
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selectively inhibiting malignant melanoma migration and invasion in an engineered skin model using actin-targeting dinuclear RuII-complexes†
Ahtasham
Raza
a,
Stuart A.
Archer
b,
Jim A.
Thomas
 *b,
Sheila
MacNeil
*a and
John W.
Haycock
*a
*b,
Sheila
MacNeil
*a and
John W.
Haycock
*a
aMaterials Science & Engineering, University of Sheffield, Mappin St, Sheffield S1 3JD, UK. E-mail: s.macneil@sheffield.ac.uk; j.w.haycock@sheffield.ac.uk
bDepartment of Chemistry, University of Sheffield, Brook Hill, Sheffield, S3 7HF, UK. E-mail: james.thomas@sheffield.ac.uk; Tel: +44 (0)114 222 9325
First published on 1st November 2022
Abstract
Due to the poor prognosis of metastatic cancers, there is a clinical need for agents with anti-metastatic activity. Here we report on the anti-metastatic effect of a previously reported Ru(II) complex [{(phen)2Ru}2(tpphz)]4+, 14+, that has recently been shown to disrupt actin fiber assembly. In this study, we investigated the anti-migratory effect of +14+ and a close structural analogue+, 24+, on two highly invasive, metastatic human melanoma cell lines. Laser scanning confocal imaging was used to investigate the structure of actin filament and adhesion molecule vinculin and results show disassembly of central actin filaments and focal adhesions. The effect of both compounds on actin filaments was also found to be reversible. As these results revealed that the complexes were cytostatic and produced a significant inhibitory effect on the migration of both melanoma cell lines but not human dermal fibroblasts their effect on 3D-spheroids and a tissue-engineered living skin model were also investigated. These experiments demonstrated that the compounds inhibited the growth and invasiveness of the melanoma-based spheroidal tumor model and both complexes were found to penetrate the epidermis of the skin tissue model and inhibit the invasion of melanoma cells. Taken together, the cytostatic and antimigratory effects of the complexes results in an antimetastatic effect that totally prevent invasion of malignant melanoma into skin tissue.
Introduction
Over the decades following the success identification and application of cisplatin as an anticancer therapeutic,1,2 several related Pt(II) complexes have moved into clinical application.3–7 Whilst the original SAR employed to identify these treatments has been exhausted,8,9 a number of PtII and PtIV leads unrelated to cisplatin are still being developed and10–12 complexes of other members of the platinum group metals have also been investigated;13–16 in particular, ruthenium compounds have attracted much attention.17–20Although the original focus was on RuIII systems,21,22 RuII complexes with promising therapeutic activity have also been identified. Predicated by the therapeutic action of cisplatin and its derivatives, the vast majority of these complexes have been designed to be classically cytotoxic.23–29 This approach has led to the identification and development of IT-139, a cytotoxic therapeutic lead that has been the subject of human trials.30,31
A small number of unconventional ruthenium leads have also been identified, perhaps the most well-known being NAMI-A.32 This compound is not cytotoxic nor is it significantly internalized into cells,33–35 yet it was the first ruthenium complex to enter clinical trials36,37 due to its in vivo anti-metastatic action.
More recently, research into the therapeutic action of kinetically inert ruthenium complexes, particularly polypyridyl RuII complexes, has rapidly evolved.38–40 By their nature, these complexes interact with biomolecules through reversible, non-covalent interactions, potentially leading to very different effects than cisplatin and analogues. They have also attracted attention because they possess photoactive excited states that can be exploited in applications such as imaging41–44 and phototherapeutics.45–50
In most of the associated therapeutic studies, it is assumed that such complexes interact with DNA and act as classical genotoxins or DNA-damaging photosensitizers. Comparatively less studies have investigated their interactions with proteins.
In pioneering work, the Meggers group carried out a program of studies on kinetically inert RuII complexes as protein kinase inhibitors.51–54 And more lately, the Martí group has shown that luminescent RuII complexes are effective probes for the formation of pathogenic protein fibrils.55,56
The possibility that this class of compounds could affect protein–protein interactions has also been explored. For example, a range of [Ru(bpy)3]2+ derivatives containing structurally complex ligands have been used to recognize extended surfaces of proteins such as cytochrome c and even inhibit the interaction between cytochrome c and cytochrome c peroxidase.57–61
Such effects can have interesting therapeutic implications; for example, recently, the MacDonnell group revealed that the mononuclear complex [Ru(DIP)3]2+ (DIP = 4,7-diphenyl-1,10-phenanthroline) promotes tubulin polymerization and stabilizes microtubules within cells through a high affinity binding interaction with assembled microtubules resulting in cellular effects that are comparable to paclitaxel.62 And in a very recent report we discussed the effect of a dinuclear RuII complex, 14+, Fig. 1, on another cytoskeletal element, actin-based microfilaments.63
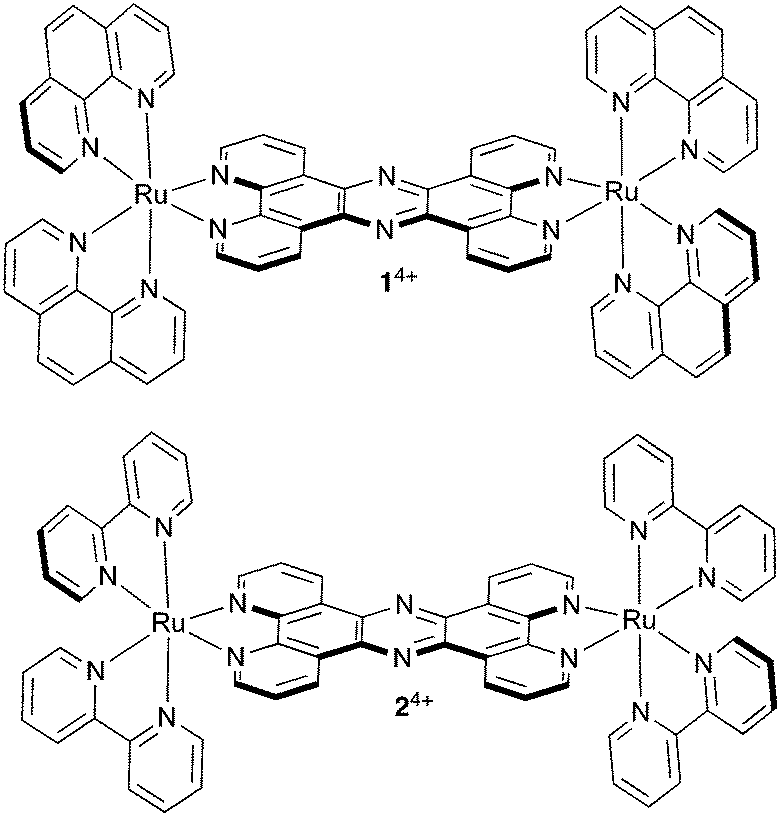 | ||
| Fig. 1 Structures of cationic complexes 14+ and 24+ studied in this report as their chloride salts. | ||
It was already established that this compound is neither classically genotoxic nor phototoxic;42,43 however, it was found that in live CP70 cells [1]Cl4 inhibits the interaction of G-actin subunits by binding to their surfaces, thus preventing their assembly into F-actin microfilaments, which leads to a late cytokinesis block and reduced cell motility.63 Ultimately, despite the absence of a DNA damage response or apoptosis signaling typically seen on treatment with genotoxic therapeutics, this interaction produces an inhibition of proliferation, an effect that has implications for the treatment of metastatic cancer.64–66
Metastases occur in later stages of tumorigenesis; when cancer cells leave a primary tumor and travel through the vascular and lymphatic system to distant sites in the body to form new colonies. This metastatic spread is closely associated with poor prognosis with up to 90% of cancer-related mortalities being associated with metastases67,68 and while the exact factors that lead to this process are not fully understood, they certainly involve degradation of the extracellular matrix (ECM), and increases in cellular migration, specific secondary seeding site, and angiogenesis.69–71 Indeed, the anti-metastatic effect of NAMI-A is thought to be caused by inhibition of angiogenesis and increasing adhesion, although how these effects occur is still not established.32,72,73 Therefore, the observed changes in cell motility and proliferation induced by 14+ suggests that it offers potential as a novel anti-metastatic therapeutic.
