
Impact of the metal centre (Al3+, Fe3+) on the post-synthetic lithiation of functionalized MIL-53s and the electrochemical properties of lithiated derivatives†
Morgane
Denis
a,
Hubert
Chevreau
b,
Pablo
Salcedo-Abraira
 a,
Philippe
Moreau
a,
Nicolas
Dupré
a,
Michael
Paris
a,
Philippe
Poizot
a and
Thomas
Devic
*a
a,
Philippe
Moreau
a,
Nicolas
Dupré
a,
Michael
Paris
a,
Philippe
Poizot
a and
Thomas
Devic
*a
aNantes Université, CNRS, Institut des Matériaux de Nantes Jean Rouxel, IMN, F-44000 Nantes, France. E-mail: thomas.devic@cnrs-imn.fr
bSynchrotron SOLEIL, L'Orme des Merisiers, BP 48, F-91192 Saint-Aubin, France
First published on 9th March 2023
Abstract
Metal–organic frameworks (MOFs) combining both organic and inorganic redox-active moieties have recently drawn interest in the field of electrochemical energy storage. Here we focused our attention on MIL-53(M) (M = Al, Fe) analogues based on 2,5-dioxo-1,4-benzenedicarboxylate, as this ligand was already found to present an interesting electrochemical activity based on the quinone/phenolate redox couple in the solid state. We described here our attempts to chemically lithiate the title solids. Various synthetic paths were explored, and the resulting solids were characterized by a broad set of techniques, including X-ray diffraction, MAS NMR spectroscopy, transmission electron microscopy, inductively coupled plasma-atomic emission spectroscopy and total X-ray scattering experiments, among others. We showed that although the lithiation was accompanied by a loss of the long-range order whatever the synthetic conditions and the trivalent cation, the reactivity strongly differed for M = Al and Fe. Eventually, the electrochemical extraction/uptake of Li+ in the lithiated derivatives was evaluated in Li-half cells. Although their storage capacities are moderate, we found that the presence of even a minor amount of M3+ cations not only impacts the working potential of the ligand but also improves their long term capacity retention.
Design, System, ApplicationOne of the key features of MOFs, together with their porosity, is the possibility to combine within a single material both organic and inorganic functional motifs, which could give rise to enhanced properties. In the present work, we are especially interested in the preparation of MOFs combining redox-active organic and inorganic moieties. Such solids could ultimately present multiple redox processes and hence lead to high energy storage capacity when used as electrode materials in Li-ion batteries. We especially aim to decipher the relationships between the nature of the building units (both organic and inorganic), their structural arrangements, and their reactivity towards chemical lithiation, as well as their solid-state electrochemical behavior. |
Introduction
Apart from traditional applications related to sorption (storage, capture, separation, controlled release, sensing, etc.…), metal–organic frameworks (MOFs) have been proposed as potential electrode materials for electrochemical energy storage.1–7 Although unable (yet?) to compete with conventional inorganic and organic electrode materials, MOFs offer interesting features, such as (i) the possibility to combine multiple redox centres (this is nevertheless sometimes achievable in inorganics through anionic redox) and (ii) their microporosity, which might favour ionic transport. When used as negative electrode materials, their reduction at low potential is accompanied by an irreversible conversion into polyphasic systems. In contrast, when used as positive electrode materials,4 a few solids can be reversibly oxidized/reduced through a conventional insertion mechanism. MIL-53(Fe) (MIL stands for Materials Institute Lavoisier), with the chemical formula FeIII(OH)(BDC) (BDC = 1,4-benzenedicarboxylate), was the first MOF studied for this purpose. This solid delivers ∼70 mA h g−1 at ∼3.0 V vs. Li+/Li, this activity being related to the reversible reduction of ca. half of FeIII to FeII.8 2,5-Dioxo-1,4-benzenedicarboxylate (known as DOBDC and p-DHT in the fields of MOFs and electrochemical energy storage, respectively) is a terephthalate derivative known to be redox active, not only in solution,9 but also in the solid state,10,11 this activity being associated with the quinone/phenolate redox couple. Noteworthily, some of us showed that, when employed as a positive electrode in an electrochemical half-cell, the redox potential could be tuned by playing with the surrounding cations, increasing from 2.55 to almost 3.5 V vs. Li+/Li when moving from Li4(DOBDC) to MgLi2(DOBDC), while maintaining a decent capacity (close to 100 mA h g−1).12With this in mind, we were interested in combining the redox activity of this organic ligand with that of a transition metal (TM) cation. MOFs containing both DOBDC and a TM are known, and they can be divided in two subclasses: (i) those containing fully deprotonated linkers (charge −4) bound to the TM through both the carboxylate and phenolate moieties,13–18 the most famous member being CPO-27/MOF-74, with the chemical formula MII2(DOBDC), and (ii) those containing ligands with a proton remaining on the phenolic oxygen (charge −2) and the TM interacting solely with the carboxylate groups,19–22 such as functionalized analogues of MIL-53(Fe), MIL-88(Fe) and UiO-66(Zr). The first ones are a priori not suitable materials for electrochemical energy storage, as their redox activity would require the displacement of multivalent cations (except if the initial solids also contain charge compensating monocations, or if the redox-activity is accompanied by anion rather than cation insertion23). For the second series, access to the quinone/phenolate redox couple in standard battery electrolytes (anhydrous, aprotic) first requires chemical lithiation (e.g. through an acid–base reaction) of the phenolic groups. This approach was very recently successfully applied by the Vlad group to a Mn-DOBDC MOF, leading to a mixed electrochemical activity centered of around 3.2 V vs. Li+/Li but with a modest reversible capacity (∼60 mA h g−1).24 Considering the already reported electrochemical activity of MIL-53(Fe), we focused our attention on its relative based on DOBDC, namely MIL-53(Fe)-(OH)2 or Fe(OH)(H2DOBDC) (Fig. 1).20 For the sake of comparison, the analogue built from the redox inert Al3+, MIL-53(Al)-(OH)2,25 was also considered.
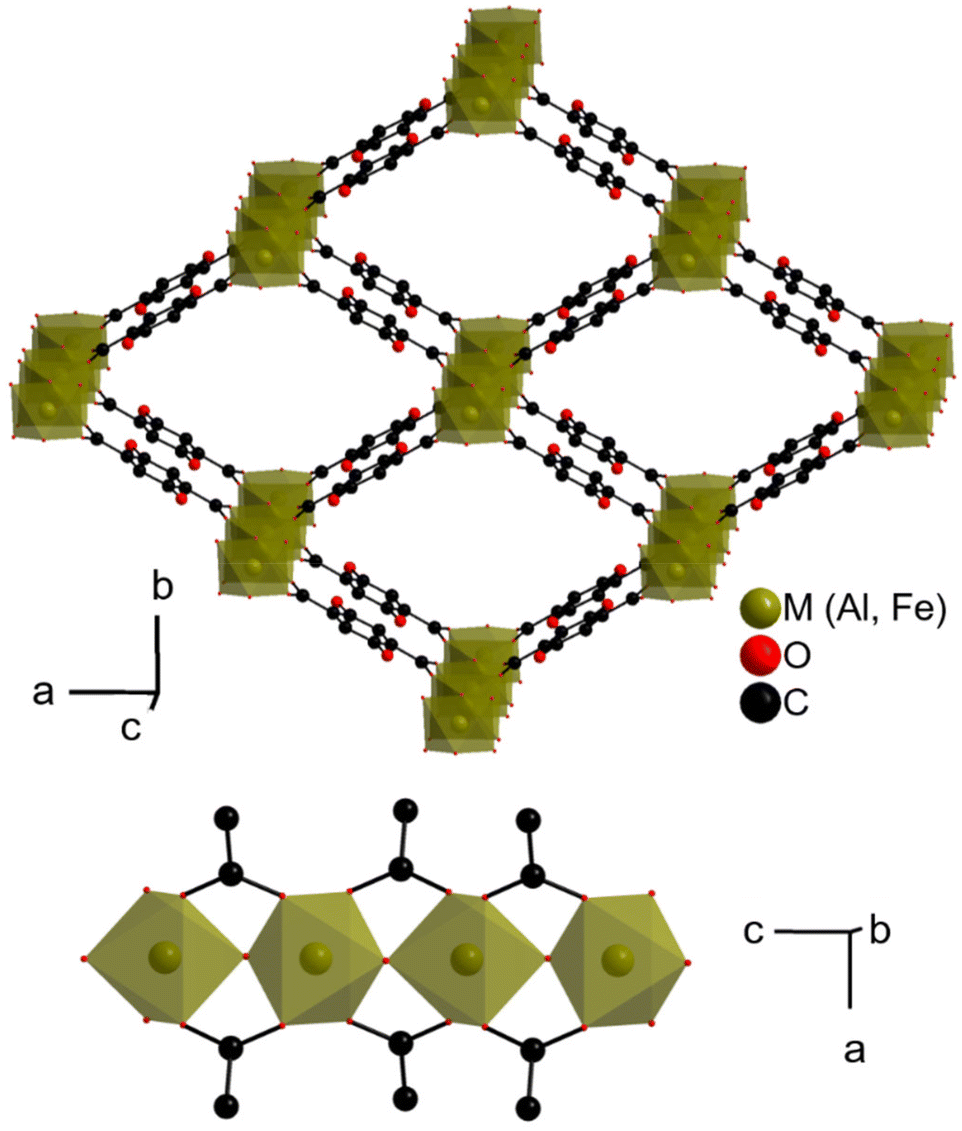 | ||
| Fig. 1 Crystal structure of MIL-53(M)-(OH)2 or M(OH)(H2DOBDC).20 Top: View along the pore axis; bottom: view of the inorganic chain. Protons (bound to C, organic O and inorganic O) are omitted, and only one of the two disordered positions of the phenolic groups is shown. | ||
We will describe here our attempts to produce fully lithiated DOBDC-based MIL-53s (Li3MO(DOBDC)) (M = Al, Fe) by post-synthetic basic treatments. Through a broad set of characterization tools, including powder X-ray diffraction (PXRD), infrared (IR) spectroscopy, 13C and 27Al solid magic angle spinning nuclear magnetic resonance (MAS NMR) spectroscopy, thermogravimetric analyses (TGA), inductively coupled plasma-atomic emission spectroscopy (ICP-AES), scanning transmission electron microscopy-energy dispersive X-ray analysis (STEM-EDX) and total X-ray scattering experiments, we will show that although lithiation is accompanied by the collapse of the structure whatever the trivalent cation, the composition of the final product of lithiation strongly differs for Al and Fe. Eventually, the electrochemical properties of the lithiated MOFs will also be presented and compared to those of the fully lithiated ligand Li4(DOBDC).
Experimental
Synthetic procedures
The syntheses of pristine MOFs were carried out in air, while lithiation experiments were conducted in a glove box filled with Ar.MIL-53(Al)-(OH)2 was prepared in N,N-diethylformamide (DEF) according to a published procedure.25 The solvent was removed from the pores in two steps (DEF–water exchange followed by thermal activation). First, the as-synthesized solid was dispersed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of water and methanol, heated at 150 °C in an autoclave for 12 hours, and recovered by filtration. This solid was further dried at 180 °C under vacuum for 16 hours, affording the activated form of MIL-53(Al)-(OH)2, as evidenced by PXRD (see Fig. S1–S4 and Table S1† for details).
:1 mixture of water and methanol, heated at 150 °C in an autoclave for 12 hours, and recovered by filtration. This solid was further dried at 180 °C under vacuum for 16 hours, affording the activated form of MIL-53(Al)-(OH)2, as evidenced by PXRD (see Fig. S1–S4 and Table S1† for details).
MIL-53(Fe)-(OH)2 was also prepared from a reported procedure, this time in N,N-dimethylformamide (DMF).20 The last step (washing with ethanol and drying in air) was sufficient to obtain the hydrated compound. This solid was dried at 150 °C under vacuum for 16 hours, which led to the partially activated form of MIL-53(Fe)-(OH)2.
:1 mixture of anhydrous tetrahydrofuran (THF) and methanol (MeOH) in a Schlenk line filled with Ar. 0.284 mL (0.63 mmol, 3 eq.) of a 2.2 M solution of LiOMe in MeOH was added. The mixture was heated at 50 °C under stirring for 16 hours. No drastic colour change was observed. The resulting solid was recovered by filtration, washed with anhydrous MeOH and THF, and dried at 180 °C under vacuum for 16 hours.
:1 mixture of anhydrous THF and methanol (MeOH) in a Schlenk line filled with Ar. 0.254 mL (0.56 mmol, 3 eq.) of a 2.2 M solution of LiOMe in MeOH was added. The mixture was heated at 50 °C under stirring for 16 hours. The resulting solid was recovered by filtration, washed with anhydrous MeOH and THF, and dried at 170 °C under vacuum for 16 hours.
Materials and methods
Characterization of the pristine MOFs was carried out under ambient conditions, whereas the dried and lithiated solids were analysed under an inert atmosphere or in sealed sample holders.Infrared spectra were collected using a Bruker Vertex 70 FTIR spectrometer in transmission mode between 400 and 4000 cm−1 on solids diluted in KBr pellets. PXRD patterns were collected either in Bragg–Brentano mode with a Bruker D8 ADVANCE diffractometer or in Debye–Scherrer mode with an INEL XRG3500 diffractometer, both equipped with a Cu anode (λ = 1.5406 Å). Le Bail refinements were carried out with the FullProf suite.26 TGA was performed under Ar at 5 °C min−1 up to 800 °C using either a Setaram SENSYS evo or a NETZSCH STA 449F3 Jupiter apparatus. For the ICP-AES experiments, solids were first dissolved in a 0.02 or 0.2 M aqueous solution of NaOH (Fe- and Al-based solids, respectively) and further analysed thanks to an iCAP 6300 radial analyser (Thermo Scientific). Scanning electron microscopy (SEM) was carried out using a JEOL JSM-7600F microscope. Samples were pasted on carbon tape and further coated with platinum to improve their surface electronic conductivity. STEM experiments were carried out using a Themis Z G3 Cs-probe corrected microscope from Thermo Fisher Scientific, operating at 80 kV and equipped with a high-angle annular dark field (HAADF) detector and a Super-X EDX detector. The solid was deposited onto a lacey carbon film supported by a copper grid. A vacuum transfer sample holder (GATAN 648) was used to prevent any air from coming into contact with the reactive sample before its introduction into the microscope. The 1H–13C cross-polarization and magic angle spinning (CP-MAS) and 27Al MAS NMR spectra were recorded on a 500 MHz Bruker Avance III spectrometer (B0 = 11.7 T) using a 2.5 mm probe head. For the 27Al spectra, the MAS rate, π/12 pulse length and recycle delay were set to 25 kHz, 3 μs and 1 s, respectively. For the 1H–13C spectra, the MAS rate and contact time were set to 11 kHz and 8 ms, respectively. 27Al and 13C chemical shifts were referenced to a 1 M Al(NO3)3 aqueous solution and to tetramethylsilane (TMS), respectively. All spectra were analysed using the DMFit software.27 X-ray total scattering experiments were carried out on the CRISTAL beamline at Synchrotron Soleil (L'Orme les Merisiers, France) at λ = 0.97015 Å, in the Debye–Scherrer configuration. The contribution of the glass sample holder (capillary) was subtracted, and the data were treated with the software PDFgetX3.28 Solid-state electrochemical experiments were carried out by using a two-electrode Swagelok®-type cell with a Li metal disc as the negative electrode and a glass fiber separator soaked with either 1 M LiClO4 in propylene carbonate (PC) or 1 M LiPF6 in 1:1 ethylene carbonate (EC) dimethylcarbonate (DMC) electrolyte. The composite positive electrodes were prepared in an argon filled glovebox by grinding the solid and a Ketjenblack® EC-600JD (Akzo Nobel) carbon conducting additive to insure proper electronic conduction (MOF:carbon ratio = 66:33 wt%, ca. 3 mg of active material per electrode). The electrochemical cells were then cycled in galvanostatic mode at a rate of 1 e−/1 Li+ exchanged per ligand in 15 hours within various potential windows starting with oxidation (charging step) by using a MPG-2 multi-channel system (Bio-Logic SAS, Seyssinet-Pariset, France).
Results and discussion
Two post-synthetic strategies were proposed in the literature to chemically insert Li+ into neutral MOFs. The first one relies on a redox reaction: a strongly reducing Li salt is added to the MOF, and the reduction of the ligand or the cation is associated with the insertion of Li+.29–32 The second one, which is of interest for us, is based on an acid–base reaction:33–36 a strong lithium base is added to the MOF, leading to the exchange of protons (organic or inorganic) with Li+. These approaches have been mainly devoted to the optimisation of the gas sorption properties (e.g., H2 storage29 or CO2 capture37) and, except in a few rare cases,32 only a minor amount of Li+ could be incorporated (typically <15 at%) without any significant loss of porosity or crystallinity. We thus here face four challenges:(i) the quantitative exchange of protons by Li+. As shown by Himsl et al. on MIL-53-(Al)-OH,34 lithiation occurs unselectively on organic and inorganic protons. Three Li+ per M3+ must then be inserted to insure that all phenolic groups are lithiated.
(ii) the base must be small enough to diffuse in the pores, strong enough to deprotonate, but not too nucleophilic to limit the degradation of the MOF.33,34 This could be an even stronger issue when quantitative exchange is targeted.
(iii) MIL-53 derivatives are flexible (the pore size and shape evolve with their content or with external stimuli).38 For example, when suspended in a liquid, MIL-53(Fe)-(OH)2 could present a close pore form (CP, no porosity), a narrow pore (NP) form (slight adsorption) or a large pore form (LP, high sorption capacity) depending on the nature and the physicochemical characteristics of the solvent.20 The solvent used for the acid–base-reaction must then obviously solubilize the base at least to some extent but must also insure the opening of the pores.
(iv) MIL-53(Fe)-(OH)2 is highly hydrophilic but also less thermally stable than other functionalized MIL-53s (decomposition starts below 200 °C, even under vacuum).20 Dehydration must thus be carried out with care, knowing that residual water in the presence of a base might lead to the complete degradation of the material.39
With this in mind, two pairs of base and solvent were selected: LiH in DMF40 and LiOMe in a mixture of THF and MeOH.10,24 The first experiments were carried out on MIL-53(Al)-(OH)2, which is easier to obtain as a fully dehydrated solid and could be studied by both 13C and 27Al MAS NMR spectroscopy. In the dehydrated state, the PXRD pattern and unit-cell parameters are indeed consistent with a CP form (Fig. 2a, S4 and Table S1,† respectively). On the IR spectrum (Fig. 2b, orange curve), signals associated with X–H (X = C, O) bonds are also well resolved, with inorganic μ2-OH, phenolic O–H and aromatic C–H vibration bands clearly distinguishable at 3690, 3352, and 3082 cm−1, respectively. Vibration bands characteristic of the ligand, notably COOasym, aromatic CC and COOsym, were also clearly identified at 1585, 1494 and 1471 cm−1, respectively. No residual DEF arising from the solvothermal synthesis is detected (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–H vibration bands at ∼1660 and ∼2900 cm−1, respectively). This solid was treated with 3 equivalents of LiH in DMF, which is known to lead to a large pore form.25 The reaction was performed at a high temperature (130 °C) to improve the otherwise low solubility of LiH in this solvent. Initially yellow, the suspension turned orange within a few hours (Fig. S8†), suggesting a change of the protonation state of the ligand and/or of its coordination mode.
O and C–H vibration bands at ∼1660 and ∼2900 cm−1, respectively). This solid was treated with 3 equivalents of LiH in DMF, which is known to lead to a large pore form.25 The reaction was performed at a high temperature (130 °C) to improve the otherwise low solubility of LiH in this solvent. Initially yellow, the suspension turned orange within a few hours (Fig. S8†), suggesting a change of the protonation state of the ligand and/or of its coordination mode.
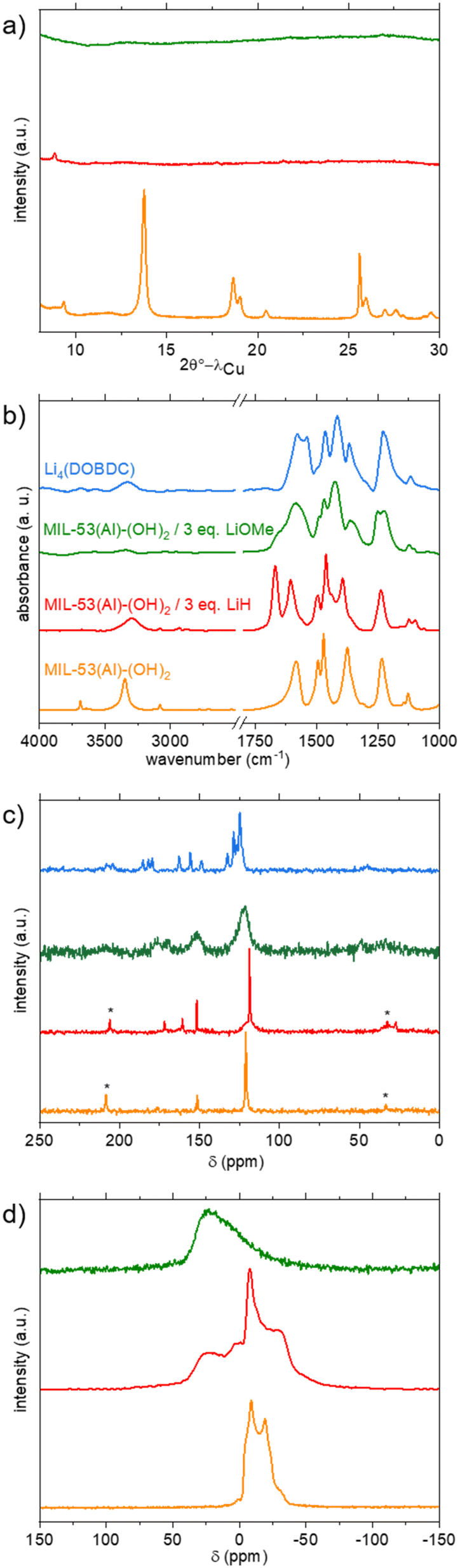 | ||
| Fig. 2 Characterization of MIL-53(Al)-(OH)2 and its lithiated products. a) PXRD patterns (λ = 1.5406 Å); b) IR spectra; c) 1H–13C CP-MAS NMR spectra (* = spinning side bands); d) 27Al MAS NMR spectra. Data related to dry MIL-53(Al)-(OH)2, Li4(DOBDC), and lithiation products from LiH–DMF and LiOMe–MeOH/THF are shown in orange, blue, red and green, respectively. | ||
The final product was thoroughly washed with THF and dried at 150 °C under vacuum prior to characterization. Note that attempts to replace THF by MeOH led to the recovery of the initial colour, suggesting that this change of coordination/protonation state is reversible. The orange product was first studied by PXRD; an almost complete loss of long-range order is observed (Fig. 2a, red curve). IR spectroscopy showed the complete disappearance of the signal belonging to inorganic μ2-OH, while the characteristics of organic OH groups remained, although with a lower intensity (Fig. 2b, red curve). Vibration bands characteristic of the ligand, notably COOasym (1602 cm−1), COOsym (1462 cm−1), and aromatic CC (1494 cm−1), were slightly shifted compared to the pristine solid, in accordance with a modification of the local environment of the ligand. The presence of residual DMF (CO and C–H vibration bands at 1666 and 2870–3940 cm−1, respectively) was also detected. Attempts to remove this solvent by extended washing with THF or dichloromethane or drying at higher temperature failed, suggesting that DMF is possibly bound, or at least strongly interacting, with the framework. This is in agreement with the thermogravimetric analysis performed under Ar, which does not exhibit a clear plateau corresponding to the desolvated solid prior to the thermal degradation (Fig. S10†). The relative amount of Li and Al was evaluated by ICP after dissolving the solid in a basic aqueous medium. A Li/Al ratio equal to 1.5 was obtained (expected value = 3), suggesting that only half of the protons were removed, in agreement with the IR analysis. On the solid-state 13C CP-MAS spectrum (Fig. 2c, red curve), residual DMF is detected (CH3 and CO at 27 and 161 ppm, respectively), again in line with the IR study. The signals characteristic of the ligand (COO, CO and Cq + CH at 172, 152 and 119 ppm, respectively) remained visible, although slightly broader and shifted compared to pristine MIL-53(Al)-(OH)2, in agreement with a change of the local surrounding of the ligand upon lithiation. This was confirmed by MAS 27Al NMR spectroscopy. The signal of the dried solid is characteristic of Al3+ in a single octahedral site.41 After lithiation, the spectrum exhibits at least two components, one similar to the pristine material although broadened, and the other one at a higher chemical shift and very broad, characteristic of disordered environments (Fig. 2d, red curve). To summarize, these synthetic conditions (LiH in DMF) lead only to a partial deprotonation, with a final composition close to Li1.5AlO(H2.5DOBDC)(DMF)x. Attempts to increase the amount of LiH in the reaction medium (up to 7 eq.) lead to similar IR spectra (Fig. S12†) suggesting that this procedure is not suitable for achieving complete lithiation of MIL-53(Al)-(OH)2.
For this reason, we switched to the second procedure (LiOMe in THF/MeOH), which was recently shown to be successful for the lithiation of a Mn-DOBDC MOF.24 The first experiments were again carried out with MIL-53(Al)-(OH)2, which adopts a large pore conformation when suspended in THF (see Fig. S5 and Table S1†). This solid was treated with 3 equivalents of LiOMe in a 1:1 mixture of THF and MeOH at 50 °C. The solid was ultimately recovered by filtration, washed and dried at 180 °C. In contrast to the previous case, no significant colour change was detected. Nevertheless, when exposed to air, the colour very rapidly turned from yellow to green (Fig. S9†). A similar behaviour was already observed for the parent Li4(DOBDC), and this was attributed to its oxidation by O2.10 This suggests that the lithiation was successful but that the oxidation potential of the final product is below that of the redox couple O2/Li2O2 (∼3.0 V vs. Li+/Li).
Again, PXRD analysis indicated a full loss of the long-range order upon lithiation (Fig. 2a, green curve). On the IR spectrum, no signal belonging to both inorganic and organic OH was detected (Fig. 2b, green curve), suggesting that complete lithiation occurred. This disappearance is accompanied by a shift of the bands characteristic of the core of the ligand (<1600 cm−1). Although not strictly identical, the spectrum showed some similarities with that of Li4(DOBDC) in this region, suggesting that both products are structurally related (Fig. 2b). No residual solvent (MeOH, THF) was detected this time, in line with the TG analysis, which showed that the solvent was removed below 130 °C, before the thermal degradation of the solid above 250 °C (Fig. S10†). The ICP analysis of the product dissolved in a basic aqueous medium indicates a Li/Al ratio close to 6–7 (Fig. S17†), suggesting that leaching of Al3+ occurred during the lithiation. This was confirmed by the analysis of the supernatant of the reaction, which was found to contain a significant amount of aluminium. On the 27Al NMR spectrum, only a broad signal, characteristic of very disordered local environments, is visible (Fig. 2d, green curve). The 1H–13C CP-MAS NMR spectrum also shows broad signals but with chemical shifts similar to those of Li4(DOBDC).
To summarize, all these characterization studies eventually indicated that the reaction of MIL-53(Al)-(OH)2 with LiOMe lead to a degradation of the MOF, accompanied by the partial release of Al3+, ultimately leading to a very disordered solid, which presents some structural similarities with Li4(DOBDC). Furthermore, the use of harsher reaction conditions such as a longer reaction time (15 days) or a higher amount of LiOMe (8 eq. instead of 3) leads eventually to the formation of Li4(DOBDC), as indicated by PXRD, ICP-AES and STEM-EDX analyses (Fig. S6, S17 and S15,† respectively). A question then arose whether the final product is simply a mixture of Li4(DOBDC) with another phase related to MIL-53(Al)-(OH)2, or a new compound. Additional characterization studies were carried out to try to answer this question. SEM imaging (Fig. 3a) first indicated that the lithiation was accompanied by a strong modification of the morphology of the particles. While the initial MIL-53(Al)-(OH)2 consists of rod-like crystals shorter than 1 μm, larger (up to a few μm) 2-D flake-like particles were finally produced, suggesting that the transformation occurred through a dissolution–reprecipitation mechanism with the formation of a layered structure. This change of morphology was confirmed by TEM analysis (Fig. 3b). As shown in Table 1, EDX analysis carried out on these fluffy particles suggested a rather homogeneous distribution of remaining Al in the solid (note however that a few particles of Li4(DOBDC) were also detected). Analysis of the relative amount of Al, C and O suggested a ligand-to-Al ratio close to 5/2. This result, combined with the ICP analysis, and taking into account the charge balance, gave rise to the tentative formula Li7Al(DOBDC)5/2.
 | ||
| Fig. 3 Characterization of the reaction product between MIL-53(Al)-(OH)2 and 3 eq. of LiOMe in MeOH/THF. a) SEM pictures of the pristine solid (left) and the lithiated solid (right); b) STEM pictures of the lithiated product. Areas used for the EDX analyses provided in Table 1 are also shown. | ||
| Area #1 | Area #2 | Area #3 | Average | Li7Al(DOBDC)5/2 | |
|---|---|---|---|---|---|
| C/Al | 29 | 21 | 20 | 23 | 20 |
| C/O | 1.4 | 1.3 | 1.5 | 1.4 | 1.3 |
| O/Al | 22 | 16 | 12.5 | 17 | 15 |
The solid was eventually studied by X-ray total scattering experiments at the CRISTAL beamline (Synchrotron Soleil, France). MIL-53(Al)-(OH)2, the lithiated product and Li4(DOBDC), all in a dried form, were compared. The reduced pair distribution function (PDF) G(r) and the radial distribution function (RDF) R(r) are shown in Fig. 4. For both G(r) and R(r), peaks represent distances between atom pairs. G(r) is reduced so that it goes down to 0 when r increases; it is particularly suitable to evaluate the order from low-range to medium-range. When comparing G(r) functions of the lithiated solid with MIL-53(Al)-(OH)2 (Fig. 4a), the almost complete disappearance of well-defined peaks above 8 Å indicates a lack of order for distances larger than the length of ligand–cation complexes. This is of course in line with the complete amorphization of the solid upon lithiation. R(r) takes into account the total number of neighbours, and hence it increases in r2. It is especially adapted to obtain quantitative information at short distances (number of atom pairs). R(r) functions were normalized on the peak at 1.4 Å, which corresponds to CC and CO intra-ligand bonds (Fig. 4b). The intensity of the peak at 1.9 Å, which is associated with both LiO and AlO bonds, then depends on the atomic number Z of the cations bound to the oxygen atoms (I α Z2) and to their number N (I α N). The intensity of this peak decreases in the following order: MIL-53(Al)-(OH)2 > lithiated product > Li4(DOBDC) bonds (Fig. 4b, inset). This indicates that less Al–O bonds are present after lithiation (MIL-53(Al)-(OH)2 > lithiated product), but that a few of these bonds remain (lithiated product > Li4(DOBDC)). The fact that the signals of the lithiated product and Li4(DOBDC) in the range 3–6 Å are not identical is another clear indication that they are truly different compounds. To summarize, the lithiation route based on LiOMe in MeOH/THF leads to the degradation of the pristine MOF and to the formation of a Li-rich layered phase similar to Li4(DOBDC), which still contains some aluminium cations. Note that attempts to prepare this solid directly from either AlCl3, LiOMe and H4DOBDC, or AlCl3 and Li4(DOBDC) failed (see Fig. S13† for the IR spectra), suggesting that the use of the MOF precursor drives, at least to some extent, the formation of Li7Al(DOBDC)5/2.
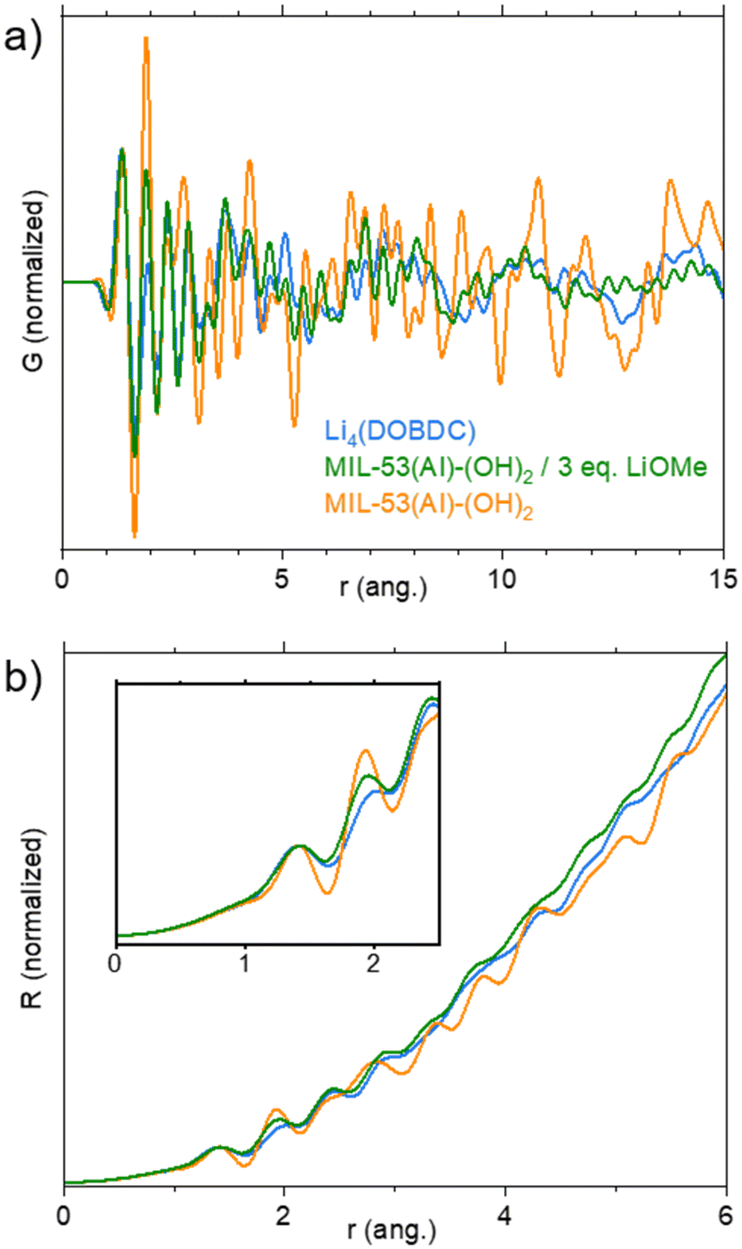 | ||
| Fig. 4 Characterisation of the reaction product between MIL-53(Al)-(OH)2 and 3 eq. of LiOMe in MeOH/THF by X-ray total scattering experiments. a) Reduced pair distribution function G(r); b) radial distribution function R(r) (inset: zoom on the 0–2.5 ang. region). Data related to dry MIL-53(Al)-(OH)2, Li4(DOBDC), and the lithiation product from LiOMe–MeOH/THF, are shown in orange, blue and green, respectively. | ||
The same lithiation strategy was eventually applied to MIL-53(Fe)-(OH)2. As mentioned earlier, this solid presents low thermal stability while being highly hydrophilic. The thermogravimetric data (Fig. S11†) indeed show that the complete water departure (up to ∼155 °C) is almost concomitant with the beginning of the degradation of the network (∼170 °C). Activation under vacuum at 155 °C affords a mixture of dehydrated (CP) and hydrated (NP) forms, as indicated by PXRD (Fig. S7†), but higher temperatures lead to the collapse of the framework. Therefore, the lithiation was carried out on the partially dehydrated solid, using experimental conditions identical to those used for the Al analogue (3 eq. of LiOMe in MeOH/THF at 50 °C for 16 hours and further drying).
Again, the lithiation reaction is accompanied by a strong change of the PXRD pattern (Fig. 5a, pink curve). Two broad peaks at 10.8 and 12.8° are nevertheless still discernible, suggesting that the Fe lithiated product presents some long-range order, in contrast to the Al analogue. On the IR spectrum (Fig. 5b, pink curve), the bands characteristic of the inorganic (3634 cm−1) and organic (3210 cm−1, likely overlapping with the band characteristic of the residual water molecules) OH groups disappear after lithiation, suggesting that the reaction is complete. The vibration bands characteristic of the ligand, notably COOasym, aromatic CC and COOsym at 1618, 1574 and 1461 cm−1, respectively, are also shifted, and their positions do not match with those found for the salt Li4(DOBDC). Again, no band characteristic of residual solvent molecules was found, in line with the TG curve, which exhibits a clear plateau between 130 and 240 °C (Fig. S10†).
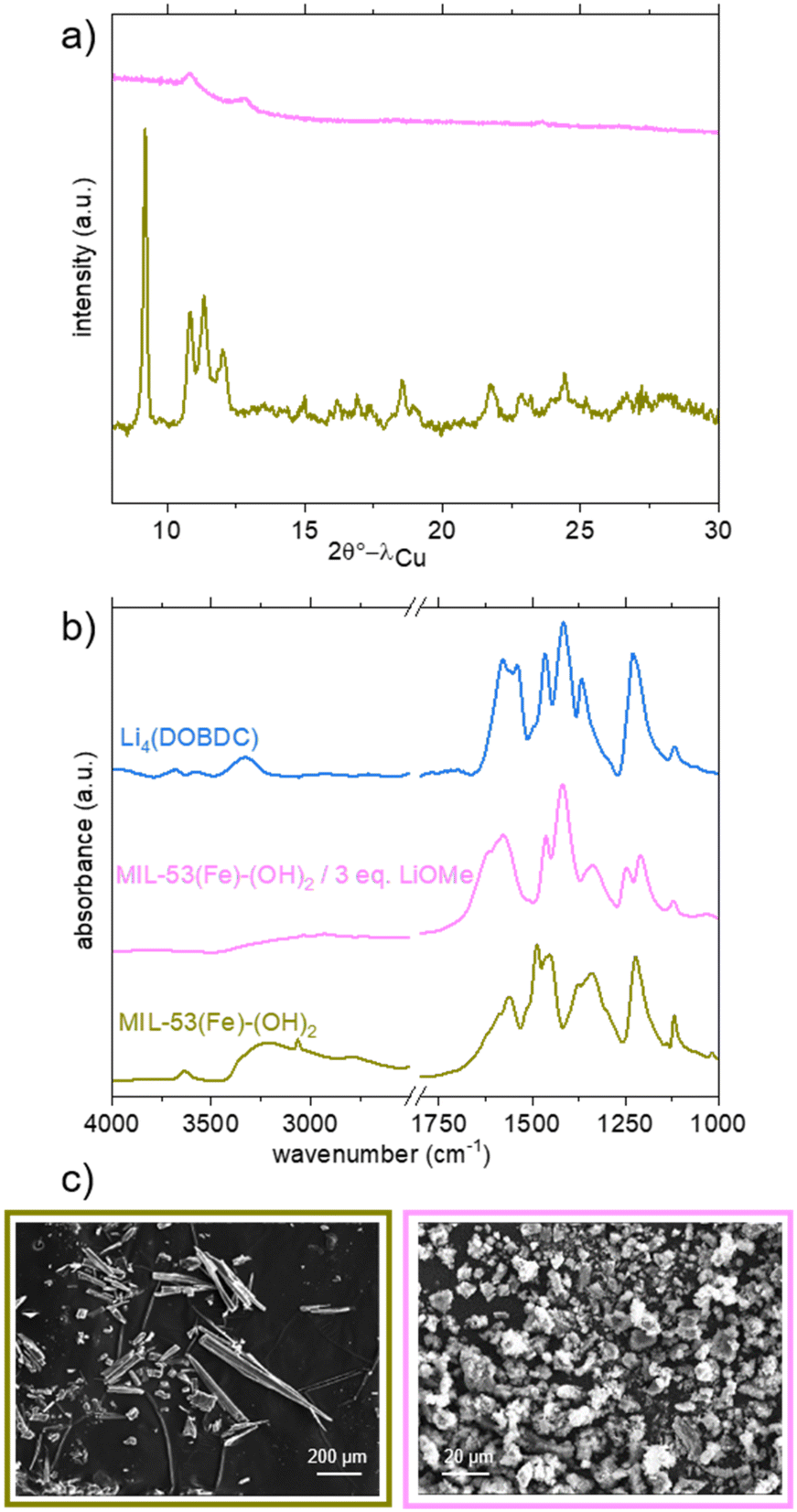 | ||
| Fig. 5 Characterisation of MIL-53(Fe)-(OH)2 and its lithiated product from LiOMe in MeOH/THF. a) PXRD patterns (λ = 1.5406 Å); b) IR spectra; c) SEM pictures. Data related to dry MIL-53(Fe)-(OH)2, Li4(DOBDC) and the lithiation product are shown in khaki, blue and pink, respectively. | ||
Eventually, the solid was also analysed by ICP analysis after dissolution in a basic aqueous medium (Fig. S18†). A Li/Fe ratio equal to 3 was measured. This result strongly differs from the one observed for the Al analogue and suggests that no Fe was released in the reaction medium, as ultimately confirmed by the analysis of the supernatant. This suggests that the lithiation of MIL-53(Fe)-(OH)2 was successful, and it occurred through a pathway different from that observed for MIL-53(Al)-(OH)2. As shown in Fig. 5c, SEM analysis showed that the initial solid is composed of long (tens to hundreds of μm) needle-like crystals. After lithiation, smaller particles (<10 μm) are observed, which could result from the breakage of crystals. In contrast to the case of the Al analogue, no evidence of dissolution–reprecipitation steps was detected, in line with the aforementioned analyses.
The solid-state electrochemical properties of the lithiated products were then investigated and compared with those of the pristine MOFs. Composite electrodes prepared by grinding the solids with conducting carbon (weight ratio ∼2:1) with a pestle and mortar were electrochemically tested in Li half-cells under galvanostatic cycling conditions at one electron exchanged per ligand in 15 hours, the electrolyte consisting first in 1 M LiClO4 in propylene carbonate. Considering the previous reports on DOBDC derived materials and MIL-53(Fe), experiments were carried out starting with oxidation, within the potential window 2.2 ≤ E ≤ 3.8 V vs. Li+/Li.
The first and fifth cycles are shown Fig. 6. For pristine MIL-53(Al)-(OH)2, no significant activity was detected, as expected (see the orange curve in Fig. 6a). For the derived product obtained upon reaction with LiH in DMF (Fig. 6a, red curve), a short plateau centered at 3.45 V is detected during the first oxidation. This redox potential could match with that of DOBDC when interacting with a cation of high ionic potential (here Al3+).12 Nevertheless, the capacity is moderate and found to be mostly irreversible from the first reduction and upon further cycling obliterating the practical interest of such a solid.
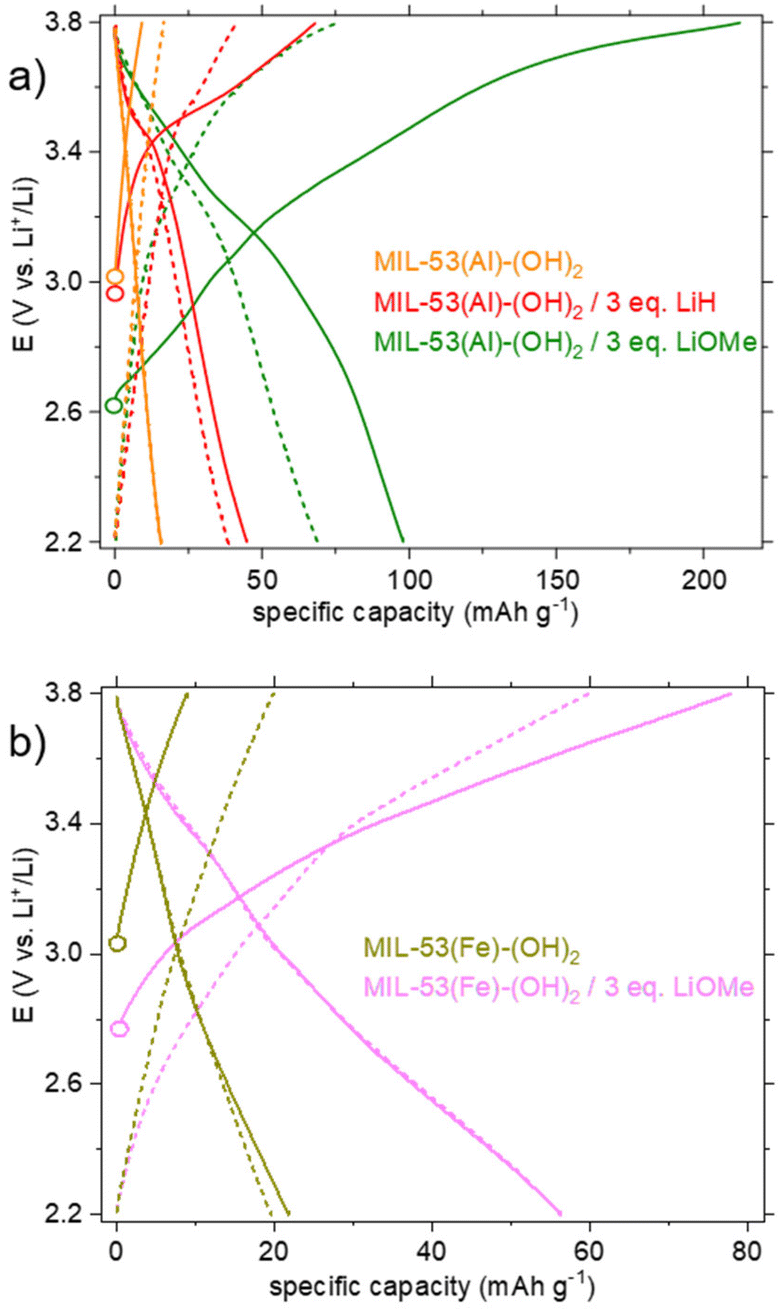 | ||
| Fig. 6 Electrochemical behaviour upon galvanostatic cycling in Li half-cells of the lithiated products compared to the pristine MOFs materials (electrolyte: 1 M LiClO4 in PC): potential vs. specific capacity curves. Plain lines correspond to the 1st cycle; dashed lines correspond to the 5th cycle. a) Data related to dry MIL-53(Al)-(OH)2 and lithiation products from LiH–DMF and LiOMe–MeOH/THF are shown in orange, red and green, respectively; b) data related to the lithiation product of MIL-53(Fe)-(OH)2 from LiOMe–MeOH/THF are shown in khaki and pink, respectively. | ||
The behavior of the product lithiated with LiOMe is different (Fig. 6a, green curve). The potential at rest (open circuit voltage, OCV) is lower (∼2.6 instead of ∼3 V vs. Li+/Li for other solids), in line with the instability of this solid in air (see above). During the first oxidation, a gradual oxidation is observed up to 3.8 V and is associated with a high capacity (>200 mA h g−1). Nevertheless, more than half of this capacity is lost during the first reduction and further decreased during the following cycles. As mentioned earlier, a question arose whether this compound is related to the salt Li4(DOBDC). The cycling behaviors of Li4(DOBDC) and this solid are compared in Fig. 7a on a narrower potential range (2.2–3.2 V vs. Li+/Li). It is clearly seen that the average redox potential of the lithiated MIL-53(Al)-(OH)2 is significantly higher (+180 mV) than that of Li4(DOBDC) (2.76 vs. 2.58 V vs. Li+/Li, respectively). As initially demonstrated for MgLi2(DOBDC), the presence of a cation with a high ionic potential close to the DOBDC redox unit dramatically increases the oxidation potential of phenolate moieties.12 This suggests that Al3+ cations remain close to the ligand, in line with other characterization results.
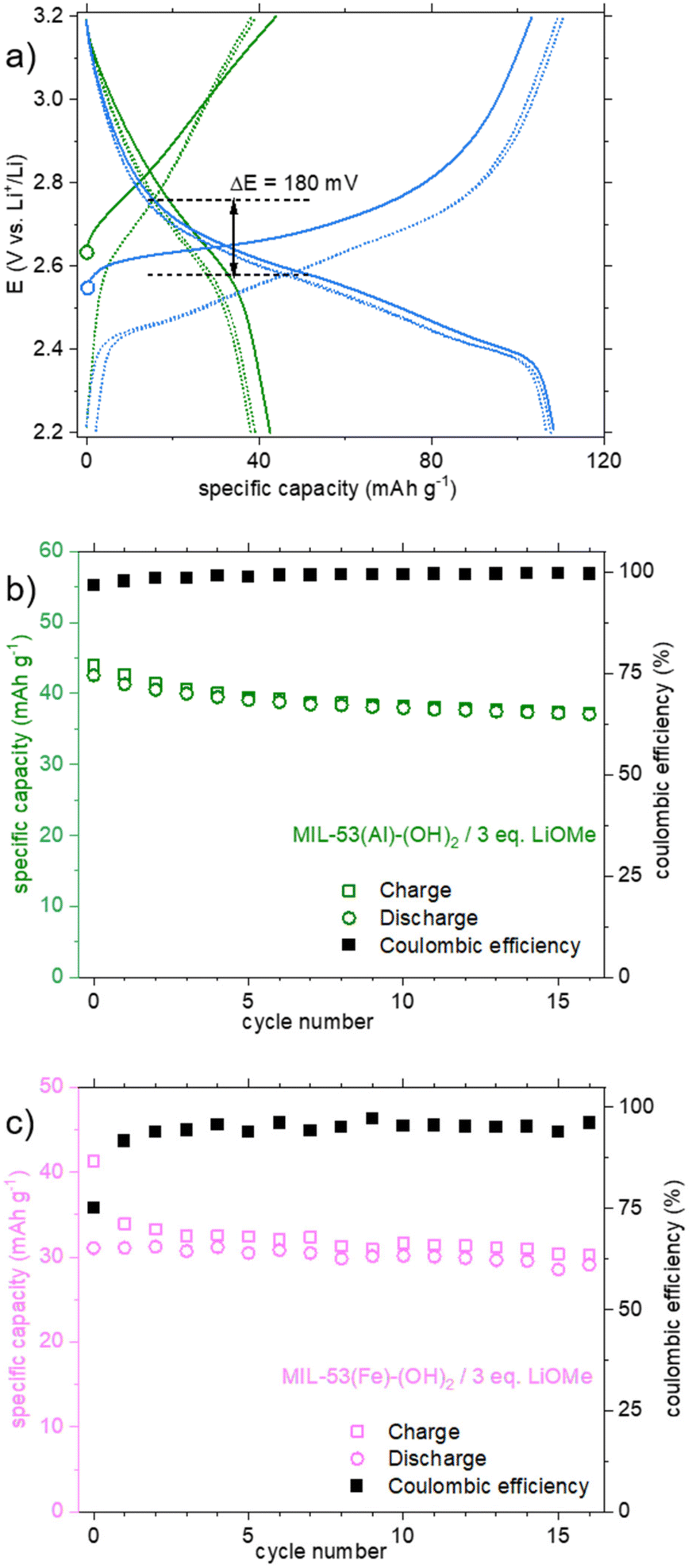 | ||
| Fig. 7 a) Comparison of potential vs. specific capacity curves for Li4(DOBDC) (blue) and the lithiation product from MIL-53(Al)-(OH)2 and LiOMe–MeOH/THF (green). Plain lines correspond to the 1st cycle; dashed lines correspond to the 5th and 10th cycles; b) capacity retention and coulombic efficiency for the lithiation product from MIL-53(Al)-(OH)2 and LiOMe–MeOH/THF (electrolyte: 1 M LiClO4 in PC; potential range: 2.2–3.2 V vs. Li+/Li); c) capacity retention and coulombic efficiency for the lithiation product from MIL-53(Fe)-(OH)2 and LiOMe–MeOH/THF (electrolyte: 1 M LiPF6 in 1:1 EC:DMC; potential range: 2.2–3.6 V vs. Li+/Li). | ||
Finally, the cyclability of this solid was evaluated within this potential range. As shown in Fig. 7b, the reversible capacity reached ca. 40 mA h g−1, with a rather good capacity retention (−12% after 15 cycles). Considering this initial result, a very-long term evaluation of the capacity retention was further carried out, this time using a solid obtained by treatment with 4 eq. of LiOMe instead of 3. Both solids present the same composition (Li7Al(DOBDC)5/2), as proven by IR, STEM-EDX and ICP-AES analyses (see Fig. S14, S16 and S17,† respectively). As shown in Fig. 8a, using identical cycling conditions (potential range and cycling rate), the capacity remained almost constant for more than 900 cycles corresponding to more than one year in operation. Such an outstanding capacity retention paired with a perfectly stable electrochemical feature upon cycling demonstrates that the small amount of Al3+ bound to the redox-active ligands is amazingly stable (Fig. 8b). Note that this lithiated product does not evolve towards Li4(DOBDC) probably due to the trivalent state of Al3+ that does not favor any ion exchange reaction with Li+ ions contained in the electrolyte.
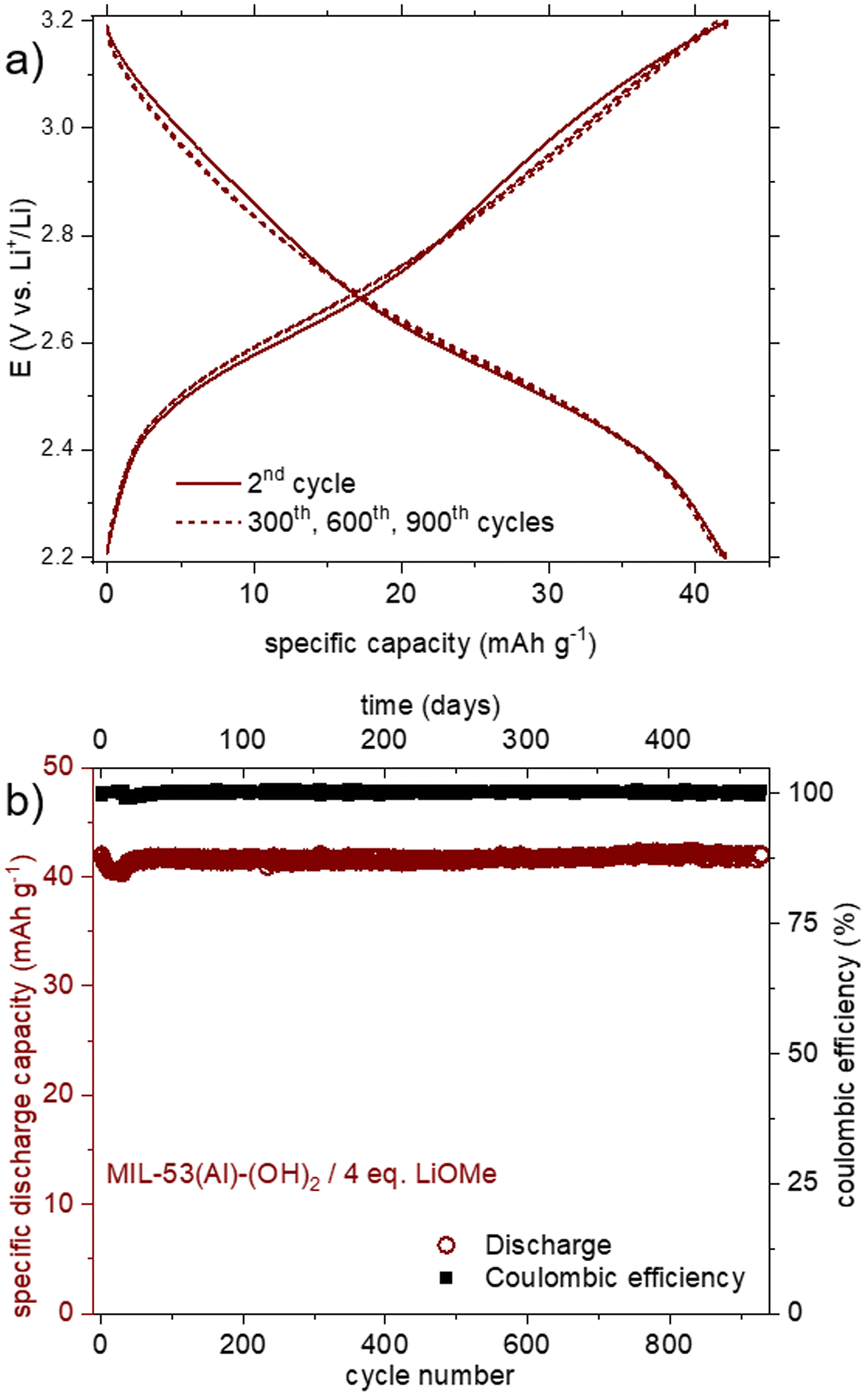 | ||
| Fig. 8 a) Potential vs. specific capacity curves for the lithiation product from MIL-53(Al)-(OH)2 and LiOMe–MeOH/THF. Plain lines correspond to the 2nd cycle; dashed lines to the 300th, 600th and 900th cycles; b) capacity retention and coulombic efficiency for the lithiation product from MIL-53(Al)-(OH)2 and 4 eq. of LiOMe (electrolyte: 1 M LiClO4 in PC; potential range: 2.2–3.2 V vs. Li+/Li). | ||
Eventually, an electrochemical study was also conducted on the lithiated product obtained from the reaction of MIL-53(Fe)-(OH)2 with LiOMe. Again, compared to the pristine MOF, a significant activity was detected during the first oxidation of the lithiated derivative (Fig. 6b, pink curve), with a continuous increase of the capacity up to ∼80 mA h g−1 at 3.8 V, likely associated with the oxidation of the ligand. The capacity is slightly lower during the first reduction (57 mA h g−1). Knowing that the reduction of Fe3+ to Fe2+ occurs at ∼3.0 V in the parent MIL-53(Fe)8 and in the pristine MIL-53(Fe)-(OH)2 (Fig. 6b, khaki curve), the absence of additional capacity upon reduction suggests that the Fe3+ ions are inactive in our experimental conditions. This might relate to the fact that the solid already contains 3 Li+ per iron and cannot accommodate the additional Li+ that would be necessary to balance the charge of Fe2+. The capacity retention of this solid was nevertheless evaluated in the potential range 2.2–3.6 V vs. Li+/Li (Fig. 7c). From the second cycle, the capacity appears rather constant (−6% after 15 cycles) but reaches only ∼30 mA h g−1, corresponding to the reversible insertion of only ∼0.3 electrons per Li3FeO(DOBDC).
Conclusions
The reactivity of functionalised MIL-53(M)-(OH)2 (M = Al, Fe) with various strong bases was studied, with the aim of fully exchanging the acidic protons (both organic and inorganic) with Li+ ions to ultimately exploit the redox activity of the phenolic ligand in the solid state. This reactivity was found to strongly depend on the nature of the base (LiH vs. LiOMe) and of the cation. More specifically, whereas it was not possible to fully deprotonate MIL-53(Al)-(OH)2 with LiH in DMF, the reaction with LiOMe in MeOH/THF led to a partial release of Al3+ and the formation of an amorphous layered phase formulated Li7Al(DOBDC)5/2. In contrast, the lithiation of the Fe analogue occurred without leaching, leading to the expected composition Li3FeO(DOBDC). In both cases, the electrochemical activity of the ligand in the solid state was studied. The associated reversible capacities were moderate, but a shift of the redox potential compared to those of the salt Li4(DOBDC) was observed, together with an outstanding capacity retention (>900 cycles, >one year), at least for one solid. Although these lithiated MOF derivatives are of limited practical interest for electrochemical energy storage, this study eventually suggests that it is possible to expand the scope of polycations (from +2 to +3) suitable for tuning the redox potential of the ligand and to use them in small amounts (as “dopants”) in a layered Li4(DOBDC)-type structure.Author contributions
MD: synthesis, XRD and IR characterization and electrochemical evaluation; HC: PDF analysis; PSA: synthesis and ICP characterizations; PM: STEM-EDX analysis; ND and MP: NMR analysis; PP: conceptualization and electrochemical analysis; TD: conceptualization, spectroscopic analysis and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding from the region Pays de la Loire (project PSR ‘MatHySE2’) and the Agence Nationale de la Recherche (project ‘ThiOMOFs’) is acknowledged (fellowships for M. D. and P. S.-A., respectively). Funding by the French Contrat Plan État-Région and the European Regional Development Fund of Pays de la Loire, the CIMEN Electron Microscopy Center in Nantes, is gratefully acknowledged. P. Deniard is thanked for his advice for the PDF analysis, and S. Grolleau is thanked for the thermal analyses. A. Vlad (UCLouvain) is acknowledged for fruitful discussions. The authors also thank the Synchrotron Soleil for providing access to the Cristal Soleil beamline. The ICP-AES analyses were performed at the LPG-UMR 6112 (C. La, M. Rivoal), Nantes Université.Notes and references
- J. Zhou and B. Wang, Chem. Soc. Rev., 2017, 46, 6927–6945 RSC
.
- Y. Xu, Q. Li, H. Xue and H. Pang, Coord. Chem. Rev., 2018, 376, 292–318 CrossRef CAS
- L. Zhang, H. Liu, W. Shi and P. Cheng, Coord. Chem. Rev., 2019, 388, 293–309 CrossRef CAS
- Z. Wang, H. Tao and Y. Yue, ChemElectroChem, 2019, 6, 5358–5374 CrossRef CAS
- J. Liu, D. Xie, W. Shi and P. Cheng, Chem. Soc. Rev., 2020, 49, 1624–1642 RSC
- A. Schneemann, R. Dong, F. Schwotzer, H. Zhong, I. Senkovska, X. Feng and S. Kaskel, Chem. Sci., 2021, 12, 1600–1619 RSC
-
T. Devic, in Metal-Organic Frameworks in Biomedical and Environmental Field, ed. P. Horcajada and S. Rojas, Springer Nature, 2021, pp. 111–154 Search PubMed
- G. Férey, F. Millange, M. Morcrette, C. Serre, M.-L. Doublet, J.-M. Grenèche and J.-M. Tarascon, Angew. Chem., Int. Ed., 2007, 46, 3259–3263 CrossRef PubMed
- C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2006, 128, 8726–8727 CrossRef CAS PubMed
- S. Renault, S. Gottis, A.-L. Barrès, M. Courty, O. Chauvet, F. Dolhem and P. Poizot, Energy Environ. Sci., 2013, 6, 2124–2133 RSC
- S. Wang, L. Wang, K. Zhang, Z. Zhu, Z. Tao and J. Chen, Nano Lett., 2013, 13, 4404–4409 CrossRef CAS PubMed
- A. Jouhara, N. Dupré, A.-C. Gaillot, D. Guyomard, F. Dolhem and P. Poizot, Nat. Commun., 2018, 9, 4401 CrossRef PubMed
- P. D. C. Dietzel, Y. Morita, R. Blom and H. Fjellvåg, Angew. Chem., Int. Ed., 2005, 44, 6354–6358 CrossRef CAS PubMed
- N. L. Rosi, J. Kim, M. Eddaoudi, B. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504–1518 CrossRef CAS PubMed
- M. Maercz, D. S. Wragg, P. D. C. Dietzel and H. Fjellvag, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, m152 CrossRef CAS PubMed
- F. Luo, C. Yan, L. Dang, R. Krishna, W. Zhou, H. Wu, X. Dong, Y. Han, T.-L. Hu, M. O'Keeffe, L. Wang, M. Luo, R.-B. Lin and B. Chen, J. Am. Chem. Soc., 2016, 138, 5678–5684 CrossRef CAS PubMed
- H. Assi, L. C. Pardo Pérez, G. Mouchaham, F. Ragon, M. Nasalevich, N. Guillou, C. Martineau, H. Chevreau, F. Kapteijn, J. Gascon, P. Fertey, E. Elkaim, C. Serre and T. Devic, Inorg. Chem., 2016, 55, 7192–7199 CrossRef CAS PubMed
- H. Chun and D. Moon, Cryst. Growth Des., 2017, 17, 2140–2146 CrossRef CAS
- P. C. Dietzel, R. Blom and H. Fjellvåg, Z. Anorg. Allg. Chem., 2009, 635, 1953–1958 CrossRef CAS
- T. Devic, P. Horcajada, C. Serre, F. Salles, G. Maurin, B. Moulin, D. Heurtaux, G. Clet, A. Vimont, J.-M. Grenèche, B. L. Ouay, F. Moreau, E. Magnier, Y. Filinchuk, J. Marrot, J.-C. Lavalley, M. Daturi and G. Férey, J. Am. Chem. Soc., 2010, 132, 1127–1136 CrossRef CAS PubMed
- P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M. Daturi, O. David, E. Magnier, N. Stock, Y. Filinchuk, D. Popov, C. Riekel, G. Férey and C. Serre, J. Am. Chem. Soc., 2011, 133, 17839–17847 CrossRef CAS PubMed
- D. Cunha, C. Gaudin, I. Colinet, P. Horcajada, G. Maurin and C. Serre, J. Mater. Chem. B, 2013, 1, 1101–1108 RSC
- M. L. Aubrey and J. R. Long, J. Am. Chem. Soc., 2015, 137, 13594–13602 CrossRef CAS PubMed
- D. Rambabu, A. E. Lakraychi, J. Wang, L. Sieuw, D. Gupta, P. Apostol, G. Chanteux, T. Goossens, K. Robeyns and A. Vlad, J. Am. Chem. Soc., 2021, 143, 11641–11650 CrossRef CAS PubMed
- S. Biswas, T. Ahnfeldt and N. Stock, Inorg. Chem., 2011, 50, 9518–9526 CrossRef CAS PubMed
- J. Rodriguez-Carvajal, Phys. B, 1993, 192, 55 CrossRef CAS
- D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calvé, B. Alonso, J. O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70 CrossRef CAS
- P. Juhás, T. Davis, C. L. Farrow and S. J. L. Billinge, J. Appl. Crystallogr., 2013, 46, 560–566 CrossRef
- K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 9604–9605 CrossRef CAS PubMed
- K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2008, 47, 7936–7938 CrossRef CAS PubMed
- K. L. Mulfort, T. M. Wilson, M. R. Wasielewski and J. T. Hupp, Langmuir, 2009, 25, 503–508 CrossRef CAS PubMed
- N. Biggins, M. E. Ziebel, M. I. Gonzalez and J. R. Long, Chem. Sci., 2020, 11, 9173–9180 RSC
- K. L. Mulfort, O. K. Farha, C. L. Stern, A. A. Sarjeant and J. T. Hupp, J. Am. Chem. Soc., 2009, 131, 3866–3868 CrossRef CAS PubMed
- D. Himsl, D. Wallacher and M. Hartmann, Angew. Chem., Int. Ed., 2009, 48, 4639–4642 CrossRef CAS PubMed
- K. Riascos-Rodríguez, S. Marks, P. G. Evans, S. P. Hernández-Rivera, J. L. Ruiz-Caballero, D. Piñero and A. J. Hernández-Maldonado, Cryst. Growth Des., 2020, 20, 3898–3912 CrossRef
- M. Kubo, H. Hagi, A. Shimojima and T. Okubo, Chem. – Asian J., 2013, 8, 2801–2806 CrossRef CAS PubMed
- Y.-S. Bae, B. G. Hauser, O. K. Farha, J. T. Hupp and R. Q. Snurr, Microporous Mesoporous Mater., 2011, 141, 231–235 CrossRef CAS
- F. Millange and R. I. Walton, Isr. J. Chem., 2018, 58, 1019–1035 CrossRef CAS
- Z. Wang, A. Bilegsaikhan, R. T. Jerozal, T. A. Pitt and P. J. Milner, ACS Appl. Mater. Interfaces, 2021, 13, 17517–17531 CrossRef CAS PubMed
- S. Renault, J. Geng, F. Dolhem and P. Poizot, Chem. Commun., 2011, 47, 2414–2416 RSC
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem. – Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Powder X-ray diffraction data and thermogravimetric analyses, infrared spectroscopy, and transmission electron microscopy results. See DOI: https://doi.org/10.1039/d3me00030c |
| This journal is © The Royal Society of Chemistry 2023 |