
Restoring susceptibility to aminoglycosides: identifying small molecule inhibitors of enzymatic inactivation
Angel J.
Magaña
a,
Jan
Sklenicka
a,
Clemencia
Pinilla
b,
Marc
Giulianotti
b,
Prem
Chapagain
 cd,
Radleigh
Santos
e,
Maria Soledad
Ramirez
a and
Marcelo E.
Tolmasky
*a
cd,
Radleigh
Santos
e,
Maria Soledad
Ramirez
a and
Marcelo E.
Tolmasky
*a
aCenter for Applied Biotechnology Studies, Department of Biological Science, College of Natural Sciences and Mathematics, California State University Fullerton, Fullerton, CA 92831, USA. E-mail: mtolmasky@fullerton.edu
bCenter for Translational Science, Florida International University, Port St. Lucie, FL 34987, USA
cDepartment of Physics, Florida International University, Miami, FL 33199, USA
dBiomolecular Sciences Institute, Florida International University, Miami, FL 33199, USA
eDepartment of Mathematics, Nova Southeastern University, Fort Lauderdale, FL 33314, USA
First published on 21st July 2023
Abstract
Growing resistance to antimicrobial medicines is a critical health problem that must be urgently addressed. Adding to the increasing number of patients that succumb to infections, there are other consequences to the rise in resistance like the compromise of several medical procedures and dental work that are heavily dependent on infection prevention. Since their introduction in the clinics, aminoglycoside antibiotics have been a critical component of the armamentarium to treat infections. Still, the increase in resistance and their side effects led to a decline in their utilization. However, numerous current factors, like the urgent need for antimicrobials and their favorable properties, led to renewed interest in these drugs. While efforts to design new classes of aminoglycosides refractory to resistance mechanisms and with fewer toxic effects are starting to yield new promising molecules, extending the useful life of those already in use is essential. For this, numerous research projects are underway to counter resistance from different angles, like inhibition of expression or activity of resistance components. This review focuses on selected examples of one aspect of this quest, the design or identification of small molecule inhibitors of resistance caused by enzymatic modification of the aminoglycoside. These compounds could be developed as aminoglycoside adjuvants to overcome resistant infections.
Introduction
Aminoglycoside antibiotics became a viable therapy to treat infections in 1944 when streptomycin was introduced for clinical use. In November of that year, a woman with tuberculosis became the first patient to be treated with an aminoglycoside. The treatment was successful, and the patient lived an active life.1 From that moment until now, new aminoglycosides, discovered or generated by modification of natural ones (semisynthetic), were used alone or in combination therapies to treat tuberculosis and numerous other infections.2–4 Aminoglycoside antibiotics are primarily used against infections caused by Gram-negative aerobic bacilli, staphylococci, and other Gram-positives.4–6 Examples of causative agents of infections that are treated with aminoglycosides include, but are not limited to, Enterobacterales, Pseudomonas, Acinetobacter, Yersinia pestis, Francisella tularensis, and Staphylococcus.4–8 Besides their utilization against bacterial infection, some aminoglycosides are also used to treat cryptosporidiosis, leishmaniasis, and other diseases caused by protozoa and cestodes.9,10 The ability of aminoglycosides to suppress premature translation-termination codon caused by missense mutations led to their use as a treatment for other conditions like cystic fibrosis, Duchenne muscular dystrophy, Ménière's disease, and others.11–18 Aminoglycosides are also being researched as gene delivery vectors for gene therapies.19,20The chemical structure of aminoglycosides consists of an aminocyclitol nucleus (streptamine, 2-deoxystreptamine, or streptidine) linked to amino sugars through glycosidic bonds. Exceptions to this rule are spectinomycin, an aminocyclitol not linked to amino sugars, or compounds that include the aminocyclitol fortamine.4,6,21,22 Initially, all aminoglycosides were of natural origin, isolated from soil bacteria belonging to the genera Streptomyces (named with the suffix “-mycin”) or Micromonospora (named with the suffix “-micin”). However, the development of resistance together with the aminoglycosides' relatively high toxicity, led to the development of semisynthetic molecules that are less toxic or refractory to one or more resistance mechanisms (see the representative referenced articles and reviews for a more extensive description).23–36 Examples of aminoglycoside structures are shown in Fig. 1.
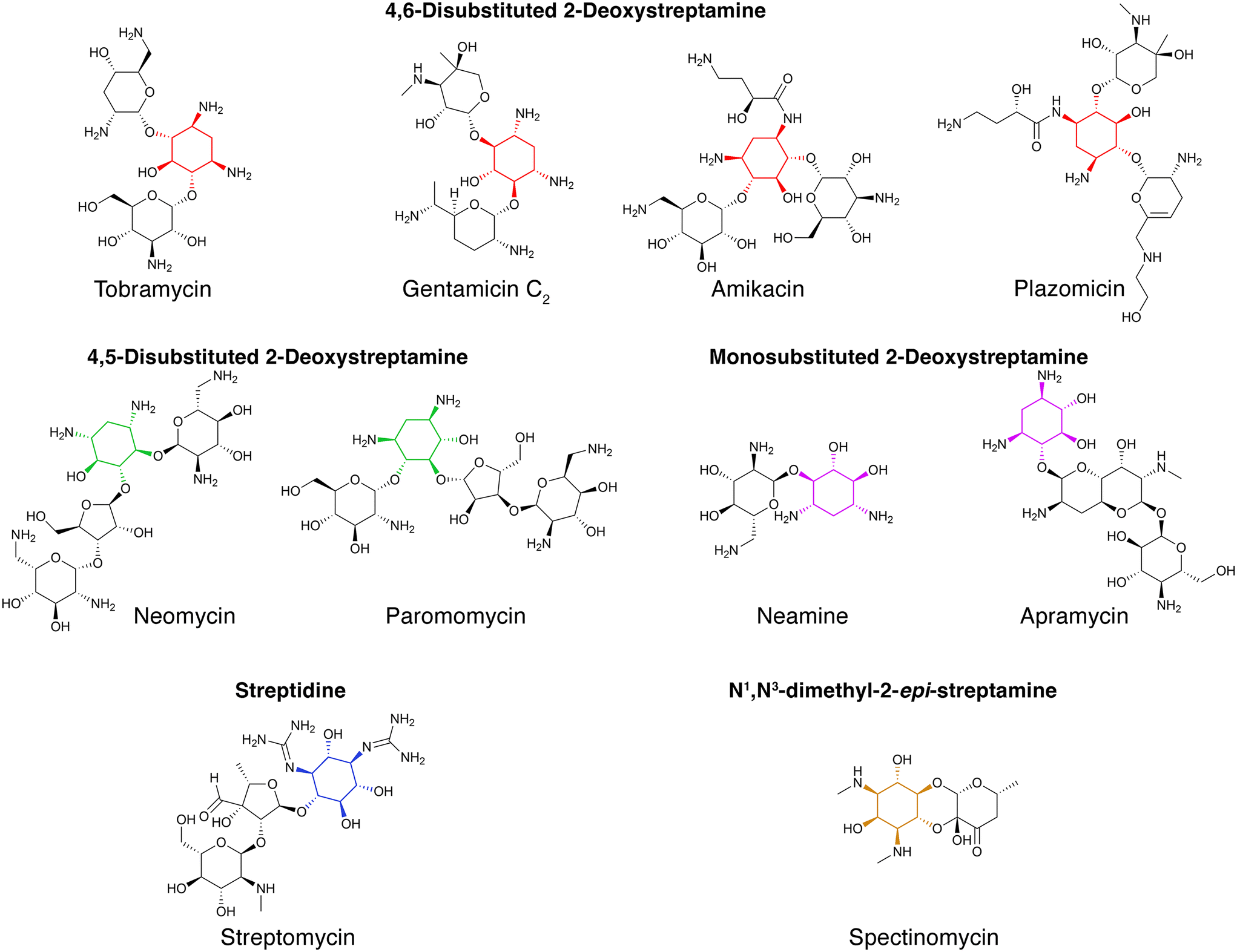 | ||
| Fig. 1 Chemical structures of representative aminoglycosides. Representative structures of aminoglycosides, which contain an aminocyclitol nucleus (colored) linked to amino sugars through glycosidic bonds.6 Spectinomycin contains an aminocyclitol not linked to amino sugars.22 | ||
When aminoglycosides are in the environment that surrounds bacterial cells, they are attracted to the negatively charged bacterial surface. The interactions between the polycationic aminoglycosides and the anionic cell surface components result in increased permeability, permitting the energy-independent internalization of a limited number of molecules into the periplasm.37 Two energy-dependent phases follow this phase. The energy-dependent phase I is characterized by the penetration to the cytoplasm of a small number of aminoglycoside molecules. This process requires a functional electron transport system.38–40 The need for a functional electron transport system determines that, generally, aminoglycosides are ineffective against anaerobic bacteria.41 Once inside the cytosol, the aminoglycoside molecules bind the ribosomes and induce mistranslation. In the energy-dependent phase II, faulty membrane proteins damage the cytoplasmic membrane integrity allowing the access of a large number of aminoglycoside molecules to the cytosol.40 As the number of aminoglycoside molecules increases within the cytosol, the number of mistranslated proteins grows to enhance the membrane's damage, resulting in a yet faster uptake that ultimately results in cell death.40,42,43
Through binding the 30S ribosome subunit, aminoglycosides induce a modification of the conformation of the A site, making it closer to that adopted after the interaction between an mRNA and its cognate tRNA. This process reduces the ribosome proofreading properties, increasing mistranslation.44–48 Although all aminoglycosides cause mistranslation, not all bind to identical sites of the 16S rRNA. Furthermore, aminoglycosides produce other effects like inhibition of assembly of the 30S ribosomal subunit (detected for the aminoglycosides neomycin and paromomycin), various disruptions of ribozyme activities, modification of transcription rates, and interference with the formation of the Z ring, these latter two effects at sublethal concentrations of the antibiotic.49–53
