze and quantify pore size distribution and physical surface area. Raman, X-Ray Photoeletron (XPS), and Fourier-transform infrared (FTIR) spectra are recorded to confirm the successful passivation of the VN by an oxide layer. Electrochemical performance was assessed using cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) of VN in a series of chloride (Cl−) and sulfate (SO42−) solutions containing Mg2+, Ca2+, Li+, Na+, and K+ cations in addition to 1 M and 0.1 M KOH for comparison. Aqueous salt systems exhibited similar voltage windows to the KOH systems indicating that they can be used in place of the corrosive alkaline media. Electrochemical performance was determined by evaluating capacitance in each electrolyte against the scan rate and number of cycles. It was found that MgSO4 at high scan rates (2000 mV s−1) outperformed 1 M KOH indicating higher capacitance for fast charge technologies. It was also found that MgSO4 based supercapacitors provide significantly greater stability when compared to the KOH systems. Finally, these data were used to hypothesize a novel pseudocapacitive mechanism of high surface area VN in Mg2+ systems.
Experimental methods
Material synthesis
Vanadium nitride was synthesized by reducing the precursor V2O5 (Sigma-Aldrich) by a temperature-programmed reaction (TPR) synthesis at 750 °C in an NH3 atmosphere. In brief, the oxide precursor was first ground and sieved to keep the particle size below 37 μm then placed on a quartz boat in a quartz tube reactor. The reactor was ramped up to 750 °C and kept at that temperature for 1 hour. After synthesis, the material was cooled to room temperature followed by flowing a mixture of 1% O2/He (Airgas) to intentionally passivate the material, which will prevent the bulk material from further oxidation when exposed to air.Physical characterization
The bulk crystalline structure of the material was characterized by X-ray diffraction (XRD) using a Rigaku Miniflex diffractometer. XRD was conducted over a 2θ range of 3° to 90° at a scan rate of 2.0° min−1. Surface area and pore size distribution were determined by N2-physisorption (Quantachrome Autosorb-iQ) with the Brunauer–Emmett–Teller (BET) method and Barrett–Joyner–Halenda (BJH) method, respectively. The material was degassed in vacuum at 350 °C for 8 hours before the measurement. Raman spectroscopy was carried out using a Renishaw inVia Qontor with a 532 nm laser, 1800 lines per mm grating, and a 50× long objective lens. Fourier Transform Infrared spectroscopy (FTIR) was conducted on a Bruker INVENIO R with a diamond ATR module installed.Electrode preparation
Electrodes were prepared via a slurry method containing 85% VN, 10% carbon black (Super P®, Alfa Aesar) and 5% polyvinyldifluoride (PVDF) in N-methyl-2-pyrrolidone (NMP). Additional NMP was added to the mixture until a slurry consistency was achieved. The slurries were then manually painted onto 18 mm diameter conductive carbon paper substrates (5.8 mΩ cm−1, MSE Supplies) and dried in a vacuum oven for 8 hours at 80 °C. Electrode mass was obtained by subtracting the substrate mass from the total mass after drying and then multiplying by 85%. Approximately 3 mg of working material (VN) was loaded to each electrode.Electrochemical setup
Electrochemical characterization was carried out in a three-electrode setup (EL-Cell PAT Series, Aqueous Core) using activated carbon on a stainless steel pseudo-reference electrode and a carbon cloth counter electrode (1000 m2 g−1, MSE Supplies) (Fig. S1, ESI†). Titanium foil acted as a single-use current collector for both the working and counter electrodes. The working and counter electrodes were separated using two porous separators (21.6 mm × 0.26 mm each) saturated with approximately 400 μL of the electrolyte. The working electrode face and counter electrode were also soaked in the electrolyte overnight prior to assembly.Electrochemical measurements
Cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and potentiostatic electrochemical impedance spectroscopy (EIS) measurements were taken in each electrolyte environment. Each experiment used a fresh electrolyte and fresh working, reference, and counter electrodes. First, open circuit potential (OCP) was recorded until a stable voltage was reached. Next, EIS was conducted at OCP using a frequency range from 200 kHz to 10 mHz at an amplitude of 10 mV. The working voltage window in each electrolyte was then determined using CV by expanding the voltage window at increments of 100 mV at a scan rate of 50 mV s−1 until H2 and O2 onset was reached indicated by a sharp increase in current magnitude; the working window was cut to just before the onset of these reactions (Fig. S2, ESI†). To begin characterizing the material, CV scans were performed at variable scan rates from 2 to 2000 mV s−1 (Fig. S3, ESI†) followed by a 1000 cycle stability test at 50 mV s−1. Next, to determine the electrochemical surface area (ECSA), CV scans were performed within a 150 mV window of the operating voltage window wherein no redox activity occurred at variable scan rates from 2 to 200 mV s−1. Finally, GCD measurements were taken at varying current densities from 0.1 to 50 mA cm−2.Capacitance calculation
Gravimetric specific capacitance (F g−1) values were calculated using:where Vcathodic (V) and Vanodic (V) represent cathodic and anodic potential limits, respectively, i (A) represents the current, m (g) represents the electrode mass, and ν (mV s−1) represents the scan rate.
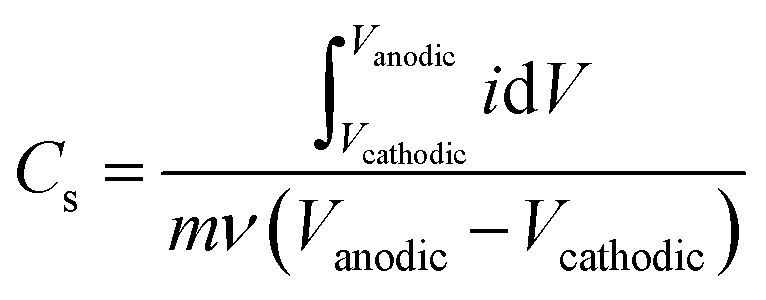
From Cs, areal specific capacitance (μF cm−2) values were calculated using
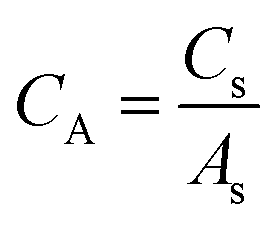
Electrochemical surface area (ECSA) calculation
Electrochemical double layer capacitance (CDL) was measured to determine the difference of ECSA in different electrolytes. Within the operating voltage window for each electrolyte, a 150 mV non-faradaic region was chosen and CVs were run at different scan rates from 2 mV s−1 to 200 mV s−1 to obtain the corresponding current. Here, the specific double layer capacitance (CSDL) was estimated to be 50 μF cm−2 based on the maximum theoretical specific charge of 50 μC cm−2 stored by a double layer capacitor operating under a 1 V operating voltage as reported previously in the literature.52,53The electrochemical surface areas (ECSAs) of VN in different electrolytes were estimated using the relationship given below.
