
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssessing the bio-stability of microRNA-146a conjugated nanoparticles via electroanalysis†
Chaimae
El Ghzaoui
a,
Craig J.
Neal
 a,
Elayaraja
Kolanthai
a,
Yifei
Fu
a,
Udit
Kumar
a,
Junyi
Hu
b,
Carlos
Zgheib
d,
Kenneth W.
Liechty
d and
Sudipta
Seal
*ac
a,
Elayaraja
Kolanthai
a,
Yifei
Fu
a,
Udit
Kumar
a,
Junyi
Hu
b,
Carlos
Zgheib
d,
Kenneth W.
Liechty
d and
Sudipta
Seal
*ac
aAdvanced Materials Processing and Analysis Center, Dept. of Materials Science and Engineering, University of Central Florida, Orlando, FL, USA. E-mail: Sudipta.Seal@ucf.edu
bDepartment of Surgery, School of Medicine, University of Colorado Denver, Anschutz Medical Camps, Aurora, Colorado, USA
cCollege of Medicine, Nanoscience Technology Center, University of Central Florida, Biionix Cluster, Orlando, FL, USA
dLaboratory for Fetal and Regenerative Biology, Department of Surgery, College of Medicine, University of Arizona, Tucson, AZ, USA
First published on 15th November 2022
Abstract
The number of diabetics is increasing worldwide and is associated with significant instances of clinical morbidity. Increased amounts of reactive oxygen species (ROS) and proinflammatory cytokines are associated with the pathogenesis of diabetic wounds and result in a significant delay in healing. Our previous studies have shown the ability of a cerium oxide nanoparticle (CNP) formulation conjugated with the anti-inflammatory microRNA miR146a (CNP-miR146a) to enhance the healing of diabetic wounds. The observed therapeutic activity exceeded the combined efficacies of the individual conjugate components (CNPs and miR146a alone), suggesting a synergistic effect. The current study evaluates whether the previously observed enhanced activity arises from increased agent delivery (simple nanocarrier activity) or is specific to the CNP-miR146a formulation (functional, bio-active nanomaterial). Comparison with miR146a conjugated gold (bioactive, metal) and silica (bioinert, oxide) nanoparticles (AuNPs and SiO2NPs) was performed in the presence of H2O2, as an analogue to the high levels of ROS present in the diabetic wound environment. Electrochemical studies, materials characterization, and chemical assays showed limited interaction of AuNP-miR146a with H2O2 and instability of SiO2NP-miR146a over time. In contrast, and in support of our prior results, CNP-miR146a displayed chemical stability and persistent ROS scavenging ability. Furthermore, it was determined that CNPs protect miR146a from oxidative damage under prolonged exposure to H2O2, whereas AuNPs and SiO2NPs were shown to be ineffective. Overall, these results reinforce the ability of CNPs to stabilize and protect miRNA while exhibiting robust antioxidant properties, suggesting that therapeutic activity observed in related earlier studies is not limited to a facile nanocarrier function.
1. Introduction
The wound healing process involves coordination between multiple complex cellular processes including cell migration, proliferation, and tissue remodelling.1 However, for individuals afflicted with diabetes, these events are dysregulated, causing impaired and delayed healing.2 In diabetic wounds, the presence of high levels of reactive oxygen species (ROS) and continuous inflammation3 have been implicated in the pathogenesis of this impairment. This problem has raised an ongoing clinical concern and gained the interest of many researchers.Micro-RNAs (miRNAs) are small non-coding RNA molecules containing 19 to 24 nucleotides and have an extensive role in controlling gene expression.4 Due to their broad range of functions across varied molecular pathways (spanning cell proliferation, metabolism, and cell death), study and therapeutic investigation of these bio-molecules have been the subjects of extensive investigation.5 MiRNA dysregulation has further been identified as contributing to the pathogenesis and progression of disease states.5 MiR146a is a miRNA which has been identified as contributing to the regulation of the inflammatory immune response during the wound healing phase, through the reduction of IL-6 and IL-8 expression leading to decreased activation of the proinflammatory NFkB pathway.6 MiR146a has been shown to be dysregulated in diabetic wounds and supplementation of this miRNA has been shown to enhance the healing of diabetic wounds.6–9 While miRNAs have shown substantial efficacy in treating certain conditions, these bio-molecules are typically short-lived in the cell environment. Specifically, the negative surface charge of bare miRNA limits cellular uptake due to electrostatic repulsion between the cell membrane and the miRNA. This hindrance is in addition to the tendency of miRNA species to undergo oxidative degradation before reaching gene targets.5,10,11 Researchers have mitigated these issues by conjugating the miRNA to different compositions of inorganic nanoparticles, enhancing delivery to target cells and stabilizing the miRNA by neutralizing its surface charge.5,12
Cerium oxide nanoparticles (CNPs) are widely used for bio-applications due to their ability to change the valence state reversibly between Ce3+ and Ce4+, enabling certain redox reactions. The equilibrium ratio of Ce3+/Ce4+ can determine the properties of the synthesized CNPs. For instance, CNPs' ROS scavenging activity was shown in a high Ce3+/Ce4+ ratio, while catalase activity has been consistently observed in the case of smaller ratios.10,13,14 This property is attributed to the presence of oxygen vacancies in the crystal structure particles, which mediate the formation of Ce3+.14,15 CNPs have been demonstrated to function as a catalytic antioxidant and to regulate oxidative stress by scavenging ROS.13 In our previous studies, we have demonstrated that the conjugation of CNPs to miR146a (CNP-miR146a) significantly enhanced diabetic wound healing in murine and porcine animal models of diabetic wound healing.7–9 The CNP-miR146a conjugate improved the extent of diabetic wound healing and reduced the overall healing time to values comparable with non-diabetic wounds.3
The success of CNP-miR146a in enhancing the wound healing process has encouraged us to further investigate the physicochemical characteristics of the conjugate which may determine its therapeutic efficacy. Thus, we hypothesize that the high surface reactivity of CNPs and improved stability of miRNA in the presence of ROS will lead to a substantially greater therapeutic effect of CNP-miR146a.
In the presented study, a CNP formulation conjugated with miR146a was characterized in the presence of hydrogen peroxide, a by-product of the wound repair process. Changes to either conjugate component were investigated via traditional materials characterization methods, electroanalytical methods, and chemical assay. Furthermore, the uniqueness of CNP-miR146a’s ability to accelerate diabetic wound healing is compared against two comparable nanomaterial compositions in all characterization methods. Gold and silica nanoparticles of comparable size and shape to the previously tested CNPs were chosen due to their prevalence in the nanomedicine literature. Silica nanoparticles were used in many drug delivery applications because of the ease of controlling the nanoparticles' size and their remarkable biocompatibility, in addition to their stability in the biological medium.16–18 SiO2NPs have been used in wound applications, in which amine-functionalized SiO2NPs showed good wound closure performance,19 and they were employed as a carrier for different types of miRNAs. Tivnan et al. have employed SiO2NPs as a vehicle for miR34a to suppress tumors by hindering neuroblastoma expansion.5,20 Gold nanoparticles are also known in the literature for their biocompatibility and facility of synthesizing with different sizes and morphologies, in addition to their antibacterial activity and ability to be easily factionalized.5,21–24 Due to these benefits, many groups have used AuNPs as a miRNA delivery vector.25 AuNPs conjugated with miR133b were employed to inhibit scar formation and repair corneal wounds by downregulating type-I collagen and α-smooth muscle actin.10,26 AuNPs were also conjugated with miRNA-145 to regulate the miRNA presence in prostate and breast cancer cells.27 Results from this study confirm that CNPs are unique, among the tested nanomaterials, in their ability to confer protection to adsorbed miR146a from oxidative damage. Thereby, we demonstrate nanozyme ROS scavenging behavior and a miRNA conjugate stabilizing functionality beyond a simple nanocarrier function. We further note that results from this study are generalizable to arbitrary miRNA compositions, suggesting that CNPs conferring stability may be utilized in any application benefitting from miRNA supplementation.
2. Experimental section
2.1 Materials
Cerium nitrate hexahydrate (Ce (NO)3, 6H2O; purity > 99.999) and 3% hydrogen peroxide (H2O2) were purchased from Sigma Aldrich. Gold and silica nanoparticles of sizes 10 nm and 08 nm, respectively, were purchased from Cytodiagnostics and nanoComposix. MiR146a was purchased from Integrated DNA Technologies IDT. Murine dermal fibroblasts (MDFs), human umbilical vein endothelial cells (HUVECs), vascular endothelial cell growth kit (VEGF kit), cell culture medium, FBS (fetal bovine serum), and other cell culture reagents were procured from ATTC (American Type Culture Collection). The live/dead assay was procured from Invitrogen. Dimethyl sulfoxide solvent, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT assay) powder, cell culture well plates and flasks were purchased from Fisher Scientific.2.2 MiR146a conjugated nanoparticles
Cerium oxide nanoparticles (CNPs) of 5 nm size were synthesized by a wet chemistry procedure according to our previous work.28 Briefly, Ce(NO)3, 6H2O was dissolved in ultrapure water, and then an excess of H2O2 was added as an oxidizer. The resulting solution was aged for 6 to 8 weeks until the samples became colorless. Silica and gold nanoparticles (SiO2NPs and AuNPs) were purchased from commercial suppliers and used without any change.For the miR146a conjugation, we used the same protocol described in our previous work7 for each CNP, AuNP, and SiO2NP. In short, 30 μL of each sample was resuspended in dimethyl sulfoxide (DMSO). Then, the chemical crosslinker 1,1′-carbonyldiimidazole (CDI) was added to activate the functional groups on the nanoparticles' surfaces towards subsequent miRNA conjugation (the hydroxyl group on the SiO2NP, CNP, and AuNP surfaces, as well as the carboxyl group on the AuNP surface since the commercial AuNPs used in our study contain both COOH and OH groups), resulting in imidazole intermediate state formation (Fig. 1). This chemical group reacts with the amine group in miR146a, leading to the conjugation of miR146a to the surfaces of CNPs, AuNPs, and SiO2NPs. The conjugated samples are named CNP-miR146a, AuNP-miR146a, and SiO2NP-miR146a. The conjugated samples were dialyzed against RNase-free water to remove DMSO and unbound miR146a. The samples were stored at −20 °C for conservation until further use.
