
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScreening and optimization of phage display cyclic peptides against the WDR5 WBM site†
Lingyu
Song
ab,
Jiawen
Cao
ab,
Lin
Chen
b,
Zhiyan
Du
b,
Naixia
Zhang
ac,
Danyan
Cao
*b and
Bing
Xiong
 *abd
*abd
aDepartment of College of Pharmacy, University of Chinese Academy of Sciences, 19A Yuquan Road, Beijing 100049, China
bDepartment of Medicinal Chemistry, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China. E-mail: s20-songlingyu@simm.ac.cn; caody@simm.ac.cn; bxiong@simm.ac.cn
cDepartment of Analytical Chemistry, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China
dState Key Laboratory of Chemical Biology, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China
First published on 17th August 2023
Abstract
Of the various WD40 family proteins, WDR5 is a particularly important multifunctional adaptor protein that can bind to several protein complexes to regulate gene activation, so it was considered as a promising epigenetic target in anti-cancer drug development. Despite many inhibitors having been discovered directing against the arginine-binding cavity in WDR5 called the WIN site, the side hydrophobic cavity called the WBM site receives rather scant attention. Herein, we aim to obtain novel WBM-targeted peptidic inhibitors with high potency and selectivity. We employed two improved biopanning approaches with a disulfide-constrained cyclic peptide phage library containing 7 randomized residues and identified several peptides with micromole binding activity by docking and binding assay. To further optimize the stability and activity, 9 thiol-reactive chemical linkers were utilized in the cyclization of the candidate peptide DH226027, which had good binding affinity. This study provides an effective method to discover potent peptides targeting protein–protein interactions and highlights a broader perspective of peptide-mimic drugs.
Introduction
Epigenetics is a growing field and has shown encouraging early outcomes in drug discovery. Epigenetic alterations have been found in a variety of human malignancies and are thought to be reversible, making them promising targets for pharmacological agents.1 The main classes of epigenetic inhibitors studied include histone deacetylase (HDAC) inhibitors,2 bromodomain-containing protein 4 (BRD4) inhibitors,3 histone lysine methyltransferase nuclear receptor-binding SET domain protein 2 (NSD2) inhibitors,4 DNA-methyltransferase (DNMT) inhibitors,5 coactivator-associated arginine methyltransferases 1 (CARM1),6 and so on. As a member of the WD40 family protein, WDR5 has many essential biological functions, especially in gene expression regulation, and is considered as a promising epigenetic target for cancer treatment.7 WDR5 participates in many protein complexes, such as the MLL complex,8 subunits of the nonspecific lethal (NSL) complex,9 nucleosome remodeling and deacetylase (NuRD) complex,10 Ada2a-containing (ATAC) complex11 and Pax transactivation domain-interacting protein (PTIP) complex.12 As an important cornerstone of these complexes, WDR5 has two confirmed interacting sites: the WDR5-interacting site (WIN site) and the WDR5-binding motif (WBM site). In the MLL complex, WDR5 interacts with MLL1 and RbBP5 through the WIN site and WBM site, respectively. Since 2010, many inhibitors targeting the WIN site that is located at the top of the central cavity and is surrounded by seven leaf β-propeller motifs13 have been developed. Compared to the WIN site, the WBM site is a more superficial hydrophobic cavity and requires aliphatic side chains from hydrophobic residues to stabilize interactions. Four partners have been reported to bind to the WBM site: RbBP5, KANSL2, MYC, and IncRNAs.9,14 Among these, RbBP5, KANSL2, and MYC share a similar core sequence named the ‘AAXDV’ motif (X = V, I, or L; A is an acidic amino acid).15 Besides, Asn225 and Gln289 are the key residues in the WBM binding pocket, responsible for the formation of H-bonds with the interacted proteins. Recently, two series of small molecular inhibitors were reported by Fesik's group,16 which were developed from the fragment hits harboring a salicylic acid scaffold. The binding affinity of these inhibitors against the WBM site of WDR5 was in the range of 30–100 nM. The co-immunoprecipitation assay showed that these compounds could disturb the interaction between WDR5 and c-MYC.17 Limited to a few numbers and types, we know poorly about the mechanism and effectivity of WBM site inhibitors applied in WDR5-associated cancers. Therefore, new types of WBM inhibitors need to be explored urgently.The discovery of ligands that selectively bind to a certain target plays a vital role in the area of clinically relevant diagnostics and therapeutics. Since it was first described in 1985,18 phage display has evolved and been known as a widely used and powerful methodology for the study of target-specific ligands and molecular biology mechanisms.19 Furthermore, the correct and efficient correlation of phenotypes and genotypes achieved by phage display allows for binder identification in only two or three iterative rounds of phage selection, which endows the technique with advantages of low cost and high efficiency. At present, phage-based genetically encoded libraries have become a major pipeline for finding lead compounds.
Importantly, given their intrinsic nature, large surface area and limited conformational flexibility, cyclic peptides possess numerous advantages, such as low toxicity, high binding affinity, great target specificity, excellent metabolic stability and cell permeability.20 Thus, cyclic peptides have emerged as a potential therapeutic modality for drug discovery and disease treatment over the past few decades, especially against challenging targets such as protein–protein interactions.21,22 In this paper, we employed biopanning methods with the disulfide-bridged peptide phage display library (Ph.D.™-C7C phage library, CX1X2X3X4X5X6X7C, where X represents any amino acid) to explore new peptidic inhibitors toward the WBM site. In general, disulfide-bridged peptides could lose bioactivity easily in vivo by reductions, so disulfide bonds inside selected peptides were further replaced with more stable chemical bonds formed by a series of thiol-reactive chemical linkers. Finally, optimized cyclic peptides with better stability and binding affinity were obtained. This article aims to provide a substantial application of phage display cyclic peptides in the WDR5-WBM site.
