
Design and synthesis of atorvastatin derivatives with enhanced water solubility, hepatoselectivity and stability†
Svetlana Yu.
Maklakova
 *a,
Anton V.
Lopukhov
a,
Alexandr D.
Khudyakov
a,
Sergey V.
Kovalev
a,
Maria P.
Mazhuga
a,
Olga E.
Chepikova
b,
Andrey A.
Zamyatnin
Jr.
bcde,
Alexander G.
Majouga
af,
Natalia L.
Klyachko
a and
Elena K.
Beloglazkina
a
*a,
Anton V.
Lopukhov
a,
Alexandr D.
Khudyakov
a,
Sergey V.
Kovalev
a,
Maria P.
Mazhuga
a,
Olga E.
Chepikova
b,
Andrey A.
Zamyatnin
Jr.
bcde,
Alexander G.
Majouga
af,
Natalia L.
Klyachko
a and
Elena K.
Beloglazkina
a
aChemistry Department, Lomonosov Moscow State University, GSP-1, Leninskie Gory 1/3, Moscow 119991, Russian Federation. E-mail: maklakovaSU@yandex.ru
bDepartment of Biotechnology, Sirius University of Science and Technology, Olympic Avenue 1, Sochi 354340, Russian Federation
cInstitute of Molecular Medicine, Sechenov First Moscow State Medical University, Trubetskaya Street 8/2, Moscow 119991, Russian Federation
dBelozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, GSP-1, Leninskie Gory, Moscow 119992, Russian Federation
eFaculty of Health and Medical Sciences, University of Surrey, Guildford GU2 7XH, UK
fDmitry Mendeleev University of Chemical Technology of Russia, Miusskaya Square 9, Moscow 125047, Russian Federation
First published on 11th October 2022
Abstract
Statins are effective 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-R) inhibitors, which are successfully used for cardiovascular disease treatment. Statins' side effects are generally attributed to poor bioavailability and hepatoselectivity, which are closely related to their high lipophilicity. Targeted delivery of statins to the liver is considered as a way to reduce unwanted side effects. Herein we report on synthesis and evaluation of atorvastatin conjugates targeting the galactose-specific hepatic asialoglycoprotein receptor (ASGPR). The prepared conjugates showed greater water solubility compared to unmodified atorvastatin. The synthesised compounds demonstrated potent binding to the ASGPR with submicromolar KD values. The conjugates with an amide bond connecting atorvastatin and the targeting moiety displayed the optimal stability under model conditions, as they underwent hydrolysis only when incubated with the intracellular protease. The hydrolysis products effectively inhibited HMG-R activity. The results suggest that the designed amide-based compounds have the potential to be further developed as orally administered prodrugs of atorvastatin.
Introduction
According to the World Health Organization, the number one cause of death is ischemic heart disease, responsible for 16% of the world's total deaths in 2019. The current therapeutic strategies for cardiovascular diseases involve the management of risk factors, especially dyslipidemia and hypertension. Statins are clinical inhibitors that target 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMG-R). The latter catalyses the conversion of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) to mevalonic acid, which is the rate-limiting step in the hepatic cholesterol biosynthesis. Although statins are generally well tolerated, some adverse reactions have been noted, for instance, patients might experience muscle aches (myalgia), weakness, stiffness, and cramps.1The mechanism by which statins cause side effects remains elusive. Passive diffusion into non-hepatic tissues could be one possible explanation.2 Tissue selectivity was proposed to be influenced by the relative lipophilicity of the drugs, as more hydrophilic statins exhibited higher liver selectivity.3 Consequently, one strategy to reduce side effects is to improve hepatic selectivity by increasing the hydrophilicity of the drug.4,5 Low solubility (especially in the gastric fluid) causes limited oral bioavailability of class II drugs in the Biopharmaceutical Classification System (including statins).6 Thus, the increased hydrophilicity results in pharmacokinetic parameter improvement.
Targeted drug delivery is another approach that holds promise to improve therapeutic outcomes by reducing side toxicity.7 As far as the liver plays a central role in cholesterol metabolism,8 this organ is considered the prime target for statin delivery. The liver consists mostly of hepatocytes. The asialoglycoprotein receptor (ASGPR) can be exploited to target liver cells since this receptor is abundantly present on hepatocyte membranes.9 The receptor belongs to the lectin group (C type) and is known to be responsible for maintaining the homeostasis of serum glycoproteins by receptor mediated endocytosis. The ASGPR exhibits high affinity for galactose (Gal) and N-acetylgalactosamine (GalNAc) residues. Using Gal or GalNAc derivatives as targeting moieties to the ASGPR significantly increases internalization of the drug conjugate in the hepatocytes.10,11 In addition, conjugation with carbohydrate residues increases hydrophilicity.12
To date, there is only one publication describing statin prodrugs with an ASGPR ligand.13 The two conjugates 1a and 2a (Fig. 1) reported in the article were designed as improved modifications of atorvastatin, the best-seller drug yet with low water solubility and bioavailability. Both compound 1a and compound 2a demonstrated enhanced cellular uptake compared to unmodified atorvastatin. The hydrolysis products of the conjugates inhibited the activity of HMG-CoA reductase.