Aminoglycosides have numerous advantages as anti-infectives. Besides being highly efficacious, their properties include broad-spectrum, rapid action, low allergies, and low interactions with other medicines. However, a combination of factors like their side effects, development of resistance, and inconvenient routes of administration diminished the interest in this kind of antimicrobials.25 Aminoglycosides' main side effects are ototoxicity, nephrotoxicity, and, less commonly, neuromuscular blockade.5,25,28,54–56 Ototoxicity may result in permanent hearing loss caused by damage to the inner ear sensory hair cells.56 Nephrotoxicity is usually reversible; the most common manifestation is nonoliguric acute kidney injury (due to decreased glomerular filtration).55 Fortunately, several studies are being conducted to find drugs to protect against these undesirable effects.57–59
Aminoglycoside resistance mechanisms, which in many cases coexist in the same bacterial cells,48,60,61 include enzymatic inactivation by modifying enzymes;5,62–64 methylation of 16S rRNA;65,66 mutation of the 16S rRNA or ribosomal proteins to prevent binding;65 reduced uptake that can occur by reduced inner membrane transport or modification of outer membrane's permeability;67 export outside the cell by efflux pumps,68 and other more rare mechanisms like active swarming or sequestration of the drug by tightly binding to an acetyltransferase of deficient activity.69 Of all these mechanisms, the antibiotic's enzymatic inactivation mediated by aminoglycoside modifying enzymes, which catalyze the transfer of acetyl, nucleotidyl, or phosphate groups to –OH or –NH2 positions of the aminocyclitol or the sugar moieties of the molecules, is the most prevalent in the clinical setting.5,28,62–64,70
The current antibiotic resistance crisis has reawakened interest in aminoglycoside antibiotics.6,25,60 There are renewed efforts to repurpose aminoglycosides not yet used in humans like apramycin70 or design new molecules refractory to resistant mechanisms that also have lower toxicity. A product of these efforts is the recent approval for human use of plazomicin.71 Another strategy to preserve aminoglycosides as part of the armamentarium against infections is the development of inhibitors of the resistance that, in combination with an antimicrobial, would be active against resistant pathogens.61 Although aminoglycoside resistance inhibitors have not progressed beyond the experimental laboratory phase, the success obtained with similar strategies in the case of inhibitors of β-lactamases, which are already part of combination therapies with β-lactams,72 strongly suggests that it is a viable option in the fight against aminoglycoside resistance. The main strategies to overcome aminoglycoside modifying enzymes-mediated resistance consist of designing compounds that prevent the expression of resistance genes, such as antisense compounds, or inhibit the enzymatic inactivation of aminoglycosides (Fig. 2). However, other possibilities like identifying compounds such as the proline-rich antimicrobial peptide OM19r, which augment the antibacterial activity of aminoglycosides turn them effective against resistant strains are also being researched.73
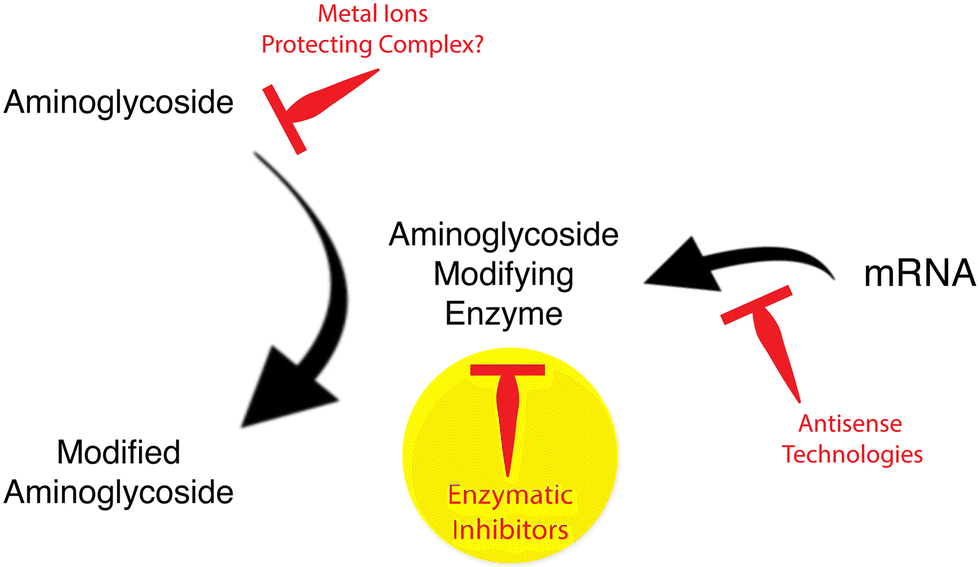 | ||
| Fig. 2 Strategies to reduce aminoglycoside modifying enzymes-mediated resistance to aminoglycoside antibiotics. The yellow circle points to the focus of this review article. | ||
Numerous antisense-mediated mechanisms of inhibition of gene expression have been explored to interfere with translation by steric hindrance or inducing degradation of the mRNA.74–82 While most attempts to use antisense in bacterial therapeutics target essential genes, i.e., the antisense would be the antibiotic, another less utilized path is to target resistance genes and combine the antisense with the cognate antibiotic. Antisense oligonucleotide analogs were used to silence genes coding for resistance to numerous antibiotics like β-lactams,83–85 carbapenems,86,87 chloramphenicol,88,89 fluoroquinolones,86,90 aminoglycosides,5,75 and others. Experiments on antisense inhibition of resistance to aminoglycosides mediated by the aminoglycoside 6′-N-acetyltransferase type Ib [AAC(6′)-Ib] produced promising results.75,91,92
Reversal of resistance mediated by aminoglycoside modifying acetyltransferases was observed in the presence of some cations (Fig. 2), which interfere with the acetylating reaction.93,94 In the cases of Cu2+ and Zn2+, the concentrations required to observe a phenotypic conversion to susceptibility were significantly reduced when coordination complexes of the cations and ionophores were added to the culture medium.94–99 The mechanism of inhibition of resistance remains to be understood. However, an attractive hypothesis is that the antibiotic in complex with the metal ion becomes a poor substrate.61
A third strategy to counter resistance mediated by aminoglycoside modifying enzymes is the design or identification of enzymatic inhibitors. Several research groups made valuable contributions, and numerous inhibitors with diverse effectiveness have been studied. This review describes representative recent advances in the quest for identifying or designing enzymatic inhibitors of aminoglycoside modifying enzymes. For more extensive descriptions of inhibitors of resistance to aminoglycosides, the reader is referred to other recent review articles.28,29,60,61,63,64,100–106
Bisubstrate prodrugs
The aminoglycoside 6′-N-acetyltransferase type Ii [AAC(6′)-Ii] is an enzyme coded for by a gene located in the chromosome of Enterococcus faecium, which makes this Gram-positive pathogen resistant to several aminoglycosides.107E. faecium, together with E. faecalis, causes the vast majority of enterococcal infections.108 Early strategies to characterize AAC(6′)-Ii led to the synthesis of aminoglycoside-CoA bisubstrates,109 valuable compounds in prior studies on other GCN5-related acetyltransferases that were also inhibitors of their cognate enzymes.110–113 Several aminoglycoside-CoA bisubstrates were not only instrumental in characterizing AAC(6′)-Ii, but they were also potent inhibitors of its activity.29,114 For example, selected neamine-containing bisubstrates (Fig. 3) can inhibit the enzymatic acetylation of aminoglycosides at nanomolar levels.115 However, these compounds cannot reach the bacterial cytosol since the bacterial cell surface is negatively charged. An ingenious way around this problem was to generate prodrugs whose chemical structures are truncated. These compounds do not show inhibitory activity but are internalized and subsequently processed by the host cell's CoA biosynthetic pathway enzymes to produce the active inhibitor.115,116 The proposed mechanism for this conversion consists of a phosphorylation step catalyzed by the pantothenate kinase followed by adenylylation and phosphorylation at the 3′ position of the ribose mediated by phosphopantetheine adenylyltransferase and dephosphocoenzyme A kinase (Fig. 3).115 Bisubstrate inhibitors were also researched for the AAC(6′)-Iy from Salmonella enterica.117 Prodrug bisubstrates are a promising alternative to design adjuvants that inhibit resistance to aminoglycosides mediated by AAC(6′)-Ii and maybe other acetyltransferases.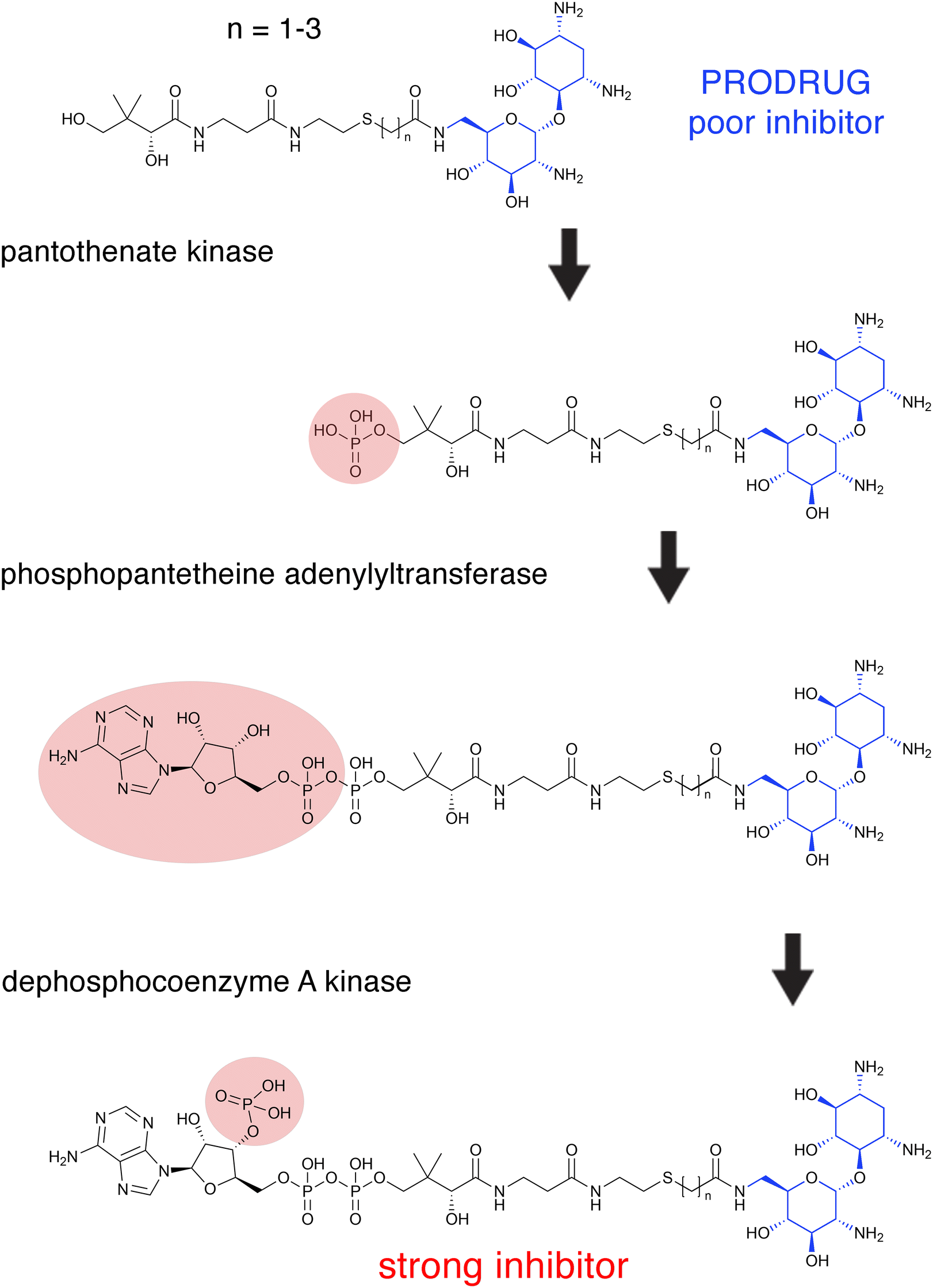 | ||
| Fig. 3 Intracellular prodrug to active bisubstrate inhibitor conversion. Proposed pathway of intracellular conversion of a neamine-containing bisubstrate prodrug (inactive) to an active bisubstrate inhibitor by enzymes of the CoA biosynthetic pathway.115 The donor substrate in all three reactions is ATP. The region of the molecules modified in each enzymatic reaction is circled. The neamine is shown in blue. | ||
Peptide inhibitors of aminoglycoside phosphotransferases and acetyltransferases
The presence of anionic locations that interact with positively charged aminoglycosides was taken advantage of to screen cationic peptides as enzymatic inhibitors.118 Clinically relevant enzymes that confer resistance to aminoglycosides in Gram-positives, APH(3′)-IIIa, AAC(6′)-Ii, and the bifunctional AAC(6′)-Ie-APH(2′′)-Ia, were used in a study to test the inhibitory capabilities of indolicidin, an antimicrobial peptide of bovine origin, and synthetic analogs. Although not all those peptides that showed an inhibitory effect were active against all enzymes tested, all three were susceptible to inhibition by one or more peptides. Determination of the mechanisms showed competitive and noncompetitive inhibition with respect to the aminoglycoside and acetyl CoA, respectively, in the case of AAC(6′)-Ii. Conversely, inhibition of the phosphotransferases occurred through noncompetitive inhibition patterns for both the antibiotic and the donor ATP. Unfortunately, none of the peptides tested in this work reversed resistance in bacterial cells in culture, probably because of their inability to penetrate the cell wall.118Inhibitors of the Mycobacterium tuberculosis Eis acetyltransferase
Tuberculosis, one of the oldest known diseases, continues to be a scourge to humans, and despite multiple efforts to control it, about 1.6 million people succumb to the disease every year.119–121 Aminoglycosides such as kanamycin or amikacin are second-line drugs for treating tuberculosis. Unfortunately, numerous M. tuberculosis strains developed resistance to these antibiotics, mainly through the acetyltransferase known as enhanced intracellular survival (Eis).122 This protein is encoded by the chromosomal gene eis, which is part of the WhiB7 regulon.123 Aminoglycoside resistance M. tuberculosis strains overexpress Eis, a phenotype caused by mutations in the eis promoter region or the 5′-untranslated segment of whiB7.124–126 Unlike most aminoglycoside modifying enzymes, the Eis enzyme catalyzes the acetylation of multiple positions on the antibiotic molecule.122,127 Over ten years of intensive research applying a high throughput screening methodology followed by optimization based on structure–activity relationship studies yielded numerous Eis inhibitors with diverse chemical structures that decrease the resistance to aminoglycosides in resistant M. tuberculosis strains at different levels.128–135 An initial high throughput study screened 123![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds, of which 617 molecules containing an isothiazole S,S-dioxide heterocyclic core (Fig. 4) were selected for further studies that ended up individualizing 17 that were potent inhibitors of resistance when tested in combination with kanamycin.128 This pioneering work showed that it is possible to identify small molecules that inhibit aminoglycoside resistance caused by acetyltransferases. A library of compounds with a sulfonamide scaffold (Fig. 4) contained a compound including a substituted aniline ring that showed strong inhibition of the Eis activity. Analogs to this compound with modifications to the aniline ring were subsequently synthesized and tested, that one consisting of the sulfonamide scaffold with a 2-naphthyl substitution was a potent inhibitor of resistance to kanamycin mediated by Eis.129 Crystal structure analysis showed that the inhibitor binds the Eis protein at the aminoglycoside-binding pocket. Further high throughput screening followed by optimization, by synthesizing compounds with different substituents, in some cases guided by structural information, led to identification of other Eis inhibitors containing the scaffolds methyl 4H-furo[3,2-b]pyrrole-5-carboxylate,130 3-(1,3-dioxolano)-2-indolinone,130 pyrrolo[1,5-a]pyrazine containing a phenyl ring (where one of the substituents is attached) adjacent to the pyrrolo[1,5-a]pyrazine moiety and an acetophenone group where another substituent binds,131 1,2,4-triazino[5,6b]indole-3-thioether,132 thieno[2,3d]pyrimidines,133 and benzyloxy-benzylamine134 (Fig. 4).
000 compounds, of which 617 molecules containing an isothiazole S,S-dioxide heterocyclic core (Fig. 4) were selected for further studies that ended up individualizing 17 that were potent inhibitors of resistance when tested in combination with kanamycin.128 This pioneering work showed that it is possible to identify small molecules that inhibit aminoglycoside resistance caused by acetyltransferases. A library of compounds with a sulfonamide scaffold (Fig. 4) contained a compound including a substituted aniline ring that showed strong inhibition of the Eis activity. Analogs to this compound with modifications to the aniline ring were subsequently synthesized and tested, that one consisting of the sulfonamide scaffold with a 2-naphthyl substitution was a potent inhibitor of resistance to kanamycin mediated by Eis.129 Crystal structure analysis showed that the inhibitor binds the Eis protein at the aminoglycoside-binding pocket. Further high throughput screening followed by optimization, by synthesizing compounds with different substituents, in some cases guided by structural information, led to identification of other Eis inhibitors containing the scaffolds methyl 4H-furo[3,2-b]pyrrole-5-carboxylate,130 3-(1,3-dioxolano)-2-indolinone,130 pyrrolo[1,5-a]pyrazine containing a phenyl ring (where one of the substituents is attached) adjacent to the pyrrolo[1,5-a]pyrazine moiety and an acetophenone group where another substituent binds,131 1,2,4-triazino[5,6b]indole-3-thioether,132 thieno[2,3d]pyrimidines,133 and benzyloxy-benzylamine134 (Fig. 4).
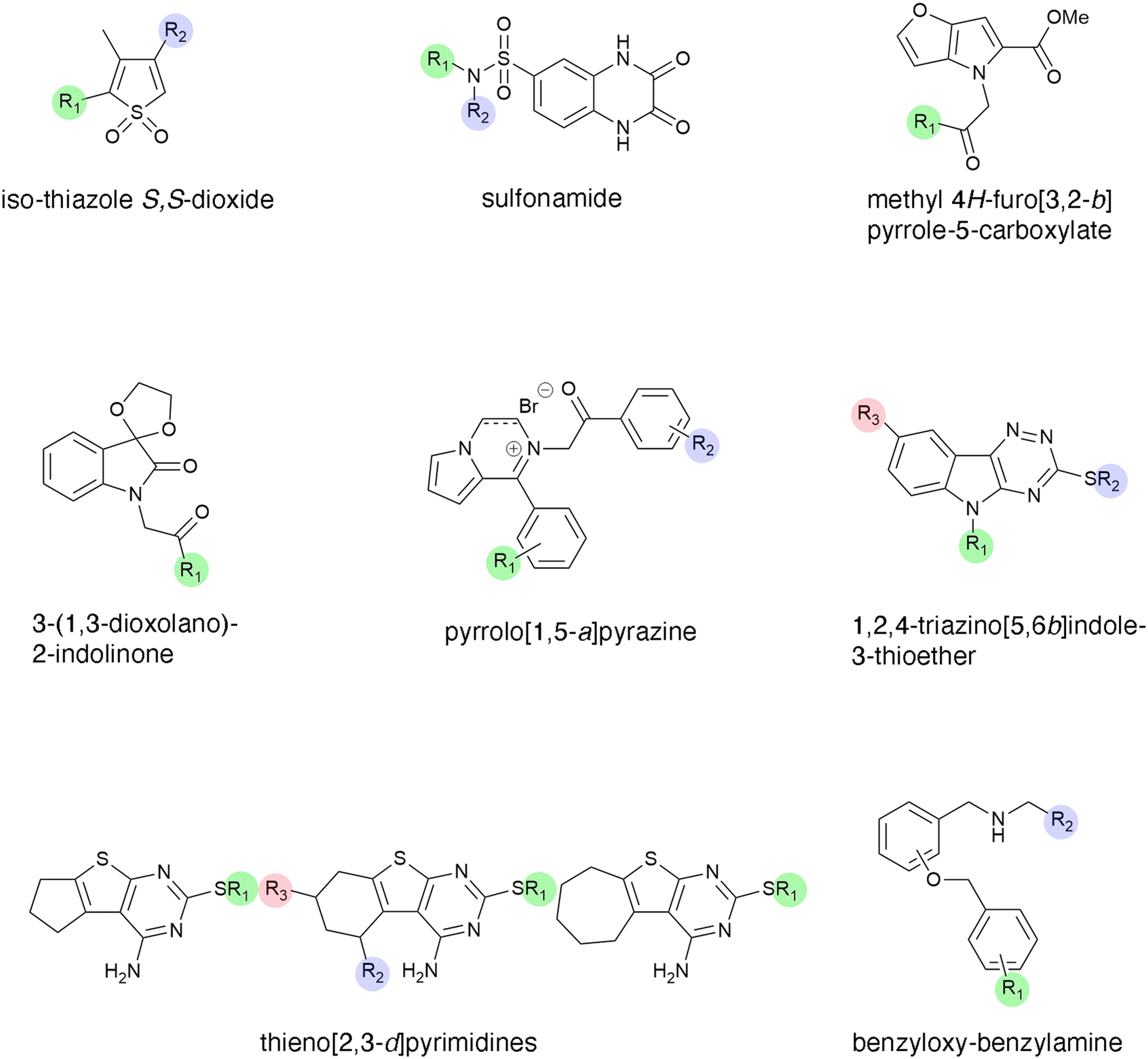 | ||
| Fig. 4 Chemical structures of scaffolds that originated Eis inhibitors.128–135 Analogs were generated changing substituents at the positions highlighted in color. | ||
The approaches described in the previous paragraph were complemented with drug repurposing studies, i.e., the search for inhibitors among compounds already approved for human use or hybrid strategies like designing and testing analogs of existing drugs that showed Eis inhibitory properties.120,135 These studies permitted the identification of inhibitors and their rational modification to generate even more potent compounds that could be used as adjuvants to aminoglycosides to treat resistant M. tuberculosis infections.135 Haloperidol (Fig. 5), an antipsychotic drug, exhibited inhibition of the reaction catalyzed by Eis with an IC50 of 0.39 ± 0.08 μM. Numerous analogs were designed on the basis of structural studies of the complex formed by haloperidol and Eis. The modifications to the haloperidol molecule were carried out at the positions indicated in colors in Fig. 5. Analysis of these analogs determined that eight also have potent inhibition properties. However, none of them was more powerful than haloperidol.135 Three of these compounds showed additive effect with spectinomycin against resistant M. tuberculosis. These studies proved the potential of repurposing existing drugs.