Results and discussion
Target proteins prepared for screening
For the subsequent biopanning experiments, we prepared both His-tagged WDR5 and biotinylated WDR5 for the screening corresponding to Ni-NTA magnetic agarose beads and streptavidin magnetic beads, respectively. After purification, 19.6 mg WDR5 protein with His-tag was obtained from 9.35 g cells. From the gel-filtration elution profile, we observed that His-tagged WDR5 protein was eluted at a single peak at 89 mL corresponding to 36.5 kDa. The protein concentration before gel filtration should not be too high; otherwise, a peak at 47 mL corresponding to 70.0 kDa appeared which is confirmed to be the aggregation of WDR5. After SDS-PAGE separation, we detected and verified the bands of protein migrating around 35.0 kDa (Fig. S1†). As for biotinylated WDR5 obtained with a biotinylation kit, a HABA assay was performed to estimate the biotinylated level of WDR5. The absorbance at 500 nm of HABA/avidin solution was 0.6, and the A500 of HABA/avidin/biotin decreased to 0.2. Then, an online HABA calculator on the ThermoFisher website was used and 3.7 biotin groups per protein molecule were obtained for the biotin ratio of WDR5.Affinity selection of cyclic peptides specific to the WBM site of WDR5
The Ph.D.™-C7C phage library of random cyclic peptides with a disulfide constrained loop was applied in the WBM-targeting biopanning. As we disclosed previously,23 peptides with arginine can easily bind to the WIN site of WDR5. So, a small molecular inhibitor (WIN-IN-4; CAS: 2407457-36-5) with sub-nanomole binding activity was employed to block the WIN site before selection. Meanwhile, the inhibitor was added to the wash buffer for additional washing of the WIN binding peptides. On the other hand, the enrichment of selection-related target-unrelated peptides (TUPs) is a major problem for phage library screening, which may contribute to false affinity toward the target and give misleading results. Furthermore, phages binding to magnetic beads may predominate during the solution-biopanning.24 Therefore, affinity selection with two different strategies was designed and carried out (Fig. 1). (A) Subtractive panning: only Ni-NTA magnetic agarose beads were used throughout three rounds of the screening, and the amplified phages were incubated with beads prior to His-tagged WDR5 in the first two cycles to deplete bead binders. Besides, an extremely stringent washing procedure was performed. The concentration of WIN-IN-4 in the wash buffer was raised from 10 μM and 50 μM, to 100 μM stepwise. (B) Alternant panning: two kinds of magnetic beads were employed alternately in three rounds without depleting, and the WIN site inhibitor was kept at 10 μM in the wash buffer under a moderate washing condition. | ||
| Fig. 1 The overview of two different panning strategies. (A) The subtractive panning with Ni-NTA magnetic agarose beads throughout the three rounds. (B) The alternant panning with two kinds of magnetic beads employed alternately. | ||
After each cycle, the phage titer was tested immediately and recovery efficiency could be obtained (Table S1†). As the results indicated, alternant panning without depleting could decrease the loss of library effectively and fold increase increased obviously after three rounds compared with subtractive panning. Then fortunately, when we picked 33 clones and 124 clones of the third round in the subtractive panning and the alternant panning separately for DNA sequencing, no histidine-rich peptides or ‘HPQ’ tripeptide motifs appeared (Fig. S2†), which were the most recognizable TUPs binding to Ni2+ and streptavidin, respectively.25–27 And it implied that the two kinds of selection methods worked well. Moreover, the ‘RT’-containing peptides decreased significantly in the proportions of total peptides sequenced although they still existed. The ‘RT’-containing sequences could not be eliminated by blocking the WIN site with a sub-nanomole compound, because the chelation interaction between the WIN site and multivalent-displayed peptides was inevitable. Despite all the precautions, it was impossible to completely avoid TUPs adsorbing non-specifically to plastic polystyrene surfaces. Thus, we utilized SAROTUP, a free online service for scanning, reporting and excluding TUPs,28 to distinguish true binders from false-positive plastic binders with PSBinder values lower than 0.7 (Tables S2 and S3†). In the end, the eligible sequences in the screen are listed in Fig. 2. Among the 35 clones in total, 8 sequences emerged more than once and the frequency of the sequence CGSLDWPHC was the highest, indicating that the favourable fold increase exists in the panning. Besides, the sequence CGEGEADVC containing the WBM site conservative ‘DV’ motif also appeared among these sequences.
 | ||
| Fig. 2 Partial picked sequences deduced from the third round. (A) 11 sequences from the subtractive panning with Ni-NTA magnetic agarose beads. (B) 24 sequences from the alternant panning with two kinds of magnetic beads. | ||
Docking study and binding affinity assay of sequenced peptides
Molecular docking is a powerful computation tool in drug discovery. Several docking programs have been available for protein–peptide complexes, such as AutoDock CrankPep (ADCP), GalaxyPepDock, MDockPeP, HPEPDOCK, etc. A comprehensive evaluation of fourteen docking programs on protein–peptide complexes was reported,29 which shows that ADCP achieves the best predictions and reaches high level success rates in local docking. ADCP has been shown to reliably dock peptides of up to 20 amino acids in length and cyclic peptides with disulfide bonds. We conducted docking study with ADCP after these screens. 35 sequences among the potential binders were picked and prepared by PyMOL for docking (Tables S2 and S3†). The highest affinity in the docking results of each sequence was compared, and peptides with an affinity value lower than −21.0 were selected for solid-phase peptide synthesis (SPPS). Finally, 7 cyclic peptides with a single disulfide bridge were synthesized and tested for binding affinity against WDR5 by HTRF assay (Table 1). To our surprise, sequence CNSLNWFWC (DH226027) derived from scoring calculation showed the best binding activity with an EC50 of 2.2 μM, so it could be a candidate peptide for the following structural optimization and modification.| Peptide sequence | Affinity (ADCP) (kcal mol−1) | HTRF assay IC50 (μM) |
|---|---|---|
| Subtractive panning with Ni-NTA magnetic agarose beads | ||
| a Sequence of the candidate peptide. | ||
| DH226015-CADTNWLVC | −22.7 | 65 |
| D210012-CPNQWKSYC | −22.0 | >100 |
| D210014-CSQAWWYQC | −22.7 | 30 |
| DH226016-CFLNVSNAC | −21.1 | >100 |
Binding mechanism between DH226027 and WDR5 indicated by ADCP and NMR experiments
After docking, ten ranked PDB files appeared as results and one of the PDB file was chosen to show the binding interactions between DH226027 and WDR5 (Fig. 3A). The data revealed that DH226027 employed both its backbone and side chain atoms when binding to WDR5, forming predominantly hydrogen bonds and cation–π stacking interactions. Six intermolecular hydrogen bonds were formed between N2 of DH226027 and K227, and Y228 of WDR5, between S3 of DH226027 and N225, and K227 of WDR5, and between W8 of DH226027 and K272, and Q289 of WDR5. Formation of cation–π stacking interactions was observed between W6 of DH226027 and P224 of WDR5. | ||
| Fig. 3 (A) The binding interactions between WDR5 and DH226027 in the molecular docking model. The peptide DH226027 and the key residues of WDR5-binding regions are presented as pink and cyan sticks, respectively. Hydrogen bonds and cation–π stacking interactions are represented as yellow and green dashes, respectively. (B) Superposition of the 1H-15N-HSQC spectra of 15N-labeled WDR5 with (blue) and without (red) DH226027. | ||
In order to map the interacting sites of the peptide DH226027 in WDR5, [1H, 15N] HSQC NMR titration experiments were performed. The residues with their chemical shifts perturbed and attenuated significantly upon the addition of DH226027 in WDR5 were identified as follows: A47, V48, S49, S50, I90, S91, W95, D107, F133, C134, F149, V174, S175, Y191, C195, L209, F219, L230, A232, T233, L234, D235, Y243, K245, E258, K259, Y260, F266, K272, W273, S276, T301, I305, and T307 for DH226027 (Fig. 3B). Compared with the key residues of WDR5-binding regions in the literature,13 the perturbed residues in WDR5 upon the binding of DH226027, particularly F266, K272, and W273, localized to the WBM site region. Therefore, the above results demonstrated that the peptide DH226027 could be chosen as a candidate ligand targeting the WBM site in WDR5.