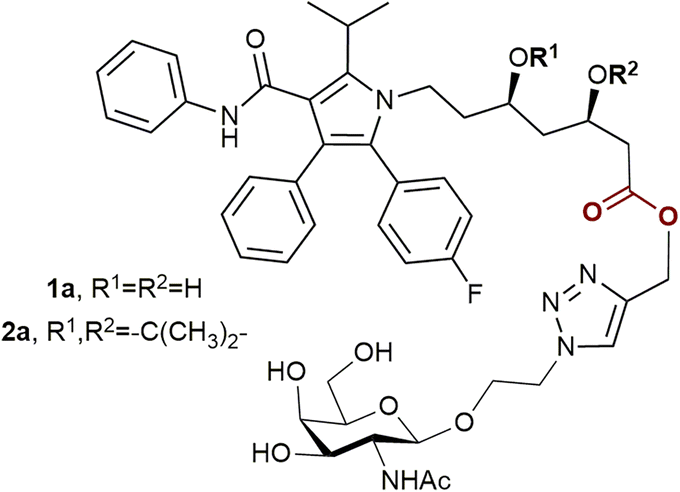 | ||
| Fig. 1 The previously reported conjugates of atorvastatin and ASGPR ligands.13 | ||
As statin therapy extends to a large population and lasts for a long period, oral administration is the most convenient and suitable for this class of drugs. In such a case, to provide all the benefits of targeted drug delivery, the intact therapeutic conjugates must traverse the harsh gastrointestinal environment. Taking into account the presence of acid-labile functional groups in 1a and 2b, it is doubtable that these conjugates will meet this condition. Considering all the facts listed above, in the present work 5 new conjugates of atorvastatin and N-acetylgalactosamine were prepared, as well as previously reported compounds 1a and 2a. Water solubility, affinity for the ASGPR and stability under physiological conditions of the obtained conjugates were studied.
Results and discussion
Design and synthesis
Two N-acetylgalactosamine derivatives A and B were used as structural blocks to produce atorvastatin conjugates (Scheme 1). Azides A and B were previously successfully applied by our group for the synthesis of several antitumor drug conjugates (doxorubicin,14 paclitaxel,15 and docetaxel16). Therefore, in this work the ligands were obtained using the earlier elaborated techniques. While compound A had already been applied by Y. Dong and coworkers to prepare atorvastatin conjugates,13 azide B was used for this purpose for the first time. Taking into consideration that galactosyl analogues with diverse substituents at the anomeric position exhibit different affinities for the ASGPR,17 the change from ligand A to ligand B may lead to increased binding to the receptor, as it was observed in the case of paclitaxel conjugates.15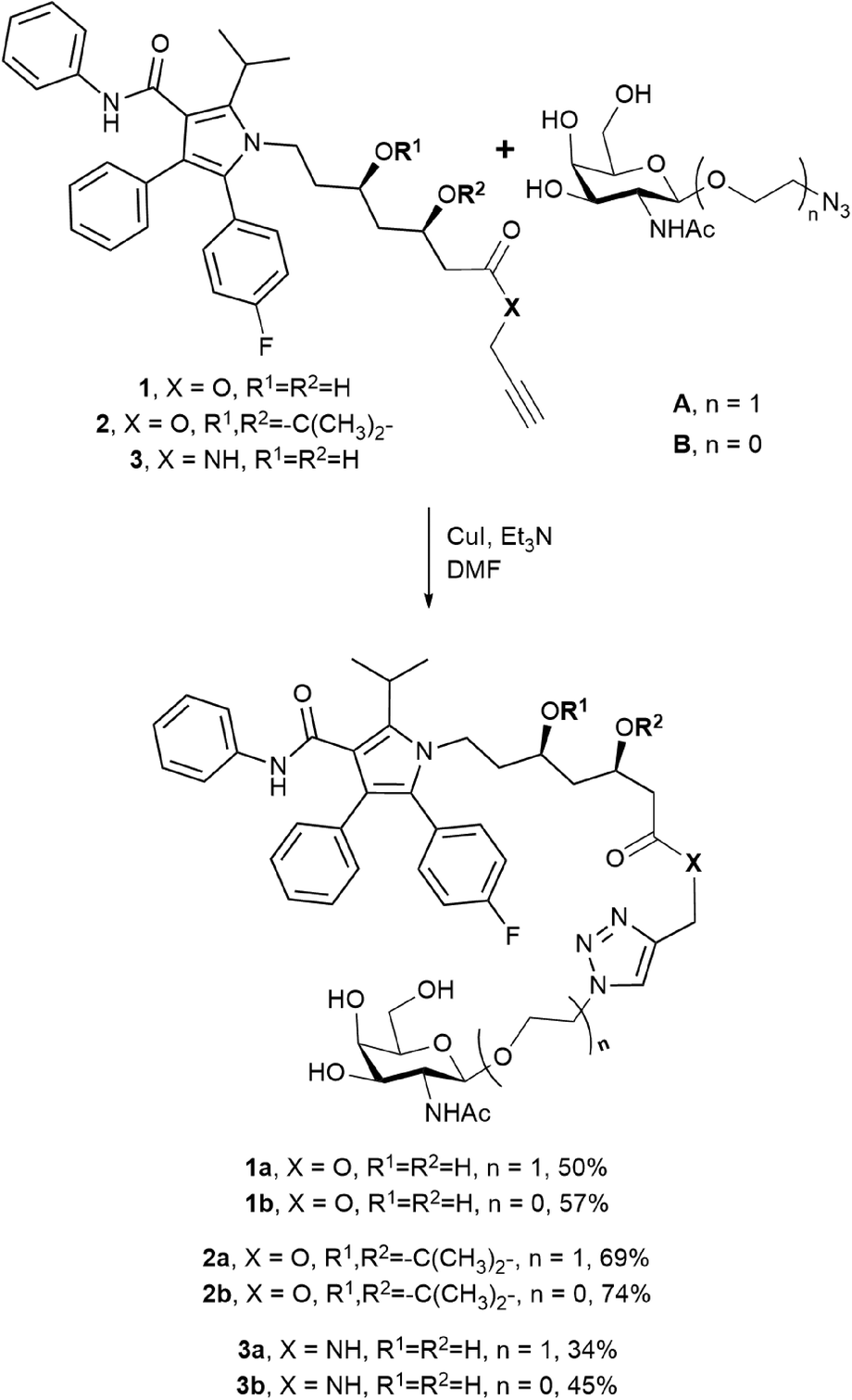 | ||
| Scheme 1 Synthesis of conjugates 1a–3b. | ||
From the chosen ASGPR ligands and three atorvastatin derivatives 1–3 (Scheme 1), we synthesised six conjugates 1a–3b by the copper(I)-catalysed azide–alkyne cycloaddition (CuAAC) with moderate to good yields. The obtained conjugates differed in the structure of the ligand and in the way the drug and the N-acetylgalactosamine derivative were connected. The type of bond, either amide or ester, should affect the ability of the resulting compounds to release the drug, and the structure of the targeting moiety should affect their affinity for the receptor. Compounds 1a and 2a were previously published, while the other conjugates were synthesised for the first time.