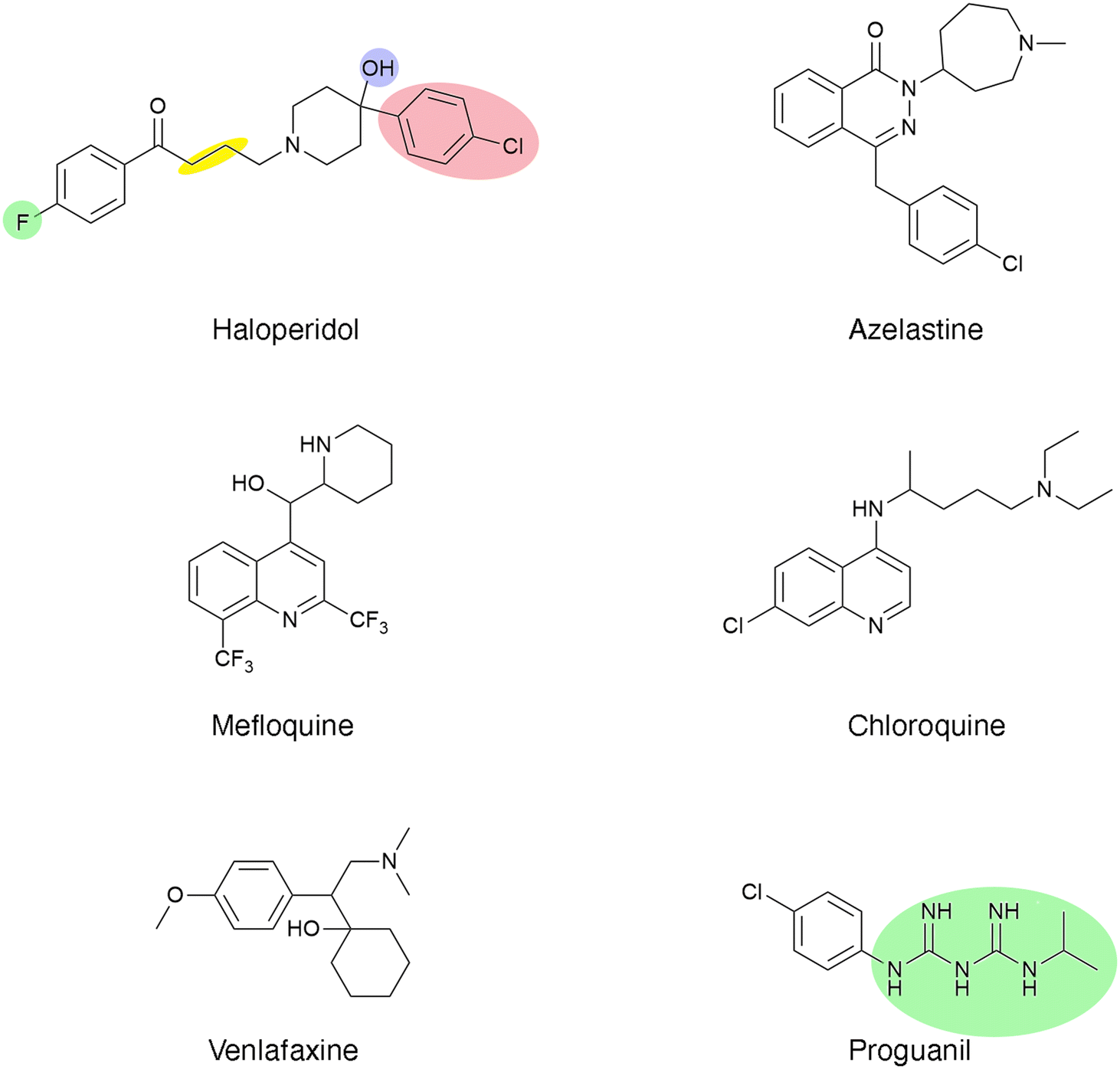 | ||
| Fig. 5 Chemical structures of haloperidol (antipsychotic), azelastine (antihistamine), venlafaxine (antidepressant), chloroquine (antimalarial), mefloquine (antimalarial), and proguanil (antimalarial). Analogs were generated changing substituents at the positions highlighted in color. The yellow highlight indicates that the number of methylene groups can be modified.135 | ||
A recent study identified five FDA-approved medicines, azelastine (antihistamine), venlafaxine (antidepressant), and the antimalarial chloroquine, mefloquine, and proguanil (Fig. 5).120 Analysis of the crystal structure of Eis in complex with these compounds found that they bind the aminoglycoside binding pocket. Based on the knowledge acquired, three new compounds were designed, and one of them, a proguanil analog, showed 3-fold more potency than the original drug.120 Unfortunately, these compounds did not show significant activity to overcome kanamycin resistance in M. tuberculosis.120
An inhibitor of the AAC(6′)-Ib enzyme identified using mixture-based combinatorial libraries
Aminoglycoside N-acetyltransferases (AAC) are a large family of aminoglycoside modifying enzymes of high clinical relevance found in Gram-negatives and Gram-positives.5,63,64,100,136 The more than sixty enzymes in this group are unequally represented in clinical strains; for example, the AAC(6′)-Ib enzyme is present in a disproportionally high number of AAC(6′)-I-producing Gram-negatives of clinical origin.137 Its high impact on complications to treat Gram-negative infections resulted in numerous efforts to find inhibitors that can restore susceptibility to aminoglycosides such as amikacin or tobramycin.91–93,95–97,138–141 Most of these numerous strategies to inhibit the expression of the aac(6′)-Ib gene or the acetylation reaction have been recently reviewed.28,61,75 Early attempts to identify small molecule inhibitors of AAC(6′)-Ib by computer docking successfully selected compounds with in vitro activity. However, they did not reverse resistance in bacterial cells in culture.139,140 An inhibitor has recently been identified using mixture-based combinatorial libraries.142 Thirty-seven combinatorial libraries, each including tens or hundreds of thousands of compounds with different scaffolds, were ranked to determine if they contained a potential inhibitor.143 The library containing pyrrolidine pentamine as a scaffold and four positions for functionalities was the most active. Upon deconvolution, a compound with S-phenyl at two positions, S-hydromethyl, and 3-phenylbutyl functionalities, was the most active inhibitor of in vitro acetylation (IC50 = 2.1 μM) (Fig. 6). This compound, in combination with amikacin, restored susceptibility of Escherichia coli, A. baumannii, and Klebsiella pneumoniae harboring aac(6′)-Ib. Furthermore, it improved survival when in combination with amikacin, it was used to treat infected Galleria mellonella, and it was not toxic when tested on HEK 293 cells.143 Structure–activity relationship analysis expanded the number of compounds studied and helped to illuminate some interactions between the inhibitor and enzyme and design analogs. Other modifications consisted of truncations of the molecule, which in all cases resulted in the loss of the inhibitory properties. Furthermore, none of those assayed to date was more active than the compound initially identified (Fig. 6).144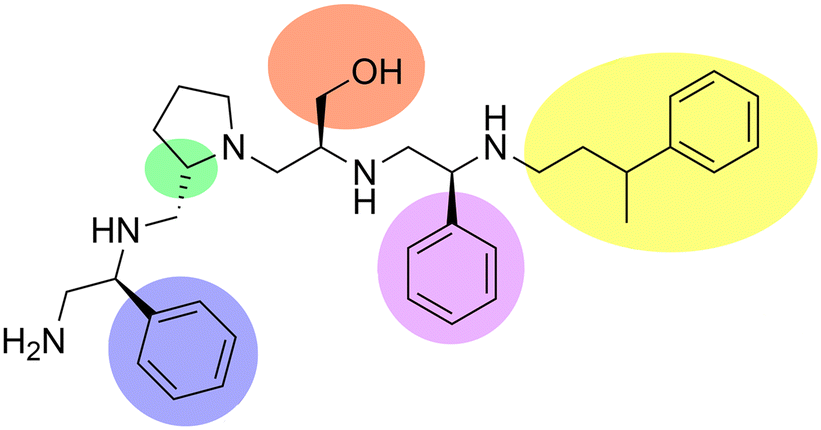 | ||
| Fig. 6 Chemical structure of the pyrrolidine pentamine scaffold compound with the most potent AAC(6′)-Ib-mediated acetylation inhibitory activity. The colored locations are those where different substituents were placed in the combinatorial library and analogs. The green region is modified in stereochemistry. | ||
Small molecule inhibitors of aminoglycoside O-phosphotransferases
Aminoglycoside O-phosphotransferases of different subtypes catalyze the inactivation of numerous aminoglycosides of clinical relevance by transferring a phosphate from ATP or GTP to a hydroxyl group in the aminoglycoside molecule.5,101,145–147 The crystal structures of many enzymes from this family have been resolved, and interestingly, they share characteristics with eukaryotic protein kinases.148 This property was exploited in early work when already known inhibitors of eukaryotic protein kinases were tested on two aminoglycoside O-phosphotransferases, APH(3′)-IIIa and the bifunctional AAC(6′)-Ie-APH(2′′)-Ia. This study identified isoquinolinesulfonamides (Fig. 7), compounds known to inhibit protein kinases by a competitive mechanism with respect to ATP that could inhibit the action of one or both enzymes. However, as happens in many cases, the chemicals failed to overcome resistance in bacterial cells in culture.149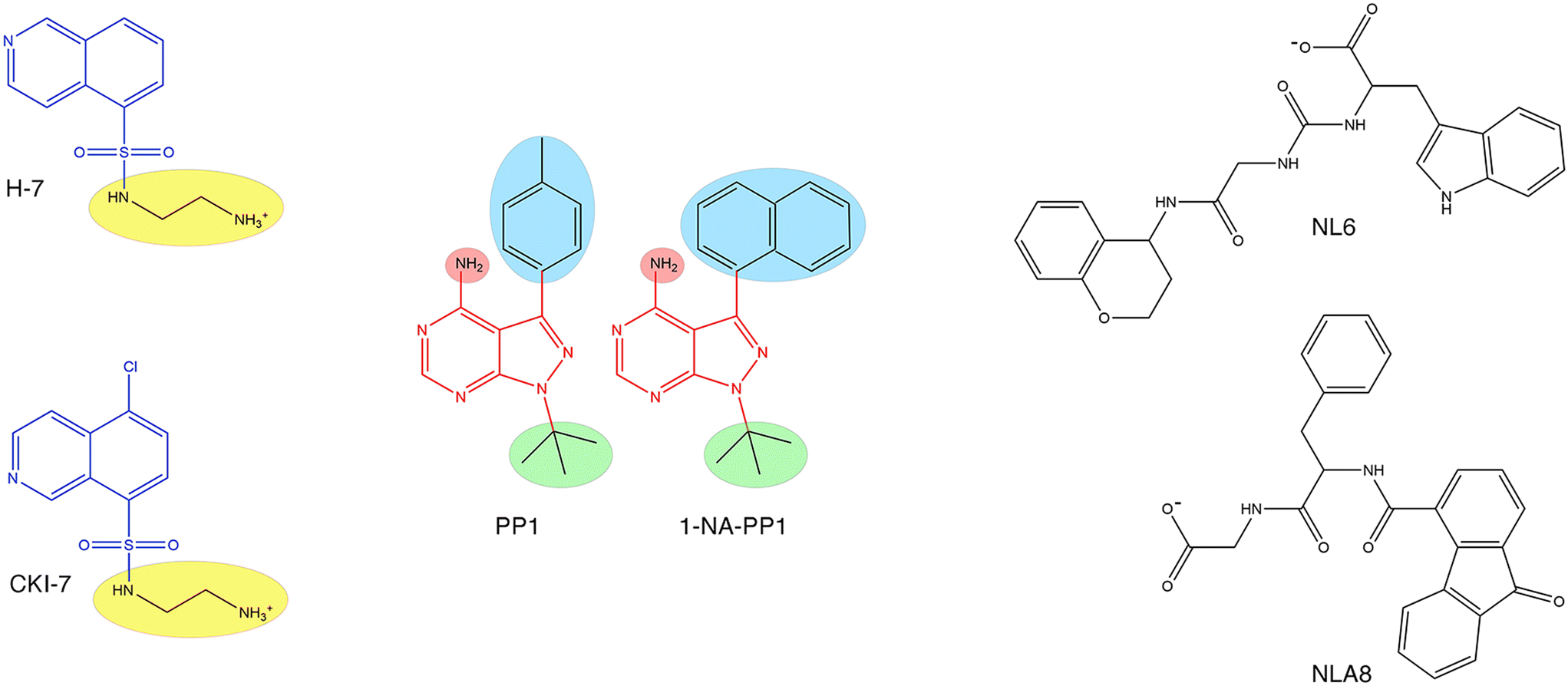 | ||