Cyclization of the candidate peptide with different chemical linkers
Disulfide-bridged peptides can be converted to linear peptides in vivo by reducing intramolecular disulfide bonds into free sulfhydryl groups, and the instability severely restricted their intracellular applicability. In order to stabilize the bioactive conformation and improve the binding affinity of the candidate peptide DH226027, one of the promising modification strategies was cysteine alkylation.30–32 The key of this method is the identification of optimal linkers. In our work, cyclization was performed with a synthesized linear peptide DH226027-L (H-CNSLNWFWC-NH2, Fig. S3†), and different chemical linkers were selected and experimentally tested in order to obtain stabilized cyclic peptides with high binding affinity. Hoping to get a cyclic peptide whose chain length is short and close to that of a disulfide bond, 9 thiol-reactive crosslinkers commonly used in peptide cyclization were employed, including benzylbromides C1–C3,33,34 allylbromides C4 and C9,35 acetone C5,36 maleimide C6,37 pyridine C7,38 and malonamide C8 (Scheme 1).39 | ||
| Scheme 1 The cyclization reactions between the peptide DH226027-L and 9 chemical linkers. | ||
As shown in Fig. 4, among reactions conducted with C1, C2, and C3 in which the 1,2-, 1,3- and 1,4-positions of the benzene ring were decorated with bromine groups, linker C3 gave relatively clean formation of the desired products and it was easy to purify. However, cross-linking reactions with linkers C1 and C2 yielded more by-products. For the rest, HPLC analysis indicated that the most efficient macrocyclizations were carried out by C4 and C7, which provided the major peak of cyclization products proved by LC-MS results. Incubation of peptide DH226027-L with C5 gave a new product with a mass of 1294.37 Da that was inconsistent with the expected mass (1226.47 Da). It was likely that linker C5 with high activity had undergone a lot of side reactions. Cyclization with maleimide linker C6 also resulted in an unknown product mixture which was further complicated because of hydrolysis of imide (observed mass = 1462.38 Da, different from the anticipated mass of 1265.44 Da). Additionally, it was worth noting that the intramolecular disulfide bond formation due to oxidation was a major side reaction. Product analysis revealed that the disulfide-bridged peptide was the main product when C8 was added, and the two peaks of alkenyl-bridged and disulfide-bridged cyclic peptides were too close to separate when DH226027-L was treated with C9.
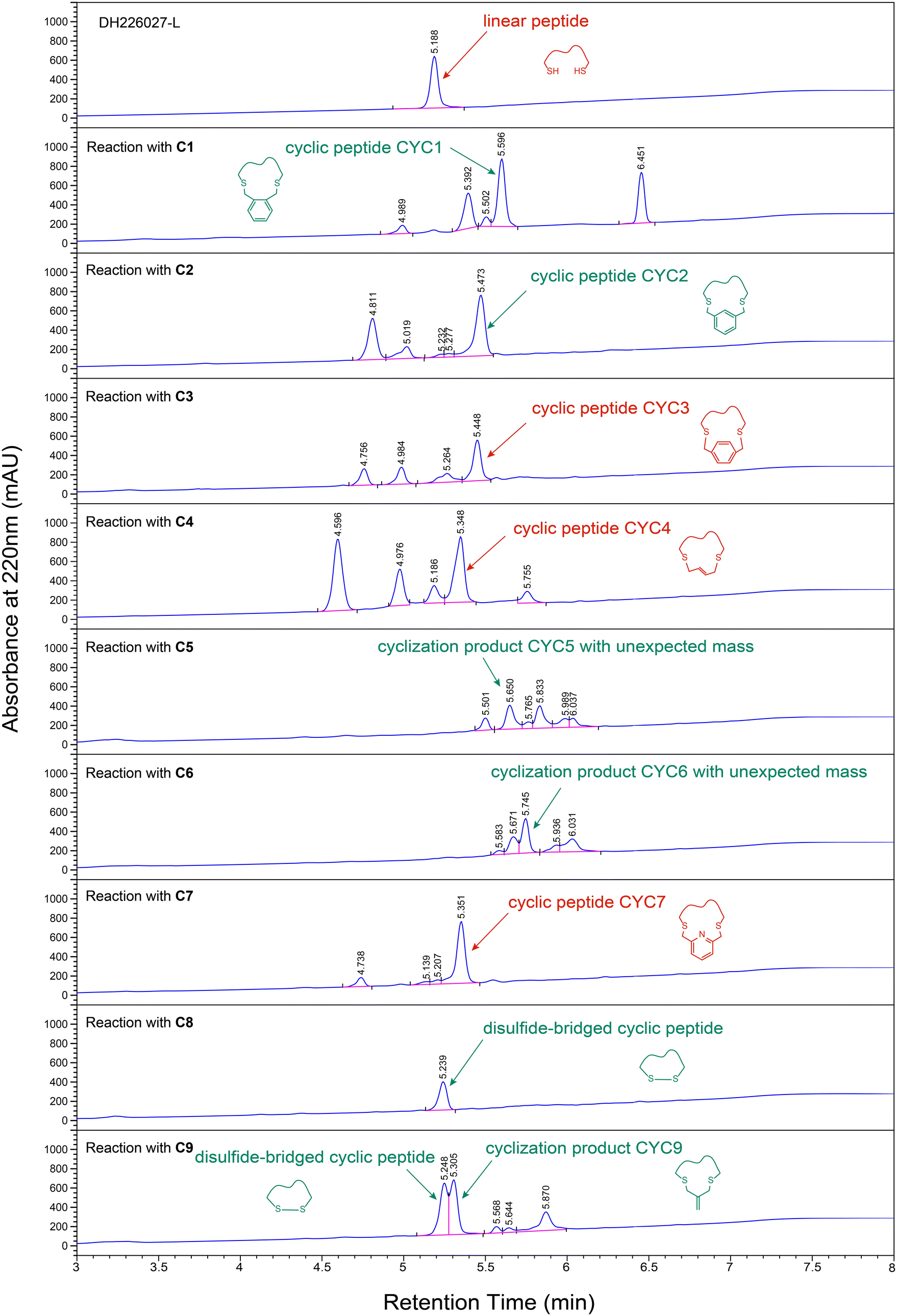 | ||
| Fig. 4 HPLC chromatograms of cyclization reactions between linear peptide DH226027-L and chemical linkers C1–C9. | ||
Above all, we employed C3, C4 and C7 as cyclization reagents to create cyclopeptides CYC3, CYC4, and CYC7 respectively with stable chemical bonds simply and cost-effectively. See the ESI† for more details about HPLC chromatograms and LC-MS spectra (Table S4 and Fig. S4–S6†).