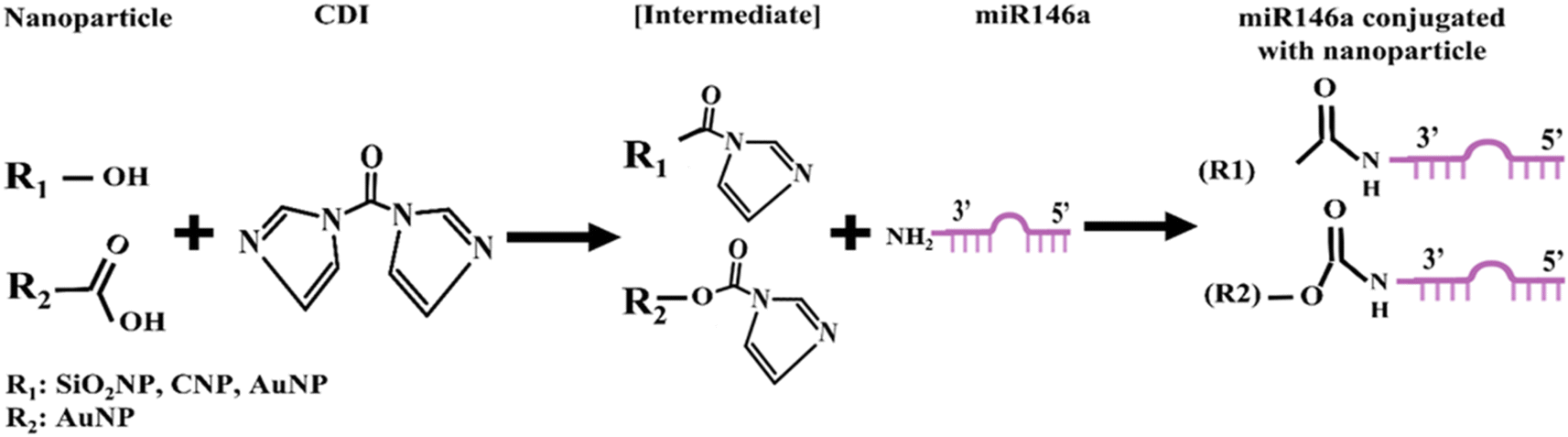 | ||
| Fig. 1 Schematic diagram of miR146a chemically conjugated with cerium oxide, gold, and silica nanoparticles by the bioconjugation method. The CDI molecule was used as a connecting agent between miR146a and nanoparticles at room temperature. | ||
2.3 Materials characterization
UV-vis spectrophotometry analysis was used to determine the concentration of bare and conjugated gold nanoparticles. A PerkinElmer spectrophotometer was employed over a range of 220–800 nm, and the concentration estimation was derived from the peak intensity at 520 nm wavelength. The concentration of cerium oxide nanoparticles was calculated using inductively coupled plasma-mass spectrometry (ICP-MS). The operated instrument is in the Trace Evidence Analysis Facility at Florida International University. Silica nanoparticles were diluted from stock concentrations provided by a commercial supplier and approximated from ICP-MS data. The synthesized nanoparticles' morphology and size were characterized using a Philips Tecnai 300 kV HRTEM. The samples were coated on the TEM grids and dried under UV light. Zeta potential values were measured by operating dynamic light scattering measurements on a Zeta Sizer Nano (Malvern Instruments), using a 633 nm wavelength laser. Quantification of the miR146a concentration in conjugated samples was performed using a molecular probe Quanti-iT microRNA assay kit (Catalog number: Q32882) and carried out as per the manufacturer's protocol. The chemical composition of all the samples was determined through X-ray photoelectron spectroscopy (XPS) analysis. XPS studies were performed using a Thermo Scientific ESCALAB 250Xi spectrometer. Experiments were performed under high vacuum conditions (Pressure ∼10−10 mbar) with Al-Kα radiation as the source. Samples were prepared by drop-casting on a silicon or gold substrate depending on the nature of the sample. XPS data were analyzed using Avantage software. The C 1s scan C–C peak at 284.6 eV was taken as the standard for peak shifting. A Fourier transform infrared (FTIR) spectrometer was used to evaluate the functional group composition of CNP-miR146a, AuNP-miR146a, and SiO2NP-miR146a, before and after 6 hours of exposure to 10 mM H2O2. The samples were mixed with KBr powder and dried in a vacuum desiccator for 12 hours. The range of the FTIR spectrum used was the 600–4000 cm−1 region with 8 cm−1 resolution in the PerkinElmer infrared spectrometer. Baseline correction for IR spectra was performed using a function in Origin software.2.4 Electrochemistry measurements
All electrochemical measurements were performed using a multi-channel potentiostat from BioLogic. A round disk glassy carbon electrode (BioLogic) was used as the working electrode in all experiments with a platinum mesh counter electrode and Ag/AgCl reference electrode (saturated NaCl electrolyte; purchased from BioLogic). Prior to any experiment, the working electrode was cleaned via slurry polishing on a polishing pad and subsequently via electrochemical cycling from −0.4 to 1.6 V at a scan rate of 1 V s−1 over 1000 cycles. The cleaned glassy carbon electrode was coated with 20 μL of 5 mM cerium oxide nanoparticles and dried under vacuum in a desiccator for 6 hours. Then, cyclic voltammetry (CV) experiments were performed over a potential range of −0.3 V to 0.7 V at a scan rate of 20 mV s−1 for ten cycles in an electrolyte solution containing a mixture of H2O2/PBS (10 mM 10 mM−1) solution.Before conducting chronoamperometry (CA) measurements, each modified GC electrode underwent two CV cycles in 10 mM PBS to inspect its performance.29 The CA measurements were performed at a fixed potential of 0.65 V with continuous stirring of the electrolyte solution, which initially contained 49 ml of 10 mM PBS. The reference electrode used is Ag/AgCl/sat.NaCl while the counter electrode was the platinum mesh. The first H2O2 addition took place after 7 min of CA measurement in PBS, ensuring the system's stability and allowing the current equilibrium. The stepwise H2O2 addition was at concentrations of 0.05, 0.1, 0.2, 0.4, 0.8, 5, 8, 12, 16, and 20 mM with 2 min time gap between each addition. The PBS concentration of the electrolyte solution was maintained at 10 mM throughout the entire CA by adding 100 μL of 50 mM PBS in each 900 μL addition of H2O2.
Potentiostatic electrochemical impedance spectroscopy (EIS) was performed versus an open circuit and versus 0.65 V potential in a frequency range of 10 MHz to 100 KHz with a sinus amplitude of 12 mV. The same protocol of EIS measurement was repeated for all samples: CNPs, CNP-miR146a, AuNPs, AuNP-miR146a, SiO2NPs, SiO2NP-miR146a, and miR146a. All the samples were deposited on the working electrode the same way as described in the CV experiments.
2.5 Enzyme–mimetic assay
Superoxide dismutase (SOD) mimetic activity was measured by using home kit assay. Reduction of WST-1 was utilized to assess the kinetics of the SOD mimetic activity of nanoparticles and nanoparticle conjugates. The protocol was slightly adjusted from that of the Peskin et al. study.30 Briefly, superoxide was generated by a 0.1 mM hypoxanthine/xanthine oxidase system. The generation rate of superoxide was controlled by adjusting the xanthine oxidase concentration, resulting in an absorbance change at 438 nm of approximately 0.025 units per minute. Each assay was conducted at room temperature for 20 min in a 96-well plate with a total volume of 240 μL containing 20 μL of sample. All reactions were buffered with 50 mM Tris buffer of pH 7.5. The SOD activity is calculated based on the below equation:
2.6 Cytotoxicity study
Human umbilical vein endothelial cells (HUVECs) were cultured with the VEGF-endothelial cell growth kit supplemented with the vascular endothelial growth medium at 37 °C in a 5% CO2 atmosphere. The cells were detached from the culture flask after 85% confluency with a 0.1% trypsin–EDTA solution, and then the cells were collected and washed by centrifugation. The washed cells of 1 × 104 per well were added to a 96 well plate and it was incubated at 37 °C under CO2 conditions for 12 h to attach the cells to the culture plate. After 12 h of incubation, the medium was removed, and two different concentrations of 0.2 ng μL−1 and 0.4 ng μL−1 of pure and miR146a conjugated (CNPs, AuNPs, SiO2NPs, CNP-miR146a, AuNP-miR146a, and SiO2-miR146a) nanoparticles containing the vascular endothelial growth medium were added to HUVECs and incubated for one and two days to study the cytotoxicity before and after conjugation of these nanoparticles. After a pre-determined time interval, 20 μL of (5 mg ml−1) 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution was added to a 200 μL growth medium containing each well and incubated for 4 h in a CO2 incubator. Then, MTT solution was removed and 200 μL of dimethyl sulfoxide solvent was added to each well to dissolve the formazan crystal. The optical density of these solutions was measured at 570 nm in a BMG Labtech multi-plate reader. Without nanoparticles, treated cells were considered the control. The experiment was conducted in triplicate for each sample. Furthermore, the live/dead assay was performed on treated cells with and without a nanoparticle concentration of 0.4 μg μL−1 to determine the cytotoxicity of CNPs, AuNPs, SiO2NPs, CNP-miR146a, AuNP-miR146a, and SiO2-miR146a. After removing the culture medium, the cells were stained using the molecular probes live/dead assay kit. A mixture of calcein and ethidium homodimer (2 mM 4 mM−1) solution was added to miR146a conjugated samples, with and without nanoparticle-treated cells, and was incubated at 37 °C in a CO2 incubator for 30 min. Live and dead images were imaged using a Nikon FL microscope. The HUVECs were used for the experiment in the third passage cells.2.7 CNP-miR146a distributed throughout the cytoplasm
Under In vitro conditions, skins from adult mice were placed dermal side down to allow fibroblast outgrowth. Dermal fibroblasts were cultured in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and antibiotics. Cultures were maintained in a humidified incubator in an atmosphere of 5% CO2 and 95% air. To examine the uptake and distribution of our CNP-miR-146a conjugate, we incubated murine dermal fibroblasts with 0.1 μg of our CNP-miR146a conjugate for 24 h and processed them for TEM analysis. We found that our conjugates were distributed throughout the cytoplasm, seen as dark black dots within the cytoplasm and indicated by the white arrows in Fig. 9.2.8 CNP-miR146a decreases ROS production in macrophages
RAW 264.7 macrophages (ATCC, USA) were maintained in DMEM (Gibco, USA) supplemented with 10% (v/v) FBS (Gibco, USA), 100 μg per ml streptomycin, and 100 U per ml penicillin, and incubated at 37 °C in an atmosphere of 5% CO2. Cells were serum-starved overnight in DMEM with 1% FBS, 100 μg per ml streptomycin, and 100 U per ml penicillin before being treated with PBS or CNP-miR146a. ROS measurements were performed using a MarkerGene Live Cell Fluorescent Reactive Oxygen Species Detection Kit from Marker Gene Technologies, Inc. These cultured murine macrophages were treated for 24 h with either PBS or 0.1 μg CNP-miR146a.2.9 Real-time qPCR