Water solubility
The concentrations of the substances in saturated solutions were determined through a UV-spectrophotometry technique. The molar solubility of the atorvastatin calcium salt was 0.11 ± 0.01 mM, whereas the molar solubilities of the obtained conjugates were from 0.19 mM to 0.54 mM with only a few exceptions (Table 1). The results indicate that the introduction of selected N-acetylgalactosamine fragments into the structure of atorvastatin generally has a favorable effect on the hydrophilicity of the drug.| Compound | Molar solubility (mmol L−1) | Clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P P |
K D (nmol L−1) |
|---|---|---|---|
| Atorvastatin | 0.11 ± 0.01 | 4.46 | 1.0 ± 0.5 × 103 |
| 1a | 0.32 ± 0.02 | 2.91 | 79 ± 3 |
| 1b | 0.20 ± 0.01 | 2.95 | 48 ± 12 |
| 2a | 0.05 ± 0.01 | 4.89 | 0.33 ± 0.03 |
| 2b | 0.03 ± 0.01 | 4.93 | 0.15 ± 0.03 |
| 3a | 0.54 ± 0.01 | 2.16 | 161 ± 6 |
| 3b | 0.45 ± 0.01 | 2.20 | 113 ± 5 |
The molar solubilities of the conjugates 3a and 3b with an amide bond were approximately 4- and 5-fold higher than that of the atorvastatin calcium salt. The best result was obtained for compound 3a. Esters 1a and 1b also showed enhanced water solubility, but the measured values were moderate. Derivatives 2a and 2b displayed the lowest molar solubilities among all atorvastatin conjugates and were less soluble than unmodified atorvastatin. It could be considered as the influence of the highly lipophilic acetal protecting group.
The obtained results were in line with logP values predicted with the help of CS ChemDraw Ultra (Cambridge Soft Corporation, Cambridge, MA, USA).
Affinity to the ASGPR
The binding affinity of the compounds to the human ASGPR was evaluated via the surface plasmon resonance spectroscopy technique. The receptor was immobilised on the surface of the CM5 sensor chip, as was reported previously.18,19 Results are presented as equilibrium dissociation constants (KD, Table 1). The obtained conjugates demonstrated greater affinity to the ASGPR compared to unmodified atorvastatin. Compounds 2a and 2b exhibited subnanomolar bindings to the receptor (KD = 0.33 and 0.15 nM, respectively) which are 4 orders of magnitude lower than those measured for atorvastatin itself and the native ASGPR ligand N-acetylgalactosamine (KD = 1.0 and 4.5 μM, respectively). We suggest that these results are associated with the nonspecific binding to the receptor. A previous report demonstrated that atorvastatin readily interacts with nonpolar amino acid residues.20 As was shown, compounds 2a and 2b were the most lipophilic among all conjugates.The equilibrium dissociation constants obtained for the remaining conjugates correspond with the molar solubility and clogP as well. Thus, more lipophilic atorvastatin esters 1a and 1b bound to the receptor with higher affinities than amides 3a and 3b did. As expected, azide B-derived conjugates exhibited slightly greater affinity to the receptor than those derived from azide A. Nevertheless, all of the obtained compounds demonstrated KD values in nanomolar and submicromolar ranges. Thus, the designed conjugates are potential prodrugs for targeted delivery of atorvastatin in hepatocytes via ASGPR-mediated endocytosis.
HMG-CoA reductase inhibition assay
The ability of the atorvastatin conjugates to inhibit 3-hydroxy-3-methylglutaryl coenzyme A reductase activity was assessed using an HMG-CoA reductase assay kit. Previous studies reported that atorvastatin esters were not able to cause a biological effect until free atorvastatin was released.13,21 Nevertheless, in our case esters 1b and 2a demonstrated slight inhibition of the HMG-CoA reductase (Fig. 2). One could account these results for partial hydrolysis of the investigated compounds in neutral aqueous media, which occurs in the absence of any hydrolases. Taking into consideration that the conjugates must be stable enough to enter the target cell before degradation, the hydrolysis kinetics of the synthesised esters was thoroughly studied.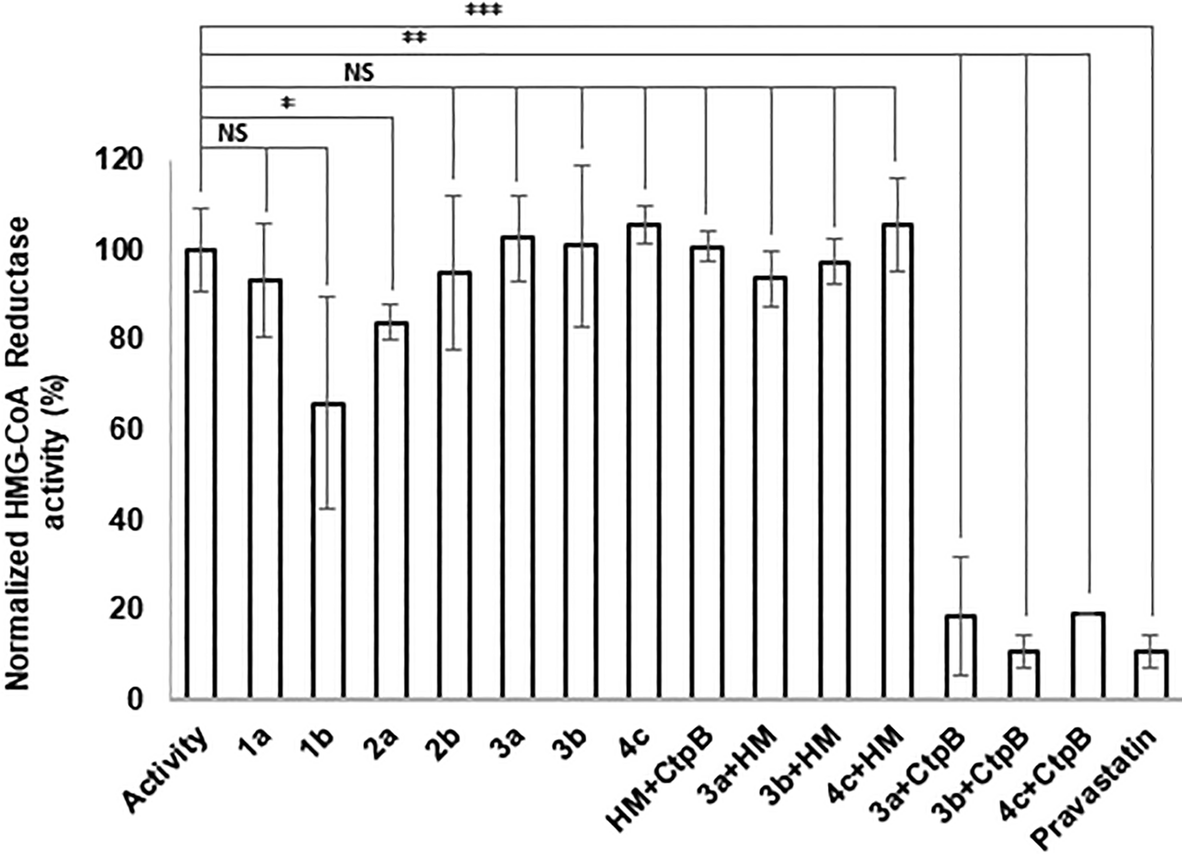 | ||