Herein, we report the effect of 14+ and its closely related analogue 24+ on highly invasive and aggressively metastatic human malignant melanoma as represented by the two cell lines, A375-SM and C8161 and compare these data with those obtained with human dermal fibroblast, HDF, cells. In particular, the effect on melanoma cell adhesion and migration after short-term and long-term exposure to the two compounds was examined.
Results and discussion
Migration assay
We first set out to investigate what effect treatment with the complexes had on the mobility of melanoma cells using a standard scratch migration assay. The distance untreated cells migrated across a cell exclusion zone was measured at 0, 3, 6, 8 and 24 hours. Both A375-SM and C8161 cell lines rapidly migrated into the cell exclusion zone, though –over the 24 hours period– C8161 migration rates were faster than those of A375-SM and HDFs, (Fig. 2A and B). After cells were 90% confluent in a monolayer, a similar study was performed in serum-free media and rapid migration of melanoma cells was again evident (see ESI,† Fig. S1). We also confirmed our previous observation74 that these rates were predominately due to genuine migration processes rather than cell doubling times.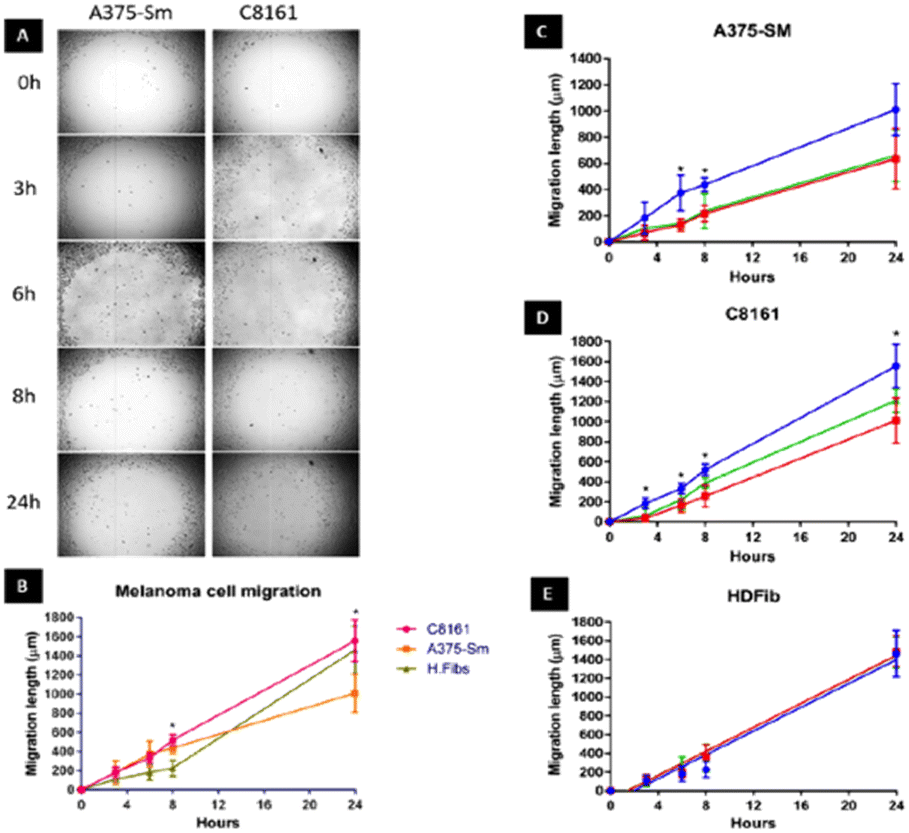 | ||
| Fig. 2 Human A375-SM and C8161 melanoma cell migration. (A) Investigation of two melanoma cell lines migration using cell exclusion zone created by cell stopper and cultured for 3,6,8 and 24 hours. (B) Comparison of distance migrated by melanoma cells and HDF over 24 hours. Inhibition of migratory distance by melanoma A375-SM (C), C8161 (D) and HDF (E) after 1 hour treatment with 14+ (red line) or 24+ (green line) compared to no treatment control (blue line) in serum free media. | ||
Remarkably, pre-incubation with the Ru(II) compounds for one hour in serum free media prior to introduction of a cell exclusion zone resulted in a pronounced decrease in cell migration in both the melanoma cell lines (Fig. 2C and D).
A375-SM cell migration rates were most inhibited (∼40%), whilst C8161 cell migration rates were slightly less affected with inhibition rates of 25–32% being observed; a similar pattern of migration inhibition was seen in serum deprived melanoma cells (see ESI,† Fig. S1). Apart from the inhibitory effect on both A375-SM and C8161 melanoma cells, treatment with either complex has no detectable effect on the migration of HDF cells – Fig. 2E. If the metal complexes were first dissolved in 10% serum media their effect on melanoma cell migration was significantly inhibited, which is consistent with previous studies75 showing that this class of complex interacts with serum proteins, inhibiting their cellular uptake.
Effect on actin filaments
To visualize actin filaments, both A375-SM and C8161 melanoma cell lines were labelled with phalloidin TRITC. To ensure that actin filaments were studied independent of interference from effects caused by mitosis, cells were starved overnight so that they were synchronized in their cell cycle.Actin filaments are the most prominent feature in stress fibers, the bundles of filaments and myosin binding proteins that are crucial for cell contractility and mechano-sensing. It has been previously observed that motile cells contain thinner and more dynamic stress fibers76,77 and at enhanced magnification melanoma cell lines showed long and thin actin filaments along the axis of cell, with emission intensities being weaker in the center of the cells compared to cell cortices (see ESI,† Fig. S2).
Initially, the short-term effects on A375-SM cells treated with either complex were investigated. They were exposed to the complexes for 1 hour in serum-free media and then fixed for phalloidin labelling, after which analysis was carried out through measurement of filament lengths within a single cell – Fig. 3. This revealed that the total length of actin filaments within these cells was significantly reduced, with the total actin filament length of the 24+ treated group (576.91 ± 209 μm) being considerably shorter than the 14+ treated group (783.41 ± 253.82 μm). However, both these measurements were both significantly shorter than the length measured in untreated cells (1139.9 ± 207.92 μm). Having observed this effect, a more detailed morphological experiment involving both melanoma cell lines treated with 14+ and 24+ for 1, 3, 6 and 24 hours was carried out.
