
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-organizing maps as a data-driven approach to elucidate the packing motifs of perylene diimide derivatives†
Francesco
Marin
,
Alessandro
Zappi
 *,
Dora
Melucci
and
Lucia
Maini
*
*,
Dora
Melucci
and
Lucia
Maini
*
Dipartimento di Chimica ‘G. Ciamician’, Università di Bologna, via Selmi 2, 40126 Bologna, Italy. E-mail: alessandro.zappi4@unibo.it; l.maini@unibo.it
First published on 4th January 2023
Abstract
The efficient classification or prediction of crystal structures into a small number of families of related structures can be extremely important in the design of materials with specific packing and properties. In this respect, the traditional way to classify the crystal packing of organic semiconductors as herringbone, sandwich-herringbone, and β- or γ-sheets by visual inspection has its limitations. Herein, we present the use of a clustering method based on a combination of self-organizing maps and principal component analysis as a data-driven approach to classify different π-stacking arrangements into families of similar crystal packing. We explored the π-stacking arrangements within the crystal structures deposited in the Cambridge Structural Database of perylene diimide (PDI) derivatives with different types and positions of the substituents. The structures were characterised by a set of descriptors that were then used for classification. Six different packing families of PDIs were identified and their characteristics are discussed here. Finally, the effects of different substituent types and positions on the resulting packing arrangement are discussed.
Design, System, ApplicationPerylene-diimide (PDI) derivatives are a promising class of organic molecules for semiconductive applications and their properties can be tuned by substitution on the imide, bay or ortho positions. Their use as semiconductors is strictly related to the packing of their crystal forms that is strongly influenced by the nature and the position of functional group. In the present work, we explore a chemometric method based on self-organizing maps (SOM) with the aim of classifying PDIs in families of structures sharing similar characteristic. The present approach is a mathematical computation that starts from the structural characteristics of the crystal forms, and shows and describes six main families, with some structures not univocally classified into any of them. Our results indicate the limitations of the traditional classification method for PDI, that is based on a visual inspection of the structures and identifies only four classes, and demonstrate the advantages of the use of a chemometric approach for crystal forms grouping. |
Introduction
Organic electronics have received an increasing amount of academic and industrial interest in recent decades in the form of research and development across various fields such as chemistry, physics, material science and engineering. The rapid growth in research related to π-conjugated materials, and in particular organic semiconductors (OSCs), has been inspired by their numerous attractive properties, including mechanical flexibility, low cost, solution processability and molecular-level tunability. These properties make OSCs highly versatile functional materials for use as the building blocks for fabricating high-performance flexible optoelectronic devices.1–5 Currently, OSCs are widely used in various organic electronic devices, such as organic light-emitting diodes (OLEDs),6,7 organic field-effect transistors (OFETs),8–10 and organic photovoltaic (OPV) cells.11–13 The performance of these devices depends critically on the efficiency of charge transport within the material. Therefore, to successfully develop and commercialise everyday devices based on OSCs, these materials must achieve high charge-carrier mobility, good air stability and solubility in common solvents to enable low-cost processing.1,3,14Since their discovery, a large number of OSCs have been developed, reported and investigated, such as oligoacenes (like pentacene and rubrene),1,8,9 oligothiophenes,1,8 thiazole derivatives,1,15 benzothienobenzothiophenes,9,16 fullerenes,1,8,15 and perylenes.1,2,8,17 Among them, perylene diimides (PDIs) have been a research topic for more than 100 years, mainly because of their use as high-performance industrial organic pigments, which are currently used in fibre applications and industrial paints.18,19 In the past 20 years the interest in PDI, as well as other dyes and pigments, has moved from the use as a traditional colourant toward applications as functional solid-state material.13,20 In fact, PDI derivatives are currently a well-known family of very promising and versatile n-type materials for organic optoelectronic applications owing to their commercial availability; low cost; excellent chemical, thermal and photostability; high electron affinity; strong absorption in the visible region; low LUMO energies and good charge-transport properties. Furthermore, their electronic, optical and charge-transport properties can be tuned over a wide range via functionalisation.2,21,22 The combination of these properties makes PDIs promising candidates for several applications, such as OFETs,2,8–10,21,23 OPV cells,11–13 laser dyes, sensors25 and bioimaging.26 PDIs are also studied for low-temperature thermoelectric generator applications.27
Because of their planar structure and peripherally rich oxygen atoms, PDIs typically arrange to form π–π stacking interactions between molecules. Nevertheless, substitution can greatly vary the extent of intermolecular π-orbital overlap among PDI derivatives and thus can significantly affect the solubility, optical and electrochemical properties, crystal packing and structural morphology.5,17,28–32 The PDI core has 10 positions that can be functionalised: the N,N′ imide positions; the 1,6,7,12 bay positions and the 2,5,8,11 ortho positions (Fig. 1). These numerous positions for introducing functional groups are the reason for the popularity of PDIs, as they enable versatile tailoring of the properties for specific applications.17,21 In general, substitution at imide positions maintains the planarity of the perylene aromatic core, and can effectively tune solubility, aggregation and solid-state molecular packing, but has only limited effects on the molecular-level optical and electronic properties of PDIs.17,19,29,33 However, the optical and electronic properties of PDIs can be significantly modified via substitution at the perylene core in the bay and ortho positions. In addition, substitution at bay positions may lead to the twisting of the two naphthalene half units in PDIs due to steric effects; however, large geometric distortions of the core may weaken the intermolecular π–π overlap and thus may decrease the charge-carrier mobility.17,21,24,34 Finally, ortho functionalisation enables modification of the optoelectronic properties while retaining the planarity of the perylene core.17,35 The adjustment of the PDI properties can be achieved through combinations of imide, bay and ortho functionalisation.
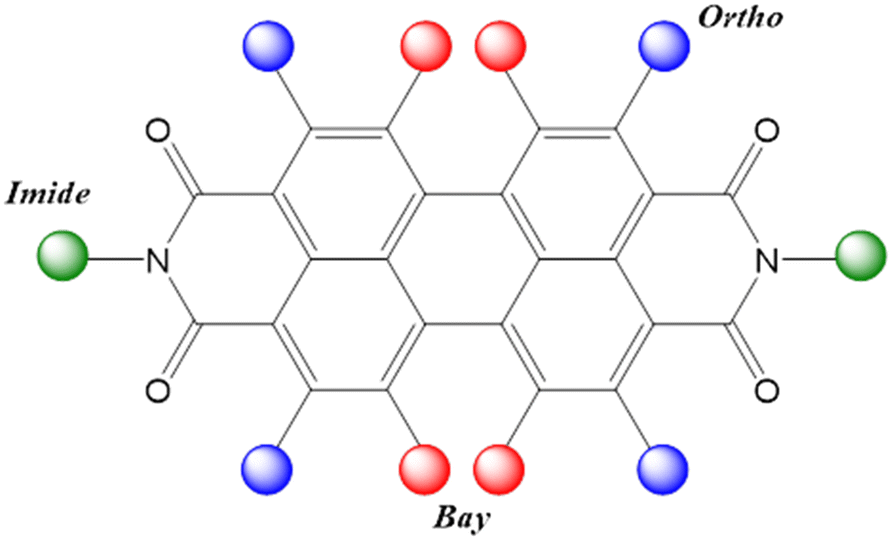 | ||
| Fig. 1 Chemical structure of a generic PDI showing the different positions for substitutions: imide positions highlighted in green, bay positions in red and ortho positions in blue. | ||
Much experimental and computational effort has been applied to a long-standing goal and challenge of OSCs materials and crystal engineering, namely, understanding the relationships among the molecular packing motifs, intermolecular interactions and properties of molecular materials for efficient design of crystalline material for specific applications. Attempts to correlate the structural arrangements to specific properties to predict solid-state materials with tailored properties have been made since the 1980s, where the crystallochromy (dependence of colour on crystal packing) of PDIs with various substituents at the imide position was investigated in depth both experimentally and theoretically, highlighting that intermolecular packing greatly affect the optical properties. In particular, an empirical model to correlate the absorption maxima with the π–π contact area between stacked molecules was developed, and a strong correlation between crystal colour and displacement along the long (x) and short (y) axes of the perylene core was predicted.19,28,36 After these pioneering works, much effort has been made to provide theoretical insight into the optoelectronic properties of PDIs to provide a deeper understanding of the structure–property relationships. For instance, the potential energy surfaces for the ground state of PDI dimers as a function of the shift along the x- and y-axes of the perylene core was evaluated as a function of the rotation (φ) between co-facially stacked dimers, and the excitation energies of PDI crystal structures were evaluated.37,38 Another study mapped the relationships between stacking geometry, binding energy and electronic coupling for dimers of 20 PDI derivatives based on density functional theory.39 The effect of the type, number and position of the substituents on the charge-transport properties of 30 imide-substituted PDIs and 7 bay- and bay/imide-substituted PDIs was investigated.31 In addition, the influence of the substituents at different positions (ortho, bay and imide) of 17 PDI derivatives on their packing, intermolecular interactions and electronic properties was studied.32 Furthermore, several other works, both experimental and theoretical, have been published on the effects of different substituents on the material performance of PDIs for specific applications, for example, the use of linear chains,3,40 fluorinated chains41 or branched chains3,42,43 at the imide position and the use of cyano substituent,44 halogenated substituents45 or other substituents at the core.24,46
Most of the works mentioned above mainly used dimer approaches based on reduced data sets of similar molecules, because analysis of the large target molecules leads to high computational costs. Recent advances in computational power and the development of smart algorithms in the field of machine learning and artificial intelligence are helping overcome the limitations of the dimeric approach. The discovery and design of OSCs materials with interesting properties and the investigation of their structure–property relationships now applies two main computational approaches that consider the entire crystal structure: multiscale approaches and data-driven searches.47 Multiscale approaches combine different methods and models for computing the reorganisation energies and electronic couplings, and evaluating disorder effects.47–49 These methods can be combined with crystal structure predictions to screen for interesting packing arrangements.50–52 Data-driven searches make use of the high amount of data present in databases such as the Cambridge Structural Database (CSD) to perform large-scale screening strategies to predict the semiconducting properties of materials from various computable descriptors and thus to explore their structure–property relationships.47,53–55
The recent advances in predicting OSCs with interesting properties make use of prior knowledge on the crystal structures, either experimental54 or calculated from CSD.51 The knowledge of the crystal packing is very important for investigating the material properties, because it gives information about the intrinsic properties of organic semiconductors, providing a powerful tool for examining structure–property relationships. Therefore, achieving a good understanding of the molecular packing features is beneficial for both the design and synthesis of OSCs and for enhancing the understanding of the structure–property relationships and the charge-transport limitations.5 Furthermore, the possibility to efficiently classify observed or predicted molecular crystals into a small number of families of related structures can be extremely important in the design of materials and crystal phases with specific packing and properties. The conventional method of classifying the crystal packing of OSCs is to describe the π-stacking interactions responsible for the charge-carrier mobility, such as herringbone, sandwich-herringbone and β- and γ-sheets by visual inspection of the structures. However, there are limitations to this method, and the classification can be insufficient.
In this work, we investigate the packing arrangements of 103 PDI-derivative crystal structures from the CSD database to test a clustering method based on a combination of self-organizing maps (SOMs) and principal component analysis (PCA) as a data-driven approach to classify the different π-stacking arrangements of PDIs. This aim of this method was to identify families of PDI stacking arrangements using a number of descriptors that were chosen to characterise each PDI crystal structure, thereby providing guidelines for predicting the most likely packing family depending on the substituents and assessing new families of PDI crystal structures. Furthermore, we believe that this method of crystal structure clustering can be applied to other types of OSCs and assist future theoretical studies in achieving efficient and data-driven clustering of structures to identify the model systems that best describe a specific family of packing arrangements. Finally, this clustering-based classification of crystal structures can be used in the future to correlate different families of structures with OSC properties, although this was beyond the scope of this work.
Results and discussion
The search for PDI-derivative structures in the CSD resulted in the selection of 103 different crystal structures which resulted in 142 independent molecules for analysis variables indicating the temperature of the data collection (T), Z (number of molecules in the cell) and Z′ (number of molecules in the asymmetric unit) were used to give information about the crystal structure. The variables used to describe the molecule within the crystal structure included the molecular volume (Vmol), substituent position, type of substituent at the imide position, type of substituent at the core, torsional angle of the perylene core resulting from the twisting of the two naphthalene half units (τ) and molecular aspect ratio (with the three variables S, S/L and M/L). Finally, the π-stacking arrangement within the crystal packing was described by considering the stacking between dimers formed by two neighbouring PDI molecules in the structure, where the variables were calculated to give the relative position of one molecule in the dimer with respect to the other. The variables describing the stacking arrangement included the distance between the centroids of the perylene cores (SV); interplanar distance between the perylene planes (dπ–π); displacement of the perylene units along the long (Δx) and short (Δy) molecular axis; directional cosine of SV (χ with the x-axis and ψ with the y axis); angles of perylene unit slipping along the x- and y-directions (P and R, respectively); tilt angle between the plane of the two perylene cores within the dimer (θ) and the degree of rotation of the molecules along the stacking direction (ρ). These variables were collected or calculated, as described in detail in the Experimental section.From the collection and calculation of the different variables, we observed two different mechanisms by which PDIs minimise repulsion between the aromatic units and achieve tighter stacking compared with the face-to-face situation: (i) shifted cofacial alignment, i.e., means non-zero Δx and Δy, and/or (ii) rotation between the stacked aromatic cores, i.e. non-zero ρ.
After collecting and calculating the variables for the 142 samples (henceforth called ‘objects’), we used the SOM method56 to identify families of packing arrangements. SOM is an unsupervised machine learning technique that does not require initial information about the samples group, and was used to identify groups of objects with similar features. The computation was performed with the R package SOMEnv,57 adapting the algorithms developed by Licen et al. for environmental problems for crystallographic analysis.
Since the SOM output and the final object grouping may depend on the map dimensionality and number of clusters in which the SOM units are divided, we trained and clustered four different maps. After training, SOMEnv enables automatic calculation of several numbers of clusters for the SOM units, and the optimal number of clusters was selected as the one that minimised the DB index.58 For all trained maps, up to eight clusters were calculated.
The first two eigenvalues calculated by PCA on the original dataset were 4.38 and 3.51. These values (together with the number of samples) were then used as guidelines to define the map dimensions, following the empirical rules of Nakagawa et al.59 The trained maps had the following dimensions and number of clusters: 5 × 3 with eight clusters; 9 × 7 with five and eight clusters (the two DB indices were very similar, making it difficult to choose between them); 10 × 6 with five clusters and 17 × 4 with six and seven clusters. Therefore, six different results of object grouping were obtained. To ensure that these grouping were reliable, the SOM results were compared, and the structures that were grouped together by all trained SOMs were considered to belong to the same family. In this way, six main families of structures were identified, describing 109 of the initial 142 objects. In general, the remaining objects were at the borders between these families and were either grouped into minor families (with fewer than five objects) or not grouped at all. To best visualise the result of SOM grouping, the PCA scores were plotted. The scores of principal components PC1 vs. PC2 are shown in Fig. 2a, where the colour of the scores corresponds to the family assigned to each structure.
 | ||
| Fig. 2 a) PC2 vs. PC1 score plot obtained by PCA, with the scores categorised by family. The six major families are: tight-long-x (green), crisscross (brown), tight-long-y (blue), nostack (red), tight-twist (orange) and verylong-x (yellow). The ‘extra’ group (purple) contains all the minor groups that do not entirely fit into any of the six major families, while the black dots are the scores that do not belong to any major or minor group. b) PCA loading plot providing a visualisation of the most-influential variables for each family. | ||
From the score plot in Fig. 2a, we observed that the main families obtained using SOM were grouped well in different areas of the PC1–PC2 plot. We named the different groups depending on the area of the score plot where they were located. The six major families were tight-long-x (purple coloured), which lies close to the origin; crisscross (red), which lies at positive values of both PC1 and PC2; tight-long-y (orange), which lies at positive PC1 and negative PC2 values; tight-twist (yellow), which lies at moderately negative PC1; verylong-x (brown), which lies at negative PC1 and positive PC2 values and nostack (green), which lies at negative values of both PC1 and PC2. The extra group (blue) contains all minor clusters found in the SOM, whereas the black points are those that did not fit unambiguously in any of the major or minor groups. Interestingly, the scores at negative PC1 tended to be more scattered than those at positive values, i.e., the families on the right side of the score plot are more homogeneous than those on the left side.