| Fig. 2 Inhibition of HMG-CoA reductase activity by pravastatin, conjugates and the hydrolysis. HM, hydrolysis media, CtpB, cathepsin B (triplicate, *, p < 0.05, **, p < 0.005, ***, p < 0.001, NS, no significance, t test, double tailed). | ||
The amide conjugates 3a and 3b showed no inhibition of the HMG-CoA reductase (Fig. 2) due to their stability in water-based solution at neutral pH, which is a desirable trait of the conjugate to reduce side toxicity. However, the prodrug should be hydrolysed by intracellular enzymes for the therapeutic effect to occur. To ensure that the designed amide conjugates are able to release functional atorvastatin compounds 3a and 3b were treated with cathepsin B, a lysosomal cysteine protease.
Cathepsin B exhibits both exopeptidase and endopeptidase activities, cleaving external and internal peptide bonds.22,23 Hydrolysis was carried out at pH 3.6 to optimise the experiment. After incubation with cathepsin B, the hydrolysis products of compounds 3a and 3b inhibited HMG-CoA reductase as well as pravastatin did (the statin recommended by the manufacturer for use as a reference inhibitor). Neither the reaction medium nor cathepsin B itself inhibited the HMG-CoA reductase. This indicates that the observed effect is due to the presence of hydrolyzed atorvastatin conjugates. The conjugates incubated in the reaction medium without cathepsin B did not exhibit inhibitory activity either. Thus, the amide conjugates are stable under acidic conditions simulating the gastric fluid. Free atorvastatin is insoluble at pH values lower than 4 and tend to crystallize at low pH.24 The main approach to enhance its bioavailability is to increase its solubility in water-based solutions, as was demonstrated in previous studies.25,26 In the current manuscript, we utilize the solubility-enhancing approach combined with targeted delivery. The result suggests that the amide conjugates could reach target cells and release atorvastatin intracellularly.
Hydrolysis assays
In order to predict the behavior of the conjugates under physiological conditions, hydrolysis of compounds 1a–1b and 3a–3b in aqueous solutions was studied. Taking into consideration that enhanced water solubility is one of the key goals of the work, lipophilic conjugates 2a and 2b were not tested. Water-based buffer solutions were used to simulate the endosome environment (pH 5.0) and blood plasma (pH 7.4). Amide-derived conjugates 3a and 3b exhibited high stability under the selected conditions: HPLC-MS analysis did not reveal significant hydrolysis in aqueous media even after 24 hours of incubation. The ester-based compounds 1a and 1b were stable in acetate buffer (pH 5.0); while at pH 7.4, the concentrations of the conjugates decreased exponentially. At the same time, the concentration of atorvastatin increased linearly alongside the conjugate degradation. Atorvastatin conjugates underwent a two-step hydrolysis process that includes lactone formation as was demonstrated by the previous report.13 Therefore, the linearity of the atorvastatin release corresponds to the pseudo zero-order intramolecular deesterification.In addition, compounds 1a and 1b were quickly transformed into atorvastatin when incubated with porcine liver esterase and non-specific enzyme blend Pronase®. The rates of the drug release during enzymatic hydrolysis were significantly higher compared to those measured in buffer solutions. Conjugates 3a and 3b turned out to be stable under these conditions.
Thus, compounds 1a and 1b seem to be too unstable for further development as orally administered drug substances, as they quickly release atorvastatin even in a neutral medium and are especially labile in the presence of enzymes. Conjugates 3a and 3b demonstrated high stability implying that amide-based atorvastatin derivatives are unlikely to degrade in a harsh gastrointestinal environment prior to absorption.