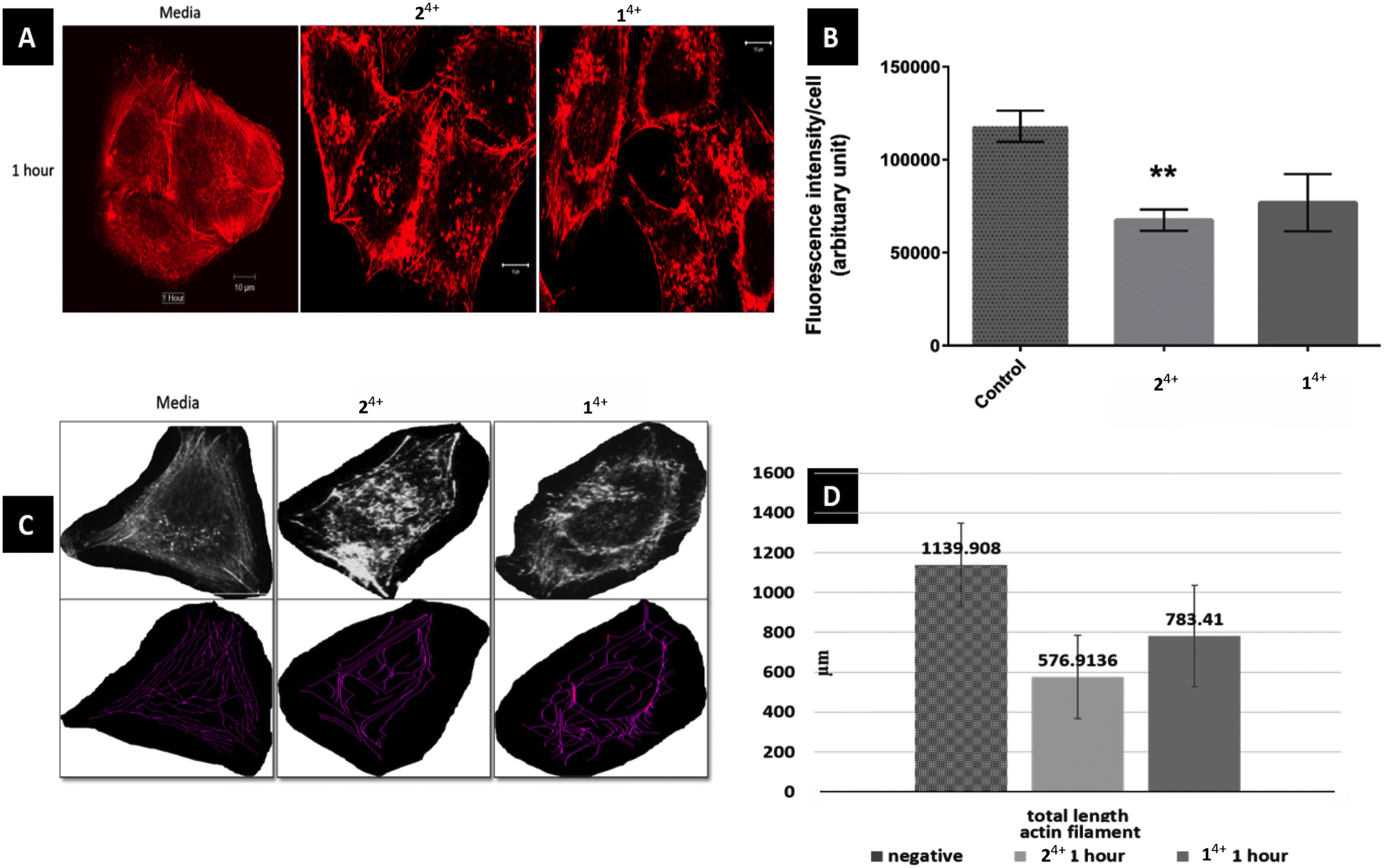 | ||
| Fig. 3 Effect of 14+ and 24+ on actin filaments of melanoma cell line A375-SM. (A) Actin filaments in cell center are more disruptive than outer filament just after 1 hour treatment of Ru-Phen and Ru-Bpy (magnification 40X, SB 10 μm) (B) corrected fluorescence intensity per cell was much reduced in Ru-Bpy than Ru-Phen as compared to normal group (p = 0.0013 and 0.051 respectively) (C) measurement of actin filaments using NeuronJ plug-in (D) total length of actin filaments has reduced significantly in treated group. | ||
After treatment and phalloidin labeling, any differences in actin filaments morphology were analyzed. Fig. 4 shows a representative sequential series of images after treatment with 24+; equivalent images for treatment with 14+ are shown in the ESI,† Fig. S3.
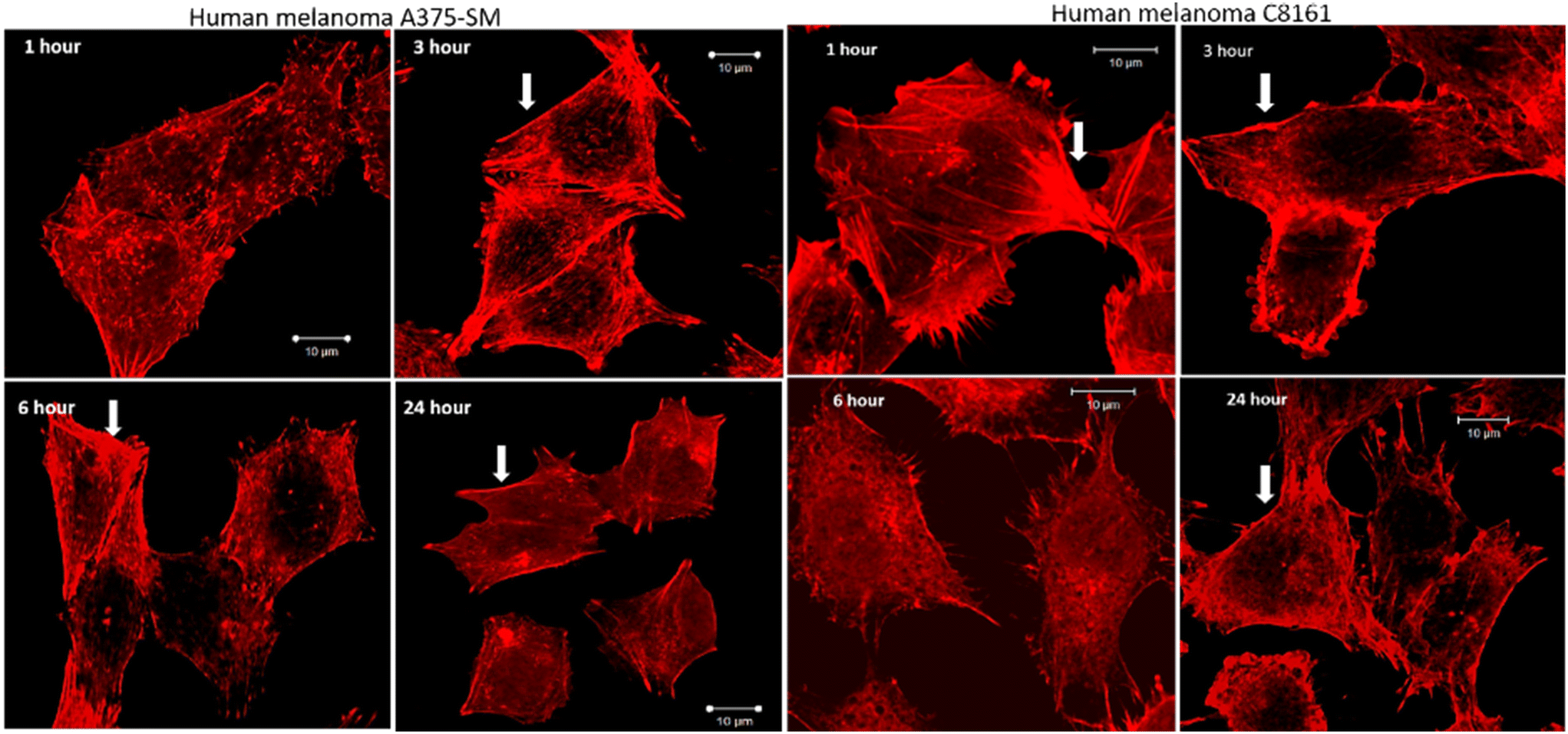 | ||
| Fig. 4 Effect of actin filaments after treatment with 24+ for 1, 3, 6, and 24 hours. Progressive loss of central actin filaments while the cortical filaments (arrowhead) remain prominently in contact. A loss of cell size after 24 hour treatment is also visible (magnification 40X, SB 10 μm). | ||
While actin filaments in non-treated cells appeared long and undisrupted throughout the entire length of the cells, treated cells of both lines displayed a progressive loss of actin filaments over time, which was more prominent in central actin filaments than peripheral filaments (Fig. 5, peripheral actin filament arrowhead). Furthermore, although peripheral actin extensions (likely lamellipodia/filopodia/invadopodia) could be observed throughout the experiment, their overall numbers dropped considerably at later time intervals; a trend that was accompanied by a substantial loss of cell size.
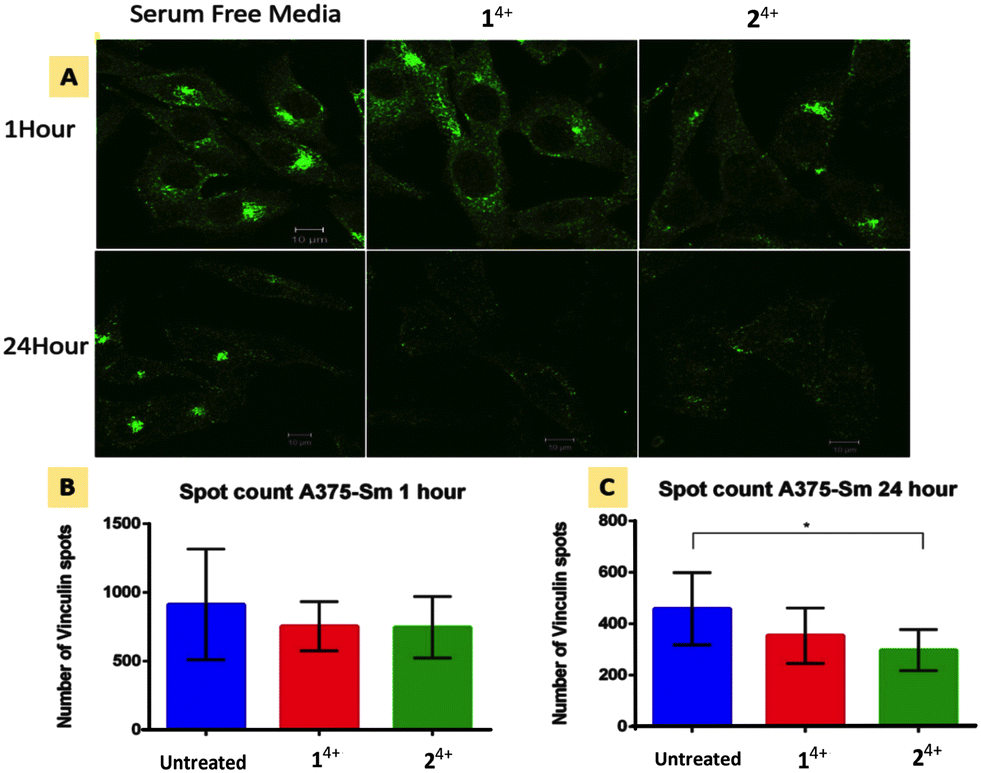 | ||
| Fig. 5 Focal adhesion response in human melanoma A375-SM cells after 1 and 24 hour treatment with Ru(II) compounds. (A) Immunofluorescence imaging of vinculin spots with in cells. (B and C) Vinculin spot count after 1 hour and 24 hour treatment (p = 0.05, significant difference between no Rx and Ru–Ru Phe or Bpy group was 0.2 and 0.05 respectively). | ||