| Fig. 7 Chemical structures of aminoglycoside O-phosphotransferase inhibitors. Left, the isoquinolinesulfamide scaffold (blue) was replaced by various chemical groups at the R position (highlighted in yellow). The compounds shown, H-7 and CKI-7 are competitive inhibitors (versus ATP) of APH(3′)-IIIa and the bifunctional AAC(6′)-Ie-APH(2′′)-Ia.149 Center, two pyrazolopyrimidine scaffolds (red) showing the R positions (highlighted in colors) used to insert various chemical groups. The compounds shown, 1-NA-PP1 and the parent compound PP1, are inhibitors of APH(3′)-Ia activity in vitro and reduce resistance in hyperpermeable E. coli.151 Right, compounds NL6 and NL8. Both compounds are inhibitors of APH(3′)-IIIa, and NL6 is also an APH(2′)-IVa inhibitor.152 | ||
An extensive screening of several aminoglycoside O-phosphotransferases against chemically diverse protein kinase inhibitors confirmed that protein kinase inhibitors could also inhibit this class of resistance enzymes. Among others, the authors identified the flavonoid quercetin as a potential inhibitor of resistance to aminoglycosides mediated by aminoglycoside O-phosphotransferases.150 Further work utilized a selected group of these inhibitors in a study of the 3D structure of the APH(3′)-Ia in complex with the inhibitors and the aminoglycoside substrate. The results of these studies concluded that inhibitors with chemical scaffold pyrazolopyrimidine (Fig. 7) not only reduced APH(3′)-Ia activity in vitro and reversed aminoglycoside resistance in APH(3′)-Ia-harboring hyperpermeable E. coli cells in culture but also demonstrated advantages over eukaryotic protein kinase inhibitors tested before.151 This latter property makes them attractive chemicals that could have low toxicity to the host.
There were other attempts to identify inhibitors by computer docking. One of them led to the finding of two inhibitors, NL6 and NL8 (Fig. 7) of the APH(3′)-IIIa, and one of them, NL6, with activity towards APH(2′)-IVa.152 The compound NL6, a noncompetitive inhibitor towards the substrates ATP and kanamycin A, was studied more thoroughly by assessing the inhibitory properties of two analogs. Only one of them showed inhibition but with lower efficiency.152 In another study, molecular docking identified an inhibitor of a Bacillus phosphotransferase.153,154 While these studies are promising, they were limited to in vitro analysis, and further work, including molecular optimization and in vivo experiments, will be required.
Concluding remarks
After a period in which the use of aminoglycosides seemed to be in decline, these antibiotics are making a comeback due to a combination of factors like the urgent need for antimicrobials and their favorable properties that outweigh the reasons they were being left aside (administration route, toxicity, resistance). Furthermore, newer aminoglycoside molecules, improved dosing schemes, and alternative administration continue minimizing the negative aspects of aminoglycosides, reinforcing the renewed interest in these drugs.4,25,60 A major cause of failure in treating infections with aminoglycosides is the development and dissemination of resistance, which can occur through numerous mechanisms, several of which can coexist in the same bacterial cell.5 Besides generating molecules refractory to the existing resistance mechanisms, designing combination therapies that include the antibiotic and an inhibitor of resistance can prolong the useful life of existing aminoglycosides. Numerous efforts are underway to counter resistance from different angles, and this review described a few examples of just one aspect of this quest. Small molecules have enormous potential to become adjuvants that overcome resistant infections in combination with aminoglycosides. Although the multitude of efforts still did not result in an adjuvant approved for its use in humans, the direction and depth of the efforts by multiple research groups present an optimistic outlook. Of course, there will be challenges to overcome, like possible toxicity issues, spectrum of action, or ability of bacteria to develop enzymes that resist the action of the inhibitors. The efficacy of combinations aminoglycoside/enzyme inhibitor will most probably depend on the relationships between the pharmacokinetics and pharmacodynamics of the inhibitor and the antibiotic. Optimizing therapeutic regimens might become a challenging proposition depending on the chemical nature of the inhibitor. It may be worth mentioning that in the case of β-lactam/β-lactamase inhibitor combinations, while still complex, the dose adjustments are expected to be simpler to determine because the pharmacokinetics and pharmacodynamics of the inhibitor and antibiotic are usually similar.155 Despite all challenges, it is possible that in the not-so-far future, a combination aminoglycoside/adjuvant that overcomes resistance caused by common aminoglycoside modifying enzymes will be used for human treatment.Author contributions
MET, conceptualization and writing of the first draft. All authors contributed to the literature search, writing, and editing.Conflicts of interest
None to declare.Acknowledgements
Cited work made by the authors was funded by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, grant number R15AI047115, and National Institute of General Medical Sciences, National Institutes of Health, grant number SC3GM125556.References
- J. H. Comroe, Jr., Am. Rev. Respir. Dis., 1978, 117, 957–968 Search PubMed.
- W. H. Organization, WHO consolidated guidelines on drug-resistant tuberculosis, Geneva, 2019 Search PubMed.
