
Ultrasmall water-stable CsPbBr3 quantum dots with high intensity blue emission enabled by zeolite confinement engineering†
Hongyue
Zhang‡
ab,
Bolun
Wang‡
ab,
Zijian
Niu
a,
Guangrui
Chen
a,
Buyuan
Guan
 a,
Jiyang
Li
*a and
Jihong
Yu
*ab
a,
Jiyang
Li
*a and
Jihong
Yu
*ab
aState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: lijiyang@jlu.edu.cn; jihong@jlu.edu.cn
bInternational Center of Future Science, Jilin University, 2699 Qianjin Street, Changchun 130012, P. R. China
First published on 25th August 2023
Abstract
Ultrasmall CsPbBr3 perovskite quantum dots (PQDs) as promising blue-emitting materials are highly desired for full-color display and lighting applications, but their inferior efficiency and poor ambient stability hinder extensive applications. Herein, a “break-and-repair” strategy has been developed to tightly confine monodispersed ultrasmall CsPbBr3 PQDs in a zeolite. In this strategy, the CsPbBr3 PQDs are introduced into the zeolite via a high temperature evaporation method, wherein the perovskite precursors break the zeolite framework, and amino acids and silane are then used to fix the damaged framework and lock the perovskite QDs within the matrix. By modulating the synthetic conditions to control the growth of CsPbBr3, PQDs with ultrasmall size of 2 nm have been obtained in the zeolite, giving emission centered at 460 nm with a high quantum yield of 76.93%. Strikingly, the PQDs@zeolite composite exhibits water-induced reversible photoluminescence promoted by the coordination between the amino acids and PQDs in a dynamic manner, achieving enhanced water stability (14 days in aqueous solution). This work provides a new perspective for the synthesis of water-stable blue-emitting perovskite composites for potential applications in lighting fields.
 Jihong Yu | Congratulations on the 10th anniversary to Materials Horizons, a leading journal for the publication of original research and breakthrough developments in materials science. I’m honored to have contributed to Materials Horizons as an advisory member since 2017 and excited to witness its ever-increasing scholarly impact. I would like to showcase our recent discovery about zeolites as an ideal matrix for the confined growth of water-stable blue-emitting perovskite composites. This work offers important potential of perovskite@zeolite composites for advanced light-emitting and light-harvesting devices. I strongly believe the journal will continue to serve as a platform to create new opportunities for the future of materials research. |
New conceptsIn this study, we have demonstrated a feasible synthetic strategy to realize the successful confinement of water-stable ultrasmall CsPbBr3 quantum dots (QDs) in a zeolite. The high-temperature evaporation approach is utilized to break the framework of the zeolite and introduce CsPbBr3 QDs, and post modification is performed to repair the matrix and passivate the occluded perovskite QDs. This encapsulation strategy achieves the size control of the CsPbBr3 QDs (2–6 nm), a high quantum yield of 76.93% for blue emission and long-term water stability. This work provides a new perspective for the synthesis of water-stable blue emitting perovskite composites for potential applications in lighting fields. |
Introduction
Metal halide perovskites have attracted considerable attention for their applications in the fields of solar cells, light-emitting diodes, lasers, photodetectors and photocatalysis.1–5 Compared with the bulk crystals, perovskite quantum dots (PQDs) exhibit unique size-dependent optoelectronic properties. The all-inorganic CsPbX3 (X = Cl, Br or I) PQDs offer promising photoelectric characteristics as opposed to the organic–inorganic hybrid perovskite, such as better optical and thermal stability and suitable bandgap.6–10 By now, all-inorganic PQDs with stable green and red fluorescence emission have achieved a PLQY of nearly 100%,11–15 but the blue light efficiency remains to be improved due to the uncontrollable phase fusion and difficult size adjustment.16 Cl–Br mixed perovskites, which are easily produced by using halide substitution to create blue perovskite emitters, are susceptible to halide segregation under an electric field, which causes spectral shifting.17 Another feasible strategy to achieve blue emissive CsPbX3 is size engineering, wherein PQDs smaller than 5 nm have proven challenging to produce using conventional colloidal synthesis. Surface ligands are easily lost in ligand exchange during assembly into semiconducting solids, leading to further increased polydispersity.18–22Recently, more reports have proven that porous materials are suitable matrices to reduce the decomposition of PQDs by moisture in the air and enhance their structural stability.19–23 More importantly, the independent nanospace in porous materials ensures that the growth of nanoparticles is restricted and separated from each other, so as to achieve the quantum confinement effect. Among porous matrices, mesoporous materials with a wide-range pore size (2–50 nm) could accommodate various PQDs and regulate their size, composition and luminescence.24 Through confinement growth of CsPbBr3 in mesoporous SiO2, a type of blue luminescent PQDs with size of 3.1 nm and PLQY of 31.8% was synthesized.25 Compared with mesoporous materials, zeolites, whose inorganic frameworks are built exclusively by TO4 tetrahedra (T denotes Si, Al, or P, etc.), have superior thermal/chemical stability to prepare ultra-stable PQDs. In addition, their narrow pore size (<2 nm) provides the possibility for confinement growth of ultrasmall PQDs to emit efficient blue emission. By now, many PQDs@zeolite composites with stable green PL have been prepared by the solution impregnation method.26,27 However, due to the low solubility of CsCl and PbCl2 in organic solvents and the mixed halogen phase separation, blue PQDs are still difficult to achieve. Furthermore, the framework of the zeolite is usually partially damaged during the encapsulation/growth of PQDs, and thereby, monodispersed PQDs with stable blue emission have not yet been achieved in zeolites.
In this work, a “break-and-repair” synthesis strategy has been proposed to limit the growth of CsPbBr3 and stabilize ultrasmall PQDs in a zeolite matrix of W-doped silicalite-1 via a high temperature evaporation method followed by amino acid passivation and silane restoration. First, the introduction of perovskite precursors would break part of the zeolite skeleton, and the formed ultrasmall CsPbBr3 QDs are confined in the interrupted pores. Then, amino acids and silane are employed to repair the broken skeleton to lock the encapsulated QDs. This strategy facilitates the confined growth of PDs in zeolite, achieving the size control of PQDs from 2 nm to 6 nm by modulating the synthetic conditions, which endows the composites with variable fluorescence emissions from deep-blue to green. Moreover, such encapsulation process not only prevents the exposure of PQDs to water, promoting the high stability of blue emission even soaking in aqueous solution, but also greatly enhances the PL performance to reach the highest PLQY of up to 76.93%, much higher than the reported blue emissive PQDs in the solid state.