Gene expression was analyzed using murine dermal fibroblasts (MDFs). MDFs were cultured in a full medium comprising Dulbecco's modified Eagle high-glucose medium (DMEM, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 20% fetal bovine serum (FBS) + 1% antibiotic–antimycotic and maintained at 37 °C in a humidified atmosphere containing 5% CO2. For further experiments, cells were seeded and cultured for 12 h. After that, cells were serum-starved for 16 h and then treated with biocompatible 0.2 ng μL−1 of CNPs, CNP-miR146a, SiO2NPs, SiO2NP-miR146a, AuNPs, and AuNP-miR146a for 6 h. Following incubation, total cellular RNA was isolated from the cells by homogenizing in a Qiazol (Qiagen) as per manufacturer’s instructions. Isolated RNA was converted to cDNA (Applied Biosystems RT kit), to analyze IL-6 gene expression, where GAPDH, a housekeeper gene, was used for normalization. Another set of RNA samples was diluted to 5 ng μL−1 by serial dilution and converted to miR146a and U6 (housekeeping miRNA) cDNA (Applied Biosystems RT kit), which was amplified by reverse transcriptase amplification. A real-time quantitative polymerase chain reaction (RT-qPCR) was performed for IL-6, GAPDH, miR146a, and U6 using a BioRad CFX-9600 thermal cycler. PCR analysis was performed in triplicate, with the average of each triplicate used for normalization. One-way ANOVA with Tukey's test was used for the statistical analyses for multiple comparisons of various nanoparticle-treated samples. Statistical differences were considered p < 0.05.3. Results and discussion
3.1 Material synthesis and miRNA quantification
The synthesis of CNPs was based on simple aqueous chemistry, as detailed in the previous section, while gold (AuNPs) and silica (SiO2NPs) nanoparticles were obtained from commercial suppliers. The conjugation of the nanoparticles to miR146a was achieved via standard conjugation chemistry using the crosslinker 1,1′-carbonyldiimidazole, as detailed in our previous publication.7 To confirm the conjugation of miR146a to each of the AuNPs, SiO2NPs, and CNPs, a commercial miRNA assay kit was used and the miR146a content was quantified. The concentration of miR146a and the loading efficiency for each type of conjugated nanoparticle were measured. The assay shows that the miR146a molecules are present in all three particle formulations and that the amounts of miR146a loaded on CNPs, SiO2NPs, and AuNPs were 7.8 ± 0.4 ng μL−1, 6.5 ± 0.8 ng μL−1, and 7.8 ± 2.3 ng μL−1, respectively. The loading efficiency for miR146a (calculated as: (massmiR146a, final/massmiR146a, initial) × 100) is similar for the AuNPs and CNPs (46%), while for SiO2NPs the value is slightly lower (38%). These results indicate a material composition-specific miR146a loading efficiency. This variation will lead to a lower dose of miR146a per particle for the SiO2NP formulation under ideal conditions. In addition, the slight difference in sizes and theoretical densities (because of different crystal structures) of AuNPs (19.3 g cm−3), SiO2NPs (2.65 g cm−3), and CNPs (7.22 g cm−3) between the control nanoparticles will also have an impact on the number of miR146a per nanoparticle. The theoretical estimation number of miR146a per nanoparticle of CNPs, SiO2NPs, and AuNPs is 112, 15, and 478, respectively (the used approach and the assumptions used are detailed in the ESI†).To determine the relative stability of conjugated miR146a given each nanomaterial composition and to consider the utility of each conjugate formulation, we next characterized the effects of the conjugation process on material surface chemistry.
3.2 Surface chemical state analysis
X-ray photoelectron spectroscopy (XPS) was performed on CNPs, CNP-miR146a, SiO2NPs, SiO2NP-miR146a, AuNPs, and AuNP-miR146a to analyze the nanoparticles' surface chemical states and determine any changes resulting from conjugation. The Ce 3d and C 1s scans for control CNPs are shown in Fig. 2a and b. The high-resolution Ce 3d spectra give information about the oxidation state at the surface of the particles and enable the assessment of the Ce3+/Ce4+ ratio.31 For the unconjugated CNP sample, the peaks corresponding to Ce3+ were situated at 882.2, 886.1, 900.1, and 904.4 eV; while the peaks corresponding to Ce4+ were found at 883.88, 888.3, 901.2, 902.6, 908.0 and 916.8 eV. For the CNP-miR146a sample, the Ce 3d scan is shown in Fig. 2c, where the peaks corresponding to Ce3+ were situated at 881.7, 885.3, 897.7, and 903.2 eV; the peaks corresponding to Ce4+ were situated at 883.5, 887.9, 899.3, 900.9, 906.3 and 916.1 eV. Measurement of the Ce3+/Ce4+ ratio is critical since it affects CNP's catalytic and biomedical properties.13,32 The value can be estimated by using the area under the curve values for peaks corresponding to a given redox state.28,32 The conjugation of CNPs to miR146a resulted in a decrease in the Ce3+/Ce4+ ratio (from 3.96 to 1.23). Since the XPS analysis provides surface-dependent estimated values, the observed shift in the Ce3+/Ce4+ ratio could occur through oxidation of Ce3+ sites as a result of the conjugation process (e.g., from the conjugation reaction occurring at pH 8.5). For AuNP-miR146a and SiO2NP-miR146a samples, the presence of gold and silica was confirmed by elemental gold (Au 4f5/2 and Au 4f7/2) (Fig. 2e) and silicon-oxide peaks (Si 2p) (Fig. 2g). The control AuNPs and SiO2NPs are presented in Fig. S1.† In C 1s scans for all the conjugated samples, we have observed peaks corresponding to C–C (284.6 eV, used as the standard), C–O (286.15 ± 0.12 eV), and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (287.73 ± 0.45 eV) as well as a π–π* satellite peak (289.38 ± 0.01 eV) in the control sample. The presence of different carbon functional group peaks in the C 1s scan for the conjugated samples confirms the presence of miRNA in each of the CNP, AuNP, and SiO2NP samples.
O (287.73 ± 0.45 eV) as well as a π–π* satellite peak (289.38 ± 0.01 eV) in the control sample. The presence of different carbon functional group peaks in the C 1s scan for the conjugated samples confirms the presence of miRNA in each of the CNP, AuNP, and SiO2NP samples.
 | ||
| Fig. 2 XPS characterization of nanoformulations. XPS scans for control ceria nanoparticles (CNPs) are shown in (a) for the Ce 3d and (b) for the C 1s binding regions. Scans for CNP-miR146a are shown in (c) for the Ce 3d and (d) for the C 1s binding regions. Similarly, scans for AuNP-miR146a and SiO2NP-miR146a are shown in (e) Au 4f and (f) C 1s and (g) Si 2p, and (h) C 1s, respectively. In the control CNP and CNP-miR146a samples’ Ce 3d scans, peaks corresponding to Ce3+ and Ce4+ oxidation states are indicated and the Ce3+/Ce4+ ratio was estimated using the area under the curve of the corresponding peaks. Comparisons between these values show a clear difference; the ratio decreased from 3.96 to 1.23 from the CNP control to the CNP-miR146a sample. In the AuNP-miR146a sample, we have observed peaks corresponding only to metallic (Au0) Au (Au 4f5/2 and Au 4f7/2). In the Si 2p scan of the SiO2NP-miR146a sample, we have observed peaks corresponding to oxide and elemental Si. In C 1s scans for all conjugated samples, we have observed peaks corresponding to C–C, C–O, and CO. The C 1s scan for control CNPs has peaks corresponding to C–C, C–O, and a satellite peak. | ||
3.3 Surface morphology and size analysis
High-resolution transmission electron microscopy (HRTEM) was used to determine the nanoparticles' morphology and average size (n = 50) before and after conjugation with miR146a. Fig. 3 indicates that the uniform spherical morphology and colloidal dispersity of AuNPs, SiO2NPs, and CNPs (Fig. 3a–c) have not changed appreciably after miR146a conjugation (Fig. 3g–i). Fig. 3d and j show that the average diameter of AuNPs has slightly increased due to the conjugation process from 7.3 ± 1.0 nm (AuNPs) to 7.6 ± 1.1 nm (AuNP-miR146a). Lattice fringes observed in the (inset image) AuNP-miR146a images demonstrate the crystallinity of the conjugated nanoparticles. Fig. 3e and k display the change in the size of SiO2NPs from 4.9 ± 1.1 to 5.2 ± 0.7 nm in the conjugated sample. Lattice fringes were not observed in SiO2NP and SiO2NP-miR146a samples due to their amorphous structures. CNP conjugation to miR146a was suggested by an observed increase in the particle size from 5.0 ± 1.2 nm (bare CNP) to 5.5 ± 0.7 nm (CNP-miR146a) after conjugation (Fig. 3c and i). The inset image (Fig. 3i) shows the crystallinity of cerium oxide. Selected area electron diffraction patterns (Fig. S2)† were collected from Fig. 3g and i to confirm the crystal structures of AuNP-miR146a and CNP-miR146a. The Debye–Scherrer diffraction rings in the SAED pattern from Fig. S2† can be indexed to the crystal planes, (111), (200), (220), and (311), of a cubic fluorite structure for cerium oxide and the face centered cubic structure of gold. We have conducted zeta potential measurements on the conjugated samples for further investigations. The results have shown a high negative charge potential of the conjugated nanoparticles (CNP-miR146a: −30.3 mV, AuNP-miR146a: −28 mV, and SiO2NP-miR146a: −25 mV), suggesting that the limited agglomeration present in TEM imaging is relatively weak (possibly due to the measurement sample preparation character) and the colloidal stability of conjugated samples is due to the electrostatic repulsion. Further relevant discussions are made in later sections. | ||
| Fig. 3 TEM characterization. HRTEM images of (a) AuNPs, (g) AuNP-miR146a, (b) SiO2NPs, (h) SiO2NP-miR146a, (c) CNPs, and (i) CNP-miR146a. (d) AuNPs exhibit an average size of 7.3 ± 1.0 nm, (j) AuNP-miR146a shows an average size of 7.6 ± 1.1 nm, (e) SiO2NPs show an average size of 4.9 ± 0.7 nm, (k) SiO2NP-miR146a displays an average diameter size of 5.2 ± 0.7 nm, (f) CNPs show an average diameter size of 5.0 ± 1.2 nm, and (l) CNP-miR146a shows an average diameter size of 5.5 ± 0.7 nm. Particles from each formulation show a comparable spherical morphology and average particle size, suggesting character in these respects and allowing for mutual comparison. Furthermore, particles appear well-dispersed and relatively uniform. (Scale bar in a, b, c, g, h, and i is 10 nm). | ||
Given the confirmation of nanoparticle presence in the conjugated samples from TEM, SAED, and XPS, we next sought to determine any changes in material chemical states upon interaction with an oxidizing agent. Specifically, we looked to identify the stability of the conjugated particles towards oxidation, as a model of the ROS-containing chemical environment of a diabetic wound.
In diabetic wounds, H2O2 is produced in excess and can be a source of other ROS molecules.33 Additionally, the lifetime of H2O2 in solution is substantially greater than most other ROS species and thereby, we suggest an appropriate surrogate molecule to analyze and, ideally, generalize ROS-dependent material behavior. Understanding the behavior and any changes in material properties for each particle formulation in oxidative environments is necessary for approximating (modelling) therapeutic efficacy in the wound environment. In determining the conjugated nanoparticle formulations' stability in harsh oxidizing environments, such as in a diabetic wound or cells, voltametric measurements in phosphate buffered saline (PBS) were produced in the presence of H2O2.
3.4 Electrochemical response of formulations to hydrogen peroxide
To interpret the extent of material interaction with H2O2, particles from each formulation were drop cast onto an electrochemically cleaned glassy carbon (GC) electrode surface and allowed to dry under vacuum. In this approach, peroxide-related redox reactions at the miRNA-particles are detected at the nanomaterial-modified electrode surface.From Fig. 4a, it was found that the onset potential of the oxidation reaction for conjugated ceria CNP-miR146a (390 mV) is lower than that of the unconjugated CNP sample (450 mV) and pure miR146a (520 mV), suggesting activation towards oxidative electron transfer. Additionally, CNP-miR146a demonstrates a higher anodic current (29 μA) compared to the two other samples (Fig. 4a), suggesting that a higher fraction of charge is transferred relative to the other samples (CNPs: 20 μA and miR146a: 13 μA). Additionally, it should be noted that the observed current density was stable over the measured cycle range. From the literature, it was found that CNPs are reactive towards H2O2 through redox cycling of cerium sites (Ce3+ ↔ Ce4+), with this behavior being analogous to that of antioxidant enzymatic reactions.13,29,34 In support of the high current density observed for the conjugated material, the Ce3+/Ce4+ ratio was seen to decrease for CNPs after conjugation with miR146a from 3.96 to 1.23 (as determined from XPS measurements, Fig. 2). It was determined from previous studies that the catalase activity of ceria possesses an inverse relationship to Ce3+/Ce4+ and a positive relationship with measured SOD activity.13 Since a higher relative fraction of Ce4+ is required to degrade H2O2 to oxygen and water (catalase activity of cerium) with fast kinetics,13 our results can be explained using the following reaction, suggested by Kosto et al.:29
| 2Ce3+ + H2O2 → Ce4+ + Ce4+ − OAds. + H2O + 2e− | (1) |
| Ce4+ + Ce4+ − OAds. + H2O2 → 2Ce3+ + H2O + O2 + 2e− | (2) |
 | ||
| Fig. 4 CV of nanomaterial-modified glassy carbon (GC) vs. Ag/AgCl in PBS/H2O2 (10 mM 10 mM−1) over the potential range −0.3–0.7 V and a scan rate of 20 mV s−1 for 10 cycles. The 10th cycle of (a) CV curves of CNP, conjugated CNP-miR146a, and bare miR146a modified GC electrodes show that the onset potential (Eonset) for oxidation of CNP-miR146a is lower than that for unconjugated CNPs or miR146a. (b) CVs of AuNP, conjugated AuNP-miR146a, and bare miR146a modified GC electrodes indicate a total current decrease and disappearance of the oxidation peak from AuNPs ascribed to the surface coverage of gold nanoparticles with miR146a. (c) CVs of SiO2NP, conjugated SiO2NP-miR146a, and bare miR146a modified GC electrodes show that the current corresponding to conjugated SiO2NP-miR146a has a higher oxidation current. Subsequent CV curves of (d) SiO2NPs show that the subsequent cycles are consistent and overlap suggesting the stability of the SiO2NP modified electrode in the electrochemical cell. (e) Conjugated SiO2NP-miR146a shows a variation over subsequent cycles, especially in the reduction reaction implying that the conjugation of miR146a to SiO2NP leads to material changes and instability. (f) Conjugated CNP-miR146a displays consistency in subsequent cycles, which implies stability of the synthesized CNP-miR146a nanoparticles in the electrochemical cell. | ||
The degradation of hydrogen peroxide may be occurring at the surface of CNP-miR146a as it does in the case of bare ceria-nanoparticles and lead to the creation of vacancies in the crystal structure of ceria.35 Alternatively, peroxide may react with miRNA moieties, allow charge transfer via a hopping mechanism preserving the miRNA composition, and result in electron transfer to the electrode via ceria redox.36
As shown in Fig. 4b, the electrode modified with AuNPs has a roughly 40 μA peak anodic current. However, conjugation of AuNPs with miR146a leads to loss of current likely due to the blockage of gold surface sites. As shown in Fig. 4c, SiO2NP-miR146a showed an anodic peak current of 21 μA, which is higher than that of both bare SiO2NP (14 μA) and miR146a (13 μA). This trend is almost similar to CNP-miR146a behavior with a slight difference in anodic peak current following conjugation (SiO2NP-miR146a: 21 μA and CNP-miR146a: 29 μA). By considering the observed performances of all conjugated particle compositions, we determine that SiO2NP-miR146a and CNP-miR146a have a more pronounced reactivity towards H2O2 over AuNP-miR146a. These results led us to investigate whether the observed reactivity was coming from the nanoparticle side (exchange of electrons between Ce3+ and Ce4+) or the miR146a side (oxidation of miRNA), as discussed in subsequent Sections (3.5 and 3.6).
Given the observed sensitivity of conjugated CNPs and silica formulations towards peroxide, we next sought to determine the stability of these reactions over time.
As shown in Fig. 4d the CV of a GC modified electrode with bare SiO2NPs shows that the system is stable over electrochemical cycling, while the CV of conjugated SiO2NP-miR146a (Fig. 4e) displays a variation in cycle character with increasing cycle number: suggesting material instability. Since bare silica was stable before conjugation (Fig. 4d), there is a possibility that miR146a in the conjugated nanoparticles becomes degraded. As shown in Fig. 4f, the subsequent cycles were consistent and stable on the CV of the modified GC with conjugated CNP-miR146a nanoparticles, suggesting stability and preservation of the material. By comparing the CVs of the three types of conjugated nanoparticles, it was found that CNP-miR146a shows higher anodic current (29 μA Vs. 21 and 10 μA for silica and gold conjugate nanoparticles) and greater system stability in the presence of hydrogen peroxide, implying a potential catalytic/regenerative surface activity towards ROS.
The sensitivity of the conjugated particle formulations towards the peroxide surface reaction was investigated using chronoamperometry (CA) measurements (Vapplied: 0.65 V vs. Ag/AgCl, potential of instability for H2O2 (ref. 29 and 37)). Material sensitivity was investigated via serial additions of H2O2 in varied quantities in the range of 0.05–20 mM in a PBS electrolyte. The electrolyte concentration was maintained at 10 mM and pH ∼7.4 to avoid the production of cerium phosphate, which could modify the particle surface and impede H2O2 oxidation due to interference with the Ce4+/Ce3+ redox reaction.29,38Fig. 5a shows the addition-current response behavior of the three conjugated nanoparticle compositions. CNP-miR146a exhibits the highest current response in each addition of H2O2, followed by SiO2NP-miR146a and then AuNP-miR146a in agreement with our observations in CV measurements supporting the high reactivity of CNP-miR146a with H2O2. The calibration curves were obtained by plotting log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) I versus log[H2O2]. CNP-miR146a shows a linear trend (Fig. S3a†) with the regression equation:
I versus log[H2O2]. CNP-miR146a shows a linear trend (Fig. S3a†) with the regression equation:
| logI = 0.65log[H2O2] + 0.98 | (3) |
 | ||
| Fig. 5 Chronoamperometry (CA) measurements. (a) CA conducted on the modified glassy carbon electrode using CNP-miR146a, AuNP-miR146a, and SiO2NP-miR146a immersed in 10 mM PBS with successive additions of H2O2, achieving total H2O2 concentrations in the electrolyte solution of 0.05, 0.1, 0.2, 0.4, 0.8, 5, 8, 12, 16, and 20 mM with an interval of 2 min between each addition. The first H2O2 addition was conducted after 7 min of CA measurement in 10 mM PBS. (b) Calibration curve plots of I vs. [H2O2] in the range between 0.05 mM and 0.8 mM for CNP-miR146a, AuNP-miR146a, and SiO2NP-miR146a. | ||
Fig. S4a† shows only a negligible difference between the high electrochemical response to H2O2 of bare CNPs and conjugate CNP-miR146a. These results suggest the surface catalytic activity of CNPs, reported in many studies,29,34,39 is preserved in conjugated CNP-miR146a. Bare miR146a has a negligible electrochemical response with successive additions of H2O2 (analogous to the bare GC electrode response, Fig. S5a†). The higher response observed in the modified CNP-miR146a GC electrode may be due to the reversible CNP redox and the adsorption of oxygen-containing species on the nanoparticles' surface.39 Furthermore, in some studies, CNP formulations have shown high protection of DNA/miRNA against oxidation from various sources of oxidative damage.33,38 One such study noted that the integrity of bound DNA molecules may be preserved through DNA phosphate groups adsorbing to the ceria surface.40 In addition, the system shows stability over successive cycles in the CV experiments, contra-indicating the occurrence of miR146a oxidation for conjugated CNP-miR146a. A high level of current fluctuations is observable for ceria samples in Fig. 5a, which is ascribed to the dynamic nature of the CNP surface throughout the H2O2 reaction with the modified electrode (e.g., formation and transformation of adsorbed intermediates and surface species). Fig. S3b and c† show that SiO2NP-miR146a and AuNP-miR146a also have linear trends with regression equations of logI = 0.61log[H2O2] + 0.81 with R2 = 0.996 and logI = 0.59log[H2O2] + 0.76 with R2 = 0.990, respectively. The increased electrochemical response observed in SiO2NP-miR146a compared to bare SiO2NP and the decreased current for AuNP-miR146a (negligible response; no significant difference from bare GC and miR146a as shown in Fig. S5b†) compared to bare AuNP (Fig. S4b and c†) supports our findings in the CV experiments. The sensitivity of CNP-miR146a in CA measurements (determined as the slope of the fitted linear regression function) is the greatest among those of the tested material compositions at 0.65, as compared with 0.61 and 0.59 for conjugate miR146a with SiO2NPs and AuNPs, respectively. The measured difference was more substantial in the low concentration region of H2O2 (from 0.05 mM to 0.8 mM). This region and trend are plotted in Fig. 5b and show that CNP-miR146a has a higher overall affinity and sensitivity toward H2O2 (slope of 8.87) compared to the other two conjugate nanoparticles (slopes of 6.36 for SiO2NP-miR146a and 4.17 for AuNP-miR146a) over the entire measurement range.