- N. F. Sabur, M. S. Brar, L. Wu and S. K. Brode, BMC Infect. Dis., 2021, 21, 254 CrossRef CAS PubMed.
- K. M. Krause, A. W. Serio, T. R. Kane and L. E. Connolly, Cold Spring Harbor Perspect. Med., 2016, 6, a027029 CrossRef PubMed.
- M. S. Ramirez and M. E. Tolmasky, Drug Resistance Updates, 2010, 13, 151–171 CrossRef CAS PubMed.
- A. W. Serio, T. Keepers, L. Andrews and K. M. Krause, EcoSal Plus, 2018, 8(1) DOI:10.1128/ecosalplus.ESP-0002-2018.
- J. Yao and R. Moellering, in Manual of Clinical Microbiology, ed. P. Murray, E. Baron, J. Jorgensen, M. Landry and M. Pfaller, American Society for Microbiology Press, Washington, DC, 2007, pp. 1077–1113 Search PubMed.
- M. Thy, J. Timsit and E. de Montmollin, Antibiotics, 2023, 12, 860 CrossRef CAS PubMed.
- R. N. Davidson, M. den Boer and K. Ritmeijer, Trans. R. Soc. Trop. Med. Hyg., 2009, 103, 653–660 CrossRef CAS PubMed.
- J. E. Rehg, J. Infect. Dis., 1994, 170, 934–938 CrossRef CAS PubMed.
- L. Vitiello, L. Tibaudo, E. Pegoraro, L. Bello and M. Canton, Int. J. Mol. Sci., 2019, 20, 6053 CrossRef CAS PubMed.
- D. P. Schoo, G. X. Tan, M. R. Ehrenburg, S. E. Pross, B. K. Ward and J. P. Carey, Curr. Otorhinolaryngol. Rep., 2017, 5, 132–141 CrossRef PubMed.
- K. M. Keeling, X. Xue, G. Gunn and D. M. Bedwell, Annu. Rev. Genomics Hum. Genet., 2014, 15, 371–394 CrossRef CAS PubMed.
- P. Martins-Dias and L. Romao, Cell. Mol. Life Sci., 2021, 78, 4677–4701 CrossRef CAS PubMed.
- R. B. V. Abreu, T. T. Gomes, T. C. Nepomuceno, X. Li, M. Fuchshuber-Moraes, G. De Gregoriis, G. Suarez-Kurtz, A. N. A. Monteiro and M. A. Carvalho, Front. Pharmacol., 2022, 13, 935995 CrossRef CAS PubMed.
- G. Chen, X. Fei and J. Ling, Clin. Appl. Thromb./Hemostasis, 2012, 18, 538–541 CrossRef PubMed.
- P. D. James, S. Raut, G. E. Rivard, M. C. Poon, M. Warner, S. McKenna, J. Leggo and D. Lillicrap, Blood, 2005, 106, 3043–3048 CrossRef CAS PubMed.
- S. Li, J. Li, W. Shi, Z. Nie, S. Zhang, F. Ma, J. Hu, J. Chen, P. Li and X. Xie, Biomolecules, 2023, 13, 988 CrossRef CAS PubMed.
- M. C. Bellucci and A. Volonterio, Antibiotics, 2020, 9, 504 CrossRef CAS PubMed.
- C. Pennetta, N. Bono, F. Ponti, M. C. Bellucci, F. Viani, G. Candiani and A. Volonterio, Bioconjugate Chem., 2021, 32, 690–701 CrossRef CAS PubMed.
- P. Veyssier and A. Bryskier, in Antimicrobial Agents, ed. A. Bryskier, ASM Press, Washington, DC, 2005, pp. 453–469 Search PubMed.
- D. Lyutzkanova, J. Distler and J. Altenbuchner, Microbiology, 1997, 143, 2135–2143 CrossRef CAS PubMed.
- N. Thamban Chandrika and S. Garneau-Tsodikova, Chem. Soc. Rev., 2018, 47, 1189–1249 RSC.
- J. Zhang, A. Litke, K. Keller, R. Rai and C. W. Chang, Bioorg. Med. Chem., 2010, 18, 1396–1405 CrossRef CAS PubMed.
- E. C. Bottger and D. Crich, ACS Infect. Dis., 2020, 6, 168–172 CrossRef CAS PubMed.
- S. R. Park, J. W. Park, Y. H. Ban, J. K. Sohng and Y. J. Yoon, Nat. Prod. Rep., 2013, 30, 11–20 RSC.
- G. Cox, L. Ejim, P. J. Stogios, K. Koteva, E. Bordeleau, E. Evdokimova, A. O. Sieron, A. Savchenko, A. W. Serio, K. M. Krause and G. D. Wright, ACS Infect. Dis., 2018, 4, 980–987 CrossRef CAS PubMed.
- M. S. Ramirez and M. E. Tolmasky, Molecules, 2017, 22, 2267 CrossRef PubMed.
- K. Vong and K. Auclair, MedChemComm, 2012, 3, 397–407 RSC.
- L. Zimmermann, I. Das, J. Desire, G. Sautrey, R. S. V. Barros, M. El Khoury, M. P. Mingeot-Leclercq and J. L. Decout, J. Med. Chem., 2016, 59, 9350–9369 CrossRef CAS PubMed.
- N. Thamban Chandrika, K. D. Green, J. L. Houghton and S. Garneau-Tsodikova, ACS Med. Chem. Lett., 2015, 6, 1134–1139 CrossRef CAS PubMed.
- S. K. Shrestha, M. Grilley, T. Anderson, C. Dhiman, J. Oblad, C. W. Chang, K. N. Sorensen and J. Y. Takemoto, Med. Mycol., 2015, 53, 837–844 CrossRef CAS PubMed.
- J. Y. Takemoto, G. A. Altenberg, N. Poudyal, Y. P. Subedi and C. T. Chang, Front. Microbiol., 2022, 13, 1000199 CrossRef PubMed.
- M. S. Butler, V. Gigante, H. Sati, S. Paulin, L. Al-Sulaiman, J. H. Rex, P. Fernandes, C. A. Arias, M. Paul, G. E. Thwaites, L. Czaplewski, R. A. Alm, C. Lienhardt, M. Spigelman, L. L. Silver, N. Ohmagari, R. Kozlov, S. Harbarth and P. Beyer, Antimicrob. Agents Chemother., 2022, 66, e0199121 CrossRef PubMed.
- N. T. Chandrika and S. Garneau-Tsodikova, MedChemComm, 2016, 7, 50–68 RSC.
- N. T. Chandrika and S. Garneau-Tsodikova, Chem. Soc. Rev., 2018, 47, 1189–1249 RSC.
- R. Vanhoof, P. Sonck and E. Hannecart-Pokorni, J. Antimicrob. Chemother., 1995, 35, 167–171 CrossRef CAS PubMed.
- M. E. Muir, R. S. van Heeswyck and B. J. Wallace, J. Gen. Microbiol., 1984, 130, 2015–2022 CAS.
- W. W. Nichols and S. N. Young, Biochem. J., 1985, 228, 505–512 CrossRef CAS PubMed.
- H. W. Taber, J. P. Mueller, P. F. Miller and A. S. Arrow, Microbiol. Rev., 1987, 51, 439–457 CrossRef CAS PubMed.
- L. E. Bryan and H. M. Van Den Elzen, Antimicrob. Agents Chemother., 1977, 12, 163–177 CrossRef CAS PubMed.
- B. Davis, J. Antimicrob. Chemother., 1988, 22, 1–3 CrossRef CAS PubMed.
- H. J. Busse, C. Wostmann and E. P. Bakker, J. Gen. Microbiol., 1992, 138, 551–561 CrossRef CAS PubMed.
- B. Davis, Microbiol. Rev., 1987, 51, 341–350 CrossRef CAS PubMed.
- S. Jana and J. K. Deb, Appl. Microbiol. Biotechnol., 2006, 70, 140–150 CrossRef CAS PubMed.
- L. S. McCoy, Y. Xie and Y. Tor, Wiley Interdiscip. Rev.: RNA, 2011, 2, 209–232 CrossRef CAS PubMed.
- J. M. Ogle and V. Ramakrishnan, Annu. Rev. Biochem., 2005, 74, 129–177 CrossRef CAS PubMed.
- S. Magnet and J. S. Blanchard, Chem. Rev., 2005, 105, 477–498 CrossRef CAS PubMed.
- R. Mehta and W. S. Champney, Curr. Microbiol., 2003, 47, 237–243 CrossRef CAS PubMed.
- M. J. Belousoff, B. Graham, L. Spiccia and Y. Tor, Org. Biomol. Chem., 2009, 7, 30–33 RSC.
- S. A. Kawamoto, C. G. Sudhahar, C. L. Hatfield, J. Sun, E. J. Behrman and V. Gopalan, Nucleic Acids Res., 2008, 36, 697–704 CrossRef CAS PubMed.
- C. Possoz, J. Newmark, N. Sorto, D. J. Sherratt and M. E. Tolmasky, Antimicrob. Agents Chemother., 2007, 51, 252–256 CrossRef CAS PubMed.
- E. B. Goh, G. Yim, W. Tsui, J. McClure, M. G. Surette and J. Davies, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 17025–17030 CrossRef CAS PubMed.
- E. A. Radigan, N. A. Gilchrist and M. A. Miller, J. Intensive Care Med., 2010, 25, 327–342 CrossRef PubMed.
- M. P. Mingeot-Leclercq and P. M. Tulkens, Antimicrob. Agents Chemother., 1999, 43, 1003–1012 CrossRef CAS PubMed.
- O. W. Guthrie, Toxicology, 2008, 249, 91–96 CrossRef PubMed.
- P. M. Uribe, A. M. Hudson, G. Lockard, M. Jiang, J. Harding, P. S. Steyger and A. B. Coffin, Hear. Res., 2023, 434, 108786 CrossRef PubMed.
- Y. R. Kim, J. I. Baek, K. Y. Lee and U. K. Kim, Free Radical Biol. Med., 2023, 204, 177–183 CrossRef CAS PubMed.
