
Recent developments in membrane targeting antifungal agents to mitigate antifungal resistance
Devashish
Mehta
 ,
Varsha
Saini
and
Avinash
Bajaj
*
,
Varsha
Saini
and
Avinash
Bajaj
*
Laboratory of Nanotechnology and Chemical Biology, Regional Centre for Biotechnology, Faridabad-121001, Haryana, India. E-mail: bajaj@rcb.res.in
First published on 26th June 2023
Abstract
Fungal infections cause severe and life-threatening complications especially in immunocompromised individuals. Antifungals targeting cellular machinery and cell membranes including azoles are used in clinical practice to manage topical to systemic fungal infections. However, continuous exposure to clinically used antifungal agents in managing the fungal infections results in the development of multi-drug resistance via adapting different kinds of intrinsic and extrinsic mechanisms. The unique chemical composition of fungal membranes presents attractive targets for antifungal drug discovery as it is difficult for fungal cells to modify the membrane targets for emergence of drug resistance. Here, we discussed available antifungal drugs with their detailed mechanism of action and described different antifungal resistance mechanisms. We further emphasized structure–activity relationship studies of membrane-targeting antifungal agents, and classified membrane-targeting antifungal agents on the basis of their core scaffold with detailed pharmacological properties. This review aims to pique the interest of potential researchers who could explore this interesting and intricate fungal realm.
1. Introduction
Fungal infections cause mild allergic reactions to severe, disfiguring, and potentially fatal invasive fungal diseases affecting over a billion people globally, and are responsible for ∼1.6 million deaths per year around the globe.1 Superficial fungal infections such as ringworm, mucosal, and vaginal infections account for 15% of fungal infections, thereby making them the most deadly among other communicable diseases.2,3 Innate immunity, also called the first line of defence, caused by the body surface and epithelial surfaces of the respiratory, genitourinary, and gastrointestinal tracts, acts as a barrier against fungi.4 Cells of innate response like neutrophils and dendritic cells can cause direct antifungal effects or can secrete microbicidal compounds to clear fungal infections.5–7 Notably, the host immune system acts as a defence army against fungal infections. However, immunocompromised patients are always at high risk as employment of immunosuppressive drugs in clinical settings and advent of HIV patients are important factors for increased number of immunocompromised individuals.8,9Fungal pathogens can be categorized into primary and opportunistic, where primary fungi are responsible for invasive fungal infections in healthy individuals, and opportunistic pathogens only affect immunodeficient individuals.10 Five different antifungal classes, including azoles, polyenes, echinocandins, allylamines, and antimetabolites, are commonly prescribed to manage topical to systemic fungal infections. Azoles like fluconazole, ketoconazole, and itraconazole are commonly used to manage mild topical and systemic infections,11 and polyenes like amphotericin B (AmB) are employed to tackle systemic fungal infections.12 Apart from these, echinocandins, allylamines, and antimetabolites are the other commonly used drugs that are usually prescribed.13 However, overprescription or misuse of these antifungal agents can lead to the development of drug-resistant fungal pathogens like C. auris.14 Therefore, antimicrobial resistance (AMR), considered the next pandemic, is responsible for 0.7 million deaths annually, and around 10 million deaths are expected by 2050.15,16 Development of multidrug-resistant microbial infections, including skin structure, nosocomial, and urinary tract infections, creates a challenge for patients undergoing chemotherapy, surgical procedures, and transplantation.17
Microbial cells, including bacteria and fungi, develop different intrinsic and extrinsic mechanisms to develop resistance against antimicrobial regimens.18 Fungal cells possess several mechanisms like overexpression of drug targets and efflux pumps, alteration in drug targets, and biofilm formation to develop drug resistance. Overexpression of efflux pumps and alteration in drug targets are mainly associated with azole resistance, whereas alteration in drug targets and biofilm formation are two mechanisms commonly adapted by fungal cells to gain resistance against every antifungal drug. Individually or collectively, these mechanisms result in drug-resistant fungal cells,19 and development of drug-resistant pathogens has accelerated the pursuit of novel antifungal agents like new azole-derivatives (luliconazole and albaconazole).20,21 Apart from this, repurposing of non-antifungal drugs, biofilm inhibitors, and molecules from natural resources has emerged as a potential antifungal approach.22–27
The fungal cell membrane presents a unique chemical composition that differs from those of other microbial and mammalian cells due to the presence of ergosterol in the lipid bilayer, chitin as the basement layer, and glucan-based molecules, thereby making it a potential target for antifungal drug discovery.28 Antimicrobial peptides (AMPs) play an essential role in the host defence system, and as a part of the innate system, AMPs disrupt microbial membranes, including fungal membranes, through electrostatic interactions to eradicate microbial infections.29 However, the stability and safety profile of AMPs limit their clinical applications which inspired researchers to design and develop mimics of AMPs to target fungal membranes.30 Here, we described different clinically used antifungal drugs with their limitations, and emphasized the emergence of antifungal resistance and its different mechanisms. We then presented recent advancements in the design and development of membrane-targeting antifungals that use fungal cell membranes as potential therapeutic targets.
2. Clinically used antifungal agents
Over the last century, several topical antimicrobial agents have been used to manage common fungal and bacterial infections. However, to target invasive mycoses that need systemic antifungal medication, five major antifungal classes, azoles, polyenes, echinocandins, antimetabolite, and allylamines are approved by the Food and Drug Administration (FDA) (Fig. 1). Azole-based drugs are commonly prescribed, and known as principal class antifungal agents to manage topical and systemic fungal infections as these agents target the lanosterol 14α-demethylase enzyme, an essential enzyme in ergosterol biosynthesis. Major advantages of azoles are that they can be taken orally and display a broad-spectrum activity against various fungal strains. On the basis of the heterocyclic ring, azole-based antifungal drugs can be further divided into three subclasses: imidazoles, triazoles, and tetrazoles (Fig. 1a–c).31 From the mid-1970s to the 1990s, various imidazole-based azoles emerged such as miconazole, clotrimazole, econazole, ketoconazole, tioconazole, and sulconazole (Fig. 1a). Different imidazole-containing antifungals have been approved recently, including sertaconazole and luliconazole.32 Starting in the 1990s, triazole-based azoles have become increasingly popular such as terconazole, fluconazole, itraconazole, voriconazole, posaconazole, efinaconazole, and isavuconazonium.33 Triazole-based azoles are thought to be more specific to the fungal cytochrome P450 enzyme than their earlier imidazole-based counterparts. In addition, two more triazoles, albaconazole and PC945, are currently in clinical trials.34 Tetrazole-based antifungal agents have shown a broad spectrum of antifungal properties against different fungal species, and also displayed good oral bioavailability as compared to other azoles (Fig. 1c). Recently, a tetrazole-based fungal cytochrome P450-inhibitor, oteseconazole (VT-1161), has completed its phase III clinical trial. Studies demonstrated that VT-1161 has greater specificity against fungal cytochrome over the human one.35,36 VT-1129 and VT-1598 are two more tetrazole-based molecules in the pre-clinical phase.37,38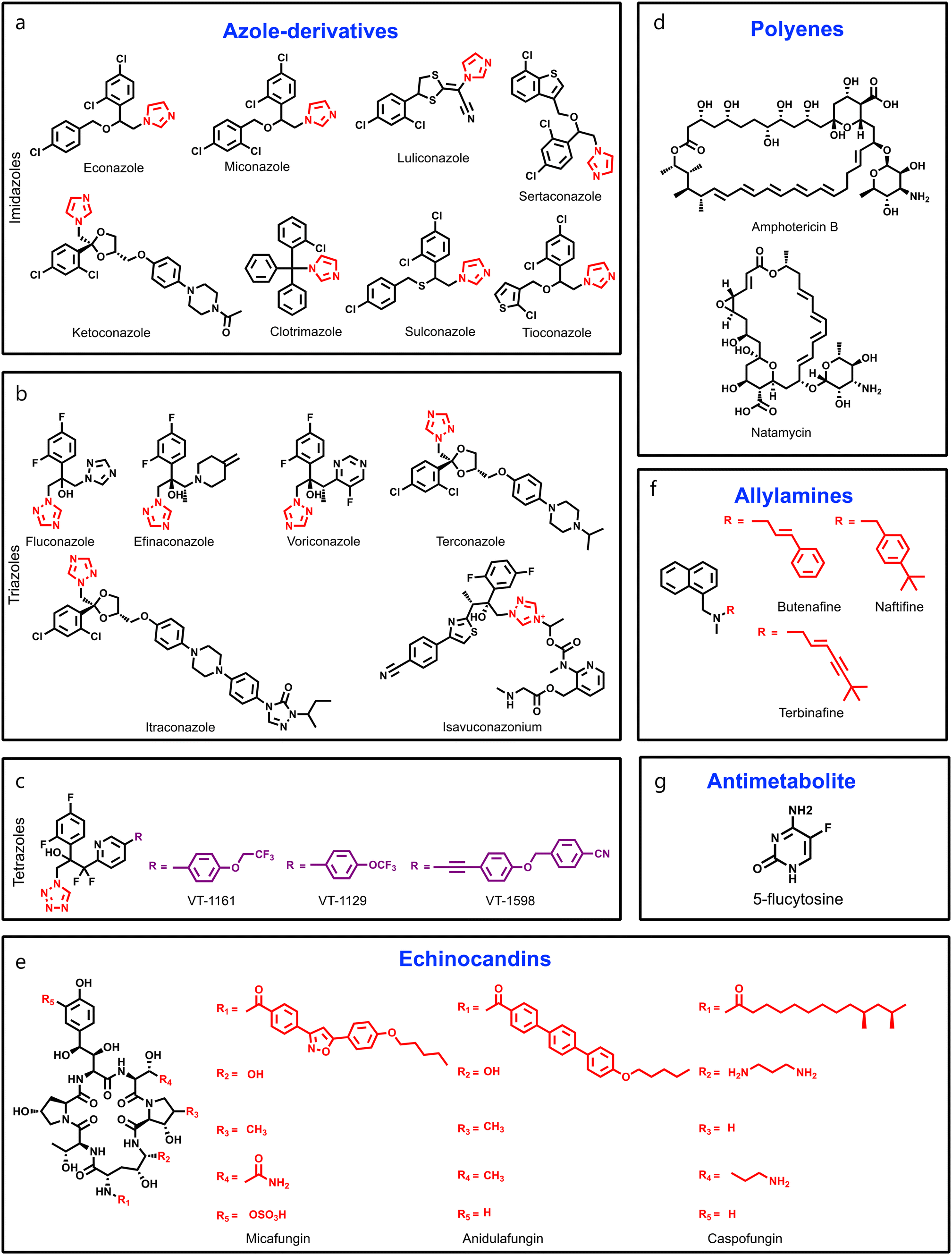 | ||
| Fig. 1 (a–c) Chemical structures of FDA-approved antifungal drugs: imidazole ring containing azoles (a), triazole ring containing azoles (b), and tetrazole ring containing azoles (c). (d) Chemical structures of amphotericin B and natamycin. (e) General chemical structures of echinocandin antifungal drugs. (f) Common chemical structures of antifungal allylamines. (g) Chemical structure of 5-flucytosine (5-FU). | ||
Apart from azoles, polyenes have also been used extensively as primary fungicidal agents against species of Aspergillus, Candida, and Cryptococcus since their discovery in the 1950s (Fig. 1d).39,40 Polyenes are amphipathic natural products of Streptomyces nodosus, a soil actinomycete,41 and are usually prescribed for systemic fungal infections such as cryptococcal meningitis, aspergillosis, and superficial fungal infections such as thrush. Among over 200 polyenes, six molecules have been used in clinical settings, including amphotericin B (AmB), nystatin, natamycin, trichomycin, candicidin, and methyl paricin,42 where natamycin is used against ophthalmic infections, and nystatin is used to manage vulvovaginal and oral fungal infections (Fig. 1d).43 AmB is the leading prototype for systemic fungal infections as it forms membrane-spanning channels during its interactions with ergosterol-containing membranes, thereby causing leakage of cellular components and cell death. A liposomal formulation of AmB has been prescribed to treat systemic mycoses.43 However, recent in-depth structural and biophysical studies revealed that polyenes bind to ergosterol, and impair its ability to carry out its normal vital cellular functions, leading to membrane permeabilization.44,45 As there is a close structural relationship between ergosterol and the mammalian membrane sterol cholesterol, the use of polyenes in medicine is constrained despite its potent killing activity.46,47
Echinocandins are antifungal drugs that treat fungal infections associated with Candida and Aspergillus, and are derived from the natural product echinocandin B, a lipopeptide produced by the fungus Glarea lozoyensis.48 Echinocandins have three structural components, a cyclic hexapeptide lactone core, a fatty acid side chain, and an amide group. The cyclic hexapeptide lactone core is composed of two amino acids, a diaminobutyric acid (Dab) and either a hydroxyphenylalanine (Hyp) or a homophenylalanine (Hph).49 A fatty acid side chain is linked to the core via an amide bond, and provides a hydrophobic environment that is essential for the drug's activity.50 In order to develop potent and better echinocandins, researchers are keen to modify the lipid side chains.51,52 Mechanistically, echinocandins act by inhibiting the synthesis of 1,3-β-D-glucan, an essential component of the fungal cell wall, and therefore, these are specific in nature, as normal cells do not possess a cell wall.53 Echinocandins like caspofungin (CFG), anidulafungin (AFG), and micafungin (MFG) are particularly useful in treating infections in immunocompromised patients,54 and a new molecule of this class, rezafungin (CD101), is in phase III clinical studies (Fig. 1e). However, the poor oral bioavailability of echinocandins affects their usage in clinical settings, and like AmB, echinocandins are administered through an intravenous route.55
Allylamines and antimetabolites are two other classes of synthetic antifungals, where the allylamines have a naphthalene group as an essential pharmacophoric feature (Fig. 1f).56 Allylamine-based drugs hinder the production of ergosterol by blocking the action of squalene epoxidase, also known as squalene monooxygenase, and this is a selective, reversible, and non-competitive inhibition.57 Drugs like terbinafine, butenafine, and naftifine are examples of allylamines, and are generally formulated as creams or powders to target topical fungal infections, and terbinafine is only used as an oral formulation and employed to treat onychomycosis.58 Apart from fungal cell wall targeting drugs, DNA/RNA targeting drugs like 5-flucytosine (5-FU) are used in managing fatal fungal infections (Fig. 1g). It is a pyrimidine-based prodrug of the active metabolite 5-fluorouracil, and targets DNA/RNA synthesis by inhibiting thymidylate synthase.59 However, the toxicity profile of 5-FU, and development of fungal resistance restrict its use in clinical practice. Therefore, it is used in combination with AmB for the treatment of severe candidiasis.60
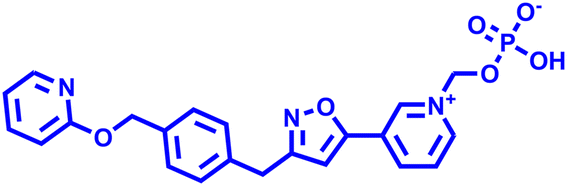
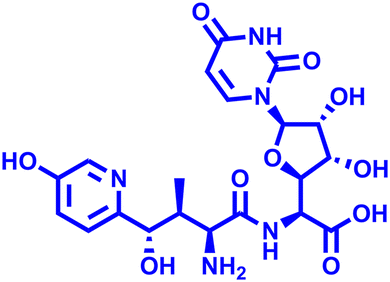
Besides this, several compounds like fosmanogepix (APX-001A), nikkomycin Z, and T-2307 are in clinical trials (Table 1). Amplyx Pharmaceuticals developed APX-001 and its N-phosphonooxymethyl prodrug that affects the glycosylphosphatidylinositol (GPI) biosynthetic pathway by inhibiting the Gwt1 enzyme. APX-001 showed good tolerance and antifungal properties in clinical phase 1 trials. Nikkomycin Z was isolated from Streptomyces tendae which is a pyrimidine nucleoside and inhibits the biosynthesis of chitin. Nikkomycin Z is a specific antifungal agent, as chitin is absent in mammalian cells. T-2307 is an arylamidine-based antifungal agent that exhibits potent fungicidal properties. It mainly disrupts the mitochondrial membrane potential of fungal cells, which leads to fungal cell death, and is well tolerated in human phase 1 studies.
| Drug candidate | Molecular target | Current status |
|---|---|---|
 Fosmanogepix (APX-001A) Fosmanogepix (APX-001A) |
Glycosyl phosphatidylinositol synthesis | Phase II completed (NCT04148287) |
 Nikkomycin Z Nikkomycin Z |
Chitin synthase | Phase I completed (NCT00834184) |
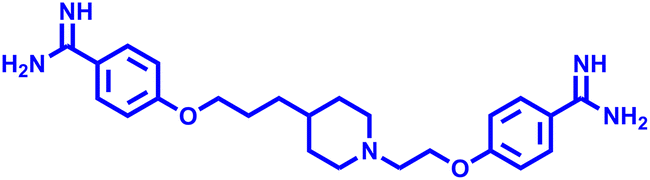 T-2307 T-2307 |
Mitochondrial membrane potential | Phase I completed (ref. 206) |
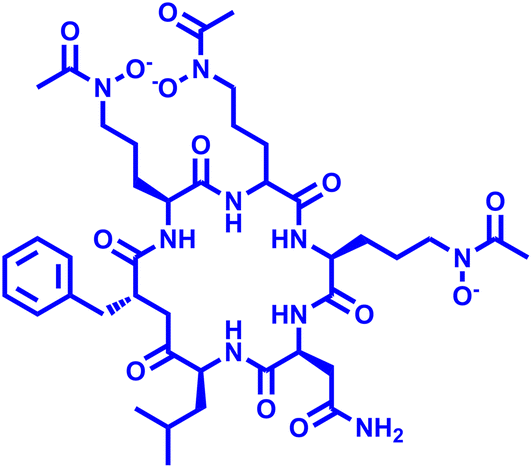 VL-2397 (ASP2397) VL-2397 (ASP2397) |
Aluminium chelating agent | Phase II terminated (NCT03327727) |
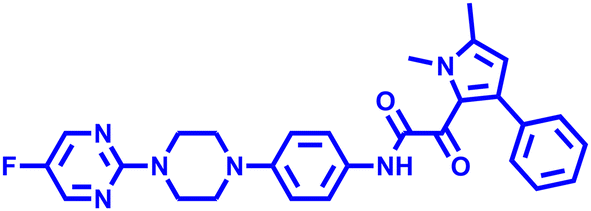 F901318 F901318 |
Dihydroorotate dehydrogenase | Phase II (NCT02856178) |
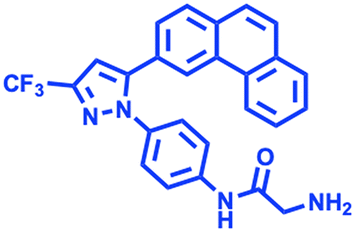 AR-12 AR-12 |
Stimulates host immunity, and inhibits fungal acetyl CoA synthase 1 | Phase I completed for oncology (NCT00978523) |
3. Antifungal resistance: an emerging problem
Fungal cells can adapt different mechanisms to evade antifungal drugs for their survival, like modification of drug targets, overexpression of multi-drug transporters, and stimulation of cellular stress (Fig. 2a).61 Fungal cells employ overexpression and mutations of ERG11, and expression of efflux pumps to develop resistance against azoles. As the ERG11 gene is a critical player in the encoding of lanosterol 14α-demethylase, fungi cells increase the transcriptional levels of ERG11 mRNA leading to overexpression of lanosterol 14α-demethylase.62 Fungi cells can also alter the azole-binding site of lanosterol 14α-demethylase as found in C. albicans. According to earlier research, three “hot spot” areas within ERG11p include several crucial allelic variants that reduce fluconazole sensitivity.63 Whaley et al. summarized different mutations found in ERG11 of C. albicans. In addition, overexpression of efflux pumps, such as ATP-binding cassette transporters, can expel intracellular azoles and cause drug resistance.64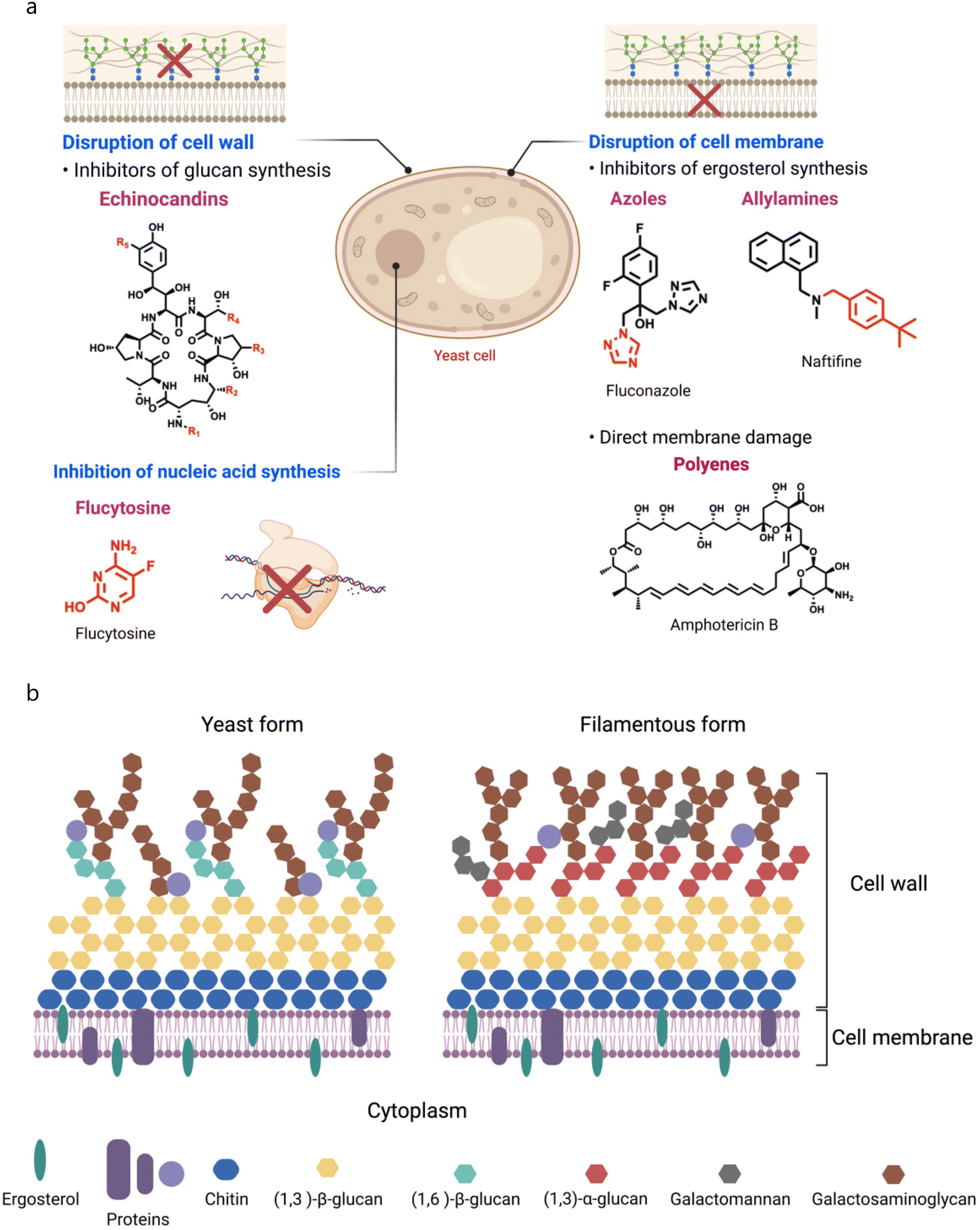 | ||
| Fig. 2 (a) Mechanism of action of antifungal drugs. (b) Structural features of the fungal cell wall and cell membrane. | ||
Even though polyenes have been utilized for many years, resistance against polyenes is still much less common than that against other antifungal drugs as polyenes target a structural component of the cell membrane instead of a vital enzyme.65 However, mutations in ergosterol biosynthesis enzymes, including ERG2, ERG3, ERG5, and ERG11, can contribute to the reduction of AmB potency against C. albicans.66 Mutations in ERG2, ERG6, and ERG11 were found in polyene-resistant C. glabrata clinical isolates,67 and in ERG1, ERG2, ERG6, and ERG13 in the case of C. auris.68 A recent investigation on C. auris emphasized the potential role of drug transporters in amphotericin B resistance, where whole-genome sequencing of polyene-resistant clinical isolates showed four non-synonymous mutations, one of which was in a potential membrane transporter.69
Chronologically, echinocandins are the newest antifungals with a narrow spectrum of activity, and only few reports have demonstrated resistance to echinocandins. Candida species develop resistance against echinocandins by mutating FKS genes.70 Echinocandins target β-1,3-glucan synthase, which is present in fungal membranes and possesses a catalytic subunit called FKS1. Fungi also possess its homologs, the FKS2 and FKS3 proteins, with low expression, and they regulate the FKS1 expression.71 FKS1 and FKS2 protein mutations majorly contribute to echinocandin resistance, and there are three hot spot regions in the FKS1 protein for mutations, where region I includes amino acid residues FLTLSLRDPI, region II includes PAIDWIRR, and region III includes WRNIFTRL.72 In addition, a few reports also suggested an increase in chitin production responsible for echinocandin resistance. However, the overexpression of efflux pumps shows a minimal role in echinocandin resistance.73
Apart from these mechanisms, fungal cells can form biofilms that act as a barrier against antifungal drugs and the host immune system. A fungal biofilm involves a community of irreversible adherent fungal cells on a surface like inert materials, living tissue, and medical devices.74 The biofilm life-cycle consists of four steps, initial attachment, proliferation, maturation, and dispersion, and among fungi strains, Candida species have the highest ability to form biofilms. Biofilm formation in terms of morphology, extracellular matrix (ECM), and ability to adapt to antifungal resistance varies from species to species, and this variability creates a hurdle in discovering an effective approach to address biofilm-associated threats.75 Fungal biofilms show intrinsic resistance against both azoles and polyene derivatives.76 However, some reports suggest that polyenes display potent antibiofilm properties, but a high toxicity profile limits their clinical applications.77,78 A liposomal formulation of AmB demonstrates potent antibiofilm activity against C. albicans biofilms.79 Apart from these drugs, several new approaches have emerged to avoid fungal biofilms, including the combination of DNAse with antifungal regimens, modulators of quorum sensing, AMPs, antifungal coatings, and photodynamic therapy.80 Drug repurposing is also an emerging approach to tackle fungal biofilms,81 and several reviews emphasized emerging new and old drug candidates against fungal biofilms.82,83
4. Fungal membrane as a therapeutic target
The fungal plasma membrane and cell wall work together to provide cells strength, which allows cells to sustain turgor pressure, and also offers protection against antifungal agents.84 Fungal cells are fundamentally different from mammalian cells since their membranes are composed of different lipids, which therefore, makes them more susceptible to certain drugs, and making it possible to create therapeutic regimens that specifically target fungal cells without impacting human cells.85 As lipids are essential components of all cells, the fungal cell membrane is also composed of different lipids such as glycerophospholipids, sphingolipids, and sterols, where glycerophospholipids constitute 55–75% of the total lipids.86 Depending upon head groups, glycerophospholipids can be phosphatidylethanolamine (PE), phosphatidylcholine (PC), phosphatidylserine (PS), and phosphatidylinositol (PI) (Fig. 2b),87 and are major partners of total phospholipids in Saccharomyces cerevisiae.88 Apart from glycerophospholipids, sphingolipids are also essential constituents of fungal membranes, and constitute 7–16% of fungal membrane lipids.89 Sphingolipids have a sphingosine backbone, linked with sphingoid long-chain aliphatic amino alcohols, and sphingoids such as sphingosine, dihydrosphingosine, and ceramides are biosynthesized from nonsphingolipid precursors.90In addition, sterols are another lipid component constituting 30–40% of fungal membranes. Sterols are amphipathic lipids with rigid and compact ring structures, and play vital roles including regulation of membranes' fluidity and permeability, and control of membrane-bound enzymes. Like cholesterol, fungal membranes are armed with a sterol-based biomolecule, ergosterol, which is also known as a fungal hormone, and maintains fungal membrane integrity and promotes growth and proliferation.91 Numerous reports described the role of ergosterol in the maintenance of mitochondrial DNA and stress adaptation as well.92,93 The biosynthetic pathway of ergosterol involves multistep biochemical reactions that occur in the endoplasmic reticulum, and numerous enzymes catalyse these biochemical conversions.94,95 Two categories of genes involved in the initial stages of ergosterol biosynthesis include essential genes (such as ERG1, ERG7, ERG9, ERG11, ERG24, ERG25, ERG26, and ERG27) and non-essential genes. For example, ERG9 encodes squalene synthase, an enzyme that catalyzes the production of squalene, a precursor for ergosterol. In addition, ERG1 and ERG7 code for squalene epoxidase and lanosterol synthase, other essential enzymes of the ergosterol synthesis pathway. ERG11, which codes for lanosterol 14α-demethylase, is also of the fungal cytochrome P450 family.96 Therefore, a majority of antifungal medications available for clinical use have been developed to target ergosterol biosynthesis due to its unique biosynthesis pathway, distinctive structural characteristics, and essential roles.97 Apart from the individual class of lipids, the fungal membrane is also armed with lipid rafts composed of sterol and sphingolipids that play an important role in cell growth and development of cell polarity, formation of hyphae, and pathogenicity.98 Besides lipids, the fungal membrane also has various proteins that serve different functions like signal transduction, and cytoskeleton and cell wall synthesis.99,100 Therefore, the fungal membrane is composed of various biomolecules, including lipids and proteins, and collectively, these biomolecules impart a negative charge on the cell membrane. Miyake et al. showed that C. albicans cells possess a negative zeta potential at pH 7.4.101 They treated the fungal cells with antifungal agents (AmB, miconazole, ketoconazole, azalomycin F, and aculeacin A) at sub-inhibitory concentration and observed that these agents affected the zeta potential of fungal cells. Moreover, the relationship obtained between the change in zeta potential and adherence suggests that decreased electric repulsive forces were responsible for enhanced adherence of fungal cells.101
C. albicans is the most common and opportunistic pathogen responsible for IFDs, and its cell wall is composed of a two-layered structure. A β-glucan–chitin skeleton is considered as the main core of C. albicans that provides strength and shape to it. Chitin is localized in the inner layer, and its chains can form strong anti-parallel H-bonded structures. β-1,3-glucans are present in the inner cell wall, and are connected with β-1,6-glucans that link the inner and outer cell walls.102 The synthesis of β-1,3-glucans is catalysed by β-1,3-glucan synthase, composed of an enzyme complex with two subunits (Fksp and Rho1p). Fksp is responsible for the transfer of sugar groups from an activated donor to a specific donor through glycosidic linkage, and it is encoded by three ortholog genes, FKS1, FKS2, and FKS3, in C. albicans.103 Apart from this, β-1,6-glucans have side chains with varying lengths and distributions that can form complex structures stabilized through interchain H-bonds. Notably, β-1,6-glucans serve as linker molecules connecting various cell wall proteins to the β-1,3-glucan–chitin core through glycosylphosphatidylinositol (GPI) proteins. In addition, β-1,6-glucan levels are high in the C. albicans cell wall compared to that in S. cerevisiae.104 Mannoproteins are the major biochemical constituents of the outer layer of the C. albicans cell wall. Chemically, these GPI-modified molecules are cross-linked to β-1,6-glucans through N- or O-linkage N-linked mannans are constituted of α-1,6-mannose, that have a backbone with α-1,2-oligomannose, and sidechains armed with β-1,2-mono to tetra mannans. In contrast, O-linked mannans are linked to glycoproteins of the cell wall. Mannans do not affect the cell shape, as they are comparatively less rigid than β-glucans and chitin.105 However, they have low permeability and porosity which contributes to the cell wall's resistance against antifungal agents and host defense mechanisms. In addition, they are also known as PAMP (pathogen-associated molecular pattern) ligands that affect host defense mechanisms.106
AMPs are part of a host's innate immune system, and bear short amino acid sequences with a net positive charge and can form α-helical and β-sheet secondary structures. These conformations provide facial amphiphilicity to AMPs, essential for their antimicrobial activity. As electrostatic differences exist, AMPs preferentially target microbial membranes over the host, and their structural features and mode of action are well documented in several reviews.107,108 Different models, including barrel-stave, carpet, toroidal-pore, and translocation models were proposed to dictate the membrane disruption properties of AMPs.109 Apart from membrane disruptions, AMPs can also impact intracellular targets via generation of ROS (reactive oxygen species), autophagy, and mitochondrial dysfunction.110,111 Cysteine-rich antifungal peptides have been classified based on their source like insects, plants, and mammals,112 and recently, Struyfs et al. summarized known peptides with antifungal properties through membrane interactions.113,114 However, the lack of stability and poor pharmacological profile of AMPs limit their clinical applications,115,116 and therefore, led to researchers developing various molecules targeting the fungal membranes.117
5. Recent fungal membrane-targeting molecules
5.1 Peptide-based molecules
Limited therapeutic applications of AMPs have inspired researchers to design and develop AMP-mimicking peptide-based molecules, as macromolecular antimicrobials like peptides and cationic polymers can target multi-drug-resistant pathogens.118,119 In this regard, Zhou et al. developed synthetic copolypeptides, and investigated their antimicrobial activities against fungal and bacterial pathogens.120 However, the presence of a hydrophobic moiety is responsible for their higher toxicity profile. Li et al. developed and tested peptidopolysaccharides as broad-spectrum antimicrobials against both fungal and bacterial strains, and found that the antimicrobial properties depend on the number of tethered lysine residues.121 A peptidopolysaccharide tethered with 16 lysine residues was found to be highly effective with membrane-disruptive properties and negligible toxicity against RBCs and human aorta smooth muscle cells (SMCs) (Fig. 3a–c). Apart from the peptidopolysaccharides, nylon-based polymers (called poly-β-peptides) also showed broad-spectrum properties against different bacterial and fungal pathogens.122–124 | ||
| Fig. 3 Peptide-based antifungal molecules. (a) Chemical structure of peptidopolysaccharide scaffold derived molecules. (b) Membrane disruption activity of a peptidopolysaccharide derivative (CS-g-K16). (c) Cytotoxicity profile of the peptidopolysaccharide derivative (CS-g-K16) at different concentrations. (d) Molecular structure of hyperbranched polylysine (HPL) scaffold containing molecules. (e) Time-dependent killing studies of HPL derivatives against fungal cells. (f) Drug resistance studies of HPLs with fluconazole. (g) Membrane lysing property of HPLs. (h) Survival efficacy of HPLs against a murine fungal infection model. (i) Molecular structure of compound 1. (j) Molecular structure with amino acid sequences of acylated short peptides. (k) Growth inhibitory rate of acylated short peptides (Fig. 3b and c reproduced from ref. 121 with permission from John Wiley and Sons, copyright 2012, Fig. 3e–h reproduced from ref. 125 with permission from John Wiley and Sons, copyright 2022, and Fig. 3k reproduced from ref. 129 with permission from Elsevier, copyright 2023). | ||
Liu et al. developed a series of hyperbranched polylysine (HPL) based antifungal agents (Fig. 3d).125 They employed condensation polymerization to get six different HPL derivatives, and amine groups of HPL3 were guanylated through 1H-pyrazole-1-carboxamidine which yielded HPL3-Gx. Among the guanylated HPLs, HPL3-G60 showed potent antifungal activity against different fungal pathogens, and HPL-Gx (x = 40–80) was found to be 32-fold more active than HPL3 against C. parapsilosis (Fig. 3e). Interestingly, fungal pathogens could not develop resistance against these HPL-Gx agents (Fig. 3f). In-depth mechanistic studies revealed that treatment with HPL-Gx can damage the cell wall and membrane (Fig. 3g) with minimal toxicity against RBCs and NIH 3T3 cells. Further, they investigated these peptides against a lethal C. albicans murine model, and among different treatment regimens, fluconazole and HPL3 through an intravenous route showed better mice survival (Fig. 3h).125
Lipopolypeptide-based amphiphiles also showed pharmacological properties against bacterial and fungal infections.126 Zoysa et al. designed and developed a series of sixteen battacin lipopolypeptides as putative antifungal agents.127 Among these molecules, the 4-methylhexanoyl tethered trimeric lipopolypeptide having ten units of D-2,4-diaminobutyric acid (D-Dab) (compound 1) exhibited potent antifungal activity against C. albicans with a MIC value of 6.25 μM without affecting RBCs (Fig. 3i). Importantly, the antifungal activity of compound 1 was maintained in an acidic environment, and showed synergism with AmB. Additionally, compound 1 demonstrated potent antibiofilm properties against pre-formed C. albicans biofilms in a dose-dependent manner. Mechanistically, compound 1 was found to disrupt the fungal membranes.127 Lu′s group reported acylated antimicrobial peptides to target bacterial pathogens where they used a CxG(IIKK)yI–NH2 backbone with varied acyl chains.128 In another report, they tested a series of CnKI3 lipopeptides against different microbes, and found that an increase in alkyl chain length can enhance the spectrum of antimicrobial activities and decrease the critical aggregation concentration (Fig. 3j). They also reported that the peptide bearing a C14 alkyl chain showed potent antifungal activity against a C. albicans strain (Fig. 3k).129
Recently, Zhang and co-workers designed and developed lipo-γ-AA peptides (oligomers of N-acylated-N-aminoethyl amino acids) having different fatty acids including palmitic acid, oleic acid, and stearic acid to target fungal infections (Fig. 4a).130 They found that MW5 (palmitic acid-tethered) showed potent broad-spectrum activity against fungal pathogens, and fungal cells do not gain any resistance against MW5 up to 25 days (Fig. 4b). Mechanistically, MW5 disrupts the fungal membrane and produces ROS (Fig. 4c), and application of MW5 enhanced the G. mellonella survival against fungal infection (Fig. 4d). Interestingly, MW5 can rejuvenate the therapeutic efficacy of fluconazole against drug-resistant C. albicans, and the combination of MW5 and fluconazole exhibited promising therapeutic potential against a mucocutaneous murine model with an ∼2 fold reduction in fungal burden as compared to the untreated group (Fig. 4e and f).
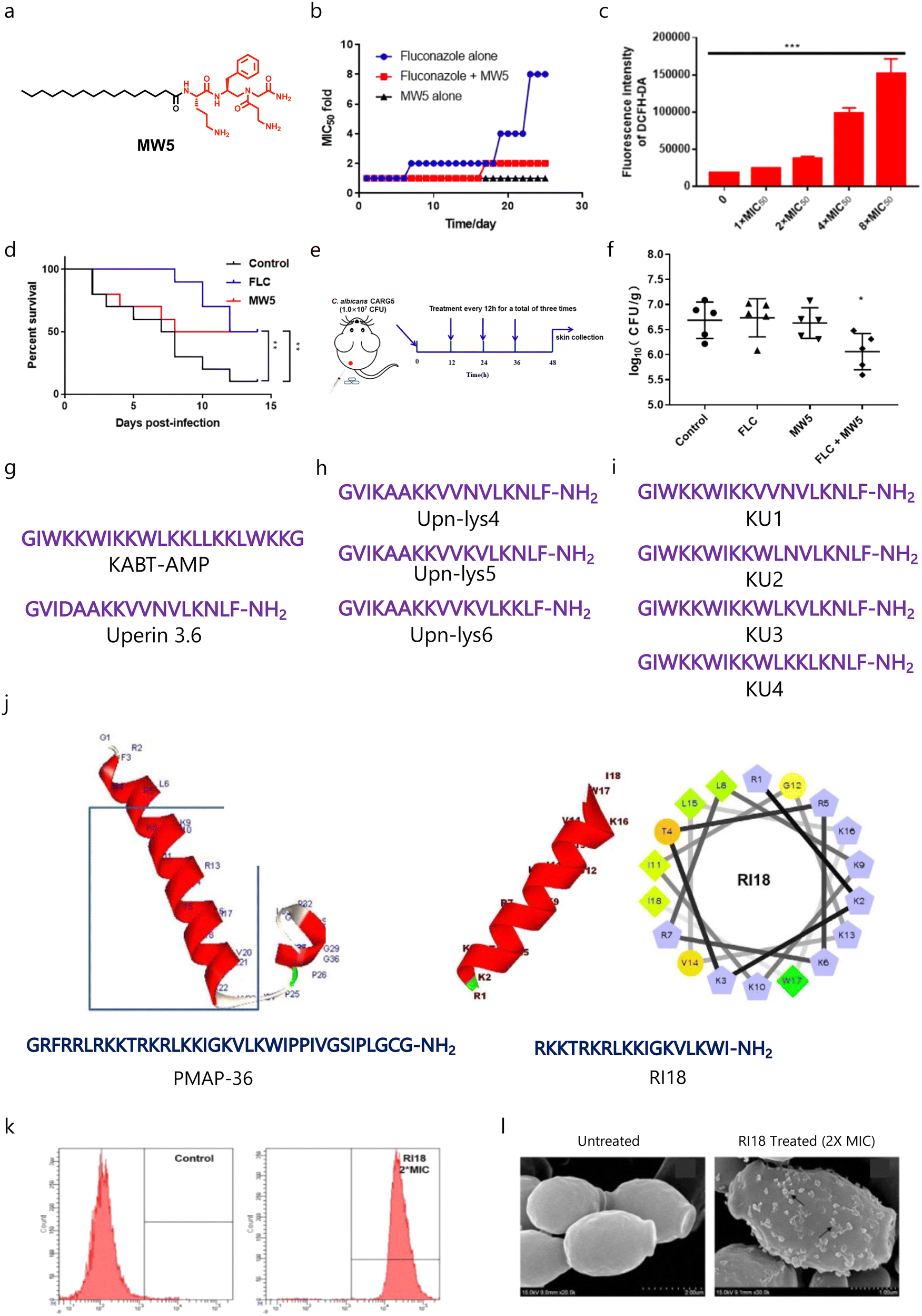 | ||
| Fig. 4 (a) Chemical structure of the MW5 molecule. (b) Drug-resistance study of the MW5 amphiphile with and without fluconazole. (c) ROS study showing increased production of ROS with increased concentration of MW5. (d) Survival studies of MW5 against a G. mellonella fungal infection model. (e) A schematic showing the experimental plan for a murine infection model. (f) Antifungal efficacy of MW5 against the murine infection model. (g–i) Amino acid sequences of parent AMPs and their derivatives. (j) Three dimensional structure and amino acid sequences of the parent peptide (PMAP-36) and developed potent short peptide (RI18). (k and l) Membrane-targeting properties of RII8 (Fig. 4b–f reproduced from ref. 130 with permission from the American Chemical Society, copyright 2022, and Fig. 4j–l reproduced from ref. 132 with permission from Springer Nature, copyright 2016). | ||
As the synthesis of polypeptides and their derivatives is tedious, therefore, short synthetic peptide-based cationic amphiphiles have gained more attention. Lum et al. employed two known AMPs, KABT-AMP and uperin 3.6 as prototypes (Fig. 4g), to design and develop a series of new antifungal peptides.131 In order to increase the therapeutic potential of uperin 3.6, the less hydrophobic amino acids were replaced with three lysine residues (Fig. 4h). Further, they fused KABT and uperin 3.6 to develop four hybrids (KU1–KU4) (Fig. 4i). In the case of uperin 3.6 analogues, replacement of two lysine residues led to improved antimicrobial activity, whereas substitution of a single lysine residue led to similar antimicrobial properties. In the case of uperin derivatives, both the parent molecule and Upn-Lys6 showed similar killing kinetics. Among the developed hybrid peptides, KU4 was found to be the most potent peptide in terms of killing kinetics, as KU4 showed an ∼5.55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log decrease in CFU mL−1 within 6 h. Among the hybrid peptides, KU4 also showed potent antibiofilm properties against C. albicans. Interestingly, Upn-lys6 showed similar antibiofilm properties to KU4, while the parent peptide and other uperin analogues were found to be less potent. Moreover, these peptides displayed synergism with conventional antifungal drugs and other AMPs against C. albicans, and displayed minimal toxicity against RBCs and human epithelial cells.131 In a similar fashion, Lyu et al. developed a series of PMAP-36-based short peptides (Fig. 4j), and tested them against different fungal and bacterial pathogens.132 SAR studies showed that a decrease in chain length enhances the antimicrobial properties and reduces the toxicity profile of the parent peptide, and RII8 showed promising antimicrobial properties (Fig. 4j). Mechanistically, RII8 can cause membrane disruptions and cellular damage in a dose-dependent manner (Fig. 4k and l).132
log decrease in CFU mL−1 within 6 h. Among the hybrid peptides, KU4 also showed potent antibiofilm properties against C. albicans. Interestingly, Upn-lys6 showed similar antibiofilm properties to KU4, while the parent peptide and other uperin analogues were found to be less potent. Moreover, these peptides displayed synergism with conventional antifungal drugs and other AMPs against C. albicans, and displayed minimal toxicity against RBCs and human epithelial cells.131 In a similar fashion, Lyu et al. developed a series of PMAP-36-based short peptides (Fig. 4j), and tested them against different fungal and bacterial pathogens.132 SAR studies showed that a decrease in chain length enhances the antimicrobial properties and reduces the toxicity profile of the parent peptide, and RII8 showed promising antimicrobial properties (Fig. 4j). Mechanistically, RII8 can cause membrane disruptions and cellular damage in a dose-dependent manner (Fig. 4k and l).132
Recently, Sharma et al. designed and developed short synthetic peptide-derived amphiphiles based on the dipeptides Trp–His(1-Bn)–OMe/NHBn and tripeptides His(1-Bn)–Trp–His(1-Bn)–OMe/NHBn, and highlighted compound 2 as an effective fungicide against different fungal pathogens which did not affect mammalian cells and RBCs (Fig. 5a).133 In-depth mechanistic studies revealed the membrane-disruptive properties of compound 2 (Fig. 5b), and it showed synergism with AmB against C. neoformans fungal cells.133Polybia-MPI was initially discovered in the venom of the social wasp species Polybia paulista, and displayed broad-spectrum antibacterial properties. Wang's group showed the antifungal properties of Polybia-MPI with a MIC80 of 8 and 16 μM against C. glabrata and C. albicans, respectively (Fig. 5c).134 Flow cytometry analysis showed that Polybia-MPI can disrupt the fungal membranes in a dose-dependent manner (Fig. 5d). They also validated these findings with confocal (Fig. 5e) and SEM imaging (Fig. 5f). In addition, they observed concentration-dependent antibiofilm properties against C. glabrata biofilms (Fig. 5g).
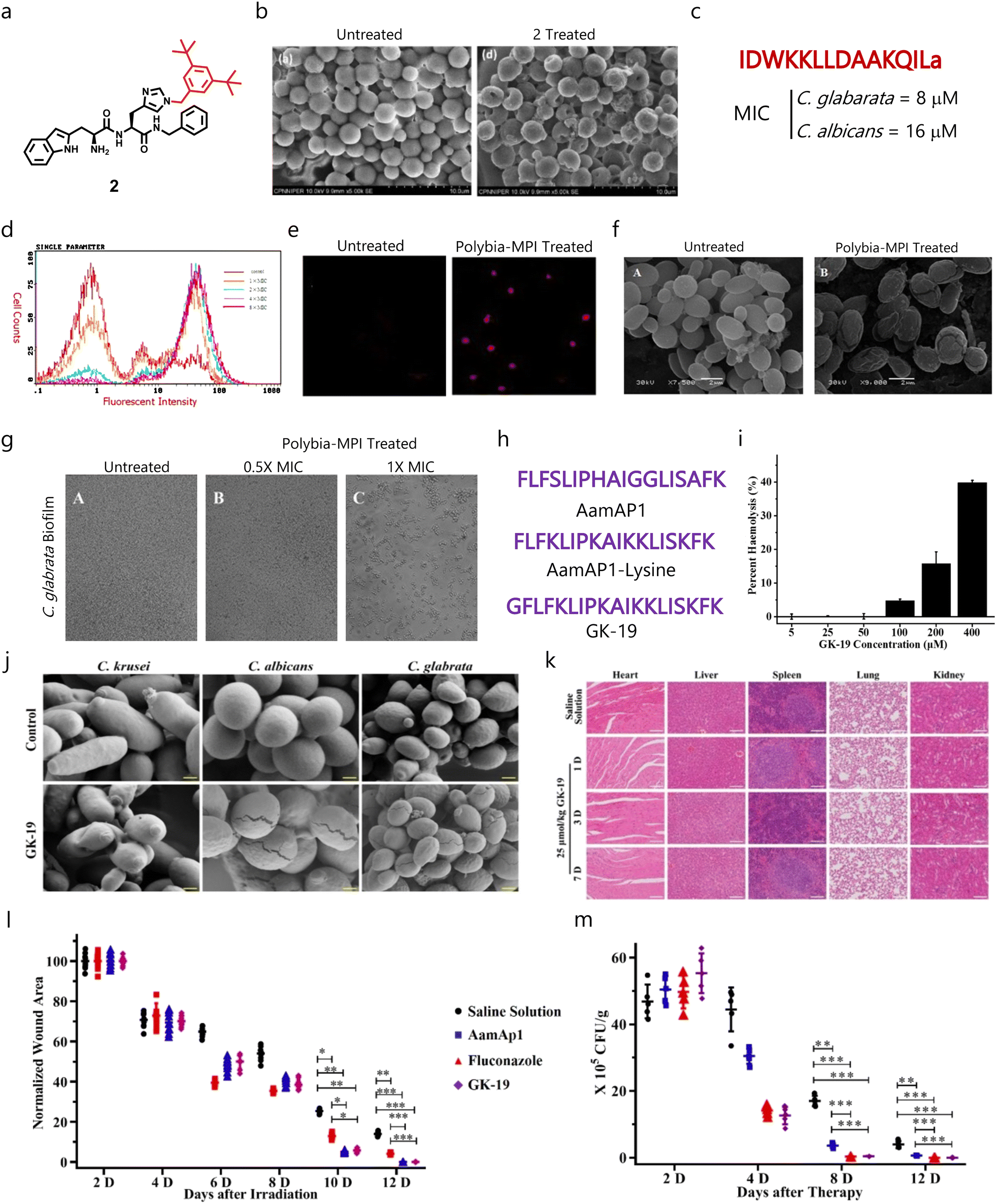 | ||
| Fig. 5 (a) Chemical structure of compound 2. (b) SEM images of untreated and treated fungal cells with compound 2 showing its membrane disruption properties. (c) Amino acid sequence and MIC of Polybia-MPI. (d) Flow cytometry studies showing PI-positive cells or membrane compromised cells after treatment with Polybia-MPI. (e) Confocal images validating the flow cytometry results. (f) SEM micrographs validating the membrane disruption mechanism of Polybia-MPI. (g) SEM images demonstrating the dose-dependent antibiofilm properties of Polybia-MPI. (h) Amino acid sequence of AamAP1, AamAP1-lysine (parent AMPs) and GK-19. (i) Hemolytic properties of GK-19 against RBCs. (j) SEM micrographs displaying the membrane-targeting properties of GK-19 against C. albicans and C. glabrata. (k) Histological analysis of major organs suggesting the non-toxic behaviour of GK-19. (l) Time-dependent wound healing study showing the ability of GK-19 to heal the wound area. (m) Change in fungal load burden after giving different treatment regimens, suggesting that GK-19 has promising antifungal therapeutic efficacy (Fig. 5b reproduced from ref. 133 with permission from Elsevier, copyright 2022, Fig. 5d–g reproduced from ref. 134 with permission from Elsevier, copyright 2014, and Fig. 5i–m reproduced from ref. 136 with permission from MDPI, copyright 2022). | ||
Venom obtained from scorpions contains AMPs like AamAP1 that displays broad-spectrum antimicrobial properties.135 As their hemolytic profile is a major challenge, different derivatives were developed to enhance the safety of these AMPs. Recently, Song et al. designed and evaluated a venom AMP-derivative called GK-19 as a putative antimicrobial agent by introducing a glycine group at the N-terminal end of AamAP1 to decrease the hydrophobicity and enhance helicity (Fig. 5h).136 Studies demonstrated that the developed peptide exhibited minimal toxicity against RBCs (Fig. 5i). Further, GK-19 showed broad-spectrum antimicrobial properties against different bacterial and fungal pathogens, and exhibited dose-dependent membrane-lysis properties (Fig. 5j). In addition, it was found to be non-toxic against major organs like the heart, liver, spleen and lungs (Fig. 5k). GK-19 also exhibited promising antimicrobial and healing properties in a murine skin and soft tissue infection model (Fig. 5l and m).136
5.2 Steroid-based amphiphiles
Bile acids (BAs) are steroidal biomolecules that play a significant role in lipid metabolism. BAs are derived from cholesterol metabolism, and the presence of a steroidal backbone and hydroxyl groups is responsible for their amphipathic nature.137 BA-derivatives have been employed in advanced drug delivery systems like hydrogels and nanoformulations.138–141 BA-derivatives also showed potent anticancer,142 anti-inflammatory,143 and antimicrobial properties,144 and can also be used as diagnostic tools.145 BA-based antimicrobials called ceragenins were introduced by Paul B. Savage, as they can act as putative antibacterial agents by targeting bacterial membranes,146–151 and have also been shown as potent antifungals.Hazra et al. reported a series of BA–chloramphenicol derivatives by conjugating chloramphenicol at the C24 position of cholic acid (CA) and deoxycholic acid (DCA) through an amide linkage.152 Among the series of seven molecules, the DCA derivative showed potent antifungal properties against Cryptococcus neoformans (Fig. 6a). In another report, they synthesized BA–fluconazole conjugates using DCA and CA through click chemistry and tested them against different fungal strains (Fig. 6b and c).153 Among these conjugates, the DCA-based fluconazole conjugate was found to be most potent with a MIC80 of 3.12 μg mL−1 against C. albicans (Fig. 6b). SAR studies showed that conjugation of fluconazole at the C24 position of BA led to better antifungal properties as compared to that at the C3 position. They also conjugated different heterocyclic groups including imidazole, benzimidazole, triazole, and benzotriazole at the C24 position of DCA. However, these heterocyclic conjugates were found to be less fungicidal in nature.153 They further installed a β-lactam moiety at the C24 position of CA and DCA through triazole linkage with varying amide linkages at the C24 position of BAs to establish SARs (Fig. 6d).154 Studies showed that incorporation of an amide linkage with para-chlorobenzene can enhance the antimicrobial properties of DCA and CA-derivatives.154 Aher et al. designed and synthesized BA-based amino sterol molecules by installing a terminal amine group through ethyleneamine and ethylamine at the C3 position of methyl esters of CA and DCA.155 SAR studies showed that molecules armed with ethylenediamine bearing an extra amino moiety showed better antifungal properties over molecules having an ethylamine group.155
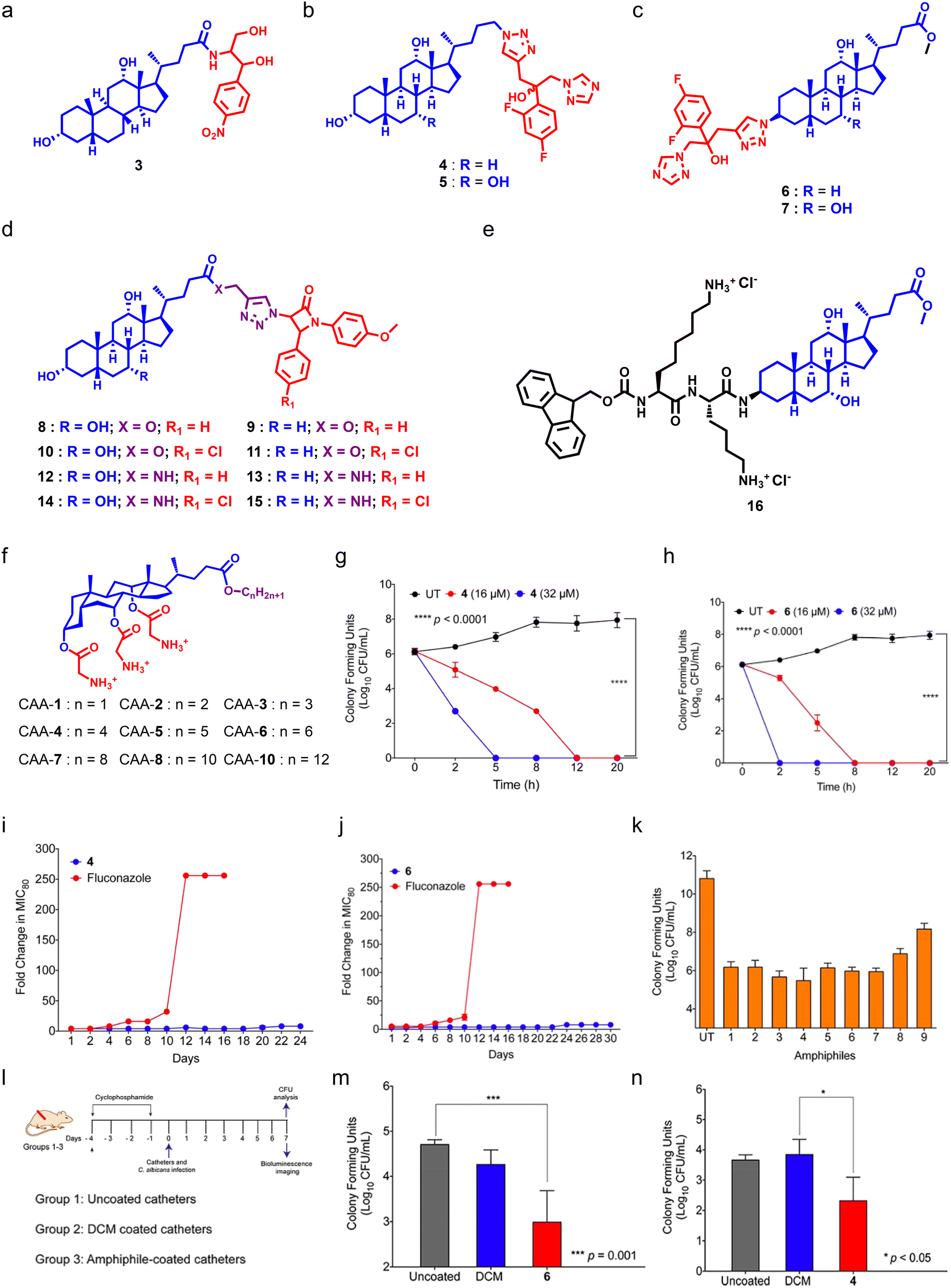 | ||
| Fig. 6 (a) Chemical structure of potent chloramphenicol-tethered bile acid molecule. (b) General chemical structure of C24 tethered fluconazole bile acid molecules. (c) General chemical structure of C3 tethered fluconazole bile acid molecules. (d) General molecular structure of β-lactam conjugated bile acid molecules. (e) Chemical structure of potent cholic acid–lysine conjugate amphiphile. (f) Chemical scaffold of cholic acid-based amphiphiles. (g and h) Time-dependent killing kinetics of CAA-4 (g) and CAA-6 (h) against C. albicans. (I and j) Drug-resistant studies of CAA-4 (i) and CAA-6 (j) against C. albicans. (k) Antibiofilm properties of cholic acid-based amphiphiles with preformed C. albicans-mediated biofilms. (l) A schematic showing the plan for animal experiments. (m and n) Antifungal efficacy of CAA-4 (m) and CAA-6 (n) against a murine wound infection model (Fig. 6g–n reproduced from ref. 158 with permission from the American Chemical Society, copyright 2021). | ||
Singla et al. designed and synthesized a series of 16 amphipathic derivatives of CA by attaching lysine to C3-β-amino cholic acid methyl ester to maintain a suitable ratio of hydrophobic to hydrophilic groups, which is necessary for antimicrobial effects (Fig. 6e).156 A set of synthesized conjugates that featured a fluorenyl-9-methoxycarbonyl moiety linked to CA via a lysine linker displayed decisive antimicrobial action against S. aureus, E. coli, and C. albicans. The efficacy of these compounds further increased with an increase in lysine residues. Moreover, the lead compounds exhibited good antimicrobial properties against drug-resistant bacterial and fungal clinical isolates, and also boosted the effectiveness of antifungal agents such as AmB and voriconazole. In addition, they were also found to damage microbial membranes while not causing any hemolytic activity or toxicity to normal cells or cancer cell lines.156
For the past decade, our group has been working on the development of CA-derived antimicrobial agents. We installed glycine groups at the C3, C7, and C12 positions of CA, and varied the C24 position with different alkyl chains (Fig. 6f).157 Among these amphiphiles, molecules having three glycine moieties with butyl (CAA-4) and hexyl chains (CAA-6) at the C24 terminal were found to be highly potent against fungal cells. Studies revealed that both amphiphiles were fungicidal in nature (Fig. 6g and h). Notably, fungal cells did not gain resistance against both amphiphiles (Fig. 6i and j), and the amphiphiles were found to be active against pre-formed Candida biofilms (Fig. 6k). Moreover, amphiphile-coated catheters displayed promising therapeutic efficacy against a C. albicans wound infection model in mice (Fig. 6l–n).158 Moreover, CAA-6 also showed potent antibacterial properties against different Gram-positive and Gram-negative pathogens.159
We also designed and synthesized CA–peptide conjugates by installing a benzyl moiety at the C24 position of CA, and 20 natural amino acids were conjugated at the hydroxyl position of CA through a glycine linker (Fig. 7a).160 Among the series, the molecule armed with a glycine–valine moiety (CAP-3) displayed potent broad-spectrum antimicrobial properties against Gram-positive bacteria, Gram-negative bacteria, and fungal pathogens. CAP-3 showed potent antibiofilm properties against Candida-mediated biofilms (Fig. 7b), and against polymicrobial biofilms (Fig. 7c and d). Mechanistically, CAP-3 targets the microbial membranes through electrostatic interactions, and can also degrade pre-formed polymicrobial biofilms on cover slips and medical devices like catheters. CAP-3 showed promising therapeutic potential against S. aureus (Fig. 7e), C. albicans (Fig. 7f) and polymicrobial (Fig. 7g) murine wound infection models.161 Recently, Hryniewicka et al. synthesized steroid-based imidazolium salts using lithocholic acid (LCA) and 3-oxo-23,24-dinorchol-4-en-22-al. First, the imidazole moiety was installed at the C24 position of LCA, and was treated with alkyl bromides or iodides with varying chain lengths (Fig. 7h).162 In a similar fashion, they have synthesized imidazolium salts of 3-oxo-23,24-dinorchol-4-en-22-al (Fig. 7i). Both series were screened against C. albicans and other bacterial strains, and the LCA-based imidazolium salts bearing methyl (compound 17) and ethyl (compound 18) showed potent fungicidal properties against C. albicans with a MIC80 value of 0.25 μg mL−1 and 0.5 μg mL−1 respectively.162
 | ||
| Fig. 7 (a) Chemical structure of CAP-3. (b) Antibiofilm studies of CAP-3 against C. albicans-mediated pre-formed biofilms. (c) Antibiofilm studies of CAP-3 against S. aureus, C. albicans, and polymicrobial (S. aureus + C. albicans) biofilms. (d) SEM images showing that CAP-3 can eradicate the pre-formed polymicrobial biofilms. (e–g) Therapeutic efficacy of CAP-3 against a S. aureus wound infection model (e), C. albicans wound infection model (f), and polymicrobial (S. aureus + C. albicans) murine infection model (g). (h) General chemical structure of lithocholic acid-based imidazolium salts. (i) General chemical structure of 3-oxo-23,24-dinorchol-4-en-22-al. (j) Images showing the dose-dependent antifungal activity of compound 18 (Fig. 7b–g reproduced from ref. 161 with permission from the American Society for Microbiology, copyright 2019, and Fig. 7j reproduced from ref. 162 with permission from Elsevier, copyright 2019). | ||
Collectively, these reports suggest that steroid-based amphiphiles display promising therapeutic applications against different microbial infections by disrupting their cell membrane. Notably, findings also demonstrated that fungal strains were unable to gain resistance against these steroid-based amphiphiles. In addition, these amphiphiles also displayed potent antifungal activity against different murine infection models. However, their cytotoxicity and pharmacokinetic profiles still remain a quest to solve. Further fine-tuning of potent amphiphiles can be done to afford lower cytotoxicity with a better pharmacokinetic profile.
5.3 Aromatic and heterocyclic amphiphiles
Aromatic and heterocyclic cationic amphiphiles are easy to synthesize, and display a wide range of therapeutic applications.163 Notably, aromatic and heterocyclic cationic amphiphiles, including naturally obtained plant secondary metabolites, display potent antimicrobial activity. Lin et al. used a xanthone scaffold and installed aliphatic amines and basic amino acids through ether linkage,164 and found that molecules bearing formamidyl (compound 25) and n-butyl (compound 26) showed potent antifungal proprieties with a MIC80 of 0.78 and 3.13 μg mL−1 respectively (Fig. 8a). Compound 25 was found to be fungicidal in nature whereas compound 26 displayed fungistatic properties. Interestingly, only a 2-fold change in the MIC80 of compound 26 was observed in multi-passage resistance studies, whereas the MIC80 of compound 25 decreased with passage. Additionally, compound 25 demonstrated high membrane-permeabilizing properties compared to compound 26, and showed potent antifungal activity against drug-resistant fungal pathogens. Moreover, both the compounds exhibited additive and synergistic effects in combination with FDA approved antifungal agents against C. albicans. Topical application of compound 25 (0.2%) showed potent antifungal efficacy compared to natamycin with a 25-fold reduction in fungal burden.164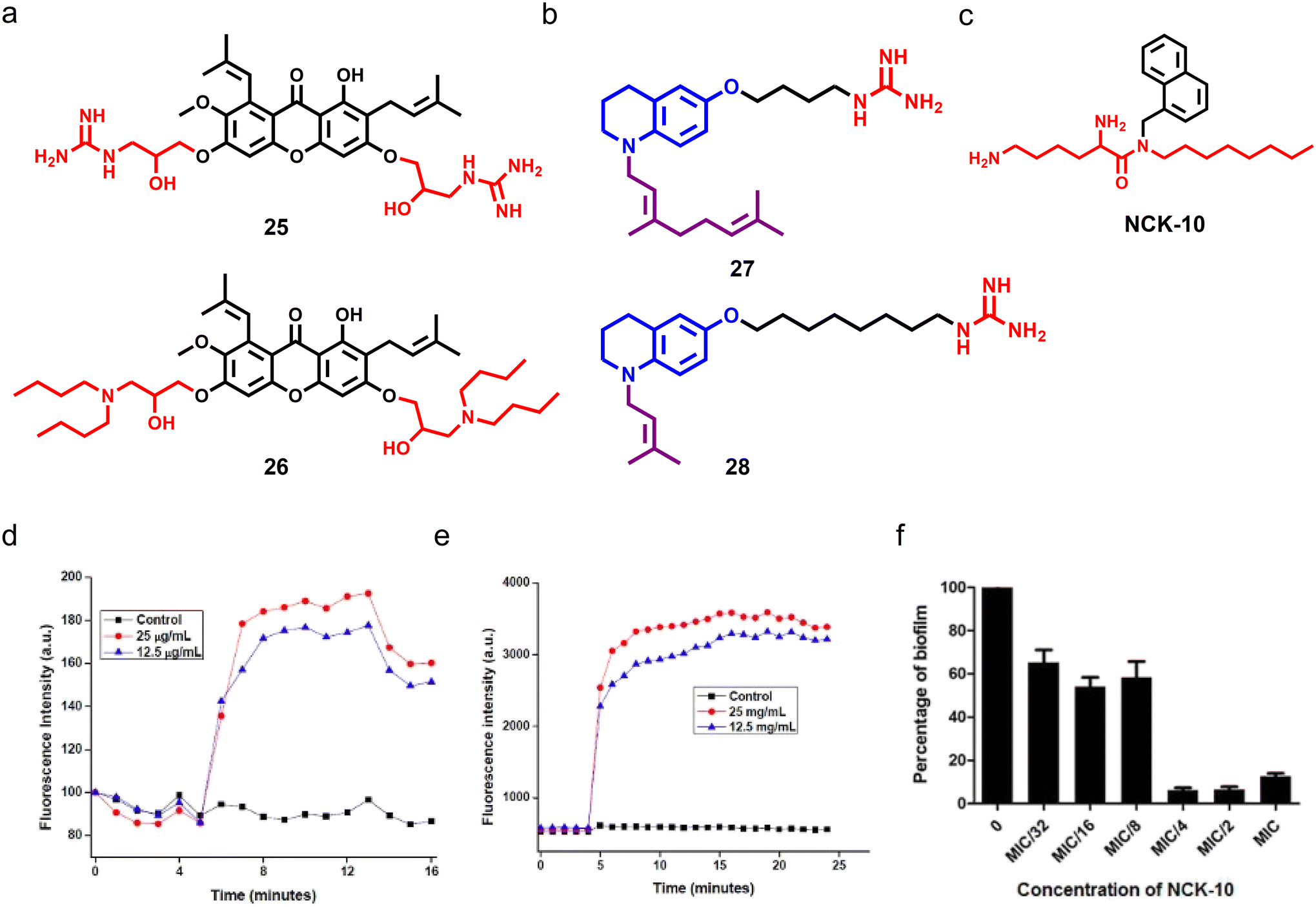 | ||
| Fig. 8 (a) Chemical structures of potent xanthone-based cationic compounds 25 and 26. (b) Molecular structures of most potent tetrahydroquinoline-based compounds 26 and 27. (c) Molecular structure of NCK-10. (d) Fluorescence-based assay showing that NCK-10 can depolarize the fungal cell membrane in a dose-dependent manner. (e) Fluorescence-based study suggesting that NCK-10 can permeabilize the fungal membrane. (f) In vitro study showing that NCK-10 can inhibit the formation of C. albicans biofilms. (Fig. 8d–f reproduced from ref. 166 with permission from the American Chemical Society, copyright 2017). | ||
In another report, a series of tetrahydroquinoline-based amphiphiles was synthesized and explored for antimicrobial properties, and both cationic and hydrophobic groups were tethered to the scaffold.165 Among the series, compounds having nonyl and isopropyl groups with a spacer length of 4 and 8 displayed potent broad-spectrum antimicrobial properties against fungi, Gram-positive and Gram-negative pathogens (compounds 27 and 28) (Fig. 8b). Both the amphiphiles showed dose-dependent fungicidal behaviour, and continued exposure to both compounds did not allow fungal cells to gain any resistance against them. Mechanistic studies demonstrated that both compounds have membrane-permeabilization activities.165 Ghosh et al. developed an aryl-based cationic amphiphile (NCK-10) by conjugating an aryl–alkyl–lysine with an alkyl chain of ten carbon atoms, which showed potent antifungal activity against C. albicans and C. neoformans with a MIC80 value of 12.5 μg mL−1 and 3.1 μg mL−1 respectively (Fig. 8c).166 Different biochemical and microscopy studies revealed the dose-dependent fungal membrane-permeabilization ability of NCK-10 (Fig. 8d and e). NCK-10 also exhibited dose-dependent antifungal properties against pre-formed C. albicans biofilms (Fig. 8f).166
As long-chain of ionic surfactants is important for amphiphilic nature and antimicrobial properties, Kashapov and colleagues developed single-chain diatonic surfactants bearing pyridinium as a core scaffold.167 Cationic surfactants armed with vinyl bipyridinium (VBP-16) and viologen (V-16) moieties as the head groups were tethered with a hexadecyl tail, and exhibited moderate antifungal and antibacterial properties (Fig. 9a).167 Kuznetsova et al. developed imidazolium-based cationic amphiphiles (called the MPI series) with self-assembly properties, where they used a methoxyphenyl fragment linked with an imidazole group, and different alkyl chains including decyl, dodecyl, tetradecyl, and hexadecyl were installed at the imidazole group (Fig. 9b).168 Transmission electron microscopy displayed spherical aggregates of MPI-10 (Fig. 9c). SAR studies showed that the antifungal properties were enhanced with an increase in alkyl chain length, and the amphiphile armed with a 16 carbon chain length group demonstrated potent antifungal properties with a MIC80 of 7.8 μg mL−1 and minimum fungicidal concentration of 125 μg mL−1.168 Stephen G. Pyne's group extensively worked on the design and synthesis of small peptidomimetic antimicrobial agents.169–173 Recently, they reported the design and synthesis of a series of biphenyl positional isomers (Fig. 9d), and among these isomers, the 4,4′-isomer (compound 36) showed potent antifungal activity against C. albicans with a MIC80 of 1 μg mL−1.174
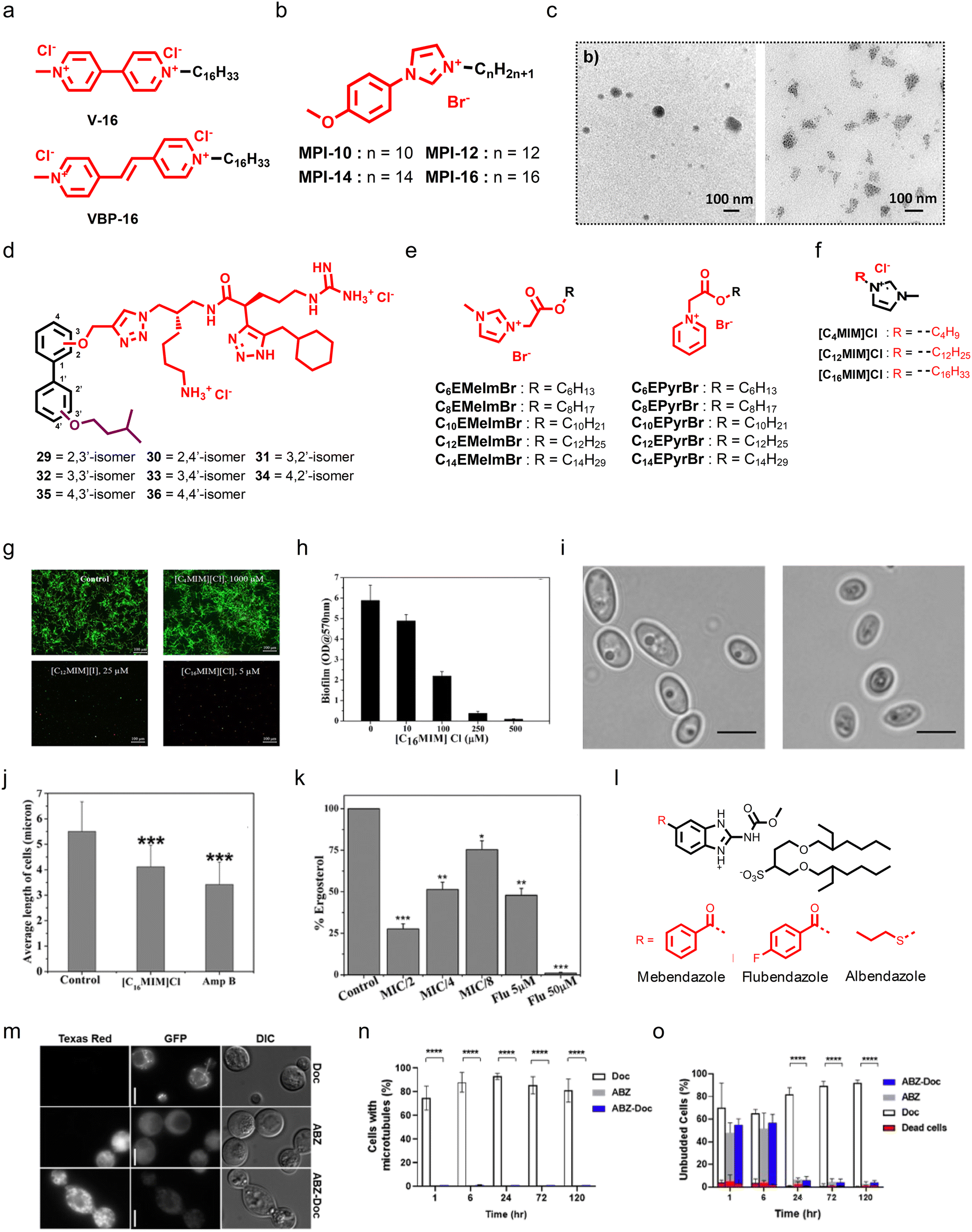 | ||
| Fig. 9 (a) Chemical structures of V-16 and VBP-16. (b) Chemistry of the MPI series. (c) TEM images displaying that MPI-10 can form aggregates. (d) Chemistry of biphenyl positional isomers. (e) Chemistry of imidazole and pyridine-based cationic esters. (f) Chemical structure of the potent imidazole-based cationic amphiphile [C16MIM]Cl. (g) Confocal microscopy images showing the antibiofilm properties of [C16MIM]Cl against pre-formed Candida biofilms. (h) Quantification of antibiofilm properties of [C16MIM]Cl. (i) Micrographs showing healthy and [C16MIM]Cl treated fungal cells, suggesting that [C16MIM]Cl can affect the morphology and length of fungal cells. (j) Quantitative analysis suggesting that [C16MIM]Cl can reduce the fungal cell length. (k) In vitro study showing that [C16MIM]Cl treatment can reduce the ergosterol level in a dose-dependent manner. (l) Chemistry of docusate-based ionic liquids from anthelmintic drugs. (m) Investigation of β-tubulin dynamics during treatment with ABZ, ABZ-Doc, and Doc against C. neoformans. (n) Quantification of visible microtubules at different time points after treatment with ABZ, ABZ-Doc, and Doc. (o) Quantification of unbudded cells at different time points after ABZ, ABZ-Doc, and Doc treatment (Fig. 9c reproduced from ref. 168 with permission from the American Chemical Society, copyright 2022, Fig. 9g–k reproduced from ref. 177 with permission from Frontiers Media S.A., copyright 2020, and Fig. 9m and n reproduced from ref. 178 with permission from the American Chemical Society, copyright 2021). | ||
Apart from AMP mimicking small molecules, ionic liquids also possess promising antifungal therapeutic potential. Inspired by previously reported quaternary ammonium-based surfactants (QACs), Garcia et al. designed and synthesized two series of cationic liquids by employing N-methylimidazole and pyridine as the core scaffold, and tethered different alkyl chains, including C6 to C14, through a cleavable ester linkage (Fig. 9e).175 In both series, the molecules having a pyridine core tethered with a C12 alkyl chain showed potent antifungal properties against C. albicans (Fig. 9e).175 Further, ionic liquids also displayed potent antifungal properties against Alternaria species, known to cause a seed-borne disease that affects plant production.
Karaman et al. tested a series of 18 imidazolium-based ionic liquids, and all the ionic liquids showed good antifungal properties that depend on the alkyl chain.176 As imidazolium is a key pharmacophoric feature for the development of antifungal ionic liquids and their exact mechanism is still an enigma, Reddy et al. investigated the antibiofilm activity of already reported alkylated imidazolium ionic liquids and performed in-depth mechanistic studies.177 They employed three different ionic liquids bearing C4, C12, and C16 alkyl chains, and demonstrated that an increment in alkyl chain length can also enhance the antifungal potency of ionic liquids. The ionic liquid having C16 displayed potent biofilm inhibition properties (Fig. 9f and g). Interestingly, both ionic liquids with C12 and C16 chains were found to be effective against clinical strains, and the ionic liquid bearing a C16 alkyl chain was found to be most potent against clinical strain-mediated biofilms (Fig. 9h). Microscopy images revealed that treatment with the C16 ionic liquid can cause shrinkage of C. albicans cells and membrane permeabilization, thereby causing the release of intracellular materials (Fig. 9i and j). In addition, treatment with the C16 ionic liquid decreased the ergosterol content in a dose-dependent manner (Fig. 9k). Moreover, treatment with the C16 ionic liquid also caused ROS generation and affected the mitochondrial membrane potential.177
Sutar and co-workers employed FDA-approved anthelmintic drugs, including albendazole (ABZ), mebendazole (MBZ), and flubendazole (FBZ), to develop docusate-based ionic liquids (Fig. 9l).178 The docusate-based ionic liquids were synthesized by treating these drugs with sodium docusate (Doc) in the presence of 1 M HCl and methanol. Antifungal studies showed that the developed ABZs were more potent than the parent drugs. Further, the treatment with the docusate-based ionic liquids inhibited C. neoformans growth by interfering with microtubule assembly (Fig. 9m–o). Notably, the docusate-based ionic liquids have greater organic solubility over the drugs, and incorporation of polymers provided a micelle-forming ability to the docusate-based ionic liquids.178
5.4 Antibiotic-derivatives as antifungal agents
Aminoglycosides like kanamycin, tobramycin (TOB), and gentamicin are naturally occurring antibiotics primarily utilized as bacteria-fighting agents. The inositol–amino sugar combination having hydroxyl and amino groups in the general structural motif of these antibiotics is essential for their interactions with RNA of the 30S subunit of ribosomes, and impedes protein translation. Long-term and excessive use of traditional aminoglycosides in medicine and agriculture has allowed resistant strains to develop, thereby making these antibiotics ineffective. Therefore, to combat this resistance, development of aminoglycosides should consider the likelihood of resistance and be flexible enough to adapt to the evolution of bacteria.179,180 Although aminoglycosides are effective antifungals at much higher concentrations, the S. Garneau-Tsodikova group designed and synthesized a series of cationic amphiphilic derivatives of TOB by conjugating different linear, branched, cyclized, and aromatic groups at the C6′ position (Fig. 10a).181 In further studies, they added a new amphiphile with a C14 alkyl chain in the series and tested it against both yeast and filamentous fungi strains.182 TOB alone showed minimal antifungal properties, while its derivatives showed moderate to high fungicidal properties (Fig. 10b and c). In contrast, the amphiphile having C14 showed potent antifungal efficacy through membrane disruption with a minimal cytotoxicity profile (Fig. 10d). Moreover, confocal microscopy studies demonstrated that treatment with the C14 TOB-derivative can induce late apoptosis or necrosis in fungal cells (Fig. 10e).182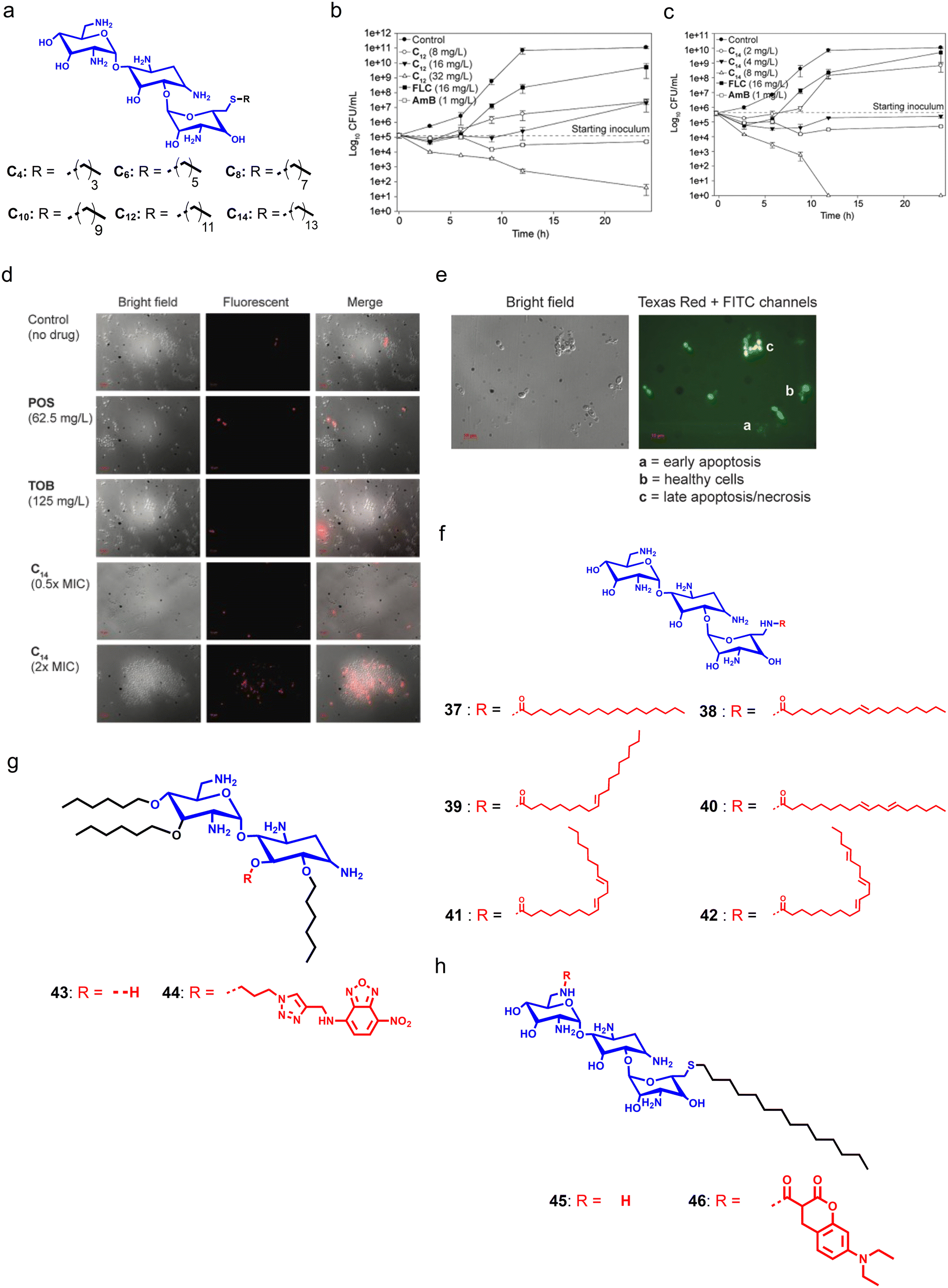 | ||
| Fig. 10 (a) Molecular structure of the C14-tethered derivative of tobramycin. (b and c) Time-dependent killing kinetics with different concentrations of tobramycin-derivatives. (d) Micrographs revealing the dose-dependent membrane-targeting properties of the C14-tethered tobramycin. (e) Confocal images showing that the C14-tethered derivative of tobramycin can induce late apoptosis or necrosis in fungal cells. (f) Chemical structure of tobramycin and its derivatives. (g) Chemical structures of neamine derivatives. (h) Chemical structures of fluorescent tobramycin derivatives (Fig. 10b–e reproduced from ref. 176 with permission from the American Society for Microbiology, copyright 2015). | ||
Steinbuch et al. designed and synthesized a series of TOB-derived cationic compounds with varying degrees of unsaturation in the lipid chain to target fungal infections. Initial antifungal screening of the synthesized compounds against different fungal strains suggested that tethering of lipid chains can enhance the antifungal activity by 32-fold as compared to TOB.183 Further, they selected two compounds having a fully saturated and unsaturated lipid group, respectively. SAR studies indicated that these compounds have poor antibacterial properties, and are specific against fungal cells. Compound 41, having the highest degree of unsaturation, showed the lowest toxicity against RBCs (Fig. 10f). Additionally, these amphiphiles can disrupt the fungal membrane and also displayed antifungal activities against intracellular fungal infection.183 To understand the antifungal mechanism of cationic aminoglycoside-derivatives, Jaber et al. designed and synthesized fluorophore–aminoglycoside conjugates (compounds 43–46) using neamine and TOB, and installed nitrobenzoxadiazole (NBD) on neamine, and 7-diethylaminocoumarin on tobramycin (Fig. 10g and h).184 The developed amphiphiles showed similar antifungal properties and showed the disappearance of mCherry fluorescence in Eno1–mCherry expressing C. albicans which demonstrated plasma membrane destruction. In addition, treatment with aminoglycoside-based cationic amphiphiles also disrupted the nuclear envelope and penetrated the nucleus where these amphiphiles strike the DNA structure.184
Logviniuk et al. synthesized and studied a series of 16 cationic amphiphiles, where they conjugated different alkyl chains with varying degrees of unsaturation at hydroxy groups of nebramine (Fig. 11a–d).185 Amphiphiles tethered with a di-O-n-hexyl alkyl chain showed moderate to poor antifungal properties. In contrast, amphiphiles bearing di-O-n-nonyl groups with varying degrees of unsaturation demonstrated better antifungal properties as they have a comparatively higher hydrophobicity than the di-O-n-hexyl group substituted molecules. SAR studies showed that the arrangement of unsaturation modestly impacted the antifungal properties of di-O-n-nonyl substituted amphiphiles, and the presence of three saturated aliphatic chains reduced the antifungal properties due to increased hydrophilicity (Fig. 11a and b). Therefore, an optimum balance of hydrophilicity and hydrophobicity should be obtained to achieve antifungal properties. To decipher membrane accumulation, they also synthesized fluorophore conjugates, compounds 47 and 48, by installing NBD through click chemistry (Fig. 11c and d).185 Apart from TOB, other aminoglycoside-derivatives like kanamycin also displayed potent antifungal properties.186 Recently, Alfindee et al. tested earlier reported C4′′,C6′′-dialkyl and diaryl kanamycin derivatives against different fungal pathogens and showed the disubstituted kanamycin-derivatives as potent membrane-permeabilizers (Fig. 11e and f).187
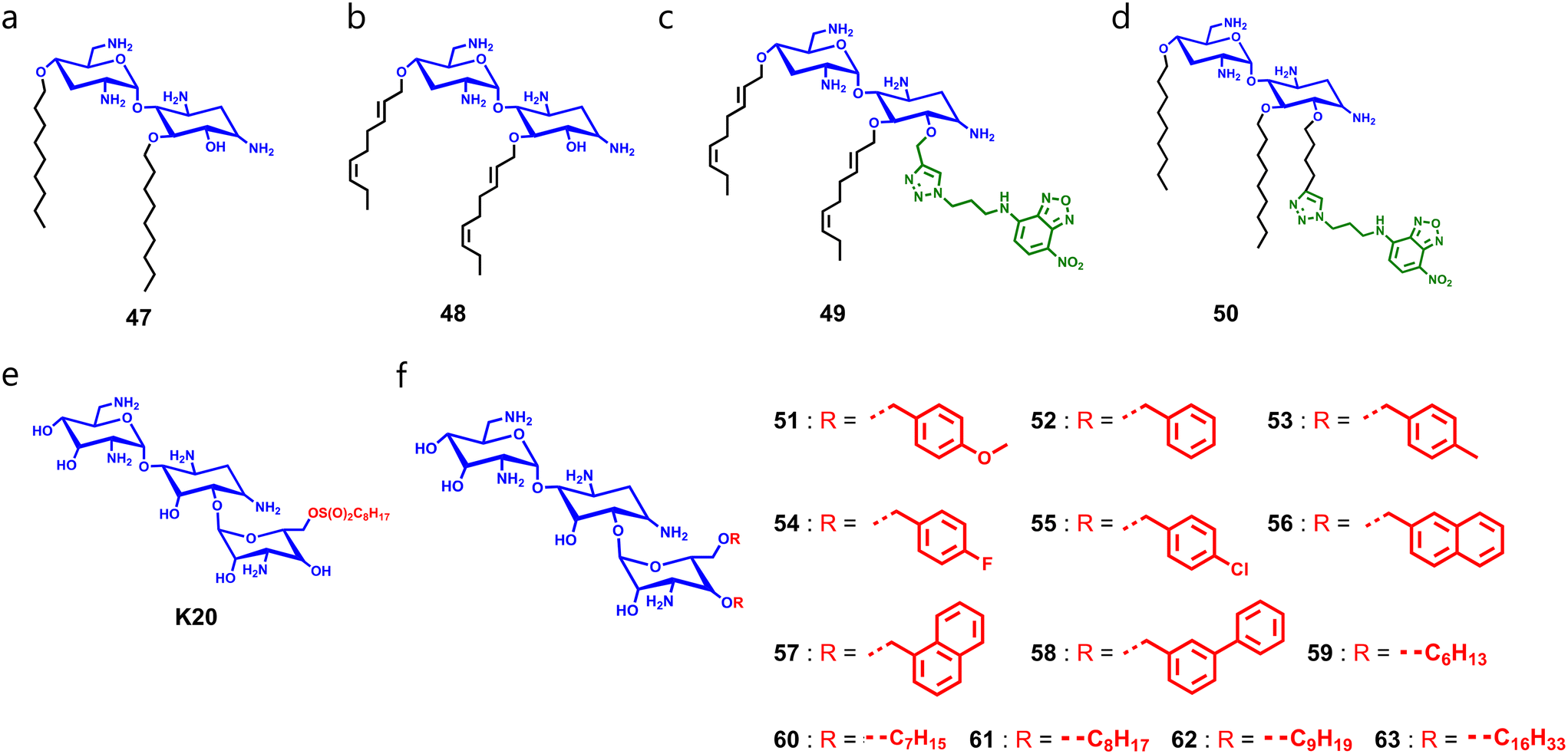 | ||
| Fig. 11 (a–d) Chemical structures of nebramine-derivatives 47 (a), 48 (b), 49 (c), and 50 (d). (e) Chemical structure of kanamycin-derivative K20. (f) General chemical structure of diaryl and dialkyl substituted kanamycin-derivatives. | ||
Collectively, these findings suggest that incorporation of amphiphilicity in available aminoglycosides can enhance their antifungal potency. In addition, developed amphiphilic aminoglycosides can disrupt the fungal membranes and eradicate pre-formed fungal biofilms. Synthesized fluorophore-labeled molecules validated the membrane-targeting abilities of amphiphilic aminoglycosides. However, development of minimally toxic antifungal amphiphilic aminoglycosides is still a mystery which may be resolved by further fine tuning of these amphiphiles.
5.5 Polymer-based amphiphiles
Antifungal polymers are important in both medical and agricultural industries, where fungal infections can cause significant damage. In the medical sector, antifungal polymers are used in drug delivery systems and wound dressings, while in the agricultural industry, they are used as antifungal coatings for fruits and vegetables. In addition, cationic polymeric materials can also be used in paint and film coatings to restrict microbial growth. Therefore, the development of antifungal polymers has been driven by the need to find effective and safe alternatives to traditional antifungal agents. One of the most common mechanisms of antifungal polymers involves the interactions of the polymer with the fungal cell membrane resulting in membrane disruptions, causing leakage of intracellular contents and ultimately leading to fungal cell death. Another mechanism involves interactions of the polymer with the fungal cell wall which can lead to cell wall damage and subsequent cell death. The exact mechanism of action of antifungal polymers can vary depending on the specific polymer used and type of fungus being targeted.188 Understanding the mechanisms of action of antifungal polymers is crucial for their effective application in both medical and agricultural settings. Several reviews summarized the applications of polymers to combat infectious diseases.189,190Guanidine is an important pharmacophoric feature that displays potent antimicrobial properties, and Baugh summarized recently reported guanidine-containing antifungal molecules.191 Oule and co-workers reported polyhexamethylene guanidine (PHMG) hydrochloride as a potent antimicrobial macromolecule that showed broad-spectrum antibacterial properties against drug-resistant pathogens with minimal toxicity against mammalian cells.192 In addition, its physiochemical properties, like water solubility, and non-corrosive and odorless nature, make it a versatile antibacterial disinfectant.192 Choi et al. demonstrated that treatment with the hydrochloride salt of PHMG can hamper the morphology of C. albicans cells, reduce the phospholipid area and disrupt the plasma membrane by altering the membrane potential without exhibiting any toxicity against human RBCs.193 Brzezinska and co-workers developed derivatives of PHMG using polylactide (PLA), polyhydroxybutyrate (PHB), and polycaprolactone (PCL) which were found to be more fungicidal in nature as compared to native PHMG.194 Further studies demonstrated that these derivatives can hamper the morphology of fungal cells, eradicate biofilm formation, and inhibit hydrolase activity in C. albicans.194 PHMG was also used to manage plant fungal diseases, where it showed potent fungicidal properties against P. digitatum by disrupting the cell wall and membrane.195 Recently, Ntow-Boahene et al. investigated the antifungal properties of the polyhexamethylene biguanide (PHMB) macromolecule against different fungal pathogens such as S. cerevisiae, C. albicans, F. oxysporum and P. glabrum. PHMB showed potent antifungal activity against fungal pathogens with a MIC90 value of 2–8 μg mL−1. Further, biochemical studies revealed that PHMB can permeabilize fungal cell membranes in a time-dependent and dose-dependent manner. After permeabilizing fungal membranes, PHMB accumulates within the cytosol and hampers the nuclear membrane leading to DNA binding and fragmentation. Collectively, these findings suggest that PHMB also targets intracellular organelles.196 Despite its antifungal properties, the aerosol form of PHMG can cause lung fibrosis by inducing epithelial–mesenchymal transition (EMT).197
Polyethyleneimines (PEIs) are polymeric compounds that bear an amine group and a spacer of two carbon atoms. As they have the ability to permeabilize the cell membrane, they are widely used in drug delivery and gene therapy applications.198 In addition, PEIs display antifungal properties through depolarizing C. albicans membranes, and also exhibit antibiofilm activity in a dose-dependent manner.199 Low molecular weight PEIs display minimal toxicity, and higher molecular weight PEIs are more toxic against mammalian cells.200 Haldar and group designed and developed PEI-based antimicrobial coating materials where they prepared two series of linear and branched colorless organo-soluble PEI (of different molecular weights) coating materials through Eschweiler–Clarke methylation, and then quaternized them with different alkyl bromides.201 Among the linear derivatives, the polymer bearing C18H37 alkyl chains with lower molecular weight showed potent antimicrobial activity, and in the case of branched derivatives, polymers armed with C12H25 alkyl chains with higher molecular weight were found to be more active against bacterial and fungal pathogens. In-depth mechanistic studies revealed that the active polymers can disrupt the bacterial and fungal cell membranes without affecting hRBCs, and fungal pathogens were unable to gain resistance against the active polymers for up to 20 passages.201
Real et al. prepared an antifungal film coating by loading econazole nitrate in a polymeric matrix composed of chitosan, Carbopol, polyethylene glycol 400, and sorbitol which displayed better antifungal properties against C. krusei and C. parapsilosis as compared to commercial creams.202 Nagaraja and co-workers developed a hydrophilic-antimicrobial polymer coating material functionalized with polymaliamides, and it was found to be more active against Gram-positive bacteria and M. smegmatis. Moreover, it displayed excellent film-forming properties and thus it can be used in the paint and food packaging industries.203 Schaefer et al. designed and developed a library of antifungal polyacrylamide polymers using the PET-RAFT polymerization technique. Among these polyacrylamide polymers, 40-LP-2030, a linear polymer with a pentyl alkyl chain, was found to be a potent antifungal agent, and non-toxic against mammalian RBCs and fibroblasts.204 Recently, Yeung et al. developed two polyethylene-derived water-soluble amphiphilic polymers that showed potent antifungal activities through depolarizing the fungal membrane potential.205
Collectively, these reports suggest that polymer-based amphiphiles can effectively target the fungal membrane, and can be used to deliver already available antifungal agents to a targeted site. Notably, antifungal polymeric materials can be used in paint and film coatings to prevent invasive fungal diseases.
6. Conclusion
Fungal infections in hospitals and clinical settings, exacerbated by drug resistance, are more challenging at the global level, and cause serious public health concern. Fungi cause diverse disease forms ranging from superficial allergic conditions to life threatening invasive diseases affecting millions of individuals annually, and ineffective detection tools hinder the timely discernment of infection. Further, overuse and misuse of antifungal drugs in clinical settings contributes to the development of AMR where fungal cells employ intrinsic and extrinsic factors to gain resistance against azoles, polyenes, echinocandins, antimetabolites, and allylamines. As the emergence of drug-resistant fungal pathogens like C. auris is a serious threat to mankind, the condition of antimicrobial resistance is dire, and it is imperative to take some potential steps to overcome this crisis. Therefore, knowledge at the ground level and policies focusing to improve appropriate diagnostic testing in humans would play a significant role.Continuous pre-clinical evaluation and clinical studies with available approaches can successfully overcome the incidences of antifungal resistance. The discoveries uncovered in screening campaigns and detailed sequencing of resistant species could lay a stepping stone to starting research on new antifungal agents having improved efficacy. Although development of new antibiotics capable to target the specific mechanism is a time consuming exercise, adoption of existing antibiotics using stewardship therapy and usage of small membrane targeting agents could complement to fill in the void of antifungal pipeline in future. Recently, a few molecules like VT-II29, VT-II6I and VT-I598 (specific inhibitors of Cyp5I), CDI0I (inhibitor of glucan synthase), F90I3I8 (inhibitor of fungal pyrimidine biosynthesis) and T-2307 (fungal mitochondrial membrane inhibitor) have come up as agents to tackle the resistance of fungal species, and could have potential to act as future antifungal agents. Moreover, repurposing of old drugs and host immune cell targeted approaches can potentially eradicate existing resistant fungal species. Personalized immune therapy, translation of sequencing techniques, awareness of resistant dermatophytosis, ability to access antifungal susceptibility testing, and antifungal vaccines can have a scope in the future antifungal field.
The fungal cell membrane presents unique drug targets as its biochemical composition is different from those of other microbes, and the presence of PC, PE, sphingolipids, and lipid rafts make it anionic in nature. Therefore, AMPs which are well-defined biomolecules of the host innate immune system can disrupt the microbial membrane through strong electrostatic interactions. However, the stability and toxicity profile of AMPs restrict their employment in clinical settings. Inspired from the unique biochemical composition of fungal cell membranes and the functions of AMPs, researchers are keen to design and develop membrane-targeting amphiphilic molecules. Moreover, these amphiphiles can act as potential adjuvants that rejuvenate the antifungal properties of available drugs.
However, their toxicity against hosts creates a huge hurdle in clinical translation as most of the antifungal amphiphiles were found to be toxic against mammalian cells. Therefore, amphiphiles with a high therapeutic index can be advanced to preclinical studies. However, some cationic amphiphiles display lower toxicity against mammalian cells but they have a narrow spectrum of activity, and the poor pharmacokinetic profile of amphiphiles hampers their usage in clinical settings. Membrane-targeting agents like daptomycin are specific against Gram-positive pathogens, and is not orally bioavailable to cure lung infections. Collectively, these factors affect the clinical translation of membrane-targeting amphiphiles, and also provide an opportunity to rationally design and develop membrane targeting cationic amphiphiles. In summary, the unique biochemical composition of fungal membranes can be employed as a therapeutic target to design and develop antifungal regimens against fungal pathogens.
Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
The support from RCB and the Department of Biotechnology (DBT), India is greatly acknowledged. AB thanks the DBT (BT/PR27264/MED/29/1277/2018) for funding. DM thanks ICMR (Myco/Fell/11/2022ECD-II) for a senior research fellowship, and VS thanks the DBT (DBT/JRF/BET-19/I/2019/AL/16) for a research fellowship. We acknowledge the support from the DBT e-Library Consortium (DeLCON) for providing access to e-resources. Fig. 2 and the TOC figure were created using https://BioRender.com.References
- A. Rokas, Nat. Microbiol., 2022, 7, 607–619 CrossRef CAS PubMed.
- L. Angiolella, Microorganisms, 2022, 10, 409 CrossRef PubMed.
- S. A. Howell, Br. J. Biomed. Sci., 2023, 80, 11314 CrossRef CAS PubMed.
- M. S. Lionakis, R. A. Drummond and T. M. Hohl, Nat. Rev. Immunol., 2023, 23, 433–452 CrossRef CAS PubMed.
- T. B. Burgess, A. M. Condliffe and P. M. Elks, J. Fungi, 2022, 8, 805 CrossRef CAS PubMed.
- M. G. Netea, L. A. Joosten, J. W. Van Der Meer, B. J. Kullberg and F. L. Van De Veerdonk, Nat. Rev. Immunol., 2015, 15, 630–642 CrossRef CAS PubMed.
- M. R. Dunne, J. Wagener, J. Loeffler, D. G. Doherty and T. R. Rogers, Clin. Immunol., 2021, 227, 108734 CrossRef CAS PubMed.
- M. M. S. Riley, Crit. Care Nurs. Clin. North Am., 2021, 33, 395–405 CrossRef PubMed.
- F. Bongomin, S. Gago, R. O. Oladele and D. W. Denning, J. Fungi, 2017, 3, 57 CrossRef PubMed.
- E. J. Polvi, X. Li, T. R. O'Meara, M. D. Leach and L. E. Cowen, Cell. Mol. Life Sci., 2015, 72, 2261–2287 CrossRef CAS PubMed.
- G. Vanreppelen, J. Wuyts, P. Van Dijck and P. Vandecruys, J. Fungi, 2023, 9, 171 CrossRef CAS PubMed.
- F. B. Cavassin, J. L. Baú-Carneiro, R. R. Vilas-Boas and F. Queiroz-Telles, Infect. Dis. Ther., 2021, 10, 115–147 CrossRef PubMed.
- F. A. Malayeri, A. A. Rezaei and O. Raiesi, J. Basic Res. Med. Sci., 2018, 5, 48–55 CrossRef.
- K. Forsberg, K. Woodworth, M. Walters, E. L. Berkow, B. Jackson, T. Chiller and S. Vallabhaneni, Med. Mycol., 2019, 57, 1–12 CrossRef PubMed.
- P. Dadgostar, Infect. Drug Resist., 2019, 12, 3903–3910 CrossRef CAS PubMed.
- L. Brown, C. Langelier, M. J. Reid, R. L. Rutishauser and L. Strnad, Clin. Infect. Dis., 2017, 64, 106–107 CrossRef PubMed.
- P. M. Dohmen, J. Hosp. Infect., 2008, 70, 15–20 CrossRef PubMed.
- N. Q. Balaban, K. Gerdes, K. Lewis and J. D. McKinney, Nat. Rev. Microbiol., 2013, 11, 587–591 CrossRef CAS PubMed.
- S. Bhattacharya, S. Sae-Tia and B. C. Fries, Antibiotics, 2020, 9, 312 CrossRef CAS PubMed.
- K. M. Chandrika and S. Sharma, Bioorg. Med. Chem., 2020, 28, 115398 CrossRef PubMed.
- G. Wall and J. L. Lopez-Ribot, Antibiotics, 2020, 9, 445 CrossRef CAS PubMed.
- C. Vallières, N. Singh, C. Alexander and S. V. Avery, ACS Infect. Dis., 2020, 6, 2950–2958 CrossRef PubMed.
- Q. Zhang, F. Liu, M. Zeng, Y. Mao and Z. Song, Appl. Microbiol. Biotechnol., 2021, 105, 5259–5279 CrossRef CAS PubMed.
- A. Izadi, S. A. Gharehbolagh, F. Sadeghi, M. Talebi, K. Darmiani, A. Zarrinnia, F. Zarei, F. Peymaeei, S. Khojasteh, A. M. Borman and S. Mahmoudi, Mycoses, 2022, 65, 784–793 CrossRef CAS PubMed.
- S. Wu, Y. Wang, N. Liu, G. Dong and C. Sheng, J. Med. Chem., 2017, 60, 2193–2211 CrossRef CAS PubMed.
- R. Mishra, A. K. Panda, S. De Mandal, M. Shakeel, S. S. Bisht and J. Khan, Front. Microbiol., 2020, 11, 566325 CrossRef PubMed.
- J. R. Perfect, Nat. Rev. Drug Discovery, 2017, 16, 603–616 CrossRef CAS PubMed.
- D. G. Sant, S. G. Tupe, C. V. Ramana and M. V. Deshpande, J. Appl. Microbiol., 2016, 121, 1498–1510 CrossRef CAS PubMed.
- J. Lei, L. Sun, S. Huang, C. Zhu, P. Li, J. He, V. Mackey, D. H. Coy and Q. He, Am. J. Transl. Res., 2019, 11, 3919–3931 CAS.
- V. Basso, D. Q. Tran, A. J. Ouellette and M. E. Selsted, J. Fungi, 2020, 6, 241 CrossRef CAS PubMed.
- H. Washton, Diagn. Microbiol. Infect. Dis., 1989, 12, 229–233 CrossRef PubMed.
- H. L. Hoffman, E. J. Ernst and M. E. Klepser, Expert Opin. Invest. Drugs, 2000, 9, 593–605 CrossRef CAS PubMed.
- L. R. Peyton, S. Gallagher and M. Hashemzadeh, Drugs Today, 2015, 51, 705–718 CrossRef CAS PubMed.
- M. Alison, L. Cass, K. Ito, N. Pagani, D. A. James, P. Dalal, A. Reed and P. Strong, J. Fungi, 2020, 6, 373 CrossRef PubMed.
- A. K. Kabi, S. Sravani, R. Gujjarappa, A. Garg, N. Vodnala, U. Tyagi, D. Kaldhi, R. Velayutham, V. Singh, S. Gupta and C. C. Malakar, Nanostruct. Biomater., 2022, 307–349 Search PubMed.
- M. G. Martens, B. Maximos, T. Degenhardt, K. Person, S. Curelop, M. Ghannoum, A. Flynt and S. R. Brand, Am. J. Obstet. Gynecol., 2022, 227, 880 CrossRef PubMed.
- G. Bouz and M. Doležal, Pharmaceuticals, 2021, 14, 1312 CrossRef CAS PubMed.
- M. M. Teixeira, D. T. Carvalho, E. Sousa and E. Pinto, Pharmaceuticals, 2022, 15, 1427 CrossRef CAS PubMed.
- H. Carolus, S. Pierson, K. Lagrou and P. Van Dijck, J. Fungi, 2020, 6, 321 CrossRef CAS PubMed.
- S. B. Zotchev, Curr. Med. Chem., 2003, 10, 211–223 CrossRef CAS PubMed.
- D. R. Worthen, M. Jay and P. M. Bummer, Drug Dev. Ind. Pharm., 2001, 27, 277–286 CrossRef CAS PubMed.
- P. Chandrasekar, J. Antimicrob. Chemother., 2011, 66, 457–465 CrossRef CAS PubMed.
- S. Qiu, G. Q. Zhao, J. Lin, X. Wang, L. T. Hu, Z. D. Du, Q. Wang and C. C. Zhu, Guoji Yanke Zazhi, 2015, 8, 597–602 Search PubMed.
- K. C. Gray, D. S. Palacios, I. Dailey, M. M. Endo, B. E. Uno, B. C. Wilcock and M. D. Burke, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2234–2239 CrossRef CAS PubMed.
- D. M. Kamiński, Eur. Biophys. J., 2014, 43, 453–467 CrossRef PubMed.
- M. Baginski, H. Resat and E. Borowski, Biochim. Biophys. Acta, Biomembr., 2002, 1567, 63–78 CrossRef CAS PubMed.
- R. J. Hamill, Drugs, 2013, 73, 919–934 CrossRef CAS PubMed.
- M. Szymański, S. Chmielewska, U. Czyżewska, M. Malinowska and A. Tylicki, J. Enzyme Inhib. Med. Chem., 2022, 37, 876–894 CrossRef PubMed.
- N. D. Grover, Indian J. Pharmacol., 2010, 42, 9–11 CrossRef CAS PubMed.
- W. Hüttel, Appl. Microbiol. Biotechnol., 2021, 105, 55–66 CrossRef PubMed.
- J. Yao, H. Liu, T. Zhou, H. Chen, Z. Miao, C. Sheng and W. Zhang, Eur. J. Med. Chem., 2012, 50, 196–208 CrossRef CAS PubMed.
- K. D. James, C. P. Laudeman, N. B. Malkar, R. Krishnan and K. Polowy, Antimicrob. Agents Chemother., 2017, 61, e01541-16 CrossRef PubMed.
- D. W. Denning, Lancet, 2003, 362, 1142–1151 CrossRef CAS PubMed.
- A. J. Sucher, E. B. Chahine and H. E. Balcer, Ann. Pharmacother., 2009, 43, 1647–1657 CrossRef CAS PubMed.
- B. R. Krishnan, K. D. James, K. Polowy, B. J. Bryant, A. Vaidya, S. Smith and C. P. Laudeman, J. Antibiot., 2017, 70, 130–135 CrossRef CAS PubMed.
- V. Ong, K. D. James, S. Smith and B. R. Krishnan, Antimicrob. Agents Chemother., 2017, 61, e01626-16 CrossRef PubMed.
- D. H. Halat, S. Younes, N. Mourad and M. Rahal, Membranes, 2022, 12, 1171 CrossRef PubMed.
- M. Nowosielski, M. Hoffmann, L. S. Wyrwicz, P. Stepniak, D. M. Plewczynski, M. Lazniewski, K. Ginalski and L. Rychlewski, J. Chem. Inf. Model., 2011, 51, 455–462 CrossRef CAS PubMed.
- W. D. Ashe Jr and D. E. Van Reken, Clin. Pediatr., 1977, 16, 364–366 CrossRef CAS PubMed.
- A. Vermes, H. J. Guchelaar and J. Dankert, J. Antimicrob. Chemother., 2000, 46, 171–179 CrossRef CAS PubMed.
- M. A. Ghannoum and L. B. Rice, Clin. Microbiol. Rev., 1999, 12, 501–517 CrossRef CAS PubMed.
- J. L. Song, J. B. Harry, R. T. Eastman, B. G. Oliver and T. C. White, Antimicrob. Agents Chemother., 2004, 48, 1136–1144 CrossRef CAS PubMed.
- P. Marichal, L. Koymans, S. Willemsens, D. Bellens, P. Verhasselt, W. Luyten, M. Borgers, F. C. Ramaekers, F. C. Odds and H. Vanden Bossche, Microbiology, 1999, 145, 2701–2713 CrossRef CAS PubMed.
- S. G. Whaley, E. L. Berkow, J. M. Rybak, A. T. Nishimoto, K. S. Barker and P. D. Rogers, Front. Microbiol., 2017, 7, 2173 CrossRef PubMed.
- B. M. Vincent, A. K. Lancaster, R. Scherz-Shouval, L. Whitesell and S. Lindquist, PLoS Biol., 2013, 11, 1001692 CrossRef PubMed.
- S. Patra, M. Raney, A. Pareek and R. Kaur, J. Fungi, 2022, 8, 875 CrossRef CAS PubMed.
- D. S. Perlin, R. Rautemaa-Richardson and A. Alastruey-Izquierdo, Lancet Infect. Dis., 2017, 17, e383–e392 CrossRef PubMed.
- M. G. Frías-De-León, R. Hernández-Castro, T. Vite-Garín, R. Arenas, A. Bonifaz, L. Castañón-Olivares, G. Acosta-Altamirano and E. Martínez-Herrera, Antibiotics, 2020, 9, 568 CrossRef PubMed.
- P. Escandón, N. A. Chow, D. H. Caceres, L. Gade, E. L. Berkow, P. Armstrong, S. Rivera, E. Misas, C. Duarte, H. Moulton-Meissner and R. M. Welsh, Clin. Infect. Dis., 2019, 68, 15–21 Search PubMed.
- D. S. Perlin, Drug Resistance Updates, 2007, 10, 121–130 CrossRef CAS PubMed.
- D. S. Perlin, Ann. N. Y. Acad. Sci., 2015, 1354, 1–11 CrossRef CAS PubMed.
- K. R. Healey and D. S. Perlin, J. Fungi, 2018, 4, 105 CrossRef CAS PubMed.
- M. D. Lenardon, C. A. Munro and N. A. Gow, Curr. Opin. Microbiol., 2010, 13, 416–423 CrossRef CAS PubMed.
- S. Fanning and A. P. Mitchell, PLoS Pathog., 2012, 8, e1002585 CrossRef CAS PubMed.
- M. Gulati and C. J. Nobile, Microbes Infect., 2016, 18, 310–321 CrossRef CAS PubMed.
- M. Cavalheiro and M. C. Teixeira, Front. Med., 2018, 5, 28 CrossRef PubMed.
- D. M. Kuhn, T. George, J. Chandra, P. K. Mukherjee and M. A. Ghannoum, Antimicrob. Agents Chemother., 2002, 46, 1773–1780 CrossRef CAS PubMed.
- R. Laniado-Laborín and M. N. Cabrales-Vargas, Rev. Iberoam. Micol., 2009, 26, 223–227 CrossRef PubMed.
- C. Faustino and L. Pinheiro, Pharmaceutics, 2020, 12, 29 CrossRef CAS PubMed.
- J. Tits, B. P. Cammue and K. Thevissen, Int. J. Mol. Sci., 2020, 21, 8873 CrossRef CAS PubMed.
- K. De Cremer, E. Lanckacker, T. L. Cools, M. Bax, K. De Brucker, P. Cos, B. P. Cammue and K. Thevissen, Antimicrob. Agents Chemother., 2015, 59, 421–426 CrossRef PubMed.
- J. Ramakrishnan, S. S. Rathore and T. Raman, RSC Adv., 2016, 6, 42387–42401 RSC.
- M. Billamboz, Z. Fatima, S. Hameed and S. Jawhara, Microorganisms, 2021, 9, 634 CrossRef CAS PubMed.
- S. Hasim and J. J. Coleman, Future Med. Chem., 2019, 11, 869–883 CrossRef CAS PubMed.
- R. Tada, J. P. Latge and V. Aimanianda, Curr. Pharm. Des., 2013, 19, 3738–3747 CrossRef CAS PubMed.
- A. I. de Kroon, P. J. Rijken and C. H. De Smet, Prog. Lipid Res., 2013, 52, 374–394 CrossRef CAS PubMed.
- L. Klug and G. Daum, FEMS Yeast Res., 2014, 14, 369–388 CrossRef CAS PubMed.
- M. E. Van der Rest, A. H. Kamminga, A. Nakano, Y. Anraku, B. Poolman and W. N. Konings, Microbiol. Rev., 1995, 59, 304–322 CrossRef CAS PubMed.
- F. C. Santos, J. T. Marquês, A. Bento-Oliveira and R. F. de Almeida, FEBS Lett., 2020, 594, 3698–3718 CrossRef CAS PubMed.
- C. R. Gault, L. M. Obeid and Y. A. Hannun, Adv. Exp. Med. Biol., 2010, 688, 1–23 CrossRef CAS PubMed.
- S. Uemura, F. Shishido, M. Tani, T. Mochizuki, F. Abe and J. I. Inokuchi, J. Lipid Res., 2014, 55, 1343–1356 CrossRef CAS PubMed.
- M. L. Rodrigues, MBio, 2018, 9, e01755-18 CrossRef PubMed.
- S. Dhingra and R. A. Cramer, Front. Microbiol., 2017, 8, 92 Search PubMed.
- Z. Hu, B. He, L. Ma, Y. Sun, Y. Niu and B. Zeng, Indian J. Microbiol., 2017, 57, 270–277 CrossRef CAS PubMed.
- L. Alcazar-Fuoli and E. Mellado, Front. Microbiol., 2013, 3, 439 Search PubMed.
- K. Kristan and T. L. Rižner, J. Steroid Biochem. Mol. Biol., 2012, 129, 79–91 CrossRef CAS PubMed.
- M. K. Kathiravan, A. B. Salake, A. S. Chothe, P. B. Dudhe, R. P. Watode, M. S. Mukta and S. Gadhwe, Bioorg. Med. Chem., 2012, 20, 5678–5698 CrossRef CAS PubMed.
- V. Wachtler and M. K. Balasubramanian, Trends Cell Biol., 2006, 16, 1–4 CrossRef CAS PubMed.
- S. W. Martin and J. B. Konopka, Eukaryotic Cell, 2004, 3, 675–684 CrossRef CAS PubMed.
- F. Mollinedo, Front. Oncol., 2012, 2, 140 Search PubMed.
- Y. Miyake, T. Tsunoda, S. Minagi, Y. Akagawa, H. Tsuru and H. Suginaka, FEMS Microbiol. Lett., 1990, 69, 211–214 CrossRef CAS.
- N. Shibata, A. Suzuki, H. Kobayashi and Y. Okawa, Biochemistry, 2007, 404, 365–372 CrossRef CAS PubMed.
- E. T. Sawistowska-Schröder, D. Kerridge and H. Perry, Echinocandin inhibition of 1, 3-β-D-glucan synthase from Candida albicans, FEBS Lett., 1984, 173, 134–138 CrossRef PubMed.
- F. M. Klis, P. D. Groot and K. Hellingwerf, Med. Mycol., 2001, 39, 1–8 CrossRef CAS PubMed.
- S. J. Free, Adv. Genet., 2013, 81, 33–82 CAS.
- I. Rubin-Bejerano, C. Abeijon, P. Magnelli, P. Grisafi and G. R. Fink, Cell Host Microbe, 2007, 2, 55–67 CrossRef CAS PubMed.
- D. Mehta, V. Saini, B. Aggarwal, A. Khan and A. Bajaj, Mol. Aspects Med., 2021, 81, 100999 CrossRef CAS PubMed.
- N. Mookherjee, M. A. Anderson, H. P. Haagsman and D. J. Davidson, Nat. Rev. Drug Discovery, 2020, 19, 311–332 CrossRef CAS PubMed.
- Y. Huan, Q. Kong, H. Mou and H. Yi, Front. Microbiol., 2020, 11, 12559 Search PubMed.
- A. H. Benfield and S. T. Henriques, Front. Med. Technol., 2020, 2, 610997 CrossRef PubMed.
- M. Acosta-Zaldívar, M. T. Andrés, A. Rego, C. S. Pereira, J. F. Fierro and M. Côrte-Real, Apoptosis, 2016, 21, 163–173 CrossRef PubMed.
- N. Hegedüs and F. Marx, Fungal Biol. Rev., 2013, 26, 132–145 CrossRef PubMed.
- C. Struyfs, B. P. Cammue and K. Thevissen, Front. Cell Dev. Biol., 2021, 9, 649875 CrossRef PubMed.
- C. Zhang and M. Yang, Antibiotics, 2022, 11, 349 CrossRef CAS PubMed.
- C. H. Chen and T. K. Lu, Antibiotics, 2020, 9, 24 CrossRef CAS PubMed.
- S. L. Regen, JACS Au, 2021, 1, 3–7 CrossRef CAS PubMed.
- S. J. Lam, N. M. O'Brien-Simpson, N. Pantarat, A. Sulistio, E. H. Wong, Y. Y. Chen, J. C. Lenzo, J. A. Holden, A. Blencowe, E. C. Reynolds and G. G. Qiao, Nat. Microbiol., 2016, 1, 1–11 Search PubMed.
- F. Nederberg, Y. Zhang, J. P. Tan, K. Xu, H. Wang, C. Yang, S. Gao, X. D. Guo, K. Fukushima, L. Li and J. L. Hedrick, Nat. Chem., 2011, 3, 409–414 CrossRef CAS PubMed.
- M. Xiong, M. W. Lee, R. A. Mansbach, Z. Song, Y. Bao, R. M. Peek Jr, C. Yao, L. F. Chen, A. L. Ferguson, G. C. Wong and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13155–13160 CrossRef CAS PubMed.
- C. Zhou, X. Qi, P. Li, W. N. Chen, L. Mouad, M. W. Chang, S. S. J. Leong and M. B. Chan-Park, Biomacromolecules, 2010, 11, 60–67 CrossRef CAS PubMed.
- P. Li, C. Zhou, S. Rayatpisheh, K. Ye, Y. F. Poon, P. T. Hammond, H. Duan and M. B. Chan-Park, Adv. Mater., 2012, 24, 4130–4137 CrossRef CAS PubMed.
- R. Liu, X. Chen, Z. Hayouka, S. Chakraborty, S. P. Falk, B. Weisblum, K. S. Masters and S. H. Gellman, J. Am. Chem. Soc., 2013, 135, 5270–5273 CrossRef CAS PubMed.
- R. Liu, X. Chen, S. P. Falk, B. P. Mowery, A. J. Karlsson, B. Weisblum, S. P. Palecek, K. S. Masters and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 4333–4342 CrossRef CAS PubMed.
- R. Liu, X. Chen, S. P. Falk, K. S. Masters, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2015, 137, 2183–2186 CrossRef CAS PubMed.
- X. Liu, Y. Yang, M. Han, J. Guo, H. Liu, Y. Liu, J. Xu, S. Ji and X. Chen, Adv. Healthcare Mater., 2022, 11, 2201091 CrossRef CAS PubMed.
- K. R. Meena and S. S. Kanwar, BioMed Res. Int., 2015, 2015, 473050 Search PubMed.
- G. H. Zoysa, H. D. Glossop and V. Sarojini, Eur. J. Med. Chem., 2018, 146, 344–353 CrossRef PubMed.
- H. Gong, M. A. Sani, X. Hu, K. Fa, J. W. Hart, M. Liao, P. Hollowell, J. Carter, L. A. Clifton, M. Campana, P. Li, S. M. King, J. R. P. Webster, A. Maestro, S. Zhu, F. Separovic, T. A. Waigh, H. Xu, A. J. McBain and J. R. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 55675–55687 CrossRef CAS PubMed.
- K. Fa, H. Liu, Z. Li, H. Gong, J. Petkov and J. R. Lu, J. Colloid Interface Sci., 2023, 630, 911–923 CrossRef CAS PubMed.
- X. Zhang, M. Wang, X. Zhu, Y. Peng, T. Fu, C. H. Hu, J. Cai and G. Liao, J. Med. Chem., 2022, 65, 8029–8039 CrossRef CAS PubMed.
- K. Y. Lum, S. T. Tay, C. F. Le, V. S. Lee, N. H. Sabri, R. D. Velayuthan, H. Hassan and S. D. Sekaran, Sci. Rep., 2015, 5, 9657 CrossRef CAS PubMed.
- Y. Lyu, Y. Yang, X. Lyu, N. Dong and A. Shan, Sci. Rep., 2016, 6, 1–12 CrossRef PubMed.
- K. Sharma, S. Aaghaz, I. K. Maurya, S. M. Rudramurthy, S. Singh, V. Kumar, K. Tikoo and R. Jain, Bioorg. Chem., 2022, 127, 106002 CrossRef CAS PubMed.
- K. Wang, J. Yan, W. Dang, J. Xie, B. Yan, W. Yan, M. Sun, B. Zhang, M. Ma, Y. Zhao and F. Jia, Peptides, 2014, 56, 22–29 CrossRef CAS PubMed.
- A. Almaaytah, S. Tarazi, A. Abu-Alhaijaa, Y. Altall, N. Alshar'i, K. Bodoor and Q. Al-Balas, Pharmaceuticals, 2014, 7, 502–516 CrossRef CAS PubMed.
- C. Song, R. Wen, J. Zhou, X. Zeng, Z. Kou, J. Zhang, T. Wang, P. Chang, Y. Lv and R. Wu, Pharmaceutics, 2022, 14, 1937 CrossRef CAS PubMed.
- M. Blanchet and J. M. Brunel, Curr. Med. Chem., 2018, 25, 3613–3636 CrossRef CAS PubMed.
- S. Pal, N. Medatwal, S. Kumar, A. Kar, V. Komalla, P. S. Yavvari, D. Mishra, Z. A. Rizvi, S. Nandan, D. Malakar, M. Pillai, A. Awasthi, P. Das, R. D. Sharma, A. Srivastava, S. Sengupta, U. Dasgupta and A. Bajaj, ACS Cent. Sci., 2019, 5, 1648–1662 CrossRef CAS PubMed.
- N. Medatwal, M. N. Ansari, S. Kumar, S. Pal, S. K. Jha, P. Verma, K. Rana, U. Dasgupta and A. Bajaj, Nanoscale, 2020, 12, 18463–18475 RSC.
- V. Sreekanth, A. Kar, S. Kumar, S. Pal, P. Yadav, Y. Sharma, V. Komalla, H. Sharma, R. Shyam, R. D. Sharma, A. Mukhopadhyay, S. Sengupta, U. Dasgupta and A. Bajaj, Angew. Chem., 2021, 60, 5394–5399 CrossRef CAS PubMed.
- V. Sreekanth, S. Pal, S. Kumar, V. Komalla, P. Yadav, R. Shyam, S. Sengupta and A. Bajaj, Biomater. Sci., 2021, 9, 5626–5639 RSC.
- M. Singh, S. Bansal, S. Kundu, P. Bhargava, A. Singh, R. K. Motiani, R. Shyam, V. Sreekanth, S. Sengupta and A. Bajaj, MedChemComm, 2015, 6, 192–201 RSC.
- H. K. Rachamalla, C. Voshavar, P. Arjunan, G. Mahalingam, R. P. Chowath, R. Banerjee, P. K. Vemula and S. Marepally, ACS Appl. Mater. Interfaces, 2022, 14, 14859–14870 CrossRef CAS PubMed.
- C. Lin, Y. Wang, M. Le, K. F. Chen and Y. G. Jia, Bioconjugate Chem., 2021, 32, 395–410 CrossRef CAS PubMed.
- S. Gupta, D. K. Mishra, M. Z. Khan, V. Saini, D. Mehta, S. Kumar, A. Yadav, M. Mitra, P. Rani, M. Singh, C. K. Nandi, P. Das, V. Ahuja, V. K. Nandicoori and A. Bajaj, Adv. Healthcare Mater., 2022, 11, 2102640 CrossRef CAS PubMed.
- C. H. Li, A. S. Peters, E. L. Meredith, G. W. Allman and P. B. Savage, J. Am. Chem. Soc., 1998, 120, 2961–2962 CrossRef CAS.
- C. Li, L. P. Budge, C. D. Driscoll, B. M. Willardson, G. W. Allman and P. B. Savage, J. Am. Chem. Soc., 1999, 121, 931–940 CrossRef CAS.
- Q. Guan, C. Li, E. J. Schmidt, J. S. Boswell, J. P. Walsh, G. W. Allman and P. B. Savage, Org. Lett., 2000, 2, 2837–2840 CrossRef CAS PubMed.
- B. Ding, Q. Guan, J. P. Walsh, J. S. Boswell, T. W. Winter, E. S. Winter, S. S. Boyd, C. Li and P. B. Savage, J. Med. Chem., 2002, 45, 663–669 CrossRef CAS PubMed.
- B. Ding, U. Taotofa, T. Orsak, M. Chadwell and P. B. Savage, Org. Lett., 2004, 6, 3433–3436 CrossRef CAS PubMed.
- X. Z. Lai, Y. Feng, J. Pollard, J. N. Chin, M. J. Rybak, R. Bucki, R. F. Epand, R. M. Epand and P. B. Savage, Acc. Chem. Res., 2008, 41, 1233–1240 CrossRef CAS PubMed.
- B. Hazra, V. Pore, S. Dey, S. Datta, M. Darokar, D. Saikia, S. P. S. Khanuja and A. Thakur, Bioorg. Med. Chem. Lett., 2004, 14, 773–777 CrossRef CAS PubMed.
- V. S. Pore, N. G. Aher, M. Kumar and P. K. Shukla, Tetrahedron, 2006, 62, 11178–11186 CrossRef CAS.
- N. S. Vatmurge, B. G. Hazra, V. S. Pore, F. Shirazi, P. S. Chavan and M. V. Deshpande, Bioorg. Med. Chem. Lett., 2008, 18, 2043–2047 CrossRef CAS PubMed.
- N. G. Aher, V. S. Pore, N. N. Mishra, P. K. Shukla and R. G. Gonnade, Bioorg. Med. Chem. Lett., 2009, 19, 5411–5414 CrossRef CAS PubMed.
- P. Singla, M. Kaur, A. Kumari, L. Kumari, S. V. Pawar, R. Singh and D. B. Salunke, ACS Omega, 2020, 5, 3952–3963 CrossRef CAS PubMed.
- K. Yadav, P. S. Yavvari, S. Pal, S. Kumar, D. Mishra, S. Gupta, M. Mitra, V. Soni, N. Khare, P. Sharma, C. V. Srikanth, A. Kapil, A. Singh, V. K. Nandicoori and A. Bajaj, Bioconjugate Chem., 2019, 30, 721–732 CrossRef CAS PubMed.
- R. Gupta, J. Thakur, S. Pal, D. Mishra, P. Rani, S. Kumar, A. Saini, A. Singh, K. Yadav, A. Srivastava, R. Prasad, S. Gupta and A. Bajaj, ACS Appl. Bio Mater., 2021, 4, 7332–7341 CrossRef CAS PubMed.
- S. Kumar, J. Thakur, K. Yadav, M. Mitra, S. Pal, A. Ray, S. Gupta, N. Medatwal, R. Gupta, D. Mishra, P. Rani, S. Padhi, P. Sharma, A. Kapil, A. Srivastava, U. D. Priyakumar, U. Dasgupta, L. Thukral and A. Bajaj, ACS Biomater. Sci. Eng., 2019, 5, 4764–4775 CrossRef CAS PubMed.
- K. Yadav, S. Kumar, D. Mishra, M. Asad, M. Mitra, P. S. Yavvari, S. Gupta, M. Vedantham, P. Ranga, V. Komalla, S. Pal, P. Sharma, A. Kapil, A. Singh, N. Singh, A. Srivastava, L. Thukral and A. Bajaj, J. Med. Chem., 2019, 62, 1875–1886 CrossRef CAS PubMed.
- S. Gupta, J. Thakur, S. Pal, R. Gupta, D. Mishra, S. Kumar, K. Yadav, A. Saini, P. S. Yavvari, M. Vedantham, A. Singh, A. Srivastava, R. Prasad and A. Bajaj, Antimicrob. Agents Chemother., 2019, 63, e00520-19 CrossRef PubMed.
- A. Hryniewicka, M. Malinowska, T. Hauschild, K. Pieczul and J. M. Morzycki, J. Steroid Biochem. Mol. Biol., 2019, 189, 65–72 CrossRef CAS PubMed.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- S. Lin, W. L. W. Sin, J. J. Koh, F. Lim, L. Wang, D. Cao, R. W. Beuerman, L. Ren and S. Liu, J. Med. Chem., 2017, 60, 10135–10150 CrossRef CAS PubMed.
- J. Liu, H. Li, Q. He, K. Chen, Y. Chen, R. Zhong, H. Li, S. Fang, S. Liu and S. Lin, Eur. J. Med. Chem., 2022, 243, 114734 CrossRef CAS PubMed.
- C. Ghosh, V. Yadav, W. Younis, H. Mohammad, Y. A. Hegazy, M. N. Seleem, K. Sanyal and J. Haldar, ACS Infect. Dis., 2017, 3, 293–301 CrossRef CAS PubMed.
- R. R. Kashapov, Y. S. Razuvayeva, A. Y. Ziganshina, R. K. Mukhitova, A. S. Sapunova, A. D. Voloshina and L. Y. Zakharova, Colloids Surf., B, 2019, 175, 351–357 CrossRef CAS PubMed.
- D. A. Kuznetsova, D. M. Kuznetsov, S. K. Amerhanova, E. V. Buzmakova, A. P. Lyubina, V. V. Syakaev, I. R. Nizameev, M. K. Kadirov, A. D. Voloshina and L. Y. Zakharova, Langmuir, 2022, 38, 4921–4934 CrossRef CAS PubMed.
- S. M. Wales, K. A. Hammer, K. Somphol, I. Kemker, D. C. Schröder, A. J. Tague, Z. Brkic, A. M. King, D. Lyras, T. V. Riley and J. B. Bremner, Org. Biomol. Chem., 2015, 13, 10813–10824 RSC.
- S. M. Wales, K. A. Hammer, A. M. King, A. J. Tague, D. Lyras, T. V. Riley, P. A. Keller and S. G. Pyne, Org. Biomol. Chem., 2015, 13, 5743–5756 RSC.
- A. J. Tague, P. Putsathit, K. A. Hammer, S. M. Wales, D. R. Knight, T. V. Riley, P. A. Keller and S. G. Pyne, Eur. J. Med. Chem., 2019, 168, 386–404 CrossRef CAS PubMed.
- A. J. Tague, P. Putsathit, M. L. Hutton, K. A. Hammer, S. M. Wales, D. R. Knight, T. V. Riley, D. Lyras, P. A. Keller and S. G. Pyne, Eur. J. Med. Chem., 2019, 170, 203–224 CrossRef CAS PubMed.
- M. K. Mahadari, S. Jennepalli, A. J. Tague, P. Putsathit, M. L. Hutton, K. A. Hammer, D. R. Knight, T. V. Riley, D. Lyras, P. A. Keller and S. G. Pyne, Antibiotics, 2021, 10, 913 CrossRef CAS PubMed.
- A. J. Tague, P. Putsathit, T. V. Riley, P. A. Keller and S. G. Pyne, ACS Med. Chem. Lett., 2021, 12, 413–419 CrossRef CAS PubMed.
- M. T. Garcia, I. Ribosa, L. Perez, A. Manresa and F. Comelles, Langmuir, 2013, 29, 2536–2545 CrossRef CAS PubMed.
- M. Karaman, M. Vraneš, A. Tot, S. Papović, D. Miljaković, S. Gadžurić and M. Ignjatov, RSC Adv., 2020, 10, 22318–22323 RSC.
- G. K. K. Reddy and Y. V. Nancharaiah, Front. Microbiol., 2020, 11, 730 CrossRef PubMed.
- Y. Sutar, S. R. Fulton, S. Paul, S. Altamirano, S. Mhatre, H. Saeed, P. Patel, S. Mallick, R. Bhat, V. B. Patravale and H. Chauhan, ACS Infect. Dis., 2021, 7, 2637–2649 CrossRef CAS PubMed.
- C. W. Chang and J. Y. Takemoto, MedChemComm, 2014, 5, 1048–1057 RSC.
- C. Dezanet, J. Kempf, M. P. Mingeot-Leclercq and J. L. Décout, Int. J. Mol. Sci., 2020, 21, 7411 CrossRef CAS PubMed.
- I. M. Herzog, K. D. Green, Y. Berkov-Zrihen, M. Feldman, R. R. Vidavski, A. Eldar-Boock, R. Satchi-Fainaro, A. Eldar, S. Garneau-Tsodikova and M. Fridman, Angew. Chem., Int. Ed., 2012, 12, 5652–5656 CrossRef PubMed.
- S. K. Shrestha, M. Y. Fosso, K. D. Green and S. Garneau-Tsodikova, Antimicrob. Agents Chemother., 2015, 59, 4861–4869 CrossRef CAS PubMed.
- K. B. Steinbuch, R. I. Benhamou, L. Levin, R. Stein and M. Fridman, ACS Infect. Dis., 2018, 4, 825–836 CrossRef CAS PubMed.
- Q. Z. Jaber, R. I. Benhamou, I. M. Herzog, B. B. Baruch and M. Fridman, Angew. Chem., Int. Ed., 2018, 57, 16391–16395 CrossRef CAS PubMed.
- D. Logviniuk and M. Fridman, ACS Infect. Dis., 2020, 6, 3212–3223 CrossRef CAS PubMed.
- Y. P. Subedi, M. N. AlFindee, J. Y. Takemoto and C. W. T. Chang, MedChemComm, 2018, 9, 909–919 RSC.
- M. N. Alfindee, Y. P. Subedi, M. M. Grilley, J. Y. Takemoto and C. W. T. Chang, Molecules, 2019, 24, 1882 CrossRef CAS PubMed.
- M. A. Velazco-Medel, L. A. Camacho-Cruz, J. C. Lugo-González and E. Bucio, Med. Devices Sens., 2021, 4, 10134 Search PubMed.
- S. Wu, W. Guo, B. Li, H. Zhou, H. Meng, J. Sun, R. Li, D. Guo, X. Zhang, R. Li and W. Qu, Front. Cell. Infect. Microbiol., 2023, 13, 231 Search PubMed.
- M. M. Konai, B. Bhattacharjee, S. Ghosh and J. Haldar, Biomacromolecules, 2018, 19, 1888–1917 CrossRef CAS PubMed.
- S. D. P. Baugh, J. Fungi, 2022, 8, 1085 CrossRef CAS PubMed.
- M. K. Oule, R. Azinwi, A. M. Bernier, T. Kablan, A. M. Maupertuis, S. Mauler, R. K. Nevry, K. Dembele, L. Forbes and L. Diop, J. Med. Microbiol., 2008, 57, 1523–1528 CrossRef PubMed.
- H. Choi, K. J. Kim and D. G. Lee, Fungal Biol., 2017, 121, 53–60 CrossRef CAS PubMed.
- M. S. Brzezinska, M. Walczak, A. Burkowska-But, M. Chylińska, A. Kalwasińska and J. Świątczak, J. Polym. Environ., 2019, 27, 1760–1769 CrossRef CAS.
- G. M. Olmedo, L. Cerioni, M. Sepulveda, J. Ramallo, V. A. Rapisarda and S. I. Volentini, Food Microbiol., 2018, 76, 128–134 CrossRef CAS PubMed.
- W. Ntow-Boahene, I. Papandronicou, J. Miculob and L. Good, Sci. Rep., 2023, 13, 2790 CrossRef CAS PubMed.
- Y. J. Park, M. H. Jeong, I. J. Bang, H. R. Kim and K. H. Chung, Inhalation Toxicol., 2019, 31, 161–166 CrossRef CAS PubMed.
- P. Vicennati, A. Giuliano, G. Ortaggi and A. Masotti, Curr. Med. Chem., 2008, 15, 2826–2839 CrossRef CAS PubMed.
- M. M. Azevedo, P. Ramalho, A. P. Silva, R. Teixeira-Santos, C. Pina-Vaz and A. G. Rodrigues, J. Med. Microbiol., 2014, 63, 1167–1173 CrossRef CAS PubMed.
- K. A. Gibney, I. Sovadinova, A. I. Lopez, M. Urban, Z. Ridgway, G. A. Caputo and K. Kuroda, Macromol. Biosci., 2012, 12, 1279–1289 CrossRef CAS PubMed.
- J. Hoque, P. Akkapeddi, V. Yadav, G. B. Manjunath, D. S. Uppu, M. M. Konai, V. Yarlagadda, K. Sanyal and J. Haldar, ACS Appl. Mater. Interfaces, 2015, 7, 1804–1815 CrossRef CAS PubMed.
- D. A. Real, M. V. Martinez, A. Frattini, M. Soazo, A. G. Luque, M. S. Biasoli, C. J. Salomon, A. C. Olivieri and D. Leonardi, AAPS PharmSciTech, 2013, 14, 4–73 CrossRef PubMed.
- A. Nagaraja, Y. M. Puttaiahgowda, A. Kulal, A. M. Parambil and T. Varadavenkatesan, Macromol. Res., 2019, 27, 301–309 CrossRef CAS.
- S. Schaefer, T. T. P. Pham, S. Brunke, B. Hube, K. Jung, M. D. Lenardon and C. Boyer, ACS Appl. Mater. Interfaces, 2021, 13, 27430–27444 CrossRef CAS PubMed.
- C. W. Yeung, M. H. Periayah, J. Y. Teo, E. T. L. Goh, P. L. Chee, W. W. Loh, X. J. Loh, R. Lakshminarayanan and J. Y. Lim, Macromolecules, 2023, 56, 815–823 CrossRef CAS.
- N. P. Wiederhold, J. Fungi, 2021, 7, 130 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |