
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen bonding to oxygen in siloxane bonds drives liquid phase adsorption of primary alcohols in high-silica zeolites†
Sambhu
Radhakrishnan
 ab,
Charlotte
Lejaegere
a,
Karel
Duerinckx
ab,
Wei-Shang
Lo
c,
Alysson F.
Morais
ab,
Dirk
Dom
ab,
C. Vinod
Chandran
ab,
Ive
Hermans
acd,
Johan A.
Martens
ab and
Eric
Breynaert
*ab
ab,
Charlotte
Lejaegere
a,
Karel
Duerinckx
ab,
Wei-Shang
Lo
c,
Alysson F.
Morais
ab,
Dirk
Dom
ab,
C. Vinod
Chandran
ab,
Ive
Hermans
acd,
Johan A.
Martens
ab and
Eric
Breynaert
*ab
aCentre for Surface Chemistry and Catalysis - Characterization and Application Team (COK-KAT), KU Leuven, Celestijnenlaan 200F Box 2461, 3001-Heverlee, Belgium. E-mail: Eric.Breynaert@kuleuven.be
bNMRCoRe - NMR/X-Ray platform for Convergence Research, KU Leuven, Celestijnenlaan 200F Box 2461, 3001-Heverlee, Belgium
cDepartment of Chemistry, University of Wisconsin-Madison, 1101 University Avenue, Madison, Wisconsin 53706, USA
dDepartment of Chemical and Biological Engineering, Wisconsin Energy Institute, University of Wisconsin-Madison, 1552 University Ave, Madison, WI 53726, USA
First published on 27th June 2023
Abstract
Upon liquid phase adsorption of C1–C5 primary alcohols on high silica MFI zeolites (Si/Al = 11.5–140), the concentration of adsorbed molecules largely exceeds the concentration of traditional adsorption sites: Brønsted acid and defect sites. Combining quantitative in situ1H MAS NMR, qualitative multinuclear NMR and IR spectroscopy, hydrogen bonding of the alcohol function to oxygen atoms of the zeolite siloxane bridges (Si–O–Si) was shown to drive the additional adsorption. This mechanism co-exists with chemi- and physi-sorption on Brønsted acid and defect sites and does not exclude cooperative effects from dispersive interactions.
New conceptsZeolites can be polar or non-polar. The highly polar ones contain lots of aluminium in their framework and cations in their pores. They are strong adsorbents entirely filling up their pores when exposed to liquids and vapours. The non-polar ones are generally considered to poorly adsorb polar compounds. Surprisingly, we discovered pore-filling adsorption of rather polar molecules such as alcohols also occurs on high-silica zeolites, yielding strongly adsorbed alcohol at concentrations largely exceeding the availability of polar sites provided by the Al atoms. So what is keeping those extra molecules in the pores? For decades, literature ascribed it to dispersive van der Waals interactions, the same mechanism responsible for the analogous adsorption of alkanes. Combining in situ NMR and IR spectroscopy, hydrogen bonding of the alcohol function to the siloxane bridges of the purely silicious section of the zeolite was identified as the culprit, a new mechanism co-existing with the traditionally accepted alcohol adsorption mechanisms like Brønsted acid site adsorption and adsorption at framework defects (i.e. silanols). Such insights are not only relevant for rationalizing adsorption but also of foundational importance to understand solvent effects in liquid phase reactions, catalysed by such microporous materials. A similar mechanism involving interaction of H of alkane C–H bonds with O in siloxane bridges was suggested theoretically to drive preferential adsorption of C2H6 over C2H4 in purely siliceous, defect-free zeolites. In a similar way as the alcohols, 17O isotope-enriched water can H-bond to the siloxane bridges and assist their reversible opening. This can finally explain the unexpected isotope exchange of 17O into zeolite frameworks, simply by equilibrating zeolites with 17O enriched water at room temperature. |
Industrial processes involving ion-exchange, gas separation and catalytic conversion often implement zeolites.1,2 Aluminium-rich zeolites are commonly used as cation exchangers,3–5 while catalytic and separation applications typically implement siliceous zeolites especially when dealing with organic molecules such as volatile organic compounds (VOC's) and water pollutants.6–8 Few applications explicitly target adsorptive separation, yet almost all zeolite applications are impacted by adsorption phenomena.9–11 Classical adsorption applications of zeolites include drying of gases and solvents, natural gas desulfurization, and gas separation, e.g. CH4/N2 and O2/N2. In the frame of renewable energy and feedstock production, adsorption of alcohols on zeolites is rapidly gaining interest, as separation of alcohol–water mixtures is an important aspect of the valorisation of bio-based chemicals produced via fermentation.12,13 Zeolites are introduced in pervaporation membranes targeting such separations.14–19 In catalytic conversion of methanol-to-olefins20,21 and catalytic dehydration of ethanol to ethylene,22 competitive adsorption between alcohols, hydrocarbons and water play a key role. Dominant factors controlling alcohol adsorption are the zeolite pore structure, pore dimensions and aluminium content,22 the latter determining the Brønsted acidity and polarity of the pore wall.
A prime example of a liquid phase catalytic process where adsorption drives reaction selectivity is the ethoxylation of β-citronellene over acid zeolite beta (*BEA framework type) catalyst.9 Ethanol is preferentially adsorbed inside the zeolite pores, while β-citronellene molecules are preferentially adsorbed at pore mouths spread over the external surface of the zeolite particles. Whereas pore mouth adsorbed β-citronellene undergoes selective ethoxylation, β-citronellene molecules occasionally adsorbed inside the zeolite pores undergo isomerization reactions to side products, thus lowering the selectivity for the desired product. By competitive adsorption of ethanol inside the zeolite pores, the concentration of β-citronellene adsorbed inside the zeolite is lowered, favouring the desired bimolecular reaction.9
Most investigations specifically addressing alcohol adsorption onto zeolites report on adsorption of alcohols from the vapor phase. At low partial pressures, vapor phase adsorption of an alcohol molecule on a zeolite predominantly occurs on Brønsted acid sites (BAS). Adsorption either occurs via chemisorption, forming a protonated alcohol, or through physisorption with the alcohol accepting the Brønsted acid proton in a strong hydrogen bonding interaction.23–27 For methanol, adsorption of up to 3 molecules per Brønsted acid site has been reported. This was explained by formation of large protonated methanol clusters held together by up to six strong hydrogen bonds.28 Adsorption on framework defects, i.e. silanol groups, has been reported as a secondary mechanism, but the affinity of water and alcohols for such sites is much lower as compared to the affinity for BAS.29
Using vapor phase adsorption, pore saturation is not often reached and most studies consequently report trends observed at low surface coverage.30 Aronson et al. achieved substantial pore filling of ZSM-5 zeolite (MFI framework type) of Si/Al ratio 140 with C1–C4 alcohol vapours.31 The majority of the adsorbed alcohol could be evacuated at room temperature, leaving 1–2 alcohol molecules adsorbed per BAS. Short chain alcohols (methanol, ethanol and 1-propanol) were removed easily, while 1-butanol required significantly longer evacuation times to attain a residual coverage of two molecules per BAS.31 The monotonous increase in the adsorption enthalpy for every carbon atom added to the alkyl chain length (methanol < ethanol < 1-propanol < 1-butanol) has been attributed to dispersive van der Waals interactions, an interaction depending on pore size and shape.25,32,33 For example, as the steric fit is closer in MFI pores (diameter 5.4 Å) as compared to *BEA channels (7.6 Å), dispersion forces for 1-butanol in MFI are suggested to be larger.34,35 Modelling of C1–C4 primary alcohols in MFI channels showed that adsorption in the zigzag channels is energetically preferred, the difference with the linear channels amounting up to 20 kJ mol−1.25 Similarly, oligomerization of furfuryl alcohol preferentially occurs in the straight channels of MFI when water is used as a solvent and in the straight channels with dioxane as the solvent.10
For liquid phase adsorption of alcohols, polarity, hydrogen bonding and adsorbate–adsorbate packing efficiency within the adsorbent pore channels modulate the adsorption process.33–38 Smaller molecules typically reach the highest packing efficiencies.33,39 Interestingly, selectivity trends observed for vapor phase adsorption may reverse when studying adsorption from the liquid phase. For a homologous series of hydrocarbons, this reversal is spectacular and explained by molecular packing effects.11