Results and discussion
Synthesis and size modulation of CsPbBr3@zeolite composites
Herein, silicalite-1 zeolite doped with tungsten (named W-S-1) has been employed as a matrix to limit the growth of CsPbBr3 QDs within its nanospaces via a simple high-temperature evaporation method taking account of the following reasons. Firstly, W-S-1 zeolite has few defects, uniform pores (5.3 × 5.6 Å) and a stable framework at high temperature (over 1000 °C).28 Secondly, as guest balanced ions, Na+ in the channels of W-S-1 provides hydrophilicity, which facilitates the entrance of polar metal salts into the zeolite pores during the high temperature evaporation process; more importantly, Na+ could promote the nucleation of 2 or 3 layer [PbBr6] octahedron connection (denoted by the octahedron layer number as n = 2 or 3) with deep blue emission in zeolite channels.29,30 Thirdly, due to the limited pore size of W-S-1, it is hard to get the target composite by solution or thermal injection methods due to the limitations of diffusion.26,31 Therefore, high temperature evaporation is the optimal method to confine all inorganic perovskite precursors into zeolite channels as the hot metal vapors are able to break the zeolite framework.14
Fig. 1a shows the schematic illustration of the synthetic procedure of CsPbBr3 QDs in W-S-1 zeolite. A small amount of CsBr and PbBr2 with a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was ground and mixed uniformly in a mortar, and laid flat on a quartz boat covered with W-S-1 zeolite separated by porous graphite paper to avoid the direct contact between them. Then, the temperature of the quartz boat was progressively raised to 700 °C and kept for 30 min. During such process, the perovskite precursors of CsBr and PbBr2 would experience the following reactions: firstly, during the grinding, reactions (1) and (2) would occur to generate CsPb2Br5 and CsPbBr3 (Fig. S1, ESI†);32,33 subsequently, reactions (3) and (4) occurred when the mixture was heated, resulting in the complete conversion of CsPb2Br5 to CsPbBr3; when the As-synthesized W-S-1 crystals possess a type of hexagonal morphology with size about 150 nm (Fig. S2a, ESI†). The powder X-ray diffraction (PXRD) pattern confirms the MFI framework topology of W-S-1 zeolite (Fig. S2b, ESI†). After the evaporation of CsPbBr3 QDs into the W-S-1 matrix, an amorphous peak at 20–25° appears in the composite, which suggests that the partially disordered structure is formed due to the breaking of the zeolite skeleton by perovskite vapors (Fig. S3 and S4, ESI†). The perovskite peak could not be observed in the PXRD pattern of the composite, indicating that there are no large perovskite crystals attached on the surface of the zeolite. The transmission electron microscopy (TEM) image reveals the existence of mesopores (more than 10 nm) in the composite (Fig. 1b), which further proves the breaking of the zeolite skeleton by perovskite fragments during the high temperature evaporation process. High resolution TEM (HRTEM) and scanning TEM (STEM) images and TEM energy spectra (Fig. 1c–e) show that ultrasmall CsPbBr3 QDs with a size distribution of 1–2.5 nm are evenly immobilized in the zeolite matrix with Cs, Pb and Br elements distributed uniformly in the composite. It should be pointed out that the larger sized QDs should be confined in the interrupted nanospaces or mesopores of W-S-1 zeolite formed by the breaking of the zeolite skeleton during the high temperature evaporation treatment. In addition, the interrupted functional groups formed in the broken zeolite can form complex hydrogen bonds with the halide anions of the perovskite, which enhances the cohesion of the perovskite with the zeolite matrix.
:1 was ground and mixed uniformly in a mortar, and laid flat on a quartz boat covered with W-S-1 zeolite separated by porous graphite paper to avoid the direct contact between them. Then, the temperature of the quartz boat was progressively raised to 700 °C and kept for 30 min. During such process, the perovskite precursors of CsBr and PbBr2 would experience the following reactions: firstly, during the grinding, reactions (1) and (2) would occur to generate CsPb2Br5 and CsPbBr3 (Fig. S1, ESI†);32,33 subsequently, reactions (3) and (4) occurred when the mixture was heated, resulting in the complete conversion of CsPb2Br5 to CsPbBr3; when the As-synthesized W-S-1 crystals possess a type of hexagonal morphology with size about 150 nm (Fig. S2a, ESI†). The powder X-ray diffraction (PXRD) pattern confirms the MFI framework topology of W-S-1 zeolite (Fig. S2b, ESI†). After the evaporation of CsPbBr3 QDs into the W-S-1 matrix, an amorphous peak at 20–25° appears in the composite, which suggests that the partially disordered structure is formed due to the breaking of the zeolite skeleton by perovskite vapors (Fig. S3 and S4, ESI†). The perovskite peak could not be observed in the PXRD pattern of the composite, indicating that there are no large perovskite crystals attached on the surface of the zeolite. The transmission electron microscopy (TEM) image reveals the existence of mesopores (more than 10 nm) in the composite (Fig. 1b), which further proves the breaking of the zeolite skeleton by perovskite fragments during the high temperature evaporation process. High resolution TEM (HRTEM) and scanning TEM (STEM) images and TEM energy spectra (Fig. 1c–e) show that ultrasmall CsPbBr3 QDs with a size distribution of 1–2.5 nm are evenly immobilized in the zeolite matrix with Cs, Pb and Br elements distributed uniformly in the composite. It should be pointed out that the larger sized QDs should be confined in the interrupted nanospaces or mesopores of W-S-1 zeolite formed by the breaking of the zeolite skeleton during the high temperature evaporation treatment. In addition, the interrupted functional groups formed in the broken zeolite can form complex hydrogen bonds with the halide anions of the perovskite, which enhances the cohesion of the perovskite with the zeolite matrix.
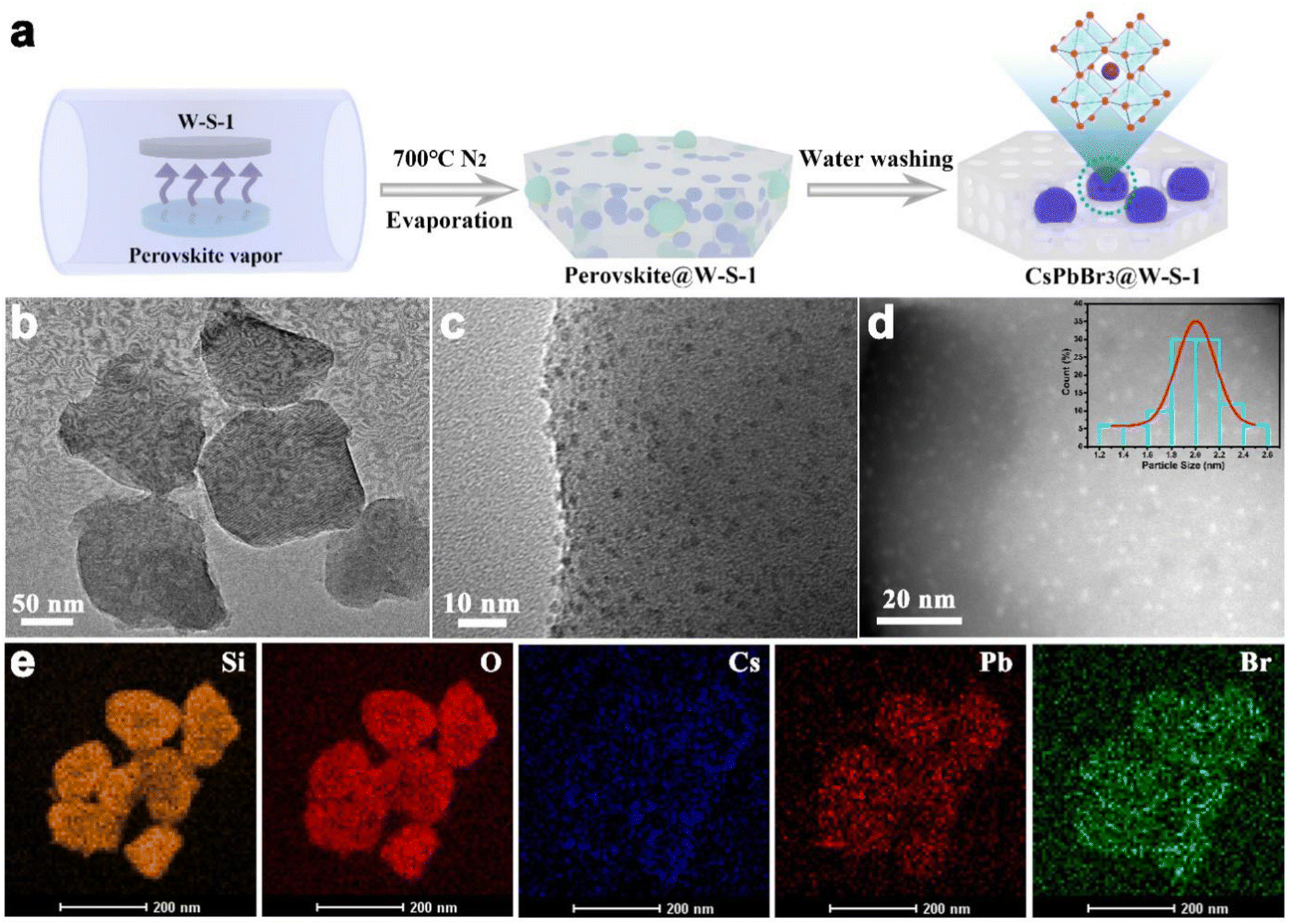 | ||
| Fig. 1 Synthetic procedure and morphology of the representative CsPbBr3@W-S-1 composite. (a) Schematic illustration of the synthetic procedure of the CsPbBr3@W-S-1 composite. (b) TEM and (c) HRTEM image of the CsPbBr3@W-S-1 composite. (d) STEM image of CsPbBr3 QDs distributed in a zeolite matrix. (e) EDX mapping images of Si, O, Cs, Pb and Br elements in the composite. | ||
By modulating the reaction conditions and the amount of perovskite precursor, the size of the CsPbBr3 QDs confined in the W-S-1 zeolite can be regulated from 2 nm to 6 nm (Fig. 2a, e, i, m). Typically, the more amount of perovskite precursors and the shorter heating time favor a large amount of perovskite precursor resided in the zeolite matrix, which may be attributed to the formation of large sized QDs in the matrix. As is known, reducing the size of the perovskite crystal to below the exciton Bohr diameter of CsPbBr3 (7 nm, n = 12) would realize blue emission due to the strong quantum confinement effect.34 Therefore, the luminescence emission of the resulting composites could be tuned from green to deep-blue relying on the quantum size effect of QDs (Table S1, ESI†). According to the previous reports, the size modulations of PQDs are mainly achieved via two types of methods: one is the ligand-assisted method in which long chain amine ligands are usually employed as capping ligands to reduce the size of PQDs; the other is the matrix-confinement method where porous matrices such as mesoporous silica and MOFs with tunable pore sizes are governed as a template to confine the growth of PQDs.17,35 However, it is still challenging to reduce the PQD size below 3 nm due to the instability of colloid chemistry and weak confinement effect resulting from the fragile framework of porous matrices. Compared to mesoporous silica with a large pore size and MOFs with soft frameworks, zeolites possess robust and rigid skeletons, which would be not collapsed during high temperature reaction, leading to a strong template confinement effect, thus allowing the growth of ultrasmall PQD particles. Furthermore, benefiting from the high temperature evaporation approach and confined environment of the zeolite matrix, precise control over the size of the CsPbBr3 QDs can be achieved by facile tuning of the amount of precursors and synthetic conditions, demonstrating the feasible strategy to synthesize tunable sized CsPbBr3 QDs by space-confined solid-state synthesis and in situ encapsulation.