After obtaining three modified cyclic peptides, we performed HTRF assay to acquire their binding affinity targeting the WBM region of WDR5 protein in vitro. Before testing the selected peptides, HTRF assay had been proven to be reliable because the positive control peptide SV-10, a peptide derived from MYC MbIIIb, had an affinity with an EC50 of 1.6 ± 0.1 μM (Fig. S7†) that was comparable with Kd = 9.3 ± 1.7 μM in the literature.15 Based on the results (Fig. 5A), we find that cyclic peptide CYC3 (Fig. 5B) has better binding activity with an EC50 of 8.1 ± 0.5 μM among all three synthetic peptides. As shown in the figure, all three cyclopeptides have relatively low inhibition at the high concentration, which is due to poor solubility in the aqueous solution.
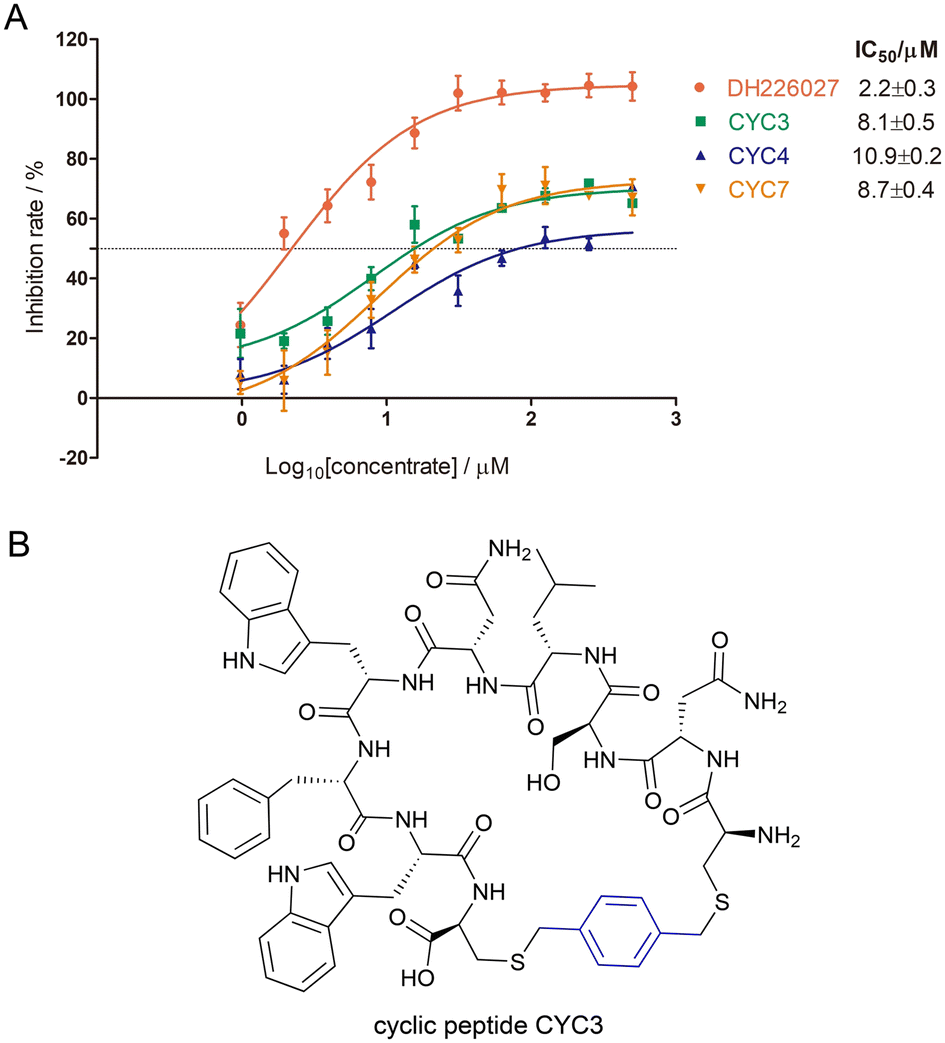 | ||
| Fig. 5 (A) Binding affinity derived from HTRF assay of three modified cyclic peptides targeting the WDR5-WBM site (the positive control peptide SV-10: EC50 = 1.6 ± 0.1 μM). (B) The structure of cyclic peptide CYC3. | ||
Furthermore, the stability of cyclic peptides with a disulfide bond or a chemical linker against a reduction environment was studied. Under the reduction conditions, both DH226027 and CYC3 were dissolved in the acidic or neutral methanol buffer with tris(2-carboxyethyl)phosphine (TCEP), and the changes of the peptide structure at different time points were detected by the HPLC system. As shown in Fig. 6, the disulfide bond in the cyclic peptide DH226027 (HPLC peak 1) was broken rapidly and the corresponding linear peptide (HPLC peak 2) was observed in 30 min under pH 3.0 and pH 7.4 conditions. However, under both conditions, chemically modified cyclic peptide CYC3 was pretty stable without any changes after two hours. The results of this experiment supported our hypothesis that cyclization with chemical linkers could improve stability against a reduction environment under acidic and neutral conditions.
 | ||
| Fig. 6 HPLC chromatograms of the stability experiments. (A) Peptide DH226027 was dissolved in pH 3.0 solution. (B) Peptide DH226027 was dissolved in pH 7.4 solution. (C) Peptide CYC3 was dissolved in pH 3.0 solution. (D) Peptide CYC3 was dissolved in pH 7.4 solution. | ||
Nevertheless, we found CYC3 has similar binding activity to the original DH226027, and could serve as a new stable cyclic peptide for further biological evaluation.
Conclusions
As an important mediator in several protein complexes, WDR5 protein plays a substantial role in anti-cancer drug development. This article focuses on new peptide-mimic inhibitors of the WBM region in WDR5, which have been seldom reported. At first, we established two different screening strategies with a disulfide-based cyclic peptide library (Ph.D.™-C7C phage library) that could reduce the proportions of TUPs, and obtained a series of specific peptides targeting the WBM binding site. And then, considering sequencing data and molecular docking ADCP energy, the candidate peptide DH226027 which has good binding activity at the μM level was identified and further modified with different cyclization chemistries. Finally, we found a phenyl-bridged cyclic peptide (CYC3) towards the WDR5-WBM pocket. It has better stability and similar binding affinity compared with the original DH226027. In summary, we believe that our selection and optimization strategies work well for developing peptide-mimic inhibitors in the protein–protein interaction areas. Besides, these opportunities and challenges present a strong case for applying widely the phage display technique in drug discovery and peptide therapeutics.Experimental
Protein expression and purification for biopanning
The WDR5 (residues 23–483) expression strain with His6-sumo tag at N-terminal was preserved in-house.23 The 500 mL terrific broth cultures supplemented with 50 μg mL−1 kanamycin were inoculated with 1/100 saturated E. coli bacterial culture for overnight growth at 37 °C until the OD600 reached 0.8. The protein was induced at 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) and cultured at 18 °C for 16 h before harvest. The cells were lysed using an ultra-high-pressure cell disruptor (UH-03, Union, China) in the lysis buffer (50 mM HEPES pH 7.5, 250 mM NaCl, 20 mM imidazole, 5% (v/v) glycerol and 5 mM TCEP). After centrifugation with 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at 4 °C for 1 h, the supernatant was loaded onto a Ni Sepharose 6 FF (Cytiva, Danaher Corporation) gravity column. The target protein was eluted with elution buffer (20 mM Hepes pH 7.5, 250 mM NaCl, 250 mM imidazole, 5 mM TCEP, and 5% (v/v) glycerol) after the washing procedure with several 30–50 mM imidazole containing buffers. The elution was concentrated with 10000 MWCO Vivaspin® 20 centrifugal concentrators (Cytiva, Danaher corporation) at 4000 g on the centrifuge 5810R (Eppendorf) at 4 °C before loading onto a Hiload 16/600 Superdex® 200 pg column (Cytiva) in the buffer (20 mM HEPES pH 7.5, 250 mM NaCl, 5 mM TECP). The fractions with an absorption peak around 89 mL were pooled and concentrated to 10 mg mL−1, and then stored in 20% glycerol at −80 °C.
000 rpm at 4 °C for 1 h, the supernatant was loaded onto a Ni Sepharose 6 FF (Cytiva, Danaher Corporation) gravity column. The target protein was eluted with elution buffer (20 mM Hepes pH 7.5, 250 mM NaCl, 250 mM imidazole, 5 mM TCEP, and 5% (v/v) glycerol) after the washing procedure with several 30–50 mM imidazole containing buffers. The elution was concentrated with 10000 MWCO Vivaspin® 20 centrifugal concentrators (Cytiva, Danaher corporation) at 4000 g on the centrifuge 5810R (Eppendorf) at 4 °C before loading onto a Hiload 16/600 Superdex® 200 pg column (Cytiva) in the buffer (20 mM HEPES pH 7.5, 250 mM NaCl, 5 mM TECP). The fractions with an absorption peak around 89 mL were pooled and concentrated to 10 mg mL−1, and then stored in 20% glycerol at −80 °C.
Biotinylation of WDR5 protein
WDR5 was biotinylated for the alternate magnetism phage biopanning with an EZ-Link™ Sulfo-NHS-LC-Biotinylation kit (Thermo Scientific). The protein was buffer-exchanged to amine-free PBS buffer with a single-use desalting column included in the kit. Then, 500 μL of 230 μM WDR5 in the PBS buffer was added to the 230 μL of freshly prepared 10 mM sulfo-NHS-LC-biotin solution. The reaction was incubated on ice for two hours before buffer exchange to remove the excess biotin reagents. Finally, a HABA assay was performed to verify the level of biotin incorporation, and the biotin/mole value of protein was detected after calculating with the HABA online calculator (ThermoFisher Scientific).Selective biopanning of phage display cyclic peptides
Phage display peptide library Ph.D.™-C7C (New England Biolabs Inc) was employed for three rounds of biopanning. To obtain specific peptides binding to the WBM site of WDR5, a positive inhibitor (WIN-IN-4; CAS: 2407457-36-5) was used to occupy the WIN site of WDR5. Two solution-phase panning strategies with both His-tagged target and biotinylated protein were adopted.In the subtractive panning, we used Pierce™ Ni-NTA magnetic agarose beads (ThermoFisher Scientific) for all three cycles. 200 μL of 4 μg His-tagged WDR5 protein was incubated with 10 μM WIN-IN-4 for 1 h at 4 °C in a 2 mL Eppendorf tube coated with the blocking buffer (PBS containing 1% BSA). 10 μL phage library (2 × 1013 pfu mL−1) was diluted in the mixture for further incubation with gentle rotation overnight. After washing and coating, 30 μL Ni-NTA magnetic agarose beads were added to the tube for capturing protein and specific phages at room temperature for 1 h. Unbound phages were washed eight times with PBS containing 0.1% Tween-20 and 20 mM imidazole. Two more washes were followed with 10 μM WIN-IN-4 added in the wash buffer. Then, the bound phages were eluted with the elution buffer (0.2 M glycine-HCl (pH 2.2), 1 mg mL−1 BSA) for 15 min at room temperature. 1 M tris (pH 9.1) was added immediately to neutralize the elution. The eluate was tested for phage titration after each round and amplified after the first two cycles as described in the manufacturer's protocol. More importantly, the amplified phages should be mixed twice for half an hour with 100 μL coated Ni-NTA magnetic agarose beads to deplete false-positive TUPs binding directly to beads before next round screening.
The alternant panning is the same as the previous panning, but the magnetic beads were replaced with Pierce™ Streptavidin magnetic beads (ThermoFisher Scientific) to prevent the enrichment of Ni-specific peptides and imidazole was omitted from the wash buffer in the second round. There was no need for the depleting procedure. Moreover, WIN-IN-4 was added in the mixture for competitive binding behind the incubation between WDR5 protein and the phage library, and 10 μM WIN-IN-4 was kept in the wash buffer throughout three rounds. In both selection experiments, the amplified phages were calculated to be 2 × 1011 pfu as an input according to the results of titration. Stringency was gradually enhanced by reducing the amount of protein stepwise (4 μg, 2 μg, 0.5 μg) while enhancing the concentration of Tween-20 in the wash buffer (0.1%, 0.3%, 0.5%, respectively).
Sequence analysis of the selected clones
The sequenced clones were acquired from a colony-PCR protocol directly instead of purifying the phage DNA for more convenience. After titer experiments, blue plaques from the titration plates were picked for a PCR reaction. The primers for the PCR reaction were:| Forward C7C-1: 5′-GTCGGCGCAACTATCGGTATC-3′; |
| Reverse C7C-R1: 5′-GCCCTCATAGTTAGCGTAACG-3′. |
The PCR reaction mixture has a total volume of 50 μL including 25 μL of 2× Hieff robust PCR master mix (Yeasen, Suzhou), 1 μL of forward and reverse primers each at 0.4 μM, and the blue plaque picked as a template. The PCR reaction was started by initial denaturation (95 °C, 30 s), followed by 30 cycles of denaturation, annealing, and extension (95 °C for 15 s, 53 °C for 15 s and 72 °C for 30 s). All steps were performed on a SimpliAmp Thermal Cycler. The 313 bp PCR products appeared above the 250 bp DNA marker at 1.5% agarose gel. After that, the target fragments were sent to Personalbio, Inc. (Shanghai China) for Sanger sequencing.
Virtual screening with AutoDock CrankPep (ADCP)
The PDB files of cyclic peptides identified from sequencing analysis were prepared with PyMOL. Docking cyclic peptides to the WBM site of WDR5 was performed using ADCP v1.1. The docking box was set to the WBM site using a ligand (MYC MbIIIb peptide) extracted from 4y7r (PDB code). After computing pockets with AutoSite 1.1, four pockets appeared, and all fills were selected to generate the target file. We performed the docking into this box, performed 50 independent searches, allowed each search to consume 2.5 million evaluations, and peptides were cyclic through cysteine option.Synthesis of the selected peptides with disulfide bonds
A total of 7 disulfide-bridged cyclic peptides were synthesized by DongHeng Biopharm Co., Ltd. (Hangzhou, China). They were prepared by solid-phase peptide synthesis on an Advanced ChemTech 348 Ω peptide synthesizer (Aapptec, Louisville, USA) using standard 9-fluorenylmethoxycarbonyl (Fmoc) chemistry and Rink amide-MBHA resin (GL Biochem, Shanghai, China). Firstly, the Fmoc protected amino-acid attached to the resin was deprotected by 20% v/v piperidine in DMF. The subsequent amino acid protected by Fmoc at N-terminal was activated by HBTU in the presence of base NMM to form an acetic ester and was coupled to the C-terminal amino acid attached to the resin forming a dipeptide. Similarly, the remaining amino acids were coupled to form the complete sequence one by one. Finally, Fmoc groups was removed by incubating the resin twice with 20% v/v pyridine in DMF. The resin was washed in DMF four times before Fmoc removal and five times after Fmoc removal.The crude peptides were deprotected and cleaved from the resin under reducing conditions by incubation in the cleavage solution (90% TFA, 2.5% H2O, 2.5% thioanisole, 2.5% phenol, 2.5% EDT) with shaking for 4 h at room temperature. And then, they were isolated and purified by preparative high-performance liquid chromatography (Pre-HPLC, Agilent, Inc., USA). The final products were lyophilized for storing. The purity of the peptides was established by analytical HPLC (Agilent 1100, Agilent, Inc., USA), and the composition of the peptides was confirmed using a mass spectrometer (Bruker Daltonics micro TOF-Q, Hamburg, Germany). See the ESI† for more details (Fig. S8–S13).
Homogeneous time-resolved fluorescence (HTRF) binding assay
The binding activity of cyclic peptides towards the WBM site of WDR5 was tested by HTRF binding assay. The competitive substrate (RBBP5) peptide AAEDEEVDVTSVD with biotinylation at N-terminal and the positive control peptide SV-10 (SEEEIDVVSV) were synthesized by DongHeng Biopharm Co., Ltd. (Hangzhou, China). The WDR5 protein with His-sumo tag at N-terminal was aliquoted from storage. The fluorescent donor MAb anti-6His Tb and the fluorescent receptor streptavidin-d2 used in the assay were purchased from Cisbio (PerkinElmer). The reaction was conducted with 20 μL final volume of terbium detection buffer (Cisbio) in an HTRF 96-well low volume plate (white). 5 μL of WDR5 protein at 20 nM and 5 μL of substrate peptide at 4 nM were mixed. 5 μL serially doubly diluted compound solution (SV-10, DH226027, CYC3, CYC4 and CYC7, final concentration from 500 μM to 976.6 nM) were followed. Finally, a 5 μL mixture of MAb anti-6His Tb and streptavidin-d2 at a 1/100 dilution was added. After incubation at room temperature for 3 h, the plate was assayed on a Biotek Synergy Neo2 multi-mode microplate reader (excitation at 330 nm, emission at both 620 nm and 665 nm, Agilent). The measurement was conducted 3 times for reproducibility. The HTRF signal was calculated by signal ratio: [intensity (665 nm)/intensity (620 nm)] × 104. The inhibition was calculated by the following formula. The results were analyzed by GraphPad Prism software using a non-linear curve-fitting method.
S max: the signal ratio of positive control without compounds; Smin: the signal ratio of negative control without protein; S: the signal ratio of each compound.
NMR spectroscopy
[1H, 15N] HSQC spectra of 15N-labeled WDR5 with or without 10-fold molar excess of DH226027 were recorded on a Bruker 800 MHz NMR spectrometer equipped with a cryogenically cooled probe at 25 °C. The samples were prepared using a HEPES buffer containing 20 mM 2-[4-(2-hydroxyethyl) piperazin-1-yl] ethanesulfonic acid, 250 mM NaCl, 5 mM TCEP and 10% D2O (pH 7.5), and the final concentration of WDR5 was 100 μM. The spectra were processed by using NMRPipe40 and Sparky.41Cyclization and purification of peptides with different chemical linkers
The chemical reagents were commercially available and used without further purification. The linear peptide DH226027-L was synthesized by DongHeng Biopharm Co., Ltd. (Hangzhou, China) as previously described. 10 mg lyophilized powder DH226027-L was dissolved in 8.5 mL methanol under the ultrasound conditions. Then, the cyclization linker (2 equiv.) and base triethylamine (3 equiv.) were added stepwise, and the mixture was stirred at 30 °C for 2 h. The reaction was quenched with 40% formic acid finally. Products were separated and purified with a C18 reversed-phase silica-gel column under methanol/water conditions. Fractions were collected and analyzed with an analytical HPLC system equipped with a C18 reversed-phase column (X-bridge peptide BEH C18 5 μm, 300 Å, 10 × 250 mm, Waters), applying a flow rate of 0.1 mL min−1 in 10 min and a mobile phase composed of solvent A (99.9% v/v H2O and 0.1% v/v TFA) and solvent B (99.9% v/v ACN and 0.1% v/v TFA). The gradient elution condition is shown in Table S5.† The absorbance at a wavelength of 220 nm was used. The desired fractions were pooled, concentrated and successively lyophilized. In the end, the purity was assessed by analytical HPLC as previously mentioned, and the mass was determined by electrospray ionization mass spectrometry (ESI-MS) in positive ion mode on a single quadrupole liquid chromatography-mass spectrometer (Thermo Finnigan LCQ Deca XP LCMS-MS system, USA).Stability experiments of cyclic peptides
The disulfide-bridged cyclic peptide DH226027 and the phenyl-bridged cyclic peptide CYC3 were chosen for the stability test. Additionally, in order to investigate the effects of pH values and reduction conditions of buffer solutions, methanol buffers with TCEP at pH 3.0 and 7.4 were applied. The pH was measured using a Sartorius PB-10 basic benchtop pH meter equipped with a pH combination gel-filling electrode. Both powdered cyclic peptides were dissolved in two kinds of buffers that had been previously filtered through a 0.22 μm filter. The solutions were brought to a final volume of 2 mL, resulting in concentrations of 0.5 mM peptides and 1.0 mM TCEP. Then, the solutions were taken at different time points and analyzed using an analytical HPLC system.Author contributions
The project was conceptualized by B. Xiong and D. Cao. Biopanning experiments were designed and performed by D. Cao and L. Song. Protein expression was completed by L. Song and Z. Du. Sequence analysis and binding assay were conducted by D. Cao and L. Song. NMR data were provided by N. Zhang. Chemical analysis and validation were interpreted by L. Song and L. Chen. The manuscript was written by B. Xiong, D. Cao and L. Song.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (82173658, 82373720).References
- B. Sadikovic, K. Al-Romaih, J. A. Squire and M. Zielenska, Curr. Genomics, 2008, 9, 394–408 CrossRef CAS PubMed.
- T. C. S. Ho, A. H. Y. Chan and A. Ganesan, J. Med. Chem., 2020, 63, 12460–12484 CrossRef CAS PubMed.
- J. P. Hu, C. Q. Tian, M. S. Damaneh, Y. L. Li, D. Y. Cao, K. K. Lv, T. Yu, T. Meng, D. Q. Chen, X. Wang, L. Chen, J. Li, S. S. Song, X. J. Huan, L. H. Qin, J. K. Shen, Y. Q. Wang, Z. H. Miao and B. Xiong, J. Med. Chem., 2019, 62, 8642–8663 CrossRef CAS PubMed.
- N. Li, H. Yang, K. Liu, L. W. Zhou, Y. T. Huang, D. Y. Cao, Y. L. Li, Y. L. Sun, A. S. Yu, Z. Y. Du, F. Yu, Y. Zhang, B. Y. Wang, M. Y. Geng, J. Li, B. Xiong, S. L. Xu, X. Huang and T. C. Liu, J. Med. Chem., 2022, 65, 9459–9477 CrossRef CAS PubMed.
- S. Valente, Y. W. Liu, M. Schnekenburger, C. Zwergel, S. Cosconati, C. Gros, M. Tardugno, D. Labella, C. Florean, S. Minden, H. Hashimoto, Y. Q. Chan, X. Zhang, G. Kirsch, E. Novellino, P. B. Arimondo, E. Miele, E. Ferretti, A. Gulino, M. Diederich, X. D. Cheng and A. Mai, J. Med. Chem., 2014, 57, 701–713 CrossRef CAS PubMed.
- Z. Q. Zhang, Z. H. Guo, X. W. Xu, D. Y. Cao, H. Yang, Y. L. Li, Q. Y. Shi, Z. Y. Du, X. B. Guo, X. Wang, D. Q. Chen, Y. Zhang, L. Chen, K. X. Zhou, J. Li, M. Y. Geng, X. Huang and B. Xiong, J. Med. Chem., 2021, 64, 16650–16674 CrossRef CAS PubMed.
- J. F. Couture, E. Collazo and R. C. Trievel, Nat. Struct. Mol. Biol., 2006, 13, 698–703 CrossRef CAS PubMed.
- J. H. Lee, C. M. Tate, J. S. You and D. G. Skalnik, J. Biol. Chem., 2007, 282, 13419–13428 CrossRef CAS PubMed.
- J. Dias, N. Van Nguyen, P. Georgiev, A. Gaub, J. Brettschneider, S. Cusack, J. Kadlec and A. Akhtar, Genes Dev., 2014, 28, 929–942 CrossRef CAS PubMed.
- L. S. Ee, K. N. McCannell, Y. Tang, N. Fernandes, W. R. Hardy, M. R. Green, F. Chu and T. G. Fazzio, Stem Cell Rep., 2017, 8, 1488–1496 CrossRef CAS PubMed.