Alternative approach to the design of atorvastatin amide conjugates
The triazole-derived conjugates 1a–3b were obtained through the azide–alkyne cycloaddition which requires copper(I)-catalysis. Copper is essential, but a potentially toxic element,27 and purification from heavy metals during synthesis remains a matter of great concern. In addition, triazoles produced via the CuAAC reaction were recently reported as ester bond isosteres, producing an inhibitory effect on hydrolases and proteases.28,29 This effect was also demonstrated in glycoside derivatives with a triazole linkage.30 Hence, we propose an alternative synthetic approach for amide-based atorvastatin conjugates.Compound 4c was synthesised by coupling atorvastatin NHS ester 4 and N-acetylgalactosamine derivative C (Scheme 2). The synthetic procedure involved the formation of intermediate product 4′c. The protecting groups were further removed to yield conjugate 4c.
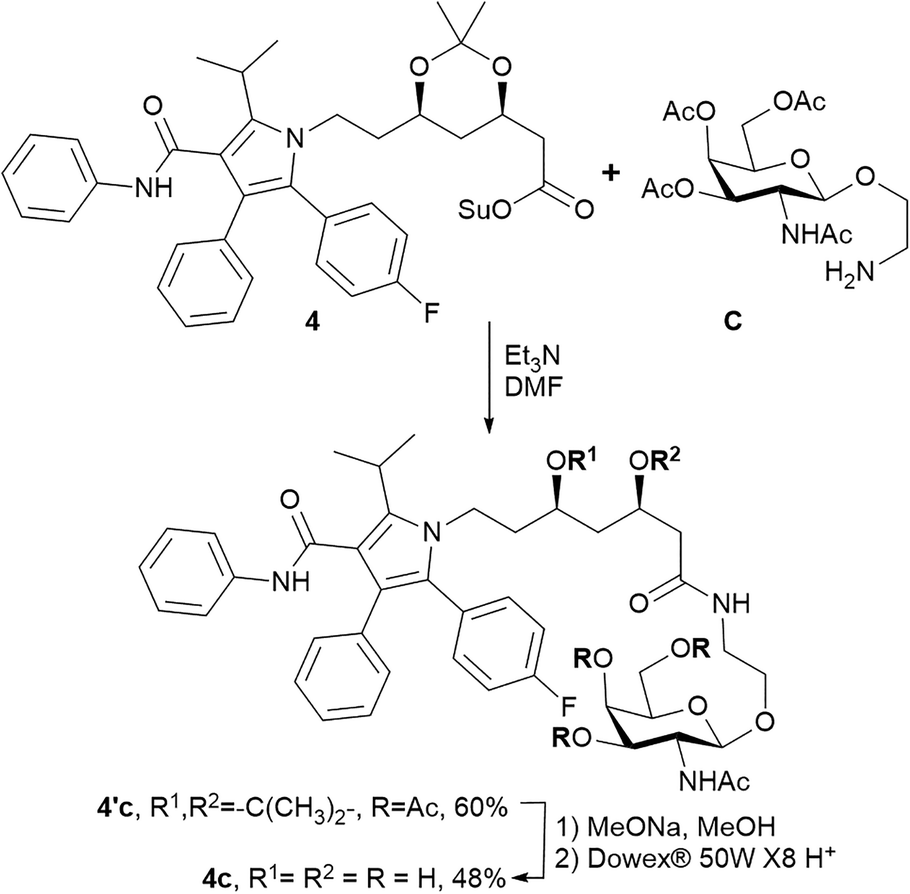 | ||
| Scheme 2 Synthesis of compound 4c. | ||
The molar solubility of the obtained conjugate 4c was approximately 4-fold higher than that of atorvastatin (0.43 ± 0.02 mmol L−1 and 0.11 ± 0.01 mmol L−1, respectively). Compound 4c exhibited high binding affinity to the ASGPR (KD = 69 ± 7 nmol L−1) and good stability in water-based buffer solutions. The release of free atorvastatin after incubation of 4c with the lysosomal protease was demonstrated. Thus, conjugate 4c is a viable alternative to triazole-containing amides 3a and 3b. A previous report suggested that metabolic activation of prodrugs depends on the conjugated side chain, revealing underlying enzyme specificity.31 Thus, conjugate 4c could be hydrolysed under different conditions in further more profound evaluations.
Conclusions
Herein we report on the synthesis and evaluation of the ASGPR-targeted atorvastatin conjugates. Several N-acetylgalactosamine derivatives were used as targeting moieties. The conjugation of the selected carbohydrate fragments to atorvastatin generally produced a favorable effect on the hydrophilicity of the drug. All of the synthesised conjugates demonstrated greater affinity to the ASGPR compared to unmodified atorvastatin.To date, there are few publications dealing with atorvastatin amides. Here we described two structural types of atorvastatin conjugates with an amide bond. The amide-based conjugates exhibited higher water solubility and stability for hydrolysis than ester-based atorvastatin conjugates. The release of atorvastatin after incubation of conjugates 3a, 3b and 4c in the presence of a lysosomal protease was demonstrated. The results suggest that the designed amide-based compounds are potential orally administered liver-targeted prodrugs of atorvastatin.
Experimental
Materials and methods
1H, 19F and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer with operating frequencies of 400 MHz for 1H and 100 MHz for 13C and 19F NMR. ESI-HRMS spectra were measured with a TripleTOF 5600+ (AB Sciex, Canada). The isolation of the conjugates by reverse phase chromatography was carried out using a puriFlash 4250 (Interchim, France). The silica gel used for column chromatography was MN Kieselgel 60 0.04–0.063 mm/230–400 mesh ASTM. TLC analysis was performed on Merck TLC silica gel 60 F254 plates. Organic chemicals with purity no less than 95% were used as purchased. Solvents were purified routinely and freshly distilled prior to use. Compounds A, B, and C were synthesised according to the previously reported methods.15,32,33 Compounds 1–4 were prepared through the procedures described in the ESI.†:MeOH 50:1 → 20:1, v:v). Pure compound 4′c was obtained as a white solid in 60% yield (62 mg). 1H NMR (400 MHz, CDCl3) δ ppm: 1.06 (q, J = 11.8 Hz, 1H), 1.25–1.42 (m, 7H), 1.53 (d, J = 7.2 Hz, 6H), 1.64–1.69 (m, 2H), 1.97 (s, 3H), 2.02 (s, 3H), 2.05 (s,3H), 2.16 (s,3H), 2.23 (dd, J = 14.8, 4.7 Hz, 1H), 2.36 (dd, J = 14.8, 7.5 Hz, 1H), 3.26–3.34 (m, 1H), 3.54–3.71 (m, 4H), 3.78–3.92 (m, 3H), 4.05–4.22 (m, 5H), 4.58 (d, J = 8.4 Hz, 1H), 5.11 (dd, J = 11.3, 3.3 Hz, 1H), 5.36 (d, J = 3.0 Hz, 1H), 5.69 (d, J = 8.7 Hz, 1H), 6.52 (t, J = 5.3 Hz, 1H), 6.89 (s, 1H), 6.96–7.04 (m, 3H), 7.07 (d, J = 7.8 Hz, 2H), 7.13–7.23 (m, 9H). 13C NMR (100 MHz, CDCl3) δ ppm: 19.7, 20.6, 21.5, 21.7, 23.4, 26.0, 29.9, 35.9, 37.9, 38.7, 40.7, 43.0, 50.8, 61.4, 65.9, 66.3, 66.5, 68.3, 70.2, 70.7, 98.8, 101.3, 115.2, 115.3 (d, 2JCF = 21.4 Hz), 119.6, 121.7, 123.5, 126.5, 128.1 (d, 4JCF = 3.2 Hz), 128.3, 128.6, 128.7, 130.4, 133.1 (d, 3JCF = 8.1 Hz), 134.5, 138.2, 141.3, 162.2 (d, 1JCF = 247.5 Hz), 164.8, 170.1, 170.4, 170.6, 170.7. 19F NMR (100 MHz, CDCl3) δ ppm: −113.65–113.58 (m). HRMS (ESI) m/z calcd for C52H64FN4O13 [M + H]+m/z 971.4448, found: 971.4464.
Quantification of water solubility
The UV-spectra were obtained on a HITACHI U-2900 spectrophotometer. To determine the solubility of the obtained compounds in water, standard curves of absorption intensity at 290 nm against concentration of atorvastatin (in a calcium salt form) and each of the conjugates' solutions were constructed (Table S1, ESI†). Excess amount of the tested substance was added to 1 ml of water and shaken strongly for 15 min. After filtration or centrifugation, 50 μL of the saturated solution was added to 1 mL of methanol and the absorption intensity at 290 nm was measured. The concentration of the substances in their saturated solutions was further determined using standard curves.Surface plasmon resonance
The experiment was performed on a Biacore X100 machine (Biacore AB, Uppsala, Sweden) using a CM5 chip. Data were analysed using BIAevaluation 3.0 software. The ASGPR was immobilised according to the default amine-coupling protocol provided by the manufacturer. The ligands were dissolved in DMSO followed by serial dilution in a running buffer (150 mM NaCl, 50 mM CaCl2, 50 mM Tris, pH 7.4) up to 5 × 10−11 M. The samples were injected at a flow rate 20 μL min−1 at 25 °C for 180 s followed by 60 s dissociation. The regeneration of the sensor chip was performed by injection of 20 μL of 20 mM EDTA. All solutions were filtered and deoxygenated. The KD values were evaluated using the 1:1 Langmuir association model.
Hydrolysis assays
A sample of the tested compound was dissolved in DMSO (C = 10 mg mL−1). After that 25 μL of the resulting solution were diluted with 975 μL of the appropriate buffer solution (pH 7.4: 0.2 M Tris-HCl or 0.2 M PBS; pH 5.0: 0.2 M AcONa–AcOH). In the case of enzymatic hydrolysis 925 μL of PBS was used and 50 μL of the solution of the appropriate enzyme (esterase from porcine liver or pronase from Streptomyces griseus, Merck) in distilled water (C = 10 mg mL−1) was added. The mixtures were incubated at 37 °C. Aliquots for analysis were prepared by diluting 10 μl of the mixture with 990 μl of cold methanol. The aliquots were taken at time intervals of 0, 0.5, 1, 2, 4, 8, and 24 hours and were further stored at −20 °C. Immediately before analysis, the samples were thawed, centrifuged at 13000g, and the collected supernatant was examined by HPLC-MS.
Quantification of HMG-CoA reductase activity
The activity of the HMG-CoA reductase was measured through an HMG-CoA reductase assay kit following the standard procedure. A solution of pravastatin (0.1 mM) was included in the reagent kit; the conjugates' solutions (0.1 mM) were prepared by diluting 10 μL of stock DMSO solutions of the compounds (0.01 M) with 990 μL of water. Experiments were conducted in a 96-well plate and 2 μL of the conjugates' solutions were added. After adding the reagents in required order, the absorption at 340 nm was immediately collected every 20 s for up to 10 min. Measurements were performed in triplicate.The activity of the HMG-CoA reductase was calculated according to the following equation:

Cathepsin B hydrolysis
Conjugate cleavage by recombinant cathepsin B was analysed using a modified method from previously published work.34 Stock solutions of conjugates in 50/50 (v/v) DMSO/H2O (12 μL, 5 mM) were added to the 1.488 mL cathepsin B containing buffer (0.1 M sodium acetate buffer pH 3.6, 100 mM NaCl, 1 mM EDTA, and 0.6% DMSO) pre-warmed at 37 °C at the final concentrations of 30 nM (cathepsin B) and 40 μM (conjugate). After 24 hours of hydrolysis, the reaction mixture (5 μL) was used to measure the inhibitory activity. Both the reaction buffer with cathepsin B (without any conjugate) and the conjugates incubated in the reaction buffer without a protease were used as controls.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the RSF grant no. 20-73-00219. The SPR spectroscopy experiment (receptor immobilization) was partially funded by the RSF grant no. 20-63-46029 and MSU Program of development.References
- E. A. Stein and F. J. Raal, Endocrinol. Metab. Clin. North Am., 2014, 43, 1007–1033, DOI:10.1016/j.ecl.2014.08.008.