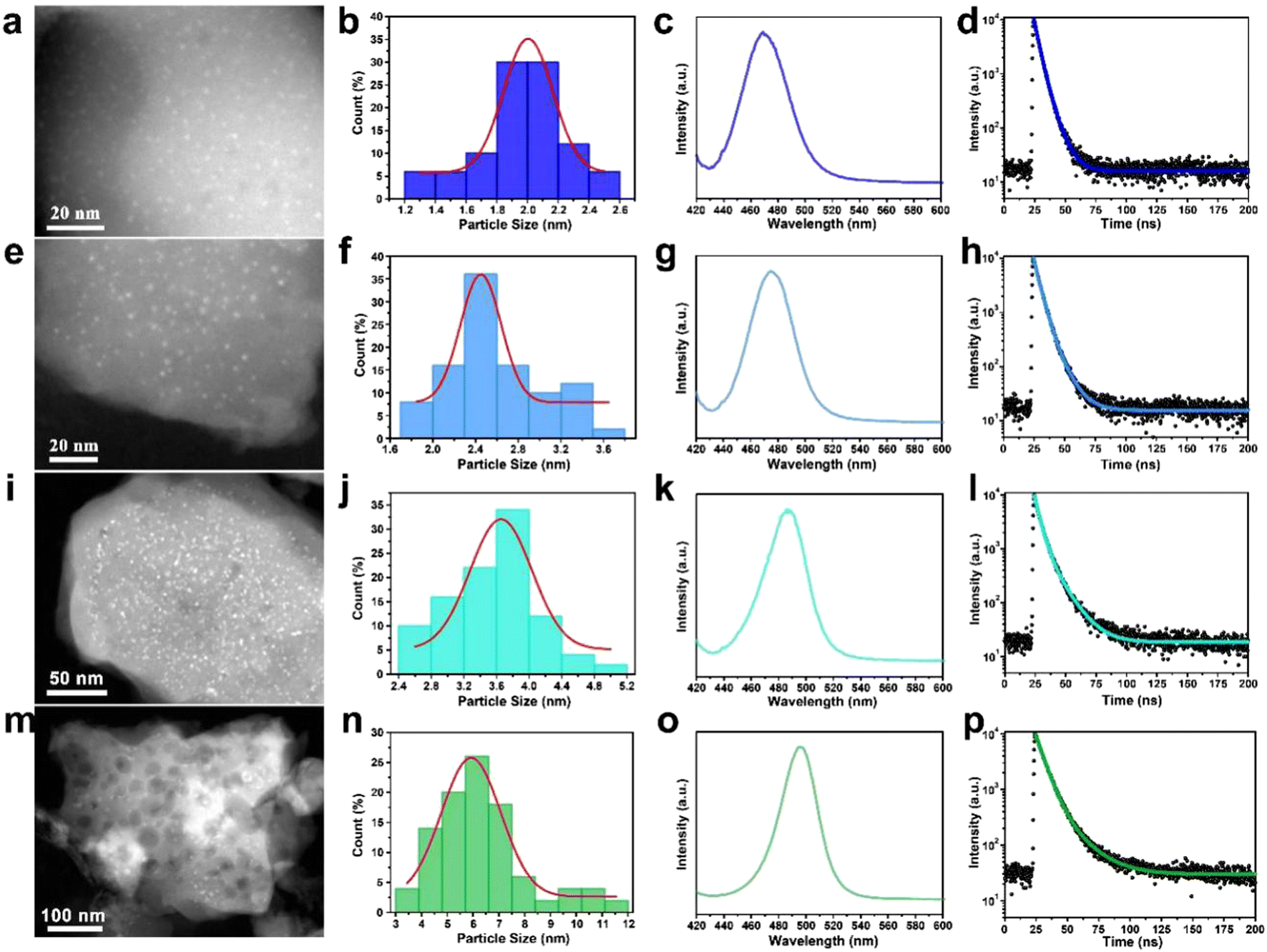 | ||
| Fig. 2 Morphology and optical properties of CsPbBr3@W-S-1 composites obtained in different conditions. (a), (e), (i) and (m) STEM images of various sized CsPbBr3 PQDs confined in the W-S-1 matrix and (b), (f), (j) and (n) the corresponding size distribution. (c), (g), (k) and (o) Fluorescence spectra of the corresponding CsPbBr3@W-S-1 composites, and (d), (h), (l) and (p) their fluorescence lifetime decay spectra, excited by 400 nm light. | ||
Photoluminescence property and water stability of PQDs@W-S-1 composites
To better understand the quantum size effect of CsPbBr3 QDs in a zeolite matrix, a series of optical investigations were performed on different size distributed CsPbBr3 in W-S-1 zeolite. The fluorescence emissions of the composites can be gradually regulated from deep-blue light of 468 nm to green light of 508 nm on increasing the QD sizes from 2 nm to 6 nm, and new UV-vis absorption peaks also appeared at about 500 nm (Fig. 2c, j, k, o and Fig. S5, ESI†). Meanwhile, the fluorescence lifetimes of the composite also prolong from 5.16 ns to 10.87 ns (Fig. 2d, h, l, p and Table S1, ESI†), and the QYs of these composites are 11.24%, 12.36%, 10.95% and 28.4%, respectively.Considering the development of three primary colors for balanced high-resolution lighting, deep-blue luminescent perovskites with high PLQY are highly desired. Therefore, the photoluminescence (PL) property of the CsPbBr3@W-S-1 composite with a QD size of 2 nm is studied as a representative example.
Under the excitation of 400 nm, such composite emits intense deep-blue light with an emission peak at around 460 nm (Fig. 3a). The absorption peak of the composite locates at around 410 to 450 nm, which is associated with the n = 2 and n = 3 phase CsPbBr3. (Fig. 3b). Compared with the normal green light emissive (525 nm) CsPbBr3 perovskite reported by previous studies,36,37 the PL peak of CsPbBr3@W-S-1 is blue shifted about 65 nm, which implies the strong band gap expansion of CsPbBr3 QDs from 2.36 eV to 2.70 eV due to the confined growth of the zeolite matrix. According to the literature, the lattice space of CsPbBr3 is 0.58 nm,38 and CsPbBr3 QDs with 460 nm fluorescence emission correspond to three or four cell sizes of 1.74–2.32 nm, which is consistent with the size of 1.8–2.2 nm (av. 2 nm) measured by STEM. The particle size of the CsPbBr3 NCs is about 2 nm, which corresponds to 2–3 unit cells, in accordance with the optical spectra. In addition, a weak fluorescence peak at 510 nm is observed (Fig. 3a), indicating that a small amount of aggregated 3D CsPbBr3 exists on the surface or in the interrupted pores of the zeolite.