- Y. L. Wang, F. Faiola, M. Xu, S. Pan and E. Martinez, J. Biol. Chem., 2008, 283, 33808–33815 CrossRef CAS PubMed.
- Y. W. Cho, T. Hong, S. Hong, H. Guo, H. Yu, D. Kim, T. Guszczynski, G. R. Dressler, T. D. Copeland, M. Kalkum and K. Ge, J. Biol. Chem., 2007, 282, 20395–20406 CrossRef CAS PubMed.
- X. Chen, J. Xu, X. Wang, G. Long, Q. You and X. Guo, J. Med. Chem., 2021, 64, 10537–10556 CrossRef CAS PubMed.
- Y. W. Yang, R. A. Flynn, Y. Chen, K. Qu, B. B. Wan, K. C. Wang, M. Lei and H. Y. Chang, eLife, 2014, 3, e02046 CrossRef PubMed.
- L. R. Thomas, Q. Wang, B. C. Grieb, J. Phan, A. M. Foshage, Q. Sun, E. T. Olejniczak, T. Clark, S. Dey, S. Lorey, B. Alicie, G. C. Howard, B. Cawthon, K. C. Ess, C. M. Eischen, Z. Zhao, S. W. Fesik and W. P. Tansey, Mol. Cell, 2015, 58, 440–452 CrossRef CAS PubMed.
- S. Chacon Simon, F. Wang, L. R. Thomas, J. Phan, B. Zhao, E. T. Olejniczak, J. D. Macdonald, J. G. Shaw, C. Schlund, W. Payne, J. Creighton, S. R. Stauffer, A. G. Waterson, W. P. Tansey and S. W. Fesik, J. Med. Chem., 2020, 63, 4315–4333 CrossRef CAS PubMed.
- J. Tian, K. B. Teuscher, E. R. Aho, J. R. Alvarado, J. J. Mills, K. M. Meyers, R. D. Gogliotti, C. Han, J. D. Macdonald, J. Sai, J. G. Shaw, J. L. Sensintaffar, B. Zhao, T. A. Rietz, L. R. Thomas, W. G. Payne, W. J. Moore, G. M. Stott, J. Kondo, M. Inoue, R. J. Coffey, W. P. Tansey, S. R. Stauffer, T. Lee and S. W. Fesik, J. Med. Chem., 2020, 63, 656–675 CrossRef CAS PubMed.
- G. P. Smith, Science, 1985, 228, 1315–1317 CrossRef CAS PubMed.
- J. Pande, M. M. Szewczyk and A. K. Grover, Biotechnol. Adv., 2010, 28, 849–858 CrossRef CAS PubMed.
- M. A. Abdalla and L. J. McGaw, Molecules, 2018, 23, 2080 CrossRef PubMed.
- A. Zorzi, K. Deyle and C. Heinis, Curr. Opin. Chem. Biol., 2017, 38, 24–29 CrossRef CAS PubMed.
- S. S. Usmani, G. Bedi, J. S. Samuel, S. Singh, S. Kalra, P. Kumar, A. A. Ahuja, M. Sharma, A. Gautam and G. P. S. Raghava, PLoS One, 2017, 12, e0181748 CrossRef PubMed.
- J. Cao, T. Fan, Y. Li, Z. Du, L. Chen, Y. Wang, X. Wang, J. Shen, X. Huang, B. Xiong and D. Cao, Molecules, 2021, 26, 1225 CrossRef CAS PubMed.
- W. D. Thomas, M. Golomb and G. P. Smith, Anal. Biochem., 2010, 407, 237–240 CrossRef CAS PubMed.
- C. F. Barbas, J. S. Rosenblum and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 6385–6389 CrossRef CAS PubMed.
- P. C. Weber, M. W. Pantoliano and L. D. Thompson, Biochemistry, 1992, 31, 9350–9354 CrossRef CAS PubMed.
- M. Vodnik, U. Zager, B. Strukelj and M. Lunder, Molecules, 2011, 16, 790–817 CrossRef CAS PubMed.
- J. Huang, B. Ru, S. Li, H. Lin and F. B. Guo, J. Biomed. Biotechnol., 2010, 2010, 101932 Search PubMed.
- G. Weng, J. Gao, Z. Wang, E. Wang, X. Hu, X. Yao, D. Cao and T. Hou, J. Chem. Theory Comput., 2020, 16, 3959–3969 CrossRef CAS PubMed.
- G. E. Mudd, A. Brown, L. Chen, K. van Rietschoten, S. Watcham, D. P. Teufel, S. Pavan, R. Lani, P. Huxley and G. S. Bennett, J. Med. Chem., 2020, 63, 4107–4116 CrossRef CAS PubMed.
- P. Diderich, D. Bertoldo, P. Dessen, M. M. Khan, I. Pizzitola, W. Held, J. Huelsken and C. Heinis, ACS Chem. Biol., 2016, 11, 1422–1427 CrossRef CAS PubMed.
- S. S. Kale, C. Villequey, X. D. Kong, A. Zorzi, K. Deyle and C. Heinis, Nat. Chem., 2018, 10, 715–723 CrossRef CAS PubMed.
- L. E. J. Smeenk, N. Dailly, H. Hiemstra, J. H. Van Maarseveen and P. Timmerman, Org. Lett., 2012, 14, 1194–1197 CrossRef CAS PubMed.
- P. Timmerman, J. Beld, W. C. Puijk and R. H. Meloen, ChemBioChem, 2005, 6, 821–824 CrossRef CAS PubMed.
- Y. Wang and D. H.-C. Chou, Angew. Chem., Int. Ed., 2015, 54, 10931–10934 CrossRef CAS PubMed.
- N. Assem, D. J. Ferreira, D. W. Wolan and P. E. Dawson, Angew. Chem., Int. Ed., 2015, 54, 8665–8668 CrossRef CAS PubMed.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- M. T. W. Lee, A. Maruani and V. Chudasama, J. Chem. Res., 2016, 1–9, DOI:10.3184/174751916x14495034614855.
- G. A. Woolley, Acc. Chem. Res., 2005, 38, 486–493 CrossRef CAS PubMed.
- F. Delaglio, S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer and A. Bax, J. Biomol. NMR, 1995, 6, 277–293 CrossRef CAS PubMed.
- D. G. Kneller and I. D. Kuntz, J. Cell. Biochem., 1993, 254–254 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00288h |
| This journal is © The Royal Society of Chemistry 2023 |