- A. H. Rageh, N. N. Atia and H. M. Abdel-Rahman, J. Pharm. Biomed. Anal., 2017, 142, 7–14, DOI:10.1016/j.jpba.2017.04.037.
- B. D. Roth, T. M. A. Bocan, C. J. Blankley, A. W. Chucholowski, P. L. Creger, M. W. Creswell, E. Ferguson, R. S. Newton and P. O'Brien, J. Med. Chem., 1991, 34, 463–466, DOI:10.1021/jm00105a071.
- L. D. Bratton, B. Auerbach, C. Choi, L. Dillon, J. C. Hanselman, S. D. Larsen, G. Lu, K. Olsen, J. A. Pfefferkorn, A. Robertson, C. Sekerke, B. K. Trivedi and P. C. Unangst, Bioorg. Med. Chem., 2007, 15, 5576–5589, DOI:10.1016/j.bmc.2007.05.031.
- W. K. C. Park, R. M. Kennedy, S. D. Larsen, S. Miller, B. D. Roth, Y. Song, B. A. Steinbaugh, K. Sun, B. D. Tait, M. C. Kowala, B. K. Trivedi, B. Auerbach, V. Askew, L. Dillon, J. C. Hanselman, Z. Lin, G. H. Lu, A. Robertson and C. Sekerke, Bioorg. Med. Chem. Lett., 2008, 18, 1151–1156, DOI:10.1016/j.bmcl.2007.11.124.
- M. A. Shaker, J. Drug Delivery Sci. Technol., 2018, 43, 178–184, DOI:10.1016/j.jddst.2017.10.003.
- B. S. Pattni and V. P. Torchilin, Targeted Drug Delivery Systems: Strategies and Challenges, in Targeted Drug Delivery: Concepts and Design, ed. P. V. Devarajan and S. Jain, Springer International Publishing, 2015, pp. 3–40, DOI:10.1007/978-3-319-11355-5.
- K. Nemes, F. Åberg, H. Gylling and H. Isoniemi, World J. Hepatol., 2016, 8, 924, DOI:10.4254/wjh.v8.i22.924.
- A. A. D'Souza and P. V. Devarajan, J. Controlled Release, 2015, 203, 126–139, DOI:10.1016/j.jconrel.2015.02.022.
- R. A. Petrov, S. R. Mefedova, E. Y. Yamansarov, S. Y. Maklakova, D. A. Grishin, E. V. Lopatukhina, O. Y. Burenina, A. V. Lopukhov, S. V. Kovalev, Y. V. Timchenko, E. E. Ondar, Y. A. Ivanenkov, S. A. Evteev, A. N. Vaneev, R. V. Timoshenko, N. L. Klyachko, A. S. Erofeev, P. V. Gorelkin, E. K. Beloglazkina and A. G. Majouga, Mol. Pharmaceutics, 2021, 18, 461–468, DOI:10.1021/acs.molpharmaceut.0c00980.
- C. A. Sanhueza, M. M. Baksh, B. Thuma, M. D. Roy, S. Dutta, C. Préville, B. A. Chrunyk, K. Beaumont, R. Dullea, M. Ammirati, S. Liu, D. Gebhard, J. E. Finley, C. T. Salatto, A. King-Ahmad, I. Stock, K. Atkinson, B. Reidich, W. Lin, R. Kumar, M. Tu, E. Menhaji-Klotz, D. A. Price, S. Liras, M. G. Finn and V. Mascitti, J. Am. Chem. Soc., 2017, 139, 3528–3536, DOI:10.1021/jacs.6b12964.
- D. Melisi, A. Curcio, E. Luongo, E. Morelli and M. Grazia Rimoli, Curr. Top. Med. Chem., 2011, 11, 2288–2298, DOI:10.2174/156802611797183258.
- Y. Zhang, X. Zhang, C. Zeng, B. Li, C. Zhang, W. Li, X. Hou and Y. Dong, Bioorg. Med. Chem., 2019, 27, 2187–2191, DOI:10.1016/j.bmc.2019.04.019.
- Y. A. Ivanenkov, A. G. Majouga, R. A. Petrov, S. A. Petrov, S. V. Kovalev, S. Y. Maklakova, E. Y. Yamansarov, I. V. Saltykova, E. V. Deyneka, G. I. Filkov, V. E. Kotelianski, T. S. Zatsepin and E. K. Beloglazkina, Bioorg. Med. Chem. Lett., 2018, 28, 503–508, DOI:10.1016/j.bmcl.2017.12.004.
- R. A. Petrov, S. Yu. Maklakova, Y. A. Ivanenkov, S. A. Petrov, O. V. Sergeeva, E. Y. Yamansarov, I. V. Saltykova, I. I. Kireev, I. B. Alieva, E. V. Deyneka, A. A. Sofronova, A. V. Aladinskaia, A. V. Trofimenko, R. S. Yamidanov, S. V. Kovalev, V. E. Kotelianski, T. S. Zatsepin, E. K. Beloglazkina and A. G. Majouga, Bioorg. Med. Chem. Lett., 2018, 28, 382–387, DOI:10.1016/j.bmcl.2017.12.032.