When melanoma cells were treated with 14+ or 24+ for an hour then washed and replenished with fresh media, after six hours actin filaments started redeveloping, reaching almost complete recovery within 24 hours. Again, it was noticed that central actin filaments were particularly disrupted and took the longest to recover (see ESI,† Fig. S4). These observations, demonstrating the reversible effect of these compounds on actin filaments, further support our hypothesize that 14+ disrupts the interaction of G-actin assembly through reversible non-covalent binding effects.63
Vinculin disruption
Cell migration is driven by the interplay between F-actin and transmembrane integrin receptors; by binding to extracellular ligands, the external domains of integrin at focal adhesions provide the first step in the transduction of adhesion into movement; whilst the linkage of integrin internal domains to stress fibers provides a conduit to couple mechanical forces to the cytoskeleton.78 This relationship suggests that treatment with 14+ or 24+ will ultimately inhibit focal adhesion function. To explore this hypothesis, we investigated the effect of treatment with the complexes on the cytoskeletal protein vinculin which functions as a linking agent between integrin and the cytoskeleton, anchoring stress fibers to focal adhesions at the cell membrane.79,80Immuno-fluorescent analysis of vinculin was performed on both melanoma lines. The cells were fixed after 1 and 24 hour treatment with either 14+ or 24+ and then labelled with rabbit monoclonal vinculin antibody. The imaging data was then analyzed to measure the vinculin spot count per cell; Fig. 5 shows the results of such an analysis for the A3375-SM line (see the ESI,† Fig. S5, for equivalent data for C8161 melanoma cells).
Whilst no effects were observed after 1 hour of treatment, after 24 hour both melanoma lines showed reductions in vinculin spot clusters. This suggests that the loosening of central actin filament observed after 1 hour treatment does not initially affect the firm anchoring of focal adhesions. After 24 hour treatment with either complex, stress fibers become so shortened and disarranged that they are no longer mechanical linked to focal adhesions leading to a concomitant drop in vinculin levels.
Effects on cell viability
In our initial study, we found that that even though 14+ is not genotoxic it has a potent cytostatic effect on breast and ovarian cancer cell lines as its interaction with actin ultimately disrupts late cytokinesis.63 As melanoma is notoriously resistant to most anti-neoplastic chemotherapeutics due to aberrant apoptotic signaling,81 we investigated the effect of 14+ or 24+ on both of aggressively invasive melanoma lines.MTT assays showed a significant reduction in cell metabolic activity after 24 hour of treatment in both cell lines compared to no treatment (in serum free media) – see ESI.† For example, the cell metabolic activities after 24 hour of treatment with 14+ (at 100 μM) in A375-SM and C8161 were reduced to 89% ± 19% and 65% ± 21% of their original activity respectively and were reduced to 89% ± 24 and 87% ± 29 on treatment with 24+ at the same concentrations. At still higher concentrations more profound effects were observed: exposure to 200 μM of 14+ resulted in a decrease in metabolic activity of 53% ± 13% in C8161 cells after 24 hours' treatment.
With the observation of both antimigratory and cytostatic effects in both melanoma lines we went on to investigate the effect of 14+ or 24+ in 3-D tumor and tissue models.
Melanoma spheroid studies
Cell spheroids provide an excellent 3D preclinical model for studying therapeutic efficacy.50 As they provide more clinically relevant cellular conditions, they can help to reduce drug attrition rates in clinical trials. With this in mind, we used spheroids to investigate the effect of 14+ and 24+ on melanoma spheroids.Using a previously described procedure, C8161 spheroids were cultured for three days until a consistent size (555 ± 50.5 μm) and quality (exponential growth) was achieved. Day 3 spheroid were then treated with 14+ or 24+ at concentrations of 10, 50, 100, 200 and 500 μM respectively in serum free media.
Exposure to 14+ at the highest concentration of 500 μM produced a 15% reduction of spheroid growth (569 ± 20 to 493 ± 180), whereas treatment with 24+ at the same concentration produced a ∼20% reduction in growth (569 ± 20 to 451 ± 40) over 7 days. The metabolic activity of the spheroids, tested on day 7 using MTT assays, also revealed that exposure to the complexes resulted in a viability decrease of ∼30% – see ESI,† Fig. S6.
Interestingly, this effect showed a threshold concentration: whilst the decrease in spheroid size and metabolic activity in response to a 200 μM treatment of 14+ or 24+ was also most identical to that at the higher dose, lower dose (10, 50, 100 μM) produced a much less pronounced effect. Having observed this distinctive effect on spheroid growth, we set out to investigate whether exposure to 14+ and 24+ altered the invasive properties of the C8161 spheroids.
Spheroids embedded in a suitable matrix also act as excellent physiological relevant 3D model for tumor invasion, as the matrix facilitates the cancer cells' ability to anchor, adhere, and migrate away from the primary spheroid site. In our migration studies a previous procedure82 was adapted to embed C8161 melanoma spheroids in rat tail collagen 1 gel which functioned as a model of the extracellular matrix, ECM. After the spheroids were exposed to a bolus dose of 14+ or 24+, the number of migrating cells and total migration area were then evaluated over the period of several days and compared to a nontreated control. This procedure resulted in some distinctive results – Fig. 6.
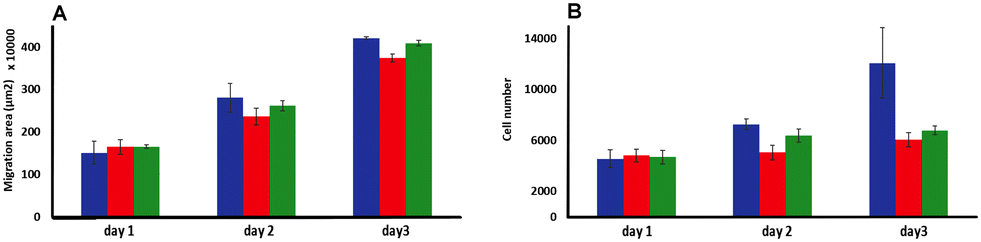 | ||
| Fig. 6 Invasion studies using C8161 melanoma spheroids embedding in a rat tail collagen 1 gel matrix. (A) Melanoma cells migrated area and (B) total number of migrated cells analyzed (over 3 days) after a bolus treating at day 1 with 500 μM of 14+ (red) or 24+ (green) compared to no treatment (blue) (n = 3). | ||
Although there was no significant difference in the extent of migration area between treated and untreated spheroids, the total number of cells migrating from the primary spheroid area was significantly reduced after treatment. Exposure to a single dose of either 14+ or 24+ led to a reduction in migrating cells of ∼30% after two days and 50% after three days compared to the untreated spheroids. These observations confirm that a bolus dose of either complex can successfully diffuse through the collagen ECM model and penetrate multicellular spheroids resulting in significant therapeutic effects.
The fact that there is a reduction in migrated tumor number from the primary area but not in the extent of spheroid migration distance is consistent with the previously discussed dose-dependent threshold effect observed in spheroid growth and suggests that the population of cells that do not receive an above-the-threshold dose of the complex are still migratory.
As both complexes affect cancer cell viability and the invasiveness of spheroid tumor model, a study to assess their efficacy in inhibiting melanoma invasion through a tissue-engineered human skin model was initiated.
Inhibition of melanoma invasion through a human skin model
The use of tissue-engineered skin is particularly valuable in studies on invasive melanoma as the live skin model used in our experiments – first reported and extensively studied by the MacNeil lab – provides physiologically relevant dermal and epidermal structures, including a keratinized stratum corneum.83,84 For the invasion studies, isolated primary human keratinocytes, fibroblasts, and C8161 melanoma spheroids were submerged in media and co-cultured on an acellular skin dermis, then raised to an air–liquid interface, ALI, culture. A model of normal skin was developed through the same protocol, but without the addition of C8161 melanoma spheroids. After 14 days, both ALI cultures were treated with 14+ or 24+ and the results were compared to an untreated skin model.Treatment of the normal skin tissue model with either complex led to observable changes in morphology; in particular, even at low treatment concentrations, some separation of the epidermal and dermal layers occurred due to disruption of the basement membrane of the dermal–epidermal junction. At higher concentrations a thickening and sloughing of the stratum corneum occurred; epidermal parakeratosis like this is seen in conditions such as psoriasis – see ESI,† Fig. S7.
When melanoma spheroids are incorporated within tissue engineered human skin, the biological architecture of this model very closely approximates that of native skin tissue suffering from melanoma metastasis. Indeed, as previously reported,85 the C8161 cell spheroids proved to be highly metastatic and rapidly showed extensive and aggressive invasion into the dermis of untreated tissues.
In contrast with these observations, treatment with either complex resulted in minimal or even no invasion of melanoma cells into surrounding tissue, confirming the anti-migration and invasion properties of 14+ and 24+. Although it should be noted that treatment also led to the formation of fluid-filled vacuoles in the epidermis characteristic of spongiosis, which is consistent with some toxicity effects in healthy tissue (Fig. 7).
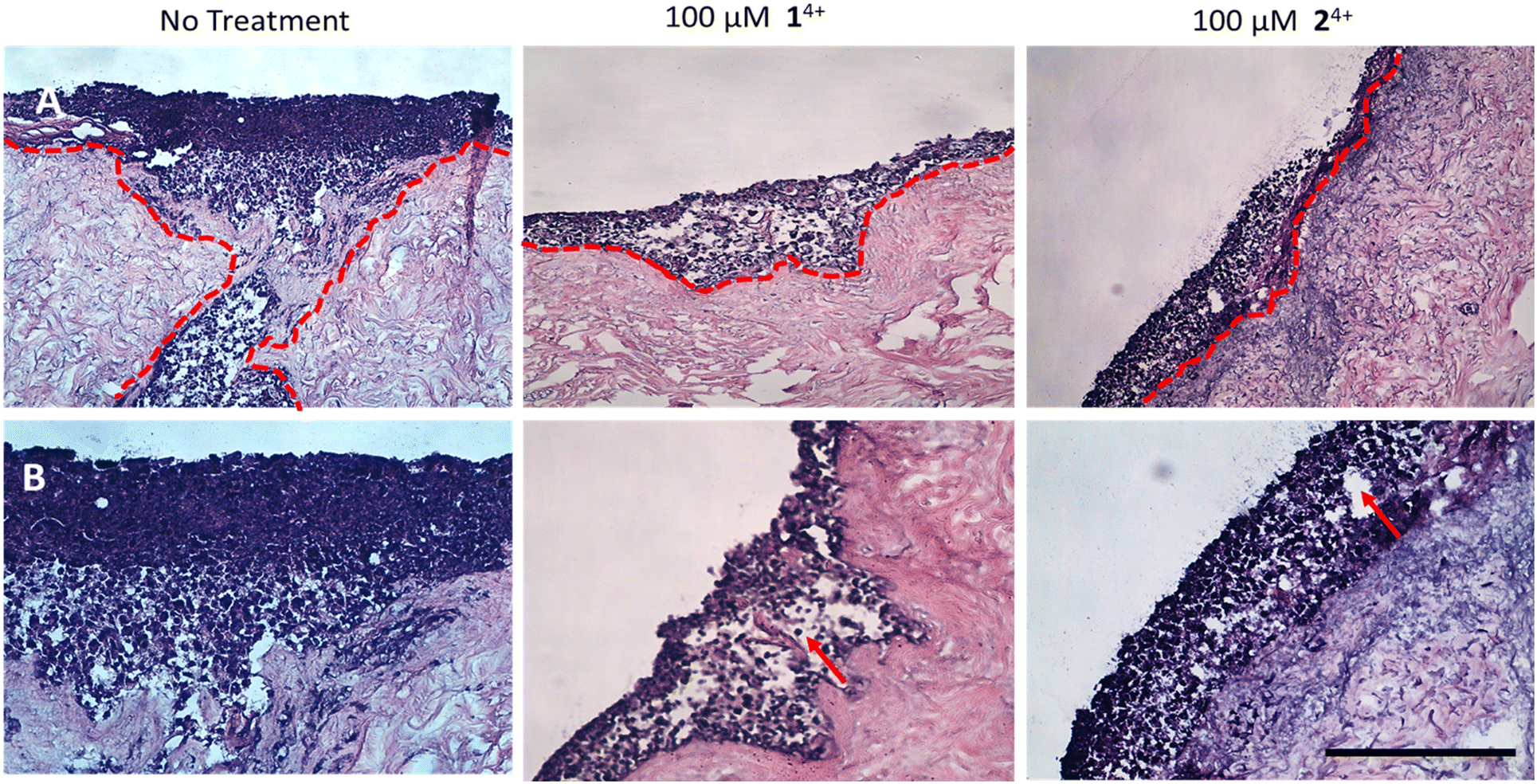 | ||
| Fig. 7 H&E-stained tissue engineered skin model. A. At ×10 magnification B. At ×20 magnification. The model was developed using melanoma spheroid (day 3) and primary cells (human isolated fibroblast and keratinocytes) cultured together on a human de-epithelialized dermis for 14 days (air–liquid interface culture conditions). The untreated control (left) was compared to the model dosed with either 14+ (middle) or 24+ (right). After 24 hours, the tissues were fixed and processed for histology. Melanoma cell migration from epidermis to dermis is observed in untreated tissues (red dotted line), while little to no cell migration was seen in the treated group. Vacuolar fluid-filled cells in the epidermis provide evidence of spongiosis. (red arrow) (SB = 500 μm). | ||
Conclusions
Taken together these results show that both 14+ and 24+ strongly disrupt actin filaments, stress fibers, and focal adhesions in highly migratory human melanoma cells. Consequently, the complexes are cytotostatic and profoundly antimigratory in 2-D cultures. More significantly, in 3-D cultures they totally inhibit the ability of an aggressive melanoma to invade the dermis of our tissue-engineered skin model, therefore meeting one of the biggest challenges for the treatment of this pernicious, hard-to-treat cancer. Given these unprecedented results, and the fact that these complexes are neither classically genotoxic nor phototoxic, but disrupt protein–protein interactions within melanoma cells, this study offers a new paradigm for the use of metallodrugs in the treatment of one of the most fatal and therapeutically intractable forms of cancer.The observation that both complexes have this effect on melanoma but no detectable effect on migration rates of HDF cells is striking. This may just be a function of the higher metabolic rates of melanoma cells,86 however previous studies in a range of cell types have demonstrated that whilst the uptake of 14+ is through an active, energy dependent, 24+ is hardly taken up at all.42
The fact that both 14+ and 24+ are active in melanoma suggests uptake is modulated in these cells. Certainly, it is known that endocytosis, and rates of endocytosis (specifically micropinocytosis) are greatly modulated in melanoma cells87,88 and it is also known that 14+ is internalized into cells if it is delivered through an endocytosis-compatible carrier system.89
The results on the tissue-engineered model reveal that that the complexes can penetrate the epidermis but do produce some deleterious effects on epidermal cells. However, if these complexes are used as a topical treatment, it is likely any off-target effects would largely be localized to the site of treatment. Given that 14+ and 24+ are cytostatic and anti-invasive, they could provide one component of a novel therapy. For example, although 14+ and 24+ are not in themselves phototoxic,42,43 we have developed close structural analogues that are highly photo-oxidizing, thus displaying cell killing effects even within hypoxic regions of tumor models. A topically applied combined regime of these two components would provide an anti-invasive and a (photo-)cytotoxic therapeutic as a targeted therapy for melanoma with reduced off-target effects compared to systemic dosing. Indeed, given the close structural similarities between 14+ and 24+ and their photo-oxidizing analogues, it seems likely that both of these functions could be provided by a single molecular architecture – studies to identify/develop such a multimodal system are already underway and will form the basis of a future report.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the EPSRC for postdoctoral funding of SAA and AR (EP/M015572/1). Waste human skin was collected from routine surgical procedures, with written informed consent, following a protocol approved by the NHS research ethics committee (15/YH/0177 & 21/NE/0115). All tissue was obtained and used on an anonymous basis from Sheffield Teaching Hospitals NHS Trust, Directorate of Plastic, Reconstructive Hand and Burns Surgery. Samples were stored at room temperature for up to 24 hours before processing.Notes and references
- B. Rosenberg, L. Van Camp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385–386 CrossRef CAS PubMed
.
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed
- T. Boulikas, A. Pantos, E. Bellis and P. Christofis, Cancer Ther., 2007, 5, 537–583 Search PubMed
-
I. A. Riddell and S. J. Lippard, in Metal Ions in Life Sciences, ed. A. Sigel, H. Sigel, E. Friesinger and R. K. O. Sigel, Walter de Gruyter GmbH, Berlin, Germany, 2018, vol. 18, pp. 1–42 Search PubMed
- T. Makovec, Radiol. Oncol., 2019, 53, 148–158 CrossRef CAS
- R. C. Todd and S. J. Lippard, Metallomics, 2009, 1, 280–291 CrossRef CAS
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC
- S. Dilruba and G. V. Kalayda, Cancer Chemother. Pharmacol., 2016, 77, 1103–1124 CrossRef CAS
- E. Wong and C. M. Giandornenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS
- Y.-R. Zheng, K. Suntharalingam, T. C. Johnstone, H. Yoo, W. Lin, J. G. Brooks and S. J. Lippard, J. Am. Chem. Soc., 2014, 136, 8790–8798 CrossRef CAS
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS
- Z. Guo and P. J. Sadler, Angew. Chem., Int. Ed., 1999, 38, 1512–1531 CrossRef CAS
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106 RSC
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Chem. Sci., 2020, 44, 930–941 Search PubMed
- E. Boros, P. J. Dyson and G. Gasser, Chem, 2020, 6, 41–60 CAS
- E. S. Antonarakis and A. Emadi, Cancer Chemother. Pharmacol., 2010, 66, 1–9 CrossRef CAS
- A. Bergamo and G. Sava, Dalton Trans., 2011, 40, 7817 RSC
- S. M. Meier-Menches, C. Gerner, W. Berger, C. G. Hartinger and B. K. Keppler, Chem. Soc. Rev., 2018, 47, 909–928 RSC
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS
- M. J. Clarke, Coord. Chem. Rev., 2002, 232, 69–93 CrossRef CAS
- A. Levina, A. Mitra and P. A. Lay, Metallomics, 2009, 1, 458–470 CrossRef CAS
- H. Chen, J. A. Parkinson, R. E. Morris and P. J. Sadler, J. Am. Chem. Soc., 2003, 125, 173–186 CrossRef CAS
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed
- E. Corral, A. C. G. Hotze, H. Den Dulk, A. Leczkowska, A. Rodger, M. J. Hannon and J. Reedijk, J. Biol. Inorg. Chem., 2009, 14, 439–448 CrossRef CAS
- V. Brabec and O. Nováková, Drug Resistance Updates, 2006, 9, 111–122 CrossRef CAS
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC
- H. K. Saeed, S. Sreedharan and J. A. Thomas, Chem. Commun., 2020, 56, 1464–1480 RSC
- V. Brabec and J. Kašpárková, Coord. Chem. Rev., 2018, 376, 75–94 CrossRef CAS
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC
- H. A. Burris, S. Bakewell, J. C. Bendell, J. Infante, S. F. Jones, D. R. Spigel, G. J. Weiss, R. K. Ramanathan, A. Ogden and D. Von Hoff, ESMO Open, 2016, 1, e000154 CrossRef
- F. Guidi, A. Modesti, I. Landini, S. Nobili, E. Mini, L. Bini, M. Puglia, A. Casini, P. J. Dyson, C. Gabbiani and L. Messori, J. Inorg. Biochem., 2013, 118, 94–99 CrossRef CAS
- A. Bergamo and G. Sava, Chem. Soc. Rev., 2015, 44, 8818–8835 RSC
- E. Alessio, Eur. J. Inorg. Chem., 2017, 2017, 1549–1560 CrossRef CAS
- A. R. Simović, R. Masnikosa, I. Bratsos and E. Alessio, Coord. Chem. Rev., 2019, 398, 113011 CrossRef
- J. M. Rademaker-Lakhai, D. Van Den Bongard, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS
- S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. Van Werkhoven, E. Alessio, G. Sava, J. H. Beijnen and J. H. M. Schellens, Invest. New Drugs, 2015, 33, 201–214 CrossRef CAS
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179–3192 RSC
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z. S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC
- A. Notaro and G. Gasser, Chem. Soc. Rev., 2017, 46, 7317–7337 RSC
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46–47 CrossRef CAS PubMed
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662–667 CrossRef CAS PubMed
- S. Sreedharan, M. R. Gill, E. Garcia, H. K. Saeed, D. Robinson, A. Byrne, A. Cadby, T. E. Keyes, C. Smythe, P. Pellett, J. Bernardino De La Serna, J. A. Thomas, J. de la Serna and J. A. Thomas, J. Am. Chem. Soc., 2017, 139, 15907–15913 CrossRef CAS PubMed
- J. Shum, P. K.-K. Leung and K. K.-W. Lo, Inorg. Chem., 2019, 58, 2231–2247 CrossRef CAS PubMed
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC
- F. Heinemann, J. Karges and G. Gasser, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed
- J. Liu, C. Zhang, T. W. Rees, L. Ke, L. Ji and H. Chao, Coord. Chem. Rev., 2018, 363, 17–28 CrossRef CAS
- S. A. Archer, A. Raza, F. Dröge, C. Robertson, A. J. Auty, D. Chekulaev, J. A. Weinstein, T. Keane, A. J. H. M. Meijer, J. W. Haycock, S. MacNeil and J. A. Thomas, Chem. Sci., 2019, 10, 3502–3513 RSC
- H. K. Saeed, P. J. Jarman, S. Archer, S. Sreedharan, I. Q. Saeed, L. K. Mckenzie, J. A. Weinstein, N. J. Buurma, C. G. W. Smythe and J. A. Thomas, Angew. Chem., Int. Ed., 2017, 56, 12628–12633 CrossRef CAS PubMed
- A. Raza, S. A. Archer, S. D. Fairbanks, K. L. Smitten, S. W. Botchway, J. A. Thomas, S. MacNeil and J. W. Haycock, J. Am. Chem. Soc., 2020, 142, 4639–4647 CrossRef CAS PubMed
- L. Zhang, P. Carroll and E. Meggers, Org. Lett., 2004, 6, 521–523 CrossRef CAS PubMed
- H. Bregman, D. S. Williams, G. E. Atilla, P. J. Carroll and E. Meggers, J. Am. Chem. Soc., 2004, 126, 13594–13595 CrossRef CAS PubMed
- J. Maksimoska, L. Feng, K. Harms, C. Yi, J. Kissil, R. Marmorstein and E. Meggers, J. Am. Chem. Soc., 2008, 130, 15764–15765 CrossRef CAS PubMed
- E. Meggers, Chem. Commun., 2009, 1001–1010 RSC
- N. P. Cook, V. Torres, D. Jain and A. A. Martí, J. Am. Chem. Soc., 2011, 133, 11121–11123 CrossRef CAS PubMed
- N. P. Cook, K. Kilpatrick, L. Segatori and A. A. Martí, J. Am. Chem. Soc., 2012, 134, 20776–20782 CrossRef CAS PubMed
- H. Takashima, S. Shinkai and I. Hamachi, Chem. Commun., 1999, 2345–2346 RSC
- J. Ohkanda, R. Satoh and N. Kato, Chem. Commun., 2009, 6949–6951 RSC
- J. Muldoon, A. E. Ashcroft and A. J. Wilson, Chem. – Eur. J., 2010, 16, 100–103 CrossRef CAS PubMed
- M. H. Filby, J. Muldoon, S. Dabb, N. C. Fletcher, A. E. Ashcroft and A. J. Wilson, Chem. Commun., 2010, 47, 559 RSC
- S. H. Hewitt, M. H. Filby, E. Hayes, L. T. Kuhn, A. P. Kalverda, M. E. Webb and A. J. Wilson, ChemBioChem, 2017, 18, 223–231 CrossRef CAS PubMed
- N. Alatrash, F. H. Issa, N. S. Bawazir, S. J. West, K. E. Van Manen-Brush, C. P. Shelor, A. S. Dayoub, K. A. Myers, C. Janetopoulos, E. A. Lewis and F. M. MacDonnell, Chem. Sci., 2020, 11, 264–275 RSC
- M. R. Gill, P. J. Jarman, V. Hearnden, M. Bassetto, S. D. Fairbanks, J. Palmer, K. F. Ayscough, J. A. Thomas and C. Smythe, Angew. Chem., Int. Ed. DOI:10.1002/anie.202117449
- S. A. Rizvi, S. Liu, Z. Chen, C. Skau, M. Pytynia, D. R. Kovar, S. J. Chmura and S. A. Kozmin, J. Am. Chem. Soc., 2010, 132, 7288–7290 CrossRef CAS PubMed
- J. R. Stehn, N. K. Haass, T. Bonello, M. Desouza, G. Kottyan, H. Treutlein, J. Zeng, P. R. B. B. Nascimento, V. B. Sequeira, T. L. Butler, M. Allanson, T. Fath, T. A. Hill, A. McCluskey, G. Schevzov, S. J. Palmer, E. C. Hardeman, D. Winlaw, V. E. Reeve, I. Dixon, W. Weninger, T. P. Cripe and P. W. Gunning, Cancer Res., 2013, 73, 5169–5182 CrossRef CAS PubMed
- M. Schenk, B. Aykut, C. Teske, N. A. Giese, J. Weitz and T. Welsch, Cancer Lett., 2015, 358, 161–169 CrossRef CAS PubMed
- C. L. Chaffer and R. A. Weinberg, Science, 2011, 331, 1559–1564 CrossRef CAS PubMed
- H. Dillekås, M. S. Rogers and O. Straume, Cancer Med., 2019, 8, 5574–5576 CrossRef PubMed
- T. Bogenrieder and M. Herlyn, Oncogene, 2003, 22, 6524–6536 CrossRef CAS PubMed
- N. Sethi and Y. Kang, Nat. Rev. Cancer, 2011, 11, 735–748 CrossRef CAS PubMed
- Y. Suhail, M. P. Cain, K. Vanaja, P. A. Kurywchak, A. Levchenko, R. Kalluri and Kshitiz, Cell Syst., 2019, 9, 109–127 CrossRef CAS PubMed
- A. Bergamo and G. Sava, Dalton Trans., 2007, 1267–1272 RSC
- J. P. C. C. Coverdale, T. Laroiya-Mccarron and I. Romero-Canelón, Inorganics, 2019, 7, 15–31 CrossRef
- N. Zhu, R. Lalla, P. Eves, T. L. H. Brown, A. King, E. H. Kemp, J. W. Haycock and S. MacNeil, Br. J. Cancer, 2004, 90, 1457–1463 CrossRef CAS PubMed
- A. Wragg, M. R. Gill, L. McKenzie, C. Glover, R. Mowll, J. A. Weinstein, X. Su, C. Smythe and J. A. Thomas, Chem. – Eur. J., 2015, 21, 11865–11871 CrossRef CAS PubMed
- S. Pellegrin and H. Mellor, J. Cell Sci., 2007, 120, 3491–3499 CrossRef CAS PubMed
- T. Hohmann and F. Dehghani, Cells, 2019, 8, 362 CrossRef CAS PubMed
- A. Livne and B. Geiger, J. Cell Sci., 2016, 129, 1293–1304 CrossRef CAS PubMed
- J. D. Humphries, P. Wang, C. Streuli, B. Geiger, M. J. Humphries and C. Ballestrem, J. Cell Biol., 2007, 179, 1043–1057 CrossRef CAS PubMed
- I. Thievessen, P. M. Thompson, S. Berlemont, K. M. Plevock, S. V. Plotnikov, A. Zemljic-Harpf, R. S. Ross, M. W. Davidson, G. Danuser, S. L. Campbell and C. M. Waterman, J. Cell Biol., 2013, 202, 163–177 CrossRef CAS PubMed
- B. Domingues, J. Lopes, P. Soares and H. Populo, ImmunoTargets Ther., 2018, 7, 35–49 CrossRef CAS PubMed
- K. M. Charoen, B. Fallica, Y. L. Colson, M. H. Zaman and M. W. Grinstaff, Biomaterials, 2014, 35, 2264–2271 CrossRef CAS PubMed
- J. Shepherd, I. Douglas, S. Rimmer, L. Swanson and S. MacNeil, Tissue Eng., Part C, 2009, 15, 475–484 CrossRef PubMed
-
S. MacNeil, J. Shepherd and L. Smith, in Methods in molecular biology, 2011, vol. 695, pp. 129–153 Search PubMed
- P. Eves, J. Haycock, C. Layton, M. Wagner, H. Kemp, M. Szabo, R. Morandini, G. Ghanem, J. C. García-Borrón, C. Jiménez-Cervantes and S. Mac Neil, Br. J. Cancer, 2003, 89, 2004–2015 CrossRef CAS PubMed
- M. Neagu, Front. Oncol., 2020, 10, 851 CrossRef PubMed
- D. Alonso-Curbelo and M. S. Soengas, Oncotarget, 2015, 6, 2583–2584 CrossRef PubMed
- S. Demirsoy, S. Martin, H. Maes and P. Agostinis, Front. Oncol., 2016, 6, 240 Search PubMed
- X. Tian, M. R. Gill, I. Cantón, J. A. Thomas and G. Battaglia, ChemBioChem, 2011, 12, 548–551 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Materials and reagents, additional figures: morphology of stress fibers in untreated human melanoma C8161 and A375-Sm cells, effect of actin filaments after treating with 14+ for 1, 3, 6 and 24 hours, recovery of actin filaments after 1 hour treatment with 14+ or 24+ in 24 hours, focal adhesion response in human melanoma C8161 cells, human melanoma spheroid growth and metabolic analysis after 14+ or 24+ treatment, effect of 14+ on normal human tissue engineered skin model. See DOI: https://doi.org/10.1039/d2md00280a |
| This journal is © The Royal Society of Chemistry 2023 |