DOI:
10.1039/D3MD00350G
(Research Article)
RSC Med. Chem., 2024,
15, 119-126
Physicochemical characterization of B-hydroxyphenyl phosphine borane derivatives and their evaluation as nuclear estrogen receptor ligands†
Received
19th July 2023
, Accepted 27th September 2023
First published on 2nd October 2023
Abstract
Increasing the structural options in medicinal chemistry is a promising approach to develop new drug candidates. In this research, we designed and synthesized a series of B-hydroxyphenyl phosphine borane derivatives and investigated their structure–property and structure–activity relationships. The synthesized B-phenylphosphine borane derivatives exhibited sufficient stability in aqueous media, weaker hydrophobicity than the corresponding alkanes and silanes, and sufficient affinity for lipid membranes to enable permeability. Several B-hydroxyphenyl phosphine borane derivatives exhibited significant estrogen receptor (ER) agonistic activity with superior ligand-lipophilicity efficiency (LLE). The phosphine borane framework appears to be a promising option for structural development in drug discovery studies.
1. Introduction
The introduction of new structural options in medicinal chemistry by applying an elements chemistry approach is considered to be an effective tool for drug discovery.1 For example, the boron atom in the anticancer agent bortezomib reversibly forms a specific covalent bond with the catalytic site of 26S proteasome.2 In addition, phosphates, phosphoramidates, phosphonates and phosphonamides have been widely used in medicinal chemistry as components of nucleic acid derivatives, as pharmacophores, and in prodrug structures to improve solubility, though other phosphorus-containing functional groups, such as phosphinic acid and related functionalities, are less familiar in drug discovery research.3 Indeed, fosinopril (1), an angiotensin converting enzyme (ACE) inhibitor, is the only phosphinate-containing drug to have been approved so far.4 There are also a few phosphine-based drugs, such as fosazepam (2) and brigatinib (3) (Fig. 1).5,6 However, phosphorus can form stable bonds with multiple elements, including sulfur and boron, as well as carbon, oxygen and nitrogen. In particular, P–B substructures can be regarded as isosteric with alkanes because of their tetrahedral sp3–sp3 character, and therefore phosphine borane derivatives might provide a variety of chemical options for structural development in drug discovery research. In the present study, we focused on phosphine boranes as novel building blocks to expand the chemical space available to medicinal chemists.
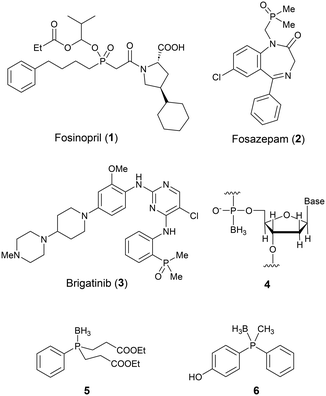 |
| | Fig. 1 Examples of pharmaceuticals bearing phosphinic acid or phosphine-related functionalities (1–3) and structures of functional molecules bearing a P–B bond (4–6). | |
Phosphine borane adducts are already utilised as synthetic intermediates of phosphines: the borane moiety functions as a protective group for oxidation-susceptive lone pairs.7 Boranophosphate-type nucleic acids (4) in which the phosphorus–oxygen double bond of the phosphate moiety is replaced by a phosphorus–boron bond have been investigated with the aim of improving the stability of nucleic acid pharmaceuticals.8 Levin and co-workers have developed phosphinoborane derivatives such as compound 5 as neuroprotective agents.9 We recently developed phosphinophenol-based estrogen receptor (ER) ligands such as 6 and found that the P–BH3 substructure functions as a less hydrophobic methyl isostere.10 In contrast, the application of B-substituted phosphine boranes to expand the chemical space in drug discovery has not been well explored, despite their synthetic accessibility and potential to provide a wide variety of analogs of alkane derivatives. Therefore, the aim of the present work was to design, synthesize and characterize a range of B-substituted phosphine borane derivatives in order to investigate their potential for expanding the medicinal chemists' toolbox for drug discovery.
2. Results and discussion
2.1. Molecular design
The phenyl group is the most common ring system in drug discovery research, and we considered that the B-phenyl phosphine borane core structure could be a versatile scaffold for development of novel drug candidates. We therefore designed a series of B-hydroxyphenyl phosphine boranes. The phenolic hydroxyl group in the designed compounds was expected to provide appropriate hydrophobicity,11 and it should be possible to investigate the electronic characteristics of the phosphinoboranyl (PR3–BH2–) substituents by monitoring the pKa of the phenolic hydroxyl group. Since 3- or 4-substituted phenol is a pharmacophore of ER ligands,11,12 we firstly designed a series of B-(4-hydroxyphenyl)phosphine boranes and B-(3-hydroxyphenyl)phosphine boranes 7–16 for evaluation of the structure–property relationship, including comparison with the corresponding alkanes and silanes. We also designed compounds 17–22 bearing bulky substituents, tri-i-propylphosphine, tri-n-butylphosphine and tricyclohexylphosphine, to evaluate these distinctive molecular structures, since preparation of the corresponding carbon analogs bearing four bulky substituents is generally difficult (Fig. 2).
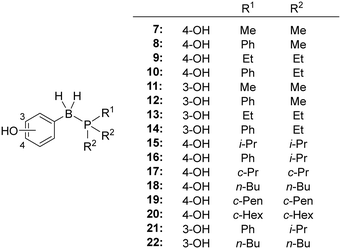 |
| | Fig. 2 Structures of the designed B-hydroxyphenyl phosphine borane derivatives. | |
2.2. Synthesis
The designed B-hydroxyphenyl phosphine borane derivatives 7–22 were synthesized from the corresponding phosphine and hydroxyphenylboronic acid under reductive conditions.13 The alkane analogue 4-(2,2-dimethylpropyl)phenol 23 is commercially available. Alkane analogue 26 was synthesized by alkylation of silyl-protected bromophenol 24 followed by removal of the silyl group. The silicon analogs 30 and 31 were synthesized from 4-benzyloxybenzyl bromide 27. Silylation of 27via Grignard reaction gave silanes 28 and 29, and removal of the protecting group afforded the desired compounds 30 and 31, respectively (Scheme 1).
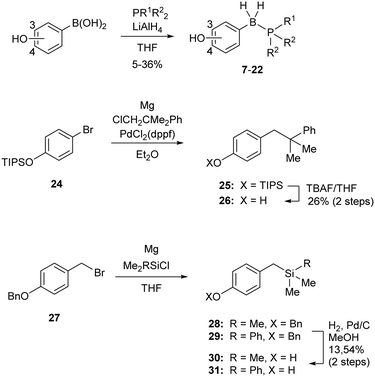 |
| | Scheme 1 Synthesis of the designed B-hydroxyphenyl phosphine borane derivatives 7–22, the corresponding alkane analogue 26 and silane analogues 30 and 31. | |
2.3. Stability in aqueous solution
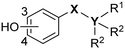
Since the stability of B-phenyl phosphine boranes in neutral aqueous solution has not been investigated, we firstly examined the stability of the compounds in pH 7.4 phosphate-buffered saline (PBS). Table 1 summarizes the stability of phosphine boranes 7–14, reference alkanes 23, 26, and silanes 30–31 in neutral PBS. All the tested phosphine borane derivatives remained more than 94% intact after 3 h in neutral PBS at 37 °C. B-(4-Hydroxyphenyl)dimethylphenylphosphine borane (8) and B-(4-hydroxyphenyl)diethylphenylphosphine borane (10) were slightly less stable than the other phosphine borane derivatives. In the stability test of phosphine borane derivative 10, a mass spectrum which appeared to be diethylphenylphosphine oxide was observed. We estimated the phosphine oxide came from the cleavage of P–B bond and oxidation. The P–B bond in those compounds might be weakened by the electron-donating character of the 4-hydroxyphenyl group and the lower coordinating ability of dialkylphenylphosphines compared with trialkylphosphines.
Table 1 Residual ratio of tested compounds determined by HPLC after 3 h in pH 7.4 PBS at 37 °C
|
|
| Cmpd |
X–Y |
|
R1 |
R2 |
Residual ratio (% remaining) |
|
7
|
BH2–P |
4-OH |
Me |
Me |
99.9 |
|
8
|
Ph |
Me |
94.5 |
|
9
|
Et |
Et |
99.3 |
|
10
|
Ph |
Et |
97.6 |
|
11
|
3-OH |
Me |
Me |
100 |
|
12
|
Ph |
Me |
99.0 |
|
13
|
Et |
Et |
99.7 |
|
14
|
Ph |
Et |
100 |
|
23
|
CH2–C |
4-OH |
Me |
Me |
92.5 |
|
26
|
Ph |
Me |
99.0 |
|
30
|
CH2–Si |
Me |
Me |
89.9 |
|
31
|
Ph |
Me |
96.2 |
2.4. Hydrophobicity
Hydrophobicity is a key determinant of the activity, pharmacokinetics and toxicity of biologically active compounds. We measured the hydrophobicity of our compounds as the octanol–water partition coefficient (P) using an HPLC method.14Table 2 summarizes the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values and the Hansch–Fujita hydrophobicity parameter π of the substituents, calculated by subtracting the logP value of phenol (1.46)15 from the observed logP value. B-(4-Hydroxyphenyl)trimethylphosphine borane (7) exhibited a logP value of 2.44, which is smaller than those of the corresponding carbon analogue 23 and silicon analogue 30 (4.36 and 4.91, respectively). In the case of dimethylphenyl analogs, phosphine borane 8 exhibited a logP value of 3.18, which is smaller than those of the corresponding carbon analogue 26 and silicon analogue 31 (5.26 and 5.74, respectively). These results indicate that the B-substituted phosphine boranes and P–BH3 derivatives are less hydrophobic than the corresponding alkanes or silanes. Thus, phosphine boranes could be useful for building sp3–sp3 frameworks with reduced hydrophobicity. Comparison of 7 and 8 or 11 and 12 indicates that switching a P-methyl group to a P-phenyl group increases the hydrophobicity with a difference in the logP values of about 0.7. This difference is similar to that in the case of alkanes and silanes.11
P values and the Hansch–Fujita hydrophobicity parameter π of the substituents, calculated by subtracting the logP value of phenol (1.46)15 from the observed logP value. B-(4-Hydroxyphenyl)trimethylphosphine borane (7) exhibited a logP value of 2.44, which is smaller than those of the corresponding carbon analogue 23 and silicon analogue 30 (4.36 and 4.91, respectively). In the case of dimethylphenyl analogs, phosphine borane 8 exhibited a logP value of 3.18, which is smaller than those of the corresponding carbon analogue 26 and silicon analogue 31 (5.26 and 5.74, respectively). These results indicate that the B-substituted phosphine boranes and P–BH3 derivatives are less hydrophobic than the corresponding alkanes or silanes. Thus, phosphine boranes could be useful for building sp3–sp3 frameworks with reduced hydrophobicity. Comparison of 7 and 8 or 11 and 12 indicates that switching a P-methyl group to a P-phenyl group increases the hydrophobicity with a difference in the logP values of about 0.7. This difference is similar to that in the case of alkanes and silanes.11
Table 2 Hydrophobicity parameters logP and π, and logKIAM of phosphine boranes 7–14 and reference compounds 23, 26, 30 and 31
| Cmpd |
X–Y |
|
R1 |
R2 |
logP |
πa |
logKIAM |
|
Hansch–Fujita hydrophobicity parameter of each substituent.
Measured using phosphate buffer as a mobile phase. Others were calculated by extrapolation using phosphate buffer and acetonitrile as a mobile phase.
|
| Phenol |
— |
— |
— |
— |
1.46 |
— |
— |
|
7
|
–BH2–P |
4-OH |
Me |
Me |
2.44 |
0.98 |
0.80b |
|
8
|
Ph |
Me |
3.18 |
1.72 |
1.91 |
|
9
|
Et |
Et |
3.28 |
1.82 |
1.81 |
|
10
|
Ph |
Et |
4.11 |
2.65 |
2.46 |
|
11
|
3-OH |
Me |
Me |
2.55 |
1.09 |
0.81b |
|
12
|
Ph |
Me |
3.27 |
1.81 |
1.71 |
|
13
|
Et |
Et |
3.47 |
2.01 |
1.59 |
|
14
|
Ph |
Et |
4.19 |
2.73 |
2.28 |
|
23
|
–CH2–C |
4-OH |
Me |
Me |
4.36 |
2.90 |
2.22 |
|
26
|
Ph |
Me |
5.26 |
3.80 |
2.73 |
|
30
|
–CH2–Si |
Me |
Me |
4.91 |
3.45 |
2.38 |
|
31
|
Ph |
Me |
5.74 |
4.28 |
3.05 |
We also investigated the calculation of logP values using two different programs familiar to medicinal chemists. As a result, the calculated values of phosphine boranes were significantly different from the experimental values (Table S1†). These results indicate that accurate estimation of the logP values of phosphine boranes is currently difficult due to the lack of actual measurements to validate the calculations. We think our results contribute to the development of more accurate algorithms.
2.5. Membrane affinity
The affinity of the synthesized compounds for the cell membrane was assessed by HPLC using an immobilized artificial membrane (IAM) column.16 The phosphine borane derivatives exhibited logKIAM values between 0.8 and 2.46 (Table 2), suggesting that the compounds have moderate membrane affinity. In general, logKIAM values positively correlate with hydrophobicity (logP), and we confirmed that this was the case for the phosphine borane derivatives (Fig. 3).17
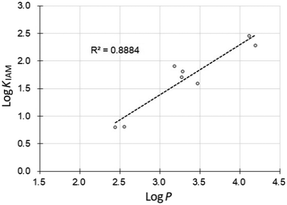 |
| | Fig. 3 Correlation between the hydrophobicity parameter logP values of phosphine boranes and the logKIAM values. | |
2.6. Acidity
The acidity of the phenolic hydroxyl group was also determined from the pH-dependent change of the absorption spectra (Table 3).18 The phosphine borane derivatives exhibited pKa values between 11.02 and 11.33, whereas the alkane and silane analogs exhibited pKa values between 10.69 and 10.89. All the tested compounds exhibited larger pKa values than that of phenol, indicating that the phosphinoboranyl groups (PR3–BH2–), like alkyl groups and silylalkyl groups, behave as electron-donating groups. The pKa values of the phosphine borane derivatives were approximately 0.5 larger than those of the alkyl and silylalkyl analogs, and therefore the phosphinoboranyl groups show a greater electron-donating character. We previously showed that boranophosphinyl groups (BH3–PR2–) behave as electron-withdrawing groups, like phosphoryl groups.10 The boron atom of phosphine boranes is electron-donating in nature, whereas the phosphorus atom is electron-withdrawing.
Table 3 pKa values of phosphine boranes 7–14 and reference compounds 23, 26, 30 and 31
| Cmpd |
X–Y |
|
R1 |
R2 |
pKa |
ΔpKa(phenol)a |
|
ΔpKa values were calculated by subtracting the pKa value of phenol (10.44) from the observed pKa.11
|
| Phenol |
— |
— |
— |
— |
10.44 |
— |
|
7
|
–BH2–P |
4-OH |
Me |
Me |
11.28 |
+0.84 |
|
8
|
Ph |
Me |
11.22 |
+0.78 |
|
9
|
Et |
Et |
11.02 |
+0.58 |
|
10
|
Ph |
Et |
11.05 |
+0.61 |
|
11
|
3-OH |
Me |
Me |
11.26 |
+0.82 |
|
12
|
Ph |
Me |
11.33 |
+0.89 |
|
13
|
Et |
Et |
11.33 |
+0.89 |
|
14
|
Ph |
Et |
11.29 |
+0.85 |
|
23
|
–CH2–C |
4-OH |
Me |
Me |
10.77 |
+0.33 |
|
26
|
Ph |
Me |
10.69 |
+0.25 |
|
30
|
–CH2–Si |
Me |
Me |
10.74 |
+0.30 |
|
31
|
Ph |
Me |
10.89 |
+0.45 |
We also investigated the calculation of pKa values using two different methods. As a result, the calculated values of phosphine boranes were also significantly different from the experimental values (Table S2†). These results indicate that accurate estimation of the pKa values of phosphine boranes is currently difficult, and that our results can contribute to the development of more accurate algorithms.
2.7. Activity toward estrogen receptor alpha (ERα)
Estrogen receptor (ER) is a member of the nuclear receptor superfamily and plays key roles in the female reproductive system, bone metabolism, and the cardiovascular system, as well as the central nervous system.19 Thus, ER ligands are attractive targets of drug discovery. We have previously reported that simple phenols bearing a bulky substituent act as ER ligands,11,12 and also that phosphine borane derivatives including 6 bearing a 4-phosphinophenol substructure function as ER ligands.10 Here, we evaluated the ligand potency of the synthesized B-hydroxyphenyl phosphine borane derivatives toward ERα by means of luciferase reporter gene assay using HEK293 cells. Table 4 summarizes the estrogenic activity and hydrophobicity of the compounds. Trimethylphosphine derivatives 7 and 11 lacked ER agonistic activity, whereas the corresponding alkane derivative 23 and silane derivative 30 exhibited significant activity. Compounds 9, 14, 15, 17 and 21 exhibited agonistic activity toward ERα, but compounds bearing a bulky phosphine moiety such as tri-n-butylphosphine (18 and 22), tricyclopentylphosphine (19) and tricyclohexylphosphine (20) did not show ERα agonistic activity. These results suggested that the trimethylphosphine borane moiety (–BH2–PMe3) is not sufficiently hydrophobic to interact with the hydrophobic receptor surface, and/or that the bulky phosphine moiety is too large to enter the ligand binding pocket of ERα.
Table 4 ER-modulating potency of the synthesized phosphine borane derivatives 7–22 and reference compounds 23 and 30
| Cmpd |
X–Y |
|
R1 |
R2 |
EC50a (μM) |
logP |
LLEd |
|
Concentration showing 50% of the maximal activity of estradiol.
No measurable ER agonistic activity.
These logP values are tentative because the hydrophobicity was outside the range of determination of logP by the HPLC method (0 < logP < 6).
Defined as LLE = pEC50 − logP.
Values not determined.
|
|
7
|
–BH2–P |
4-OH |
Me |
Me |
n.a.b |
2.44 |
n.d.e |
|
8
|
Ph |
Me |
n.a.b |
3.18 |
n.d.e |
|
9
|
Et |
Et |
4.18 |
3.28 |
2.10 |
|
10
|
Ph |
Et |
n.a.b |
4.11 |
n.d.e |
|
11
|
3-OH |
Me |
Me |
n.a.b |
2.55 |
n.d.e |
|
12
|
Ph |
Me |
>10 |
3.27 |
n.d.e |
|
13
|
Et |
Et |
>10 |
3.47 |
n.d.e |
|
14
|
Ph |
Et |
4.09 |
4.19 |
1.20 |
|
15
|
4-OH |
i-Pr |
i-Pr |
1.82 |
4.56 |
1.18 |
|
16
|
Ph |
i-Pr |
>10 |
4.79 |
n.d.e |
|
17
|
c-Pr |
c-Pr |
2.68 |
3.80 |
1.77 |
|
18
|
n-Bu |
n-Bu |
n.a.b |
6.32c |
n.d.e |
|
19
|
c-Pen |
c-Pen |
n.a.b |
6.23c |
n.d.e |
|
20
|
c-Hex |
c-Hex |
n.a.b |
7.22c |
n.d.e |
|
21
|
3-OH |
Ph |
i-Pr |
3.45 |
4.93 |
0.53 |
|
22
|
n-Bu |
n-Bu |
n.a.b |
6.43c |
n.d.e |
|
23
|
–CH2–C |
4-OH |
Me |
Me |
1.53 |
4.36 |
1.46 |
|
30
|
–CH2–Si |
Me |
Me |
1.41 |
4.91 |
0.94 |
We investigated the SAR from the viewpoint of Taft steric parameter. Although the Taft steric parameters of the bulky phosphine moieties are not available, values of the individual functional group are available, namely, i-propyl group (−1.60), n-butyl group (−1.63), cyclopentyl group (−1.75), and cyclohexyl group (−2.03).20 Our results indicated that there is a threshold between isopropyl group and n-butyl group. Among the tested phosphine borane derivatives, tri-i-propylphosphine derivative (15) showed the most potent activity with an EC50 value of 1.82 μM, and tricyclopropylphosphine derivative (17) was the next most potent, indicating that the combination of three C3 units is favorable for ER agonistic activity. These results are consistent with the previous structure–activity relationship study, and support the idea that phosphine boranes could be a versatile hydrophobic pharmacophore of ER ligands.
Next, in order to investigate the lipophilicity of the compounds, we calculated the ligand-lipophilicity efficiency (LLE). LLE is a parameter proposed by Leeson and Springthorpe, defined by the equation LLE = pIC50 − logP.21 A large LLE value indicates good lipophilic efficiency, and would generally imply that adventitious compound-related toxicity is less likely to be a major issue.22 The LLE values of phosphine boranes 9 and 17 are 2.10 and 1.77, respectively, exceeding those of alkane 17 (LLE = 1.46) and silane 19 (LLE = 0.94). Though the phosphine boranes do not show greater ER agonistic potency than alkanes and silanes, their large LLE values suggest that they could be useful options as less hydrophobic scaffolds for structural development.
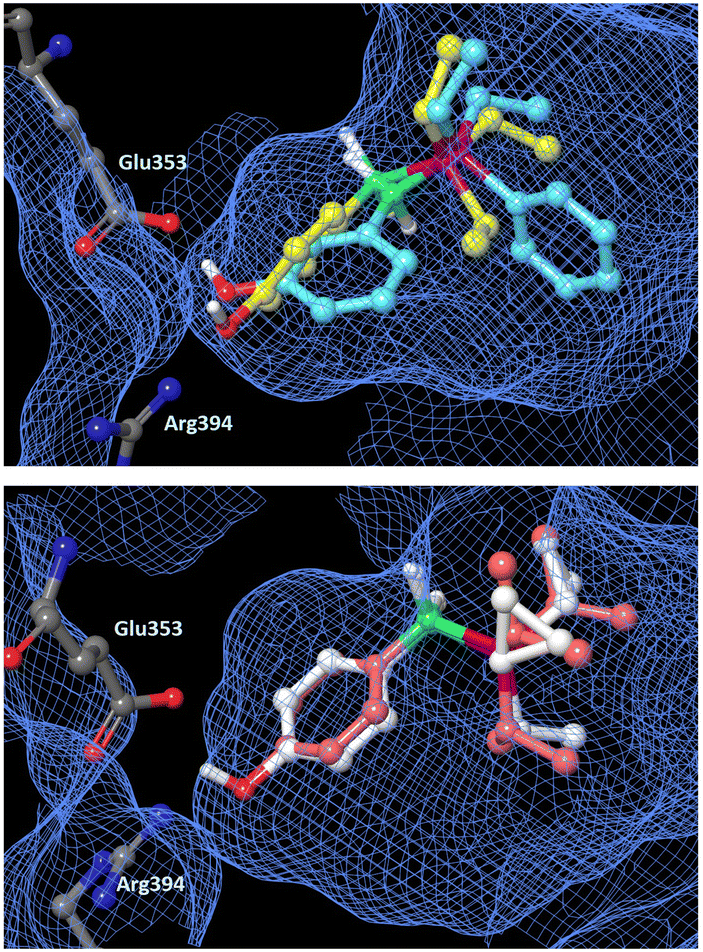
2.8. Docking simulation
In order to estimate the binding mode of the phosphine borane derivatives that showed agonistic activity toward ERα, we conducted docking simulations of compounds 9, 14, 15 and 17 with the X-ray crystal structure of the antagonist-bound form of the hERα ligand-binding domain (LBD) (PDB ID: 1ERE),23 using AutoDock 4.2.24Fig. 4 shows the docking models of the phosphine boranes in the hERα LBD as a superimposition of compounds 9 and 14, 15 and 17, respectively. In the calculated structures, the phenolic hydroxyl group of all four compounds interacts with Glu353 and Arg394, with which estradiol forms key hydrogen bonds. Furthermore, the phosphine borane moiety of each compound occupies the hydrophobic cavity of the ligand-binding pocket. In the docked structure of B-(3-hydroxyphenyl) diethylphenylphosphine borane (14), the two phenyl groups on the P–B axis adopt a synclinal conformation and the 3-hydroxyl group is located at a suitable position for hydrogen bonding. Among the P,B-diphenyl derivatives, i.e., compounds 8, 10, 12, 14, 16 and 21, the 3-hydroxy derivatives were more potent than the corresponding 4-hydroxy derivatives, and the accessibility of the synclinal conformation could be one reason for this. Regarding compounds 15 and 17, their trialkylphosphine substructure occupies the hydrophobic pocket in a similar manner, and the small difference in hydrophobic volume between these compounds may account for the difference in the activity. This result supports the idea that the wide variety of available phosphine structures could enable fine tuning of the hydrophobic interaction, indicating that phosphine boranes could be a versatile option for structural optimization of biologically active compounds.
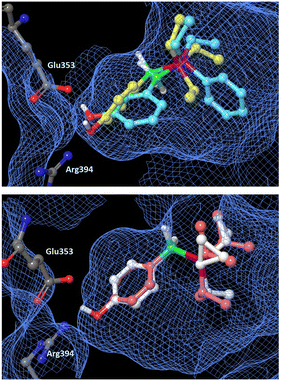 |
| | Fig. 4 Docking models of phosphine boranes 9, 14, 15 and 17 with the hERα LBD (PDB ID: 1ERE) obtained with AutoDock 4.2. The protein surface is indicated as a blue mesh. Upper: Superimposition of docking models of 9 (yellow) and 14 (light blue) in the hERα LBD. Lower: Superimposition of docking models of 15 (pink) and 17 (white). | |
3. Conclusion
In order to investigate the utility of phosphine boranes in drug discovery, we designed and synthesized a series of B-hydroxyphenyl phosphine borane derivatives, and investigated their structure–property relationship and their biological activity toward ERα. The synthesized B-hydroxyphenyl phosphineborane derivatives exhibited sufficient stability in aqueous media, lower hydrophobicity than the corresponding alkanes and silanes, and adequate affinity for lipid membranes to enable permeability. These results suggest that introduction of phosphine borane moieties into biologically active compounds would not adversely impact on membrane permeability. Phosphinoboranyl groups exhibited a greater electron-donating character than alkyl groups, whereas the boranophosphinyl groups function as electron-withdrawing substituents. Several B-hydroxyphenyl phosphine borane derivatives exhibited significant ER agonistic activity with superior ligand-lipophilicity efficiency (LLE). To our knowledge, this is the first report of biologically active B-substituted phosphine borane derivatives. Our results suggest that phosphine boranes could be versatile structural options in medicinal chemistry for expanding the chemical space available for drug discovery. The substituent parameters and structure–activity relationship reported here may be helpful for this purpose.
4. Experimental
Chemistry
All reagents were purchased from TCI Chemicals, Fujifilm Wako Pure Chemical Industries, or Kanto Kagaku Co. Inc., and used without further purification: thin-layer chromatography (TLC) was performed using silica gel coated with a fluorescent indicator F254 (Merck, #1.05715.0001). Silica gel column chromatography was performed using neutral silica gel (60 Å, 40–50 μm) purchased from Kanto Kagaku Co. Inc. NMR spectra were recorded on Bruker Avance 400 (1H: 400 MHz, 13C: 100 MHz, 11B: 128 MHz and 31P: 161 MHz) and Bruker Avance 500 (1H: 500 MHz and 13C: 125 MHz) spectrometers. High-resolution mass (HRMS) spectra were taken on a Bruker Daltonics micrOTOF-2 focus using the electron spray ionization time-of-flight (ESI-TOF) method.
General procedure for synthesis of phosphine borane derivatives 7–22
A dry round-bottomed flask, equipped with a magnetic stirring bar, sealed with a septum and protected with an Ar balloon, was charged with 4- or 3-hydroxyphenylboronic acid (2.0 mmol) in 10 mL of THF. LiAlH4 (6.0 mmol) was added at 0 °C. The mixture was stirred and the appropriate phosphine was added. Stirring was continued at room temperature with monitoring by TLC. When the reaction was complete, the mixture was quenched by the addition of sodium sulfate hydrate and filtered through Celite. The obtained solution was evaporated, and the residue was purified by silica gel column chromatography to give the desired phosphine borane derivatives. Details of the characterization and spectra of compounds are presented in the ESI.†
Determination of stability in aqueous solution
The residual ratio of the compounds was calculated from the peak area on HPLC. A COSMOSIL Packed Column 5C18-MS-II (5 μm, 150 mm × 4.6 mm id, Nacalai Tesque, Inc., Japan) column was fitted on an HPLC instrument (Multiwavelength Detector, MD-2010 Plus. JASCO) equipped with a pump (PU-2080, JASCO) and oven (SSC-2120, Senshu Scientific Co., Ltd.). The mobile phase was a methanol–water system. The concentration of the mobile phase and the flow rate were adjusted so that the retention time of the measured compound was in the range of about 7 to 13 minutes. The injection volume was 25 μL. Compounds were detected by measuring UV absorption at the peak wavelength between 220 and 240 nm. The temperature of the column was kept at 37.0 (±0.1) °C. Test compounds (0.1 mM) were dissolved in acetonitrile/PBS (5:95 v/v) and incubated at 37 °C for 3 h. The residual ratio was calculated in according to the following formula: residual ratio (%) = (peak area after 3 h/peak area at t0) × 100. Each measurement was performed in triplicate and the mean value was calculated.
Determination of hydrophobicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P)
P)
The 1-octanol/water partition coefficient, P, was determined by means of an HPLC method based on the OECD Guideline for Testing Chemicals. The HPLC instruments were the same as for the determination of logP. The injection volume was 20 μL, and the flow rate was 1.0 mL min−1 in all cases. Compounds were detected by measuring UV absorption at 254 nm. The temperature of the column was kept at 40.0 (±0.1) °C during the measurement. The mobile phase was a methanol–water system. The methanol concentration was changed between 80% (v/v) and 60% (v/v) in 5% (v/v) steps, conducted in at least three different methanol concentrations to calculate logkw. The dead time t0 was measured with thiourea as the unretained compound. Each measurement was performed in triplicate, and the mean value was calculated. The capacity factor of each compound in 100% aqueous eluent, logkw, was calculated by linear extrapolation from the measured logk values in the methanol-aqueous mobile phase. The calculated logkw values of the reference compounds (phenol, 4-chlorophenol, 4-phenylphenol, diphenylether, fluoranthene, dichlorodiphenyltrichloroethane) were plotted against logP values to prepare a calibration graph (logP = 1.0548logkw + 0.2627, R2 = 0.9920).
Determination of acidity (pKa)
The pKa values were determined by measurement of the change of absorbance at λmax in the pH-dependent UV spectra of ionic species in 20:80 (w/w) methanol-phosphate buffer (pH range of 6.0–12.0). The values of pH obtained with the pH meter were corrected by using the equation: pH* = pH (recorded) − d (d = 0.01). Each measurement was performed in triplicate.
Determination of membrane affinity (logKIAM)
The immobilized artificial membrane (IAM) partition coefficients, logKIAM, were determined by HPLC using an immobilized artificial membrane column. The IAM.PC.DD2 column (10 μm, 300 Å, 30 mm × 4.6 mm, Regis Technologies, Inc.) was fitted on an HPLC instrument (Multiwavelength Detector, MD-2010 Plus. JASCO) equipped with a pump (PU-2080, JASCO) and oven (SSC-2120, Senshu Scientific Co., Ltd.). The injection volume was 20 μL, and the flow rate was 1.0 mL min−1 in all cases. The compounds were detected by measuring the UV absorption at 240 nm. The temperature of the column was kept at 37.0 (±0.1) °C. The mobile phase consisted of 0.01 M phosphate-buffered saline or acetonitrile and 0.01 M phosphate-buffered saline, adjusted to pH 7.4. The acetonitrile concentration was changed from 40% (v/v) to 25% (v/v) in 5% (v/v) steps. The dead time t0 was measured with methanol as the unretained compound. Each measurement was performed in triplicate, and the mean value was calculated. The capacity factor of each compound in 100% aqueous eluent, logKIAM, was calculated by linear extrapolation of the measured logKIAM values in the acetonitrile–phosphate-buffered saline mobile phase.
Reporter gene assay
The ER-agonistic activities of the synthesized compounds were evaluated by means of reporter gene assay using a Gal4-human ERα reporter system described previously. Briefly, a fragment of human ERα was inserted into the pCMX-GAL4N vector to obtain pCMX-GAL4N-hER (pCMX-flag vector to generate pCMX-ERα). GAL4N-responsive MH100 (USA)x4-tk-LUK reporter was used. Human embryonic kidney (HEK 293) cells, which was obtained from RIKEN Cell Bank (RIKEN BRC, Tsukuba, Japan), were cultivated in Dulbecco's modified Eagle's medium (DMEM without phenol red) containing 10% fetal bovine serum (FBS) and antibiotic–antimycotic mixture at 37 °C in a humidified atmosphere of 5% CO2 in air. Transfections were performed by the calcium phosphate coprecipitation method. Test compounds were added at 24 h after transfection. Cells were harvested 24 h later, and luciferase and β-galactosidase activities were assayed using an Infinite M200 PRO multimode microplate reader (TECAN). Luciferase data were normalized to an internal β-galactosidase control, and reported values are means of triplicate assays. Antagonist activity was measured in the presence of 0.3 nM 17β-estradiol.
Docking simulation
The structure of the LBD of hERα was prepared from the Protein Data Bank accession 1ERE. Polar hydrogens and partial atomic charges were assigned using AutoDockTools (ADT). Molecular docking was performed using AutoDock 4.2 with the Genetic Algorithm. AutoDock parameters for boron atoms were Rii = 4.08 and eii = 0.180.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by Grants in-Aid for Scientific Research from JSPS (KAKENHI Grant No. 23K06046 (S. F.)), Takeda Science Foundation (S. F.), Hoansha Foundation (S. F.), and the Research Center for Biomedical Engineering (S. F. and T. N.-Y.). This research was also partially supported by AMED under Grant Number JP23ama121043 (Research Support Project for Life Science and Drug Discovery, BINDS).
Notes and references
-
(a) B. R. Smith, C. M. Eastman and J. T. Njardarson, J. Med. Chem., 2014, 57, 9764–9773 CrossRef CAS PubMed;
(b) S. Fujii, Med. Chem. Commun., 2016, 7, 1082–1092 RSC.
-
(a) P. G. Richardson, C. Mitsiades, T. Hideshima and K. C. Anderson, Annu. Rev. Med., 2006, 57, 33–47 CrossRef CAS PubMed;
(b) J. Adams and M. Kauffman, Cancer Invest., 2004, 22, 304–311 CrossRef CAS.
-
(a) J. B. Rodriguez and C. Gallo-Rodriguez, ChemMedChem, 2019, 14, 190–206 CrossRef CAS PubMed;
(b) H. Yu, H. Yang, E. Shi and W. Tang, Med. Drug Discovery, 2020, 8, 100063 CrossRef CAS.
- J. M. DeForrest, T. L. Waldron, C. Harvey, B. Scalese, B. Rubin, J. R. Powell, E. W. Petrillo and D. W. Cushman, J. Cardiovasc. Pharmacol., 1989, 14, 730–736 CrossRef CAS PubMed.
-
(a) S. Allen and I. Oswald, Br. J. Clin. Pharmacol., 1976, 3, 165–168 CrossRef CAS PubMed;
(b) A. N. Nicholson and C. M. Wright, Br. J. Clin. Pharmacol., 1977, 4, 494–496 CrossRef CAS PubMed.
- W.-S. Huang, S. Liu, D. Zou, M. Thomas, Y. Wang, T. Zhou, J. Romero, A. Kohlmann, F. Li, J. Qi, L. Cai, T. A. Dwight, Y. Xu, R. Xu, R. Dodd, A. Toms, L. Parillon, X. Lu, R. Anjum, S. Zhang, F. Wang, J. Keats, S. D. Wardwell, Y. Ning, Q. Xu, L. E. Moran, Q. K. Mohemmad, H. G. Jang, T. Clackson, N. I. Narasimhan, V. M. Rivera, X. Zhu, D. Dalgarno and W. C. Shakespeare, J. Med. Chem., 2016, 59, 4948–4964 CrossRef CAS PubMed.
-
(a) A. Staubitz, A. P. M. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023–4078 CrossRef CAS PubMed;
(b) J. M. Brunel, B. Faure and M. Maffei, Coord. Chem. Rev., 1998, 178, 665–698 CrossRef;
(c) R. T. Paine and H. Noth, Chem. Rev., 1995, 95, 343–379 CrossRef.
-
(a) P. Li, Z. A. Sergueeva, M. Dobrikov and B. R. Shaw, Chem. Rev., 2007, 107, 4746–4796 CrossRef CAS PubMed;
(b) J. S. Summers and B. R. Shaw, Curr. Med. Chem., 2001, 8, 1147–1155 CrossRef CAS PubMed.
-
(a) C. R. Schlieve, A. Tam, B. L. Nilsson, C. J. Lieven, R. T. Raines and L. A. Levin, Exp. Eye Res., 2006, 83, 1252–1259 CrossRef CAS PubMed;
(b) R. Remtulla, S. K. Das and L. A. Levin, Molecules, 2021, 26, 2505 CrossRef CAS PubMed.
- H. Saito, Y. Matsumoto, Y. Hashimoto and S. Fujii, Bioorg. Med. Chem., 2020, 28, 115310 CrossRef CAS PubMed.
- S. Fujii, Y. Miyajima, H. Masuno and H. Kagechika, J. Med. Chem., 2013, 56, 160–166 CrossRef CAS.
- Y. Yamakoshi, Y. Otani, S. Fujii and Y. Endo, Biol. Pharm. Bull., 2000, 23, 259–261 CrossRef CAS PubMed.
-
(a) J. A. Baban and B. P. Roberts, J. Chem. Soc., Perkin Trans. 2, 1988, 7, 1195–1200 RSC;
(b) Y. Kawano, K. Yamaguchi, S. Miyake, T. Kakizawa and M. Shimoi, Chem. – Eur. J., 2007, 13, 6920–6931 CrossRef CAS PubMed.
-
(a)
OECD Guideline for the Testing of Chemicals, 117; partition coefficient (n-octanol/water), HPLC method, Organisation for Economic Co-operation and Development (OECD), 2022 Search PubMed;
(b) W. Klein, W. Kördel, M. Weiß and H. J. Poremski, Chemosphere, 1988, 17, 361–386 CrossRef CAS;
(c) G. Cimpan, M. Hadaruga and V. J. Miclaus, J. Chromatogr. A, 2000, 869, 49–55 CrossRef CAS PubMed.
- T. Fujita, J. Iwasa and C. Hansch, J. Am. Chem. Soc., 1964, 86, 5175–5180 CrossRef CAS.
- C. Pidgeon, S. Ong, H. Liu, X. Qiu, M. Pidgeon, A. H. Dantzig, J. Munroe, W. J. Hornback, J. S. Kasher, L. Glunz and T. Szczerba, J. Med. Chem., 1995, 38, 590–594 CrossRef CAS PubMed.
- S. Ong, H. Liu and C. Pidgeon, J. Chromatogr. A, 1996, 728, 113–128 CrossRef CAS PubMed.
- R. G. Bates and A. K. Vijh, J. Electrochem. Soc., 1973, 120, 263C CrossRef.
-
(a) B. J. Deroo and K. S. Korach, J. Clin. Invest., 2006, 116, 561–570 CrossRef CAS;
(b) N. Heldring, A. Pike, S. Andersson, J. Matthews, G. Cheng, J. Hartman, M. Tujague, A. Ström, E. Treuter, M. Warner and J.-Å. Gustafsson, Physiol. Rev., 2007, 87, 905–931 CrossRef CAS PubMed.
-
Steric Effects in Organic Chemistry, ed. M. S. Newman, Wiley, New York, 1956, pp. 598–604 Search PubMed.
-
(a) P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881–890 CrossRef CAS PubMed;
(b) T. W. Johnson, R. A. Gallego and M. P. Edwards, J. Med. Chem., 2018, 61, 6401–6420 CrossRef CAS PubMed.
- M. P. Edwards and D. A. Price, Annu. Rep. Med. Chem., 2010, 45, 380–391 Search PubMed.
- A. M. Brzozowski, A. C. W. Pike, Z. Dauter, R. E. Hubbard, T. Bonn, O. Engström, L. Öhman, G. L. Greene, J.-Å. Gustafsson and M. Carlquist, Nature, 1997, 389, 753–758 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |

 *a
*a