- R. A. Petrov, S. R. Mefedova, E. Yu. Yamansarov, S. Yu. Maklakova, D. A. Grishin, E. V. Lopatukhina, O. Y. Burenina, A. V. Lopukhov, S. V. Kovalev, Y. V. Timchenko, E. E. Ondar, Y. A. Ivanenkov, S. A. Evteev, A. N. Vaneev, R. V. Timoshenko, N. L. Klyachko, A. S. Erofeev, P. V. Gorelkin, E. K. Beloglazkina and A. G. Majouga, Mol. Pharmaceutics, 2021, 18, 461–468, DOI:10.1021/acs.molpharmaceut.0c00980.
- S. K. Mamidyala, S. Dutta, B. A. Chrunyk, C. Préville, H. Wang, J. M. Withka, A. McColl, T. A. Subashi, S. J. Hawrylik, M. C. Griffor, S. Kim, J. A. Pfefferkorn, D. A. Price, E. Menhaji-Klotz, V. Mascitti and M. G. Finn, J. Am. Chem. Soc., 2012, 134, 1978–1981, DOI:10.1021/ja2104679.
- G. S. Reshitko, E. Yu. Yamansarov, S. A. Evteev, E. V. Lopatukhina, D. O. Shkil', I. V. Saltykova, A. V. Lopukhov, S. V. Kovalev, A. N. Lobov, I. V. Kislyakov, O. Y. Burenina, N. L. Klyachko, A. S. Garanina, O. A. Dontsova, Y. A. Ivanenkov, A. S. Erofeev, P. V. Gorelkin, E. K. Beloglazkina and A. G. Majouga, Bioconjugate Chem., 2020, 31, 1313–1319, DOI:10.1021/acs.bioconjchem.0c00202.
- E. Yu. Yamansarov, E. V. Lopatukhina, S. A. Evteev, D. A. Skvortsov, A. V. Lopukhov, S. V. Kovalev, A. N. Vaneev, D. O. Shkil', R. A. Akasov, A. N. Lobov, V. A. Naumenko, E. N. Pavlova, O. O. Ryabaya, O. Yu. Burenina, Y. A. Ivanenkov, N. L. Klyachko, A. S. Erofeev, P. V. Gorelkin, E. K. Beloglazkina and A. G. Majouga, Bioconjugate Chem., 2021, 32, 763–781, DOI:10.1021/acs.bioconjchem.1c00042.
- Q. Wang, C. R. Huang, M. Jiang, Y. Y. Zhu, J. Wang, J. Chen and J. H. Shi, Spectrochim. Acta, Part A, 2016, 156, 155–163, DOI:10.1016/J.SAA.2015.12.003.
- Y. Zhang, X. Zhang, C. Zeng, B. Li, C. Zhang, W. Li, X. Hou and Y. Dong, Bioorg. Med. Chem., 2019, 27, 2187–2191, DOI:10.1016/j.bmc.2019.04.019.
- D. Musil, D. Zucic, D. Turk, R. A. Engh, I. Mayr, R. Huber, T. Popovic, V. Turk, T. Towatari and N. Katunuma, EMBO J., 1991, 10, 2321–2330, DOI:10.1002/j.1460-2075.1991.tb07771.x.
- D. Keppler and B. F. Sloane, Enzyme Protein, 1996, 49, 94–105, DOI:10.1159/000468619.
- Y. Y. Lau, H. Okochi, Y. Huang and L. Z. Benet, Drug Metab. Dispos., 2006, 34, 1175–1181, DOI:10.1124/dmd.105.009076.
- A. Choudhary, A. C. Rana, G. Aggarwal, V. Kumar and F. Zakir, Acta Pharm. Sin. B, 2012, 2, 421–428, DOI:10.1016/j.apsb.2012.05.002.
- M. S. Rodde, G. T. Divase, T. B. Devkar and A. R. Tekade, BioMed Res. Int., 2014, 2014, 463895, DOI:10.1155/2014/463895.
- L. M. Gaetke and C. K. Chow, Toxicology, 2003, 189, 147–163, DOI:10.1016/S0300-483X(03)00159-8.
- E. Bonandi, M. S. Christodoulou, G. Fumagalli, D. Perdicchia, G. Rastelli and D. Passarella, Drug Discovery Today, 2017, 22, 1572–1581, DOI:10.1016/j.drudis.2017.05.014.
- A. Adibekian, B. R. Martin, C. Wang, K.-L. Hsu, D. A. Bachovchin, S. Niessen, H. Hoover and B. F. Cravatt, Nat. Chem. Biol., 2011, 7, 469–478, DOI:10.1038/nchembio.579.
- L. Li, K.-C. Chang, Y. Zhou, B. Shieh, J. Ponder, A. D. Abraham, H. Ali, A. Snow, J. M. Petrash and D. V. LaBarbera, J. Med. Chem., 2014, 57, 71–77, DOI:10.1021/jm401311d.
- K. Mizoi, M. Takahashi, S. Sakai, T. Ogihara, M. Haba and M. Hosokawa, Xenobiotica, 2020, 50, 261–269, DOI:10.1080/00498254.2019.16250839.
- M. Manoharan, K. G. Rajeev, J. Nair and M. Maier, US Pat., US2016/0199513A1, 2016 Search PubMed.
- S. B. Salunke, N. S. Babu and C.-T. Chen, Chem. Commun., 2011, 47, 10440, 10.1039/c1cc13370e.
- S. Boinapally, H.-H. Ahn, B. Cheng, M. Brummet, H. Nam, K. L. Gabrielson, S. R. Banerjee, I. Minn and M. G. Pomper, Sci. Rep., 2021, 11, 7114, DOI:10.1038/s41598-021-86551-1.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, assays details, and NMR spectra. See DOI: https://doi.org/10.1039/d2md00119e |
| This journal is © The Royal Society of Chemistry 2023 |