- M. C. Wagner, R. M. Sandoval, S. P. S. Yadav, S. B. Campos, G. J. Rhodes, C. L. Phillips and B. A. Molitoris, Kidney360, 2023, 4, 591–605 Search PubMed.
- J. L. Houghton, K. D. Green, W. Chen and S. Garneau-Tsodikova, ChemBioChem, 2010, 11, 880–902 CrossRef CAS PubMed.
- M. E. Tolmasky, in Frontiers in Clinical Drug Research-Anti Infectives, ed. Atta-ur-Rhaman, Bentham Books, Sharjah, UAE, 2017, vol. 1, pp. 1–27 Search PubMed.
- M. E. Tolmasky, in Enzyme-Mediated Resistance to Antibiotics: Mechanisms, Dissemination, and Prospects for Inhibition, ed. M. E. Tolmasky and R. A. Bonomo, ASM Press, Washington, DC, 2007, pp. 35–52 Search PubMed.
- S. Garneau-Tsodikova and K. J. Labby, MedChemComm, 2016, 7, 11–27 RSC.
- M. S. Ramirez, N. Nikolaidis and M. E. Tolmasky, Front. Microbiol., 2013, 4, 121 Search PubMed.
- M. Galimand, T. Lambert and P. Courvalin, Euro Surveill., 2005, 10, 2626 Search PubMed.
- Y. Doi and Y. Arakawa, Clin. Infect. Dis., 2007, 45, 88–94 CrossRef CAS PubMed.
- D. L. MacLeod, L. E. Nelson, R. M. Shawar, B. B. Lin, L. G. Lockwood, J. E. Dirk, G. H. Miller, J. L. Burns and R. L. Garber, J. Infect. Dis., 2000, 181, 1180–1184 CrossRef CAS PubMed.
- S. Magnet, P. Courvalin and T. Lambert, Antimicrob. Agents Chemother., 2001, 45, 3375–3380 CrossRef CAS PubMed.
- S. Magnet, T. A. Smith, R. Zheng, P. Nordmann and J. S. Blanchard, Antimicrob. Agents Chemother., 2003, 47, 1577–1583 CrossRef CAS PubMed.
- P. J. Stogios, E. Bordeleau, Z. Xu, T. Skarina, E. Evdokimova, S. Chou, L. Diorio-Toth, A. W. D'Souza, S. Patel, G. Dantas, G. D. Wright and A. Savchenko, Commun. Biol., 2022, 5, 263 CrossRef CAS PubMed.
- L. D. Saravolatz and G. E. Stein, Clin. Infect. Dis., 2020, 70, 704–709 CAS.
- K. M. Papp-Wallace, Expert Opin. Pharmacother., 2019, 20, 2169–2184 CrossRef CAS PubMed.
- Q. Cui, H. D. Yu, Q. J. Xu, Y. Liu, Y. T. Wang, P. H. Li, L. C. Kong, H. P. Zhang, X. Y. Jiang, A. M. Giuliodori, A. Fabbretti, C. G. He and H. X. Ma, Front. Microbiol., 2023, 14, 1144946 CrossRef PubMed.
- R. Pifer and D. E. Greenberg, Transl. Res., 2020, 223, 89–106 CrossRef CAS PubMed.
- S. Jani, M. S. Ramirez and M. E. Tolmasky, Biomedicines, 2021, 9, 416 CrossRef CAS PubMed.
- E. K. Sully and B. L. Geller, Curr. Opin. Microbiol., 2016, 33, 47–55 CrossRef CAS PubMed.
- C. Davies-Sala, A. Soler-Bistue, R. A. Bonomo, A. Zorreguieta and M. E. Tolmasky, Ann. N. Y. Acad. Sci., 2015, 1354, 98–110 CrossRef CAS PubMed.
- L. M. Streicher, J. Global Antimicrob. Resist., 2021, 24, 285–295 CrossRef CAS PubMed.
- K. E. Darrah, S. Albright, R. Kumbhare, M. Tsang, J. K. Chen and A. Deiters, ACS Chem. Biol., 2023 DOI:10.1021/acschembio.3c00027.
- M. Iubatti, I. M. Gabas, L. M. Cavaco, E. H. Mood, E. Lim, F. Bonanno, N. Yavari, C. Brolin and P. E. Nielsen, ACS Infect. Dis., 2022, 8, 1098–1106 CrossRef CAS PubMed.
- K. E. da Silva, S. M. Ribeiro, L. Rossato, C. P. Dos Santos, S. E. Preza, M. H. Cardoso, O. L. Franco, L. Migliolo and S. Simionatto, Res. Microbiol., 2021, 172, 103837 CrossRef CAS PubMed.
- K. Dhuri, C. Bechtold, E. Quijano, H. Pham, A. Gupta, A. Vikram and R. Bahal, J. Clin. Med., 2020, 9, 2004 CrossRef CAS PubMed.
- M. J. Beha, J. S. Ryu, Y. S. Kim and H. J. Chung, Mater. Sci. Eng., C, 2021, 126, 112167 CrossRef CAS PubMed.
- J. B. Readman, G. Dickson and N. G. Coldham, Front. Microbiol., 2016, 7, 373 Search PubMed.
- L. Good and P. E. Nielsen, Nat. Biotechnol., 1998, 16, 355–358 CrossRef CAS PubMed.
- H. Wang, J. Meng, M. Jia, X. Ma, G. He, J. Yu, R. Wang, H. Bai, Z. Hou and X. Luo, FEMS Immunol. Med. Microbiol., 2010, 60, 275–282 CrossRef CAS PubMed.
- E. K. Sully, B. L. Geller, L. Li, C. M. Moody, S. M. Bailey, A. L. Moore, M. Wong, P. Nordmann, S. M. Daly, C. R. Sturge and D. E. Greenberg, J. Antimicrob. Chemother., 2017, 72, 782–790 CAS.
- M. Y. Gao, C. R. Xu, R. Chen, S. G. Liu and J. N. Feng, World J. Gastroenterol., 2005, 11, 7368–7373 CrossRef CAS PubMed.
- N. Shen, J. H. Ko, G. Xiao, D. Wesolowski, G. Shan, B. Geller, M. Izadjoo and S. Altman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 8163–8168 CrossRef CAS PubMed.
- S. Ghosh, K. B. Cotta, A. A. Hande, M. Fernandes and S. Mehra, Microb. Pathog., 2021, 151, 104737 CrossRef CAS PubMed.
- A. J. Soler Bistue, F. A. Martin, N. Vozza, H. Ha, J. C. Joaquin, A. Zorreguieta and M. E. Tolmasky, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13230–13235 CrossRef CAS PubMed.
- C. Lopez, B. A. Arivett, L. A. Actis and M. E. Tolmasky, Antimicrob. Agents Chemother., 2015, 59, 5798–5803 CrossRef CAS PubMed.
- D. L. Lin, T. Tran, J. Y. Alam, S. R. Herron, M. S. Ramirez and M. E. Tolmasky, Antimicrob. Agents Chemother., 2014, 58, 4238–4241 CrossRef PubMed.
- Y. Li, K. D. Green, B. R. Johnson and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2015, 59, 4148–4156 CrossRef CAS PubMed.
- K. Chiem, B. A. Fuentes, D. L. Lin, T. Tran, A. Jackson, M. S. Ramirez and M. E. Tolmasky, Antimicrob. Agents Chemother., 2015, 59, 5851–5853 CrossRef CAS PubMed.
- K. Chiem, F. Hue, J. Magallon and M. E. Tolmasky, Int. J. Antimicrob. Agents, 2018, 51, 271–273 CrossRef CAS PubMed.
- J. Magallon, K. Chiem, T. Tran, M. S. Ramirez, V. Jimenez and M. E. Tolmasky, PLoS One, 2019, 14, e0217602 CrossRef CAS PubMed.
- J. Magallon, P. Vu, C. Reeves, S. Kwan, K. Phan, C. L. Oakley-Havens, M. S. Ramirez and M. E. Tolmasky, Int. J. Antimicrob. Agents, 2021, 58, 106442 CrossRef CAS PubMed.
- J. Magallon, P. Vu, C. Reeves, S. Kwan, K. Phan, C. L. Oakley-Havens, K. Rocha, V. Jimenez, M. S. Ramirez and M. E. Tolmasky, Sci. Rep., 2022, 12, 285 CrossRef PubMed.
- K. J. Labby and S. Garneau-Tsodikova, Future Med. Chem., 2013, 5, 1285–1309 CrossRef CAS PubMed.
- K. Shi, S. J. Caldwell, D. H. Fong and A. M. Berghuis, Front. Cell. Infect. Microbiol., 2013, 3, 22 Search PubMed.
- C. Hobson, A. N. Chan and G. D. Wright, Chem. Rev., 2021, 121, 3464–3494 CrossRef CAS PubMed.
- C. El-Khoury, E. Mansour, Y. Yuliandra, F. Lai, B. A. Hawkins, J. J. Du, E. J. Sundberg, N. Sluis-Cremer, D. E. Hibbs and P. W. Groundwater, RSC Med. Chem., 2022, 13, 1276–1299 RSC.
- B. O. Costa, M. H. Cardoso and O. L. Franco, Curr. Protein Pept. Sci., 2020, 21, 1011–1026 CrossRef CAS PubMed.
- M. Tyers and G. D. Wright, Nat. Rev. Microbiol., 2019, 17, 141–155 CrossRef CAS PubMed.
- V. Yarlagadda, R. Medina and G. D. Wright, Sci. Rep., 2020, 10, 8134 CrossRef CAS PubMed.
- Y. Costa, M. Galimand, R. Leclercq, J. Duval and P. Courvalin, Antimicrob. Agents Chemother., 1993, 37, 1896–1903 CrossRef CAS PubMed.
- M. Garcia-Solache and L. B. Rice, Clin. Microbiol. Rev., 2019, 32, e00058-18 CrossRef PubMed.
- F. Gao, X. Yan, O. M. Baettig, A. M. Berghuis and K. Auclair, Angew. Chem., Int. Ed., 2005, 44, 6859–6862 CrossRef CAS PubMed.
- A. I. Salah Ud-Din, A. Tikhomirova and A. Roujeinikova, Int. J. Mol. Sci., 2016, 17, 1018 CrossRef PubMed.
- C. M. Kim and P. A. Cole, J. Med. Chem., 2001, 44, 2479–2485 CrossRef CAS PubMed.
- A. N. Poux, M. Cebrat, C. M. Kim, P. A. Cole and R. Marmorstein, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14065–14070 CrossRef CAS PubMed.
- R. M. Burckhardt and J. C. Escalante-Semerena, Microbiol. Mol. Biol. Rev., 2020, 84, e00090-19 CrossRef PubMed.
- F. Gao, X. Yan and K. Auclair, Chemistry, 2009, 15, 2064–2070 CrossRef CAS PubMed.
- K. Vong, I. S. Tam, X. Yan and K. Auclair, ACS Chem. Biol., 2012, 7, 470–475 CrossRef CAS PubMed.
- J. Guan, K. Vong, K. Wee, J. Fakhoury, E. Dullaghan and K. Auclair, ChemBioChem, 2018, 19, 2107–2113 CrossRef CAS PubMed.
- M. L. Magalhaes, M. W. Vetting, F. Gao, L. Freiburger, K. Auclair and J. S. Blanchard, Biochemistry, 2008, 47, 579–584 CrossRef CAS PubMed.
- D. D. Boehr, K. A. Draker, K. Koteva, M. Bains, R. E. Hancock and G. D. Wright, Chem. Biol., 2003, 10, 189–196 CrossRef CAS PubMed.
- G. Kumar and C. Amrutha, Drug Dev. Res., 2023 DOI:10.1002/ddr.22063.
- A. H. Pang, K. D. Green, A. Punetha, N. Thamban Chandrika, K. C. Howard, S. Garneau-Tsodikova and O. V. Tsodikov, Biochemistry, 2023, 62, 710–721 CrossRef CAS PubMed.
- G. B. Migliori, D. Dowdy, J. T. Denholm, L. D'Ambrosio and R. Centis, Eur. Respir. J., 2023 DOI:10.1183/13993003.00499-2023.
- W. Chen, T. Biswas, V. R. Porter, O. V. Tsodikov and S. Garneau-Tsodikova, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 9804–9808 CrossRef CAS PubMed.
- J. Burian, G. Yim, M. Hsing, P. Axerio-Cilies, A. Cherkasov, G. B. Spiegelman and C. J. Thompson, Nucleic Acids Res., 2013, 41, 10062–10076 CrossRef CAS PubMed.
- M. A. Zaunbrecher, R. D. Sikes, Jr., B. Metchock, T. M. Shinnick and J. E. Posey, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20004–20009 CrossRef CAS PubMed.
- P. J. Campbell, G. P. Morlock, R. D. Sikes, T. L. Dalton, B. Metchock, A. M. Starks, D. P. Hooks, L. S. Cowan, B. B. Plikaytis and J. E. Posey, Antimicrob. Agents Chemother., 2011, 55, 2032–2041 CrossRef CAS PubMed.
- A. Z. Reeves, P. J. Campbell, R. Sultana, S. Malik, M. Murray, B. B. Plikaytis, T. M. Shinnick and J. E. Posey, Antimicrob. Agents Chemother., 2013, 57, 1857–1865 CrossRef CAS PubMed.
- J. L. Houghton, T. Biswas, W. Chen, O. V. Tsodikov and S. Garneau-Tsodikova, ChemBioChem, 2013, 14, 2127–2135 CrossRef CAS PubMed.
- M. J. Willby, K. D. Green, C. S. Gajadeera, C. Hou, O. V. Tsodikov, J. E. Posey and S. Garneau-Tsodikova, ACS Chem. Biol., 2016, 11, 1639–1646 CrossRef CAS PubMed.
- A. Garzan, M. J. Willby, K. D. Green, C. S. Gajadeera, C. Hou, O. V. Tsodikov, J. E. Posey and S. Garneau-Tsodikova, J. Med. Chem., 2016, 59, 10619–10628 CrossRef CAS PubMed.
- A. Garzan, M. J. Willby, K. D. Green, O. V. Tsodikov, J. E. Posey and S. Garneau-Tsodikova, ACS Med. Chem. Lett., 2016, 7, 1219–1221 CrossRef CAS PubMed.
- A. Garzan, M. J. Willby, H. X. Ngo, C. S. Gajadeera, K. D. Green, S. Y. Holbrook, C. Hou, J. E. Posey, O. V. Tsodikov and S. Garneau-Tsodikova, ACS Infect. Dis., 2017, 3, 302–309 CrossRef CAS PubMed.
- H. X. Ngo, K. D. Green, C. S. Gajadeera, M. J. Willby, S. Y. L. Holbrook, C. Hou, A. Garzan, A. S. Mayhoub, J. E. Posey, O. V. Tsodikov and S. Garneau-Tsodikova, ACS Infect. Dis., 2018, 4, 1030–1040 CrossRef CAS PubMed.
- A. Punetha, H. X. Ngo, S. Y. L. Holbrook, K. D. Green, M. J. Willby, S. A. Bonnett, K. Krieger, E. K. Dennis, J. E. Posey, T. Parish, O. V. Tsodikov and S. Garneau-Tsodikova, ACS Chem. Biol., 2020, 15, 1581–1594 CrossRef CAS PubMed.
- A. H. Pang, K. D. Green, N. T. Chandrika, A. Garzan, A. Punetha, S. Y. L. Holbrook, M. J. Willby, J. E. Posey, O. V. Tsodikov and S. Garneau-Tsodikova, Eur. J. Med. Chem., 2022, 242, 114698 CrossRef CAS PubMed.
- A. Punetha, K. D. Green, A. Garzan, N. Thamban Chandrika, M. J. Willby, A. H. Pang, C. Hou, S. Y. L. Holbrook, K. Krieger, J. E. Posey, T. Parish, O. V. Tsodikov and S. Garneau-Tsodikova, RSC Med. Chem., 2021, 12, 1894–1909 RSC.
- G. D. Wright, Curr. Opin. Microbiol., 1999, 2, 499–503 CrossRef CAS PubMed.
- S. B. Vakulenko and S. Mobashery, Clin. Microbiol. Rev., 2003, 16, 430–450 CrossRef CAS PubMed.
- T. Lombes, G. Begis, F. Maurice, S. Turcaud, T. Lecourt, F. Dardel and L. Micouin, ChemBioChem, 2008, 9, 1368–1371 CrossRef CAS PubMed.
- K. Chiem, S. Jani, B. Fuentes, D. L. Lin, M. E. Rasche and M. E. Tolmasky, MedChemComm, 2016, 7, 184–189 RSC.
- D. L. Lin, T. Tran, C. Adams, J. Y. Alam, S. R. Herron and M. E. Tolmasky, Bioorg. Med. Chem. Lett., 2013, 23, 5694–5698 CrossRef CAS PubMed.
- S. Ahmed, S. A. Sony, M. B. Chowdhury, M. M. Ullah, S. Paul and T. Hossain, Sci. Rep., 2020, 10, 19381 CrossRef CAS PubMed.
- R. A. Houghten, C. Pinilla, M. A. Giulianotti, J. R. Appel, C. T. Dooley, A. Nefzi, J. M. Ostresh, Y. Yu, G. M. Maggiora, J. L. Medina-Franco, D. Brunner and J. Schneider, J. Comb. Chem., 2008, 10, 3–19 CrossRef CAS PubMed.
- T. Tran, K. Chiem, S. Jani, B. A. Arivett, D. L. Lin, R. Lad, V. Jimenez, M. B. Farone, G. Debevec, R. Santos, M. Giulianotti, C. Pinilla and M. E. Tolmasky, Int. J. Antimicrob. Agents, 2018, 51, 752–761 CrossRef CAS PubMed.
- K. Rocha, J. Magallon, C. Reeves, K. Phan, P. Vu, C. L. Oakley-Havens, S. Kwan, M. S. Ramirez, T. LaVoi, H. Donow, P. Chapagain, R. Santos, C. Pinilla, M. A. Giulianotti and M. E. Tolmasky, Biomedicines, 2021, 9, 1218 CrossRef CAS PubMed.
- P. Aris, M. A. Boroumand and M. Douraghi, BMC Microbiol., 2019, 19, 221 CrossRef PubMed.
- T. Shakya and G. D. Wright, Antimicrob. Agents Chemother., 2010, 54, 1909–1913 CrossRef CAS PubMed.
- K. Shi and A. M. Berghuis, J. Biol. Chem., 2012, 287, 13094–13102 CrossRef CAS PubMed.
- W. C. Hon, G. A. McKay, P. R. Thompson, R. M. Sweet, D. S. Yang, G. D. Wright and A. M. Berghuis, Cell, 1997, 89, 887–895 CrossRef CAS PubMed.
- D. M. Daigle, G. A. McKay and G. D. Wright, J. Biol. Chem., 1997, 272, 24755–24758 CrossRef CAS PubMed.
- T. Shakya, P. J. Stogios, N. Waglechner, E. Evdokimova, L. Ejim, J. E. Blanchard, A. G. McArthur, A. Savchenko and G. D. Wright, Chem. Biol., 2011, 18, 1591–1601 CrossRef CAS PubMed.
- P. J. Stogios, P. Spanogiannopoulos, E. Evdokimova, O. Egorova, T. Shakya, N. Todorovic, A. Capretta, G. D. Wright and A. Savchenko, Biochem. J., 2013, 454, 191–200 CrossRef CAS PubMed.
- N. Leban, E. Kaplan, L. Chaloin, S. Godreuil and C. Lionne, Biochim. Biophys. Acta, Gen. Subj., 2017, 1861, 3464–3473 CrossRef CAS PubMed.
- R. S. Parulekar and K. D. Sonawane, J. Cell. Biochem., 2018, 119, 9444–9461 CrossRef CAS PubMed.
- R. S. Parulekar, S. S. Barale and K. D. Sonawane, Braz. J. Microbiol., 2019, 50, 887–898 CrossRef CAS PubMed.
- G. Luci, F. Mattioli, M. Falcone and A. Di Paolo, Antibiotics, 2021, 10, 769 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |