
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTilt grain boundaries in WS2 from low to high misorientation angles†
Da
Ke
,
Jinquan
Hong
and
Yubo
Zhang
 *
*
Minjiang Collaborative Center for Theoretical Physics, College of Physics and Electronic Information Engineering, Minjiang University, Fuzhou, 350108, China. E-mail: yubo.drzhang@mju.edu.cn
First published on 17th April 2023
Abstract
Grain boundaries (GBs) with low misorientation angles are interfacing lines connecting sparsely distributed dislocation cores, but high-angle GBs could have amorphous atomic arrangements with merged dislocations. Tilt GBs in two-dimensional materials frequently emerge in large-scale specimen production. In graphene, a critical value for differentiating low and high angles is quite big because of its flexibility. However, understanding transition-metal-dichalcogenide GBs meets additional complexities regarding the three-atom thickness and the rigid polar bonds. We construct a series of energetic favorable WS2 GB models using coincident-site-lattice theory with periodic-boundary conditions. The atomistic structures of four low-energy dislocation cores are identified, consistent with the experiments. Our first-principles simulations reveal an intermediate critical angle of θc ≈ 14° for WS2 GBs. Structural deformations are effectively dissipated via W–S bond distortions especially along the out-of-plane direction, instead of the prominent mesoscale buckling in one-atom-thick graphene. The presented results are informative in studies of the mechanical properties of transition metal dichalcogenide monolayers.
1. Introduction
Grain boundaries (GBs) are narrow atomic walls interfacing two crystalline grains with different orientations in a polycrystalline material. GBs are line defects in nature, and the atomistic structures of dislocation cores play a vital role in determining the physical properties of GB systems. The atoms at GBs were initially believed to be highly amorphous. Nowadays, it is generally accepted that low-energy GBs usually have relatively regular atomic arrangements inherited from their parent structure. Strong disordering occurs in the case of high misorientation angles and non-stoichiometry. It is therefore convenient to classify GB systems into two categories according to misorientation angles: low-angle and high-angle GBs. Low-angle GBs, as established by W. Read and W. Shockley,1 are sparsely distributed arrays of dislocation cores with well-defined local distortion patterns and well-organized long-range periodicity. At elevated misorientation angles, the cores get closer and quickly start to interact with each other and finally merge together, because dislocation strains can extend to several nanometers away.The critical value for differentiating low and high misorientation angles is material-dependent, and crystal dimensionality can be the most fundamental structural determinant. GBs in three-dimensional (3D) bulk materials have been extensively studied, and a small angle of θc ≥ 5–10° is generally cited.2 The smallness of the critical angle is an indication of strong mutual interactions among the nearby dislocation cores. At the microscopical level, these interactions are bridged by long-range stress fields (with mechanical energies exerted on the underline material), which cannot be effectively dissipated in a 3D confined environment. The GB-associated long-range effects in 3D bulk materials can significantly impact material properties, such as sinks for dopants and vacancies.3
Understanding GBs in atomic thin 2D materials is driven by large-scale specimen production via chemical-vapor-deposition techniques, from which GBs frequently emerge. They are tilt GBs since the two misorientated grains are placed on the same plane. 2D materials are generally mechanically soft and have an additional pathway for dissipating stress fields owing to the open geometry along the out-of-plane direction. As a result, graphene GBs were predicted to have a much higher critical angle, which was attributed to the significant buckling induced by the GBs.4 In addition, dislocation structures are significantly reconstructed due to compelling attraction between dislocation cores, resulting in unique asymmetric hillocks. The mesoscale buckling and local reconstruction make a particular high-angle system extraordinarily stable, which changes the overall landscape of GB formation energies.4,5 GB-induced buckling has been experimentally confirmed in graphene6 and hexagonal BN.7
Tilt GBs in transition-metal dichalcogenides (TMDs) have shown various interesting functions relating to the thermodynamic,8,9 mechanical,10 electrical,11 magnetic,12,13 and optoelectronic properties14,15 if rationally engineered.16 Since 2D TMD systems have three atomic layers and polar bonds, they have intermediate flexibility between 3D bulk materials and one-atom-thick graphene. Therefore, a medium critical angle for TMD-GBs is expected from a straightforward geometric analysis. This was previously inferred from an experimental study on their electrical transport properties11 and also suggested by a density-functional-theory simulation.17 However, comparative investigations on TMD-GBs covering both low-angle and high-angle systems are still lacking. Particularly, previous simulations of low-angle systems usually relied on either over-simplified or inappropriate structural models. To contain GBs within a manageable cell size, GB models were incorporated into non-periodic nanoribbons with undesired dangling bonds at the cell edge.
In this work, we carry out density-functional-theory calculations on WS2 GBs that are constructed by using coincident-site-lattice theory with periodic-boundary conditions. Twenty-three misorientation angles are considered, from the lowest angle θ = 1.297° to a high angle of θ = 42.103°. The energy-favorable GB configurations are carefully located. Systematic investigations reveal a critical value of θc ≈ 14° for separating WS2 GBs into low- and high-angle families.
2. Grain boundary in periodic models
GB structural models with periodic-boundary conditions can be generated using coincidence site lattice (CSL) theory. We construct [0001] tilt GB models for 2H-phase WS2 following a similar procedure proposed for graphene,4 both of which are hexagonal lattices. We here briefly describe the key CSL parameters and refer to ref. 4 for other details. The first step is to select two symmetric supercells, the CSL cells, to represent the two pieces of misorientated grains. There are two types of edge structures for the hexagonal lattices: the armchair line and the zigzag line. The armchair line passes through the lattice points, and, for simplicity, is chosen as the mirror plane in fixing the CSL points.4 Correspondingly, the so-obtained structures are named armchair GB models. The two grains are then stitched together side by side, and boundary atoms are removed if they are too close or even overlapped. At the same time, periodic-boundary conditions both parallel to and perpendicular to the GB direction are enforced. Fig. 1 shows the GB models with θ = 7.341°. It is clear that each model has two antisymmetric GBs with inverted elements. When using a zigzag line as the mirror plane (not shown here), atomic structures at the GBs are distinctly different and more complicated. However, those zigzag GB structures have not been identified experimentally. Theoretically, they are also energetically unstable and automatically transform to armchair motifs.17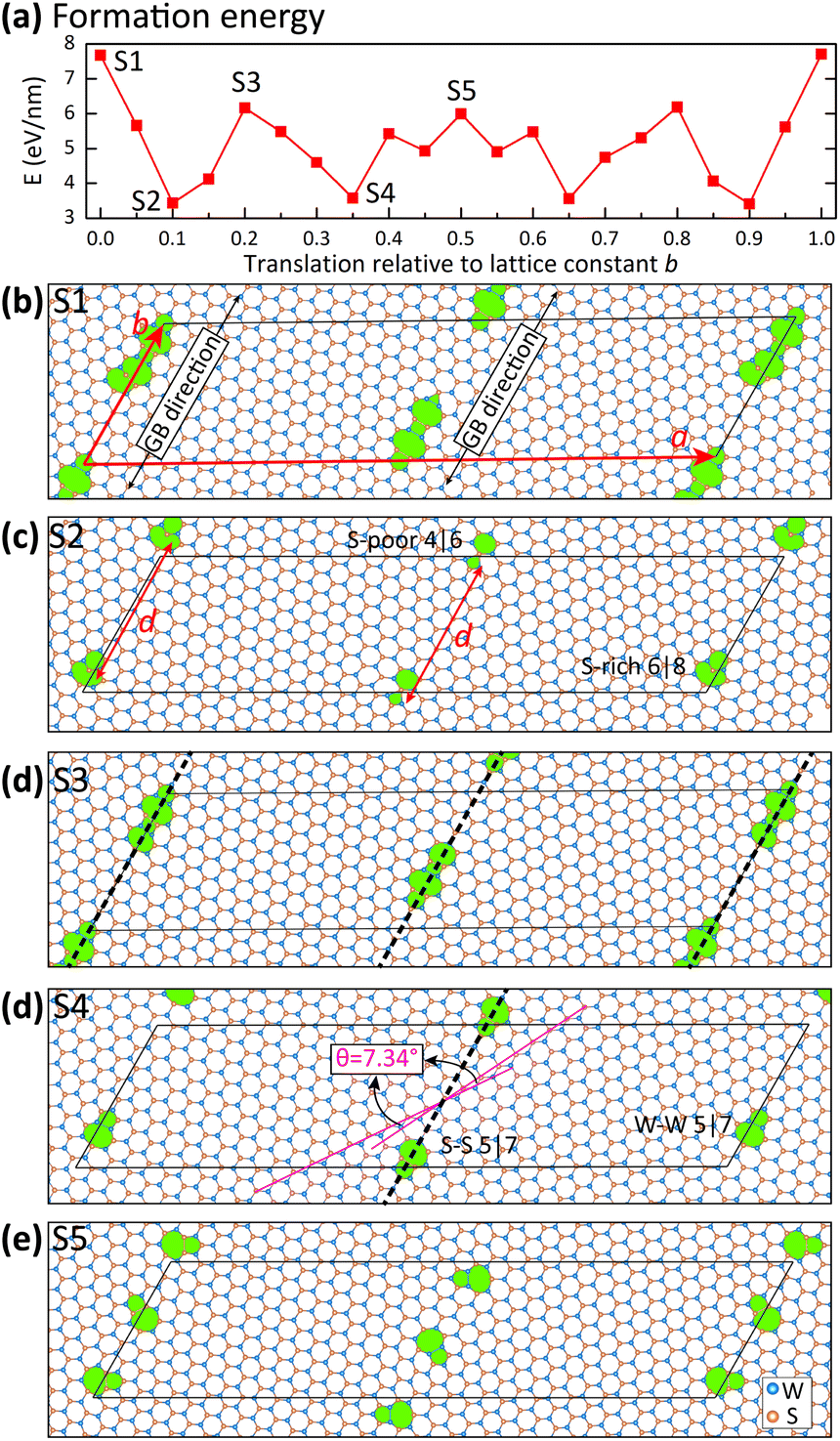 | ||
| Fig. 1 Translational degree-of-freedom in the model with a misorientation angle θ = 7.341°. (a) The GB formation energy for various translation states between the two grains. (b–e) Relaxed crystal structures of five selected translations. Bold dashed lines represent GB walls, which separate WS2 into different domains with two misfitted orientations. The misorientation angle is defined in subplot (d). The parallelograms emphasize the simulation cell satisfying the periodic-boundary conditions along directions both parallel and perpendicular to the GB direction. The non-hexagonal rings are highlighted in green color, and four low-energy types are labeled. Note: the dislocation cores rotate away from the GB direction in subplot (e). | ||
Second, the misorientation angles θ are the only input parameter for generating the models. According to CSL theory, the hexagonal lattice has 26 possible misorientation angles (0° < θ ≤ 60°). Table 1 collects 25 models with 23 misorientation angles up to 42.103°, missing the other three systems having even higher angles. All the structures are fully relaxed and shown in Fig. S1 in the ESI.†
| Dislocation cores nd | Misorientation angle θ (degree) | CSL cell size Σ | Formula account N | Lattice constant a (Å) | Lattice constant b (Å) | Dislocation rings |
|---|---|---|---|---|---|---|
| a The structural relaxation is not well converged because there are too many atoms involved. b The model difference is due to the different thresholds for removing the overlapped atoms. c The model difference is due to the relative translations between the grains. | ||||||
| n d = 1 | 1.297a | 1951 | 3853 | 277.35 | 139.81 | 4|6 |
| 2.134 | 721 | 1430 | 169.08 | 85.01 | 5|7 | |
| 3.481b | 271 | 537 | 103.75 | 52.10 | 4|6, 6|8 | |
| 3.481b | 271 | 540 | 104.15 | 52.10 | 5|7 | |
| 5.086 | 127 | 507 | 142.75 | 35.65 | 4|6, 4|5|7 | |
| 7.341c | 61 | 243 | 98.92 | 24.74 | 4|6, 6|8 | |
| 7.341c | 61 | 243 | 98.60 | 24.75 | 5|7 | |
| 9.430 | 37 | 147 | 76.83 | 19.26 | 4|6, 6|8 | |
| 13.174 | 19 | 113 | 82.58 | 13.81 | 4|6, 6|8 | |
| 21.787 | 7 | 56 | 67.55 | 8.38 | 5|7 | |
| n d = 2 | 4.723 | 589 | 1162 | 152.44 | 76.83 | 5|7 |
| 6.609 | 301 | 596 | 109.10 | 54.89 | 4|6, 6|8 | |
| 8.256 | 193 | 376 | 86.45 | 44.02 | 4|6, 6|8 | |
| 10.993 | 109 | 430 | 131.18 | 33.08 | 4|6, 6|8 | |
| 16.426 | 49 | 192 | 87.74 | 22.19 | 5|7 | |
| 32.204 | 13 | 102 | 90.42 | 11.41 | 4|6, 6|8 | |
| n d = 3 | 8.613 | 399 | 518 | 145.32 | 63.25 | 4|6, 5|7, 6|8 |
| 11.635 | 219 | 284 | 107.77 | 46.89 | 4|6, 5|7, 6|8 | |
| 15.178 | 129 | 164 | 82.37 | 36.01 | 4|6, 5|7, 6|8 | |
| 17.897 | 93 | 118 | 69.97 | 30.59 | 4|6, 5|7, 6|8 | |
| 27.796 | 39 | 74 | 67.97 | 19.76 | 4|6, 5|7, 6|8 | |
| 38.213 | 21 | 80 | 99.66 | 14.49 | 4|6, 5|7, 6|8 | |
| n d = 4 | 18.734 | 151 | 294 | 76.79 | 38.60 | 4|6, 5|7, 6|8 |
| 26.008 | 79 | 310 | 111.38 | 28.13 | 4|6, 6|8 | |
| 42.103 | 31 | 182 | 104.65 | 17.62 | 4|6, 5|7, 6|8 | |
Third, GB models are classified into four families that are characterized by the number of dislocation cores nd along the period length d of the supercell [Fig. 1(b–e)]. The parameter d is equivalent to the lattice constant b, and they are used interchangeably in this work. The separation of the dislocation cores along the boundary direction equals b/nd, a quantity reflecting the coupling tendency between the dislocations.
Fourth, a unitless parameter Σ is defined to measure the size of each grain (Ωgrain) but is normalized by the WS2 unit cell (Ωunitcell), i.e.,
| Σ = Ωgrain/Ωunitcell. | (1) |
The formula account N is in principle twice of Σ (i.e., N = 2Σ), since the GB model contains two grains. The tiny deviations found in Table 1 (e.g., N = 1.975Σ for θ = 1.297°) occur when removing the overlapped atoms between the two grains. Coupling between dislocations perpendicular to the GB direction is a spurious effect in our models. It is prevented by increasing the CSL cell size perpendicular to the GB direction, which leads to more complicated relationships between N and Σ (e.g., N = 5.947Σ for θ = 13.174°).
The relative translation of the two CSL grains along the boundary direction is unfixed by CSL theory. Fig. 1(b–e) show five GB models with the same misorientation angle of θ = 7.341° but different translations. The misorientation angle defined in Fig. 1(d) is the angle between S–S bonding directions in each grain. All the crystal structures are fully relaxed, and their formation energies (defined in eqn (2)) are compared in Fig. 1(a). Obviously, translation is a key degree-of-freedom in determining the dislocation structures, which in turn locates the energetically favorable configurations. For example, dislocation cores in S1 and S3 models are composed of many homo-elemental bonds, i.e., W–W bonds and S–S bonds, which leads to high energies of the corresponding GB models. S2 and S4 models have much lower energies because their dislocation cores are less distorted. In fact, the non-hexagonal distorted rings in S2 and S4 models are the most stable structures found for all the misorientation angles, as shown in the last column of Table 1. It is worth noting that S5 has high formation energy, although it is solely composed of low-energy rings. This is because two dislocation cores are found along the GB direction in S5, indicating that the θ = 7.341° model is accidently pushed from the anticipated family nd = 1 to the family of nd = 2 under a peculiar translation.
We discuss the dislocation structures in greater detail since they play a vital role in determining the physical properties of GB systems including stability. The distorted dislocations in the S4 model (Fig. 1) have two antisymmetric structures in the two GB walls: an S–S 5|7 ring in an S-rich environment and an element-inverted W–W 5|7 ring in a W-rich environment. One may argue that the element-inverted motifs are an artificial effect when enforcing the periodic-boundary conditions along the perpendicular direction. However, the antisymmetric patterns are absent in the S2 model, where the lateral GB wall has a 6|8 ring but the central GB wall has a 4|6 ring. To understand the differences of S2 and S4 models, the dislocations before structural optimization are shown in the left panels of Fig. 2. The 5|7 rings [Fig. 2(a and d)] can keep their overall appearance in the structural relaxation. By contrast, the W-rich 6|8 ring [Fig. 2(b)] can easily transform to an S-poor 4|6 ring [Fig. 2(c)] via dislocation climbing, indicating that the former configuration is an energetic saddle-point. Similar instability is also found for the W-poor 4|6 ring [Fig. 2(f)]. Therefore, we identify four low-energy dislocation motifs, i.e., S–S 5|7 ring, W–W 5|7 ring, S-poor 4|6 ring, and S-rich 6|8 ring, confirmed experimentally (see the right panels of Fig. 2). It is worth noting that most of these non-hexagonal rings have been theoretically reported earlier.5,9,13,17,18 Our construction approach provides an intuitive understanding of the translational degree of freedom in determining the low-energy structures.
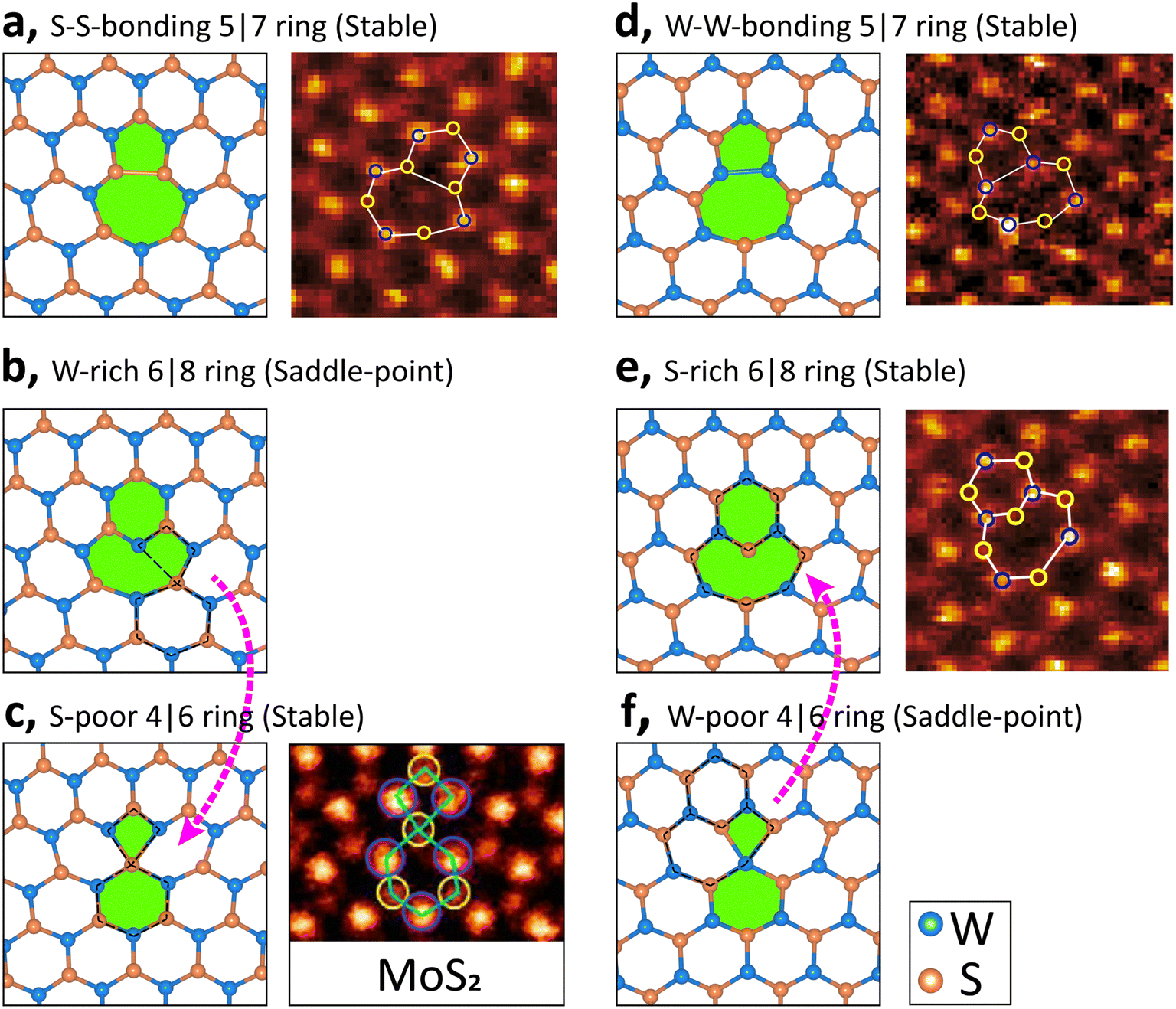 | ||
| Fig. 2 Energetically favorable dislocation rings in WS2 GB models. The left plots (with a white background) are theoretical structures before optimization. W atoms with dangling bonds in subplot (b) and 4-fold coordinates in subplot (f) are energetic saddle-points, and the structural transformations are denoted by pink arrows. The experimental HAADF-STEM images (with a black background) are shown on the right for subplots (a, d, and e), cited from ref. 15. Note: on the right of subplot (c) is the STEM-ADF image of MoS2.13 | ||
Our identified structures are the ideal least-distorted low-energy configurations of WS2 GBs, especially with low misorientation angles. In addition, stoichiometry is assumed for simplicity. Since real GBs are complicated nonequilibrium defects, energetics is not the only decisive factor for their formation. Nevertheless, our finding provides valuable insights for understanding the considerable inconsistency. For example, it was reported that 6|8 rings are the dominant type over 5|7 cores in WS2, especially under S-rich conditions.9 In contrast, a study of MoS2 GBs only observed 5|7 dislocation cores.11 Third, all the 4|6, 5|7, and 6|8 rings were found in ref. 13.
The ring types may have a direct consequence on the electronic properties, and one notable example is magnetic instability. Among the four rings, we find that the W–W 5|7 ring (specifically, the W atoms in the W–W homoelemental bond) is most likely to develop magnetic moment, as shown in the low-angle system θ = 7.341° in Fig. S4.† However, the magnetic moment is quite small. We carry our further calculation in a more defective high-angle GB with θ = 42.103°. In this case, the highest magnetic moment is about 0.5 μB. A previous report on the significant magnetization is in the presence of anti-site defects.12 We conclude that the magnetic instability is quite weak in low-energy GBs.
3. Systematic trends of formation energies and electronic properties
The procedure of locating low-energy GBs enables us to reveal the systematic trends of formation energies from low to high angles. In first-principles calculations, the GB formation energy Ef(θ) is defined as:4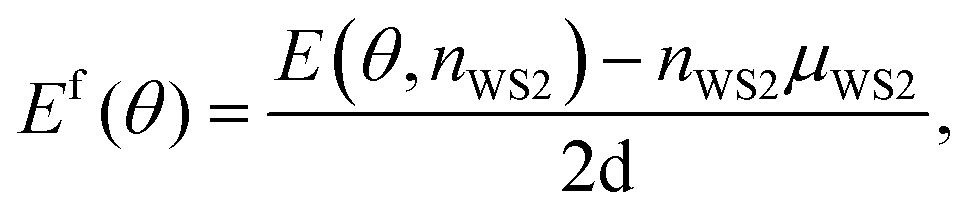 | (2) |
E(θ, nWS2) is the internal energy of the WS2 GB model with a misorientation angle θ and a formula number nWS2. μWS2 is the internal energy of pristine WS2. The parameter d in the denominator is equivalent to the lattice constant b. Note that 3D bulk materials' GB formation energy is averaged onto the grain interfacing area. By contrast, the atomic layer thickness of WS2 is not well defined, and the formation energy is the energy density over the boundary length. The factor of 2 in the denominator is due to two parallel dislocation walls in each model. Ef(θ) can have two representations, i.e., measured with respect to the GB length (eV nm−1) or dislocation core numbers (eV per core). Empirically, the Ef(θ) (in the unit of eV nm−1) of low-angle GBs follows the Read–Shockley relationship,1,2
| Ef(θ) = θE0[A − ln(θ)]. | (3) |
E 0 depends only on the GB orientation and the macroscopic elastic constants. The parameter A depends upon both the misorientation angle and the atomic energy at the dislocation core. Equivalently, the formation energy per dislocation core is,4
Ef(θ) = C − D![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(θ), ln(θ), | (4) |
The first-principles results of 23 WS2-GB models are shown in Fig. 3(a), compared with that of graphene reproduced from ref. 4. Compared with graphene, WS2 GBs not only have higher formation energies at each angle but also lack a V-shaped energy dip at the high-angle end. This can be qualitatively understood, bearing in mind that GB formation energy is composed of (1) local atomistic distortions around the dislocation cores and (2) elastic strain associated with the long-range lattice deformation.4 Graphene has only one atomic layer and is mechanically flexible. GBs in graphene release strains through the local dislocation reconstruction and the prominent out-of-plane buckling (with a height of ∼5 Å). Moreover, dislocation cores of graphene combine into pairs because of their mutual attractions, leading to an energy dip at θ ≈ 32.2°.4 The three-atom-thick WS2 is more rigid than graphene and resembles 3D materials to a large extent. WS2, in the presence of distorted dislocations, has a high resistance to being buckled at the nanoscale. Strains of dislocations are primarily released from the stretch or compression of W–S bonds [Fig. 3(b)]. Due to the out-of-plane freedom, the bond variations are more effective than 3D bulk materials. This finding agrees well with the experimental observation of slight warping in WS2.9
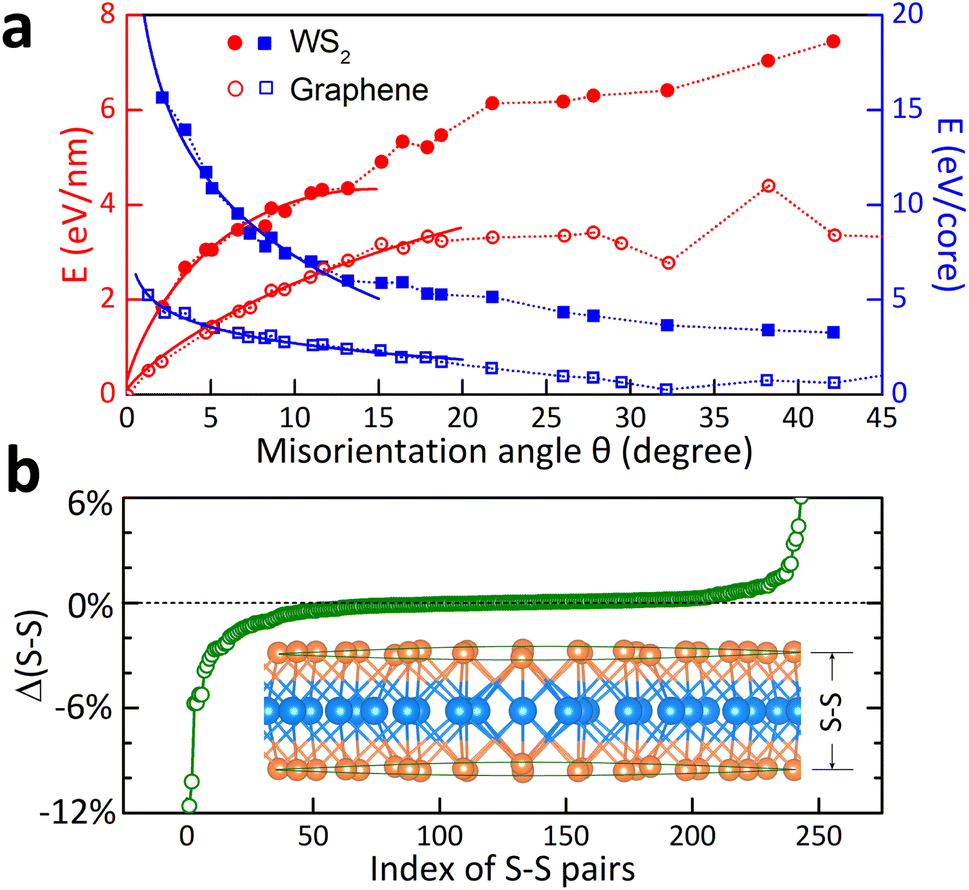 | ||
| Fig. 3 Electronic properties of symmetrical [0001] tilt grain boundaries. (a) Formation energies as a function of misorientation angles. The energy densities are represented along the dislocation-core-line (eV nm−1, the left y-axis) or averaged to the dislocation core (eV per core, the right y-axis). The solid symbols are DFT results for WS2 systems, and the open marks are for graphene calculated using the force-field approach.4 The solid lines for the low-angles are fitted to eqn (3) or (4). The dotted lines are the direct connection of the data points. (b) Changes of the vertical S–S distances in the system with θ = 7.34°. The inset shows the cut view of the crystal structure, and the green lines mark the out-of-plane warping around the dislocation core. | ||
The critical value for differentiating low and high-angle GBs is defined as the position, where formation energy deviates from the empirical Read–Shockley relationship (eqn (3) and (4)). This predicts θc ≈ 14° for WS2 and θc ≈ 20° for graphene [Fig. 3(a) and S5†]. Above these angles, the Read–Shockley relationship ceases to work because other energies (due to dislocation coupling) besides the linear-elastic energy come into play. Interestingly, the critical angle can also be used for organizing the electronic properties of WS2 GBs. Fig. 4(c–f) shows the electronic density-of-states of a few systems. Localized in-gap states enhance with respect to the misorientation angles. A quasi-linear relationship holds up to the critical angle [Fig. 4(a)]. Above the critical angle, the massive in-gap states become delocalized, resulting in a notable metallic behavior18 along the GB directions [e.g., Fig. 4(f) for θc ≈ 32.204°].
 | ||
| Fig. 4 Defective in-gap states induced by the grain boundaries. (a) The electronic density-of-states integrated within the intrinsic band gap. The dots are calculated results, and the dashed line is a linear fit. (b) Band-edge density-of-states of pristine WS2, without in-gap states. (c and d) Density-of-states of the GB systems with four angles of 3.48°, 7.34°, 13.17°, and 32.20°. To better show the defective in-gap states (blue lines), local density-of-states on atoms far from the GB lines is used as reference (shaded areas). The green dashed rectangles show the in-gap states used for the in-gap state integration in subplot (a). | ||
4. Conclusions
In summary, we conduct systematic first-principles investigations on tilt WS2 GBs from low to high misorientation angles. The energy-favorable ideal GB configurations are composed of four basic types of dislocations. The low-energy structures provide valuable clues for clarifying the experimental inconsistencies. An intermediate critical value of θc ≈ 14° is derived for classifying WS2 GBs into low-angle and high-angle categories. WS2 GBs release distortions via W–S bond distortions especially along the out-of-plane direction, lacking the prominent mesoscale buckling in one-atom-thick graphene.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Y. Z. is supported by the National Natural Science Foundation of China (11904156), Guangdong Natural Science Foundation (2021A1515010049), and Shenzhen Natural Science Foundation (JCYJ20190809153401651). D. K. is supported by the Fujian Natural Science Foundation (2022J011127).References
- W. T. Read and W. Shockley, Dislocation Models of Crystal Grain Boundaries, Phys. Rev., 1950, 78, 275 CrossRef CAS.
- D. Hull and D. J. Bacon, Introduction to Dislocations, ed. D. Hull and D. J. Bacon, Butterworth-Heinemann, Oxford, 5th edn, 2011 Search PubMed.
- A. R. Symington, M. Molinari, J. Statham, J. Wu and S. C. Parker, The role of dopant segregation on the oxygen vacancy distribution and oxygen diffusion in CeO2 grain boundaries *, J. Phys.: Energy, 2019, 1, 042005 CAS.
- J. M. Carlsson and L. M. Ghiringhelli, Annalisa Fasolino, Theory and hierarchical calculations of the structure and energetics of [0001] tilt grain boundaries in graphene, Phys. Rev. B, 2011, 84, 165423 CrossRef.
- Y. Liu, X. Zou and B. I. Yakobson, Dislocations and Grain Boundaries in Two-Dimensional Boron Nitride, ACS Nano, 2012, 6, 7053 CrossRef CAS PubMed.
- J. Coraux, A. T. N‘Diaye, C. Busse and T. Michely, Structural Coherency of Graphene on Ir(111), Nano Lett., 2008, 8, 565 CrossRef CAS PubMed.
- A. L. Gibb, N. Alem, J.-H. Chen, K. J. Erickson, J. Ciston, A. Gautam, M. Linck and A. Zettl, Atomic Resolution Imaging of Grain Boundary Defects in Monolayer Chemical Vapor Deposition-Grown Hexagonal Boron Nitride, J. Am. Chem. Soc., 2013, 135, 6758 CrossRef CAS PubMed.
- T. H. Ly, M.-H. Chiu, M.-Y. Li, J. Zhao, D. J. Perello, M. O. Cichocka, H. M. Oh, S. H. Chae, H. Y. Jeong, F. Yao, L.-J. Li and Y. H. Lee, Observing Grain Boundaries in CVD-Grown Monolayer Transition Metal Dichalcogenides, ACS Nano, 2014, 8, 11401 CrossRef CAS PubMed.
- A. Azizi, X. Zou, P. Ercius, Z. Zhang, A. L. Elías, N. Perea-López, G. Stone, M. Terrones, B. I. Yakobson and N. Alem, Dislocation motion and grain boundary migration in two-dimensional tungsten disulphide, Nat. Commun., 2014, 5, 4867 CrossRef CAS PubMed.
- L. Huang, F. Zheng, Q. Deng, Q. Huy Thi, L. W. Wong, Y. Cai, N. Wang, C.-S. Lee, S. P. Lau, T. H. Ly and J. Zhao, Anomalous fracture in two-dimensional rhenium disulfide, Sci. Adv., 2020, 6, eabc2282 CrossRef CAS PubMed.
- T. H. Ly, D. J. Perello, J. Zhao, Q. Deng, H. Kim, G. H. Han, S. H. Chae, H. Y. Jeong and Y. H. Lee, Misorientation-angle-dependent electrical transport across molybdenum disulfide grain boundaries, Nat. Commun., 2016, 7, 10426 CrossRef CAS PubMed.
- N. Gao, Y. Guo, S. Zhou, Y. Bai and J. Zhao, Structures and Magnetic Properties of MoS2 Grain Boundaries with Antisite Defects, J. Phys. Chem. C, 2017, 121, 12261 CrossRef CAS.
- W. Zhou, X. Zou, S. Najmaei, Z. Liu, Y. Shi, J. Kong, J. Lou, P. M. Ajayan, B. I. Yakobson and J.-C. Idrobo, Intrinsic Structural Defects in Monolayer Molybdenum Disulfide, Nano Lett., 2013, 13, 2615 CrossRef CAS PubMed.
- A. M. van der Zande, P. Y. Huang, D. A. Chenet, T. C. Berkelbach, Y. M. You, G.-H. Lee, T. F. Heinz, D. R. Reichman, D. A. Muller and J. C. Hone, Grains and grain boundaries in highly crystalline monolayer molybdenum disulphide, Nat. Mater., 2013, 12, 554 CrossRef CAS PubMed.
- F. Hou, Y. Zhang, D. Li, L. Che and J. Lin, Deciphering the structure-photoluminescence correlation at small-tilt-angle grain boundaries in monolayer WS2, Appl. Phys. Lett., 2022, 121, 051104 CrossRef CAS.
- P. Man, D. Srolovitz, J. Zhao and T. H. Ly, Functional Grain Boundaries in Two-Dimensional Transition-Metal Dichalcogenides, Acc. Chem. Res., 2021, 54, 4191 CrossRef CAS PubMed.
- X. Zou, Y. Liu and B. I. Yakobson, Predicting Dislocations and Grain Boundaries in Two-Dimensional Metal-Disulfides from the First Principles, Nano Lett., 2013, 13, 253 CrossRef CAS PubMed.
- X. Zou and B. I. Yakobson, Metallic High-Angle Grain Boundaries in Monolayer Polycrystalline WS2, Small, 2015, 11, 4503 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2na00709f |
| This journal is © The Royal Society of Chemistry 2023 |