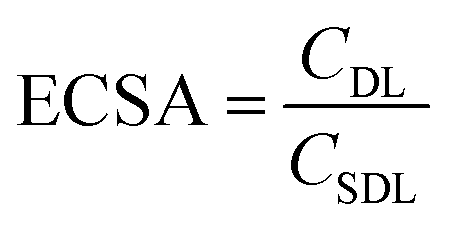
Results and discussion
Crystalline VN powder was synthesized by the reduction of an oxide precursor V2O5 through a temperature-programmed reaction at 750 °C in an NH3 atmosphere. During the reaction, the initial oxide powder changes color from a light orange characteristic of the oxide to black which visually indicates the successful reduction to VN. Following the reaction, the synthesized VN was then cooled to room temperature and passivated with an oxide layer. While it has been shown that passivation of the VN material does affect the electrochemical performance,54 the surface was oxidized to better mimic the situations that would be practically utilized for device construction. XRD spectra of both V2O5 and the passivated VN are provided in Fig. 1a. The lack of oxide peaks in the VN spectrum (red) which are present in that of V2O5 (black) strongly suggests the successful reduction of the bulk material. Moreover, the emergent peaks in the spectrum of VN belong to crystallographic planes characteristic of the material as reported in previous studies.55 The surface morphology was investigated via scanning electron microscopy (SEM) (Fig. S4, ESI†). From the SEM images, the porous nature of the material and the surface passivation layer can be clearly seen. Pore size distribution obtained from N2 physisorption is shown in Fig. 1b. The average pore size determined by the BJH method was 9.6 nm. The physical surface area calculated by the BET method was 32 m2 g−1, characteristic of high surface area VN.56 The adsorption isotherm and the BET linear regression are reported (Fig. S5, ESI†). Raman spectroscopy was then performed on VN and V2O5 to characterize the material surface, with both materials producing identical Raman spectra (Fig. 1c). The matching profile of the spectra, combined with the strict surface analysis of Raman spectroscopy, confirms the presence of a surface oxide layer on the material.57 These peaks are detected at laser powers as high as 50% indicating that a thick passivation layer was created over the material's surface. Nonetheless, the XRD spectrum of VN proves that the oxide is present on the surface only and does not extend into the bulk of the material. FTIR spectra for both V2O5 and VN were obtained to characterize the oxide bands present in the passivation layer (Fig. 1d). The transition from the bulk oxide to the surface oxide is easily traced through the disappearance of the V–O–V and V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peaks (615, 827, and 1020 cm−1).58 Upon surface reduction via amination, the peaks corresponding to the bulk oxide fade drastically in intensity, leaving minimal transmittance in the VN material. By combining the bulk characterization results from XRD spectra, surface characterization results from Raman and FTIR spectra, and the surface area and pore size distribution from N2 physisorption, there is strong evidence that high surface area VN was successfully synthesized and passivated.
O peaks (615, 827, and 1020 cm−1).58 Upon surface reduction via amination, the peaks corresponding to the bulk oxide fade drastically in intensity, leaving minimal transmittance in the VN material. By combining the bulk characterization results from XRD spectra, surface characterization results from Raman and FTIR spectra, and the surface area and pore size distribution from N2 physisorption, there is strong evidence that high surface area VN was successfully synthesized and passivated.
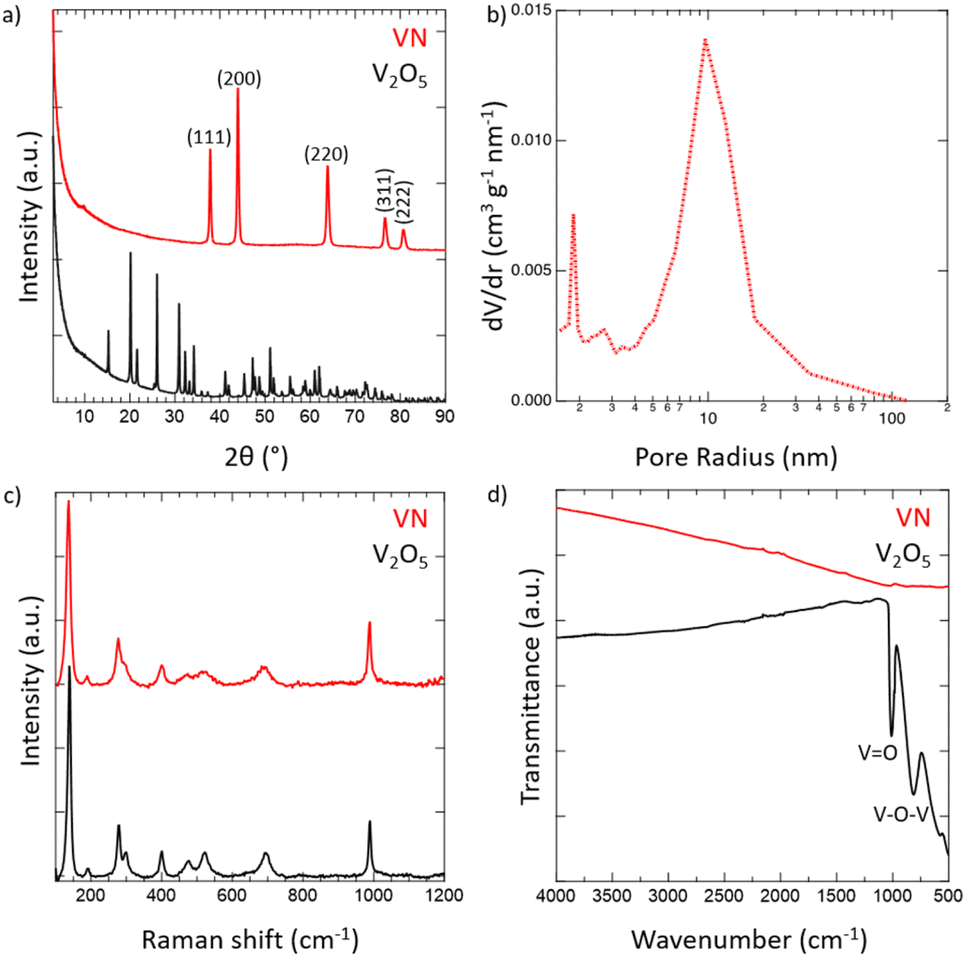 | ||
| Fig. 1 Physical characterization of high surface area passivated VN powder using (a) XRD for bulk characterization, (b) N2-physisorption for pore size distribution, and (c) Raman and (d) FTIR spectroscopies for surface characterization. | ||
To further confirm the oxide being limited to the surface of the material, X-ray photoelectron spectroscopy (XPS) was performed (Fig. 2). Taking a look at the V 2p spectrum, the peaks at 517.13 eV and 524.46 eV correspond to the 2p3/2 and 2p1/2 of the V–O bonding in V2O5,59–61 which is in accordance with our Raman spectrum showing the passivated layer. Additionally, the peaks at 515.53 eV and 522.86 eV correspond to the 2p3/2 and 2p1/2 of the VNO bonding from VNxOy,55,62 further indicating the presence of the passivated layer. The O 1s spectrum shows corroboration, where the peak at 530.18 eV corresponds to V2O5 and the remaining peaks, while unidentified, are likely due to the slight surface oxidation of VN,63 which is in accordance with the VNxOy structure that is present. Finally, the peaks at 514.23 eV and 521.83 eV correspond to the 2p3/2 and 2p1/2 of the VN.55,63 This is also in accordance with the peak at 397.17 eV in the N 1s spectrum, which corresponds to the metal–nitride bonding, in this case VN. In contrast to the Raman spectrum, the presence of the VN structure in the surface from XPS is considerable, which can be attributed to the difference in the absorptivity of the material towards visible light and X-rays.
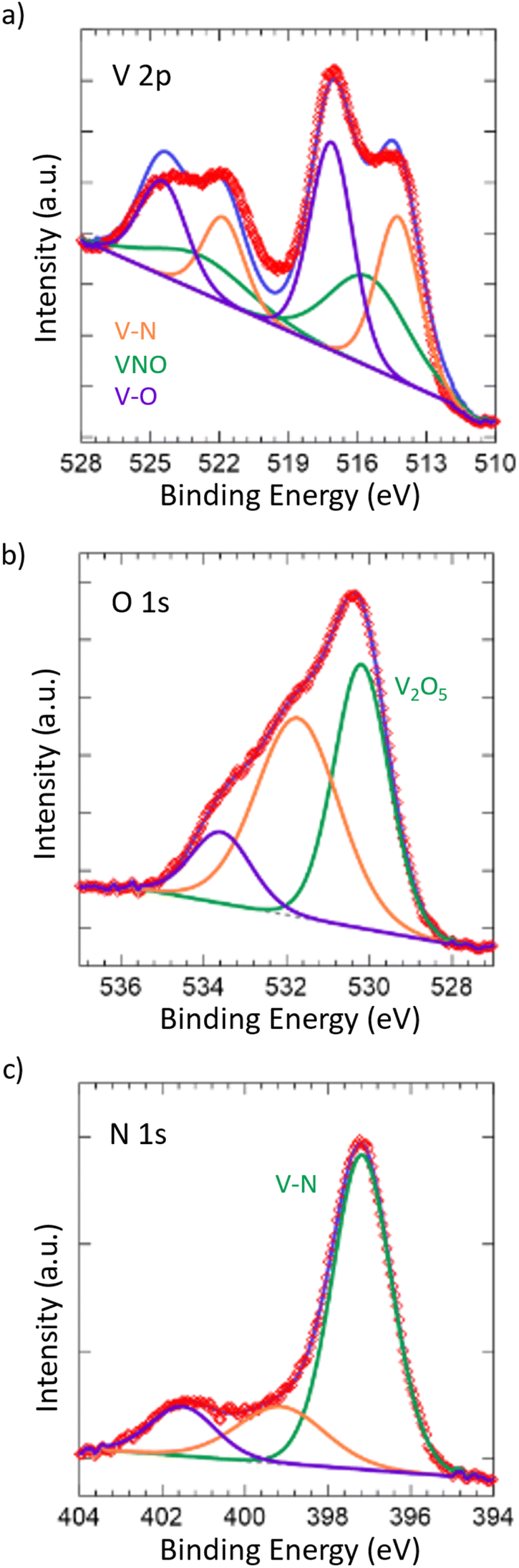 | ||
| Fig. 2 XPS spectra of (a) V 2p, (b) O 1s, and (c) N 1s for the synthesized VN. All spectra were plotted with respect to reference values. The degree of surface oxidation and amount of pristine vanadium nitride structure were tracked according to the V 2p, O 1s, and N 1s spectra. | ||
After confirming the successful synthesis of VN, its performance as a supercapacitor electrode was assessed first in aqueous 1 M and 0.1 M KOH and then in multiple aqueous salts, specifically chlorides and sulfates of varying cations (Mg2+, Ca2+, Li+, K+, Na+). For each salt, a 1 M solution was prepared with the exceptions of K2SO4, CaSO4, and CaCl2 which have solubilities below 1 M at room temperature. As such, solutions of 0.5 M K2SO4, 0.02 M CaSO4, and 0.06 M CaCl2 were prepared. Each electrolyte received a fresh VN electrode as the working electrode and was conditioned by running OCP and EIS centered at OCP (Fig. S6, ESI†). After conditioning, the operating voltage window for VN in each electrolyte was determined by running CVs at a scan rate of 50 mV s−1. Fig. 3 provides initial operating voltage windows for aqueous chlorides (Fig. 3a) and sulfates (Fig. 3b) compared against 1 M (red) and 0.1 M (pink) KOH (Fig. 3c) at 50 mV s−1. Among both sulfates and chlorides, Mg2+ (green) and Li+ (black) environments possessed the largest voltage windows. MgCl2 and MgSO4 achieved respective windows of 1.3 V and 1.35 V while LiCl and Li2SO4 achieved respective windows of 1.35 V and 1.4 V which are the closest to the characteristic 1.4 V window found in 1 M KOH both experimentally and in previous studies.64 Moreover, the voltage windows for Mg2+ and Li+ environments exist in low cathodic potentials compared to that of KOH which may contribute to their quicker charge and discharge times (Fig. S7, ESI†).
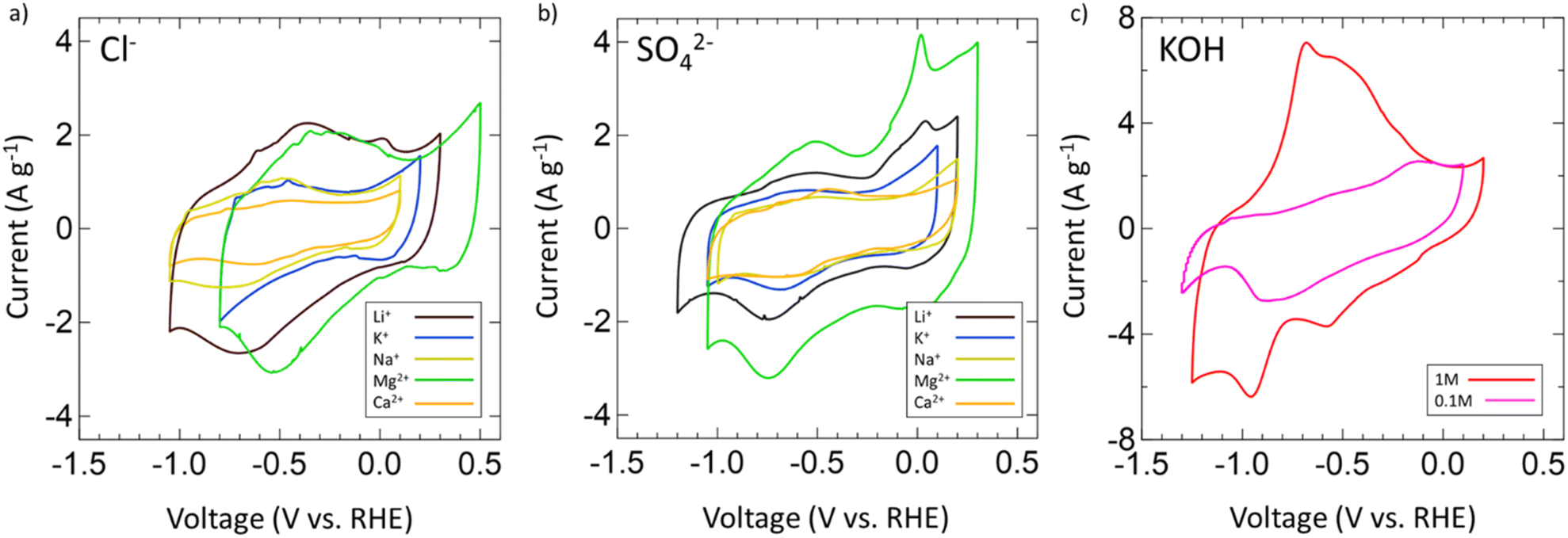 | ||
| Fig. 3 CVs of operating voltage windows for (a) chlorides, (b) sulfates of Mg2+ (green), Li+ (black), K+ (blue), Na+ (yellow), and Ca2+ (orange) at 50 mV s−1. (c) CVs for 1 M (red) and 0.1 M (pink) KOH. All chlorides and sulfates are 1 M concentration except for K2SO4 (0.5 M), CaSO4 (0.06 M), and CaCl2 (0.02 M) due to low solubility at room temperature. | ||
After the operating voltage was found for each electrolyte, CVs at variable scan rates from 2 to 2000 mV s−1 were taken and both gravimetric and areal specific capacitances were calculated for Mg2+ (green triangles), Li+ (black hourglasses), K+ (blue squares), Na+ (yellow stars), and Ca2+ (orange circles) chlorides (Fig. 4a) and sulfates (Fig. 4b) compared to 1 M (red diamonds) and 0.1 M (pink diamonds) KOH (Fig. 4c). Areal specific capacitances are based on the physical surface area obtained by N2 physisorption and were obtained by dividing the gravimetric capacitance by the physical surface area. In each electrolyte, capacitance decreased logarithmically with increasing scan rate indicative of pseudocapacitive charge storage mechanisms for every environment. Despite high capacitances at low scan rates in 1 M and 0.1 M KOH, from 2 to 2000 mV s−1 only 16% and 7% of the capacitances were retained, respectively. In 1 M KOH, the specific capacitance dropped from 1816 μF cm−2 (236 F g−1) to 285 μF cm−2 (37 F g−1) and in 0.1 M KOH from 877 μF cm−2 (114 F g−1) to 63 μF cm−2 (8 F g−1), which is only a slightly better performance than double layer capacitance (50 μF cm−2). However, in Mg2+ systems, despite lower capacitance at low scan rates compared to KOH, the retained capacitances when moving to higher scan rates are substantially better with 33% in MgCl2 and 36% in MgSO4. In fact, at 2000 mV s−1, MgSO4 achieves a specific capacitance of 294 μF cm−2 (38 F g−1), which is higher than that of 1 M KOH at the same scan rate. This outperformance of 1 M MgSO4 compared to 1 M KOH both in terms of capacitance and retention suggests that Mg2+ environments are preferable for high-rate charging and discharging.
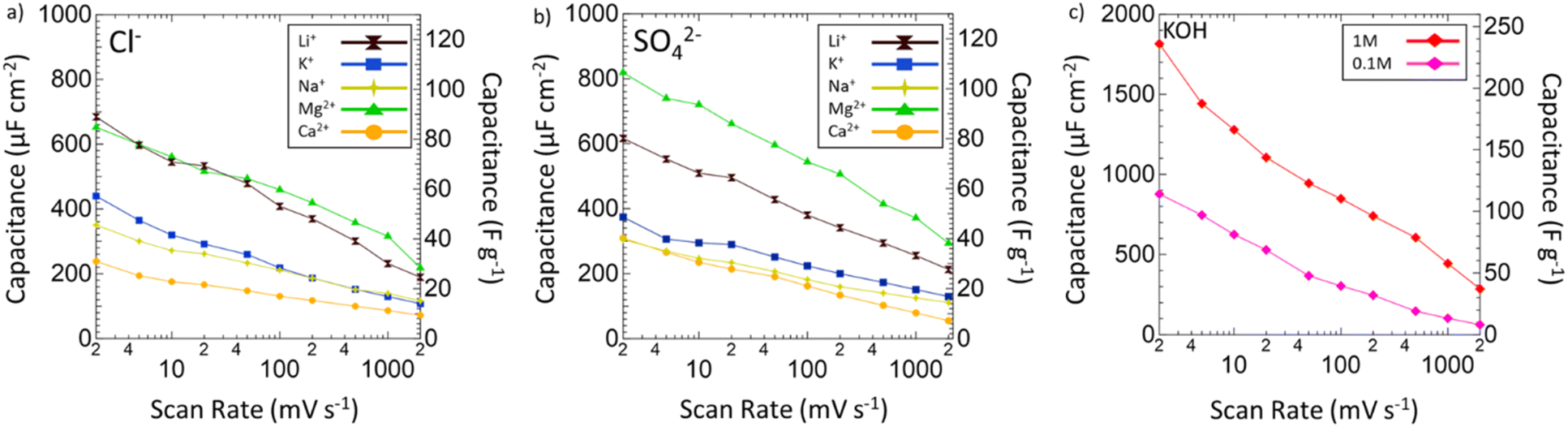 | ||
| Fig. 4 Specific capacitance plotted against scan rate from 2 to 2000 mV s−1 of VN in various (a) chlorides and (b) sulfates. Areal specific capacitance (μF cm−2) normalized by physical surface area is displayed on the left axis. Gravimetric specific capacitance (F g−1) is displayed on the right axis. Capacitances are shown for Li+ (black hourglasses), K+ (blue squares), Na+ (yellow stars), Mg2+ (green triangles), and Ca2+ (orange circles) systems. (c) Specific capacitance against scan rate for 1 M (red diamonds) and 0.1 M (pink diamonds) KOH. | ||
Next, the electrochemical surface area of VN in each system was evaluated by selecting 150 mV subsections of the operating window CVs where there was no significant redox activity and by running additional CV scans in these regions from 2 to 200 mV s−1 (Fig. S8, ESI†). Regions of low redox activity represent low pseudocapacitive contribution better representing double layer capacitive behavior, which can be used to model the electrochemical surface area (Fig. S9, ESI†). When evaluating the electrode surface area for electrochemical applications, BET analysis has limited capabilities. Due to dead pores, the physical surface area of the powder does not directly correspond to the ECSA, which can more accurately predict the electrochemically active and accessible sites, which is an important parameter for selecting supercapacitor electrodes and electrolytes. The ECSAs of VN in the different electrolytes are listed in Table 1.
| Electrolyte | Electrochemical surface area (m2 g−1) |
|---|---|
| 0.5 M K2SO4 | 17.7 |
| KCl | 11.7 |
| Na2SO4 | 12.6 |
| NaCl | 18.4 |
| Li2SO4 | 16.3 |
| LiCl | 22.6 |
| MgSO4 | 11.8 |
| MgCl2 | 25.8 |
| 0.02 M CaSO4 | 10 |
| 0.06 M CaCl2 | 9.7 |
| 1 M KOH | 31.1 |
| 0.1 M KOH | 11.5 |
Then, the electrochemical stability of VN was investigated by cycling 1000 CVs in each electrolyte at 50 mV s−1 (Fig. 5). These stability tests assessed both capacitance values over time and percent retention versus initial capacitance. After 1000 cycles, capacitance in 1 M KOH drops from 983 μF cm−2 (128 F g−1) to just 219 μF cm−2 (29 F g−1) displaying only 22% retention. In 0.1 M KOH, capacitance retention increased to 65% but at the cost of a lower overall capacitance throughout from 401 μF cm−2 (52 F g−1) to 262 μF cm−2 (34 F g−1) after 1000 cycles. Meanwhile, capacitance retention in salt environments after 1000 cycles does not drop below 73% (1 M LiCl) and even grows as seen in 1 M KCl (113%), 0.02 M CaSO4 (102%), 1 M MgCl2 (109%), 1 M Na2SO4 (peaks at 121% and ends at 108%), and 1 M MgSO4 (peaks at 126% and ends at 100%). Moreover, Mg2+ environments once again provide the best performance overall in terms of capacitance value and retention with MgCl2 surpassing 1 M KOH in capacitance after 400 cycles and MgSO4 doing the same after just 300 cycles.
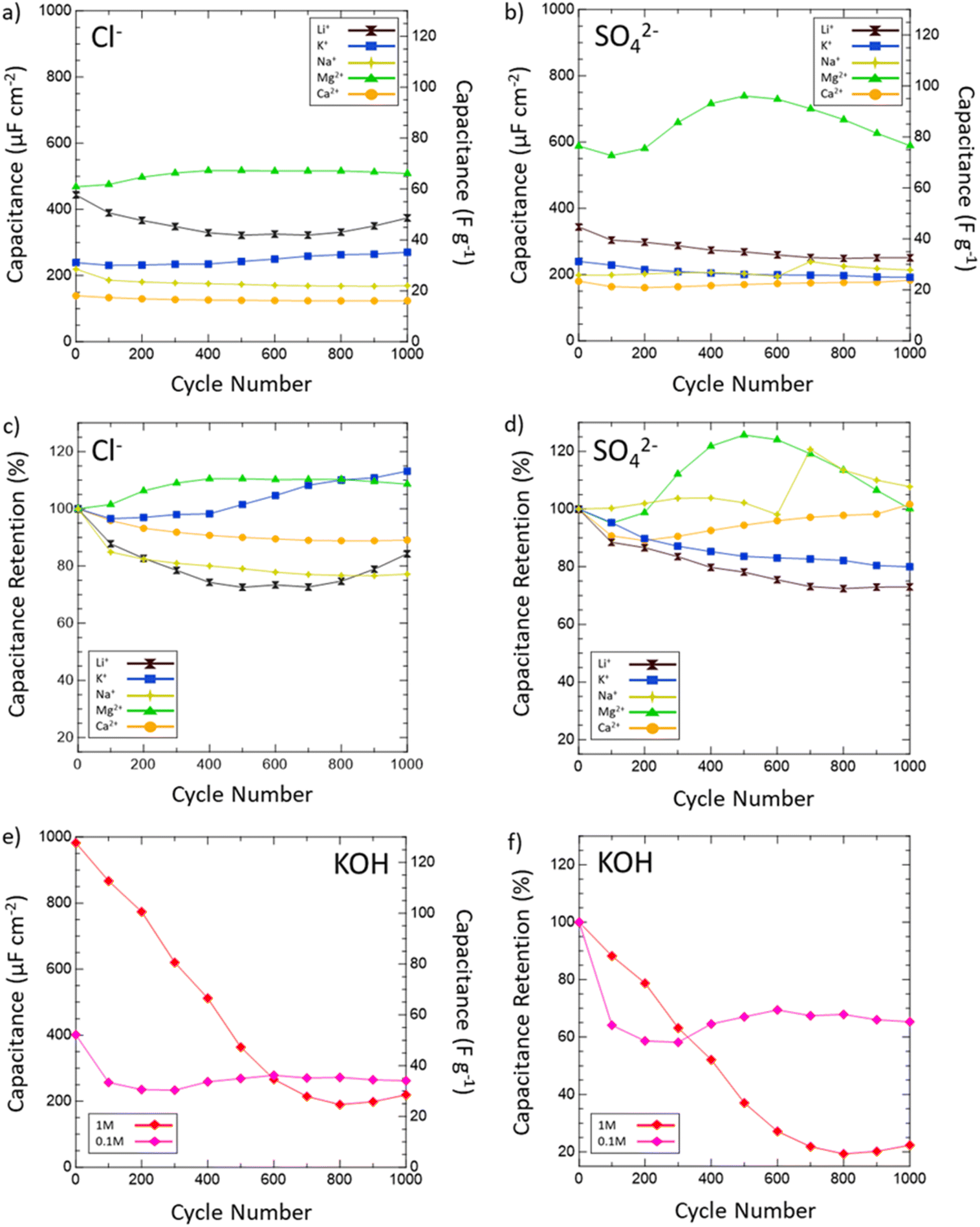 | ||
| Fig. 5 Capacitance values in various salt (a) chlorides and (b) sulfates and retention in (c) chlorides and (d) sulfates of VN after 1000 cycles at 50 mV s−1. Areal capacitances are normalized by the physical surface area obtained from N2 physisorption. Capacitances are shown for Li+ (black hourglasses), K+ (blue squares), Na+ (yellow stars), Mg2+ (green triangles), and Ca2+ (orange circles) systems. (e) Capacitance values and (f) retention values in 1 M (red diamonds) and 0.1 M (pink diamonds) KOH. Retention values are determined by dividing the capacitance after every 100 cycles by the 1st cycle capacitance and multiplying by 100. | ||
From these scan rate and stability capacitance data, the effect of different anions and cations can also be analyzed between different salt solutions. The decision to investigate chlorides and sulfates was influenced by the different oxidation states of the chloride (−1) and sulfate (−2) anions and their different sizes. A wide range of cations were selected to also investigate the influence of cation charge, size, and solubility on the pseudocapacitive mechanism of VN. From the capacitance data obtained, for most cations there seems to be no distinct effect or pattern from changing the anion oxidation state or size on capacitance values or retention. However, among sulfates and chlorides there is a consistent trend in performance influence by the cation used in the order Mg2+ > Li+ > K+ > Na+ > Ca2+. The high performance of Mg2+ systems at higher scan rates and after extensive cycling is substantial enough to outperform KOH systems of equal concentrations over time despite lower initial charge transfer from conditioning EIS data prior to cycling (Fig. 6a). In KOH solutions, the pseudocapacitive mechanism has been extensively studied and reported as shown below:
| VNxOy + OH− ↔ VNxOy‖OH− + VNxOy − OH | (1) |
| VNxOy + Mg2+ + 2e− ↔ Mg[VNxOy] | (2) |
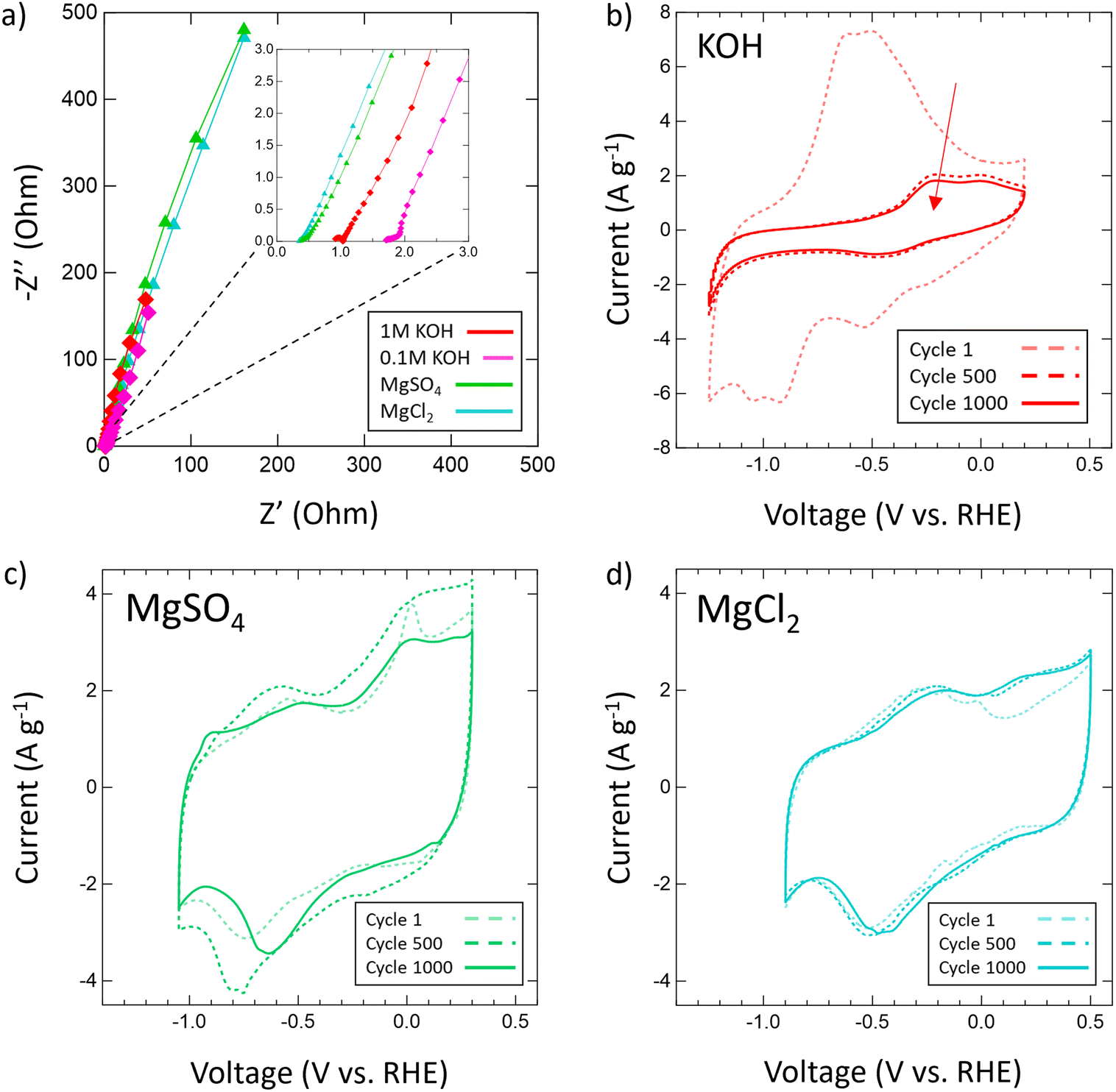 | ||
| Fig. 6 (a) EIS spectra for 1 M KOH (red), 0.1 M KOH (pink), 1 M MgSO4 (green) and 1 M MgCl2 (cyan) with a frequency range of 200 kHz to 10 mHz and an amplitude of 10 mV; (b) stability CVs of VN in (b) 1 M KOH, (c) MgSO4 and (d) MgCl2 at 50 mV s−1 at cycle 1, cycle 500, and cycle 1000. | ||
Conclusion
We report the synthesis of interstitial VN powder from a precursor V2O5 powder through a temperature-programmed reaction. XRD analysis suggests successful reduction of the oxide into VN while N2 physisorption analysis verifies a large physical surface area (32 m2 g−1). Raman spectra reveal the presence of a surface oxide layer indicating successful oxide passivation after synthesis. Electrochemical characterization of the VN was carried out in 1 M and 0.1 M KOH solutions as well as a variety of aqueous salt chlorides and sulfates of Mg2+, Ca2+, K+, Na+, and Li+. Although VN reports higher capacitance at low scan rates in KOH, a steep drop in capacitance occurs when moving to higher scan rates of 2000 mV s−1 leading to low retentions of 16% in 0.1 M KOH (114 to 8 F g−1) and only 7% in 1 M KOH (236 to 37 F g−1) from 2 to 2000 mV s−1. Yet, in aqueous salt environments, specifically Mg2+ systems, there is improved capacitance retention of 33% in MgCl2 (85 to 28 F g−1) and 36% in MgSO4 (106 to 38 F g−1) when moving to higher scan rates. Capacitance in 1 M KOH was also found to decline sharply after 1000 cycles from 128 to 28 F g−1 giving a 22% retention. On the other hand, salt environments provided improved capacitance retention with percent retentions >100% in 1 M KCl (113%), 0.02 M CaSO4 (102%), 1 M MgCl2 (109%), 1 M Na2SO4 (peaks at 121% and ends at 108%), and 1 M MgSO4 (peaks at 126% and ends at 100%). Mg2+ environments also provide the best performance overall in terms of capacitance value and retention with MgCl2 surpassing 1 M KOH in capacitance after 400 cycles and MgSO4 doing the same after just 300 cycles. Analyzing the capacitance trends across different salt electrolytes led to the conclusion that changes in the anion selection (SO42− and Cl−) produced no noticeable effect on performance while changes in cation produced varying performance in the order Mg2+ > Li+ > K+ > Na+ > Ca2+. The remarkable performance of VN in Mg2+ systems is attributed to a 2 e− transfer pseudocapacitive mechanism between Mg2+ and the VN oxynitride surface which is a faster and more reversible mechanism than the redox system found in KOH. Current work is underway to unveil the surface behavior and pseudocapacitive mechanism of VN in Mg2+ and KOH systems via in situ/operando spectroelectrochemical methods. A comprehensive understanding of these two systems can potentially be used to optimize the electrochemical environment of VN to incorporate both the high initial capacitance of KOH systems and the more stable and less corrosive behavior of Mg2+ systems.Author contributions
James Kasten: conceptualization, methodology, investigation Cheng-Che Hsiao: conceptualization, methodology, resources; Denis Johnson: verification, formal analysis, investigation; Abdoulaye Djire: formal analysis, visualization, project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the Department of Chemical Engineering at Texas A&M University and the College of Engineering for their financial support. This work was funded by Texas A&M University (TAMU), Texas A&M Engineering Experiment Station (TEES), and the Governor's University Research Initiative (GURI).References
- P. Thounthong, S. Rael and B. Davat, IEEE Trans. Ind. Electron., 2007, 54, 3225–3233 Search PubMed.
- F. Díaz-González, A. Sumper, O. Gomis-Bellmunt and R. Villafáfila-Robles, Renewable Sustainable Energy Rev., 2012, 16, 2154–2171 CrossRef.
- C. Arbizzani, M. Mastragostino and F. Soavi, J. Power Sources, 2001, 100, 164–170 CrossRef CAS.
- J. Cao and A. Emadi, IEEE Trans. Power Electron., 2012, 27, 122–132 Search PubMed.
- A. S. Aricò, P. Bruce, B. Scrosati, J.-M. Tarascon and W. van Schalkwijk, Nat. Mater., 2005, 4, 366–377 CrossRef PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- B. K. Kim, S. Sy, A. Yu and J. Zhang, in Handbook of Clean Energy Systems, pp. 1–25, DOI:10.1002/9781118991978.hces112.
- L. Borchardt, M. Oschatz and S. Kaskel, Mater. Horiz., 2014, 1, 157–168 RSC.
- D. K. Pathak and H. C. Moon, Mater. Horiz., 2022, 9, 2949–2975 RSC.
- S. Najib and E. Erdem, Nanoscale Adv., 2019, 1, 2817–2827 RSC.
- J. Huang, S. Peng, J. Gu, G. Chen, J. Gao, J. Zhang, L. Hou, X. Yang, X. Jiang and L. Guan, Mater. Horiz., 2020, 7, 2768–2769 RSC.
- X. Tong, G. Sheng, D. Yang, S. Li, C.-W. Lin, W. Zhang, Z. Chen, C. Wei, X. Yang, F. Shen, Y. Shao, H. Wei, Y. Zhu, J. Sun, R. B. Kaner and Y. Shao, Mater. Horiz., 2022, 9, 383–392 RSC.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- J. Kim, J. Lee, J. You, M.-S. Park, M. S. A. Hossain, Y. Yamauchi and J. H. Kim, Mater. Horiz., 2016, 3, 517–535 RSC.
- A. Das, M. Ojha, P. Subramanyam and M. Deepa, Nanoscale Adv., 2020, 2, 2925–2942 RSC.
- M. Laipan, J. Yu, R. Zhu, J. Zhu, A. T. Smith, H. He, D. O'Hare and L. Sun, Mater. Horiz., 2020, 7, 715–745 RSC.
- V. Augustyn, P. Simon and B. Dunn, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
- M. Jia, C. Cheng, L. Cui, Y. Li and X.-J. Jin, RSC Adv., 2020, 10, 3544–3553 RSC.
- D. Majumdar, T. Maiyalagan and Z. Jiang, ChemElectroChem, 2019, 6, 4343–4372 CrossRef CAS.
- X. Xia, D. Chao, C. F. Ng, J. Lin, Z. Fan, H. Zhang, Z. X. Shen and H. J. Fan, Mater. Horiz., 2015, 2, 237–244 RSC.
- G. Hasegawa, T. Sato, K. Kanamori, K. Nakano, T. Yajima, Y. Kobayashi, H. Kageyama, T. Abe and K. Nakanishi, Chem. Mater., 2013, 25, 3504–3512 CrossRef CAS.
- C. An, Y. Zhang, H. Guo and Y. Wang, Nanoscale Adv., 2019, 1, 4644–4658 RSC.
- K. Samdhyan, P. Chand, H. Anand and S. Saini, J. Energy Storage, 2022, 46, 103886 CrossRef.
- M. Barazandeh and S. H. Kazemi, Sci. Rep., 2022, 12, 4628 CrossRef CAS PubMed.
- R. Barik and P. P. Ingole, Curr. Opin. Electrochem., 2020, 21, 327–334 CrossRef CAS.
- S. M. Mane, A. M. Teli and J. C. Shin, J. Alloys Compd., 2022, 925, 166626 CrossRef CAS.
- C. Wang, Z. Song, P. Shi, L. Lv, H. Wan, L. Tao, J. Zhang, H. Wang and H. Wang, Nanoscale Adv., 2021, 3, 5222–5239 RSC.
- T. Chen, M. Li, S. Song, P. Kim and J. Bae, Nano Energy, 2020, 71, 104549 CrossRef CAS.
- K. Krishnamoorthy, P. Pazhamalai, S. Sahoo and S.-J. Kim, J. Mater. Chem. A, 2017, 5, 5726–5736 RSC.
- T. Zhang, K. Matthews, A. Vahidmohammadi, M. Han and Y. Gogotsi, ACS Energy Lett., 2022, 3864–3870, DOI:10.1021/acsenergylett.2c01508.
- M. R. Lukatskaya, S. Kota, Z. Lin, M.-Q. Zhao, N. Shpigel, M. D. Levi, J. Halim, P.-L. Taberna, M. W. Barsoum, P. Simon and Y. Gogotsi, Nat. Energy, 2017, 2, 17105 CrossRef CAS.
- P. K. Panda, A. Grigoriev, Y. K. Mishra and R. Ahuja, Nanoscale Adv., 2020, 2, 70–108 RSC.
- B. Pal, S. Yang, S. Ramesh, V. Thangadurai and R. Jose, Nanoscale Adv., 2019, 1, 3807–3835 RSC.
- C. Chen, D. Zhao, D. Xu and X. Wang, Mater. Chem. Phys., 2006, 95, 84–88 CrossRef CAS.
- S. Sharma, R. Adalati, M. Sharma, S. Jindal, A. Kumar, G. Malik and R. Chandra, Ceram. Int., 2022, 48, 34678–34687 CrossRef CAS.
- L. Zhang, S. Wang, Y. Shao, Y. Wu, C. Sun, Q. Huo, B. Zhang, H. Hu and X. Hao, Sci. Rep., 2017, 7, 44063 CrossRef PubMed.
- Y.-J. B. Ting, H. Wu, N. P. Kherani and K. Lian, Mater. Chem. Phys., 2015, 154, 118–124 CrossRef CAS.
- Y. Liu, Q. Wu, L. Liu, P. Manasa, L. Kang and F. Ran, J. Mater. Chem. A, 2020, 8, 8218–8233 RSC.
- D. Choi, G. E. Blomgren and P. N. Kumta, Adv. Mater., 2006, 18, 1178–1182 CrossRef CAS.
- X. Jiang, W. Lu, X. Yu, S. Song and Y. Xing, Nanoscale Adv., 2020, 2, 3865–3871 RSC.
- A. Djire, P. Pande, A. Deb, J. B. Siegel, O. T. Ajenifujah, L. He, A. E. Sleightholme, P. G. Rasmussen and L. T. Thompson, Nano Energy, 2019, 60, 72–81 CrossRef CAS.
- H.-h. Liu, H.-l. Zhang, H.-b. Xu, T.-p. Lou, Z.-t. Sui and Y. Zhang, J. Electrochem. Soc., 2018, 165, A97 CrossRef CAS.
- X. Zhou, H. Chen, D. Shu, C. He and J. Nan, J. Phys. Chem. Solids, 2009, 70, 495–500 CrossRef CAS.
- P. J. Hanumantha, M. K. Datta, K. S. Kadakia, D. H. Hong, S. J. Chung, M. C. Tam, J. A. Poston, A. Manivannan and P. N. Kumta, J. Electrochem. Soc., 2013, 160, A2195 CrossRef CAS.
- P. J. Hanumantha, M. K. Datta, K. Kadakia, C. Okoli, P. Patel and P. N. Kumta, Electrochim. Acta, 2016, 207, 37–47 CrossRef CAS.
- B. Wang, Z. Chen, G. Lu, T. Wang and Y. Ge, Mater. Res. Bull., 2016, 76, 37–40 CrossRef CAS.
- Z. Hou, K. Guo, H. Li and T. Zhai, CrystEngComm, 2016, 18, 3040–3047 RSC.
- F. Cheng, C. He, D. Shu, H. Chen, J. Zhang, S. Tang and D. E. Finlow, Mater. Chem. Phys., 2011, 131, 268–273 CrossRef CAS.
- E. Rondero-Daniel, L. F. Díaz-Ballote, L. Maldonado-López and A. Contreras, Rev. Mex. Fis., 2009, 55, 72–75 CAS.
- J. M. Wang, J. B. Wang, H. B. Shao, X. X. Zeng, J. Q. Zhang and C. N. Cao, Mater. Corros., 2009, 60, 977–981 CrossRef CAS.
- R. Kapoor and S. T. Oyama, J. Solid State Chem., 1992, 99, 303–312 CrossRef CAS.
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, Kluwer Academic/Plenum Publishers, 1999 Search PubMed.
- A. Djire, O. Ajenifujah and L. T. Thompson, Batteries Supercaps, 2018, 1, 171–175 CrossRef CAS.
- A. Djire, O. T. Ajenifujah, A. E. S. Sleightholme, P. Rasmussen and L. T. Thompson, J. Power Sources, 2015, 275, 159–166 CrossRef CAS.
- K. Huang, K. Bi, C. Liang, S. Lin, R. Zhang, W. J. Wang, H. L. Tang and M. Lei, Sci. Rep., 2015, 5, 11351 CrossRef CAS PubMed.
- H. Kwon, S. Choi and L. T. Thompson, J. Catal., 1999, 184, 236–246 CrossRef CAS.
- C. L. Londoño Calderón, C. Vargas hernandez and J. Jurado, Rev. Mex. Fis., 2010, 56, 411–415 Search PubMed.
- D. Shu, C. Lv, F. Cheng, C. He, K. Yang, J. Nan and L. Long, Int. J. Electrochem. Sci., 2013, 8, 1209–1225 CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- G. Silversmit, D. Depla, H. Poelman, G. B. Marin and R. De Gryse, J. Electron Spectrosc. Relat. Phenom., 2004, 135, 167–175 CrossRef CAS.
- K. Dewangan, G. P. Patil, R. V. Kashid, V. S. Bagal, M. A. More, D. S. Joag, N. S. Gajbhiye and P. G. Chavan, Vacuum, 2014, 109, 223–229 CrossRef CAS.
- N. Zhang, L. Cao, L. Feng, J. Huang, K. Kajiyoshi, C. Li, Q. Liu, D. Yang and J. He, Nanoscale, 2019, 11, 11542–11549 RSC.
- X. Peng, L. Wang, L. Hu, Y. Li, B. Gao, H. Song, C. Huang, X. Zhang, J. Fu, K. Huo and P. K. Chu, Nano Energy, 2017, 34, 1–7 CrossRef CAS.
- H. Xu, W. He, Z. Li, J. Chi, J. Jiang, K. Huang, S. Li, G. Sun, H. Dou and X. Zhang, Adv. Funct. Mater., 2022, 32, 2111131 CrossRef CAS.
- A. Djire, A. Bos, J. Liu, H. Zhang, E. M. Miller and N. R. Neale, ACS Appl. Nano Mater., 2019, 2, 2785–2795 CrossRef CAS.
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00810f |
| This journal is © The Royal Society of Chemistry 2023 |