Once the different families were established, we investigated the principal characteristics and typical structure of each family in detail. A visualisation of the variables most influencing each family is shown by the PCA loading plot in Fig. 2b. Indeed, there exists a quadrant correspondence between the score and loading plots. The variables in a certain quadrant of the loading plot are those that most directly influence the scores in the same quadrant. For example, the families tight-twist and verylong-x had higher values of Δx and P, while the crisscross family had higher ρ and Z values. However, to better visualise important insights into the variation of the variables in each group, boxplots of the most important variables were created and are shown in Fig. 3. Boxplots of the other variables are reported in the ESI† (Fig. S1–S10). In the boxplots, the variation range of each variable is plotted for each family, which gives information about the range and distribution of each variable. The use of boxplots, coupled with the inspection of the scores in each family allowed us to identify the most important characteristics of each family.
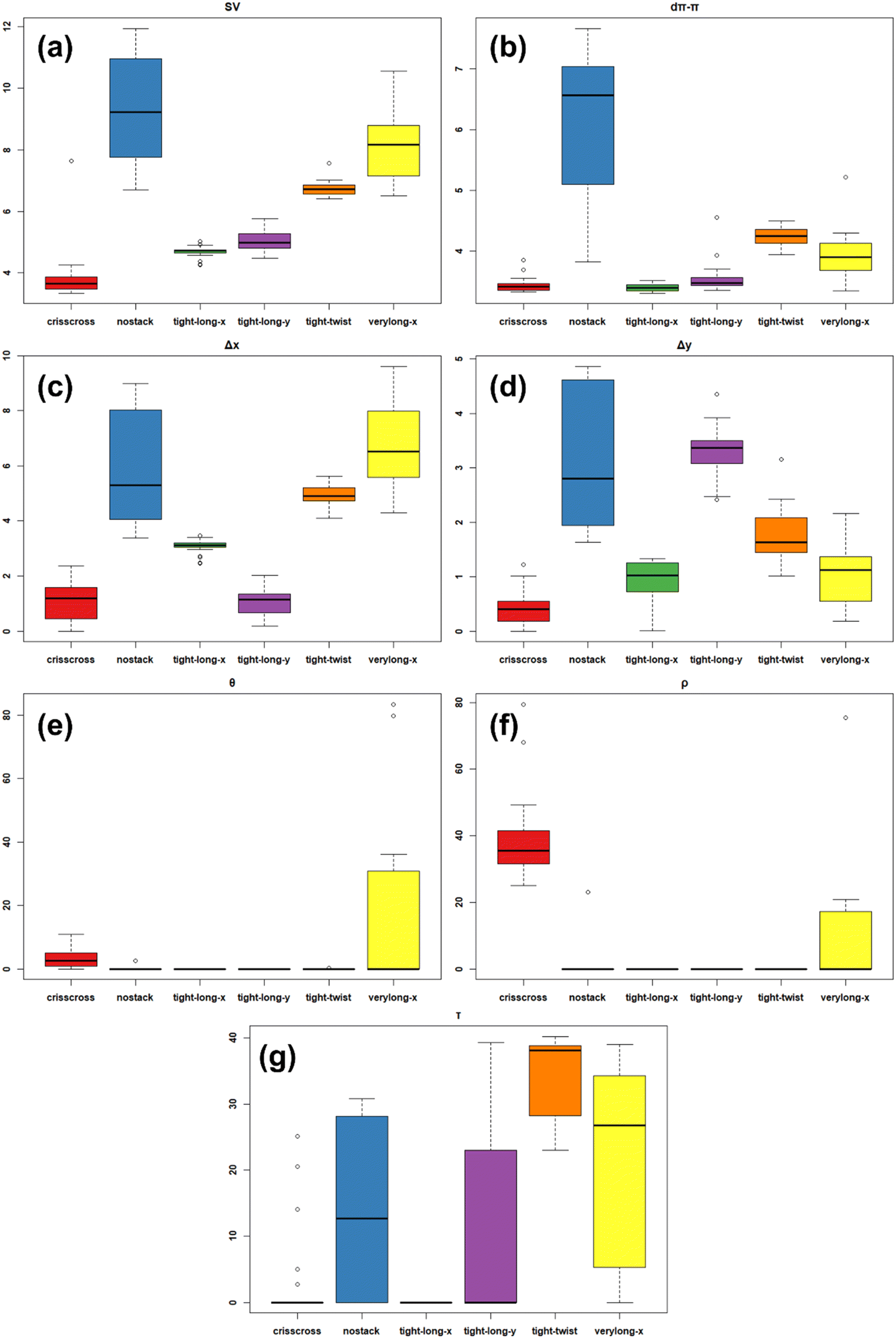 | ||
| Fig. 3 Boxplots of the distribution of variables a) SV; b) dπ–π; c) Δx; d) Δy; e) θ; f) ρ and g) τ among the identified families. | ||
The tight-long-x group comprised 31 of the initial 142 objects and was characterised by a very narrow distribution of all variables (Fig. 3). This group is characterised by a short dπ–π distance, in the range 3.31–3.52 Å, typical of effective π–π interactions between aromatic cores.60 Similar dπ–π distances were found in the crisscross (3.32–3.55 Å) and tight-long-y (3.36–3.70 Å) families. The groups with short dπ–π distances are those on the right side of the score plot, at positive PC1, apart from two with small negative PC1. The other variables characteristic of tight-long-x are Δx and Δy. For tight-long-x, Δx = 2.45–3.46 Å and Δy = 0–1.33 Å, which means that this family is characterised by a displacement with rather high Δx and small Δy (Δx ≫ Δy). Finally, no molecules with non-zero τ were present in this family, and it is the only group with such characteristics.
The tight-long-y family comprised 17 of the initial 142 objects. Apart from the short dπ–π distances (3.36–3.70 Å) already mentioned, similar to the tight-long-x family, the tight-long-y family was characterised by a displacement along the x- and y-directions to achieve tighter packing. However, in this family, the displacement was higher along the y-direction than along the x-direction, with Δx = 0.18–2.03 Å and Δy = 2.41–4.35 Å. Nevertheless, as a consequence of their similar dπ–π distances and their displacement (although in different directions), tight-long-x and tight-long-y had similar SV values. Finally, 5 of the 17 objects in this family have non-zero τ.
The crisscross family comprised 23 of the initial 142 objects. The principal characteristics of this group are highlighted by Fig. 3e and f, which shows that crisscross is one of only two families (with verylong-x) with non-zero ρ and θ. In particular, crisscross is the only group with all structures in criss-cross (non-zero ρ). Furthermore, crisscross is the family with the shortest SV, which is a consequence of the crisscross arrangement. Since rotation between the aromatic cores allows a closer packing, a displacement such as that observed for the tight-long-x or tight-long-y families is not necessary for close packing in the crisscross family. Thus, Δx (0–2.4 Å) and Δy (0–0.75 Å) were both very small in this family, resulting in short dπ–π distances (3.32–3.55 Å) and small SV. Even in this group, some objects present torsion, but only 5/23, so it is not a common feature of the group.
The tight-twist family comprised 16 of the initial 142 objects. This group lies in an area of the score plot between tight-long-x, verylong-x and nostack, at small negative PC1 and between small positive and small negative PC2. Therefore, this family has intermediate characteristics between these other three families. However, a unique characteristic of the tight-twist is that it is the only one with all structures having a twisted perylene core and the highest τ. Furthermore, in this family, the distribution of the variables are quite narrow, as for the other families. This family is characterised by rather large dπ–π distances (3.94–4.50 Å). Furthermore, Δx (4.1–5.64 Å) and Δy (1–3.16 Å) are larger than for tight-long-x, although the Δy values were not as high as those in the tight-long-y family. Indeed, considering the large values of dπ–π, Δx and Δy, this family is characterised by a higher SV than for other families where very small dπ–π values were observed.
The verylong-x family consisted of 14 of the initial 142 objects. This family is characterised by very high SV (6.51–10.55 Å) and Δx (4.29–9.62 Å), but small Δy (0.17–2.16 Å). The dπ–π values of this family were slightly lower than those in the tight-twist family (3.34–4.29 Å), apart from one object with a high value (5.22 Å). Although some of the objects within this group had non-zero ρ and/or θ, they are a minority (4/14 with θ and 6/14 with ρ). The objects were not assigned to the crisscross group because some other parameters differ.
The last major family, nostack, contained 8 of the initial 142 objects. As Fig. 3 clearly shows, this group had the highest SV, Δx, Δy and dπ–π, indicating that intermolecular interactions are more important for the structures in this family than π-stacking interactions. Furthermore, despite the low number of objects in this family, it had the largest distributions of variables. Therefore, it seems that these objects were grouped together not because they share real common traits, but because they have significantly different characteristics compared to the other families.
Among the 33 objects that did not fit into any of the major families, 12 were not assigned to any family, and 21 were assigned to the extra group, which was divided into five minor families (see Fig. S11†). These five extra families did not fit unambiguously in any of the six major families, because some of their parameters fit in one family, while other parameters fit into other families. The four extra1 objects were assigned to tight-twist, nostack or none of the major families by the various SOMs. This was because they have high dπ–π (4.05–4.09 Å), a non-zero τ and intermediate Δy values that could fit in these groups, but their Δx and SV are too small. The three extra2 objects could fit into tight-twist, nostack or none of the major families according to the different SOMs, depending on the chosen parameters. They have high SV, Δy and dπ–π values (close to nostack values), but small Δx that do not fit in this family, resulting in high R values (>38°, higher than P) that are only typical of the tight-long-y family. The three extra3 objects lied between the crisscross and tight-long-y families, and in various SOMs, they were assigned to one of these two families. These objects had small SV, dπ–π and Δx values that could fit in the crisscross family, but they did not have a non-zero ρ or θ characteristic of this family, and their Δy was slightly too high. Furthermore, their Δy and SV were too low to fit in the tight-long-y family, despite their similar dπ–π and Δx values. The extra4 family (eight objects) and extra5 family (three objects) had parameters that could fit into the tight-long-x and tight-twist families. However, these objects had dπ–π values higher than those for the tight-long-x family and lower than those in the tight-twist family, some of them were twisted and their SV values were similar to those of the tight-long-x family (except for a couple of extra4 objects). In addition, their Δx and Δy values were similar to those of the tight-long-x family, but not those of the tight-twist family.
A visual representation of the typical stacking in the crystal structures of the major families that best summarises the characteristics of each group is given in Fig. 4.
 | ||
| Fig. 4 Stacking of the perylene core for the major families viewed along the z (top) and x (bottom) axis. a) tight-long-x, CSD reference code (refcode) DICNIM; b) tight-long-y, refcode SAGWEC; c) crisscross, refcode MIWHEF; d) tight-twist, refcode NIXWIC02; e) verylong-x, refcode KUWXOR and f) nostack, refcode USAFEB01. | ||
Fig. 4 shows that the overlap between perylene cores in the tight-long-x and crisscross families (Fig. 4a and c) is high. The structures within these families are characterised by a well-defined 1D packing motif along the π-stacking direction. In contrast, the structures in the tight-long-y family (Fig. 4b) have large displacement along the y direction, leading to a minor overlap of the perylene cores. However, the large displacement leads to a 2D packing motif, which can be beneficial for charge mobility.32 The structure of the tight-twist family (Fig. 4d) clearly resembles the characteristics described previously, such as large τ, Δx and Δy that result in a low degree of core overlap and a high dπ–π. Nevertheless, some degree of π-stacking is still important in this structure, although it is clear that the packing is also driven by other interactions. Finally, although the objects within the nostack and verylong-x families were always clustered by the SOMs, they differed from each other, especially those assigned to the nostack family. Thus, the structures given in Fig. 4e and f are meant to show the main stacking characteristics that are similar for all objects in the same family. In the nostack family, there is high variability in the parameters, although some objects have torsions or rotation, whereas others do not. In contrast, the objects in verylong-x were similar, apart from the few with high ρ and/or twist. In particular, two objects (CSD reference codes YIWMEY and XAPRIQ) had extremely high θ values (∼80°). In these cases, there was no π-stacking interactions between the perylene cores, because they were almost perpendicular to each other. Instead, a dipole–π interaction was observed between the perylene core and carbonyl oxygen of the closest molecule (Fig. S12†).
Some examples of common molecular packing motifs of the PDI crystal structures are shown in Fig. 5. The packing motifs in Fig. 5a and b were common for the tight-long-x family, and there was two ways in which their well-defined 1D stacking could be achieved in the structure, i.e. β-sheets (Fig. 5a) and γ-sheets (Fig. 5b). Another form of 1D stacking is shown in Fig. 5c, where the packing of a crisscross structure shows the crisscross of the molecules along the π-stacked columns. Fig. 5d shows a packing from the tight-long-y family, where its large Δy resulted in 2D packing, where the structure can be also be described as a β-sheet packing motif. Fig. 5e shows a typical 1D packing motif with a twisted core that was observed in the tight-twist family.
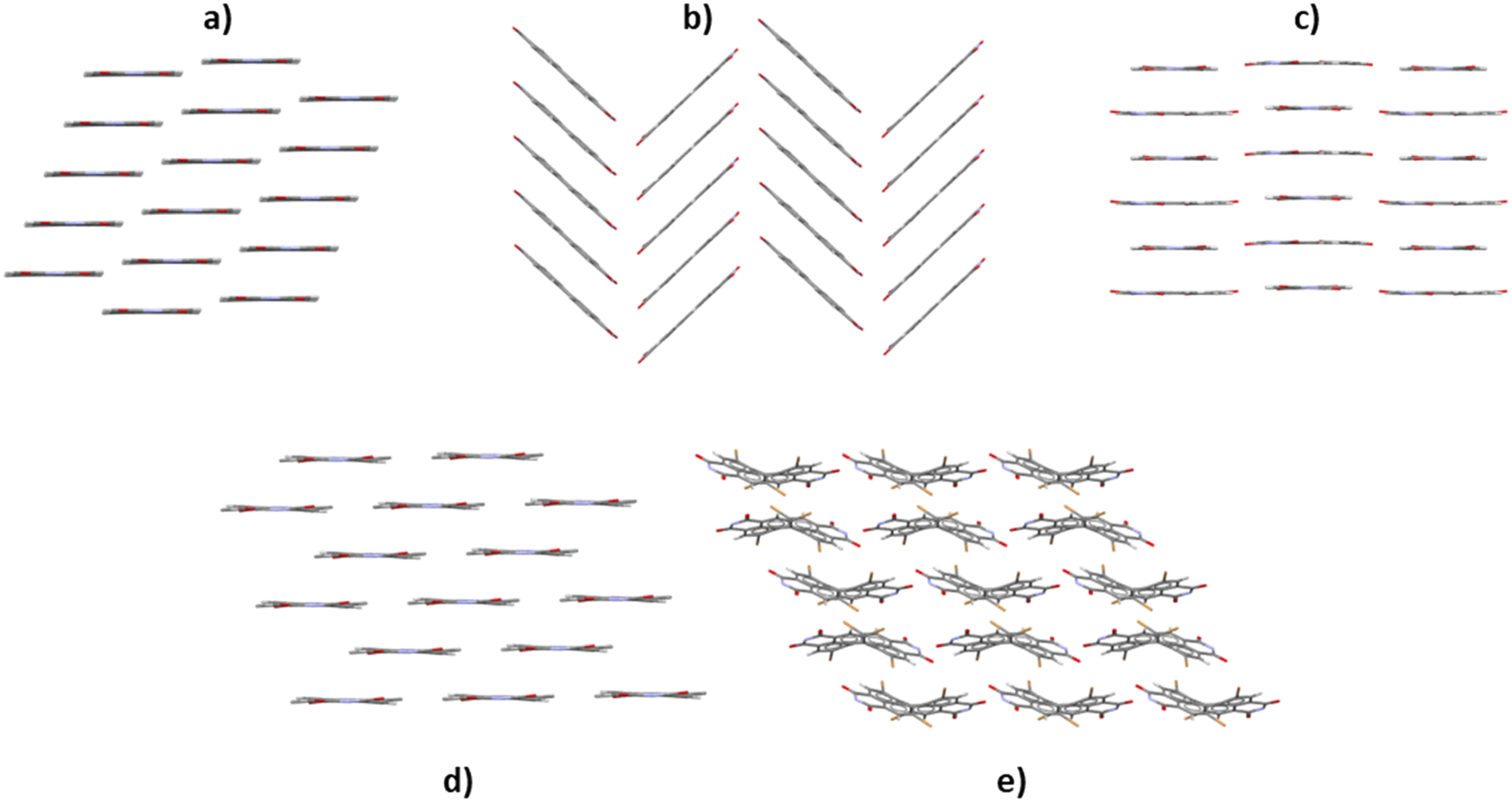 | ||
| Fig. 5 Common molecular packing motifs for the PDI crystal structures of each family: a) DICMUX, tight-long-x; b) DICNIM, tight-long-x; c) FEDPOU, crisscross; d) FEHROA, tight-long-y and e) OWOMEU, tight-twist. | ||
A limitation of this work is that the descriptors for the structures used to investigate the stacking interactions were calculated by considering the dimer of one molecule with the closest molecule. Therefore, few cases within the considered PDIs showed good π-stacking interactions within the dimer but weaker interactions with other neighbouring molecules owing to steric hindrance effects or other interactions caused by the presence of substituents. An example of such structures is shown in Fig. 6.
 | ||
| Fig. 6 Example of a crystal structure with good π-stacking interactions within the dimer, but weaker interactions with other neighbouring molecules. The crystal structure of BAMCAV is viewed along two perpendicular slices of the packed structure, described by three different objects that were clustered into the: a) verylong-x; b) crisscross or c) extra2 family. Substituents were omitted to aid readability. | ||
These results clearly show that for the studied PDIs, there are many different structures that can be achieved through different types of functionalisation, leading to a very rich landscape of possible packing arrangements and stacking interactions. The simple conventional description of the packing motif cannot fully describe such complexity. For example, Fig. 5a, c and d show structures that can all be described as β-sheets even though the stacking is quite different.
To investigate the role of different types of functionalisation in the packing of different PDI derivatives, we first investigated how different positions for substitution (i.e. imide, bay and ortho positions) can influence the packing and then how different types of substituents can determine into which group they are more likely to be classified.
First, we considered the PDIs substituted only in the imide position (68/142 objects). In these PDIs, two of the major families (tight-long-x and crisscross) are strongly preferred over the others. From these 68 objects, 29 were classified into the tight-long-x family and another 20 were in the crisscross family. This means that almost all the objects within these families (apart from two objects in tight-long-x and three in crisscross) were PDIs with only imide substitution. The third most important family for imide-substituted PDIs was the tight-long-y family with 7 objects, while the 12 remaining objects were divided between the other groups. Therefore, most of the imide objects were found in the top-right area of the scores plot where tight-long-x is located (Fig. 7) where the clusters are characterised by closer stacking between perylene cores.
 | ||
| Fig. 7 PC1–PC2 score plot obtained by PCA, with the imide-substituted objects categorised by the family they were assigned to colour. The core-substituted objects are coloured in grey. Most of the imide objects are at positive PC1 values, where the objects are characterised by closer stacking between perylene cores. | ||
We observed some patterns between the type of substituents at the imide position and the resulting stacking arrangement, and thus, the family they were assigned to. When the substituents are linear chains, especially long chains, they are most likely to have a packing arrangement characteristic of the tight-long-x group. Few PDIs with linear chains were assigned to different groups, and all of them have short chains (with one, two, three or five atoms). The structures with five atoms are various polymorphs of DICMUX that instead belong to the tight-long-x group. Therefore, we concluded that short chains allow more possible packing motifs than longer chains, maybe because the higher volume occupied by longer chains limits the interactions between PDI cores. For the same reason, short chains may be more prone to polymorphism.63 The outcome is different when a branched chain is used. It has been reported in the literature that the presence of branched chains causes the molecules to rotate along the stacking direction to reduce the steric hindrance between the chains.42,43 In fact, most of the imide objects substituted with branched chains had non-zero ρ, apart from one where the branching is not on the carbon atom bonded to the imide nitrogen. However, only five structures with branched chains are published, three of them were classified into in crisscross, one in verylong-x and the non-rotated one in the extra4 family. When an aromatic ring is present on the imide-substituted group, the possible structure depends on whether this ring is directly bonded to the imide nitrogen or there are atoms between them and the ring substitution. If the aromatic ring is directly linked to the nitrogen, typically it has a nearly 90° torsion angle with the PDI core; such conformation results in steric hindrance that enables the perylene cores to stack one above the other. Therefore, two outcomes driven by the tendency of the perylene unit to form close π-stacking interactions are possible: (1) the PDI molecules rotate along the stacking direction, similar to that for branched chains, resulting in the structure typical of the crisscross family or (2) the molecules are stacked with a higher Δy, resulting in the structure typical of the tight-long-y family (or extra3). This occurs except in the case in which the aromatic core is substituted at the ortho position with respect to the nitrogen, where steric hindrance does not allow close packing and the structure results in the nostack family or no classification. When the aromatic ring is not directly bonded to the hydrogen, there is no more steric effect and the resultant packing is usually that of the tight-long-x family, except when the aromatic ring is substituted with halogens (which in some cases produce interactions that result in other structures packing), or when the atom between the nitrogen and aromatic ring has other substituents (which produces steric hindrance that results in the structures of the tight-long-y or crisscross families). In conclusion, the most likely stacking arrangement can be predicted when substitution only in the imide position is concerned. However, substituents in this position are not able to tune the HOMO and LUMO energies of PDIs.31
In contrast, substituents at the core can efficiently tune HOMO and LUMO energies. However, when the substitution at the core positions is considered, the situation becomes more complicated. First, core substitution usually introduces functional groups in the PDIs that can result in different interactions that can compete with the π–π interactions and induce twisting of the perylene core. Furthermore, this is usually accompanied by substitution at the imide position, resulting in a wider variety of possible PDI structures. Of the core-substituted PDI structures studied here, 55 were bay-, 11 bay- and ortho- and 8 ortho-substituted. The PC1–PC2 score plot with only the core-substituted PDIs highlighted is shown in Fig. S13.†
Out of the 55 bay-substituted structures, 27 had substitution at only two positions, whereas 28 had substitution at all four bay positions. Most of the 28 objects (21/28) with four substituents at the bay positions had halogen atoms as functional groups, because bulky groups do not allow full bay functionalisation owing to steric effects. This substitution always causes increased τ in the core. In fact, the most likely family for these objects was tight-twist, which comprised 10/28 objects that had halogens at the core (F, Cl or Br), with linear chains or aromatic substituents at the imide position. The other objects with four halogens at the bay positions were assigned to the extra1 family (four with branched chains at the imide position) and tight-long-y family (4).
When only two bay positions are occupied, there is more possible variability of the substituent type, and for some substituents, the planarity of the core could be maintained. For these reasons, structures substituted in this way can be found in all major families; thus, it is difficult to predict the outcome of this type of substitution. Although the number of structures with these characteristics is too small to make strong hypotheses, some patterns between the objects were observed. First, the crisscross objects of this type are mostly structures that show this packing within the dimer and not in all the structure, so it is not a likely family. When the substituents allow the planarity to be maintained, e.g. halogens or cyano groups, the structures formed were typical of the tight-long-x, tight-long-y or other families dependent on imide substitution. Otherwise, when the core is twisted, objects with tight-twist and verylong-x characteristics were obtained.
Unfortunately, there were only few structures with ortho functionalisation in the CSD with the characteristics included in this investigation. Nevertheless, we observed that the few structures substituted in both the bay and ortho positions result mostly in tight-twist and verylong-x families, as expected from their highly twisted cores and the different competing interactions provided by the substituents. In contrast, when only ortho-substituted PDIs are considered, the planarity of the core is generally maintained, or a slight twist is observed. Therefore, the few objects with only ortho substitution do not belong to either the tight-twist or verylong-x families but were assigned to other families such as tight-long-y, extra4 or extra5.
Considering the stacking results with different positions and types of substituents, in the case of only imide functionalisation, the most likely stacking arrangement of the PDI molecules in the crystal structure can be predicted to some extent, because the molecular packing is mainly driven by the formation of closed π–π stacking interactions. However, considering substitution in the bay and ortho positions, the formation of π–π stacking interactions must compete with the formation of other interactions resulting from the substituents at the core. Thus, the high variety of different substituent and interaction combinations makes clustering (and hence structural predictions) more difficult and less efficient. Moreover, the number of structures deposited in the CSD of similar PDI derivatives is not yet sufficient to perform efficient data-driven clustering of core-substituted structures, especially in the case of ortho substitution.
Apart from the different substitutions, polymorphism can result in different stacking arrangements. However, despite it being known that PDIs can exhibit polymorphism, information about different PDI polymorphs in the CSD is very limited. Only five PDIs investigated in this study have polymorphs in the CSD, giving a total of 12 structures (of the 103 structures investigated). Nevertheless, two of the polymorphic PDIs had objects for different polymorphs that were assigned to different major families, showing that it is possible to obtain a new stacking arrangement via polymorphism. Investigating polymorphic PDI derivatives, and in general polymorphism of OSCs, could be beneficial to both experimental and theoretical work to identify OSCs with good semiconducting properties, design novel semiconductors and increase the efficiency of calculating the structure and properties of semiconducting materials.
Conclusions
In this work, we successfully applied a chemometric method based on SOMs to PDI-derivative crystal structures with similar stacking arrangements deposited in the CSD. We observed that if PDIs are substituted only at the imide position, the packing is mainly driven by the formation of closed π–π stacking interactions, where the most-probable stacking arrangement can be predicted to some extent. However, in the case of bay and ortho substitution, the observed packing is the result of the competition between the interactions due to the substituents and the formation of π–π stacking interactions with the perylene core. Therefore, introducing substituents at the core, despite being beneficial for tuning HOMO and LUMO energies, makes it more difficult to predict the possible packing outcome and can be detrimental to achieving stacking interactions appropriate for efficient charge transport. Unfortunately, the lack of crystal structures in the CSD, especially considering polymorphs, hinder the efficiency of data-driven methods and the subsequent design of novel materials with desired packing and properties. However, respect to the traditional visual classification in four families, the proposed method was able to identify six major families and several structures that cannot univocally be classified in one of these, demonstrating that the classification of PDI-derivatives is not trivial and requires mathematical tools to be carried out. The SOM method, although beyond the scope of the present work, can be also used to predict the family of a new PDI-derivative by calculating the variables used to create the model and projecting the new object onto the model. Therefore, the presented model can help in future developments by indicating to which, if any, family would pertain a new synthetized, or predicted in silico, PDI structures.Experimental section
Dataset
The search for PDI derivatives structures in CSD resulted in more than 300 structures. From these structures, only organic ones were selected. Furthermore, we discarded solvates, polymers, duplicated structures (for structures collected under different conditions, the structure with the best R1 factor and/or lower temperature of data collection was chosen) and PDIs with extended fused aromatic cores. Furthermore, we considered PDIs substituted at the imide, bay or ortho positions. Since the focus of this work was the π-stacking interactions between the perylene cores of the PDI molecule, we limited the type of substituent considered in this study. At imide positions, we considered only linear chains, branched chains (even though very bulky groups were excluded), chains containing small cyclic groups and aromatic rings, and halogen-substituted aromatics and chains. The choice of substituents at the core (ortho and bay positions) was even more strict, and only halogens, cyano groups, short chains and small cyclic groups and aromatic rings were considered.In this way, a total of 103 structures of PDI derivatives were selected, out of which 53 were substituted only at the imide position (apart from one with no substitution) and 50 were substituted at the perylene core. Of the 50 core-substituted PDIs, 35 were substituted at the bay position, 6 at the ortho position and 9 at both the bay and ortho positions. Of these 50 core-substituted PDI structures, only 3 did not have substitution at the imide position.
From the 103 PDI crystal structures evaluated, 142 different molecules (objects) were obtained for analysis. The difference in the number of crystal structures and objects is due to the presence of structures with Z′ > 1 (more than one molecule in asymmetric unit), or with Z′ = 1 but with two halves of PDI molecules in the asymmetric unit. In these cases, the PDI molecules form at least two different dimers which have been independently described.
Descriptors
The descriptors or variables used for the analysis were either numerical or categorical variables. Numerical variables are used by the algorithm to perform clustering, while categorical variables do not have a numeric value but are used to describe specific characteristics of the structure and the molecule in detail to find patterns between the clustering and object characteristics. The numerical variables included Z, Z′, Vmol, τ, S, S/L, M/L, SV, dπ–π, χ, ψ, Δx, Δy, P, R, θ and ρ (Fig. 8). The category variables included temperature, substituent position, substituent at the imide position, substituent at the core and packing type. Out of these variables, temperature, Z and Z′ were used to give information about the crystal structures and were easily retrieved from information in the CSD. The other variables were not available as simple information from CSD and were hence calculated using the software Mercury61 of the CCDC package. The variables Vmol, substituent position, substituent at the imide position, substituent at the core, τ, S, S/L and M/L are descriptors used to give information about the molecule, whereas SV, dπ–π, χ, ψ, Δx, Δy, P, R, θ, ρ and packing type are variables that give information about the packing of the PDIs and were calculated by considering the stacking between a dimer formed by two neighbouring PDI molecules. | ||
| Fig. 8 Visual representation of some of the descriptors used to describe the crystal packing: a) rotation angle ρ; b) SV, Δx and Δy; c) tilt angle θ; d) torsion angle τ; e) pitch angle P; f) roll angle R. | ||
 | (1) |
Principal component analysis
PCA65 is one of the most important chemometric methods. The mathematical procedure can be summarised as a rotation of the original variables of the dataset to convert them into new variables, called principal components (PCs). The PCs are orthogonal to each other and oriented in the most informative directions for the data. In this way, the coordinates of the first two or three PCs of objects and variables can be used to describe, with good approximation, the entire behaviour of the data. Each PC carries a percentage of information (explained variance) that can be used to check if enough information is maintained in the considered PCs. Therefore, the two main results of PCA are a score plot and loading plot. The score plot shows the behaviour of the objects, indicating possible groups (objects close to each other) and outliers (objects far from all others), while the loading plot shows the behaviour of variables, indicating correlation (variable loadings close to each other) or anti-correlation (loadings at the opposite sides of the graph).Self-organising maps
SOMs56 were used in the present work to find patterns in the crystal structures. The SOM method is a multivariate analysis technique based on artificial neural networks57 that calculates clusters from the dataset objects in an iterative way. The output of the SOM computation can be viewed as a 2D rectangular map divided into several adjacent circles (or hexagons), each of which represents a SOM unit in which some objects of the starting dataset are aggregated. The starting point of SOM computation is the definition of the number of units and the size of the map. Some empirical rules have been proposed to define such parameters, such as that reported by Nakagawa et al.59 In that work, the number of SOM units was chosen as that closest to five times the square root of the number of objects, while the dimensions of the rectangle were chosen in proportion to the first two eigenvalues of a PCA performed on the original data.The SOM computation starts from a random point once the map dimensions are defined. Each SOM unit is actually a vector with length equal to the number of original variables and the first computation step has random values for these vectors. Each object is presented to each vector, the Euclidean distance between object and vector is calculated, and the object is assigned to the closest unit. Once all objects have been assigned, the second step starts using the mean of the unit vectors from the previously assigned objects and repeats the same procedure of presenting the objects to the units and assigning them to the closest one. This procedure is repeated for a pre-determined number of steps (called epochs) or until a convergence is reached, which means that the results do not change for two or three consecutive epochs. Finally, each unit of the map represents a ‘cluster’ of starting objects. The units can be further grouped by performing a cluster analysis66 on the final vectors. In this way, a lower number of clusters is calculated (generally five to eight clusters starting from at least dozens of units), simplifying the further considerations.
However, there does not exist a general rule to define the map dimensions and optimal number of clusters, and the final result may depend on such choices. Therefore, for the present work, we performed six SOM computations with different map dimensions and using the Davies–Bouldin index58 to determine the optimal number of clusters. The results of the SOMs were then compared to evaluate the best families of crystal structures that can be derived from the dataset. SOM computations were performed using the package SOMEnv57 of the R environment (R Core Team, Vienna, Austria). A deepen description of SOM procedure and an example of the results obtained are reported in the ESI† (Fig. S14–S17).
Author contributions
Conceptualization, L. M., D. M.; methodology, F. M., A. Z.; software, F. M., A. Z.; validation, L. M.; formal analysis F. M., A. Z.; investigation, F. M.; resources, L. M., D. M.; data processing, A. Z.; writing – original draft preparation, F. M.; writing – review & editing, A. Z., L. M.; visualization, D. M.; supervision, L. M., D. M.; project administration, L. M.Conflicts of interest
There are no conflicts to declare.References
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Olivier, R. Silbey and J. L. Brédas, Charge Transport in Organic Semiconductors, Chem. Rev., 2007, 926–952 CrossRef CAS.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, N-Type Organic Semiconductors in Organic Electronics, Adv. Mater., 2010, 22(34), 3876–3892 CrossRef CAS PubMed.
- L. Ferlauto, F. Liscio, E. Orgiu, N. Masciocchi, A. Guagliardi, F. Biscarini, P. Samorì and S. Milita, Enhancing the Charge Transport in Solution-Processed Perylene Di-Imide Transistors via Thermal Annealing of Metastable Disordered Films, Adv. Funct. Mater., 2014, 24(35), 5503–5510 CrossRef CAS.
- O. Ostroverkhova, Organic Optoelectronic Materials: Mechanisms and Applications, Chem. Rev., 2016, 116(22), 13279–13412 CrossRef CAS PubMed.
- C. Wang, H. Dong, L. Jiang and W. Hu, Organic Semiconductor Crystals, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- C. W. Tang and S. A. Vanslyke, Organic Electroluminescent Diodes, Appl. Phys. Lett., 1987, 51(12), 913–915 CrossRef CAS.
- W. Brütting and J. Frischeisen, Device Efficiency of Organic Light-Emitting Diodes, in Physics of Organic Semiconductors, 2nd edn, 2013, vol. 210, pp. 497–539 Search PubMed.
- A. Facchetti, Semiconductors for Organic Transistors, Mater. Today, 2007, 10(3), 28–37 CrossRef CAS.
- J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, Integrated Materials Design of Organic Semiconductors for Field-Effect Transistors, J. Am. Chem. Soc., 2013, 6724–6746 CrossRef CAS PubMed.
- S. Chen, Z. Li, Y. Qiao and Y. Song, Solution-Processed Organic Semiconductor Crystals for Field-Effect Transistors: From Crystallization Mechanism towards Morphology Control, J. Mater. Chem. C, 2021, 1126–1149 RSC.
- B. Kippelen and J. L. Brédas, Organic Photovoltaics, Energy Environ. Sci., 2009, 251–261 RSC.
- J. E. Anthony, Small-Molecule, Nonfullerene Acceptors for Polymer Bulk Heterojunction Organic Photovoltaics, Chem. Mater., 2011, 23(3), 583–590 CrossRef CAS.
- C. Li and H. Wonneberger, Perylene Imides for Organic Photovoltaics: Yesterday, Today, and Tomorrow, Adv. Mater., 2012, 24(5), 613–636 CrossRef CAS.
- J. E. Anthony, Organic Electronics: Addressing Challenges, Nat. Mater., 2014, 773–775 CrossRef CAS PubMed.
- S. Ando, R. Murakami, J. I. Nishida, H. Tada, Y. Inoue, S. Tokito and Y. Yamashita, N-Type Organic Field-Effect Transistors with Very High Electron Mobility Based on Thiazole Oligomers with Trifluoromethylphenyl Groups, J. Am. Chem. Soc., 2005, 127(43), 14996–14997 CrossRef CAS PubMed.
- K. Takimiya, I. Osaka, T. Mori and M. Nakano, Organic Semiconductors Based on [1]Benzothieno[3,2- B ][1]Benzothiophene Substructure, Acc. Chem. Res., 2014, 47(5), 1493–1502 CrossRef CAS PubMed.
- C. Huang, S. Barlow and S. R. Marder, Perylene-3,4,9,10-Tetracarboxylic Acid Diimides: Synthesis, Physical Properties, and Use in Organic Electronics, J. Org. Chem., 2011, 2386–2407 CrossRef CAS.
- M. Kardos, German Pat., DE276357, 1913 Search PubMed.
- P. M. Kazmaier and R. Hoffmann, A Theoretical Study of Crystallochromy. Quantum Interference Effects in the Spectra of Perylene Pigments, J. Am. Chem. Soc., 1994, 116(21), 9684–9691 CrossRef CAS.
- D. Bialas, E. Kirchner, M. I. S. Röhr and F. Würthner, Perspectives in Dye Chemistry: A Rational Approach toward Functional Materials by Understanding the Aggregate State, J. Am. Chem. Soc., 2021, 4500–4518 CrossRef CAS PubMed.
- B. A. Jones, A. Facchetti, M. R. Wasielewski and T. J. Marks, Tuning Orbital Energetics in Arylene Diimide Semiconductors. Materials Design for Ambient Stability of n-Type Charge Transport, J. Am. Chem. Soc., 2007, 129(49), 15259–15278 CrossRef CAS PubMed.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Rylene and Related Diimides for Organic Electronics, Adv. Mater., 2011, 23(2), 268–284 CrossRef CAS PubMed.
- C. Huang, M. M. Sartin, N. Siegel, M. Cozzuol, Y. Zhang, J. M. Hales, S. Barlow, J. W. Perry and S. R. Marder, Photo-Induced Charge Transfer and Nonlinear Absorption in Dyads Composed of a Two-Photon-Absorbing Donor and a Perylene Diimide Acceptor, J. Mater. Chem., 2011, 21(40), 16119–16128 RSC.
- I. F. A. Mariz, S. Raja, T. Silva, S. Almeida, É. Torres, C. Baleizão and E. Maçôas, Two-Photon Absorption of Perylene-3,4,9,10-Tetracarboxylic Acid Diimides: Effect of Substituents in the Bay, Dyes Pigm., 2021, 193, 109470 CrossRef CAS.
- Q. Al-Galiby, I. Grace, H. Sadeghi and C. J. Lambert, Exploiting the Extended π-System of Perylene Bisimide for Label-Free Single-Molecule Sensing, J. Mater. Chem. C, 2015, 3(9), 2101–2106 RSC.
- T. Ribeiro, S. Raja, A. S. Rodrigues, F. Fernandes, J. P. S. Farinha and C. Baleizão, High Performance NIR Fluorescent Silica Nanoparticles for Bioimaging, RSC Adv., 2013, 3(24), 9171–9174 RSC.
- L. M. Cowen, J. Atoyo, M. J. Carnie, D. Baran and B. C. Schroeder, Review—Organic Materials for Thermoelectric Energy Generation, ECS J. Solid State Sci. Technol., 2017, 6(3), N3080–N3088 CrossRef CAS.
- G. Klebe, F. Graser, E. Hädicke and J. Berndt, Crystallochromy as a Solid-state Effect: Correlation of Molecular Conformation, Crystal Packing and Colour in Perylene-3,4:9,10-bis(Dicarboximide) Pigments, Acta Crystallogr., Sect. B: Struct. Sci., 1989, 45(1), 69–77 CrossRef.
- H. Langhals, Cyclic Carboxylic Imide Structures as Structure Elements of High Stability. Novel Developments in Perylene Dye Chemistry, Heterocycles, 1995, 40(1), 477–500 CrossRef CAS.
- K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao and L. Zang, Effect of Side-Chain Substituents on Self-Assembly of Perylene Diimide Molecules: Morphology Control, J. Am. Chem. Soc., 2006, 128(22), 7390–7398 CrossRef CAS PubMed.
- M. Carmen Ruiz Delgado, E. G. Kim, D. A. Da Silva Filho and J. L. Bredas, Tuning the Charge-Transport Parameters of Perylene Diimide Single Crystals via End and/or Core Functionalization: A Density Functional Theory Investigation, J. Am. Chem. Soc., 2010, 132(10), 3375–3387 CrossRef PubMed.
- Y. Geng, H.-B. Li, S.-X. Wu and Z.-M. Su, The Interplay of Intermolecular Interactions, Packing Motifs and Electron Transport Properties in Perylene Diimide Related Materials: A Theoretical Perspective, J. Mater. Chem., 2012, 22(39), 20840 RSC.
- F. Würthner, Perylene Bisimide Dyes as Versatile Building Blocks for Functional Supramolecular Architectures, Chem. Commun., 2004,(14), 1564–1579 RSC.
- B. A. Jones, M. J. Ahrens, M. H. Yoon, A. Facchetti, T. J. Marks and M. R. Wasielewski, High-Mobility Air-Stable n-Type Semiconductors with Processing Versatility: Dicyanoperylene-3,4:9,10-Bis(Dicarboximides), Angew. Chem., Int. Ed., 2004, 43(46), 6363–6366 CrossRef CAS PubMed.
- S. Nakazono, S. Easwaramoorthi, D. Kim, H. Shinokubo and A. Osuka, Synthesis of Arylated Perylene Bisimides through C - H Bond Cleavage under Ruthenium Catalysis, Org. Lett., 2009, 11(23), 5426–5429 CrossRef CAS.
- F. Graser and E. Hädicke, Kristallstruktur Und Farbe Bei Perylen-3,4:9,10-Bis(Dicarboximid)-Pigmenten, Liebigs Ann. Chem., 1980, 1980(12), 1994–2011 CrossRef.
- R. F. Fink, J. Seibt, V. Engel, M. Renz, M. Kaupp, S. Lochbrunner, H. M. Zhao, J. Pfister, F. Würthner and B. Engels, Exciton Trapping in π-Conjugated Materials: A Quantum-Chemistry-Based Protocol Applied to Perylene Bisimide Dye Aggregates, J. Am. Chem. Soc., 2008, 130(39), 12858–12859 CrossRef CAS PubMed.
- H. M. Zhao, J. Pfister, V. Settels, M. Renz, M. Kaupp, V. C. Dehm, F. Würthner, R. F. Fink and B. Engels, Understanding Ground- and Excited-State Properties of Perylene Tetracarboxylic Acid Bisimide Crystals by Means of Quantum Chemical Computations, J. Am. Chem. Soc., 2009, 131(43), 15660–15668 CrossRef CAS PubMed.
- J. Vura-Weis, M. A. Ratner and M. R. Wasielewski, Geometry and Electronic Coupling in Perylenediimide Stacks: Mapping Structure - Charge Transport Relationships, J. Am. Chem. Soc., 2010, 132(6), 1738–1739 CrossRef CAS.
- A. V. Mumyatov, L. I. Leshanskaya, D. V. Anokhin, N. N. Dremova and P. A. Troshin, Organic Field-Effect Transistors Based on Disubstituted Perylene Diimides: Effect of Alkyl Chains on the Device Performance, Mendeleev Commun., 2014, 24(5), 306–307 CrossRef CAS.
- V. Belova, B. Wagner, B. Reisz, C. Zeiser, G. Duva, J. Rozbořil, J. Novák, A. Gerlach, A. Hinderhofer and F. Schreiber, Real-Time Structural and Optical Study of Growth and Packing Behavior of Perylene Diimide Derivative Thin Films: Influence of Side-Chain Modification, J. Phys. Chem. C, 2018, 122(15), 8589–8601 CrossRef CAS.
- M. R. Hansen, R. Graf, S. Sekharan and D. Sebastiani, Columnar Packing Motifs of Functionalized Perylene Derivatives: Local Molecular Order despite Long-Range Disorder, J. Am. Chem. Soc., 2009, 131(14), 5251–5256 CrossRef CAS.
- F. May, V. Marcon, M. R. Hansen, F. Grozema and D. Andrienko, Relationship between Supramolecular Assembly and Charge-Carrier Mobility in Perylenediimide Derivatives: The Impact of Side Chains, J. Mater. Chem., 2011, 21(26), 9538–9545 RSC.
- R. Ahmed and A. K. Manna, Theoretical Insights on Tunable Optoelectronics and Charge Mobilities in Cyano-Perylenediimides: Interplays between -CN Numbers and Positions, Phys. Chem. Chem. Phys., 2021, 23(27), 14687–14698 RSC.
- Y. Geng, J. Wang, S. Wu, H. Li, F. Yu, G. Yang, H. Gao and Z. Su, Theoretical Discussions on Electron Transport Properties of Perylene Bisimide Derivatives with Different Molecular Packings and Intermolecular Interactions, J. Mater. Chem., 2011, 21(1), 134–143 RSC.
- X. Shang, J. Ahn, J. H. Lee, J. C. Kim, H. Ohtsu, W. Choi, I. Song, S. K. Kwak and J. H. Oh, Bay-Substitution Effect of Perylene Diimides on Supramolecular Chirality and Optoelectronic Properties of Their Self-Assembled Nanostructures, ACS Appl. Mater. Interfaces, 2021, 13(10), 12278–12285 CrossRef CAS PubMed.
- G. Gryn’Ova, K. H. Lin and C. Corminboeuf, Read between the Molecules: Computational Insights into Organic Semiconductors, J. Am. Chem. Soc., 2018, 140(48), 16370–16386 CrossRef.
- J. Cornil, S. Verlaak, N. Martinelli, A. Mityashin, Y. Olivier, T. Van Regemorter, G. D'Avino, L. Muccioli, C. Zannoni, F. Castet, D. Beljonne and P. Heremans, Exploring the Energy Landscape of the Charge Transport Levels in Organic Semiconductors at the Molecular Scale, Acc. Chem. Res., 2013, 46(2), 434–443 CrossRef CAS PubMed.
- A. Landi and A. Troisi, Rapid Evaluation of Dynamic Electronic Disorder in Molecular Semiconductors, J. Phys. Chem. C, 2018, 122(32), 18336–18345 CrossRef CAS.
- J. E. Campbell, J. Yang and G. M. Day, Predicted Energy-Structure-Function Maps for the Evaluation of Small Molecule Organic Semiconductors, J. Mater. Chem. C, 2017, 5(30), 7574–7584 RSC.
- J. Yang, S. De, J. E. Campbell, S. Li, M. Ceriotti and G. M. Day, Large-Scale Computational Screening of Molecular Organic Semiconductors Using Crystal Structure Prediction, Chem. Mater., 2018, 30(13), 4361–4371 CrossRef CAS.
- F. Musil, S. De, J. Yang, J. E. Campbell, G. M. Day and M. Ceriotti, Machine Learning for the Structure-Energy-Property Landscapes of Molecular Crystals, Chem. Sci., 2018, 9(5), 1289–1300 RSC.
- C. Schober, K. Reuter and H. Oberhofer, Virtual Screening for High Carrier Mobility in Organic Semiconductors, J. Phys. Chem. Lett., 2016, 7(19), 3973–3977 CrossRef CAS PubMed.
- T. Nematiaram, D. Padula, A. Landi and A. Troisi, On the Largest Possible Mobility of Molecular Semiconductors and How to Achieve It, Adv. Funct. Mater., 2020, 30(30), 2001906 CrossRef CAS.
- D. W. M. Hofmann and L. N. Kuleshova, Data Mining in Crystallography, Springer Publishing Company, Incorporated, 1st edn, 2009 Search PubMed.
- T. Kohonen, The Self-Organizing Map, Neurocomputing, 1998, 21(1), 1–6 CrossRef.
- S. Licen, M. Franzon, T. Rodani and P. Barbieri, SOMEnv: An R Package for Mining Environmental Monitoring Datasets by Self-Organizing Map and k-Means Algorithms with a Graphical User Interface, Microchem. J., 2021, 165, 106181 CrossRef CAS.
- D. L. Davies and D. W. Bouldin, A Cluster Separation Measure, IEEE Trans. Pattern Anal. Mach. Intell., 1979, PAMI-1(2), 224–227 Search PubMed.
- K. Nakagawa, Z.-Q. Yu, R. Berndtsson and T. Hosono, Temporal Characteristics of Groundwater Chemistry Affected by the 2016 Kumamoto Earthquake Using Self-Organizing Maps, J. Hydrol., 2020, 582, 124519 CrossRef CAS.
- C. A. Hunter and J. K. M. Sanders, The Nature of π-π Interactions, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- C. F. MacRae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, Mercury 4.0: From Visualization to Analysis, Design and Prediction, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- L. Fábián, Cambridge Structural Database Analysis of Molecular Complementarity in Cocrystals, Cryst. Growth Des., 2009, 9(3), 1436–1443 CrossRef.
- S. Milita, F. Liscio, L. Cowen, M. Cavallini, B. A. Drain, T. Degousée, S. Luong, O. Fenwick, A. Guagliardi, B. C. Schroeder and N. Masciocchi, Polymorphism in N , N ′-Dialkyl-Naphthalene Diimides, J. Mater. Chem. C, 2020, 8(9), 3097–3112 RSC.
- M. D. Curtis, J. Cao and J. W. Kampf, Solid-State Packing of Conjugated Oligomers: From π-Stacks to the Herringbone Structure, J. Am. Chem. Soc., 2004, 126(13), 4318–4328 CrossRef CAS PubMed.
- R. Bro and A. K. Smilde, Principal Component Analysis, Anal. Methods, 2014, 6(9), 2812–2831 RSC.
- B. S. Everitt, S. Landau and M. Leese, Cluster Analysis, Wiley Publishing, 5th edn, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2me00240j |
| This journal is © The Royal Society of Chemistry 2023 |