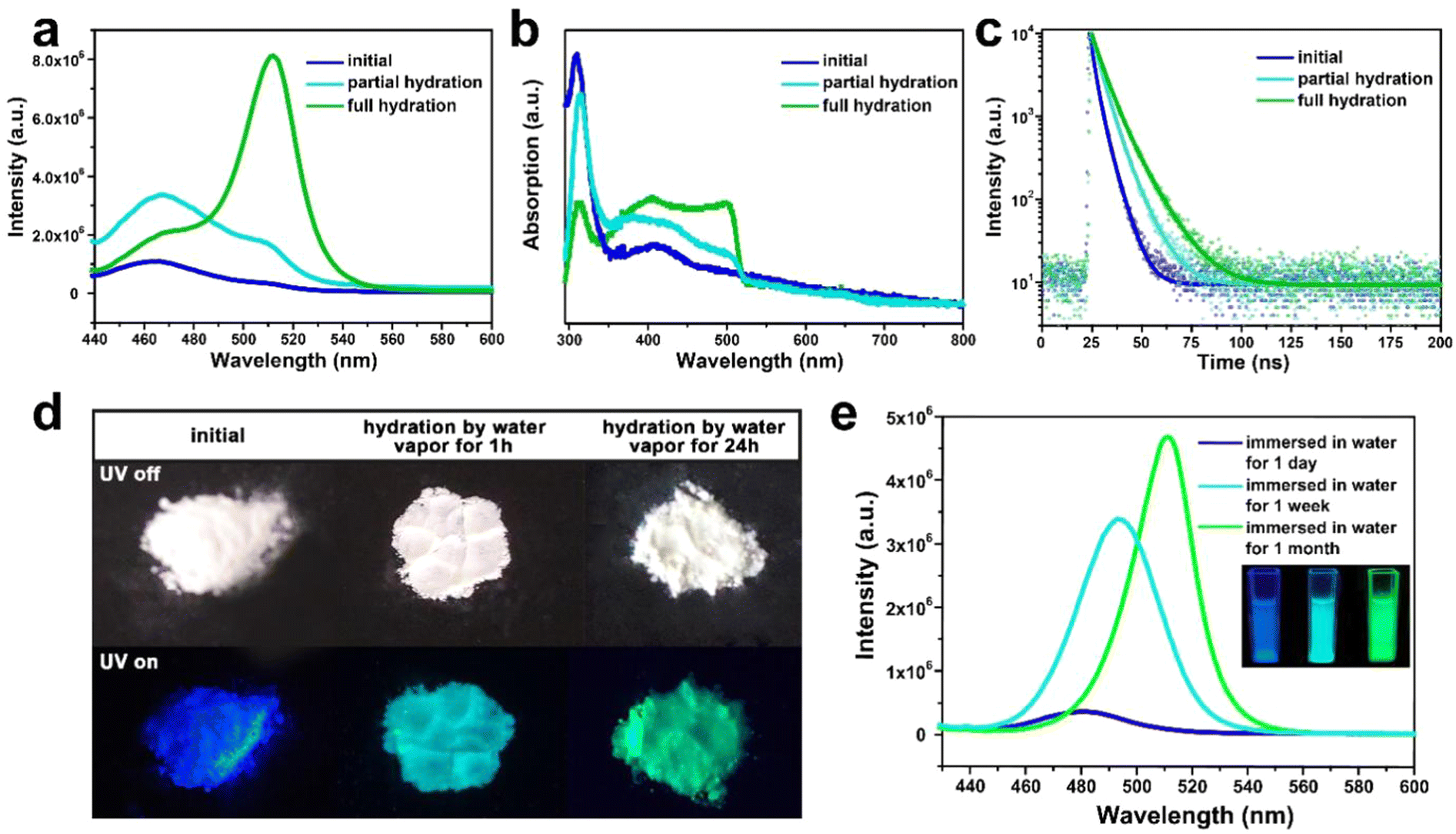 | ||
| Fig. 3 Optical properties and hydration process of the representative CsPbBr3@W-S-1 composite (2 nm sized QDs). (a) Fluorescence spectra, (b) UV-vis spectra and (c) fluorescence lifetime decay spectra of CsPbBr3@W-S-1 composites with different hydration levels. (d) The photographs of CsPbBr3@W-S-1 composites placed in hot water vapor for different times under the naked eye (top) and under UV light (bottom). (e) Fluorescence spectra of the CsPbBr3@W-S-1 composite immersed in water for different times. Inset: photographs of CsPbBr3@W-S-1 composites immersed in water for a day, a week and a month under UV light (from left to right). | ||
The stability of CsPbX3 QDs in moisture and oxygen is very crucial for practical application. To investigate the water stability of the CsPbBr3@W-S-1 composite, the sample was placed in an airtight container containing hot water vapor. Strikingly, the dry CsPbBr3@W-S-1 powder displays white color by the naked eye, which gradually changes to yellowish after being placed in the water vapor container for a while. Under the excitation of UV light, the emission of the CsPbBr3@W-S-1 powder changes from blue to blue-green (1 hour in hot water vapor), and then to green (24 hours in hot water vapor) (Fig. 3d). Fluorescence spectra clearly show that an emission peak centered at 510 nm appears in partial hydrated CsPbBr3@W-S-1 (1 hour in hot water vapor). On prolonging the exposure time in water vapor, the green fluorescence peak gradually increases, and reaches the maximum in fully hydrated CsPbBr3@W-S-1 (Fig. 3a). At the same time, the absorption peak centered at 500 nm emerges in partial hydrated CsPbBr3@W-S-1, which obviously increases in the fully hydrated sample (Fig. 4b). The above results imply that CsPbBr3 QDs in a zeolite matrix would undergo crystal merging induced by water vapor and become large sized QDs, resulting in red-shifted luminescence. To prove this inference, STEM was performed on a partially hydrated CsPbBr3@W-S-1 composite (Fig. S6, ESI†). It can be clearly seen that large-sized perovskite particles around 7 nm aggregate on the W-S-1 zeolite. It is noted that such moisture induced aggregation phenomenon of the QDs has been found in CdS QDs previously. Such increase of the particle size results in a rise of the fluorescence lifetime of CsPbBr3 due to the decreased electron binding energy and defect traps of the QDs (Fig. 3c).39
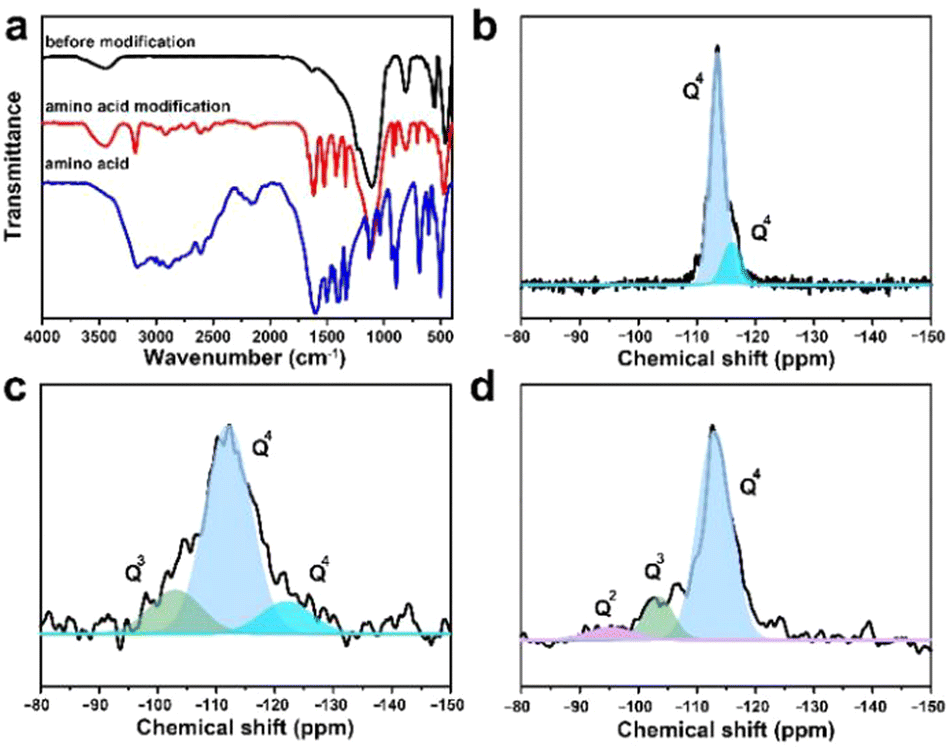 | ||
| Fig. 4 Amino acid modification and silanization repaired CsPbBr3@W-S-1 composite. (a) FTIR spectra of the CsPbBr3@W-S-1 composite, modified CsPbBr3@W-S-1 composite and amino acid. 29Si MAS NMR spectra of (b) W-S-1, (c) the CsPbBr3@W-S-1 composite and (d) the modified CsPbBr3@W-S-1 composite. | ||
The fluorescence variation of the CsPbBr3@W-S-1 composite immersed in water was studied by PL spectra. As shown in Fig. 3e, the fluorescence emission of the CsPbBr3@W-S-1 composite immediately redshifts to 480 nm when immersed in water, and further redshifts to 490 nm accompanied by the greatly enhanced fluorescence intensity after soaking in water for one week. Finally, the emission of the composite shifts to 510 nm with further improved fluorescence intensity after immersion in water for one month. Such variation is completely different from the above fluorescence changes of the composite in water vapor, and thus we speculate that under the inducement of water molecules, two or more small sized CsPbBr3 QDs would gradually aggregate in the interpreted pores of the zeolite while not on the surface of the zeolite. Meanwhile, the hydrogen bonding interactions between the Si-OH groups in the interrupted zeolite framework and occluded QDs are formed to prevent the decomposition of the QDs in water, so as to achieve enhanced fluorescence emission in aqueous solution due to the increased QD size.
Encapsulation enhanced water stability of CsPbBr3@zeolite composites
It was found that small-sized CsPbBr3 QDs in the zeolite matrix would aggregate under the action of the H2O molecule, resulting in red-shifted fluorescence emission of the composite. For practical applications, the achievement of the CsPbBr3@W-S-1 phosphor with stable blue fluorescence emission under a water environment is crucial. Inspired by the previous works that amino groups were usually used as a passivating agent to coordinate with Pb atoms for stabilizing the perovskite structure,11,40 here we selected a type of small sized neutral amino acid glycine loaded into the pores of zeolite to stabilize the PQDs. Considering the good water solubility of glycine, the zeolite pores were further silanized via the hydrolysis of tetramethyl orthosilicate to avoid the loss of glycine. As shown in the FTIR spectra (Fig. 4a), compared with the initial CsPbBr3@W-S-1 composite, there are obvious vibration peaks of amino acids in the modified composite (named modified CsPbBr3@W-S-1), indicating the presence of amino acids in the sealed composite.To analyze the silanization degree of the zeolite matrix, the 29Si MAS NMR spectra were measured. As shown in Fig. 4b, the skeleton silicon atoms exist in the form of Q4 in the initial W-S-1 zeolite. When loading CsPbBr3 QDs, some Q3 emerged in the zeolite due to the breaking of the zeolite framework by perovskite precursors. After restoration by silane reagent, Q3 decreases obviously and a small amount of Q2 appears, which is caused by incomplete cross-linked silane reagent, proving the successful repair of the zeolite framework by the silane reagent.
Impressively, the modified CsPbBr3@W-S-1 composite exhibits a slightly redshifted emission, but a remarkably enhanced QY compared to the initial CsPbBr3@W-S-1 composite (Fig. 5a and Table S2, ESI†). Among the four CsPbBr3@W-S-1 composites with different size of QDs, a deep-blue emissive composite with a PLQY of 39.3% and a sky-blue emissive composite with a QY as high as 76.9% is achieved, which is superior to most of the reported blue emissive CsPbBr3 PQDs.16,41 On the other hand, the fluorescence lifetime of the modified CsPbBr3@W-S-1 is slightly longer than that of the initial composite (Fig. 5b). The above result indicates that amino acids can passivate surface defects of CsPbBr3 QDs by coordination with halogen vacancies, thus increasing the radiation recombination rate and improving the PL efficiency.