Literature suggests ZSM-5 as a suitable zeolite for selective liquid phase adsorption of alcohol from dilute alcohol–water mixtures.40–42 However, not much is known about the adsorption mechanism. Defect-free all-silica zeolites hardly adsorb liquid water at ambient conditions.43 Liquid water forms a strongly hydrogen bonded network preventing it to fill micropores of hydrophobic zeolites.44,45 At increasing pressure, water is forced to enter the hydrophobic pores, restructuring its hydrogen bonding network and most likely forming chain-like clusters to optimize the interaction with the zeolite surface.44,46,47 Water clusters intruded at high pressure have lower density than bulk water and may even provoke framework hydrolysis, facilitating water uptake.47–49 Framework defects and BAS make zeolites less hydrophobic, enabling water to enter the framework also at ambient pressure.45,46,50 Defects typically are silanol nests, distinctly different from BAS generated by isomorphic substitution of framework silicon atoms by aluminium. Humplik et al. concluded that pure silica MFI zeolite, with a theoretical pore filling adsorption capacity of up to 35 water molecules per unit cell, could only adsorb 4 molecules in the defect-free case and up to 27 molecules in a defect-rich sample.29 The limited interaction between hydrophobic zeolites and water molecules results in a high selectivity for alcohol adsorption from alcohol–water mixtures.45,46,50 This is also reflected in the heat of adsorption of alcohols being much higher than that of water.46 Upon adsorption from water-alcohol mixtures, oxygen atoms of the zeolite framework overtake alcohol hydration with dispersive forces, resulting in an even more selective adsorption of the alcohols.29 Following initial adsorption of alcohol, water can however co-adsorb. From an aqueous solution of primary short-chain alcohols, more water molecules are co-adsorbed onto MFI zeolite as compared to adsorption of pure water onto the same zeolite.46,51 Water molecules interact more strongly with the alcohol group via hydrogen bonding than with the zeolite pore wall, thus forming alcohol–water clusters and enabling the co-adsorption.42,51,52 In case of adsorption from mixtures, again molecular packing and pore blocking need to be taken into account. In cases where entropy effects are the dominating factor driving adsorption, smaller molecules appear to be preferred. In mixtures containing larger molecules, larger compounds can block the access to channels and pores for smaller molecules.53
To understand adsorption on a molecular level, quantification of the adsorption typically needs to be combined with spectroscopy to reveal the interactions between adsorbate and adsorbent. For investigating alcohol and water adsorption, nuclear magnetic resonance spectroscopy (NMR) is one of the preferred techniques, as it enables absolute quantification while simultaneously providing insight in the molecular organization of the adsorbate inside the zeolite pores and its interactions with the pore wall.54–56
This manuscript reports on the liquid phase adsorption of C1–C5 alcohols on ZSM-5 zeolites with Si/Al ratio in the range of 11.5–140. The crystallinity and microporosity of the different zeolites were ensured by PXRD57 (Fig. S4, ESI†) and N2 physisorption experiments (Table 1 and Fig. S5, ESI†). The alcohols were adsorbed as pure components from the liquid phase. In situ quantitative direct excitation 1H MAS NMR,54 was combined with qualitative 1H–13C CPMAS, 1H–29Si heteronuclear correlation and IR spectroscopy. This combination not only enabled to quantify the adsorption, but also revealed hydrogen bonding of alcohol groups with siloxane bonds of the zeolite framework as a previously undocumented adsorption mechanism.
| Zeolite | Si/Ala | 27Al [mmol g−1] | 1Hb [mmol g−1] | BET analysis | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Totala | Tetra | Distorted tetra | Penta | Hexa | BAS | SiOH | AlOH | S BET [m2 g−1] | V micro c [cm3 g−1] | V meso d [cm3 g−1] | ||
| a Provided by the supplier. b Determined via quantitative 1H NMR on samples dried under vacuum 0.75 torr at 200 °C. | ||||||||||||
| MFI-11.5 | 11.5 | 1.34 | 0.58 | 0.33 | 0.16 | 0.27 | 1.05 | 0.14 | 0.47 | 375 | 0.16 | 0.05 |
| MFI-15 | 15 | 1.04 | 0.56 | 0.21 | 0.11 | 0.16 | 0.64 | 0.15 | 0.22 | 354 | 0.14 | 0.08 |
| MFI-25 | 25 | 0.64 | 0.47 | 0.13 | 0.0 | 0.05 | 0.60 | 0.20 | 0.04 | 367 | 0.15 | 0.09 |
| MFI-40 | 40 | 0.41 | 0.35 | 0.04 | 0.0 | 0.02 | 0.37 | 0.15 | 0.08 | 399 | 0.17 | 0.09 |
| MFI-140 | 140 | 0.12 | 0.09 | 0.02 | 0.0 | 0.01 | 0.07 | 0.04 | 0.01 | 371 | 0.17 | 0.06 |
In direct excitation MAS NMR, the presence of non-adsorbed, mobile species is indicated by the appearance of sharp resonances on top of broadened resonances associated with adsorbed molecules. The broadening of the resonances of adsorbed species is due to two factors. Immobilised species exhibit significantly less rotational freedom, leading to a less efficient averaging of the dipolar interactions. This shortens the transverse relaxation times (T2) and gives rise to peak broadening. In the absence of other broadening mechanisms (e.g. chemical shift distribution), the full width at half maximum (FWHM) of a peak is proportional to (πT2)−1.58 Additional broadening results from the heterogeneity of the adsorption sites, introducing slightly different chemical shifts for species adsorbed at different sites and thus introducing chemical shift broadening.
Single alcohol saturation capacities were determined by dosing aliquots of alcohol liquid onto MFI zeolite vacuum dried in an NMR rotor. Dosing was continued until mobile alcohol molecules were detected in the quantitative 1H MAS NMR spectra. Fig. 1a shows the evolution of the 1H MAS NMR spectra within a single 1-pentanol adsorption experiment recorded in a quantitative manner.54 Spectral decomposition of the 1H spectra was performed to quantify adsorbed and liquid-like alcohol fractions. Assignment of the 1H resonances was assisted by 1H–13C HMQC (Fig. S6, ESI†) and 1H–13C HETCOR (Fig. 1a inset, Fig. S7, ESI†) correlation spectroscopy. Assignment of the adsorbed and mobile fractions was experimentally verified by evacuation of 1-pentanol from MFI-140 at 0.75 torr for ca. 20 h. This treatment exclusively removed 1H MAS NMR resonances resulting from 1-pentanol molecules assigned as mobile, thus confirming the strong adsorption of the fraction assigned as adsorbed (Fig. S8, ESI†). Interestingly, a 1-pentanol to BAS ratio amounting to 23.2 was observed even after this evacuation step. An example of the spectral decomposition is shown in Fig. 1b for MFI-140 with the highest 1-pentanol loading. Quantification of adsorbed and mobile species was performed using, respectively, the integrated areas of the broad and sharp spectral components of the non-exchangeable methyl–methylene protons, observed in the 0 to 3 ppm range. For each zeolite, the saturation capacity expressed as number of alcohol molecules adsorbed per unit cell or per BAS for different alcohols (C1–C5) is presented in Table 2. Experimentally determined adsorption capacities were compared to the estimated theoretical maximum alcohol adsorption capacity per unit cell calculated by considering the geometry of the two-channel type MFI framework (Table 2, Table S5 and Fig. S9, ESI†). One unit cell contains four linear segments of 4.5 Å length, four sinusoidal segments of 6.5 Å length and four intersections, all of cross section 5.5 Å.59 The total channel length considered for adsorption was 18, 26.6 and 22.4 Å for the straight channels, the sinusoidal segments, and the intersections respectively. Adsorbed alcohol molecules were assumed to exhibit a critical diameter of ∼4.6 Å and to orient along the pore axis.60 As expected from a geometrical perspective, increasing chain length leads to a decrease in the maximum number of adsorbed molecules. For most alcohols, the NMR-determined number of alcohols per unit cell was comparable to the theoretically estimated capacities (Table 2).61–64 The number of alcohol molecules per unit cell of MFI changed from 23.8 for methanol to 9.5 for 1-pentanol. Smaller molecules, such as methanol and ethanol, exhibited a slightly higher ratio of the experimentally versus theoretically determined maximum capacity. As the length of these small molecules is below the critical channel diameter, they potentially can orient sideways in the channels, leading to the disparity in the estimations. Considering known liquid alcohol densities, the maximum adsorbed capacity can be converted to volume and subsequently compared to the pore volume determined by N2 physisorption, again confirming complete pore filling (Tables 1 and 2).
 | ||
| Fig. 1 (a) 1H NMR spectra of MFI-140 exposed to different amounts of liquid 1-pentanol. 1H–13C CP-HETCOR spectrum (inset) aiding assignment of the resonances. (b) Spectral decomposition of the 1H NMR spectrum at a 1-pentanol dosage of 0.19 mL g−1 on MFI-140. Adsorbed and mobile fractions are represented by the green and blue traces, respectively. All the spectra were acquired at 295 K. | ||
| 1-Alcohol | Theory | Experiment | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MFI | H-MFI-140 | H-MFI-11.5 | |||||||||
| N alcohol per UC | Adsorbed amount [mmol g−1] | Adsorbed volumea [mL g−1] | N alcohol per UC | N alcohol per BAS | N alcohol per BAS + defect sites | Adsorbed amount [mmol g−1] | Adsorbed volumea [mL g−1] | N alcohol per UC | N alcohol per BAS | N alcohol per BAS + defect sites | |
| a Assuming liquid-like packing of molecules. | |||||||||||
| Methanol | 23.9 | 4.07 | 0.17 | 23.8 | 58.2 | 33.9 | 4.25 | 0.17 | 24.8 | 4 | 2.6 |
| Ethanol | 16.5 | 3.12 | 0.18 | 18.2 | 44.5 | 26 | 3.00 | 0.18 | 17.6 | 2.9 | 1.8 |
| 1-Propanol | 12.6 | 2.34 | 0.18 | 13.7 | 33.5 | 19.5 | 1.96 | 0.15 | 11.4 | 1.9 | 1.2 |
| 1-Pentanol | 9.5 | 1.62 | 0.18 | 9.5 | 23.2 | 13.5 | 1.31 | 0.14 | 7.6 | 1.2 | 0.8 |
Exclusively considering alcohol adsorption mechanisms derived in gas phase adsorption studies,25,32i.e., chemi- and physi-sorption on BAS and framework defects, alcohol adsorption on the high silica MFI-140 should be nearly indifferent to the identity of the alcohol for short chain 1-alcohols (C1–C5). The ratio of adsorbed alcohol molecules per BAS, however, varied gradually from 58 for methanol to 23 for 1-pentanol (Table 2), values similar to those reported by Aronson et al. for pore-filling alcohol adsorption.31 These values readily exclude adsorption onto BAS as the dominant mechanism for alcohol adsorption in MFI-140. Even considering strongly hydrogen-bonded clusters of molecules associated with protonated BAS, a situation previously reported for methanol,28 for steric reasons, the maximum capacity of 58 methanol molecules per BAS cannot be explained by a mechanism based on BAS.
Next to BAS, also defect sites (SiOH and AlOH) and aluminol groups (AlOH) on extra-framework Al species can act as adsorption sites for alcohols.65 The presence of defect sites was confirmed and quantified by quantitative direct excitation 1H MAS NMR spectroscopy performed on the dehydrated zeolites. The presence of extra-framework Al was assessed using 27Al MAS NMR (Table S4 and Fig. S3, ESI†). Extra-framework Al often is created by calcination of zeolites and has been shown to generate AlOH species.66,67 Considering BAS, framework defects and extra framework aluminols as adsorption sites, the number of adsorbed molecules per adsorption site (BAS + AlOH + SiOH) becomes 33.9 for methanol and 13.5 for 1-pentanol (Table 2). These values are still unfeasible for steric reasons and confirm the adsorption mechanism for alcohols in MFI-140 not to be dominated by adsorption onto BAS, AlOH and SiOH. While not dominating the adsorption of alcohols in MFI-140, the occurrence of a 1H resonance at 8–8.5 ppm indicates chemisorption on BAS does occur on MFI-140.24,68,69
To evaluate if the dominant adsorption mechanism of alcohols is different in high silica versus in low silica zeolites, the adsorption experiments performed on MFI-140 (Si/Al = 140) were repeated on MFI-11.5 (Si/Al = 11.5) (Table 2). In this case, the number of alcohol molecules adsorbed per BAS ranged from 4 for methanol to 1.2 for 1-pentanol. Including defect sites and extra framework aluminols in the calculation, these numbers become 2.6 for methanol and 0.8 for 1-pentanol. This highly contrasts the situation observed for MFI-140 and indicates alcohol adsorption on Brønsted acid and defect sites can be the predominant sorption mechanism in MFI-11.5, in line with what has been documented for gas phase adsorption.2,24,26 Indeed, the presence of a significant fraction of downfield shifted 1H resonances (8–10 ppm) in the 1H MAS spectrum of 1-pentanol adsorbed to MFI-11.5, a resonance associated with protonated alcohol molecules, spectroscopically confirms chemisorption on BAS as the dominant adsorption mechanism in these low silica samples. Close inspection of the spectra further allows to discriminate between chemisorbed (protonated alcohols) and physisorbed alcohol molecules, retained through hydrogen bonding (Fig. S10, ESI†).24,68,69 1-Pentanol adsorption capacity was determined for 5 MFI zeolites with Si/Al ratios in the range of 11.5 to 140. The adsorption capacity of 1-pentanol increases with increasing Si/Al ratio (Table S6, ESI†), with pore-filling adsorption observed for MFI-40 and MFI-140. This also points towards a change in adsorption pathway with increase in Si/Al ratio.
Adsorption mechanism
The experimental results indisputably demonstrate the dominant mechanism of alcohol adsorption on MFI zeolites to depend on the Si/Al ratio and thus on the BAS density and surface polarity of the zeolite. In low Si/Al zeolites, chemisorption and hydrogen bonding to BAS dominate the interaction. In high silica MFI, alternative mechanisms are at play. Alternative mechanisms potentially increasing the alcohol adsorption capacity of high silica zeolites beyond the capacity of 1 or 2 molecules per BAS or defect site are limited. Linear alcohols could potentially be retained by dispersion forces, similar to the mechanism observed for adsorption of linear alkanes.70 The only alternative possibility is hydrogen bonding, either concerted to already adsorbed guest molecules (i.e. alcohol and/or water), thus forming clusters of adsorbed molecules, or to siloxane bridges of the zeolite framework, similar to what has been suggested to drive adsorption of alcohols onto pristine quartz surfaces.71 Either of these options can however be assessed from a combination of NMR and IR spectroscopy. The occurrence of hydrogen bonding is always reflected in the 1H chemical shift of the alcohol proton, as well as in a red-shift of the OH vibration in IR spectroscopy. What the alcohol group hydrogen-bonds to can always be assessed from NMR and in some cases from IR spectroscopy. In case the alcohol hydrogen-bonds to the OH group of another alcohol molecule, to a water molecule or to a silanol group, the alcohol proton should exhibit a double-quantum correlation respectively either with itself, with water or with silanol in a 1H–1H double quantum–single quantum (DQ–SQ) correlation NMR spectrum. Absence of such DQ–SQ correlations in combination with the occurrence of hydrogen bonding implicates isolated hydrogen bonding to siloxane bridges in the wall of the zeolite pores as the sorption mechanism. In case adsorption of the alcohol is driven by dispersion forces, mediated by the alkane chain of the linear alcohol, the C-1 carbon should exhibit a higher mobility as compared to the rest of the chain, especially for alcohols with a chain length of three or more carbons. It also must be noted that hydrogen bonding and adsorption via dispersion forces can occur together as cooperative mechanisms of adsorption.To gain insight into the mechanism responsible for pore filling adsorption in high silica zeolites and assess the mechanistic options described above, quantitative 1H direct excitation and 1H–13C CPMAS (Fig. 2) measurements were performed on MFI-140 and MFI-11.5 zeolite adsorbed with 1-pentanol. To assist with the assignment and interpretation of these 1-dimensional spectra, also 1H–1H RFDR, 1H–1H DQ–SQ and 1H–13C CP-HETCOR correlation NMR spectra were acquired. In addition, 1H direct excitation, 1H–13C INEPT, 1H–13C HMQC NMR spectra were recorded for 1-pentanol dissolved in CDCl3 and in D2O (Fig. 2 and Fig. S6, ESI†). CDCl3 is a non-hydrogen bonding solvent. Consequently, dilute solutions of 1-pentanol in CDCl3 (2 vol%) provides access to the chemical shift of the alcohol group of non-hydrogen bonded isolated molecules (Fig. 2A-a). At higher concentration (17.5 vol%), hydrogen bonded clusters are formed, shifting the OH resonance to a higher chemical shift (Fig. 2A-c). Dissolving 1-pentanol in D2O, a strongly hydrogen bonding solvent, provides access to the hydroxyl chemical shift of a strongly hydrogen bonded 1-pentanol molecule (Fig. 2A,f). Correct assignment of the OH resonance in the solution state 1H spectra was verified using 1H–13C HMQC (Fig. S6, ESI†).
Decomposition of the 1H NMR spectra (Fig. 2A-d,e and Fig. S10, ESI†) for zeolite adsorbed 1-pentanol revealed a significant downfield shift of the C-1 CH2 and –OH resonances with increasing hydrogen bonding strength. Correct assignment of the 1H resonances of the zeolite adsorbed 1-pentanol was aided by 1H–13C CP-HETCOR and 1H–1H RFDR NMR spectra (Fig. S7 and S12, ESI†). Apart from the difference in chemical shifts of the C-1 CH2 and OH resonances, significant differences in the integrated areas were observed between MFI-11.5 and MFI-140. As the alkyl protons are non-exchangeable, the ratio of integrated areas between the resonances of the nine protons on the C-2 to C-5 carbons at 0–3 ppm and the two protons on the C-1 carbon at 3.7 ppm (MFI-140) and 3.9 ppm (MFI-11.5) should ideally be 4.5. In the absence of chemical exchange, the ratio between the OH and C-1 CH2 resonances should be 0.5. For the mobile fractions, the observed ratios were close to the ideal values, being 4.5 and 0.49 for MFI-140 and 4.4 and 0.44 for MFI-11.5 (Fig. S10, ESI†). For the immobile fractions, significantly lower ratios of 4.1 and 0.33 were observed for MFI-140 and close to ideal 4.6 and 0.45 for MFI-11.5. The deviation from the ideal values for MFI-140 indicates that at least part of the OH resonances is appearing at a lower chemical shift than what is expected for chemisorbed alcohols leading to an overlap with the CH2-1 resonance. This is partly attributed to chemical exchange occurring between the defect protons on the zeolite and the alcohol proton, and partly due to the presence of OH resonances with an up-field shift with respect to the chemisorbed alcohol proton. A related up-field shift is observed when H-bonding molecules such as water and alcohols are dissolved in solvents with different H-bonding capacities. The OH resonance of 2 vol% 1-pentanol in D2O appears at 4.85 ppm, while it appears at 1.4 ppm when dissolved in CDCl3. This difference is due to the higher extent of H-bonding occurring when dissolved in D2O, compared to isolated molecules without any or with a limited number of H-bonds in CDCl3. Even in CDCl3, the OH resonance shifts downfield from 1.4 to 2.2 to 2.9 ppm with increase in 1-pentanol concentration from 2 to 9.1 to 16.7 vol%. This is due to the increased extent of H-bonding between 1-pentanol molecules to form clusters with the increase in concentration. For the immobile fraction adsorbed on MFI-11.5, the observed ratios were 4.6 and 0.45, respectively (Fig. S10, ESI†). The slight increase in the ratio between the alkyl resonances can be explained by the contribution of protons at defect sites (Si–OH, Al–OH) occurring between 1 and 3 ppm and thus contributing to the area assigned to the C-2 to C-5 alkyl protons. Comparing both MFI samples, the different up field shift observed for the OH resonance of 1-pentanol in MFI-140 as compared to MFI-11.5 can be explained by a varying degree of H-bonding.72 In MFI-11.5, hydrogen bonding predominantly occurs to the BAS, with the alcohol fulfilling both the role of donor and acceptor. The smaller shift observed for MFI-140 indicates a weaker H-bonding. H-bonding is highly sensitive to temperature and lower temperature is known to induce stronger H-bonds.73 Lowering the temperature of the MFI-140 sample to 253 K led to a downfield shift in the OH resonance of 1-pentanol and thus an increase in its hydrogen bonding strength. At the same time, the ratio between the area of the OH and C-1 CH21H resonances increased from 0.33 at 295 K to 0.48 at 253 K, the latter value being close to the expected value of 0.5 (Fig. S11, ESI†). The downfield shift of the OH resonance and change in the ratio between the integrated areas to theoretical values with temperature verify the NMR assignments and confirms that the alcohol proton takes part in hydrogen bonding.
Hydrogen bonding is also reflected in the 1H–13C CPMAS spectra (Fig. 2B-d, e) respectively. In the 13C NMR spectra of the 1-pentanol adsorbed zeolites, the resonance at ca. 63.8–65.6 ppm corresponds to the C-1 methylene moiety, also carrying the –OH group. Evaluating the linewidths in the spectra, both for MFI-11.5 and MF140, the FWHM of the methylene carbons (C2–C4) decreases with decreasing chemical shift and thus with increasing distance from the –OH group. The terminal methyl group (C-5) is observed at ca. 13.5–14 ppm. The increase in FWHM is induced by a difference in mobility, implying that for both samples the alcohol attaches to the zeolite framework through its C1–CH2OH moiety. The peak broadening behaviour also supports the absence of an adsorption mechanism dominated by dispersion-forces, as in this case, C-1 should become the most mobile carbon in the chain. Spectral decomposition further revealed predominantly one contribution to all resonances in the case of MFI-11.5, while two sets of resonances were required to describe the 13C spectrum of 1-pentanol adsorbed in MFI-140. The 13C chemical shift of the C-1 methylene moiety showed a downfield shift to 65.6 ppm for MFI-11.5 compared to respectively 64.6 and 63.7 ppm for the broad and sharp components in 1-pentanol adsorbed MFI-140. Complimentary to the liquid state adsorption of 1-pentanol on MFI-11.5 and MFI-140, in situ FTIR spectroscopy was performed on these zeolites in dehydrated form (Fig. S15, ESI†) and during gas adsorption as a function of the equilibrium pressure (Fig. S16, ESI†), with a similar T–O–T overtone area (1950–1750 cm−1) of the parent zeolite. In addition, FTIR spectra of liquid 1-pentanol, gas phase 1-pentanol and 5 and 10 vol% 1-pentanol in dichloromethane, a non-hydrogen bonding solvent,74 were recorded (Fig. S17, ESI†). Fig. S16 (ESI†) shows the IR spectra of 1-pentanol adsorbed on MFI-140 and MFI-11.5 with increasing dosage of 1-pentanol at 30 °C under vacuum. Adsorption of 1-pentanol was characterized by the appearance of the symmetric and asymmetric C–H stretching modes (3000–2800 cm−1) of 1-pentanol. Concurrently, a decrease in the intensity of the OH stretching mode of SiOH, AlOH (3743 cm−1) and of the BAS (3613 cm−1) was observed for both zeolites, the intensities of both modes corresponding to the concentration of unbound defects and BAS sites (Fig. S16, ESI†). The dampening of the BAS stretching mode already occurs at the lowest dosages of 1-pentanol, indicating a preferential adsorption at these sites, in line with what has been described in literature.26,75 In MFI-11.5, increasing the equilibrium coverage of 1-pentanol increasingly dampens the stretching mode associated with the BAS and also the mode associated with SiOH and AlOH groups at defect sites and at extra-framework Al (3743 cm−1). In the dataset for MFI-140, the same evolution can be observed albeit with intensities not dominating the spectra, in line with the NMR results obtained for liquid phase adsorption (supra). The dominant features occurring in the spectral series for MFI-140 are the appearance of O–H stretching modes centered around ∼3640, ∼3625, ∼3508, ∼3429 and ∼3330 cm−1. The modes at ∼3640 and ∼3625 cm−1 correspond to isolated 1-pentanol occurring in the gas phase and in a non-hydrogen bonding hydrophobic environment. Similar resonances indeed appear in gas-phase 1-pentanol (3672 cm−1) and for low concentrations of 1-pentanol dissolved in dichloromethane (3615 cm−1) (Fig. S17, ESI†).74 With increasing equilibrium concentration of 1-pentanol, new dominant features appear in the spectral series for MFI-140 (Fig. S16a, ESI†). For increasing 1-pentanol doses, a band centered at ∼3508 cm−1 first appears and, with higher dosage, the FTIR spectra become dominated by bands centered around ∼3429 and 3330 cm−1. In gas-phase FTIR studies, similar bands have previously been attributed to H-bond formation in alcohol dimers (∼3500 cm−1) and in polymeric networks (∼3400 cm−1), respectively.37,72 The presence of bands between 3200 and 3500 cm−1 are indicative of the presence of 1-pentanol occurring in a hydrogen bonded state, but FTIR spectra alone do not allow to make a more detailed assignment of the species involved, as recently, similar frequencies and frequency shifts also have been attributed to hydroxyl protons hydrogen bonding to siloxane bridges.76 In dry zeolites, the IR band associated with free BAS protons (Al–OH–Si) typically occurs around 3610 cm−1. Hydrogen bonding of this proton to oxygen in siloxane bridges results in a red shift of 360 ± 175 cm−1.76 The width of this range of shifts is associated to the variety of local geometries, with T-sites occurring in different types of channels (straight, zigzag) or at intersections. Hydrogen bonding of the hydroxyl proton of 1-pentanol to siloxane bridges, can cause a similar red-shift and could explain the frequencies observed in the FTIR spectra of 1-pentanol adsorbed MFI zeolite (Fig. S16, ESI†). NMR spectroscopy however remains essential to discriminate between all possible solutions as hydrogen bonded BAS protons give rise to 1H resonance around 7 ppm, while the resonance for hydrogen bonded alcohol protons is expected between 4 and 5 ppm (Fig. 2).
 | ||
| Fig. 2 1H NMR spectrum (A, left) and 13C NMR spectrum (B, right) of 1-pentanol dissolved in CDCl3 at 2 vol% (a), 9 vol% (b) and 16.7 vol% (c), 1-pentanol dissolved in D2O at 2 vol% (f), 1-pentanol adsorbed in H-MFI-140 (d) and H-MFI-11.5 (e). Adsorption was performed with pure 1-pentanol liquid until saturation was achieved. Samples were further evacuated at room temperature to remove the mobile species. All the spectra were acquired at 295 K. 13C NMR spectrum of pentanol adsorbed on zeolites was acquired with cross-polarization (CPMAS) NMR. | ||
Combining the FTIR results with the NMR data recorded for liquid phase adsorption of 1-pentanol on MFI-140 indeed allows to exclude certain assignments. From a geometrical perspective, the occurrence of polymeric networks of 1-pentanol in MFI-140 can be excluded. Hydrogen bonded dimers and even tetramers would potentially fit into respectively the channels and channel intersections of the framework, but their presence can be excluded as the occurrence of such species would imply that a self-correlation should be visible at coordinates (∼5, ∼10) ppm in 1H-1H DQ–SQ NMR spectra for this family of samples. Such correlation is clearly absent while the DQ self-correlations for the methylene protons ((1.5, 3) and (3.7, 7.4) ppm), the cross-correlations between C-1 and C-2 methylene protons ((1.5, 5.2) and (3.7, 5.2) ppm) and even the weak autocorrelation of the OH group of protonated alcohols (OH2+) ((∼8, ∼16) ppm) chemisorbed on the BAS, can be observed in the 1H–1H DQ–SQ spectrum acquired at 253 K (Fig. 3b). The observation of autocorrelation for the C-1 methylene 1H's at 3.6 ppm and the resonance of the protonated hydroxyl group (OH2+) of the alcohol at ca. 7–8 ppm in the DQ–SQ spectrum of 1-pentanol adsorbed MFI-140 (Fig. 3b) confirms the efficiency of the DQ–SQ experiment in identifying nuclei in spatially proximity. Also, in the 1H–1H DQ–SQ spectrum of dehydrated and partially hydrated H-MFI-140 (Fig. S14, ESI†) all expected correlations are present, further confirming the correct operation and efficacy of the pulse sequence. FTIR, direct excitation 1H NMR, 1D 1H–13C CPMAS and 1H–13C HETCOR NMR spectroscopy all clearly identify hydrogen bonding via the alcohol group as the dominant mechanism for adsorption of 1-pentanol in MFI-140. As the ratio between adsorbed 1-pentanol molecules and BAS + defect sites in the sample is 13.5, these observations reemphasize the question: What is the alcohol proton H-bonding to? Adsorption at BAS and defect sites clearly cannot explain the results. Also, the formation of hydrogen bonded dimers, trimers and tetramers has been excluded. The final possibility explaining all results is H-bonding to the siloxane bridges. Si–O bonds are more polar than a C–O bond due to the larger electronegativity differences (ENAllred-Rochow: C 2.50, Si 1.74 and O 3.50).77 Hence, the O-atom of the siloxane bridge with the enhanced electron density is a potential H-bond acceptor depending on the Si–O–Si bond angle.77,78 Higher bond angles result in delocalization of the electron density via π-back donation from O to Si. Lowering bond angles from linearity to values near tetrahedral bond angles have been shown to enhance the basicity, rendering the O as potential H-bonding sites.78 DFT study on the potential of siloxane bridges in MFI zeolites to serve as acceptor for hydrogen bonding with hydroxyl protons has been recently reported.76 If hydrogen bonding to siloxane bridges is the dominant mechanism driving the adsorption of 1-pentanol on MFI, the 1H–29Si CP-HETCOR NMR spectrum should reveal correlations between all methylene protons of 1-pentanol and Q4(0) Si sites. The 1H–29Si CP-HETCOR NMR spectrum (Fig. 3d) for 1-pentanol adsorbed MFI-140 recorded 253 K indeed reveals the correlation of all the 1-pentanol protons, viz. alkyl protons and the OH protons, to the Q4(0) Si. Spatial proximity of the 1-pentanol OH protons to the Si Q4(0Al), as well as the absence of hydrogen bonded 1-pentanol dimers, trimers or tetramers has thus been demonstrated. Hence it can now be concluded that the dominant mechanism for adsorption of 1-pentanol in high silica MFI involves H-bonding of the OH moiety of the alcohol to the siloxane (Si–O–Si) bridges of the zeolite (purple highlight, Fig. 4). This adsorption mechanism, H-bonding to the siloxane bridges, could be the first step in the alkoxylation of silica surfaces, a process described in literature.71,79 A similar mechanism involving the interaction of H in alkane C–H bonds with O in siloxane bridges have been proposed to drive the selective adsorption of ethane over ethylene on defect-free, purely-siliceous zeolites.80 Also, unexpected isotope exchange of oxygen (17O) into zeolite frameworks via equilibration of the zeolite with 17O enriched water at room temperature81–83 could be explained by a similar hydrogen bonding mechanism. H-Bonding of the isotope-enriched water to the siloxane bridges could assist in the reversible opening and closure of the siloxane bridges, thus enabling the exchange of framework 16O with 17O from water.
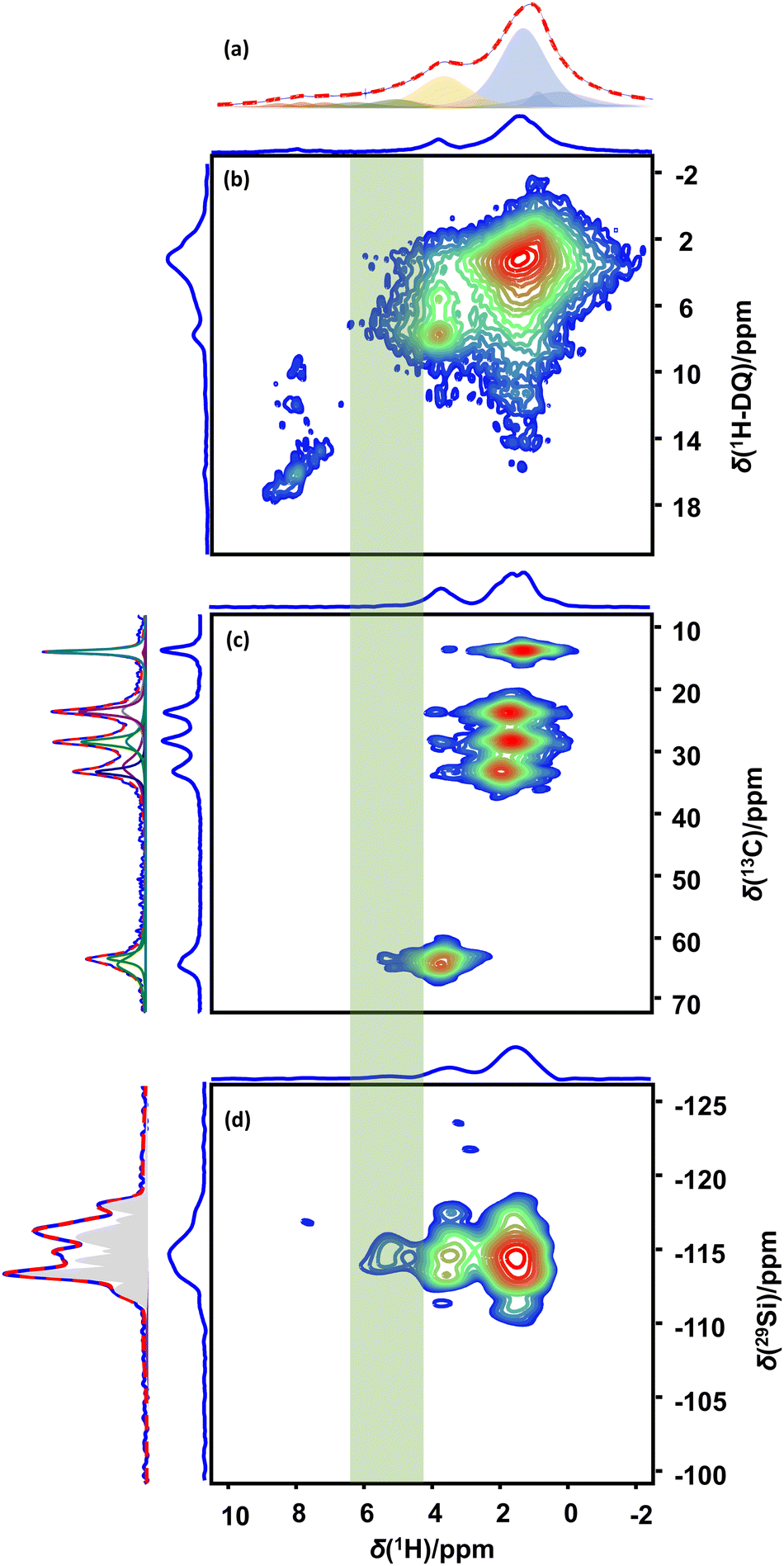 | ||
| Fig. 3 Direct excitation 1H MAS NMR (a), 1H–1H DQ–SQ (b), 1H–13C CP-HETCOR (c), 1H–29Si CP-HETCOR (d) spectra of 1-pentanol adsorbed H-MFI-140. All the spectra were acquired at 253 K. Spectral decomposition of 1H, 1H–13C CPMAS spectra and 29Si direct excitation spectra are presented along with the projections. | ||
 | ||
| Fig. 4 Summary of the different modes of alcohol adsorption in zeolites namely physisorption (blue highlight) and chemisorption (green highlight) around a BAS, H-bonding to siloxane bridges (purple highlight) and adsorption at defect sites (yellow highlight). HA, HB, and HD respectively represent the 1H corresponding to the alcohol, Brønsted acid and defect sites (SiOH here, but can also be AlOH). | ||
Conclusions
In situ NMR spectroscopy is an elegant method to quantify maximum adsorption capacities of zeolites exposed to aliquots of liquid alcohol. Sharp NMR resonances owing to mobile molecules in liquid state can be distinguished from superimposed broad signals of adsorbed molecules. Using liquid adsorbates, pore-filling adsorption indicating liquid like packing in channels was observed for linear alcohols with increasing alkyl chain length until 1-pentanol, leading to high alcohol per BAS ratios. The experimentally observed maximum adsorption capacities were in good agreement with theoretical models of the pore filling. The effect of enhanced hydrophobicity of alcohols by the increase in the alkyl chain length leading to lower adsorption capacities was observed for MFI zeolites with high aluminium content. Close to pore-filling, adsorption was observed for 1-pentanol in an MFI zeolite with Si/Al ratio 140. 1H–13C CPMAS and 1H–29Si CP-HETCOR spectra revealed the existence of a new adsorption mechanism where 1-pentanol molecules undergo hydrogen bonding to siloxane bridges. This newly proposed mechanism can co-exist with the traditionally accepted alcohol adsorption mechanisms like Brønsted acid site adsorption and adsorption at framework defects. The observed increase in 1-pentanol capacity with increasing Si/Al ratio indicates 1-pentanol as a suitable probe molecule to experimentally evaluate hydrophobic properties of zeolites and other classes of adsorbents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. B. acknowledges joint funding by the Flemish Science Foundation (FWO; G083318N) and the Austrian Science Fund (FWF) (funder ID 10.13039/501100002428, project ZeoDirect I 3680-N34) and thanks J. Lercher and R. Zhao for the interesting discussions on alcohol adsorption in MFI zeolites. J. A. M acknowledges the Flemish government for long term structural funding (Methusalem) and the European Research Council (ERC) for an Advanced Research Grant under the European Union's Horizon 2020 research and innovation program under grant agreement No. 834134 (WATUSO). NMRCoRe acknowledges the Flemish government, department EWI for infrastructure investment via the Hermes Fund (AH. 2016.134) and for financial support as International Research Infrastructure (I001321N: Nuclear Magnetic Resonance Spectroscopy Platform for Molecular Water Research). The authors acknowledge Gina Vanbutsele, Loes Verheyden and Wauter Wangermez for assistance with material characterization.References
- E. M. Flanigen, Stud. Surf. Sci. Catal., 2001, 137, 11–35 CrossRef CAS.
- Y. Li and J. Yu, Nat. Rev. Mater., 2021, 6, 1156–1174 CrossRef CAS.