3.5 Electroanalysis of peroxide and nanoparticle conjugate interaction by impedance spectroscopy
Potentiostatic electrochemical impedance spectroscopy (EIS) measurements were performed next to more precisely identify processes contributing to the electrochemical behaviors of test samples observed in CV and CA measurements. EIS measurements were conducted under the same conditions for all the samples. Data from these measurements (Fig. 6a–c) were fitted with a modified Randles circuit using Z-view software (inset images in Fig. 6). Rs is the solution resistance, representing measurement of IR drop. CPE1 is the constant phase element and represents an imperfect or non-ideal capacitor that models the complexity of the double-layer formation process at the surface of the modified electrode. R1 is the charge transfer resistance, representing charge transfer at the electrode surface. Finally, W is the Warburg diffusion element, and it models linear diffusion toward the electrode surface.41 The values of charge transfer and solution resistances obtained are represented in the inset images of Fig. 6. The solution resistance of the three different modified GC electrodes has nearly the same value, which confirms the similarity of electrolyte solution and conditions of the experiment. For charge transfer resistance (R1), the modified GC with AuNP-miR146a nanoparticles has the largest R1 (38Ω) value which is in accordance with the CV results. The R1 of CNP-miR146a is 13.48 Ω, while that of SiO2NP-miR146a is 0.38 Ω. Although the R1 of SiO2NP-miR146a is much lower than that of CNP-miR146a, the anodic current of these two samples is almost similar (Fig. 4). A potential explanation for these findings may be related to the low current evolved by OCP-fixing reactions for the various particle formulations. At OCP, current is largely transient, leading to negligible, and unstable, charge transfer across the electrode–electrolyte interface. SiO2NP-miR146a demonstrated less miRNA loading, relative to the other tested nanomaterial formulations, and, therefore, may produce lower resistance to electrolyte diffusion to the electrode or particle surface. | ||
| Fig. 6 Nyquist plots of a modified glassy carbon (GC) electrode in PBS/H2O2 (10 mM 10 mM−1) solution at OCP and 0.65 V. Nyquist plots at OCP of (a) modified GC with cerium oxide nanoparticles (CNPs) and conjugated CNP-miR146a. (b) Modified GC with AuNPs and conjugated AuNP-miR146a. (c) Modified GC with SiO2NPs and conjugated SiO2NP-miR146a. An equivalent electrical modified Randles circuit model (inset image) was used to fit the Nyquist plot data of the modified glassy carbon electrode with different miR146a conjugated nanoparticles at the OCP, where Rs is the solution resistance, R1 is the charge transfer resistance, W is the Warburg diffusion element, and CPE1 is the constant phase element. EIS measurements have also been conducted at 0.65 V (potential of instability for H2O2), and Nyquist plots were obtained for (d) CNP-miR146a, (e) AuNP-miR146a, and (f) SiO2NP-miR146a. Equivalent circuits for each sample are presented in the inset images. | ||
In addition to EIS performed at OCP, we have induced H2O2 degradation at 0.65 V (the potential of H2O2 degradation and instability) and investigated the corresponding behavior of the three different miR146a conjugated nanoparticles. The Nyquist plot of CNP-miR146a (Fig. 6d) displays a different behavior than those of SiO2NP-miR146a and AuNP-miR146a (Fig. 6e and f). In the case of CNP-miR146a, the semicircle at high frequency has a smaller diameter, implying low charge transfer resistance at the modified electrode surface. In addition, the EIS spectrum of CNP-miR146a has exhibited, at low frequency, a straight line with an angle of roughly 45° implying limited mass transfer of active electrolyte components,42 while the other two conjugate nanoparticles did not show the same character.
The three conjugate nanoparticle spectra displayed a second semicircle that is not clear in the Nyquist plot due to its overlap with the first one. This graph behavior can be explained by the presence of electrochemical processes with similar time constants.43 This justification is supported by Bode diagrams (Fig. S6†), where the phase angle plots show the presence of two peaks, suggesting the presence of two interfaces (RC components).43,44 The first interface can be attributed to the conjugate miR146a nanoparticles/electrolyte, while the second may be related to the oxidized material due to the degraded H2O2. The inset images in Fig. 6d–f represent the equivalent circuit of the complex impedance spectra. For CNP-miR146a and SiO2NP-miR146a, the first semicircle is represented by the charge transfer resistance R1 in parallel with the constant phase element CPE1 of the double-layer, while the second semicircle is represented by the R2//CPE2 circuit embedded within the first one. For AuNP-miR146a, the R1//CPE1 and R2//CPE2 are in series with each other. These differences in the equivalent circuits are also supported by the different behavior of AuNP-miR146a in the two other conjugated nanoparticles discussed before in CV measurements. As we can see from the values of R1, CNP-miRNA has exhibited a lower value (3379 Ω compared with 9716 Ω and 10947 Ω for AuNP-miR146a and SiO2NP-miR146a, respectively). This difference could be explained by the CNPs' high reactivity towards H2O2 due to their catalase activity and Ce3+/Ce4+ redox reactions, as supported by CA experiments. While SiO2NP-miR146a and AuNP-miR146a have shown a high resistance to electron transfer to the modified GC electrode at high frequencies,45 for R2, we can see only slight differences in values between the samples.
To study the influence of the accumulation of chemical changes on the conjugated nanoparticles due to excessive H2O2 exposure (at H2O2 instability potential), we have conducted four continuous successive repeats of the EIS measurements in the same electrolyte for each sample, and the results are presented in Fig. S5 and S6.† We can see clearly that the complex impedance spectrum in the CNP-miR146a case has conserved its shape, although there is a shift in peak frequencies in the Bode diagram (Fig. S6†) and an increase in the charge transfer resistance through the four spectral repeats (Fig. S7†). In contrast, the spectrum of each AuNP-miR146a and SiO2NP-miR146a measurement was significantly altered in shape with each successive measurement, suggesting substantial chemical changes at the modified electrode surface. The equivalent circuit of the fourth spectrum, provided in Fig. S7,† shows that CNP-miR146a has retained the same equivalent circuit across successive measurements. These results lead us to consider the possibility of complete oxidation of miRN146a and/or the irreversible modification of AuNP and SiO2NP surfaces, while in the case of CNPs, the Ce4+/Ce3+ redox reaction and catalase activity have protected the conjugate particles from severe oxidation and damage.
The detailed electrochemistry measurements have supported the unique and stable character of CNP-miR146a in the presence of hydrogen peroxide. To characterize the peroxide-nanomaterial conjugate interaction further, we conducted Fourier transform infrared spectroscopy (FTIR) and XPS to compare the effect of ROS on conjugated SiO2NP-miR146a, AuNP-miR146a, and CNP-miR146a chemical compositions following peroxide treatment.
3.6 Investigation of miR146a damage in the conjugated nanoparticles due to hydrogen peroxide exposure
The increased ROS in diabetic wounds can cause an intractable barrier to deliver miRNAs, which could undergo breakdown and inactivation in this high oxidative stress environment.33 To investigate between CNP, SiO2NP, and AuNP protection against miR146a damage in the presence of H2O2, which can degrade to highly reactive radicals, we have performed FTIR and XPS measurements on the miR146a conjugate nanoparticles (CNP-miR146a, AuNP-miR146a, and SiO2NP-miR146a) in the presence and absence of hydrogen peroxide.O, CC, and Si–H2O bonds has shifted to 1647 cm−1 indicating modifications in the miR146a bases.47 These changes in the band area at around 1526–1836 cm−1 (peak center at 1648.5 cm−1) and the observed shift can also be a sign of oxidation and reduction of the bases of miRNA due to the presence of ROS.48 For AuNP-miR146a, we can see (Fig. 7b) that the peak around the band from 976–1176 cm−1, centered at 1019 cm−1, has changed after H2O2 incubation. A new peak at 1096 cm−1 has appeared and was assigned to the symmetric stretching vibration of the PO2− group, indicating an alteration of chemical bonding in this region. In addition, C–H and CO for amide stretching were observed to undergo a +3 cm−1 shift in wavenumber; with a 61.7% increase in peak area. The peaks at around 1653.5 cm−1 of N–H, CO, and CC were also observed to shift by −3 cm−1. Compared to SiO2NP-miR146a and AuNP-miR146a, CNP-miR146a exhibited a slight change in the PO2− groups related to the miR146a backbone after H2O2 addition (shift by 3 cm−1 at the 1092.5 cm−1 peak center) suggesting better backbone stability of miR146a. The peak at around 1351.5 cm−1 of (C–N and CO) amide stretch and the peak center at 1654.5 of N–H, CO, and CO have shifted toward lower wavenumber by 5 and 7 cm−1, respectively, with an increase in the peak integral. These results could imply the interaction of ROS with miR146a bases or alterations to amide bonds in the conjugates.
 | ||
| Fig. 7 The miR146a conjugated nanoparticles were incubated with a total concentration of 10 mM of hydrogen peroxide for 6 hours to investigate oxidative damage and the ability of CNPs, AuNPs, and SiO2NPs to protect miR146a from modification. FTIR spectra of conjugated miR146a nanoparticles before and after H2O2 incubation of (a) SiO2NP-miR146a, (b) AuNP-miR146a, and (c) CNP-miR146a. The FTIR spectrum used in a 600–4000 cm−1 region with 8 cm−1 resolution in a PerkinElmer infrared spectrometer. (d) C 1s XPS scans of CNP-miR146a, AuNP-miR146a, and SiO2NP-miR146a after 6 hours of exposure to 10 and 500 mM H2O2 exhibiting changes in C–H/C–C, C–O/C–N, O–CO, and satellite peaks due to miR146a oxidation. | ||
Because of the close location of Si–H2O vibration to N–H, CO, and CC (at around 1648 cm−1 peak center), we could not separate the influence of H2O2 on the SiO2NP and the miR146a bases. Therefore, XPS has been performed with different H2O2 concentrations to support our findings.
3.7 Enzyme-mimetic activity chemical assay
Superoxide dismutase assay was conducted on CNP-miR146a, SiO2NP-miR146a, and AuNP-miR146a to measure the antioxidant activity of the nanoparticles after miR146a conjugation (Fig. 8a). We observed that CNP-miR146a maintained the highest SOD activity, 90.5%, after conjugation, compared to 1.7% for AuNP-miR146a and 4.8% for SiO2NP-miR146a. However, SOD activity of CNP-miR146a did decrease slightly (16.21 μM by ICP-MS) compared to the bare CNP control sample, 91%, before conjugation (33 μM). To compare the SOD activity between CNP-miR146a and CNP control more accurately, we have performed the SOD activity assay on CNP-miR146a and the CNP control sample in which the CNP concentration of CNP-miR146a was calculated empirically to be 15.6 μM. With the CNP concentration in CNP-control normalized to the CNP-miR146a-15.6 μM, a difference in SOD activity between these two samples was observed, 88.0 − 83.3 = 4.7% (Fig. 8b). To further study the SOD activity difference between CNP-miR146a and CNP control and to compare their SOD activities more clearly, we have repeated the SOD activity assay on both samples with 2X dilution. With a relatively lower concentration, the reaction between WST-1 and O*-can be inhibited to a different extent, respectively, by CNP control and CNP-miR146a, providing a more substantial difference in SOD activity between the CNP control and CNP-miR146a, 80.3 − 72.2 = 8.1%, as shown in Fig. 8b. As the batch of the CNP-miR146a sample used in the repeated SOD assay mentioned above was different from the previous CNP-miR146a (16.21) batch whose concentration was calculated by ICP-MS, the exact CNP concentration of the CNP-miR146a sample remained unknown. Based on our previous working experience, we have empirically calculated the CNP concentration in CNP-miRNA146a-15.6 μM, noted to be 15.6 μM, and the CNP-control sample was deliberately diluted to the same concentration as the CNP concentration in CNP-miRNA146a-15.6 μM in order to mimic the same SOD-mimetic component concentration. The SOD activity difference between the sample pair CNP-miR146a-15.6μm (83.3%)/CNP-control-15.6μM (88.0%) or CNP-miR146a-7.8μm (72.2%)/CNP-control-7.8 μM (80.3%) implied that the miR146a conjugated to the nanoparticles' surface could potentially block surface reaction sites and contribute to a decrease in SOD activity. Besides this, the decrease in SOD activity could also be ascribed to small concentration differences between CNP-miR146a and CNP-control. | ||
| Fig. 8 Superoxide dismutase-like activity of miR146a conjugated nanoparticles. (a) Calculated SOD activity and absorbance of sample, respectively, AuNP-miR146a (1.7%), SiO2NP-miR146a (4.8%), CNP-miR146a (16.21 μM) (90.4%), and bare SOD blank at 438 nm are plotted as a function of assay reaction time. The CNP concentration in CNP-miR146a (16.21 μM) was measured and calculated based on ICP-MS. (b) Calculated SOD activity and absorbance of samples, respectively, CNP-control-7.8 μM (80.3%), CNP-control-15.8 μM (88.0%), CNP-miR146a-7.8 μM (72.2%), CNP-miR146a-15.6 μM (83.3%) and SOD blank at 438 nm are plotted as a function of assay reaction time. The CNP concentration in CNP-miR146a-15.6 μM was calculated empirically based on previous experimental experience. The CNP-miR146a-7.8 μM sample was 2X diluted from CNP-miR146a-15.6 μM. | ||
Given the observed relative ROS scavenging activities and varied NP-miR146a formulation characters demonstrated in the above sections, we lastly looked to characterize material behavior in biological systems. The studied formulations were evaluated for their effects on cell viability and on expression of miR146a-related genes.
3.8 Cell viability studies on conjugated samples
The cytotoxicity of pure and miR146a conjugated CNPs, AuNPs, and SiO2NPs was examined using human umbilical vein endothelial cell (HUVEC) lines via MTT and live/dead assay. Fig. 9a and b show the results of the cells treated with two different concentrations (0.2 μg μL−1 and 0.4 μg μL−1) of CNPs, AuNPs, SiO2NPs, CNP-miR146a, AuNP-miR146a and SiO2NP-miR146a. Pure and miR146a conjugated CNPs, AuNPs, and SiO2NPs showed no significant changes in cell viability compared to control cells. These results suggested that there is no significant cytotoxicity for the conjugated samples. HUVECs were cultured with 0.4 μg μL−1 of CNPs, AuNPs, SiO2NPs, CNP-miR146a, AuNP-miR146a, and SiO2NP-miR146a for two days. After two days, a live/dead assay was performed with and without nanoparticle-treated cells as shown in Fig. 9c. No significant change in the number of live cells compared to control cells was observed. These results are consistent with the MTT results and provide further evidence of formulation cytocompatibility. We, therefore, consider that variation in specific biological activity should result from specific differences in formulation character and not from any general toxicity induction in cell cultures. These results are also consistent with the pervasive use of silica and gold as nanocarriers in the nanomedicine literature. Fig. 9d and e show results from cell internalization studies and ROS induction, respectively. The internalization demonstrates successful and robust uptake of the ceria conjugate, confirming the ability of the nano-therapeutic ceria conjugate to permeate the cell membrane to allow miRNA utilization. ROS studies in a macrophage line demonstrate the materials ability to confer radical scavenging therapeutic action on target cells. Finally, we looked to compare the transfection efficiencies of each formulation and related, consequent expression of IL-6.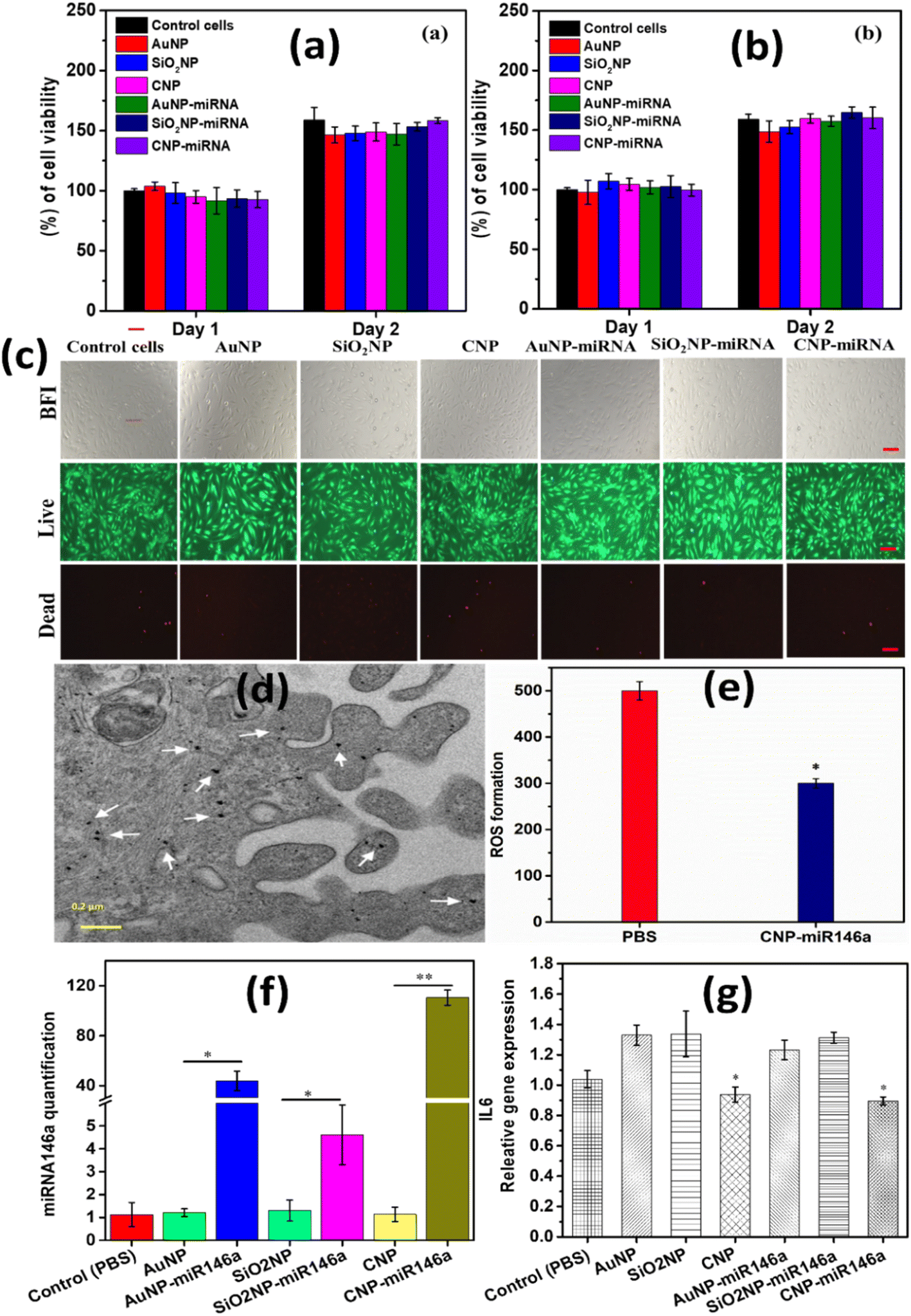 | ||
| Fig. 9 Cell viability and live/dead assay results of pure and miR146a conjugated CNP, AuNP, and SiO2NP samples. (a) and (b) show the percentage of viable cells treated with pure and miR146a conjugated CNPs, AuNPs and SiO2NPs at 0.2 μg μL−1 and 0.4 μg μL−1. 0.4 μg μL−1 of control cells (nanoparticle untreated cells), pure and miR146a conjugated CNP, AuNP, and SiO2NP treated cells’ bright field and live/dead images are shown in the images (c). The scale bar is 100 μm. The TEM image of murine dermal fibroblasts incubated with 0.1 μg CNP-miR146a for 24 h (scale bar 0.2 μm). This image shows uniform distribution of CNPs which is shows as block dots marked with white arrows (d). ROS production by macrophages with PBS and with CNP-miR146a treatment. CNP-miR146a showed less ROS production compared to PBS (control) which indicates that CNP-miR146a scavenges the ROS production in the cells (e). MiR146a and IL-6 gene expression following treatment of MDFs with pure and miR146a conjugated CNPs, AuNPs, and SiO2NPs. MiR146a levels were significantly high in CNP-miR146a compared to the control (PBS) treated sample (f). CNP and CNP-miR146a-treated cells significantly reduced IL-6 gene expression compared to the control (g). Bars indicate statistical significance with P < 0.05 and P < 0.0001**. | ||
3.9 Gene and miR146a expression
MiR146a and IL-6 gene expression were analyzed using MDF cells treated with pure and conjugated samples. Based on the cytotoxicity study, we have chosen 0.2 ng μL−1 of biocompatible pure and miR146a conjugated CNP, SiO2NP, and AuNP to treat the MDF cells for 6 h. After treatment, the cells were processed and the relative miR146a expression was quantified, which is shown in Fig. 9f. Sodium phosphate buffer (PBS) was considered a control. CNP-miR146a sample treatment resulted in the highest levels of miR146a expression compared to other miR146a conjugated nanoparticles. These results clearly show that CNPs are highly capable carrier materials for miR146a in cellular delivery. RNA was isolated from the treated (pure and miR146a conjugated nanoparticles) cells followed by cDNA synthesis to evaluate for relative gene expression of pro-inflammatory gene IL-6 (Fig. 9g). 6 h of treatment of pure and CNP-miR146a samples resulted in decreased IL-6 gene expression compared to control. This result suggested that CNPs are also capable of reducing expression of pro-inflammatory cytokines with further reduction of IL-6 gene expression observed for the conjugate CNP-miR146a sample. AuNP-miR146a and SiO2NP-miR146a samples showed no appreciable changes in relative IL-6 gene expression. This could be due to the fact that AuNPs and SiO2NPs do not produce significant modulation of ROS concentrations present in the cellular environment as CNPs have been demonstrated to do. High levels of ROS can result in and lead to high inflammation through induction of immune responses.7 CNP-miR146a breaks the vicious cycle of inflammation and oxidative stress. These results are consistent with those of our more detailed, previous study and are consistent with the physicochemical characterization studies including detailed electrochemical analysis presented in earlier sections of the current work. To investigate and eliminate the possibility of the impact of the conjugated samples' surface charges and the small observed AuNP-miR146a agglomeration (Fig. 3g) on the therapeutic properties, we have used zeta potential measurements and the cytotoxicity results. From the literature, it was found that the surface charge of nanoparticles can mainly impact the cytotoxicity and the cellular uptake mechanism due to the cell walls' surface charge (penetrability).49,50 Agglomeration also affects cellular uptake and cytotoxicity and adds to its ability to decrease the loading efficacy.51,52 CNP-miR146a, SiO2NP-miR146a, and AuNP-miR146a have exhibited a surface zeta charge of −30.3 mV, −25 mV, and −28 mV, respectively. Since the surface charges of all conjugated samples are strongly negative with slight differences in values, demonstrating colloid stability, and the cytotoxicity study exhibited similarity between the samples, we believe that the surface charge and the agglomeration are not issues for our study and that the distinctions in formulation characters maybe the reasons behind the observed differences in biological behavior (miR146a and IL6 expression).The physicochemical, materials, and biological studies performed demonstrate that CNP-miR146a presents a unique bio-active character beyond its simple function as a nanocarrier. By comparing gold and silica formulations, in particular, we are able to discount possible metal oxide (silica) or charge transfer-based nanoscale effects such as surface acid–base characters or surface plasmon mediated reactions, respectively. Furthermore, the known therapeutic ROS-scavenging character of the un-modified particles is conserved following conjugation and likely contributes to the demonstrated suppression of immune activation as IL-6 expression.
4. Conclusions
In the current study, we sought to identify physicochemical processes which may contribute to previously identified therapeutic activity of a CNP-miR146a conjugate formulation in diabetic wound healing models. Therefore, this formulation was characterized with respect to its ability to confer radical oxygen species (ROS; present at high concentrations in the diabetic wound environment) scavenging activity, while preserving miRNA chemical composition and integrity in the presence of ROS species. Specific material compositions were chosen for comparison against the ceria formulation, silica (SiO2NP) and gold (AuNP) NPs, to evidence any nanoscale effects arising from the oxide character (e.g., acid–base mediated surface chemistry) or enhanced charge transfer-related properties from quantum confinement (e.g., surface plasmon resonance).Conjugated formulations of each composition were produced. Electroanalytical measurements demonstrated a stable catalytic response towards H2O2 for CNP-miR146a, while AuNP- and SiO2-miR146a conjugates produced no or an unstable (decreasing faradaic current over time) response. These trends in ROS sensitivity were further supported by the SOD assay results. FTIR and XPS measurements suggested oxidative damage to miRNA for both SiO2NP- and AuNP-miR146a conjugates, while CNP-miR146a measurements showed negligible change following H2O2 exposure. Measurements of intracellular miR146a levels and IL-6 expression upon treatment with the varied nanomaterial conjugate formulations further recommend CNP-miR146a over the other formulations with neither AuNP- nor SiO2NP-miR146a producing a significant attenuation of IL-6 expression. In conclusion, the therapeutic efficacy of CNP-miR146a is unique to the material formulation and not limited to general nanoscale effects or to simple nanocarrier activity. Furthermore, given the ROS-scavenging therapeutic modality of this formulation is unrelated to the composition of miRNA, the observed advantages of CNP conjugation are likely not limited to miR146a and can benefit arbitrary forms of miRNA across biological systems and molecular pathways. Therefore, the use of CNPs in areas such as genetic and immuno-engineering could lead to improved efficacy, especially in pathologies which evolve a highly oxidizing chemical environment.
Author contributions
C. E., C. N., K. E., and S. S. designed the outline of the manuscript. C. E., C. N., and S. S. designed the experiments. K. E., J. H., C. Z, and K. L performed the biological studies. Formal analysis, visualization, and writing: C. E., C. N. K. E., Y. F., and U. K. All authors contributed to writing, reviewing, and editing the manuscript. All authors approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to acknowledge the NIH (National Institutes of Health) (1R01DK118793-01A1) for partial funding the nanotechnology/wound healing research. C. N. and K. E. acknowledge the UCF Preeminent Postdoctoral Program (P3) for funding support and NSF MRI ECCS (Grant No. 1726636) funding for XPS measurements. S. S. thanks the UCF Trustee Chair Professorship.References
- P. Martin, Wound healing--aiming for perfect skin regeneration, Science, 1997, 276(5309), 75–81 CrossRef CAS PubMed.
- D. M. Bermudez, B. J. Herdrich, J. Xu, R. Lind, D. P. Beason and M. E. Mitchell, et al., Impaired biomechanical properties of diabetic skin: implications in pathogenesis of diabetic wound complications, Am. J. Pathol., 2011, 178(5), 2215–2223 CrossRef CAS PubMed.
- L. C. Dewberry, S. M. Niemiec, S. A. Hilton, A. E. Louiselle, S. Singh and T. S. Sakthivel, et al., Cerium oxide nanoparticle conjugation to microRNA-146a mechanism of correction for impaired diabetic wound healing, Nanomedicine, 2022, 40, 102483 CrossRef CAS PubMed.
- V. N. Kim, MicroRNA biogenesis: coordinated cropping and dicing, Nat. Rev. Mol. Cell Biol., 2005, 6(5), 376–385 CrossRef CAS PubMed.
- S. W. L. Lee, C. Paoletti, M. Campisi, T. Osaki, G. Adriani and R. D. Kamm, et al., MicroRNA delivery through nanoparticles, J. Controlled Release, 2019, 313, 80–95 CrossRef CAS PubMed.
- J. Xu, W. Wu, L. Zhang, W. Dorset-Martin, M. W. Morris and M. E. Mitchell, et al., The role of microRNA-146a in the pathogenesis of the diabetic wound-healing impairment: correction with mesenchymal stem cell treatment, Diabetes, 2012, 61(11), 2906–2912 CrossRef CAS PubMed.
- C. Zgheib, S. A. Hilton, L. C. Dewberry, M. M. Hodges, S. Ghatak and J. Xu, et al., Use of cerium oxide nanoparticles conjugated with microRNA-146a to correct the diabetic wound healing impairment, J. Am. Coll. Surg., 2019, 228(1), 107–115 CrossRef PubMed.
- S. M. Niemiec, A. E. Louiselle, S. A. Hilton, L. C. Dewberry, L. Zhang and M. Azeltine, et al., Nanosilk increases the strength of diabetic skin and delivers CNP-miR146a to improve wound healing, Front. Immunol., 2020, 11, 590285 CrossRef CAS PubMed.
- S. M. Niemiec, S. A. Hilton, A. Wallbank, M. Azeltine, A. E. Louiselle and H. Elajaili, et al., Cerium oxide nanoparticle delivery of microRNA-146a for local treatment of acute lung injury, Nanomedicine, 2021, 34, 102388 CrossRef CAS PubMed.
- E. Kolanthai, Y. Fu, U. Kumar, B. Babu, A. K. Venkatesan and K. W. Liechty, et al., Nanoparticle mediated RNA delivery for wound healing, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2021, e1741 Search PubMed.
- G. D. Gray, S. Basu and E. Wickstrom, Transformed and immortalized cellular uptake of oligodeoxynucleoside phosphorothioates, 3′-alkylamino oligodeoxynucleotides, 2′-O-methyl oligoribonucleotides, oligodeoxynucleoside methylphosphonates, and peptide nucleic acids, Biochem. Pharmacol., 1997, 53(10), 1465–1476 CrossRef CAS PubMed.
- P. Velpurisiva, A. Gad, B. Piel, R. Jadia and P. Rai, Nanoparticle design strategies for effective cancer immunotherapy, J. Biomed., 2017, 2(2), 64 CrossRef PubMed.
- T. Pirmohamed, J. M. Dowding, S. Singh, B. Wasserman, E. Heckert and A. S. Karakoti, et al., Nanoceria exhibit redox state-dependent catalase mimetic activity, Chem. Commun., 2010, 46(16), 2736–2738 RSC.
- E. G. Heckert, A. S. Karakoti, S. Seal and W. T. Self, The role of cerium redox state in the SOD mimetic activity of nanoceria, Biomaterials, 2008, 29(18), 2705–2709 CrossRef CAS PubMed.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh and P. Fornasiero, et al., Electron localization determines defect formation on ceria substrates, Science, 2005, 309(5735), 752–755 CrossRef CAS PubMed.
- P. Yang, Z. Quan, Z. Hou, C. Li, X. Kang and Z. Cheng, et al., A magnetic, luminescent and mesoporous core–shell structured composite material as drug carrier, Biomaterials, 2009, 30(27), 4786–4795 CrossRef CAS PubMed.
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu and S. Jeftinija, et al., A mesoporous silica nanosphere-based carrier system with chemically removable CdS nanoparticle caps for stimuli-responsive controlled release of neurotransmitters and drug molecules, J. Am. Chem. Soc., 2003, 125(15), 4451–4459 CrossRef CAS PubMed.
- S.-A. Yang, S. Choi, S. M. Jeon and J. Yu, Silica nanoparticle stability in biological media revisited, Sci. Rep., 2018, 8(1), 1–9 Search PubMed.
- S. Quignard, T. Coradin, J. J. Powell and R. Jugdaohsingh, Silica nanoparticles as sources of silicic acid favoring wound healing in vitro, Colloids Surf., B, 2017, 155, 530–537 CrossRef CAS PubMed.
- A. Tivnan, W. S. Orr, V. Gubala, R. Nooney, D. E. Williams and C. McDonagh, et al., Inhibition of neuroblastoma tumor growth by targeted delivery of microRNA-34a using anti-disialoganglioside GD2 coated nanoparticles, PLoS One, 2012, 7(5), e38129 CrossRef CAS PubMed.
- A. J. Mieszawska, W. J. Mulder, Z. A. Fayad and D. P. Cormode, Multifunctional gold nanoparticles for diagnosis and therapy of disease, Mol. Pharmaceutics, 2013, 10(3), 831–847 CrossRef CAS PubMed.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity, Small, 2005, 1(3), 325–327 CrossRef CAS PubMed.
- P. K. Jain, K. S. Lee, I. H. El-Sayed and M. A. El-Sayed, Calculated absorption and scattering properties of gold nanoparticles of different size, shape, and composition: applications in biological imaging and biomedicine, J. Phys. Chem. B, 2006, 110(14), 7238–7248 CrossRef CAS PubMed.
- M. M. Mihai, M. B. Dima, B. Dima and A. M. Holban, Nanomaterials for wound healing and infection control, Materials, 2019, 12(13), 2176 CrossRef CAS PubMed.
- Z. Meng, D. Zhou, Y. Gao, M. Zeng and W. Wang, miRNA delivery for skin wound healing, Adv. Drug Delivery Rev., 2018, 129, 308–318 CrossRef CAS PubMed.
- X. Zhao, W. Song, Y. Chen, S. Liu and L. Ren, Collagen-based materials combined with microRNA for repairing cornea wounds and inhibiting scar formation, Biomater. Sci., 2019, 7(1), 51–62 RSC.
- A. Ekin, O. F. Karatas, M. Culha and M. Ozen, Designing a gold nanoparticle-based nanocarrier for microRNA transfection into the prostate and breast cancer cells, J. Gene Med., 2014, 16(11–12), 331–335 CrossRef CAS PubMed.
- C. J. Neal, C. R. Fox, T. S. Sakthivel, U. Kumar, Y. Fu and C. Drake, et al., Metal-mediated nanoscale cerium oxide inactivates human coronavirus and rhinovirus by surface disruption, ACS Nano, 2021, 15(9), 14544–14556 CrossRef CAS PubMed.
- Y. Kosto, A. Zanut, S. Franchi, Y. Yakovlev, I. Khalakhan and V. Matolin, et al., Electrochemical activity of the polycrystalline cerium oxide films for hydrogen peroxide detection, Appl. Surf. Sci., 2019, 488, 351–359 CrossRef CAS.
- A. V. Peskin and C. C. Winterbourn, Assay of superoxide dismutase activity in a plate assay using WST-1, Free Radicals Biol. Med., 2017, 103, 188–191 CrossRef CAS PubMed.
- A. Gupta, T. S. Sakthivel, C. J. Neal, S. Koul, S. Singh and A. Kushima, et al., Antioxidant properties of ALD grown nanoceria films with tunable valency, Biomater. Sci., 2019, 7(7), 3051–3061 RSC.
- S. Seal, A. Jeyaranjan, C. J. Neal, U. Kumar, T. S. Sakthivel and D. C. Sayle, Engineered defects in cerium oxides: tuning chemical reactivity for biomedical, environmental, & energy applications, Nanoscale, 2020, 12(13), 6879–6899 RSC.
- H. Wu, F. Li, W. Shao, J. Gao and D. Ling, Promoting angiogenesis in oxidative diabetic wound microenvironment using a nanozyme-reinforced self-protecting hydrogel, ACS Cent. Sci., 2019, 5(3), 477–485 CrossRef CAS PubMed.
- C. J. Neal, A. Gupta, S. Barkam, S. Saraf, S. Das and H. J. Cho, et al., Picomolar detection of hydrogen peroxide using enzyme-free inorganic nanoparticle-based sensor, Sci. Rep., 2017, 7(1), 1–10 CrossRef CAS PubMed.
- C. J. Neal, S. Das, S. Saraf, L. Tetard and S. Seal, Self-assembly of PEG-coated ceria nanoparticles shows dependence on PEG molecular weight and ageing, ChemPlusChem, 2015, 80(11), 1680 CrossRef CAS PubMed.
- A. Paul, R. M. Watson, E. Wierzbinski, K. L. Davis, A. Sha and C. Achim, et al., Distance dependence of the charge transfer rate for peptide nucleic acid monolayers, J. Phys. Chem. B, 2010, 114(45), 14140–14148 CrossRef CAS PubMed.
- M. Pourbaix, Atlas of electrochemical equilibria in aqueous solution, NACE J., 1974, 307 Search PubMed.
- Y. Xue, Y. Zhai, K. Zhou, L. Wang, H. Tan and Q. Luan, et al., The vital role of buffer anions in the antioxidant activity of CeO2 nanoparticles, Chem.–Eur. J., 2012, 18(35), 11115–11122 CrossRef CAS PubMed.
- N. P. Sardesai, D. Andreescu and S. Andreescu, Electroanalytical evaluation of antioxidant activity of cerium oxide nanoparticles by nanoparticle collisions at microelectrodes, J. Am. Chem. Soc., 2013, 135(45), 16770–16773 CrossRef CAS PubMed.
- D. Yang, M. Fa, L. Gao, R. Zhao, Y. Luo and X. Yao, The effect of DNA on the oxidase activity of nanoceria with different morphologies, Nanotechnology, 2018, 29(38), 385101 CrossRef PubMed.
- A. J. Bard and L. R. Faulkner, Fundamentals and applications, Electrochem. Methods, 2001, 2(482), 367–386 Search PubMed.
- A. J. Bard and L. R. Faulkner, Fundamentals and applications, Electrochem. Methods, 2001, 2(482), 100–104 Search PubMed.
- M. Y. Elahi, S. Bathaie, S. Kazemi and M. Mousavi, DNA immobilization on a polypyrrole nanofiber modified electrode and its interaction with salicylic acid/aspirin, Anal. Biochem., 2011, 411(2), 176–184 CrossRef PubMed.
- Electrochemical impedance study of fresh and passivated macroporous silicon for sensors, 2017 32nd Symposium on Microelectronics Technology and Devices (SBMicro), ed. R. P. Toledo, C. E. Dias, D. R. Huanca and W. J. Salcedo, IEEE, 2017 Search PubMed.
- G. Mula, M. V. Tiddia, R. Ruffilli, A. Falqui, S. Palmas and M. Mascia, Electrochemical impedance spectroscopy of oxidized porous silicon, Thin Solid Films, 2014, 556, 311–316 CrossRef CAS.
- M. H. Kuchma, C. B. Komanski, J. Colon, A. Teblum, A. E. Masunov and B. Alvarado, et al., Phosphate ester hydrolysis of biologically relevant molecules by cerium oxide nanoparticles, Nanomedicine, 2010, 6(6), 738–744 CrossRef CAS PubMed.
- G. I. Dovbeshko, N. Y. Gridina, E. B. Kruglova and O. P. Pashchuk, FTIR spectroscopy studies of nucleic acid damage, Talanta, 2000, 53(1), 233–246 CrossRef CAS PubMed.
- C. Prakash, V. K. Kamboj, P. Ahlawat and V. Kumar, Structural and molecular alterations in arsenic-induced hepatic oxidative stress in rats: a FTIR study, Toxicol. Environ. Chem., 2015, 97(10), 1408–1421 CrossRef.
- M. Ferrari, Beyond drug delivery, Nat. Nanotechnol., 2008, 3(3), 131–132 CrossRef CAS PubMed.
- A. Asati, S. Santra, C. Kaittanis and J. M. Perez, Surface-charge-dependent cell localization and cytotoxicity of cerium oxide nanoparticles, ACS Nano, 2010, 4(9), 5321–5331 CrossRef CAS PubMed.
- K. W. Powers, M. Palazuelos, B. M. Moudgil and S. M. Roberts, Characterization of the size, shape, and state of dispersion of nanoparticles for toxicological studies, Nanotoxicology, 2007, 1(1), 42–51 CrossRef CAS.
- J. Jiang, G. Oberdörster and P. Biswas, Characterization of size, surface charge, and agglomeration state of nanoparticle dispersions for toxicological studies, J. Nanopart. Res., 2009, 11(1), 77–89 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Selected area electron diffraction patterns, calibration curve plots, and chronoamperometry measurements; impedance data in Bode and Nyquist plots of a modified glassy carbon (GC) electrode; C 1s binding region XPS measurements. See DOI: https://doi.org/10.1039/d2na00600f |
| This journal is © The Royal Society of Chemistry 2023 |