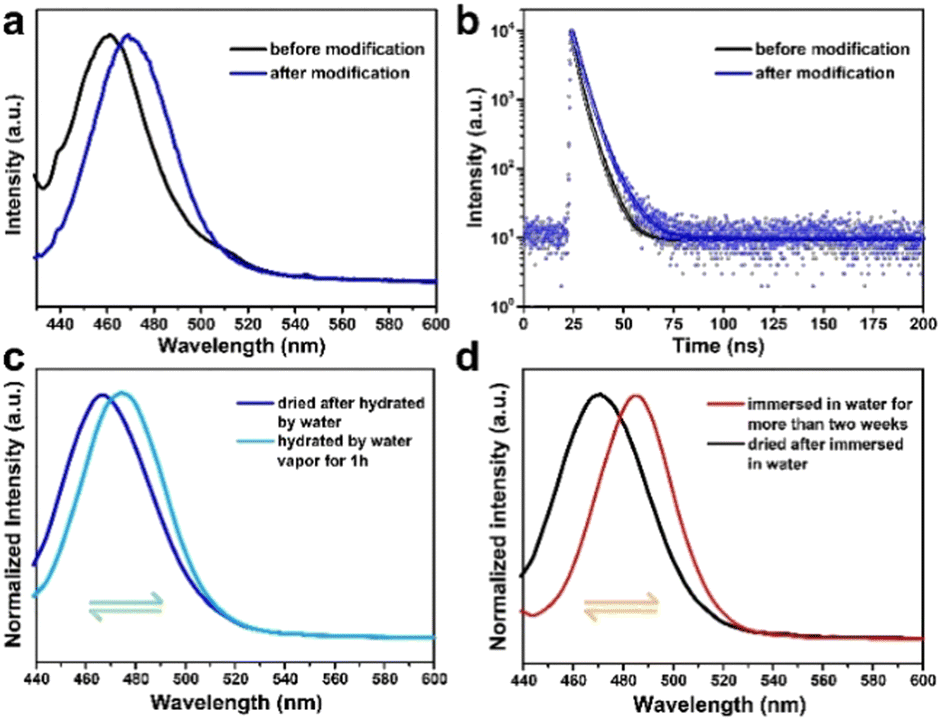 | ||
| Fig. 5 Water stability of the modified CsPbBr3@W-S-1 composite. (a) Fluorescence spectra and (b) fluorescence lifetime decay of the CsPbBr3@W-S-1 composite and modified CsPbBr3@W-S-1 composite. Reversible PL properties of the modified CsPbBr3@W-S-1 composite (c) in water steam and (d) immersed in water. | ||
To further prove the superior water stability of the modified CsPbBr3@W-S-1 composite, the experiment was performed in both water vapor and water solution. Only a 10 nm redshift of emission occurs after the modified CsPbBr3@W-S-1 was placed in hot water vapor for one hour (Fig. 5c). Strikingly, the emission could shift back to the initial position of 468 nm after the sample was placed in a room environment with 20% humidity for about 5 minutes, revealing the reversible fluorescence behavior of the modified composite. Moreover, after being immersed in water for 24 hours, the fluorescence emission of the modified CsPbBr3@W-S-1 composite red shifts to 484 nm, which keeps unchanged when prolonging the immersion time for more than 14 days. Also, the initial blue fluorescence emission centered at 468 nm returns after the removal of water from the composite (Fig. 5d). In contrast, the redshifted fluorescence of the unmodified CsPbBr3@W-S-1 composite is irreversible after water treatment. The reason for this reversible fluorescence phenomenon may be that when water molecules enter into the zeolite matrix, the amino acids in the pores would be hydrated, which weakens the hydrogen bonds formed between the amino acids and CsPbBr3 QDs, leading to partial aggregation of the small-sized perovskite in the pores and the redshifted fluorescence; while once the water molecules are removed from the zeolite, the amino acids encapsulated in the pores will be re-coordinated with QDs and separate the partial gathered CsPbBr3, resulting in the recovery of the blue fluorescence. Thus, the amino acid in the zeolite pores is crucial to passivate the defects of CsPbBr3 QDs, and to coordinate with perovskite QDs through H-bonds to regulate the emission behavior with a different environment. The synergy of nanoconfinement and trap passivation promotes the high-performance and water reversible blue luminescent perovskite phosphor.
Conclusions
This work presents the successful confinement growth of perovskite QDs into a zeolite matrix based on the “break-and-repair” strategy via the high-temperature evaporation method followed by amino acid passivation and silane restoration. In W-S-1 zeolite, the size of the CsPbBr3 QDs can be controlled from 2 nm to 6 nm by varying the synthetic conditions; as a result, the CsPbBr3@W-S-1 composites show 460 nm deep-blue to 510 nm green emission due to the strong quantum confinement. Furthermore, amino acids encapsulated in the zeolite pores can significantly passivate the defects of the CsPbBr3 QDs and silanization dynamically regulates their aggregation to promote high-efficiency, water-stable and reversible blue fluorescence emission. A deep-blue fluorescence with a QY of 39.3% and a sky-blue fluorescence with a QY of 76.9%, as well as high-stability in aqueous solution for more than 14 days are achieved. This work proves that zeolite is an ideal matrix for the growth of ultrasmall inorganic PQDs with stable luminescence via confinement engineering, which will offer significant potential for advanced light-emitting and light-harvesting devices.Author contributions
Hongyue Zhang and Bolun Wang: experiment design, experiment, investigation, data analysis, writing-original draft. Zijian Niu, Buyuan Guan: validation, investigation, data analysis. Guangrui Chen: TEM investigation. Jihong Yu: conceiving the project. Jiyang Li and Jihong Yu: supervision, revising manuscript, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (Grant No. 22288101, 21920102005 and 21835002) and the 111 Project (B17020) for supporting this work.Notes and references
- J. T. Michael, M. Lee, T. Miyasaka, T. N. Murakami and H. J. Snaith, Science, 2012, 338, 643–646 CrossRef PubMed.
- J. Burschka, N. Pellet, S. J. Moon, R. Humphry-Baker, P. Gao, M. K. Nazeeruddin and M. Gratzel, Nature, 2013, 499, 316–319 CrossRef CAS PubMed.
- C. Otero-Martinez, J. Z. Ye, J. Sung, I. Pastoriza-Santos, J. Perez-Juste, Z. G. Xia, A. Rao, R. L. Z. Hoye and L. Polavarapu, Adv. Mater., 2022, 34, 2107105 CrossRef CAS PubMed.
- N. Li, Y. H. Jia, Y. W. Guo and N. Zhao, Adv. Mater., 2022, 34, 2108102 CrossRef CAS PubMed.
- H. W. Huang, D. Verhaeghe, B. Weng, B. Ghosh, H. W. Zhang, J. Hofkens, J. A. Steele and M. B. J. Roeffaers, Angew. Chem., Int. Ed., 2022, 61, e202203261 CrossRef CAS PubMed.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- Y. Li, X. Zhang, H. Huang, S. V. Kershaw and A. L. Rogach, Mater. Today, 2020, 32, 204–221 CrossRef CAS.
- L. L. Yang, J. H. Huang, Y. K. Tan, W. Lu, Z. W. Li and A. L. Pan, Mater. Horiz., 2023, 10, 1969–1989 RSC.
- A. Dutta, M. K. Vyas, K. Bhowmik, J. Ota, S. K. Hait, K. Chandrasekaran, D. Saxena and S. S. V. Ramakumar, ACS Mater. Lett., 2022, 4, 2106–2124 CrossRef CAS.
- Y. Liu, S. Tang, Z. Gao, X. Shao, X. Zhu, J. Ràfols Ribé, T. Wågberg, L. Edman and J. Wang, Nano Res., 2023, 16, 10626–10633 CrossRef CAS.
- Y. Cao, N. Wang, H. Tian, J. Guo, Y. Wei, H. Chen, Y. Miao, W. Zou, K. Pan, Y. He, H. Cao, Y. Ke, M. Xu, Y. Wang, M. Yang, K. Du, Z. Fu, D. Kong, D. Dai, Y. Jin, G. Li, H. Li, Q. Peng, J. Wang and W. Huang, Nature, 2018, 562, 249–253 CrossRef CAS PubMed.