- L. Van Tendeloo, W. Wangermez, M. Kurttepeli, B. De Blochouse, S. Bals, G. Van Tendeloo, J. A. Martens, A. Maes, C. E. A. Kirschhock and E. Breynaert, Environ. Sci. Technol., 2015, 49, 2358–2365 CrossRef CAS PubMed.
- L. Van Tendeloo, B. De Blochouse, D. Dom, J. Vancluysen, R. Snellings, J. A. Martens, C. E. A. Kirschhock, A. Maes and E. Breynaert, Environ. Sci. Technol., 2015, 49, 1729–1737 CrossRef CAS PubMed.
- L. Van Tendeloo, W. Wangermez, A. Vandekerkhove, T. Willhammar, S. Bals, A. Maes, J. A. Martens, C. E. A. Kirschhock and E. Breynaert, Chem. Mater., 2017, 29, 629–638 CrossRef CAS.
- L. Zhu, D. Shen and K. H. Luo, J. Hazard. Mater., 2020, 389, 122102 CrossRef CAS PubMed.
- S. K. P. Veerapandian, N. De Geyter, J.-M. Giraudon, J.-F. Lamonier and R. Morent, Catalysts, 2019, 9, 98 CrossRef.
- M. V. Chandak and Y. S. Lin, Environ. Technol., 1998, 19, 941–948 CrossRef CAS.
- S. Radhakrishnan, P.-J. Goossens, P. C. M. M. Magusin, S. P. Sree, C. Detavernier, E. Breynaert, C. Martineau, F. Taulelle and J. A. Martens, J. Am. Chem. Soc., 2016, 138, 2802–2808 CrossRef CAS PubMed.
- A. V. Kubarev, E. Breynaert, J. Van Loon, A. Layek, G. Fleury, S. Radhakrishnan, J. Martens and M. B. J. Roeffaers, ACS Catal., 2017, 7, 4248–4252 CrossRef CAS PubMed.
- J. F. M. Denayer, R. A. Ocakoglu, W. Huybrechts, B. Dejonckheere, P. Jacobs, S. Calero, R. Krishna, B. Smit, G. V. Baron and J. A. Martens, J. Catal., 2003, 220, 66–73 CrossRef CAS.
- A. Bušić, N. Marđetko, S. Kundas, G. Morzak, H. Belskaya, M. I. Šantek, D. Komes, S. Novak and B. Šantek, Food Technol. Biotechnol., 2018, 56, 289–311 Search PubMed.
- C. Fu, Z. Li, C. Jia, W. Zhang, Y. Zhang, C. Yi and S. Xie, Energy Convers. Manag. X, 2021, 10, 100059 CAS.
- D. Shah, D. Bhattacharyya, A. Ghorpade and W. Mangum, Environ. Prog., 1999, 18, 21–29 CrossRef CAS.
- Y. R. Shah and D. J. Sen, Int. J. Curr. Sci. Res., 2011, 1, 57–62 Search PubMed.
- O. Becerra-Pérez, S. Georgopoulos, M. Lanara, H. E. Reynel-Ávila, M. Papadaki, A. Bonilla-Petriciolet and D. I. Mendoza-Castillo, Adsorpt. Sci. Technol., 2021, 6615766 Search PubMed.
- D. Sharma and A. Saini, Lignocellulosic Ethanol Production from a Biorefinery Perspective, Springer, 2020 Search PubMed.
- A. Singh and G. P. Rangaiah, Ind. Eng. Chem. Res., 2017, 56, 5147–5163 CrossRef CAS.
- G. D. Mehta, J. Memb. Sci., 1982, 12, 1–26 CrossRef CAS.
- J. Lefevere, S. Mullens, V. Meynen and J. Van Noyen, Chem. Pap., 2014, 68, 1143–1153 CAS.
- E. Kianfar, S. Hajimirzaee, S. Mousavian and A. S. Mehr, Microchem. J., 2020, 156, 104822 CrossRef CAS.
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505–9514 CrossRef CAS.
- M. Hunger, Catal. Rev.: Sci. Eng., 1997, 39, 345–393 CrossRef CAS.
- M. Hunger and T. Horvath, J. Am. Chem. Soc., 1996, 118, 12302–12308 CrossRef CAS.
- C. M. Nguyen, M.-F. Reyniers and G. B. Marin, Phys. Chem. Chem. Phys., 2010, 12, 9481 RSC.
- K. Alexopoulos, M. S. Lee, Y. Liu, Y. Zhi, Y. Liu, M. F. Reyniers, G. B. Marin, V. A. Glezakou, R. Rousseau and J. A. Lercher, J. Phys. Chem. C, 2016, 120, 7172–7182 CrossRef CAS.
- A. V. Kubarev, E. Breynaert, J. Van Loon, A. Layek, G. Fleury, S. Radhakrishnan, J. Martens and M. B. J. Roeffaers, ACS Catal., 2017, 7, 4248–4252 CrossRef CAS PubMed.
- G. Mirth, J. A. Lercher, M. W. Anderson and J. Klinowski, J. Chem. Soc., Faraday Trans., 1990, 86, 3039–3044 RSC.
- T. Humplik, R. Raj, S. C. Maroo, T. Laoui and E. N. Wang, Langmuir, 2014, 30, 6446–6453 CrossRef CAS PubMed.
- R. Krishna, Phys. Chem. Chem. Phys., 2015, 17, 39–59 RSC.
- M. T. Aronson, R. J. Gorte and W. E. Farneth, J. Catal., 1986, 98, 434–443 CrossRef CAS.
- R. J. Costa, E. A. S. Castro, J. R. S. Politi, R. Gargano and J. B. L. Martins, J. Mol. Model., 2019, 25, 34 CrossRef PubMed.
- J. F. Denayer, A. Bouyermaouen and G. V. Baron, Ind. Eng. Chem. Res., 1998, 37, 3691–3698 CrossRef CAS.
- J. F. M. Denayer, I. Daems and G. V. Baron, Oil Gas Sci. Technol., 2006, 61, 561–569 CrossRef CAS.
- J. F. M. Denayer, S. Van Der Beken, K. M. A. De Meyer, J. A. Martens and G. V. Baron, Stud. Surf. Sci. Catal., 2004, 154 B, 1944–1949 CrossRef.
- I. Daems, P. Leflaive, A. Méthivier, J. F. M. Denayer and G. V. Baron, Stud. Surf. Sci. Catal., 2005, 158 B, 1177–1184 CrossRef.
- J. R. Di Iorio, B. A. Johnson and Y. Román-Leshkov, J. Am. Chem. Soc., 2020, 142, 19379–19392 CrossRef CAS PubMed.
- J. J. Gutiérrez-Sevillano, S. Calero and R. Krishna, J. Phys. Chem. C, 2015, 119, 3658–3666 CrossRef.
- T. Remy, J. Cousin Saint Remi, R. Singh, P. A. Webley, G. V. Baron and J. F. M. Denayer, J. Phys. Chem. C, 2011, 115, 8117–8125 CrossRef CAS.
- N. B. Milestone and D. M. Bibby, J. Chem. Technol. Biotechnol. Chem. Technol., 1984, 34 A, 73–79 Search PubMed.
- W.-D. Einicke, U. Messow and R. Schöllner, J. Colloid Interface Sci., 1988, 122, 280–282 CrossRef CAS.
- R. Krishna and J. M. Van Baten, ACS Omega, 2020, 5, 28393–28402 CrossRef CAS PubMed.
- B. C. Bukowski, J. S. Bates, R. Gounder and J. Greeley, Angew. Chem., Int. Ed., 2019, 58, 16422–16426 CrossRef CAS PubMed.
- S. Calero and P. Gómez-Álvarez, RSC Adv., 2014, 4, 29571–29580 RSC.
- R. Gounder and M. E. Davis, AIChE J., 2013, 59, 3349–3358 CrossRef CAS.
- P. Gómez-Álvarez, E. G. Noya, E. Lomba, S. Valencia and J. Pires, Langmuir, 2018, 34, 12739–12750 CrossRef PubMed.
- E. Breynaert, M. Houlleberghs, S. Radhakrishnan, G. Grübel, F. Taulelle and J. A. Martens, Chem. Soc. Rev., 2020, 49, 2557–2569 RSC.
- T. Karbowiak, M. A. Saada, S. Rigolet, A. Ballandras, G. Weber, I. Bezverkhyy, M. Soulard, J. Patarin and J. P. Bellat, Phys. Chem. Chem. Phys., 2010, 12, 11454–11466 RSC.
- I. Khay, T. J. Daou, H. Nouali, A. Ryzhikov, S. Rigolet and J. Patarin, J. Phys. Chem. C, 2014, 118, 3935–3941 CrossRef CAS.
- J. M. Castillo, D. Dubbeldam, T. J. H. Vlugt, B. Smit and S. Calero, Mol. Simul., 2009, 35, 1067–1076 CrossRef CAS.
- R. M. Madero-Castro, S. Calero and A. O. Yazaydin, J. Mol. Liq., 2021, 340, 117297 CrossRef CAS.
- J. Kuhn, J. M. Castillo-Sanchez, J. Gascon, S. Calero, D. Dubbeldam, T. J. H. Vlugt, F. Kapteijn and J. Gross, J. Phys. Chem. C, 2010, 114, 6877–6878 CrossRef CAS.
- K. M. A. De Meyer, S. Chempath, J. F. M. Denayer, J. A. Martens, R. Q. Snurr and G. V. Baron, J. Phys. Chem. B, 2003, 107, 10760–10766 CrossRef CAS.
- M. Houlleberghs, A. Hoffmann, D. Dom, C. E. A. Kirschhock, F. Taulelle, J. A. Martens and E. Breynaert, Anal. Chem., 2017, 89, 6940–6943 CrossRef CAS PubMed.
- H. Vanderschaeghe, M. Houlleberghs, L. Verheyden, D. Dom, C. V. Chandran, S. Radhakrishnan, J. A. Martens and E. Breynaert, Anal. Chem., 2022, 95, 1880–1887 CrossRef PubMed.
- S. Radhakrishnan, H. Colaux, C. V. Chandran, D. Dom, L. Verheyden, F. Taulelle, J. Martens and E. Breynaert, Anal. Chem., 2020, 92, 13004–13009 CrossRef CAS PubMed.
- MFI: XPD Plot (verified syntheses), https://europe.iza-structure.org/IZA-SC/pow_plot_VerifSyn.php?STC=MFI&patternFN=MFI_KPL_Sydch.csv, (accessed 7 June 2023).
- M. H. Levitt, Spin Dynamics: Basics of Nuclear Magnetic Resonance, John Wiley & Sons, 2013, p. 744 Search PubMed.
- J. F. M. Denayer, K. De Meyer, J. A. Martens and G. V. Baron, Angew. Chem., Int. Ed., 2003, 42, 2774–2777 CrossRef CAS PubMed.
- Z. Song, Y. Huang, W. L. Xu, L. Wang, Y. Bao, S. Li and M. Yu, Sci. Rep., 2015, 5, 13981 CrossRef PubMed.
- E. García-Pérez, S. K. Schnell, J. M. Castillo, S. Calero, S. Kjelstrup, D. Dubbeldam and T. J. H. Vlugt, J. Phys. Chem. C, 2011, 115, 15355–15360 CrossRef.
- B. Claessens, J. Cousin-Saint-Remi and J. F. M. Denayer, New Developments in Adsorption/Separation of Small Molecules by Zeolites, 2020, vol. 184, pp. 85–119 Search PubMed.
- S. Chempath, J. F. M. Denayer, K. M. A. De Meyer, G. V. Baron and R. Q. Snurr, Langmuir, 2004, 20, 150–156 CrossRef CAS PubMed.
- H. Liu, Z. Zhang, B. H. Chen and Y. Zhao, J. Porous Mater., 2008, 15, 119–125 CrossRef CAS.
- Z. Li, C. Rieg, A.-K. Beurer, M. Benz, J. Bender, C. Schneck, Y. Traa, M. Dyballa and M. Hunger, Adsorption, 2021, 27, 49–68 CrossRef CAS.
- X. Yi, K. Liu, W. Chen, J. Li, S. Xu, C. Li, Y. Xiao, H. Liu, X. Guo, S. Bin Liu and A. Zheng, J. Am. Chem. Soc., 2018, 140, 10764–10774 CrossRef CAS PubMed.
- O. Lisboa, M. Sánchez and F. Ruette, J. Mol. Catal. A: Chem., 2008, 294, 93–101 CrossRef CAS.
- M. W. Anderson, P. J. Barrie and J. Klinowski, J. Phys. Chem., 1991, 95, 235–239 CrossRef CAS.
- F. Haase and J. Sauer, J. Am. Chem. Soc., 1995, 117, 3780–3789 CrossRef CAS.
- R. A. Ocakoglu, J. F. M. Denayer, G. B. Marin, J. A. Martens and G. V. Baron, J. Phys. Chem. B, 2003, 107, 398–406 CrossRef CAS.
- T. Luo, R. Zhang, W. Zeng, C. Zhou, X. Yang and Z. Ren, J. Phys. Chem. C, 2021, 125, 8638–8646 CrossRef CAS.
- R. Schenkel, A. Jentys, S. F. Parker and J. A. Lercher, J. Phys. Chem. B, 2004, 108, 15013–15026 CrossRef CAS.
- S. L. Wallen, B. J. Palmer, B. C. Garrett and C. R. Yonker, J. Phys. Chem., 1996, 100, 20173 CrossRef.
- A. V. Stuart and G. B. B. M. Sutherland, J. Chem. Phys., 2004, 24, 559 CrossRef.
- S. Eckstein, P. H. Hintermeier, R. Zhao, E. Baráth, H. Shi, Y. Liu and J. A. Lercher, Angew. Chem., Int. Ed., 2019, 58, 3450–3455 CrossRef CAS PubMed.
- H. Windeck, F. Berger and J. Sauer, Angew. Chem., Int. Ed., 2023, 62, e202303204 CrossRef CAS PubMed.
- F. Dankert and C. von Hänisch, Eur. J. Inorg. Chem., 2021, 2907–2927 CrossRef CAS.
- S. Grabowsky, M. F. Hesse, C. Paulmann, P. Luger and J. Beckmann, Inorg. Chem., 2009, 48, 4384–4393 CrossRef CAS PubMed.
- S. Björklund and V. Kocherbitov, Sci. Rep., 2017, 7, 1–11 CrossRef PubMed.
- J. Park, K. H. Cho, J. Kim, R. Ryoo, J. Park, Y. Lee and M. Choi, Chem. Mater., 2023, 35(5), 2078–2087 CrossRef CAS.
- S. M. Pugh, P. A. Wright, D. J. Law, N. Thompson and S. E. Ashbrook, J. Am. Chem. Soc., 2020, 142, 900–906 CrossRef CAS PubMed.
- C. J. Heard, L. Grajciar, C. M. Rice, S. M. Pugh, P. Nachtigall, S. E. Ashbrook and R. E. Morris, Nat. Commun., 2019, 10, 4690 CrossRef PubMed.
- S. E. Ashbrook, Z. H. Davis, C. M. Rice and R. E. Morris, Chem. Sci., 2021, 12, 5016–5036 RSC.
Footnote |
| † Electronic Supplementary Information (ESI) available: Experimental section; details of spectral decomposition of 1H and 27Al NMR of different MFI zeolites; estimation of different 1H speciation; 1H-13C HMQC spectrum of liquid pentanol; 1H-13C CP-HETCOR NMR spectrum of 1-pentanol adsorbed on MFI-11.5; determination of theoretical maximum adsorption capacity; FT-IR investigations; Spectral decomposition of 1H NMR spectra under variable temperature conditions. DOI: https://doi.org/10.1039/d3mh00888f |
| This journal is © The Royal Society of Chemistry 2023 |