- Q. Chen, J. Wu, X. Ou, B. Huang, J. Almutlaq, A. A. Zhumekenov, X. Guan, S. Han, L. Liang, Z. Yi, J. Li, X. Xie, Y. Wang, Y. Li, D. Fan, D. B. L. Teh, A. H. All, O. F. Mohammed, O. M. Bakr, T. Wu, M. Bettinelli, H. Yang, W. Huang and X. Liu, Nature, 2018, 561, 88–93 CrossRef CAS PubMed.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang, C. Yan, W. Li, X. Liu, Y. Lu, J. Kirman, E. H. Sargent, Q. Xiong and Z. Wei, Nature, 2018, 562, 245–248 CrossRef CAS PubMed.
- Q. Zhang, W. Zheng, Q. Wan, M. Liu, X. Feng, L. Kong and L. Li, Adv. Opt. Mater., 2021, 9, 2002130 CrossRef CAS.
- Y. J. Wu, R. J. Jia, J. Xu, L. Song, Y. X. Liu, Y. M. Zhang, S. Ullah and J. Dai, Front. Mater., 2022, 9, 845977 CrossRef.
- X.-Y. Qian, Y.-Y. Tang, W. Zhou, Y. Shen, M.-L. Guo, Y.-Q. Li and J.-X. Tang, Small Sci., 2021, 1, 2000048 CrossRef CAS.
- Z. W. Ren, K. Wang, X. W. Sun and W. C. H. Choy, Adv. Funct. Mater., 2021, 31, 2100516 CrossRef CAS.
- H. He, S. Mei, Z. Wen, D. Yang, B. Yang, W. Zhang, F. Xie, G. Xing and R. Guo, Small, 2021, 18, 2103527 CrossRef PubMed.
- V. Malgras, S. Tominaka, J. W. Ryan, J. Henzie, T. Takei, K. Ohara and Y. Yamauchi, J. Am. Ceram. Soc., 2016, 138, 13874–13881 CrossRef CAS PubMed.
- M. Anaya, A. Rubino, T. Cristina Rojas, J. Francisco Galisteo-Lopez, M. Ernesto Calvo and H. Miguez, Adv. Opt. Mater., 2017, 5, 1601087 CrossRef.
- R. J. Kashtiban, C. E. Patrick, Q. Ramasse, R. I. Walton and J. Sloan, Adv. Mater., 2022, e2208575 Search PubMed.
- Y. C. Zhang, L. Han, B. H. Li and Y. Xu, Chem. Eng. J., 2022, 437, 135290 CrossRef CAS.
- H. Tsai, H. H. Huang, J. Watt, C. H. Hou, J. Strzalka, J. J. Shyue, L. Wang and W. Y. Nie, Adv. Sci., 2022, 9, 202105850 Search PubMed.
- D. N. Dirin, L. Protesescu, D. Trummer, I. V. Kochetygov, S. Yakunin, F. Krumeich, N. P. Stadie and M. V. Kovalenko, Nano Lett., 2016, 16, 5866–5874 CrossRef CAS PubMed.
- V. Malgras, J. Henzie, T. Takei and Y. Yamauchi, Angew. Chem., Int. Ed., 2018, 57, 8881–8885 CrossRef CAS PubMed.
- P. Wang, B. Wang, Y. Liu, L. Li, H. Zhao, Y. Chen, J. Li, S. F. Liu and K. Zhao, Angew. Chem., Int. Ed., 2020, 59, 23100 CrossRef CAS PubMed.
- J. Y. Kim, K. I. Shim, J. W. Han, J. Joo, N. H. Heo and K. Seff, Adv. Mater., 2020, 32, e2001868 CrossRef PubMed.
- J. Grand, S. N. Talapaneni, A. Vicente, C. Fernandez, E. Dib, H. A. Aleksandrov, G. N. Vayssilov, R. Retoux, P. Boullay, J. P. Gilson, V. Valtchev and S. Mintova, Nat. Mater., 2017, 16, 1010–1015 CrossRef CAS PubMed.
- Z. Guo, Y. Zhang, B. Wang, L. Wang, N. Zhou, Z. Qiu, N. Li, Y. Chen, C. Zhu, H. Xie, T. Song, L. Song, H. Xue, S. Tao, Q. Chen, G. Xing, L. Xiao, Z. Liu and H. Zhou, Adv. Mater., 2021, 33, 2102246 CrossRef CAS PubMed.
- W. Cai, M. U. Ali, P. Liu, M. He, C. Zhao, Z. Chen, Y. Zang, M.-C. Tang, H. Meng, H. Fu, G. Wei and H.-L. Yip, Adv. Sci., 2022, 9, 2200393 CrossRef CAS PubMed.
- J. Sun, F. T. Rabouw, X.-F. Yang, X.-Y. Huang, X.-P. Jing, S. Ye and Q.-Y. Zhang, Adv. Funct. Mater., 2017, 27, 704371 Search PubMed.
- Q. Zhang, B. Wang, W. Zheng, L. Kong, Q. Wan, C. Zhang, Z. Li, X. Cao, M. Liu and L. Li, Nat. Commun., 2020, 11, 31 CrossRef CAS PubMed.
- H. Bloom and R. G. Anthony, Aust. J. Chem., 1971, 24, 2001–2004 CrossRef CAS.
- M. C. Jacek Jasieniak and Scott E. Watkins, ACS Nano, 2011, 5, 5888–5902 CrossRef PubMed.
- C. Zhang, W. Li and L. Li, Angew. Chem., Int. Ed., 2021, 60, 7488–7501 CrossRef CAS PubMed.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- Y. Bekenstein, B. A. Koscher, S. W. Eaton, P. Yang and A. P. Alivisatos, J. Am. Chem. Soc., 2015, 137, 16008–16011 CrossRef CAS PubMed.
- R. Ahmad, J. Y. Kim, G. B. Park, N. H. Heo and K. Seff, J. Phys. Chem. C, 2021, 125, 5904–5918 CrossRef CAS.
- W. Xu, Q. Hu, S. Bai, C. Bao, Y. Miao, Z. Yuan, T. Borzda, A. J. Barker, E. Tyukalova, Z. Hu, M. Kawecki, H. Wang, Z. Yan, X. Liu, X. Shi, K. Uvdal, M. Fahlman, W. Zhang, M. Duchamp, J.-M. Liu, A. Petrozza, J. Wang, L.-M. Liu, W. Huang and F. Gao, Nat. Photonics, 2019, 13, 418–424 CrossRef CAS.
- H. Shi, X. Zhang, X. Sun and X. Zhang, J. Phys. Chem. C, 2019, 124, 1617–1622 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3mh01092a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |