
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectron/hole piezocatalysis in chemical reactions
Shadi
Asgari
ab,
Ghodsi
Mohammadi Ziarani
 *a,
Alireza
Badiei
*b and
Siavash
Iravani
*c
*a,
Alireza
Badiei
*b and
Siavash
Iravani
*c
aDepartment of Organic Chemistry, Faculty of Chemistry, Alzahra University, Tehran, P.O. Box 1993893973, Iran. E-mail: gmohammadi@alzahra.ac.ir
bSchool of Chemistry, College of Science, University of Tehran, Tehran, Iran. E-mail: abadiei@ut.ac.ir
cIndependent Researcher, W Nazar ST, Boostan Ave, Isfahan, Iran. E-mail: siavashira@gmail.com
First published on 1st November 2023
Abstract
The emergence of piezocatalysts has led to advancements in catalyst design through the development of new technologies. Electron/hole piezocatalysis, a novel form of catalysis, utilizes piezoelectric materials that are stimulated by mechanical energy. This stimulation generates electrons and holes, which prove highly effective in promoting inefficient reactions, designing new reactions, and facilitating challenging chemical reactions and transformations. This review provides a concise historical account of electron/hole catalysis and the concept of piezoelectricity. Additionally, it explores various types of piezocatalysts, including piezoceramics (both lead-free and lead-based), piezopolymers, and piezocomposites. Notably, lead-free piezoceramics such as barium titanate and zinc oxide, as well as piezopolymers like polyvinylidene fluoride in β-phase and polyacrylonitrile in planar zigzag conformation, have gained significant recognition. The piezoelectric coefficient of polymers can be enhanced by adjusting the processing parameters and employing techniques such as blending, filler addition, stretching, and composite formation with piezo/non-piezo additives. Furthermore, piezoelectricity has been observed in unique materials such as ionic liquids, metal–organic frameworks, graphitic carbon nitride, black phosphorus, and MXenes. This review also presents recent research findings on the utilization of piezoelectric materials in various applications, including water splitting, water remediation, organic synthesis, and polymerization/crosslinking.
1. Introduction
Electron/hole catalysis is divided into different main categories: (1) piezocatalysis,1 (2) photocatalysis,2 (3) chemocatalysis,3 (4) electrocatalysis,4 and (5) pyrocatalysis,5 where electrons and holes are generated through various types of external stimuli including mechanical energy, light source, chemical redox agents, electrical current, and thermal energy, respectively, to promote chemical reactions. In all of these states, electron transfer plays the main role in encouraging chemical transformations.3,4 Amongst the different categories of electron/hole catalysis, electron/hole piezocatalysis is a recent concept of catalysis relying on piezoelectric materials. The origin of piezoelectricity is a non-centrosymmetric distribution of positive and negative electric charges in a unit cell of piezoelectric material.6 Upon mechanical force, a piezoelectric material will be polarized temporarily, resulting in separated electrons and holes. Electron catalysis comprises the reduction of substrates by inserting electrons into reactants, while holes oxidize substrates by removing electrons. After redox initiation, radical-mediated chain propagation will be followed to produce the final products.3,7 More separation of electrons and holes results in more piezoelectric responses. To explain accurately, a piezoelectric material remains in equilibrium with occupiable electronic states when no external mechanical force is applied; when subjected to mechanical force, the polarization of the material changes, resulting in the development of an electric field across it. This electric field causes the reorientation of electric charge carriers towards different ends of the material. These separated carriers then migrate to the material surface and engage in oxidation and reduction reactions, generating active species for chemical processes. Once the accumulated electric charges on the material surface balance out the built-in electric field, the system returns to its equilibrium state.6,8 This review provides an overview of different piezoelectric materials and the latest studies regarding piezoelectrically mediated organic synthesis, polymerization/crosslinking, water splitting, and water remediation. The basic concept of piezocatalysis in piezoelectrically mediated reactions is illustrated in Fig. 1.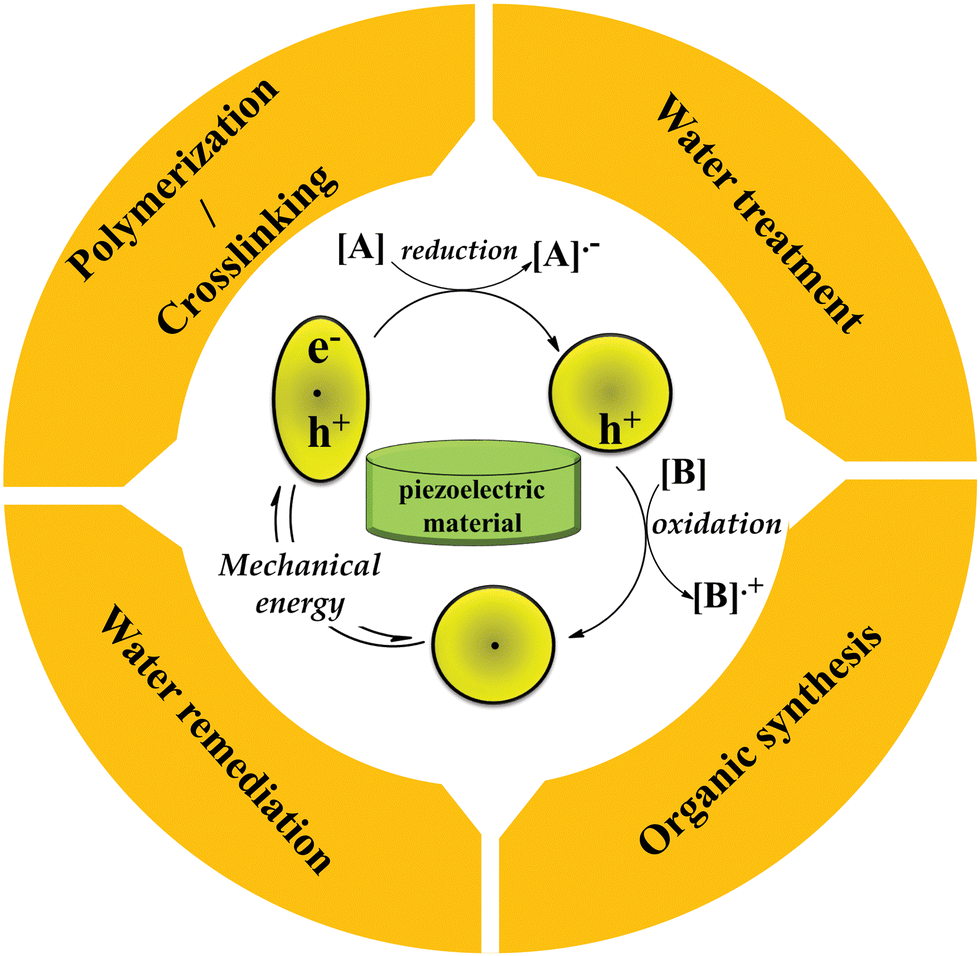 | ||
| Fig. 1 The basic principle of electron/hole piezocatalysis in piezoelectrically mediated reactions (adapted from ref. 9 with permission from John Wiley and Sons). | ||
2. Piezoelectric materials
There are four main categories of piezoelectric materials: single crystals, ceramics, polymers, and composites.10 Some less common types of piezoelectric materials are also explained subsequently. Apart from the most common natural piezoelectric crystals (such as Rochelle salt (potassium sodium tartrate), cane sugar, quartz, tourmaline, and topaz11,12), lithium niobate (LiNbO3), lead magnesium niobate–lead titanate (PMN–PT), and lead zinc niobate–lead titanate (PZN–PT) are known as the piezoelectric single crystals.13 Piezoceramics are polycrystalline materials consisting of abundant single crystals with similar chemical composition and different orientations.10 Piezoceramics display large piezoelectric coefficients but their structure and mechanical features restrict their applications.14 Piezoceramics are classified into lead-based and lead-free piezoceramics, as described below.2.1. Lead-based piezoceramics
The most common lead-based piezoceramic is lead zirconate titanate (PZT), which is a polycrystalline ferroelectric ceramic with a perovskite crystal structure.15 Another is La-modified lead zirconate titanate (PLZT) which can be produced by planetary ball milling of PbO and titanium dioxide (TiO2).16 Due to the toxicity of lead, there is a pressing need to develop environmentally benign lead-free piezoelectric materials.17,182.2. Lead-free piezoceramics
The lead-free piezoceramics have attracted much attention because of their excellent features of eco-friendliness and high inherent piezoelectric coefficients. These types of piezoceramics are divided into (1) perovskite-type ceramics such as barium titanate (BaTiO3),19 barium zirconium titanate (Ba(Ti/Zr)O3, BZT) (e.g., Ba(Ti0.95Zr0.05)O3, Ba(Ti0.9Zr0.10)O3, and Ba(Ti0.85Zr0.15)O320,21), bismuth sodium titanate ((Bi0.5Na0.5)TiO3, BNT),22 Bi0.5(Na0.5,K0.5)TiO3,23 Bi(Na,K,Li)TiO3,24 potassium niobate (KNbO3),25 and sodium tantalate (NaTaO3)26 and (2) non-perovskite type ceramics such as bismuth layer-structured ferroelectrics (BLSF) and tungsten–bronze type ferroelectrics.17 Perovskite-type ceramics display a relatively large piezoelectric coefficient but their poling is difficult.27 The most famous of the perovskite-type ceramics is BaTiO3 which shows the maximum piezoelectric coefficient in the crystal phases of tetragonal and orthorhombic.28 BaTiO3 nanocubes can also show piezoelectric properties when their morphology and structure are controlled well.29 The BaTiO3 piezoelectric properties can be further promoted by doping and forming solid solutions, or controlling the structural features by changing the processing parameters. Doping of calcium (Ca), zirconium (Zr), strontium (Sr), and samarium (Sm) in BaTiO3 increases its piezoelectric activity.30,31 The solid solution of BaTiO3–BaZrO3 is also of great interest due to its dielectric response which can be adjusted by their composition.32 The other most common lead-free piezoceramic is zinc oxide (ZnO), which is crystallized in a non-centrosymmetric wurtzite structure.33 Lanthanum substituted BaZr0.1Ti0.9O3 (Ba1−3x/2LaxZr0.1Ti0.9O3 with x = 0.01–0.05) is also known as a lead-free piezoceramic.34 Niobium-based35 and bismuth-based36 piezoceramics are also two types of effective lead-free piezoceramics but their high price and processing challenges limit their large-scale production.37 BiFeO3 is an example of bismuth-based lead-free piezoceramics, showing large spontaneous polarization and high piezoelectric coefficient at room temperature.38,39 It can be combined with BaTiO3 to make a binary BiFeO3–BaTiO3 ceramic to overcome the limitations of BiFeO3 such as weak charge mobility and a mild piezoelectric effect.19,40 A binary BiFeO3–BaTiO3 ceramic, 0.67BiFeO3–0.33BaTiO3, was prepared by Ferrero et al.41 in 2023 through two different ways of (1) mechanochemical activation of all the constituent oxides and carbonates for BiFeO3 and BaTiO3 (synthesis without BT seeds) and (2) mechanochemical activation of a combination of the constituent oxides for BiFeO3 with the previously formed BaTiO3 particles (synthesis with BT seeds). Pb2BO3X (X = Cl, Br, I) ceramics reported by Tang et al.42 are also known as lead-free piezoceramics. Indeed, a combination of the halogen atoms and metal atoms with a borate makes it a better candidate for constructing non-centrosymmetric materials. Other lead-free piezoceramics worthy of mention are Ba0.7Sr0.3TiO3,43 molybdenum disulfide (MoS2),44 molybdenum diselenide (MoSe2),45 and lithium tantalate (LiTaO3).462.3. Piezopolymers
Although piezoceramics have more piezoelectric activity compared to the other types of piezoelectric materials, they exhibit high brittleness which significantly influences their performance. Hence, polymers with higher breakdown thresholds have emerged.47,48 Piezoelectric polymers are much more durable under deformations and electric fields than ceramics.49 Flexibility, ease of processability, and biocompatibility of polymers made them better candidates for piezoelectric applications. Nevertheless, piezoelectric polymers are not an alternative to piezoceramics, and these two types of materials complement each other. Unlike the piezoelectric crystals and ceramics, where the non-centrosymmetric nature of the crystal structure is a requirement for piezoelectricity, this property in polymers arises from the dispersal of polymer chains and molecular alignment in the solid state.50Polylactic acid (PLA) has two stereoisomers of poly-D-lactic acid (PDLA) and poly-L-lactic acid (PLLA) which show the same piezoelectricity in the reverse values of piezoelectric coefficients.69,70 The carbonyl groups existing in the PLA structure induce polarity in the crystalline structure without additional poling processes.71 Because of the less piezoelectric characteristics of PLAs, their incorporation with fillers or their blending with the other polymers is suggested to improve the piezoelectric response. For instance, the piezoelectric constant of PLLA increased by two times upon blending with poly(methyl methacrylate)-b-poly(butyl acrylate)-b-poly(methyl methacrylate) (PMMA-b-PBA-b-PMMA).72
Polyurea films show piezoelectric behavior after poling treatment. Aromatic polyureas are the first introduced piezoelectric polyurea. The odd-numbered aliphatic polyurea, such as polyurea-9 and polyurea-5, gained a residual polarization after poling.10 In addition, polypropylene (PP),73 polyethylene oxide (PEO),74 and vinylidene cyanide copolymers75 are listed in the category of polymers showing piezoelectric characteristics alone or in combination with fillers. Cellulose-based materials such as wood, amylase, chitin, and starch have also been distinguished as piezoelectric polymers. The existence of dipolar alignment is mostly responsible for their piezoelectric behavior.10 Some biopolymers such as collagen76 and silk77 also show piezoelectricity.
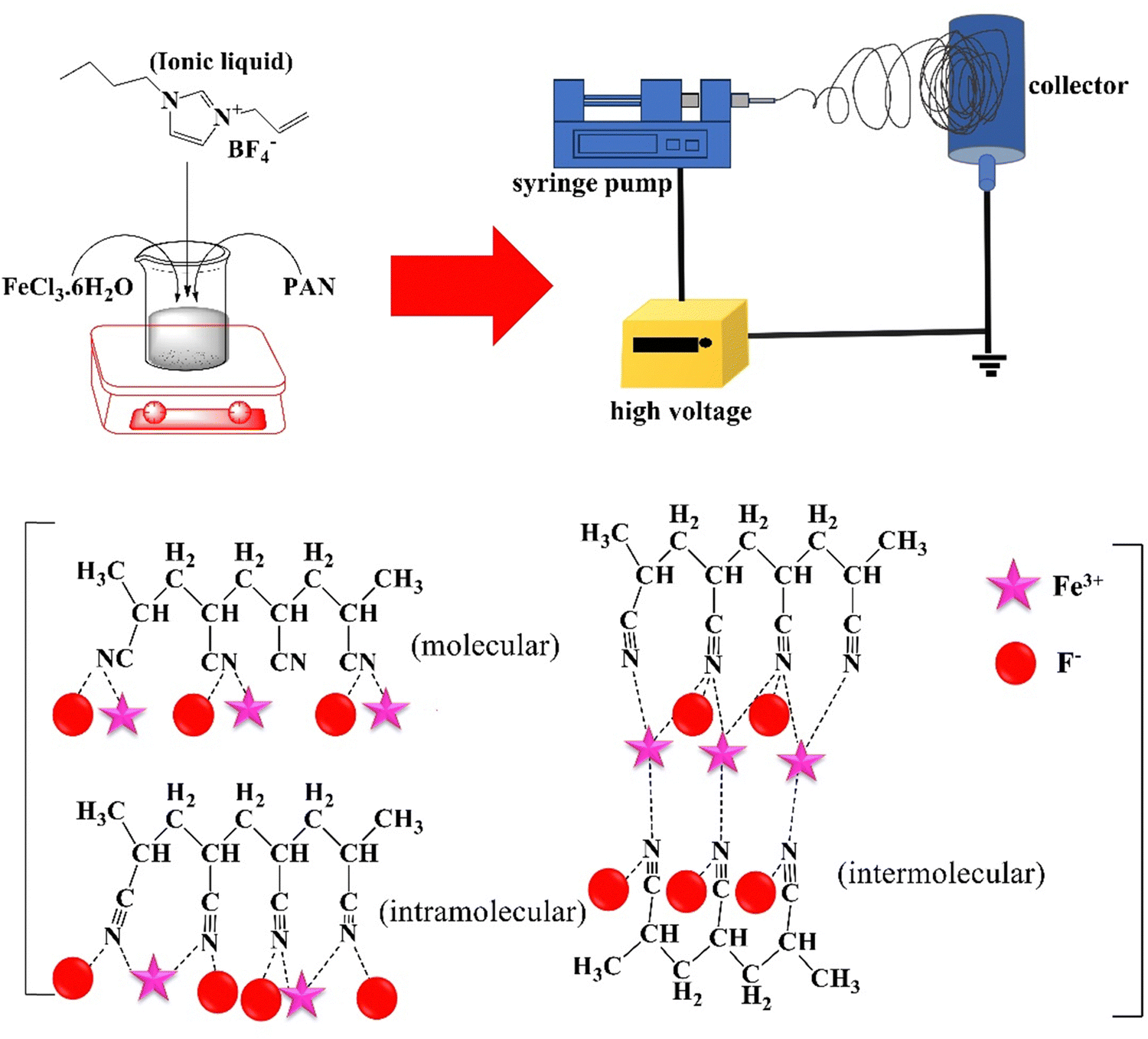 | ||
| Fig. 2 Improved piezoelectricity of PAN NFs by their incorporation with 1-allyl-3-butylimidazolium tetrafluoroborate and FeCl3 (adapted from ref. 99 with permission from John Wiley and Sons). | ||
The impact of a hydrated salt, nickel(II) chloride hexahydrate (NiCl2·6H2O), has been discovered to enhance the β-phase content of PVDF NFs by about 30%.100 Electrospinning and electrospraying are two processing techniques that apply mechanical stretching and poling simultaneously, leading to enhanced β-phase content.101 Electrospinning under the conditions of low temperature, fast evaporation of the solvent, high voltage, and using a rotating collector favors PVDF crystallization in β-phase. The electrostatic force exerted on the jet is more effective in the formation of β-phase compared to the mechanical force applied by rotation of the collector.102
Annealing (heat treatment), drawing (stretching), and poling (applying an electric field) are important techniques for enhancing piezoelectricity,103,104 which are summarized in Fig. 3. Annealing increases the crystalline regions of a polymer, drawing creates stretching or elongation leading to high alignments in polymeric chains and a large degree of anisotropy, and elongation will also re-orientate the crystalline districts in the polymeric amorphous matrix, leading to increased piezoelectricity. For instance, drawing applied by electrospinning enhances the β-phase content of PVDF and its derivatives. Poling aligns the dipole moments within a ferroelectric material by applying an electric field leading to the elimination of the center of symmetry and increasing piezoelectricity.103 Besides, the construction of nanostructures from piezoelectric polymers is an effective way to increase their piezoelectric performance. Electrospinning and template wetting are two common nanostructuring techniques.100,103
 | ||
| Fig. 3 A schematic of annealing (A), poling (B), and drawing (C) for increasing piezoelectricity. | ||
2.4. Piezocomposites
Piezoelectric composites can be prepared by adding either piezoelectric or non-piezoelectric additives into piezoelectric polymeric matrices.103 Carbon nanotubes (CNTs),105 GO,106,107 layered silicate nanoclays,108 metallic salts,100 ILs,95 and metallic NPs109 are examples of non-piezoelectric additives used to encourage the production of β and γ phases in PVDF and its copolymers. In one study, Ti3C2Tx MXene nanosheets were added to samarium doped Pb(Mg1/3Nb2/3)O3–PbTiO3/PVDF NFs and the hydrogen bonding established between the nanosheets and the fluoropolymer matrix was responsible for inducing all-trans conformation in the PVDF polymer and improving piezoelectricity.101 The piezoelectric coefficient of composites increases with the relative fraction of piezoelectric additives. Incorporation of conductive particles into the polymeric matrices is also suggested for improvement of the dielectric permittivity of composites.15–22 For instance, in a three-phase PA11/PZT/CNT composite, a synergistic effect was observed between PZT particles used as piezoelectric fillers and the CNT selected as a conductive filler.110 In another study, a ternary nanocomposite of PVDF/BaTiO3/CNT was prepared based on the homogeneous dispersion of BaTiO3 and CNT fillers in the PVDF matrix.111 BaTiO3 and the CNT acted as high dielectric particles and conductive particles, respectively. The CNT both reinforced the PVDF composite, resulting in an increased thermal, mechanical, and electrical feature compared to the pure PVDF, and induced the β-phase in PVDF. The nanocomposite films of rGO–PVDF containing 0.0 wt% to 0.2 wt% rGO were also prepared and β-phase was induced in them by drawing and poling techniques.107 rGO was employed as a filler to promote the piezoelectricity of PVDF and drawing and poling were used to further promote piezoelectricity. A binary nanocomposite of PVDF/multiwalled carbon nanotubes (MWCNTs) was also fabricated via electrospinning with various contents of MWCNTs, and the β-phase of the PVDF component was increased further via drawing and poling. The interfacial interactions between the CF2 dipole of PVDF and the functional groups of MWCNTs enhance the β-phase content.105 PVDF–clay nanocomposites were also produced using unmodified clay of montmorillonite and different organically modified clays of phosphonium-based, pyridinium-based, and ammonium-based clays.112 These clays served as nucleating agents for the PVDF matrix in order to raise the β-phase content. The phosphonium clay was more efficient in forming β-phase than the other clays. In another study, PVDF/polyaniline (PANI)/graphitic carbon nitride nanosheet (g-C3N4) nanocomposite fibers were fabricated as piezoelectric nanogenerators in order to harvest energy from human motions in different modes.113 The addition of the PANI/g-C3N4 nanocomposite into PVDF and electrospinning synergistically increased the β-phase content of PVDF and the nanogenerator could light up 70 commercial LEDs. A lightweight, low-cost, and flexible rGO/PVDF nanohybrid was also prepared as a self-powered nanogenerator through the dispersion of rGO at various contents in the PVDF matrix.106 A power generation of 14 μW cm−3 was calculated for the nanohybrid in 1 wt% rGO, which makes it a suitable candidate in low-power consuming electronic devices and wireless sensors.In piezocomposites, the matrix and additive geometry will affect the piezoelectric coefficients significantly. For instance, the particle form of fillers shows poor piezoelectricity, while the fibrous form and laminate form of fillers exhibit great piezoelectricity when their longitudinal direction is parallel to the poling direction.114 Making composites from piezoceramics and piezopolymers combines the advantages of high piezoelectricity of piezoceramics with the strength, flexibility, easy processability, levity, relatively high dielectric permittivity, and breakdown strength of polymers.115–117
2.4. Ionic liquids (ILs)
To date, piezoelectricity has been reported only for materials in solid state, while Hossain Md. Iqbal and Gary Blanchard118 reported the existence of direct piezoelectricity in bulk liquid-phase materials, termed room-temperature ionic liquids (RTILs), for the first time. They reported piezoelectricity for two ionic liquids of 1-butyl-3-methyl imidazolium bis(trifluoromethyl-sulfonyl) imide (BMIM+ TFSI−) and 1-hexyl-3-methyl imidazolium bis(trifluoromethyl-sulfonyl) imide (HMIM+ TFSI−). The value of piezoelectricity in these ILs was observed to be an order of magnitude less than that of quartz. Unlike solid-phase piezoelectric materials, bulk liquids are centrosymmetric, and their high piezoelectricity is attributed to the force-induced macroscopic lifting of the center of inversion.1182.5. Metal organic frameworks (MOFs)
MOFs are a class of highly crystalline porous materials with highly tailored and customizable structures, which owe their piezoelectricity to these characteristics.119,120 Nevertheless, the piezoelectric properties of MOFs are rarely reported. The piezo/ferroelectricity of zeolite imidazolate framework (ZIF), which is an important type of MOF, has been recently reported.121 Amongst ZIFs, ZIF-8 not only shows an ultra-high specific surface area, high nitrogen content, open pore structure, and high thermal and chemical stability but also displays great potential for application in catalysis.122,123 Due to its ultra-high specific surface area, it is expected to be used in dye wastewater treatment.124 Yao Sun et al. (2019)125 demonstrated the piezo/ferroelectric properties of UiO-66 (Zr) and UiO-66 (Hf) for the first time. They showed that UiO-66(Hf)-type crystals have a stronger piezo/ferroelectric response than UiO-66(Zr)-type crystals. The piezo/photocatalytic properties of Zr-based and Hf-based UiO-66-NH2 MOFs, named UiO-66-NH2 (Zr) and UiO-66-NH2 (Hf) respectively, were confirmed by Chenxi Zhang et al. in 2021.126 Both the MOFs showed the same hydrogen production under light irradiation while the hydrogen production over UiO-66-NH2 (Hf) was approximately two times more than that of UiO-66-NH2 (Zr) under simultaneous light irradiation and ultrasound (US) stress which is attributed to the more intrinsic piezoelectric activity of UiO-66-NH2 (Hf). NUS-6 (Hf) and MIL-53(Cr) also showed piezoelectricity.1202.6. Other piezoelectric materials
g-C3N4 is a two-dimensional (2D) semiconductor possessing a high piezoelectric coefficient in addition to high photocatalytic activity. Simple preparation procedures, eco-friendliness, and ability for large-scale production are the advantages of g-C3N4 but increasing the mobility of charge carriers to improve its piezoelectricity is still a challenge. The combination of carbon quantum dots (CQDs) with g-C3N4 nanosheets merges the piezoelectric property of g-C3N4 with the excellent charge transfer ability of CQDs, leading to increased piezoelectricity of g-C3N4.127 Another 2D piezoelectric material is MXene which is a type of transition metal carbide/nitride with a non-centrosymmetric lattice structure.128 The first studies on the piezoelectricity of MXenes were completed by Dongchen Tan et al. on the monolayer Ti3C2Tx MXene128 and by Jie Tan et al. on oxygen-containing MXene (M2CO2, M = Sc, Y, and La).129 Recently, a PVDF/Ag/MXene composite nanofibrous film with enhanced piezoelectric activity was fabricated through near-field electrospinning. The presence of MXene and Ag NPs increased the electrical conductivity of PVDF and improved piezoelectricity.130 Another category of piezoelectric materials are inorganic semiconductors including wurtzite families of ZnO, indium nitride (InN), gallium nitride (GaN), cadmium sulfide (CdS), and zinc sulfide (ZnS).131,132 Of these, ZnO has developed as the most popular semiconductor because of uniaxially oriented variants of ZnO nanostructures with multi-dimensions, including nanowires, nanorods, nanosheets, NPs, nanodiscs, thin films, etc.133 Applying mechanical force along the c-axis of ZnO causes displacement of the positive and negative charge centers, leading to the generation of dipole moment and induction of piezoelectricity. If the piezoelectric and photocatalytic properties of ZnO are coupled, the photocatalytic efficiency is probably enhanced by the piezoelectrically encouraged separation of the photogenerated electron–hole carriers.132,134 The nanostructured materials of TiO2, CdS, and Ag3PO4135 as well as multilayer black phosphorus (BP)136,137 and single-atomic-layer molybdenum disulfide (MoS2)138,139 also show piezoelectricity. BP is a mono-elemental 2D material that shows piezoelectricity because of its extremely directional properties and non-centrosymmetric structure. Although piezoelectricity is barely reported in mono-elemental materials because of their lack of ionic polarization, the piezoelectric property of BP is attributed to its non-centrosymmetric structure.136 Ma et al.136 fabricated BP devices as nanogenerators composed of multilayer Te-doped BP flakes with thicknesses of 10–30 nm exfoliated onto a polyethylene terephthalate (PET) substrate. The BP devices showed an intrinsic current output as large as 4 pA under a compressive strain of −0.72%. The highly efficient piezoelectric activity of MoS2 nanoflowers was discovered by Wu et al.140 Wood exhibits the strongest piezoelectricity when it is under mechanical force at the angle of 45° to the direction of the wood fibers.141 A piezoelectric paper fabricated from BaTiO3-decorated wood cellulose fibers showed the most piezoelectric coefficient at the BaTiO3 loading of 48 wt%. BaTiO3 NPs were attached to the surface of fibers through electrostatic binding.11 Other materials that show piezoelectricity are collagenous tissues (e.g., bone, tendon, and DNA),76,142,143 viral proteins,144,145 and amino acids.1463. Piezoelectrically mediated chemical reactions
These reactions use mechanical forces provided in most of the cases by either ball milling (BM) or US to activate the piezoelectric material and achieve highly spontaneous polarization. However, the US is limited to laboratory-scale applications because its waves are harshly weakened in the cavitation zone with increasing distance from the vibrating surface, which restricts its potential in industrial-scale applications.147 BM is a clean, green, and simple technique that has received increasing attention due to its wide applications in environmentally benign and non-thermal solid-state reactions.16 The other advantages of BM can include no use or negligible use of solvent and the use of incompatible and immiscible reagents.148 Under the continual impact, compression, shearing, and friction of BM, the solid reactants experience irreversible distortion and produce new active sites, providing more contact to react.149,150 A ball mill can create an enormous amount of instant mechanical energy at the local situations of piezoelectric materials, leading to their polarization, production of surface defects and increasing surface energy, and creation of crystal lattice distortion, resulting in a substantial increase in catalytic activity.151 Both dry BM and wet BM can be used. Although wet BM is faster than dry BM, it needs the elimination of liquid. A vibratory mill might be used instead of a conventional ball mill. The frequency of milling, size and components of balls and jars, and grinding agents are the important parameters affecting the product yield of mechanochemical reactions.9 The common chemical reactions promoted by electron/hole piezocatalysis include small-molecule organic reactions, polymerization and polymer crosslinking, water splitting, and water remediation which are described below along with the latest published research works.3.1. Water splitting
Hydrogen has great potential as an alternative energy source to chemical fuels. Because of its practical use, greenhouse gas emissions would be severely reduced.152 The production of hydrogen from water is considered as a promising solution to address the growing energy demands and environmental concerns.153 Piezocatalysts are novel types of green catalysts that can use mechanical energy for the production of hydrogen and oxygen. Hong et al. (2010)154 developed piezoelectric ZnO microfibers (∼0.4 μm) and BaTiO3 microdendrites for hydrogen and oxygen production under US vibration. The BaTiO3 dendrites were synthesized hydrothermally through the reaction of Ti(OH)4, Ba(OH)2·8H2O, and NaOH precursors at 200 °C. A hydrothermal reaction was also considered for the synthesis of ZnO fibers by heating a mixture of hexamethylenetetramine and Zn(NO3)2·6H2O solutions at 95 °C. A hydrogen production rate of 1.25 × 10−2 ppm s−1 was observed within 0–50 min over BaTiO3 dendrites and 3.4 × 10−3 ppm s−1 over ZnO fibers within 0–40 min. Su et al. (2019)155 prepared BaTiO3 nanocubes (10 nm) through the hydrothermal reaction of a mixture of an aqueous solution of Ba(NO3)2, NaOH, oleic acid, and oleylamine and 1-butanol solution of tetrabutyl titanate (Ti(OBu)4) at 150 °C. The nanocubes (10 nm) showed a high piezocatalytic coefficient compared to BaTiO3 nanocubes (200 nm) and BaTiO3 nanowires (200 nm) because the piezoelectric potential does not surpass the free energy of water splitting in the larger particles. A hydrogen production rate of 655 μmol g−1 h−1 and an oxygen production rate of 316 μmol g−1 h−1 were obtained under 60 kHz of US vibration over BaTiO3 nanocubes with a size of 10 nm. The yield of the products was very low at low concentrations of the nanocubes (<10 wt%) and it quickly increased with the increasing concentration of the nanocubes. At BaTiO3 concentration of more than 10 wt%, the evolution rate of hydrogen and oxygen decreased, which can correspond to the aggregation of NPs. Wang et al. (2021)156 prepared ultrasmall tetragonal 1,2-distearoyl-sn-glycero-3-phosphorylethanolamine–polyethylene glycol 2000 (DSPE–PEG2000) coated BaTiO3 NPs (P-BTO) by two steps of (1) synthesis of ultrasmall oleic acid modified BaTiO3 through the hydrothermal reaction of a mixture of an aqueous solution of Ba(NO3)2 and NaOH and 1-butanol solution of oleic acid and tetrabutyl titanate (Ti(OBu)4), at 135 °C and (2) its assembly with DSPE–PEG2000 in order to improve water dispersion. Under US agitation, an imbalanced charge state was induced on the surface of P-BTO, which subsequently could react with water molecules to produce oxygen or combine with the water molecules or oxygen to produce reactive oxygen species (ROS). Zhang et al. (2021)157 provided the first example of the preparation of a nanofluidic BaTiO3 suspension containing both cubic and tetragonal phases that produced hydrogen and oxygen in the ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 under US (40 kHz, 60 W, 90 min). For the synthesis, the BaTiO3 samples were prepared by mixing BaCO3 and TiO2 powders under high-speed BM, calcinating at 800 °C, ball milling, and further calcinating at 1200 °C. The BaTiO3 particle size decreased from 400 nm to 150 nm under US agitation. A repeatable hydrogen evolution was obtained within 4 days and a hydrogen evolution rate of 270 mmol h−1 g−1 was obtained for 5 mg L−1 of BaTiO3. BiFeO3 is a ferroelectric and piezoelectric material with a piezoelectric coefficient of 100 pm V−1.158 You et al. (2019)159 prepared BiFeO3 square nanosheets through a hydrothermal reaction of a pH-adjusted solution (pH = 10–11) containing Bi(NO3)3·5H2O solution in ethylene glycol and FeCl3·6H2O solution in water, which was further added to NaOH solution and stirred at 180 °C. A hydrogen production rate of 124.1 μmol g−1 was obtained under mechanical vibration (100 W) for 1 h. The combination of BiFeO3 and BaTiO3 piezoelectric materials could form a solid solution with suitable piezoelectricity which could limit the band gap and tune the conductive band edge position near the redox potential of the hydrogen evolution reaction for effective hydrogen production with less energy input. Sun et al. (2021)160 synthesized hydrothermally 0.7BiFeO3–0.3BaTiO3 (BF–BT) NPs (spherical shape, 67.4 nm) from the precursors Bi(NO3)3·5H2O, Fe(NO3)3·9H2O, Ba(NO3)2, and Ti(OC4H9)4, and used them for hydrogen production under US vibration (100 W, 40 kHz) with the production rate of 1.322 mmol g−1 within 1 h. Ranjan et al. (2022)161 synthesized rhombohedral R3c bismuth sodium titanate particles, Bi1/2Na1/2TiO3, through sol–gel reaction in various sizes, termed S400–S800 samples, obtained at different calcination temperatures of 400, 500, 600, 700, and 800 °C. The hydrogen production rates of 1520.10, 731.22, 556.12, 328.79, and 108.6 μmol g−1 were obtained for S400, S500, S600, S700, and S800, respectively, after 3 h of US (100 W, 40 kHz) agitation, showing the highest piezocatalytic efficiency for S400 because of its smaller particles, more surface area, and a greater number of oxygen vacancies. Piezoelectricity coupled with photocatalytic semiconductors, piezo/photocatalysis, has been extensively studied to tune or enhance optoelectronic activities. Profiting from the synergistic effect between the polarized electric field and photogenerated charges, the carrier separation is improved. In this regard, a common strategy is to construct heterojunctions by coupling semiconductors of covalent organic frameworks (COFs) and piezoelectric materials. Xu et al. (2022)162 constructed a BiFeO3@TpPa-1-COF core–shell hybrid, BFO@COF20-C, through the controlled growth of the excellent visible-light photocatalyst of TpPa-1-COF, prepared by Schiff base reaction, on amino-functionalized BiFeO3 nanosheets (APTES-BiFeO3) for high-efficiency water splitting. The optimum BiFeO3@TpPa-1-COF revealed hydrogen and oxygen production rates of 1416.4 and 708.2 μmol·h−1 g−1, respectively, under simultaneous US (100 W, 40 kHz) and visible light irradiation (λ ≥ 420 nm). TpPa-1-COF traps electrons, while BiFeO3 tends to gather holes. The plausible charge transfer mechanism for BiFeO3@TpPa-1-COF is shown in Fig. 4. Fig. 4A shows the photocatalysis mechanism under light irradiation. In brief, the photogenerated electrons in the conductive band (CB) of BiFeO3 are transferred to the valence band (VB) of TpPa-1-COF for combining with the photogenerated holes but part of electrons and holes are recombined, preventing the photocatalytic reaction. Under US (Fig. 4B), a built-in piezoelectric field (P0) is generated inside Bi-FeO3@TpPa-1-COF, which drives the transfer of electrons and holes in the opposite direction. Nevertheless, some electrons and holes will recombine, which greatly restricts the piezocatalysis. Under both light irradiation and US, BiFeO3@TpPa-1-COF shows piezo-photocatalytic activity. The enhanced piezoelectric potential of BiFeO3@TpPa-1-COF further accelerates the separation of electron–hole carriers, thereby enhancing the photocatalytic redox performance. The polarization direction is different in Fig. 4C and D.
:1 under US (40 kHz, 60 W, 90 min). For the synthesis, the BaTiO3 samples were prepared by mixing BaCO3 and TiO2 powders under high-speed BM, calcinating at 800 °C, ball milling, and further calcinating at 1200 °C. The BaTiO3 particle size decreased from 400 nm to 150 nm under US agitation. A repeatable hydrogen evolution was obtained within 4 days and a hydrogen evolution rate of 270 mmol h−1 g−1 was obtained for 5 mg L−1 of BaTiO3. BiFeO3 is a ferroelectric and piezoelectric material with a piezoelectric coefficient of 100 pm V−1.158 You et al. (2019)159 prepared BiFeO3 square nanosheets through a hydrothermal reaction of a pH-adjusted solution (pH = 10–11) containing Bi(NO3)3·5H2O solution in ethylene glycol and FeCl3·6H2O solution in water, which was further added to NaOH solution and stirred at 180 °C. A hydrogen production rate of 124.1 μmol g−1 was obtained under mechanical vibration (100 W) for 1 h. The combination of BiFeO3 and BaTiO3 piezoelectric materials could form a solid solution with suitable piezoelectricity which could limit the band gap and tune the conductive band edge position near the redox potential of the hydrogen evolution reaction for effective hydrogen production with less energy input. Sun et al. (2021)160 synthesized hydrothermally 0.7BiFeO3–0.3BaTiO3 (BF–BT) NPs (spherical shape, 67.4 nm) from the precursors Bi(NO3)3·5H2O, Fe(NO3)3·9H2O, Ba(NO3)2, and Ti(OC4H9)4, and used them for hydrogen production under US vibration (100 W, 40 kHz) with the production rate of 1.322 mmol g−1 within 1 h. Ranjan et al. (2022)161 synthesized rhombohedral R3c bismuth sodium titanate particles, Bi1/2Na1/2TiO3, through sol–gel reaction in various sizes, termed S400–S800 samples, obtained at different calcination temperatures of 400, 500, 600, 700, and 800 °C. The hydrogen production rates of 1520.10, 731.22, 556.12, 328.79, and 108.6 μmol g−1 were obtained for S400, S500, S600, S700, and S800, respectively, after 3 h of US (100 W, 40 kHz) agitation, showing the highest piezocatalytic efficiency for S400 because of its smaller particles, more surface area, and a greater number of oxygen vacancies. Piezoelectricity coupled with photocatalytic semiconductors, piezo/photocatalysis, has been extensively studied to tune or enhance optoelectronic activities. Profiting from the synergistic effect between the polarized electric field and photogenerated charges, the carrier separation is improved. In this regard, a common strategy is to construct heterojunctions by coupling semiconductors of covalent organic frameworks (COFs) and piezoelectric materials. Xu et al. (2022)162 constructed a BiFeO3@TpPa-1-COF core–shell hybrid, BFO@COF20-C, through the controlled growth of the excellent visible-light photocatalyst of TpPa-1-COF, prepared by Schiff base reaction, on amino-functionalized BiFeO3 nanosheets (APTES-BiFeO3) for high-efficiency water splitting. The optimum BiFeO3@TpPa-1-COF revealed hydrogen and oxygen production rates of 1416.4 and 708.2 μmol·h−1 g−1, respectively, under simultaneous US (100 W, 40 kHz) and visible light irradiation (λ ≥ 420 nm). TpPa-1-COF traps electrons, while BiFeO3 tends to gather holes. The plausible charge transfer mechanism for BiFeO3@TpPa-1-COF is shown in Fig. 4. Fig. 4A shows the photocatalysis mechanism under light irradiation. In brief, the photogenerated electrons in the conductive band (CB) of BiFeO3 are transferred to the valence band (VB) of TpPa-1-COF for combining with the photogenerated holes but part of electrons and holes are recombined, preventing the photocatalytic reaction. Under US (Fig. 4B), a built-in piezoelectric field (P0) is generated inside Bi-FeO3@TpPa-1-COF, which drives the transfer of electrons and holes in the opposite direction. Nevertheless, some electrons and holes will recombine, which greatly restricts the piezocatalysis. Under both light irradiation and US, BiFeO3@TpPa-1-COF shows piezo-photocatalytic activity. The enhanced piezoelectric potential of BiFeO3@TpPa-1-COF further accelerates the separation of electron–hole carriers, thereby enhancing the photocatalytic redox performance. The polarization direction is different in Fig. 4C and D.
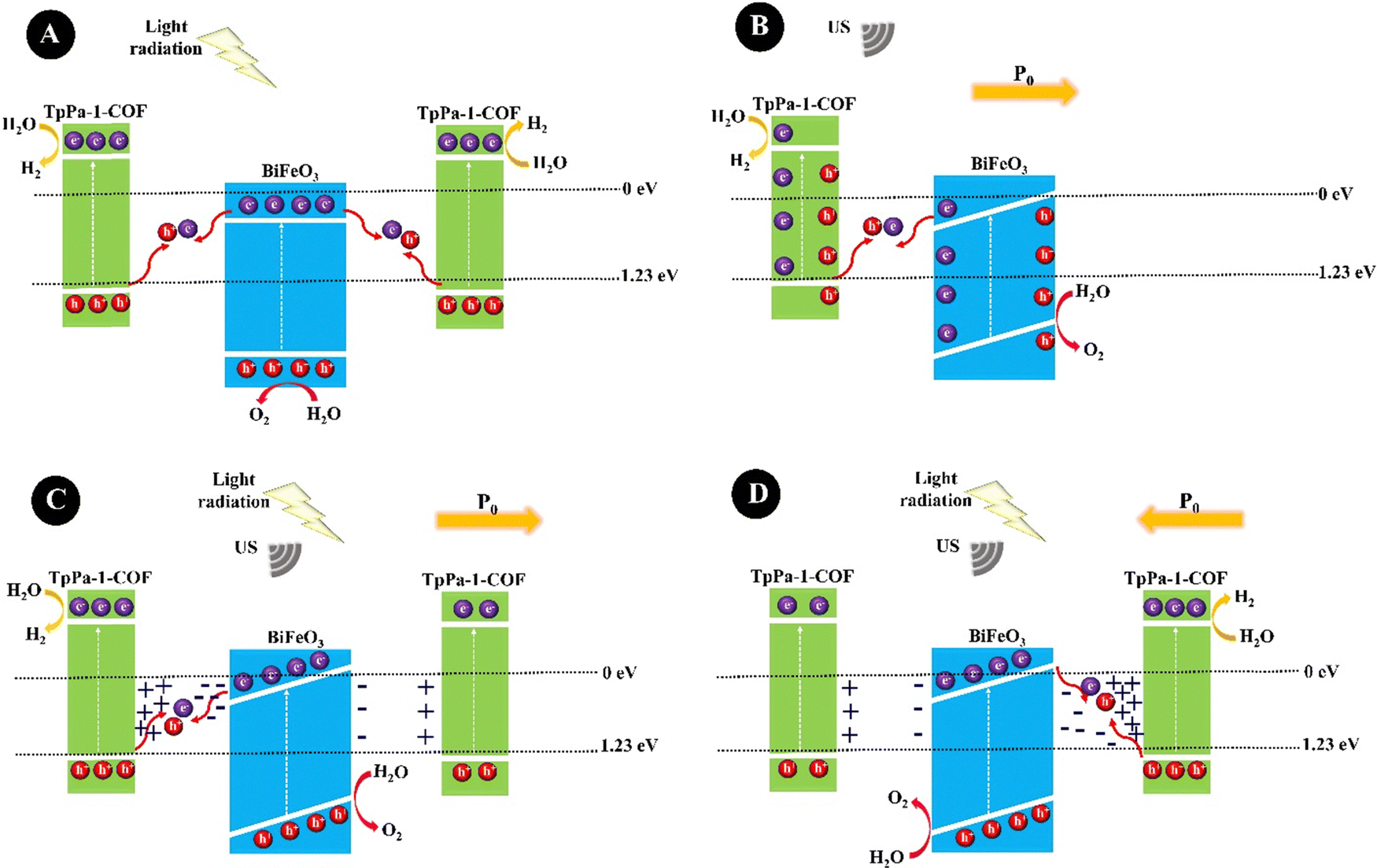 | ||
| Fig. 4 The representative diagrams of photocatalysis (A), piezocatalysis (B), and piezo/photocatalysis (C) and (D) for BiFeO3@TpPa-1-COF (adapted from ref. 162 with permission from John Wiley and Sons). | ||
To summarize, the production rate and yield of hydrogen and oxygen significantly depend on the type, size, morphology, and concentration of the piezoelectric material, the type and intensity of the mechanical source, and the time of applying mechanical force to the piezoelectric material. Piezo/photocatalysis has been introduced as an efficient strategy to increase the rate and efficiency of hydrogen and oxygen production.
3.2. Water remediation
The removal of toxic compounds from water, termed water treatment or water remediation, has been acknowledged using piezoelectric materials. When a mechanical source is applied to a piezocatalyst, a potential difference is formed on the surface of the piezocatalyst owing to the generation of separated electrons and holes, which further participate in redox reactions. The electrons react with dissolved oxygen in water and produce O2˙−, while holes produce ˙OH, thereby disintegrating pollutants existing in water.7,163 As general water contaminants, organic dyes have attracted much attention due to their carcinogenic effects.164 Lan et al. (2017)165 investigated the piezodegradation and piezodechlorination of 4-chlorophenol (4-CP) as a model of non-dye pollution over hydrothermally synthesized tetragonal BaTiO3 (tet-BaTiO3) particles under US (40 kHz, 110 W). A degradation efficiency of 71.1% was observed after 120 min of US. It was confirmed that degradation and dechlorination of 4-CP were mainly attributed to the ˙OH species. Xu et al. (2018)166 fabricated BaTiO3 NFs (200 nm) through electrospinning merged with a sol–gel process. Briefly, a solution of titanium isopropoxide mixed with 2-methoxy ethanol and acetylacetone was added to a solution of barium acetate in acetic acid (Ba to Ti molar ratio of 1:1), followed by dropwise addition of an ethanolic polyvinyl pyrrolidone (PVP) solution to obtain the BaTiO3 precursor. The precursor was loaded into a syringe for electrospinning to obtain PVP-based electrospun NFs. The BaTiO3 NFs were achieved after annealing the PVP-based NFs at 750 °C. The BaTiO3 NFs degraded 97.5% of Rhodamine B (RhB) within 60 min under US vibration. The ˙OH species were distinguished as the main species in the RhB degradation. The hydrothermally synthesized BaTiO3 NFs were prepared by Yao et al. (2022).167 In brief, Na2Ti3O7 was first synthesized hydrothermally in an autoclave with the precursor TiO2 in a sodium hydroxide solution. H2Ti3O7, which was obtained after soaking Na2Ti3O7 in an acidic solution, was dispersed in a solution of Ba(OH)2·8H2O and heated in an autoclave at 210 °C to obtain BaTiO3 NFs. RhB degradation of 94% was achieved under 20 min of US vibration over the BaTiO3 NFs exposed to mechanical milling within 30 min. Mechanical milling probably increases the piezoelectric property by both enhancing the specific surface area of BaTiO3 NFs and enhancing the ferroelectric polarization strength of BaTiO3 NFs leading to diminished recombination of the electron–hole carriers in the catalytic process. The radical trapping experiments displayed that the ˙OH species increase with the increase of the vibration time from 0 min to 60 min. Wu et al. (2018)168 synthesized BaTiO3 nanowires (100 nm) and BaTiO3 NPs (quadrilateral shape, 200 nm) in a tetragonal perovskite crystalline structure, through the hydrothermal reaction of the aqueous solutions of H2Ti3O7 and Ba(OH)2·8H2O at 210 °C and the hydrothermal reaction of the aqueous solutions of H2Ti3O7 and Ba(OH)2·8H2O at 240 °C, respectively. These piezoelectric BaTiO3 nanomaterials degraded methyl orange (MO) under US (∼40 kHz, 80 W), showing the more piezoelectric behavior of BaTiO3 nanowires compared to BaTiO3 NPs, which can be attributed to the easy deformation of the nanowires. O2˙− and ˙OH species were distinguished as the main species in the degradation of MO. Zhou et al. (2022)169 used tet-BaTiO3 NPs (200 nm and 500 nm) in the presence of CuBr2 and H2O2 for degradation of RhB under BM. Without using CuBr2 and H2O2, 16.7% of RhB was degraded over BaTiO3 NPs (200 nm) after 30 min of milling at 30 Hz and RhB degradation efficiencies of only 21.5% and 30.1% were realized within 30 min with the use of CuBr2 alone and H2O2 alone, respectively. The dye degradation reached 97.5% in 10 min and 99.6% in 30 min when a mixture of CuBr2 and H2O2 was used, showing a synergistic effect of piezoelectric NPs, CuBr2, and H2O2 in the degradation of the RhB dye. Degradation efficiencies of 99.2% and 43.4% were achieved over 200 nm and 500 nm BaTiO3, respectively, confirming the more effectiveness of smaller particles in dye degradation. Dye degradation of 46.2% and 31% was observed within 10 min at 20 Hz and 10 Hz, respectively, versus 97.8% degradation at 30 Hz. The BaTiO3 piezocatalyst remained stable after three cycles of use and O2˙− species were suggested to be the main species in dye degradation. Gaur et al. (2023)150 observed methylene blue (MB) degradation of ∼36%, ∼61%, and ∼54% over tet-BaTiO3 ceramics (∼0.5 μm) under a BM speed of 200 rpm using 5, 10, and 15 numbers of Zr balls (10 mm in diameter), respectively. The BaTiO3 ceramics were synthesized through calcination of a homogeneous mixture of BaCO3 and TiO2 at 1200 °C. Upon increasing the BM speed and number of balls, the dye degradation increased. A degradation efficiency of 77% was achieved using 15 balls at 300 rpm within 60 min. The ˙OH species were detected as the main species for MB degradation. Gaur et al. (2023)170 introduced porous BaTiO3 ceramics for MB degradation under US (150 W, 40 kHz) and investigated the dependence of the porosity level on piezocatalysis activity. The BaTiO3 ceramic powder was synthesized through (1) calcinating a mixture of BaCO3 and TiO2 at 1200 °C, (2) mixing with PMMA (0–30 wt%) as a pore former and 4 wt% PVA as a binder to make the pellets, and (3) sintering. MB degradation efficiencies of 55% and 88% were observed, respectively, for non-porous and 30% porous BaTiO3 ceramic pellets under US within 330 min. Thus, by introducing 30% of porosity, piezoelectricity increased by 1.6 times. O2˙− species were distinguished as the main species for MB degradation. Thus, the porosity in bulk BaTiO3 ceramic pellets has provided both significant enhancement in piezocatalytic activity and easy recovery after treatment. Qian et al. (2019)171 introduced a BaTiO3–polydimethylsiloxane (PDMS) composite porous foam prepared through mixing and heating of PDMS, the as-prepared BaTiO3 powder, granulated sugar, and a curing agent. The porous foam showed ∼94% RhB degradation under US (40 kHz, 400 W) within 120 min and excellent stability after 12 cycles of use when the amount of BaTiO3 was 25.0 wt%. O2˙− and ˙OH species were detected as the main species for the dye degradation. Ranjan et al. (2022)161 used Bi1/2Na1/2TiO3 particles synthesized in various sizes (S400–S800) for degradation of MB with high efficiency (up to k = 0.039 min−1) under US (40 kHz, 100 W). S400 showed a degradation efficiency of 98.5% within 80 min for 20 ppm of MB. ˙OH and O2˙− generated in water played the main roles in the dye degradation, as shown by the equations listed below: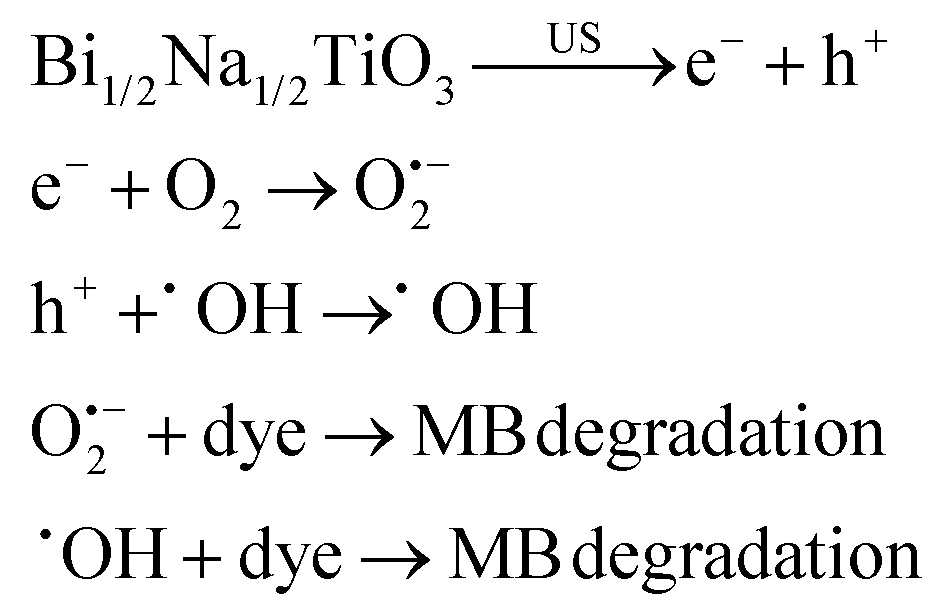
You et al. (2019)159 prepared BiFeO3 square nanosheets for RhB degradation. The decomposition yield of RhB was up to 94.1% after mechanically vibrating (∼45 kHz) for 50 min. Under low-frequency mechanical stirring (600 rpm of magnetic stirring) within 10 h, 76.4% RhB degradation was achieved.
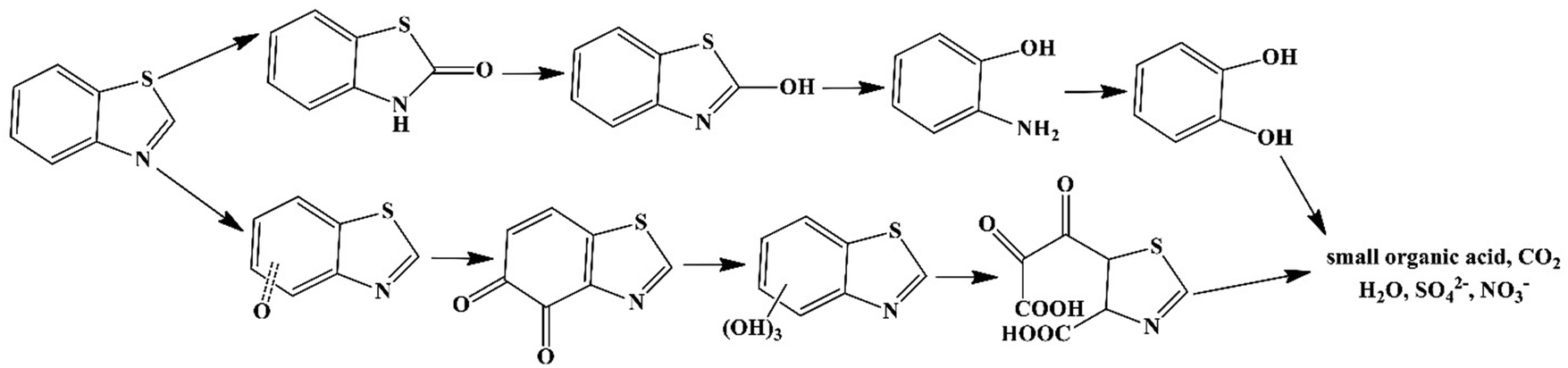
Considering the points that the piezocatalysis and photocatalysis mechanisms are the same and photocatalytic activity increases by making heterojunctions, it is expected that heterojunctions formed by two piezoelectric materials improve piezoelectricity.172 A stronger piezoelectric field can be produced in the piezoelectric heterojunctions compared to the pure piezoelectric materials, further accelerating the migration of free charge carriers for effective redox reactions.173 Since the energy band gap of Wurtzite CdS with a non-centrosymmetric structure is well matched with the energy band gap of BiFeO3, their heterojunction is of high interest. Long et al. (2022)174 fabricated the optimal BiFeO3@CdS-10% NFs for removing bisphenol A (BPA) at 99.7% within 60 min under US (40 kHz, 300 W) with the degradation rate constant of 0.098 min−1. The excellent piezocatalytic activity is probably due to the construction of the heterojunction and the synergistic effect of the two piezoelectric components. BiFeO3 NFs were fabricated through electrospinning and annealing and BiFeO3@CdS with 10 wt% CdS was prepared by regulating the quantity of C2H5NS and Cd(NO3)2·4H2O through a hydrothermal process. The ˙OH species participated mainly in the piezocatalysis process. 98.9% of BPA was degraded after 5 cycles of use, revealing a minor decrease in the catalytic activity. Lan et al. (2022)175 used ambient water motions that existed in urban drainage systems as a mechanical source for the degradation of benzothiazole (BTH) over MoS2. MoS2 was synthesized through a hydrothermal reaction of an acidic solution of Na2MoO4·2H2O and thiourea (1:5 ratio) (pH < 1) at 200 °C. The degradation efficiency of BTH reached 94.8% after 24 cycles of use, which was 8.8 and 4.9 times more than that of the quiescent solution of MoS2 and non-piezoelectric commercial MoS2, respectively. The BTH degradation reached up to 99.3% in deionized (DI) water, 94.7% in rainwater, 92.9% in saline water, and 81.3% in sewage after 24 cycles of use of MoS2 at a concentration of 1 g L−1. MoS2 showed high stability after four cycles of use. O2˙− and h+ species played the main roles in BTH degradation. The degradation pathways of BTH, as shown in Fig. 5, involved eight possible intermediate products. A decreased toxicity was observed for these intermediates compared to the pristine BTH.
 | ||
| Fig. 5 The proposed pathways of BTH degradation over MoS2 (adapted from ref. 175 with permission from American Chemical Society). | ||
Yang et al. (2023)176 introduced a hexagonal boron nitride for the degradation of solid per- and polyfluoroalkyl compounds (PFAS) under BM. The successful decomposition and defluorination of the solid perfluorooctanesulfonate (PFOS) and perfluorooctanoic acid (PFOA) were observed after 2 h of treatment. 80% of 21 targeted PFAS were treated within 6 h in a PFAS-contaminated sediment.
The degradation efficiency of dyes can be promoted by using both piezo- and photocatalysis simultaneously. A substantial enhancement in the generation, transportation, separation, and recombination time of charge carriers can be detected due to the simultaneous coupling of piezo- and photocatalysis.177 In this way, the piezoelectric polarization charges and the induced piezopotential enable the migration of photoinduced charges toward specific directions promoting their separation and diminishing their recombination.6 Mushtaq et al. (2018)178 synthesized BiFeO3 nanosheets (rhombic, 2–3 μm) and BiFeO3 nanowires (200–700 nm) hydrothermally and used them for water treatment. BiFeO3 nanowires degraded 97% and BiFeO3 nanosheets degraded 71% of RhB within 1 h under simultaneous visible light and mechanical vibration. The h+ and ˙OH species were demonstrated to be the main active species in the piezo/photocatalysis mechanism. Sharma et al. (2020)179 investigated the existing effect of Ce on the piezo-photocatalytic activity of Ba0.9Ca0.1TiO3 in the degradation of RhB, MB, and diclofenac under both US and visible light (420 nm) for 120 min. Ba0.9Ca0.1Ce0.15Ti0.85O3 with the highest piezocatalytic activity could be used for up to 5 cycles. The piezo-photocatalytic activity of Ba0.85Ca0.15Ti0.9Zr0.1O3 ceramics (the average particle size of the pellets was 5 μm) was also confirmed by Sharma et al. (2020).31 These ceramics were prepared through the solid-state reaction of the precursors of BaCO3, CaCO3, ZrO2, and TiO2 by mixing in a mortar, followed by calcinating at 1190 °C, re-milling, and pressing into disks. The degradation efficiencies of 89% and 81% were achieved for RhB and ciprofloxacin (CIP), respectively. The ceramics were successfully recycled five times. Sharma et al. (2021)180 investigated the piezo/pyro/photocatalytic (multicatalytic) behavior of Ba0.85Ca0.15Ti0.9Zr0.1O3 (BCZTO) and Ba0.85Ca0.15(Ti0.9Zr0.1)1−xFexO3 (x = 0, 0.5, and 1%) (BCZTO-Fe) ceramics for MB degradation under US (40 kHz, 70 W) and visible light irradiation (15 W) during temporal temperature change of 10 °C and 45 °C. BCZTO-Fe was prepared via solid-state reaction, as BaCO3, CaCO3, ZrO2, TiO2, and Fe2O3 were mixed by BM, calcinated at 1250 °C, pressed to make pellets with polyvinyl alcohol (PVA), and finally sintered in a furnace at 1450 °C. Fe substitution decreased the piezocatalysis performance of BCZTO. The BCZTO-Fe samples were piezocatalytically recyclable for five cycles. The trapping experiments confirmed the critical presence of ˙OH in the dye degradation. Qifeng et al. (2019)177 investigated the piezo/photocatalytic behaviour of Ba0.875Ca0.125Ti0.95Sn0.05O3 (BCT-Sn) in the degradation of MB under both the US and solar simulator (UV, 375 nm). This piezo/photocatalyst was synthesized by mixing BaCO3, CaO, TiO2, and SnCl2 in a ball mill and further calcinating at 1200 °C. The calcinated powder was mixed with PVA to fabricate pellets, which were then sintered at 1400 °C. The poled BCT-Sn showed a piezo/photodegradation efficiency of 98% for MB in the first cycle of use and 96% after five cycles of use. A piezocatalytic degradation of ∼94.5% was reported by Ruan et al. (2022) for RhB124 over ZIF-8 NPs (dodecahedron,∼2 μm) under US within 90 min. A liquid phase technique was used for the synthesis of ZIF-8, in which an aqueous solution of zinc nitrate and 2-methylimidazole was left to stand for 24 h, centrifuged and dried, and then calcinated at 350 °C for 2 h in air. O2˙−, positive charges (q+) and negative charges (q−) were identified as the main species for RhB degradation. The piezoelectric materials Pb2BO3X (X = Cl, Br, I) were introduced by Tang et al. (2022)42 for the degradation of RhB and the antibiotics CIP and norfloxacin (NOR) under BM. Their piezoresponses were in the order of Pb2BO3I > Pb2BO3Br >Pb2BO3Cl. Pb2BO3I showed a degradation efficiency of 99.0% for RhB within 30min and 90.0% for CIP and NOR within 15min. ZrO2 balls led to higher efficiency compared to SiO2 balls. The O2˙− species were determined as the main active species in the degradation of the pollutants.
In summary, a high rate of dye degradation was observed within a short time (>90% within ≤60 min in most of the cases), and the degradation efficiency increased on decreasing the size of the piezoelectric particles and on increasing the US intensity, BM speed, and the number of balls. A high recyclability was observed for the piezoelectric materials and in most of the studies, O2˙− and ˙OH species were identified as the main species participating in the degradation of pollutants. Piezo/photocatalysis promoted degradation efficiency because of the influence of both piezocatalysis and photocatalysis processes.
3.3. Controlled polymerization/crosslinking
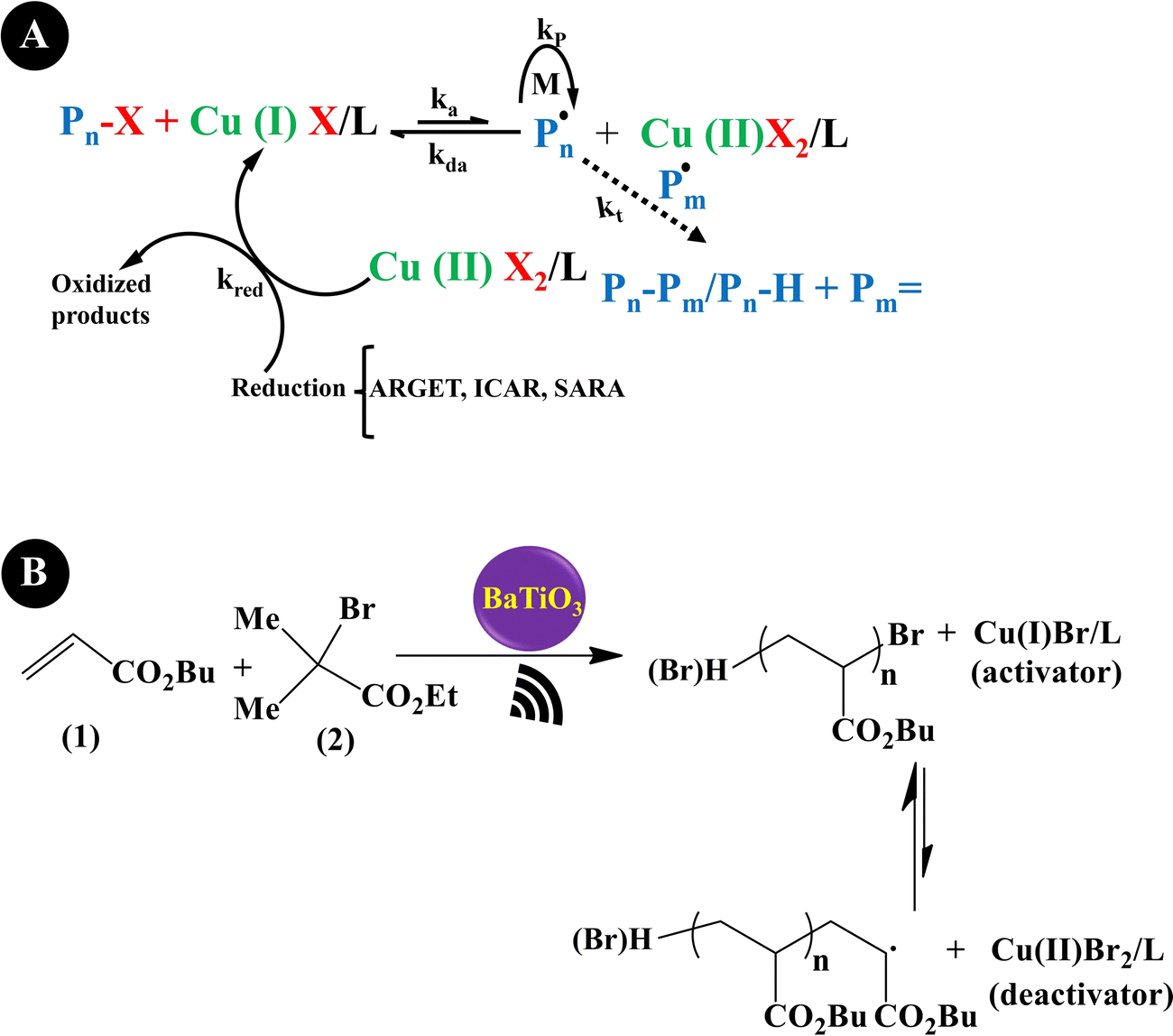
Reversible-deactivation radical polymerization (RDRP) is a controlled radical polymerization that allows the synthesis of polymers with controlled molecular weight, low dispersity, and diverse functionality. Inclusion of mechanical stimuli in RDRP allows temporal or spatial control of the composition and functionality of polymers.181 Three main RDRP techniques, nitroxide-mediated polymerization (NMP), atom transfer radical polymerization (ATRP), and reversible addition–fragmentation chain transfer (RAFT), have been recognized.182 Because of the restrictions of the three techniques in high conversions, in the synthesis of multi-block copolymerization, and in controlled polymerization on living cells, the new RDRP methods have appeared to perform under different stimuli conditions, such as chemical, light, electrical, force, etc.183 Actually, piezopolymerization is a novel approach that uses mechanical energy for controlled polymerization and cross-linking reactions.184Conventional ATRP needs a high loading of the catalyst to simplify the control equilibrium between radicals and inactive species, leading to a decreased rate of polymerization. Advanced ATRP involves the use of chemical reducing agents and external stimuli to produce an extra redox cycle. Such a system reduces the concentration of the catalyst to ppm, while its disadvantage is by-products generated by the chemical reducing agent. Moreover, the removal of the chemical reducing agents is very difficult after polymerization termination. Compared with the conventional ATRP and advanced ATRP systems, US-mediated ATRP has many advantages: (1) control of polymerization easily by switching US on/off; (2) no usage of chemical reducing agents, thereby no production of by-products; (3) simple, mild, and easy scale-up operation; and (4) suitability for organic and water medium-based polymerization.185 Generally, the ATRP technique is mediated by a redox equilibrium between two oxidation states of copper (Cu) complexes.186 The in situ generation of a Cu(I)-based polymerization activator has previously been achieved using chemical reducing agents,187 light,188 and electrochemical potential.189 Indeed, ATRP is governed by redox-active transition metal complexes in their lower oxidation state that activate the inactive species and complexes in their higher oxidation state.190 The dead chains and activation/deactivation cycles determine the molecular weight distribution in ATRP.191Fig. 6A indicates the general probable mechanism of ATRP performed with a ppm of catalyst. It is identified that the advanced ATRP produces non-living chains from radical initiators, which increases dispersity, while piezo-ATRP regenerates the activator.191 As observed in Fig. 6A, the US-mediated reduction of Cu(II) to Cu(I) causes the controlled polymerization. Mohapatra et al. (2016)192 described a US-mediated ATRP for n-butyl acrylate (BA) polymerization over tet-BaTiO3 NPs (200 nm, 4.5 wt%) under pulse mode operation of 4s on and 4s off using the pre-catalyst solution of Cu(OTf)2/Me6TREN/Bu4NBr. Fig. 6B shows the mechanism of piezopolymerization. In brief, the ATRP activator is produced through the piezoelectric reduction of the Cu(II) catalyst under US, and polymeric chain growth is started from the alkyl halide initiator (2) and consecutive addition of monomer 1. The final polymer is obtained after chain termination.
 | ||
| Fig. 6 The proposed mechanism of piezo-ATRP. ARGET: activator regenerated by electron transfer; ICAR: initiator for continuous activator regeneration; SARA: supplemental activator and reducing agent (A) (adapted from ref. 191 with permission from American Chemical Society). The US-mediated ATRP polymerization of BA (B). | ||
Wang et al. (2017)193 prepared poly(methyl acrylate) (PMA) with high end-group fidelity, predetermined molecular weight, low dispersity, and 90% of monomer conversion through the piezo-ATRP technique using ZnO piezoelectric NPs (18 nm) under US agitation (40 kHz) for 8 h. Unlike the previous work reported by Mohapatra et al.192 on the piezo-ATRP of n-butyl acrylate (BA) using a high amount of BaTiO3, 4.5 wt%, a low amount of ZnO particles, even 0.06 wt%, was used in this work, which may be attributed to the ease of electron transfer between the ZnO NPs and Cu(II)/L compared to that between BaTiO3 and Cu(II)/L. As in the previous work, the reduction of Cu(II) into Cu(I) activates polymerization. This piezo-ATRP technique was also applied for the polymerization of other acrylate monomers including ethyl acrylate, BA, and tert-butyl acrylate (t-BA). All polymerization reactions were well-controlled with appropriate molecular weights and low dispersity. Wang et al. (2017)194 prepared PMA successfully with a molecular weight of up to 20000 g mol−1 and a dispersity of 1.05–1.18 through piezo-ATRP polymerization of methyl acrylate (MA) over BaTiO3 agitated by US (40 kHz) in the presence of a low content of the Cu/tris(2-pyridylmethyl)amine catalyst (Cu/TPMA, 75 ppm) and the ethyl α-bromoisobutyrate (EBiB) initiator. Since the US disrupts metal–ligand interactions, TPMA was chosen as a ligand to produce stable Cu complexes. Polymerization with cubic BaTiO3 (50 nm, 4.5 wt%) led to a monomer conversion of 67%, a molecular weight of ∼10800 g mol−1, and a dispersity of 1.06 after 8 h. The polymerization using tet-BaTiO3 (200 nm) achieved approximately the same results. The usage of PMMA-coated BaTiO3 increased the rate of polymerization because of its better dispersion in the reaction medium. Zhou et al. (2018)191 obtained PMA with more than 90% monomer conversion, well-controlled molecular weight, and low dispersity (1.03–1.09) through piezo-ATRP polymerization of MA over tet-BaTiO3 NPs (200 nm, 4.5 wt%) in the presence of 50% v/v DMSO (1:1 volume ratio of DMSO:MA) under US agitation (40 kHz, 70 W, 50 °C). No polymerization occurred in the presence of 33.3% v/v DMSO, confirming the critical role of DMSO in the polymerization. A monomer conversion of 94% was achieved within 3 h by increasing the concentration of the catalyst from 100 ppm to 200 ppm. While the RAFT polymerization has been generally used for the modification of non-magnetic compounds,195 the surface-initiated ATRP has been well predicted to modify the magnetic compounds.196 Cvek et al. (2021)197 modified magnetic NPs with PMA by the piezo-ATRP technique, for the first time, resulting in NPs@PMA hybrids. The ATRP initiator-decorated magnetic NPs were added into a piezo-ATRP mixture, generating surface-initiated growth of the PMA chains under US (35 kHz, 45 °C) in the presence of hexagonal micro-ZnO and cubic-phase BaTiO3. Micro-ZnO was produced via annealing the as-prepared zinc oxalate dihydrate (ZnC2O4·2H2O) precursor and BaTiO3. The reaction progressed with a low amount of CuBr2, thus giving PMA with low dispersity and high monomer conversion. When using 5 wt% micro-ZnO, a monomer conversion of 75%, a molecular weight of 14000 g mol−1, and a dispersity of 1.26 were obtained after 8 h, and when using 9 wt% BaTiO3, a monomer conversion of 74%, a molecular weight of 13300 g mol−1, and a dispersity of 1.09 were obtained after 8 h.
Liu et al. (2022)198 reported piezo-ATRP polymerization of methyl methacrylate (MMA) over one-dimensional (1D) ZnO nanorods (NRs) with three different dimensions (termed NR-1, L = 1.9 μm, D = 300 nm, L/D = 6; NR-2, L = 4.2 μm, D = 420 nm, L/D = 10; NR-3, L = 6.8 μm, D = 530 nm, L/D = 13) and zero-dimensional (0D) ZnO NPs (240 nm) under US agitation (40 kHz, 50 W) within 4 h. Ethyl α-bromoisobutyrate (EBiB), CuBr2, and TPMA were used as the initiator, copper salt, and ligand, respectively, and DMSO was used as the solvent. The ZnO NRs were synthesized by chemical bath deposition using zinc nitrate hexahydrate and hexamethylenetetramine, while ZnO NPs were purchased. NR-2 and NR-3 showed more catalytic activity compared to the NPs because of their larger aspect ratios. The weakest activity was related to NR-1 owing to its small aspect ratio and specific surface area. In the presence of 1.2 wt% NR-3, a monomer conversion of 70% and a molecular weight of 7200 g mol−1 were achieved. Remarkably, the US-mediated ATRP produced PAN with a high molecular weight of 23800 g mol−1 and a low dispersity of 1.19 after only 6 h. Xu et al. (2022)185 synthesized the ZnO/BaTiO3 heterostructure by first in situ deposition of the zinc precursor on BaTiO3 particles and then annealing, and used it in US-mediated ATRP of MMA. PMMA with a monomer conversion of 63.1%, a molecular weight of 6600 g mol−1, and a dispersity of 1.30 was achieved in the presence of 400 ppm CuBr2/TPMA and1.2 wt% ZnO/BaTiO3 under US (40 kHz, 500 W) for 4 h. The ZnO/BaTiO3 piezocatalyst showed a high rate of US-mediated ATRP after ten cycles, displaying the excellent recyclability of ZnO/BaTiO3. Zhou et al. (2023)199 prepared high molecular weight PMMAs ranging from 33000 g mol−1 to 74000 g mol−1 through DMSO-assisted BM over tet-BaTiO3 (200 nm) in the presence of the CuBr2/TPMA complex (1/2). The monomer conversion reached 59–69% after 4 h in all the experiments, while dispersity increased from 1.44 to 1.75. Under a vibration frequency of 20 Hz, a monomer conversion of 46.8% was achieved after 6 h. Increasing the milling frequency to 25 Hz and 30 Hz resulted in an enhanced polymerization rate. With 250 ppm and 150 ppm of Cu(II)/L, a monomer conversion of 90% was achieved with no more grinding time at 30 Hz. 2-Hydroxylethyl acrylate (HEA) was successfully polymerized in the presence of water instead of DMSO as the liquid-assisted grinding (LAG) additive. At 30 Hz, a monomer conversion of 72%, a molecular weight of 43100 g mol−1, and a dispersity of 1.43 were achieved within 1 h of BM.
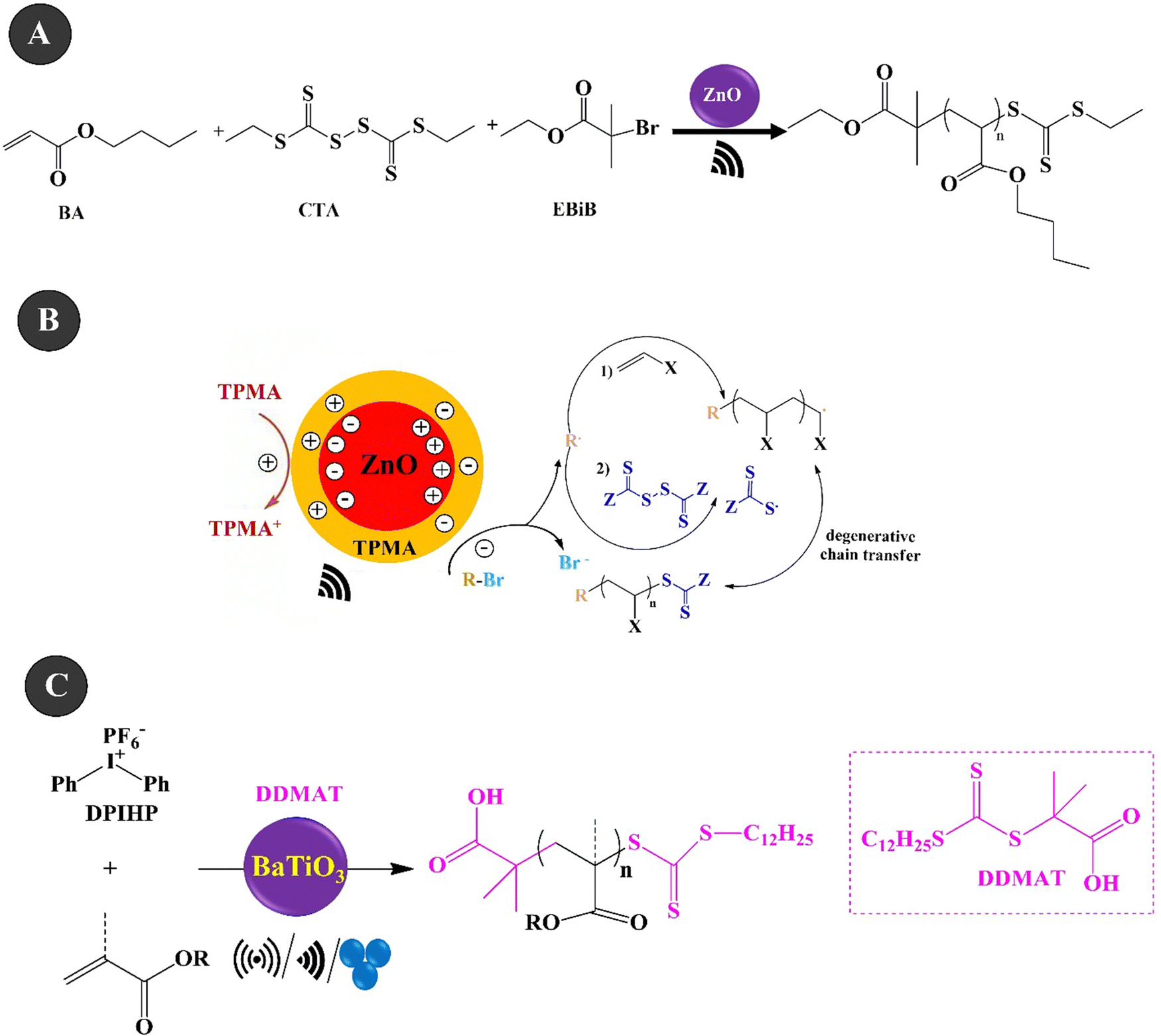
Reversible addition–fragmentation chain transfer (RAFT) polymerization is one of the most famous RDRP techniques that displays high control over molecular weight and end-group fidelity.200 The polymerization is a living and accessible polymerization technique with wide functional group tolerance.201 Ding et al. (2022)202 investigated the piezo-RAFT of BA under US agitation of 40 kHz over ZnO NPs (10–30 nm, 4.4 wt%). Fig. 7A and B present the piezo-RAFT process and the mechanism of BA. Ethyl α-bromoisobutyrate (EBiB) and bis(trithiocarbonate) bisulfide (CTA) were used as the initiator and chain transfer agent, respectively. The adsorption of TPMA on the surface of ZnO could enable electron transfer to alkyl bromides and the piezoelectrically mediated reduction of alkyl bromides can both initiate the polymerization and cleave the S–S bond of bis(trithiocarbonate) bisulfide to produce the RAFT agent in situ. A monomer conversion of 85.9% and a molecular weight of 25200 g mol−1 were achieved within 8 h under US agitation. Chakma et al. (2023)203 studied the RAFT polymerization of (meth)acrylate monomers over BaTiO3 under three agitation conditions of US (40 kHz, 70 W), BM (1.5 mL jars, 5 mm ball, 30 Hz), and vortex (1.5 mL vial, two 5 mm balls, 3000 rpm), as shown in Fig. 7C. The mechanically polarized BaTiO3 (cubic, <100 nm) could reduce diphenyl iodonium hexafluorophosphate (DPIHP) to form phenyl radicals which can start the RAFT equilibrium. The polymerization was carried out with BA over 7 wt% BaTiO3 under US for 24 h in the presence of various organic solvents and 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT) as a chain transfer agent. A monomer conversion of more than 65% and a dispersity of 1.05 were observed in the presence of DMF and DMAc solvents. The piezo-RAFT of t-BA resulted in a monomer conversion of more than 95% in the presence of BaTiO3 (15 wt%), DPIHP, and DDMAT after 3 h of BM and a small amount of DMF as the LAG solvent. The polymerization was also performed in a vortex using similar BM conditions for 8 h. In this case, a monomer conversion of 70% was achieved for BA within 8 h and the use of one milling ball. Under the same conditions, by using two balls, the monomer conversion increased to 89%.
 | ||
| Fig. 7 The piezo-RAFT process (A) and mechanism (B) of BA (adapted from ref. 202 with permission from American Chemical Society). The piezo-RAFT polymerization of (meth)acrylate monomers over BaTiO3 under the three agitation conditions of US/BM/vortex (C) (adapted from ref. 203 with permission from John Wiley and Sons). | ||
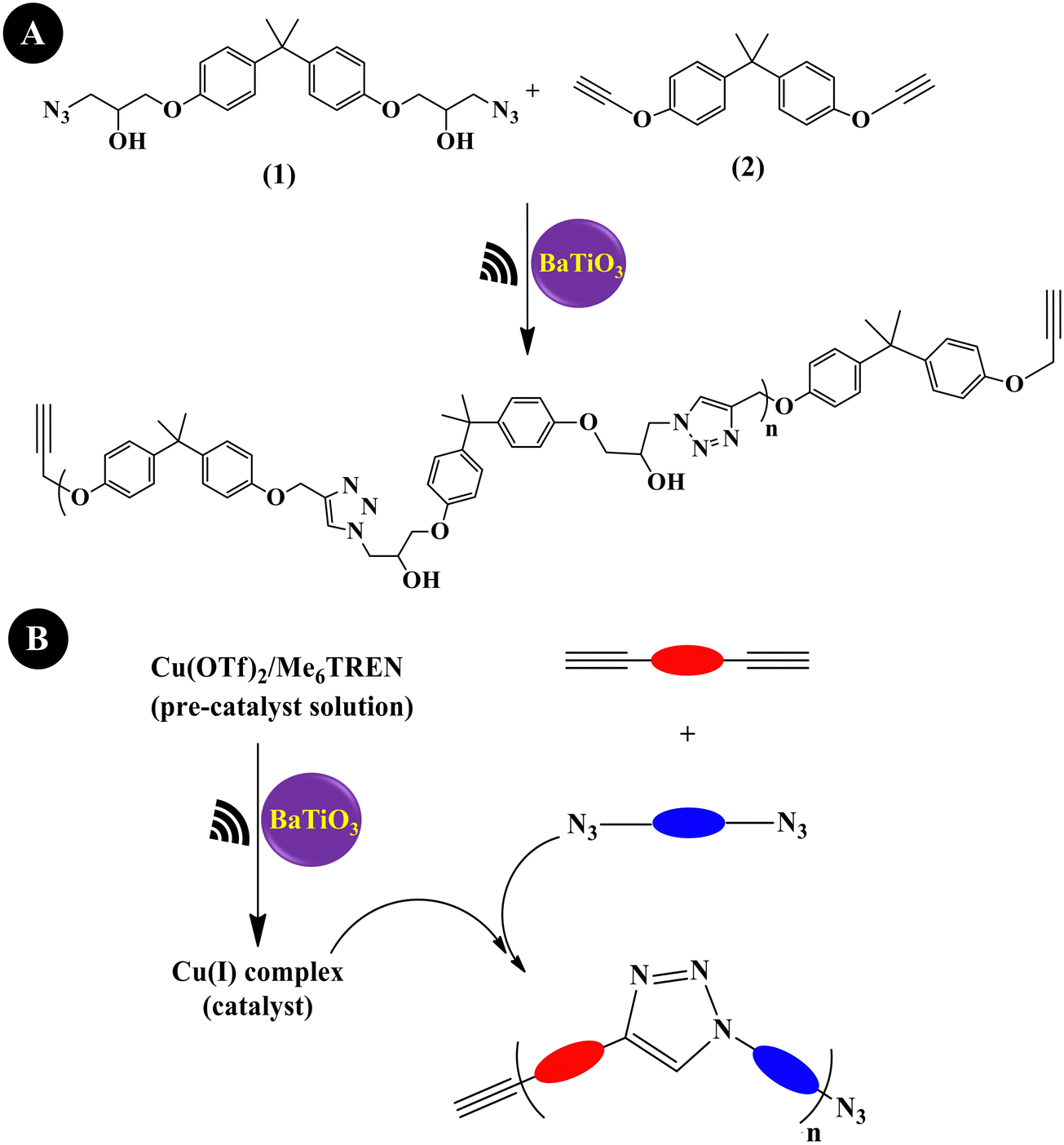
Mohapatra et al. (2018)204 prepared a linear polytriazole through step-growth polymerization via the copper-catalyzed azide–alkyne cycloaddition (CuAAC) reaction, commonly denoted as the copper ‘Click’ reaction or mechano-click reaction. As shown in Fig. 8A, equimolar amounts of monomer 1 (1 M) and monomer 2 (1 M) were used along with (Cu(OTf)2/Me6TREN) (50/55 mM) and tet-BaTiO3 (200 nm, 10 wt%). Under US agitation (40 kHz,24 h), a monomer conversion of >95% was observed. This is the first instance of US-agitated copper ‘Click’ polymerization through a piezo-mediated reaction. A general illustration of ‘Click’ polymerization catalyzed by Cu(I) produced through the US reduction of the Cu(II) precursor (Cu(OTf)2/Me6TREN) is shown in Fig. 8B. Crosslinking was also performed under the conditions of a mixture containing divalent azide monomer, tripropargyl amine crosslinker, tet-BaTiO3 (10 wt%), Cu(OTf)2/Me6TREN (5/5.5 mol%), and DMF to obtain a brittle gel.
 | ||
| Fig. 8 Piezoelectrically mediated copper ‘Click’ polymerization of monomer 1 and monomer 2 (A) and a general depiction of Cu(I)-catalyzed Click polymerization (B) (adapted from ref. 204 with permission from Johns Wiley and Sons). | ||
Wang et al. (2019)205 reported a mechanically controlled free-radical (mechano-radical) polymerization and polymer crosslinking of acrylate monomers using ZnO NPs. They indicated the mechano-radical polymerization of the monomers via cleavage of alkyl halides under low-frequency US agitation (40 kHz, 70 W), as shown in Fig. 9A. A Fe(III) complex composed of FeCl3·6H2O and tris[2-(2-methoxyethoxy)ethyl]amine (TDA-1) was used to facilitate the free radical transfer during the agitation. ZnO (20 nm, 9.0 wt%) allowed the polymerization of BA with a monomer conversion of 74% and a molecular weight of ∼241000 g mol−1. t-BA showed a monomer conversion of 80% and a molecular weight of ∼341000 g mol−1, while MMA showed a monomer conversion of 52% and a molecular weight of ∼49000 g mol−1 after 21 h of US agitation. In addition, the polymerization of 2-ethylhexyl methacrylate (HEMA) resulted in the formation of a persistent gel without the addition of a crosslinking agent. A lower monomer conversion was achieved by polymerization in DMAc and NMP solvents compared to that in DMF solvent and no polymerization was detected in DMSO. Ayarza et al. (2021)184 reported the first examples of the piezo-electrochemical synthesis of disulfide bonds to form reversible organogels from polymers containing thiol side groups. This reaction was carried out via piezo-oxidation of thiol to disulfide over ZnO NPs (18 nm, 5.0 wt%, 2.5 wt%, and 1.0 wt%) and iodide anions under US (40 kHz) or shaking agitation (2 kHz) for 6 h. Fig. 9B presents the chemical structures of mercapto-polymers and Fig. 9C shows the reaction schematic of the mechanically mediated cross-linking of mercapto-polymers. The gels of mercapto-PMMA, mercapto-PMA, and mercapto-PS showed gel fractions of 93%, 80%, and 83%, respectively, over 5.0 wt% ZnO NPs under 40 kHz of US frequency in the presence of KI base and DMF solvent. Zeitler et al. (2022)148 presented the iodonium-initiated free-radical polymerization of (meth)acrylate monomers under US and BM. They studied the polymerization of t-BA in DMF by using diphenyl iodonium hexafluorophosphate (DPIHP) as the initiator and BaTiO3 (cubic, 7 wt%, <100 nm) or ZnO (18 nm, 7 wt%) as the piezocatalyst. A monomer conversion of 92% and 68% was achieved over BaTiO3 and ZnO, respectively, after 20 h of US agitation (40 kHz, 70 W). Under BM (1.5 mL stainless steel (SS) jar, 5 mm SS grinding ball, 30 Hz) with the use of minimal DMF, a monomer conversion of >95% was observed within only 3 h compared to that of 20 h under US agitation. Unlike the US-mediated polymerization, BM did not need the removal of oxygen.
 | ||
| Fig. 9 The mechano-radical polymerization of acrylate monomers using ZnO NPs (A) (adapted from ref. 205 with permission from John Wiley and Sons). The chemical structures of mercapto-polymers (B) and mechanically promoted cross-linking of mercapto-polymers (C) (adapted from ref. 184 with permission from American Chemical Society). | ||
Considering the above-mentioned points, all of the polymerization reactions produced polymers with high molecular weights, low dispersity, and high monomer conversions in the range of 52–95% depending on the monomer. Polymer crosslinking was also carried out with the formation of high gel fractions.
3.4. Organic synthesis
For organic synthesis, solid-state reactions (using BM proceeding the reactions) are preferred for the substrates with poor solubility which can even change the chemical selectivity, providing products which are difficult or impossible to produce via the liquid-phase reactions.206,207 The piezocatalyzed organic synthesis is also preferred for conventional solid-state reactions to improve the yield and selectivity of products. Kubota et al. (2019)208 reported mechanoredox arylation and borylation reactions proceeded through the reduction of aryl diazonium salts over BaTiO3. The radicals generated from the polarized BaTiO3 under BM started the reactions. The arylation product was obtained in a yield of 40% under BM conditions of 1.5 mL milling jar, milling balls of diameter 5 mm, 20 Hz, and 1 h under air. The yield of the arylation product reached 81% at 30 Hz and reached 82% using a bigger jar (5.0 mL) and a bigger ball (7.5 mm). The highest yield of the borylation product (89%) was afforded using acetonitrile (MeCN) as the LAG additive within 3 h. The bigger jar (5.0 mL) and ball (7.5 mm) did not have a significant effect on the yield of the borylation product (86%). Schumacher et al. (2020)209 reported tet-BaTiO3 NPs (500 nm, 20 wt%) as the optimized piezocatalyst in electric-assisted BM-induced copper-catalyzed atom transfer radical cyclization (ATRC). In comparison with standard BM, electric-assisted BM uses high-voltage electrical impulses throughout BM to accelerate chemical reactions. In this mechanoredox reaction, the electrons provided by the polarization of BaTiO3 reduce inactive Cu(II) into active Cu(I), as shown in Fig. 10A. The Cu(I) complexes generate radicals (2) from alkyl halides (1) via a reversible redox process. The produced carbon-centered radicals (2) experienced intramolecular cyclization through the creation of new C–C bonds (3). To conclude, the in situ generated Cu(II) complex transfers the halide atom back to C giving the ATRC product (4), as shown in Fig. 10B. Fig. 10C shows a model of the ATRC reaction which used monobromoacetamide (a), Cu(OTf)2, and tris(2-pyridylmethyl)amine (TPMA). The reaction was performed by using tet-BaTiO3 (500 nm, 20 wt%), argon atmosphere, 25 Hz, and 90 min to obtain the ATRC product with a yield of 97%. A ZrO2 milling jar with 8 ZrO2 balls with a diameter of 5 mm favored the ATRC product. Under the same conditions, 40 wt% cub-BaTiO3 (100 nm) produced the ATRC product in 97% yield. The reaction afforded only 6% and 5% of the ATRC product in the presence of non-piezoelectric materials TiO2 (anatase) and Al2O3 (gamma), respectively.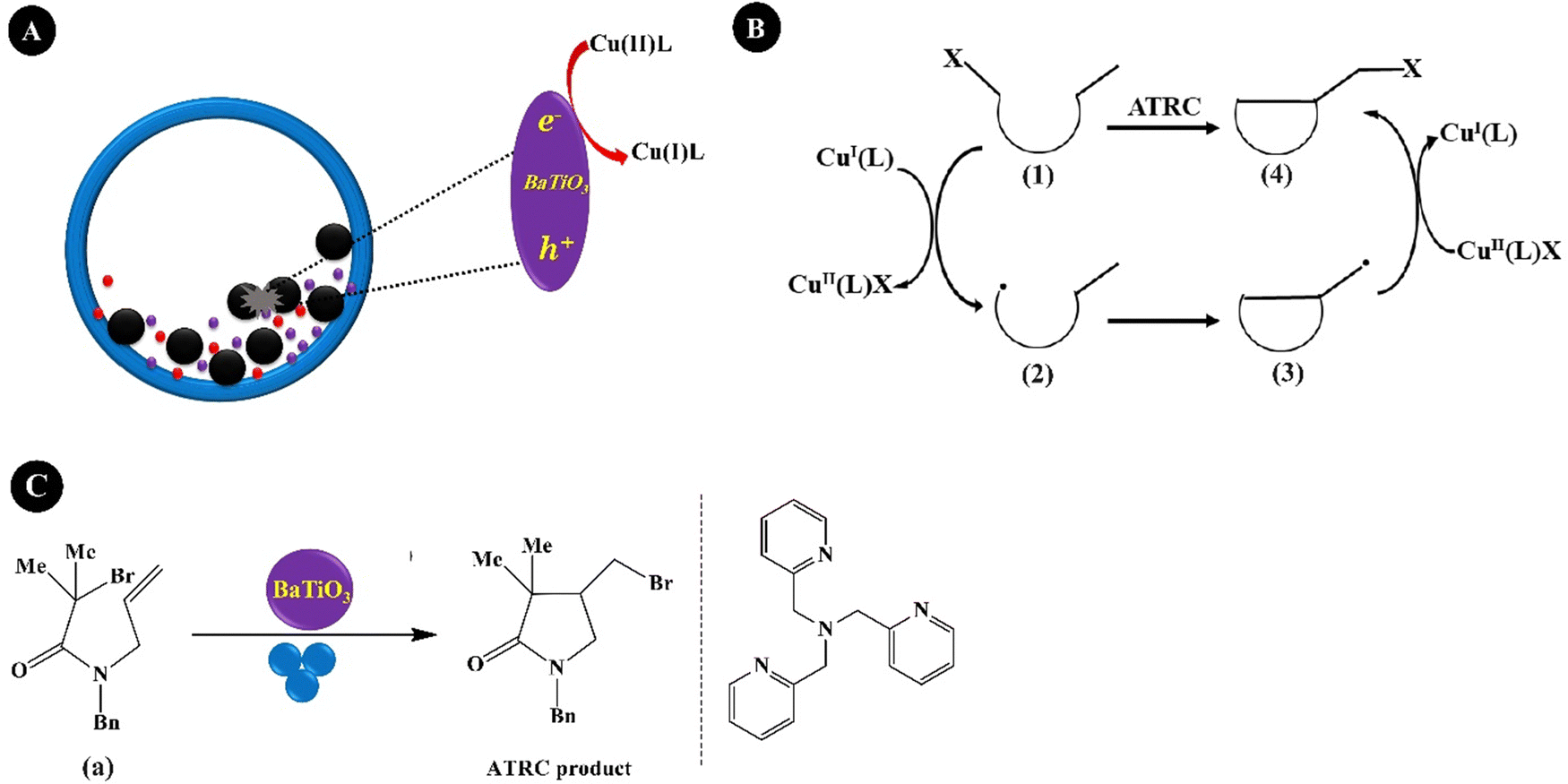 | ||
| Fig. 10 The piezo-reduction of the Cu(II) complex to Cu(I) complex (A), mechanism of copper-catalyzed ATRC reaction (B), and ATRC reaction of monobromoacetamide (C) (adapted from ref. 209 which is distributed under the terms of the Creative Commons CC BY license, Wiley's Open Access Terms and Conditions). | ||
For the first time, Pang et al. (2020)210 reported the mechano-agitated C–H trifluoromethylation of aromatic compounds to produce various trifluoromethylated N-heterocycles and peptides. The piezo-generated CF3 radical is added to the aromatic compound and produces the trifluoromethylated intermediate, which is then oxidized by the hole generated by the agitated BaTiO3, and finally, deprotonation gives the trifluoromethylated product. The product yields of 58%, 58%, and 62% were obtained through trifluoromethylation of 3-methyl indole using DMF, MeCN, and acetone, respectively, over tet-BaTiO3 (<3 μm) in a milling jar of 1.5 mL with balls of 5 mm at 30 Hz for 1.5 h under air. With a larger ball (7.5 mm) and a larger jar (5 mL) and increasing the time of reaction to 3 h and the number of balls to two, a product yield of 80% was achieved when the LAG additive was acetone.
Wang et al. (2021)211 prepared 1,2-diketoindolizine derivatives from indolizines and epoxides using BaTiO3 (cubic, <3μm) in the presence of the pivalic acid (PivOH) additive under BM (milling jar of 20 mL, 30 balls of 6 mm diameter) for 6 h, as observed in Fig. 11A. Under optimum conditions, a product yield of 87% was obtained. The plausible mechanism of the reaction is illustrated in Fig. 11B. Briefly, a radical intermediate (2) and a superoxide radical anion (O2˙−) were generated by the interaction of polarized BaTiO3 with indolizine (1) under oxygen. The intermediate (2) was converted to its resonance form (3). Then again, the ring-opening and oxidation of styrene oxide (a) produced (b), followed by oxidation by O2˙− to provide the intermediate (4). Then, the radical intermediate (3) was combined with (4) to form the 1,2-diketoindolizine cation (5). Finally, deprotonation of the cation (5) offered the final product (6). Wang et al. (2022)212 used BaTiO3 for the aerobic oxidation of thiols to disulfide bonds. The agitation of BaTiO3 under BM activated molecular oxygen to produce thiyl radicals subsequently producing disulfide bonds. In detail, O2˙− was generated during BM, which further reacted with the oxidized thiol substrate, R–SH˙+, and produced thiyl radicals, R–S˙, which finally coupled together and formed disulfide bonds (R–S–S–R). As a model reaction, oxidation of 4-methoxybenzenethiol was continued with a yield of more than 99% in a 2 mL PE milling jar equipped with 4 mm SS balls on BaTiO3 (<3 μm) under air within 6 min. A yield of 99% was obtained on BaTiO3 (200 nm) and BaTiO3 (0.6–1 mm), showing no significant effect of size on the reaction yield. Only 20% of disulfide was obtained when BaTiO3 (<3 μm) was agitated by US instead of BM. BaTiO3 could be recycled 20 times with no loss of reactivity. He et al. (2023)213 synthesized C3-acylated quinoxalin-2(1H)-ones through piezo-catalyzed decarboxylative acylation of quinoxalin-2(1H)-ones with α-oxo carboxylic acids, as a green acylating reagent, under BM over tet-BaTiO3 particles (0.6–1 μm) (Fig. 11C). The reaction of 1-methylquinoxalin-2(1H)-one and 2-oxo-2-phenylacetic acid was used as a model and (NH4)2S2O8 was used as an external oxidant. The highest yield of the product (98%) was obtained from the reaction in a 2 mL PE milling jar with three 5 mm SS balls within 18 min under 3800 rpm with the use of NaHCO3 base and 0.5 mmol of BaTiO3. The reaction under air without using (NH4)2S2O8 gave no product. BaTiO3 could be recycled three times without a substantial loss in the yield of the product. As a plausible mechanism as shown in Fig. 11D, the agitated BaTiO3 reduced (NH4)2S2O8 and formed SO4˙−, which then received a single electron from the 2-oxo-2-phenylacetic acid anion (1) and produced SO42− and the corresponding benzoyl radical (2) along with CO2. The benzoyl radical was added to quinoxalin-2(1H)-one to form a nitrogen radical intermediate (a), which underwent a 1,2-hydrogen shift to produce the carbon radical intermediate (b). Further oxidation of (b) by holes provided the carbon cation intermediate (c). Finally, the final product (d) was achieved after deprotonation of (c).
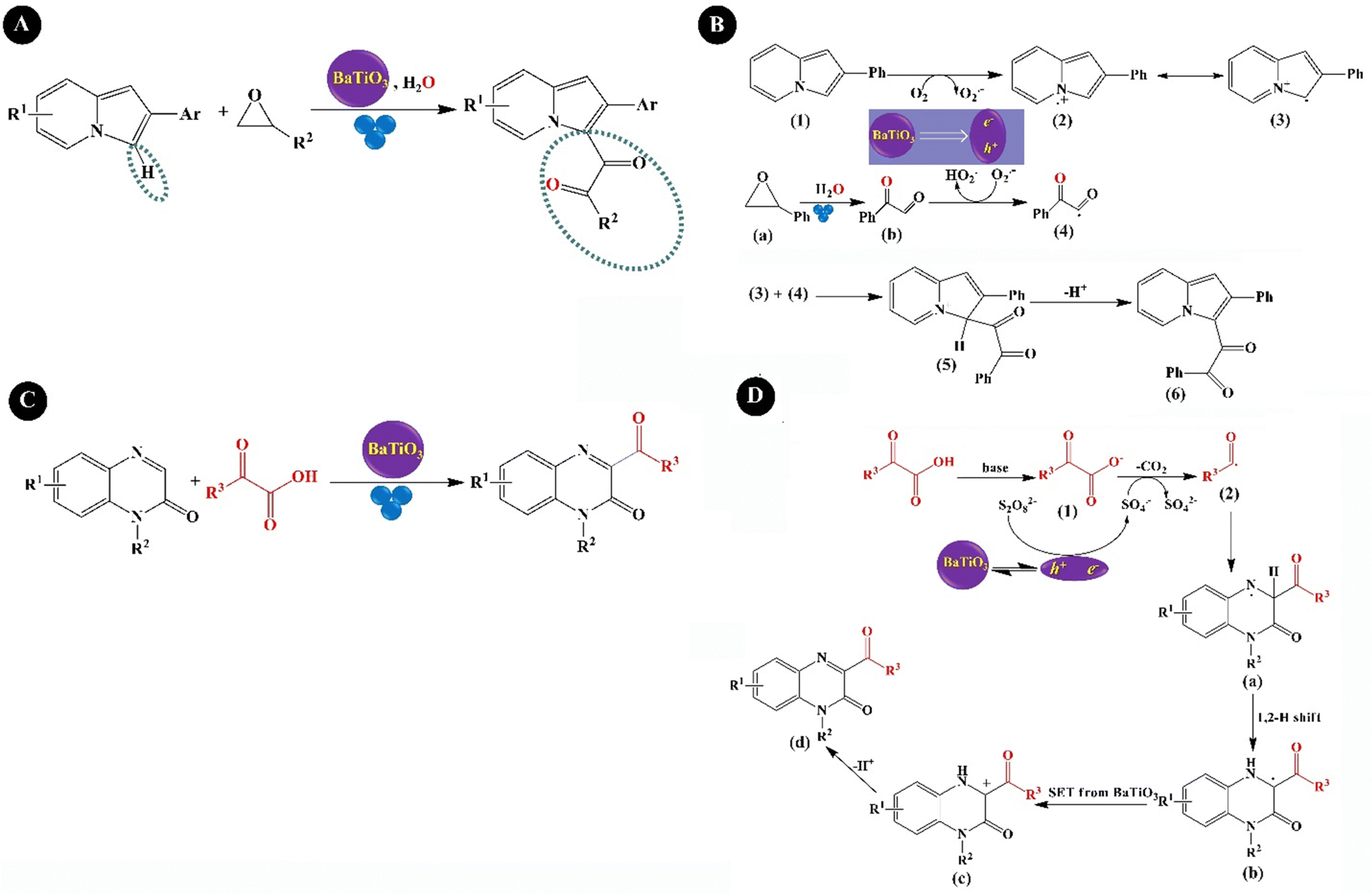 | ||
| Fig. 11 The mechano-induced synthesis procedure (A) and mechanism (B) of 1,2-diketoindolizine from indolizines and epoxides over BaTiO3 (adapted from ref. 211 with permission from American Chemical Society). The mechano-induced synthesis (C) and mechanism (D) of C3-acylation quinoxalin-2(1H)-ones (adapted from ref. 213 with permission from American Chemical Society). | ||
Lv et al. (2022)214 presented, for the first time, a highly regioselective divergent synthesis of α-arylacylamides over BaTiO3 (3 μm) with a yield of 45–89% and oxindoles over PbTiO3 (2 μm) with a yield of 71–92% in the presence of the Cu(II) precatalyst under BM agitation (30 Hz) (Fig. 12A). Jiang et al. (2022)215 reported, for the first time, the mechano-induced C(sp2)–H arylation of quinoxalin(on)es with supporting diaryliodonium salts and BaTiO3 under BM (8 balls of 10 mm size, 20 mL jar, 35 Hz) (Fig. 12B). The highest yield of production was obtained under the optimum conditions of BaTiO3 (<4 μm, 6.0 equiv.), NEt3 (LAG solvent, 3.0 equiv.), and BM agitation (2 h). The plausible mechanism is shown in Fig. 12C. The highly polarized BaTiO3 reduced diaryliodonium salts (2) to produce the aryl radical (I), which was subsequently added to quinoxalin(on)es providing a radical addition intermediate (II). Then, intermediate (II) experienced a 1,2-hydrogen shift, and finally, the target products were obtained by deprotonation of the carbocation (III).
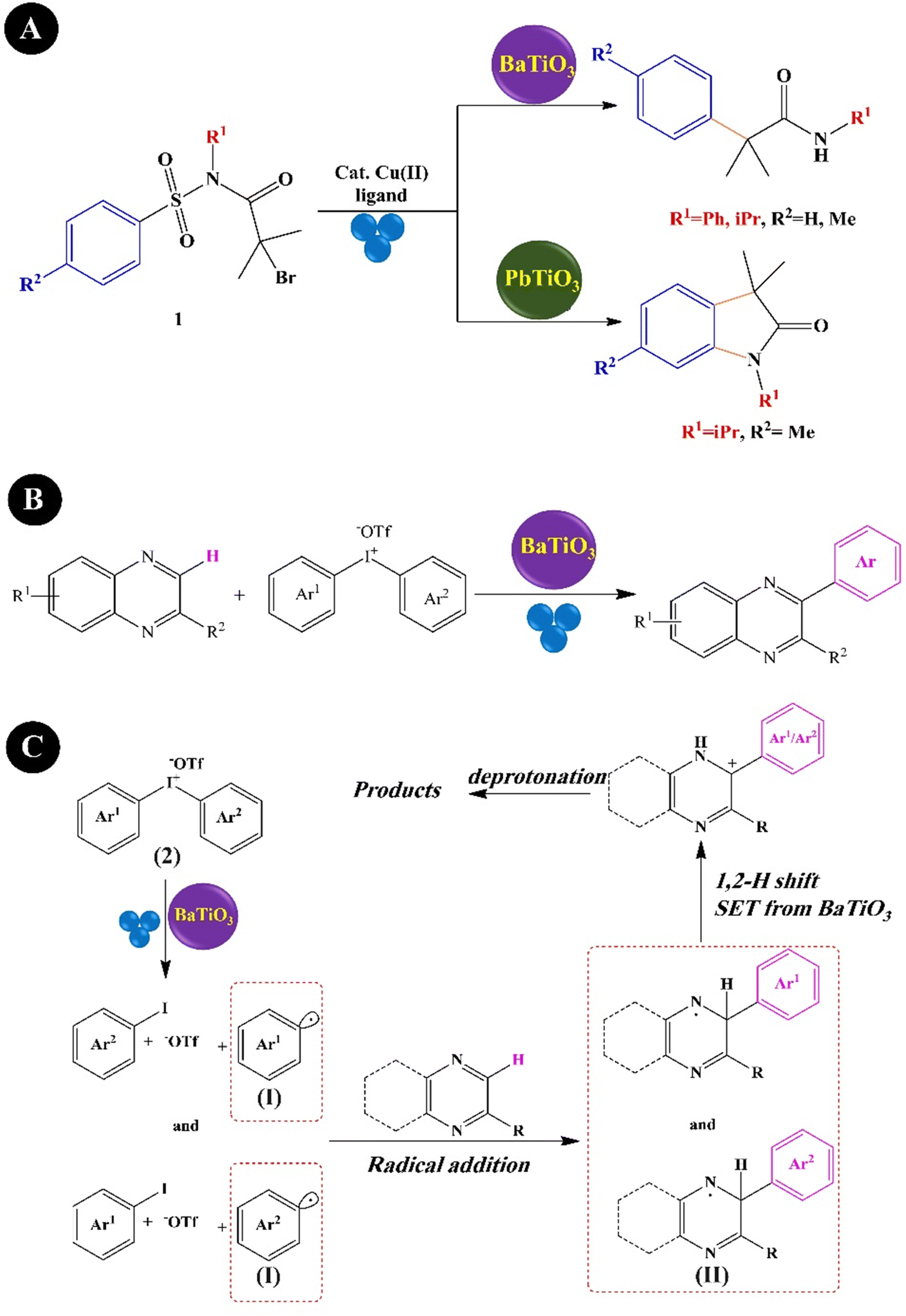 | ||
| Fig. 12 Mechano-induced synthesis of α-arylacylamides and oxindoles (A) (adapted from ref. 214 with permission from John Wiley and Sons). The procedure (B) and mechanism (C) of mechano-induced C(sp2)–H arylation of quinoxalin(on)es (adapted from ref. 215 with permission from Elsevier). | ||
Song et al. (2022)151 reported the piezocatalytic oxidation of toluene to phenol in a one-step method without excessive oxidation, over barium strontium sulfate (Ba0.75Sr0.25SO4, hexagonal crystal structure, 1–3 μm) agitated by BM under air. The highest yield of phenol production was 55.6% with 50 mg of the piezocatalyst. Under a pH of 5, the yield of phenol production reached 59.4%. At a rotating speed of 150 rpm, no phenol formation was observed, and at 600 rpm, the yield of production reached 55.6%. At 300 rpm and 450 rpm, the phenol production yields were 5.6% and 20.6%, respectively. The yield of production reached 55.6% under a BM time of 3 h and partial conversion of phenol to benzoquinone was observed in more than 3 h. At a toluene concentration of 0.1 mol mL−1, the yield of phenol production was 55.6% and at concentrations higher or lower than 0.1 mol mL−1, the yield of production decreased. O2˙− and electrons were determined as the main species for the oxidation reaction. Ding et al. (2022)120 reported the first example of piezoelectric MOFs for organic redox reactions. They synthesized structurally stable, inexpensive, and easily handled Zr-based UiO-66 and UiO-66-NH2 MOFs (octahedral, 180–220 nm) hydrothermally through the precursors ZrCl4 and terephthalic acid/2-amino terephthalic acid and used them for the reaction of diazonium salts and heteroarenes/bis(pinacolato)diboranes under BM (5 ml milling jar, 7.5 mm diameter milling balls). UiO-66 (Zr)-NH2 showed more piezoelectric activity than UiO-66 (Zr). The borylation reaction between 4-bromobenzenediazonium tetrafluoroborate and bis(pinacolato)diborane as well as the arylation reaction between 4-bromobenzenediazonium tetrafluoroborate and furan were investigated as the models. The highest yields of the borylated product (67% over UiO-66 and 78% over UiO-66-NH2) were received under the optimized conditions of 5 mL jar, DMF solvent, and 1 h reaction. The yields of the arylated products were 64% over UiO-66 and 68% over UiO-66-NH2 under the same conditions. All of the piezo-catalyzed organic reactions were carried out under BM with a high yield of production. Using the LAG additive and increasing the size of the jar and balls and the number of balls increased the yield of production significantly in most of the cases. Overall, piezocatalysis, a cutting-edge field in chemistry, has gained significant attention due to its potential for accelerating chemical reactions through mechanical forces. One intriguing aspect within piezocatalysis is the phenomenon of electron/hole piezocatalysis, which holds great promise for revolutionizing chemical reactions. The interplay of various factors, such as crystal structures, surface defects, and lattice vibrations, influences the generation and migration of charge carriers. This intricate dance of electrons and holes creates a fascinating environment for chemical reactions to unfold. Table 1 provides a summary of up-to-date studies of various piezoelectrically mediated chemical reactions.
| Piezoelectric material(s) | Preparation technique(s) | Mechanical energy source | Application and efficiency | Ref. |
|---|---|---|---|---|
| tet-BaTiO3 particles | Hydrothermal synthesis | US (40 kHz, 110 W, 120 min) | – 4-CP degradation of 71.1% | 165 |
| tet-BaTiO3 ceramics (∼0.5 μm) | Calcination of a mixture of BaCO3 and TiO2 (solid oxide reaction) | BM (Teflon-lined milling jar of 125 mL, 15 Zr balls, 300 rpm, 60 min) | – MB degradation of 77% | 150 |
| – BaTiO3 nanowires (100 nm) (BTO-1) | – Hydrothermal reaction | US (40 kHz, 80 W, 160 min) | – MO degradation in the sequences of BTO-1 > BTO-2 >BTO-3 | 168 |
| – BaTiO3 NPs (quadrilateral shape, 200 nm) (BTO-2) | – Hydrothermal reaction | |||
| – Commercial BaTiO3 NPs (BTO-3) | – Purchased from Aladdin Industrial Corporation | |||
| tet-BaTiO3 NPs (500 nm) | Purchased from US Research Nanomaterials | BM (ZrO2 milling jar, 8 ZrO2 balls with a diameter of 5 mm, 25 Hz, 90 min, argon) | – Electric-assisted ball milling-induced copper-catalyzed ATRC with the yield of 97% | 209 |
| BaTiO3 NPs (cubic crystalline phase, <75 μm) | Purchased from Sigma-Aldrich | – BM (jar volume 5.0 mL, ball diameter 7.5 mm, 30 Hz, 1 h, air) | – The arylation product yield of 82% | |
| – BM (jar volume 1.5 mL, ball diameter 5 mm, 30 Hz, 3 h, air, MeCN) | – The borylation product yield of 89% | 208 | ||
| tet-BaTiO3 particles (<3 μm) | Purchased from Sigma-Aldrich | BM (jar volume 5 mL, ball diameter 7.5 mm, 30 Hz, 3 h, acetone) | – The yield of 80% for C–H trifluoromethylation of aromatic compounds | 210 |
| – BaTiO3 (<3 μm) | Commercially available | BM (2 mL PE milling jar, steel balls with a diameter of 4 mm, 6 min, air) | – The reaction yield of >99% for aerobic oxidation of thiols to disulfides | 212 |
| – BaTiO3 (200 nm) | – The reaction yield of 99% for aerobic oxidation of thiols to disulfides | |||
| – BaTiO3 (0.6–1 mm) | – The reaction yield of 99% for aerobic oxidation of thiols to disulfides | |||
| tet-BaTiO3 particles (0.6–1 μm) | Commercially available | BM (2 mL PE milling jar, three SS balls with a diameter of 5 mm, 18 min, 3800 rpm, NaHCO3) | – The yield of 98% for synthesis of C3-acylation quinoxalin-2(1H)-ones | 213 |
| BaTiO3 particles (cubic, <3 μm) | — | BM (SS milling jar of 20 mL, 30 SS balls of 6 mm diameter, 550 rpm, 6 h) | – The yield of 87% for synthesis of 1,2-diketoindolizine | 211 |
| BaTiO3 (<4 μm) | Purchased | BM (20 mL jar, 8 balls of size 10 mm, 35 Hz, 2 h, NEt3) | – The yield of 78% for C(sp2)–H arylation of quinoxalin(on)es | 215 |
| BaTiO3 nanocubes (10 nm) | Hydrothermal synthesis | US (60 kHz) | – H2 production rate of 655 μmol g−1 h−1 and O2 production rate of 316 μmol g−1 h−1 | 155 |
| tet-BaTiO3 NPs (200 nm) | Purchased from US Research Nanomaterials, inc. | US (20 kHz, 2 h, pulse mode operation of 4 s on – 4 s off) | – Ultrasound-mediated atom-transfer radical polymerization (ATRP) of BA | 192 |
| tet-BaTiO3 NPs (200 nm, 4.5 wt%) | Purchased from US Nano | US (40 kHz, 70 W, 50 °C) | – Mechano-ATRP polymerization of MA with > 90% of monomer conversion | 191 |
| BaTiO3 (cubic 50 nm and tetragonal 200 nm, 4.5 wt%) | Purchased from US Nano | US (40 kHz, 8 h) | – Mechano-ATRP polymerization of MA with monomer conversion of 67% over cubic and 61% over tetragonal | 194 |
| tet-BaTiO3 (200 nm, 10 wt%) | Purchased from US Research Nanomaterials Inc. | US (40 kHz, 24 h) | – Production of a linear polytriazole with high azide conversion (>95%) | 204 |
| – Cubic BaTiO3 (<100 nm size, 7 wt%) | Purchased from US Research Nanomaterials | – US (40 kHz, 70 W, 24 h) | – RAFT polymerization of BA with monomer conversion of >65% | 203 |
| – Cubic BaTiO3 (<100 nm size, 15 wt%) | – BM (1.5 mL jar, 5 mm diameter ball, 30 Hz, 3 h) | – RAFT polymerization of tBA with monomer conversion of >95% | ||
| – Cubic BaTiO3 (<100 nm size, 15 wt%) | – Vortex (1.5 mL vial, two balls with a diameter of 5 mm, 3000 rpm, 8 h) | – RAFT-polymerization of BA with monomer conversion of 70% with one ball and 89% with two balls | ||
| – BaTiO3 (3 μm) | Purchased from Sigma-Aldrich | BM agitation (30 Hz) | – The yield of 45%-89% for divergent synthesis of α-arylacylamides | |
| – PbTiO3 (2 μm) | Purchased from Meyer | – The yield of 71–92% for divergent synthesis of oxindoles | 214 | |
| Porous BaTiO3 ceramics | Calcination of a mixture of BaCO3 and TiO2 at 1200 °C, followed by mixing with PMMA and PVA, making the pellets, and finally sintering | US (150 W, 40 kHz, 330 min) | – MB degradation of 88% over 30% porous BaTiO3 ceramic pellets | 170 |
| Nanofluidic BaTiO3 suspension (cubic and tetragonal phases, 400 nm, 5 mg L−1) | Mixing of BaCO3 and TiO2 powders under high-speed BM, calcinating at 800 °C, again BM, and further calcinating at 1200 °C | US (40 kHz, 60 W, 90 min) | – Hydrogen evolution rate of 270 mmol h−1 g−1 | 157 |
| Ultrasmall tetragonal DSPE–PEG2000 coated BaTiO3 NPs (P-BTO) | Hydrothermal synthesis of oleic acid modified BTO NPs, followed by coating with DSPE–PEG2000 | US (1 MHz) | – Oxygen production through water splitting | 156 |
| Milled BaTiO3 NFs (30 min) | Hydrothermal technique | US (20 min) | – RhB degradation of 94% | 167 |
| BaTiO3 NFs (200 nm) | Electrospinning combined with sol–gel process | US (40 kHz, 60 min) | – RhB degradation of 97.5% | 166 |
| BaTiO3-PDMS composite porous foam (25.0 wt% BaTiO3) | Mixing and heating of PDMS, as-prepared BaTiO3 powder, granulated sugar, and curing agent | US (40 kHz, 400 W, 120 min) | – RhB degradation of 94% | 171 |
| Ba0.85Ca0.15Ti0.9Zr0.1O3 (BCZTO) and Ba0.85Ca0.15(Ti0.9Zr0.1)1−xFexO3 (x = 0, 0.5, 1%) (BCZTO-Fe) ceramics | Solid-state method | US (40 kHz, 70 W) | – MB degradation in the sequences of BCZTO > BCZTO-Fe | 180 |
| Poled Ba0.875Ca0.125Ti0.95Sn0.05O3 (BCT-Sn) | Solid-state reaction | US and UV light (375 nm) | – MB degradation of 98% | 177 |
| Ba0.9Ca0.1CexTi1−xO3 ceramics (x = 0–15%) | Solid-state reaction | US and visible light (420 nm) | – The most RhB, MB, and diclofenac degradation for Ba0.9Ca0.1Ce0.15Ti0.85O3 | 179 |
| Ba0.85Ca0.15Ti0.9Zr0.1O3 ceramics | Solid-state reaction | US and UV light (365 nm) | – RhB degradation of 89% and CIP degradation of 81% | |
| – Antibacterial activity | 31 | |||
| Bi1/2Na1/2TiO3 particles (rhombohedral R3c, S400–S800) | Sol–gel reaction | US (100 W, 40 kHz, 3 h) | – Hydrogen production rate of 1520.10 μmol g−1 for S400, 731.22 μmol g−1 for S500, 556.12 μmol g−1 for S600, 328.79 μmol g−1 for S700, 108.6 μmol g−1 for S800 | |
| US (100 W, 40 kHz, 80 min) | – MB degradation of 98.5% | 161 | ||
| Barium strontium sulfate (Ba0.75Sr0.25SO4, hexagonal crystal structure, 1–3 μm) | Purchased | BM (600 rpm, 3 h, pH = 5) | – The yield of 55.6% for oxidation of toluene to phenol | 151 |
| BiFeO3 square nanosheets (380 nm) | Hydrothermal synthesis | – US (100 W, 1 h) | – Hydrogen production rate of ∼124.1 μmol g−1 | |
| – US (45 kHz, 50 min) | – Decomposition ratio of RhB up to ∼94.1% | 159 | ||
| BiFeO3@CdS-10% NFs with heterostructure | Hydrothermal reaction | US (40 kHz, 300 W, 60 min) | – BPA degradation of 99.7% | 174 |
| BiFeO3@TpPa-1-COF core–shell hybrid | Controlled growth of the TpPa-1-COF APTES-BiFeO3 nanosheets | light irradiation (visible light, λ ≥ 420 nm) and US (100 W, 40 kHz) | – Hydrogen and oxygen production rates of 1416.4 and 708.2 μmol h−1 g−1, respectively | 162 |
| ZnO NPs (10–30 nm) | Purchased from J&K Scientific LTD | US (40 kHz, 8 h) | – Piezo-RAFT polymerization of BA with monomer conversion of 85.9% | 202 |
| ZnO NPs (18 nm) | Purchased from US Nano | US (40 kHz, 8 h) | – Mechano-ATRP polymerization of MA with monomer conversion of 90% | 193 |
| ZnO NPs (18 nm, 5.0 wt%, 2.5 wt%, 1.0 wt%) | Purchased from US Research Nanomaterials Inc. | US (40 kHz, 110 W, 6 h) and low frequency shaking (2 kHz, 130 W, 8 h) | – Synthesis of reversible organogels by forming disulfide bonds | 184 |
| ZnO NPs (20 nm, 9.0 wt%) | Purchased from US Research Nanomaterials Inc. | US (40 kHz, 70 W, 21 h) | – Monomer conversion of 74%, 80%, and 52% for polymerization of BA, t-BA, and MMA, respectively | 205 |
| ZnO NRs (1.2 wt%) | Chemical bath deposition | US (40 kHz, 50 W, 4 h) | – Mechano-ATRP polymerization of MMA with monomer conversion of 70% over NR-3 | 198 |
| – Cubic BaTiO3 (<100 nm size, 7 wt%) | – Purchased from Sigma-Aldrich | – US (40 kHz, 70 W, 20 h) | – t-BA polymerization with monomer conversion of 92% | |
| – ZnO (18 nm, 7 wt%) | – US Research Nanomaterials | – BM (3 h) | – t-BA polymerization with monomer conversion of >95% | |
| – BM (1.5 mL SS jar, SS balls with a diameter of 5 mm, 30 Hz, 20 h) | – t-BA polymerization with monomer conversion of 68% | 148 | ||
| ZnO/BaTiO3 heterostructure | In situ deposition of the zinc precursor on BaTiO3 followed by annealing | US (40 kHz, 500 W, 4 h) | – ATRP polymerization of MMA with monomer conversion of 63.1% | 185 |
| – Hexagonal micro-ZnO (5 wt%) | – Annealing of the ZnC2O4·2H2O precursor | US (35 kHz, 45 °C, 8 h) | – Polymerization of MA with monomer conversion of 75% | |
| – Cubic-phase BaTiO3 (9 wt%) | Purchased | – Monomer conversion of 74% for MA polymerization | 197 | |
| Zr-based UiO-66 and UiO-66-NH2 (octahedral, 180–220 nm) | Hydrothermal synthesis | BM (5 mL SS jar, SS balls with a diameter of 7.5 mm, 30 Hz) | – The borylation yield of 67% over UiO-66 and 78% over UiO-66-NH2 | |
| – The arylation yield of 64% over UiO-66 and 68% over UiO-66-NH2 | 120 | |||
| ZIF-8 NPs (dodecahedron shape with an average diameter of ∼2 μm) | Liquid phase method | US (180 W, 40 kHz, 90 min) | – RhB degradation of 94.5% | 124 |
| MoS2 | Hydrothermal reaction of an acidic solution of Na2MoO4·2H2O and thiourea (1:5 ratio) (pH < 1) at 200 °C |
Water movements of urban drainage | – BTH degradation (after 24 cycles) up to 99.3% in DI water, 94.7% in rainwater, 92.9% in saline water, and 81.3% in sewage | 175 |
4. Challenges and outlook
While electron/hole piezocatalysis is a promising method for enhancing chemical reactions, it is important to note that in certain instances, electrons or holes may function as reagents or auxiliaries rather than catalysts. Despite the great advances and the exciting potential of piezoelectric materials as charge-transfer catalysts for organic reactions, their application in the synthesis of small organic molecules is still highly limited. The cost, toxicity, stability, and scalability concerns regarding piezocatalysts, especially for large-scale applications and in ambient environments, are very challenging. For scaling up the reaction, optimizing the process parameters such as mechanical source frequency and the concentration of piezocatalysts is critical. The current progress in electron/hole catalysis is limited, but further advancements in this field have the potential to revolutionize chemical reactions on a global scale. Besides, much effort still must be devoted to determining the accurate mechanism of piezocatalysis.Fully comprehending the intricate mechanisms involved presents a significant challenge in the realm of electron/hole piezocatalysis. The generation and migration of charge carriers under mechanical stress depend on various factors, such as crystal structures, defects, and vibrations. Understanding these intricate processes is crucial for optimizing catalytic materials and enhancing the efficiency of reactions. Developing efficient and stable catalytic materials for electron/hole piezocatalysis presents another challenge. Identifying materials that can withstand mechanical stress, exhibit high piezoelectric properties, and efficiently generate and transport charge carriers is essential. Ensuring selectivity and control over chemical reactions in electron/hole piezocatalysis is another challenge. The generation of charge carriers can lead to a complex network of reactions, making it challenging to achieve specific product formation. Developing strategies to manipulate and direct the charge carriers to desired reaction pathways is crucial for practical applications. In addition, maintaining the stability of electron/hole piezocatalytic systems over extended periods poses a challenge. The materials used for catalysis must exhibit long-term stability under mechanical stress, temperature variations, and other environmental conditions. Ensuring the durability and performance of these systems is essential for their commercial viability.
5. Conclusion
Electron/hole piezocatalysis is an emerging concept in catalysis that relies on the temporary polarization of piezoelectric materials under mechanical agitation. This polarization leads to the generation of electrons and holes, which can then actively participate in a variety of redox reactions, ultimately resulting in the production of valuable and highly efficient products. By unifying this concept, a new era of catalysis technology will be established, enabling the acceleration of chemical reactions in a more environmentally benign and efficient manner. This advancement will also facilitate the design of novel reactions and aid in overcoming the challenges associated with complex reactions. This study provides an overview of electron/hole catalysis, piezoelectricity, piezoelectric materials, and the different types of piezoceramics (lead-free and lead-based), piezopolymers, and piezocomposites. BaTiO3, ZnO, PVDF, and PAN were described as the most famous piezocatalysts, and MOFs, g-C3N4, BP, ILs, and MXenes were introduced as the recently developed piezoelectric materials. Improving piezoelectricity of polymers was also explained by applying different techniques. Piezocatalysis-mediated reactions and up-to-date studies regarding hydrogen and oxygen production caused by water splitting, degradation of organic dyes existing in water and wastewater, organic synthesis, and controlled polymerization/crosslinking were also discussed. The high yields of oxygen and hydrogen production, dye degradation, and organic reactions, as well as well-controlled polymerization/crosslinking with high molecular weights, low dispersity, and high monomer conversions, were achieved under optimum conditions and optimum piezocatalysts.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful for the support of the Research Council of Alzahra University and University of Tehran in this research.References
- W. Qian, W. Yang, Y. Zhang, C. R. Bowen and Y. Yang, Nanomicro Lett., 2020, 12, 1–39 Search PubMed.
- R. C. McAtee, E. J. McClain and C. R. Stephenson, Trends Chem., 2019, 1, 111–125 CrossRef CAS PubMed.
- Y. Jiao and J. F. Stoddart, Tetrahedron, 2022, 126, 133065 CrossRef CAS.
- R. Francke and R. D. Little, ChemElectroChem, 2019, 6, 4373–4382 CrossRef CAS.
- X. Xu, S. Chen, Z. Wu, Y. Jia, L. Xiao and Y. Liu, Nano Energy, 2018, 50, 581–588 CrossRef CAS.
- N. Meng, W. Liu, R. Jiang, Y. Zhang, S. Dunn, J. Wu and H. Yan, Prog. Mater. Sci., 2023, 138, 101161 CrossRef CAS.
- A. Sharma, U. Bhardwaj and H. Kushwaha, Mater. Adv., 2021, 2, 2649–2657 RSC.
- H. Xia and Z. Wang, Science, 2019, 366, 1451–1452 CrossRef CAS PubMed.
- J. A. Leitch and D. L. Browne, Chem. – Eur. J., 2021, 27, 9721–9726 CrossRef CAS PubMed.
- S. Mishra, L. Unnikrishnan, S. K. Nayak and S. Mohanty, Macromol. Mater. Eng., 2019, 304, 1800463 CrossRef.
- S. K. Mahadeva, K. Walus and B. Stoeber, ACS Appl. Mater. Interfaces, 2014, 6, 7547–7553 CrossRef CAS PubMed.
- M. C. Sekhar, E. Veena, N. S. Kumar, K. C. B. Naidu, A. Mallikarjuna and D. B. Basha, Cryst. Res. Technol., 2023, 58, 2200130 CrossRef CAS.
- Q. Zhou, K. H. Lam, H. Zheng, W. Qiu and K. K. Shung, Prog. Mater. Sci., 2014, 66, 87–111 CrossRef CAS PubMed.
- X. h Du, J. Zheng, U. Belegundu and K. Uchino, Appl. Phys. Lett., 1998, 72, 2421–2423 CrossRef CAS.
- L. Fan, J. Chen, Y. Ren and X. Xing, Inorg. Chem., 2018, 57, 3002–3007 CrossRef CAS PubMed.
- K. Takagi, J. F. Li and R. Watanabe, KONA Powder Part. J., 2003, 21, 234–241 CrossRef CAS.
- P. Panda, J. Mater. Sci., 2009, 44, 5049–5062 CrossRef CAS.
- D. R. Baldwin and W. J. Marshall, Ann. Clin. Biochem., 1999, 36, 267–300 CrossRef CAS PubMed.
- P. Dulian, W. Bąk, K. Wieczorek-Ciurowa and C. Kajtoch, Mater. Sci.-Pol., 2013, 31, 462–470 CrossRef CAS.
- F. Moura, A. Z. Simões, B. Stojanovic, M. Zaghete, E. Longo and J. A. Varela, J. Alloys Compd., 2008, 462, 129–134 CrossRef CAS.
- O. Thakur, C. Prakash and A. James, J. Alloys Compd., 2009, 470, 548–551 CrossRef CAS.
- Z. T. Li, B. Han, J. T. Li, M. Li, J. Zhang, J. Yin, L. Y. Lou, S. Chen, J. Chen and J. F. Li, J. Alloys Compd., 2019, 774, 948–953 CrossRef CAS.
- A. Sasaki, T. Chiba, Y. Mamiya and E. Otsuki, Jpn. J. Appl. Phys., 1999, 38, 5564 CrossRef CAS.
- G. J. Lee, B. H. Kim, S. A. Yang, J. J. Park, S. D. Bu and M. K. Lee, J. Am. Ceram. Soc., 2017, 100, 678–685 CrossRef CAS.
- E. Hollenstein, EPFL, 2007, 1–53 Search PubMed.
- Y. Liu, Y. Huang, H. Li, H. Du, H. Tang and G. Zhang, Ceram. Int., 2011, 37, 1959–1965 CrossRef CAS.
- T. Takenaka and H. Nagata, J. Eur. Ceram., 2005, 25, 2693–2700 CrossRef CAS.
- A. Marino, G. G. Genchi, E. Sinibaldi and G. Ciofani, ACS Appl. Mater. Interfaces, 2017, 9, 17663–17680 CrossRef CAS PubMed.
- K. i Mimura and K. Kato, Appl. Phys. Express, 2014, 7, 061501 CrossRef CAS.
- M. Sharma and R. Vaish, Mater. Today Commun., 2020, 25, 101592 CrossRef CAS.
- M. Sharma, V. P. Singh, S. Kumar and R. Vaish, J. Appl. Phys., 2020, 127, 135103 CrossRef.
- P. Kumar, S. Singh, M. Spah, J. Juneja, C. Prakash and K. Raina, J. Alloys Compd., 2010, 489, 59–63 CrossRef CAS.
- W. Rahman, S. Garain, A. Sultana, T. R. Middya and D. Mandal, Mater. Today: Proc., 2018, 5, 9826–9830 CAS.
- S. B. Reddy, K. P. Rao and M. R. Rao, J. Alloys Compd., 2009, 481, 692–696 CrossRef CAS.
- Q. Liu, J. F. Li, L. Zhao, Y. Zhang, J. Gao, W. Sun, K. Wang and L. Li, J. Mater. Chem. C, 2018, 6, 1116–1125 RSC.
- T. Zheng and J. Wu, J. Mater. Chem. C, 2015, 3, 11326–11334 RSC.
- D. Wang, G. Wang, S. Murakami, Z. Fan, A. Feteira, D. Zhou, S. Sun, Q. Zhao and I. M. Reaney, J. Adv. Dielectr., 2018, 8, 1830004 CrossRef CAS.
- J. Xu, T. Qin, W. Chen, J. Lv, X. Zeng, J. Sun, Y. Y. Li and J. Zhou, Nano Energy, 2021, 89, 106317 CrossRef CAS.
- X. Ren, H. Fan, Y. Zhao and Z. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 26190–26197 CrossRef CAS PubMed.
- T. Rojac, A. Bencan, B. Malic, G. Tutuncu, J. L. Jones and J. E. Daniels, J. Am. Ceram. Soc., 2014, 97, 1993–2011 CrossRef CAS.
- G. Ferrero, K. Astafiev, E. Ringgaard, L. S. de Oliveira, B. R. Sudireddy, A. B. Haugen, K. Žiberna, B. Malič and T. Rojac, J. Eur. Ceram. Soc., 2023, 43, 350–361 CrossRef CAS.
- Y. Tang, X. Chen, M. Zhu, X. Liao, S. Hou, Y. Yu and X. Fan, Nano Energy, 2022, 101, 107545 CrossRef CAS.
- X. Xu, L. Xiao, Y. Jia, Z. Wu, F. Wang, Y. Wang, N. O. Haugen and H. Huang, Energy Environ. Sci., 2018, 11, 2198–2207 RSC.
- J. H. Lin, Y. H. Tsao, M. H. Wu, T. M. Chou, Z. H. Lin and J. M. Wu, Nano Energy, 2017, 31, 575–581 CrossRef CAS.
- M. H. Wu, J. T. Lee, Y. J. Chung, M. Srinivaas and J. M. Wu, Nano Energy, 2017, 40, 369–375 CrossRef CAS.
- A. Verma, N. Panayanthatta, A. Ichangi, T. Fischer, L. Montes, E. Bano and S. Mathur, J. Am. Ceram. Soc., 2021, 104, 1966–1977 CrossRef CAS.
- Y. Su, W. Zhang, J. Lan, G. Sui, H. Zhang and X. Yang, ACS Appl. Nano Mater., 2020, 3, 7005–7015 CrossRef CAS.
- Y. Zheng, W. Wang, J. Niu, X. Jin, Y. Sun, L. Peng, W. Li, H. Wang and T. Lin, Nano Energy, 2022, 95, 106995 CrossRef CAS.
- Y. Bar-Cohen, T. Xue, M. Shahinpoor, J. Simpson and J. Smith, Robotocs, 1998, 98, 15–21 Search PubMed.
- A. Arnau and D. Soares, Piezoelectric Transducers Appl., 2008, 1–38 Search PubMed.
- N. Meng, X. Ren, G. Santagiuliana, L. Ventura, H. Zhang, J. Wu, H. Yan, M. J. Reece and E. Bilotti, Nat. Commun., 2019, 10, 4535 CrossRef PubMed.
- R. G. Kepler, Plastics Eng., 1995, 28, 183 CAS.
- B. Zaarour, L. Zhu, C. Huang, X. Jin, H. Alghafari, J. Fang and T. Lin, J. Ind. Text., 2021, 51, 297–340 CrossRef CAS.
- R. Tao, J. Shi, M. Rafiee, A. Akbarzadeh and D. Therriault, Mater. Adv., 2022, 3, 4851–4860 RSC.
- A. J. Lovinger, Science, 1983, 220, 1115–1121 CrossRef CAS PubMed.
- S. Yu, J. Milam-Guerrero, Y. Tai, S. Yang, Y. Y. Choi, J. Nam and N. V. Myung, AACS Appl. Polym. Mater., 2021, 4, 635–644 CrossRef.
- Q. Shi, S. He, Y. He, Y. Wu and R. Liu, J. Appl. Polym. Sci., 2023, 140, e53824 CrossRef CAS.
- Q. Y. Wu, L. S. Wan and Z. K. Xu, J. Membr. Sci., 2012, 409, 355–364 CrossRef.
- E. Fukada, Jpn. J. Appl. Phys., 1998, 37, 2775 CrossRef CAS.
- L. Lebrun, D. Guyomar, B. Guiffard, P. J. Cottinet and C. Putson, Sens. Actuators, A, 2009, 153, 251–257 CrossRef CAS.
- K. S. Ramadan, D. Sameoto and S. Evoy, Smart Mater. Struct., 2014, 23, 033001 CrossRef CAS.
- S. Mathur, J. Scheinbeim and B. Newman, J. Appl. Phys., 1984, 56, 2419–2425 CrossRef CAS.
- A. Kholkin, N. Amdursky, I. Bdikin, E. Gazit and G. Rosenman, ACS Nano, 2010, 4, 610–614 CrossRef CAS PubMed.
- E. Fukada, Biorheology, 1995, 32, 593–609 CAS.
- E. Fukada, IEEE Trans. Dielectr. Electr. Insul., 1992, 27, 813–819 CrossRef CAS.
- P. Frubing, A. Kremmer, W. Neumann, R. Gerhard-Multhaupt and I. L. Guy, IEEE Trans. Dielectr. Electr. Insul., 2004, 11, 271–279 CrossRef.
- J. Harrison, Z. Ounaies and D. M. Bushnell, 2001.
- G. Wu, O. Yano and T. Soen, Polym. J., 1986, 18, 51–61 CrossRef CAS.
- T. Yoshida, K. Imoto, T. Nakai, R. Uwami, T. Kataoka, M. Inoue, T. Fukumoto, Y. Kamimura, A. Kato and Y. Tajitsu, Jpn. J. Appl. Phys., 2011, 50, 09ND13 CrossRef.
- T. Ochiai and E. Fukada, Jpn. J. Appl. Phys., 1998, 37, 3374 CrossRef CAS.
- C. Zhao, J. Zhang, Z. L. Wang and K. Ren, Adv. Sustain. Syst., 2017, 1, 1700068 CrossRef.
- Y. Shiomi, K. Onishi, T. Nakiri, K. Imoto, F. Ariura, A. Miyabo, M. Date, E. Fukada and Y. Tajitsu, Jpn. J. Appl. Phys., 2013, 52, 09KE02 CrossRef.
- N. Wu, X. Cheng, Q. Zhong, J. Zhong, W. Li, B. Wang, B. Hu and J. Zhou, Adv. Funct. Mater., 2015, 25, 4788–4794 CrossRef CAS.
- H. Khanbareh, S. Van Der Zwaag and W. Groen, Smart Mater. Struct., 2015, 24, 045020 CrossRef.
- S. Miyata, M. Yoshikawa, S. Tasaka and M. Ko, Polym. J., 1980, 12, 857–860 CrossRef CAS.
- E. Fukada and I. Yasuda, Jpn. J. Appl. Phys., 1964, 3, 117 CrossRef CAS.
- T. Yucel, P. Cebe and D. L. Kaplan, Adv. Funct. Mater., 2011, 21, 779–785 CrossRef CAS PubMed.
- L. T. Vo and E. P. Giannelis, Macromolecules, 2007, 40, 8271–8276 CrossRef CAS.
- V. Cauda, S. Stassi, K. Bejtka and G. Canavese, ACS Appl. Mater. Interfaces, 2013, 5, 6430–6437 CrossRef CAS PubMed.
- V. Sencadas, R. Gregorio Jr and S. Lanceros-Méndez, J. Macromol. Sci., 2009, 48, 514–525 CAS.
- C. Park, Z. Ounaies, K. E. Wise and J. S. Harrison, Polymer, 2004, 45, 5417–5425 CrossRef CAS.
- J. Y. Ren, Q. F. Ouyang, G. Q. Ma, Y. Li, J. Lei, H. D. Huang, L. C. Jia, H. Lin, G. J. Zhong and Z. M. Li, Macromolecules, 2022, 55, 2014–2027 CrossRef CAS.
- M. Benz, W. B. Euler and O. J. Gregory, Macromolecules, 2002, 35, 2682–2688 CrossRef CAS.
- S. Chen, K. Yao, F. E. H. Tay and C. L. Liow, J. Appl. Phys., 2007, 102, 104108 CrossRef.
- C. M. Wu and M. H. Chou, Polymorphism, Compos. Sci. Technol., 2016, 127, 127–133 CrossRef CAS.
- S. Cheon, H. Kang, H. Kim, Y. Son, J. Y. Lee, H. J. Shin, S. W. Kim and J. H. Cho, Adv. Funct. Mater., 2018, 28, 1703778 CrossRef.
- P. Thakur, A. Kool, B. Bagchi, S. Das and P. Nandy, Phys. Chem. Chem. Phys., 2015, 17, 1368–1378 RSC.
- L. Priya and J. Jog, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 1682–1689 CrossRef CAS.
- R. Bhunia, S. Gupta, B. Fatma, Prateek, R. K. Gupta and A. Garg, ACS Appl. Mater. Interfaces, 2019, 11, 38177–38189 CrossRef CAS PubMed.
- A. A. Issa, M. A. Al-Maadeed, A. S. Luyt, D. Ponnamma and M. K. Hassan, C, 2017, 3, 30 Search PubMed.
- C. Wu and M. Chou, eXPRESS Polym. Lett., 2020, 14, 103–114 CrossRef CAS.
- Y. Feng, Q. Deng, C. Peng, J. Hu, Y. Li, Q. Wu and Z. Xu, J. Mater. Chem. C, 2018, 6, 13283–13292 RSC.
- Y. Feng, Q. Deng, C. Peng and Q. Wu, Ceram. Int., 2019, 45, 7923–7930 CrossRef CAS.
- L. He, J. Sun, X. Wang, C. Wang, R. Song and Y. Hao, Polym. Int., 2013, 62, 638–646 CrossRef CAS.
- D. M. Correia, C. M. Costa, E. Lizundia, R. Sabater i Serra, J. A. Gómez-Tejedor, L. T. Biosca, J. M. Meseguer-Dueñas, J. L. Gomez Ribelles and S. Lanceros-Méndez, J. Phys. Chem. C, 2019, 123, 27917–27926 CrossRef CAS.
- T. Nakanishi, Functional organic liquids, John Wiley & Sons, 2019 Search PubMed.
- J. Barbosa, D. Correia, R. Gonçalves, V. de Zea Bermudez, M. M. Silva, S. Lanceros-Mendez and C. Costa, J. Colloid Interface Sci., 2021, 582, 376–386 CrossRef CAS PubMed.
- G. Moni, A. Mayeen, A. Mohan, J. J. George, S. Thomas and S. C. George, Eur. Polym. J., 2018, 109, 277–287 CrossRef CAS.
- Q. Shi, S. He, Y. He, Y. Wu and R. Liu, J. Appl. Polym. Sci., 2023, 140, e53824 CrossRef CAS.
- D. Dhakras, V. Borkar, S. Ogale and J. Jog, Nanoscale, 2012, 4, 752–756 RSC.
- Y. Su, W. Li, X. Cheng, Y. Zhou, S. Yang, X. Zhang, C. Chen, T. Yang, H. Pan and G. Xie, Nat. Commun., 2022, 13, 4867 CrossRef CAS PubMed.
- F. Huang, Q. Wei, Y. Cai and N. Wu, Int. J. Polym. Anal. Charact., 2008, 13, 292–301 CrossRef CAS.
- M. Smith and S. Kar-Narayan, Int. Mater. Rev., 2022, 67, 65–88 CrossRef CAS.
- S. S. Dani, A. Tripathy, N. R. Alluri, S. Balasubramaniam and A. Ramadoss, Mater. Adv., 2022, 3, 8886–8921 RSC.
- Y. Ahn, J. Y. Lim, S. M. Hong, J. Lee, J. Ha, H. J. Choi and Y. Seo, J. Phys. Chem. C, 2013, 117, 11791–11799 CrossRef CAS.
- C. Kumar, A. Gaur, S. K. Rai and P. Maiti, Nano-Struct., Nano-Obj., 2017, 12, 174–181 CrossRef CAS.
- J. Xue, L. Wu, N. Hu, J. Qiu, C. Chang and S. Atobe, Nanoscale, 2012, 4, 7250–7255 RSC.
- S. Tiwari, A. Gaur, C. Kumar and P. Maiti, Energy, 2019, 171, 485–492 CrossRef CAS.
- D. Mandal, K. J. Kim and J. S. Lee, Langmuir, 2012, 28, 10310–10317 CrossRef CAS PubMed.
- D. Carponcin, E. Dantras, J. Dandurand, G. Aridon, F. Levallois, L. Cadiergues and C. Lacabanne, Adv. Eng. Mater., 2014, 16, 1018–1025 CrossRef CAS.
- J. González-Benito, D. Olmos, J. M. Martínez-Tarifa, G. González-Gaitano and F. A. Sanchez, J. Appl. Polym. Sci., 2019, 136, 47788 CrossRef.
- T. U. Patro, M. V. Mhalgi, D. Khakhar and A. Misra, Polymer, 2008, 49, 3486–3499 CrossRef CAS.
- M. Khalifa and S. Anandhan, ACS Appl. Nano Mater., 2019, 2, 7328–7339 CrossRef CAS.
- F. Narita and M. Fox, Adv. Eng. Mater., 2018, 20, 1700743 CrossRef.
- Z. M. Dang, Y. Q. Lin, H. P. Xu, C. Y. Shi, S. T. Li and J. Bai, Adv. Funct. Mater., 2008, 18, 1509–1517 CrossRef CAS.
- J. Li, B. Seok SIl Chu, F. Dogan, Q. Zhang and Q. Wang, Adv. Mater., 2009, 21, 217–221 CrossRef CAS.
- B. Ploss, B. Ploss, F. Shin, H. Chan and C. Choy, Appl. Phys. Lett., 2000, 76, 2776–2778 CrossRef CAS.
- M. I. Hossain and G. Blanchard, J. Phys. Chem. Lett., 2023, 14, 2731–2735 CrossRef CAS PubMed.
- J. Nyakuchena, S. Ostresh, D. Streater, B. Pattengale, J. Neu, C. Fiankor, W. Hu, E. D. Kinigstein, J. Zhang and X. Zhang, J. Am. Chem. Soc., 2020, 142, 21050–21058 CrossRef CAS PubMed.
- R. Ding, Q. Liu and L. Zheng, Chem. – Eur. J., 2023, 29, e202203792 CrossRef CAS PubMed.
- C. P. Raptopoulou, Materials, 2021, 14, 310 CrossRef CAS PubMed.
- S. Li, J. Jiang, S. H. Ho, F. Li and W. Zeng, Sep. Purif. Technol., 2021, 276, 119259 CrossRef CAS.
- Y. Huang, Y. Zhang, X. Chen, D. Wu, Z. Yi and R. Cao, Chem. Commun., 2014, 50, 10115–10117 RSC.
- L. Ruan, Y. Jia, J. Guan, B. Xue, S. Huang, Z. Wu, G. Li and X. Cui, Sep. Purif. Technol., 2022, 283, 120159 CrossRef CAS.
- Y. Sun, J. Gao, Y. Cheng, Y. W. Zhang and K. Zeng, J. Phys. Chem. C, 2019, 123, 3122–3129 CrossRef CAS.
- C. Zhang, D. Lei, C. Xie, X. Hang, C. He and H. L. Jiang, Adv. Mater., 2021, 33, 2106308 CrossRef CAS PubMed.
- C. Yan, Z. Zhao, W. Jin, Q. Yu, J. Yu, J. Ran, S. Bi, D. Cheng, D. Li and G. Cai, Compos. Commun., 2023, 37, 101466 CrossRef.
- D. Tan, C. Jiang, N. Sun, J. Huang, Z. Zhang, Q. Zhang, J. Bu, S. Bi, Q. Guo and J. Song, Nano Energy, 2021, 90, 106528 CrossRef CAS.
- J. Tan, Y. Wang, Z. Wang, X. He, Y. Liu, B. Wang, M. I. Katsnelson and S. Yuan, Nano Energy, 2019, 65, 104058 CrossRef CAS.
- C. T. Pan, K. Dutt, A. Kumar, R. Kumar, C. H. Chuang, Y. T. Lo, Z. H. Wen, C. S. Wang and S. W. Kuo, Int. J. Bioprinting, 2021, 9, 647 CrossRef PubMed.
- Z. L. Wang and J. Song, Science, 2006, 312, 242–246 CrossRef CAS PubMed.
- X. Xue, W. Zang, P. Deng, Q. Wang, L. Xing, Y. Zhang and Z. L. Wang, Nano Energy, 2015, 13, 414–422 CrossRef CAS.
- R. K. Pandey, J. Dutta, S. Brahma, B. Rao and C. P. Liu, JPhys: Mater., 2021, 4, 044011 CAS.
- F. Güell, A. Galdámez-Martínez, P. R. Martinez-Alanis, A. C. Catto, L. F. da Silva, V. R. Mastelaro, G. Santana and A. na Dutt, Mater. Adv., 2023, 4, 3685–3707 RSC.
- B. Lu, N. Ma, Y. Wang, Y. Qiu, H. Hu, J. Zhao, D. Liang, S. Xu, X. Li and Z. Zhu, J. Alloys Compd., 2015, 630, 163–171 CrossRef CAS.
- W. Ma, J. Lu, B. Wan, D. Peng, Q. Xu, G. Hu, Y. Peng, C. Pan and Z. L. Wang, Adv. Mater., 2020, 32, 1905795 CrossRef CAS PubMed.
- M. Batmunkh, M. Bat-Erdene and J. G. Shapter, Adv. Mater., 2016, 28, 8586–8617 CrossRef CAS PubMed.
- W. Wu, L. Wang, Y. Li, F. Zhang, L. Lin, S. Niu, D. Chenet, X. Zhang, Y. Hao and T. F. Heinz, Nature, 2014, 514, 470–474 CrossRef CAS PubMed.
- J. Qi, Y. W. Lan, A. Z. Stieg, J. H. Chen, Y. L. Zhong, L. J. Li, C. D. Chen, Y. Zhang and K. L. Wang, Nat. Commun., 2015, 6, 7430 CrossRef CAS PubMed.
- J. M. Wu, W. E. Chang, Y. T. Chang and C. K. Chang, Adv. Mater., 2016, 28, 3718–3725 CrossRef CAS PubMed.
- E. Fukada, Wood Sci. Technol., 1968, 2, 299–307 CrossRef CAS.
- M. H. Shamos and L. S. Lavine, Nature, 1967, 213, 267–269 CrossRef CAS PubMed.
- E. Fukada and Y. Ando, J. Polym. Sci., Polym. Phys., 1972, 10, 565–567 CrossRef.
- K. Tao, B. Xue, Q. Li, W. Hu, L. J. Shimon, P. Makam, M. Si, X. Yan, M. Zhang and Y. Cao, Mater. Today, 2019, 30, 10–16 CrossRef CAS PubMed.
- B. Y. Lee, J. Zhang, C. Zueger, W. J. Chung, S. Y. Yoo, E. Wang, J. Meyer, R. Ramesh and S. W. Lee, Nat. Nanotechnol., 2021, 7, 351–356 CrossRef PubMed.
- S. Guerin, A. Stapleton, D. Chovan, R. Mouras, M. Gleeson, C. McKeown, M. R. Noor, C. Silien, F. M. Rhen and A. L. Kholkin, Nat. Mater., 2018, 17, 180–186 CrossRef CAS PubMed.
- S. Komarov, T. Yamamoto, Y. Fang and D. Hariu, Ultrason. Sonochem., 2020, 68, 105236 CrossRef CAS PubMed.
- S. M. Zeitler, P. Chakma and M. R. Golder, Chem. Sci., 2022, 13, 4131–4138 RSC.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- A. Gaur, V. S. Chauhan and R. Vaish, Environ. Sci. Adv., 2023, 2, 462–472 CAS.
- L. Song, T. Zhang, S. Zhang, J. Wei and E. Chen, ACS Sustainable Chem. Eng., 2022, 10, 5129–5137 CrossRef CAS.
- F. Fuso Nerini, J. Tomei, L. S. To, I. Bisaga, P. Parikh, M. Black, A. Borrion, C. Spataru, V. Castán Broto and G. Anandarajah, Nat. Energy, 2018, 3, 10–15 CrossRef.
- A. Kubacka, M. Fernandez-Garcia and G. Colon, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- K. S. Hong, H. Xu, H. Konishi and X. Li, J. Phys. Chem. Lett., 2010, 1, 997–1002 CrossRef CAS.
- R. Su, H. A. Hsain, M. Wu, D. Zhang, X. Hu, Z. Wang, X. Wang, F. T. Li, X. Chen and L. Zhu, Angew. Chem., Int. Ed., 2019, 58, 15076–15081 CrossRef CAS PubMed.
- P. Wang, Q. Tang, L. Zhang, M. Xu, L. Sun, S. Sun, J. Zhang, S. Wang and X. Liang, ACS Nano, 2021, 15, 11326–11340 CrossRef CAS PubMed.
- Y. Zhang, H. Khanbareh, S. Dunn, C. R. Bowen, H. Gong, N. P. H. Duy and P. T. T. Phuong, Adv. Sci., 2022, 9, 2105248 CrossRef CAS PubMed.
- J. M. Park, S. Nakashima, M. Sohgawa, T. Kanashima and M. Okuyama, Jpn. J. Appl. Phys., 2012, 51, 09MD05 CrossRef.
- H. You, Z. Wu, L. Zhang, Y. Ying, Y. Liu, L. Fei, X. Chen, Y. Jia, Y. Wang and F. Wang, Angew. Chem., 2019, 131, 11905–11910 CrossRef.
- Y. Sun, X. Li, A. Vijayakumar, H. Liu, C. Wang, S. Zhang, Z. Fu, Y. Lu and Z. Cheng, ACS Appl. Mater. Interfaces, 2021, 13, 11050–11057 CrossRef CAS PubMed.
- A. Ranjan, K. Y. Hsiao, C. Y. Lin, Y. H. Tseng and M. Y. Lu, ACS Appl. Mater. Interfaces, 2022, 14, 35635–35644 CrossRef CAS PubMed.
- M. L. Xu, M. Lu, G. Y. Qin, X. M. Wu, T. Yu, L. N. Zhang, K. Li, X. Cheng and Y. Q. Lan, Angew. Chem., Int. Ed., 2022, 61, e202210700 CrossRef CAS PubMed.
- F. Bößl and I. Tudela, Curr. Opin. Green Sustain. Chem., 2021, 32, 100537 CrossRef.
- N. K. Amin, J. Hazard. Mater., 2009, 165, 52–62 CrossRef CAS PubMed.
- S. Lan, J. Feng, Y. Xiong, S. Tian, S. Liu and L. Kong, Environ. Sci. Technol., 2017, 51, 6560–6569 CrossRef CAS PubMed.
- X. Xu, Z. Wu, L. Xiao, Y. Jia, J. Ma, F. Wang, L. Wang, M. Wang and H. Huang, J. Alloys Compd., 2018, 762, 915–921 CrossRef CAS.
- Y. Yao, Y. Jia, Q. Zhang, S. Li, G. Li, X. Cui and Z. Wu, J. Alloys Compd., 2022, 905, 164234 CrossRef CAS.
- J. Wu, N. Qin and D. Bao, Nano Energy, 2018, 45, 44–51 CrossRef CAS.
- S. Zhou, J. Hao, M. Zhou, X. Qiao and X. Pang, Appl. Catal., A, 2022, 629, 118406 CrossRef CAS.
- A. Gaur, V. S. Chauhan and R. Vaish, Surf. Interfaces, 2023, 36, 102497 CrossRef CAS.
- W. Qian, K. Zhao, D. Zhang, C. R. Bowen, Y. Wang and Y. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 27862–27869 CrossRef CAS PubMed.
- Y. Chen, S. Lan and M. Zhu, Chin. Chem. Lett., 2021, 32, 2052–2056 CrossRef CAS.
- X. Zhou, S. Wu, C. Li, F. Yan, H. Bai, B. Shen, H. Zeng and J. Zhai, Nano Energy, 2019, 66, 104127 CrossRef CAS.
- J. Long, T. Ren, J. Han, N. Li, D. Chen, Q. Xu, H. Li and J. Lu, Sep. Purif. Technol., 2022, 290, 120861 CrossRef CAS.
- S. Lan, C. Yu, E. Wu, M. Zhu and D. D. Dionysiou, ACS ES&T Eng., 2021, 2, 101–109 Search PubMed.
- N. Yang, S. Yang, Q. Ma, C. Beltran, Y. Guan, M. Morsey, E. Brown, S. Fernando, T. M. Holsen and W. Zhang, Environ. Sci. Technol. Lett., 2023, 10, 198–203 CrossRef CAS PubMed.
- L. Qifeng, M. Jingjun, M. Sharma and R. Vaish, J. Am. Ceram. Soc., 2019, 102, 5807–5817 CrossRef CAS.
- F. Mushtaq, X. Chen, M. Hoop, H. Torlakcik, E. Pellicer, J. Sort, C. Gattinoni, B. J. Nelson and S. Pané, iscience, 2018, 4, 236–246 CrossRef CAS PubMed.
- M. Sharma, A. Halder and R. Vaish, Mater. Res. Bull., 2020, 122, 110647 CrossRef CAS.
- M. Sharma and R. Vaish, J. Am. Ceram. Soc., 2021, 104, 45–56 CrossRef CAS.
- Z. Wang, Z. Wang, X. Pan, L. Fu, S. Lathwal, M. Olszewski, J. Yan, A. E. Enciso, Z. Wang and H. Xia, ACS Macro Lett., 2018, 7, 275–280 CrossRef CAS PubMed.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 199–210 CrossRef CAS PubMed.
- J. Ayarza, Z. Wang, J. Wang and A. P. Esser-Kahn, ACS Macro Lett., 2021, 10, 799–804 CrossRef CAS PubMed.
- S. Xu, W. Zhang, C. Wang, W. Peng, G. Shi, Z. Cui, P. Fu, M. Liu, Y. He and X. Qiao, Polymer, 2022, 252, 124949 CrossRef CAS.
- K. Matyjaszewski and J. Spanswick, Mater. Today, 2005, 8, 26–33 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Macromolecules, 2005, 38, 4139–4146 CrossRef CAS.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 2271–2275 CrossRef CAS.
- A. J. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- P. Chmielarz, M. Fantin, S. Park, A. A. Isse, A. Gennaro, A. J. Magenau, A. Sobkowiak and K. Matyjaszewski, Prog. Polym. Sci., 2017, 69, 47–78 CrossRef CAS.
- Y. N. Zhou, J. J. Li, D. Ljubic, Z. H. Luo and S. Zhu, Macromolecules, 2018, 51, 6911–6921 CrossRef CAS.
- H. Mohapatra, M. Kleiman and A. P. Esser-Kahn, Nat. Chem., 2017, 9, 135–139 CrossRef CAS.
- Z. Wang, X. Pan, L. Li, M. Fantin, J. Yan, Z. Wang, Z. Wang, H. Xia and K. Matyjaszewski, Macromolecules, 2017, 50, 7940–7948 CrossRef CAS.
- Z. Wang, X. Pan, J. Yan, S. Dadashi-Silab, G. Xie, J. Zhang, Z. Wang, H. Xia and K. Matyjaszewski, ACS Macro Lett., 2017, 6, 546–549 CrossRef CAS PubMed.
- L. Wu, U. Glebe and A. Böker, Polym. Chem., 2015, 6, 5143–5184 RSC.
- M. Cvek, M. Mrlík, M. T. Ilčíková, J. Mosnáček, L. S. Münster and V. Pavlínek, Macromolecules, 2017, 50, 2189–2200 CrossRef CAS.
- M. Cvek, J. Kollar, M. Mrlik, M. Masar, P. Suly, M. Urbanek and J. Mosnacek, Polym. Chem., 2021, 12, 5093–5105 RSC.
- K. Liu, W. Zhang, L. Zong, Y. He, X. Zhang, M. Liu, G. Shi, X. Qiao and X. Pang, J. Phys. Chem. Lett., 2022, 13, 4884–4890 CrossRef CAS PubMed.
- M. Zhou, Y. Zhang, G. Shi, Y. He, Z. Cui, X. Zhang, P. Fu, M. Liu, X. Qiao and X. Pang, ACS Macro Lett., 2022, 12, 26–32 CrossRef PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- M. R. Hill, R. N. Carmean and B. S. Sumerlin, Macromolecules, 2015, 48, 5459–5469 CrossRef CAS.
- C. Ding, Y. Yan, Y. Peng, D. Wu, H. Shen, J. Zhang, Z. Wang and Z. Zhang, Macromolecules, 2022, 55, 4056–4063 CrossRef CAS.
- P. Chakma, S. M. Zeitler, F. Baum, J. Yu, W. Shindy, L. D. Pozzo and M. R. Golder, Angew. Chem., Int. Ed., 2023, 62, e202215733 CrossRef CAS PubMed.
- H. Mohapatra, J. Ayarza, E. C. Sanders, A. M. Scheuermann, P. J. Griffin and A. P. Esser-Kahn, Angew. Chem., Int. Ed., 2018, 57, 11208–11212 CrossRef CAS PubMed.
- Z. Wang, J. Ayarza and A. P. Esser-Kahn, Angew. Chem., 2019, 131, 12151–12154 CrossRef.
- G. W. Wang, Chin. J. Chem., 2021, 39, 1797–1803 CrossRef CAS.
- R. K. Fang, Z. C. Yin, J. S. Chen and G. W. Wang, Green Chem. Lett. Rev., 2022, 15, 519–528 CrossRef CAS.
- K. Kubota, Y. Pang, A. Miura and H. Ito, Science, 2019, 366, 1500–1504 CrossRef CAS PubMed.
- C. Schumacher, J. G. Hernández and C. Bolm, Angew. Chem., Int. Ed., 2020, 59, 16357–16360 CrossRef CAS PubMed.
- Y. Pang, J. W. Lee, K. Kubota and H. Ito, Angew. Chem., Int. Ed., 2020, 59, 22570–22576 CrossRef CAS PubMed.
- Y. Wang, Z. Zhang, L. Deng, T. Lao, Z. Su, Y. Yu and H. Cao, Org. Lett., 2021, 23, 7171–7176 CrossRef CAS PubMed.
- G. Wang, J. Jia, Y. He, D. Wei, M. Song, L. Zhang, G. Li, H. Li and B. Yuan, RSC Adv., 2022, 12, 18407–18411 RSC.
- Y. He, G. Wang, W. Hu, D. Wei, J. Jia, H. Li and B. Yuan, ACS Sustainable Chem. Eng., 2023, 11, 910–920 CrossRef CAS.
- H. Lv, X. Xu, J. Li, X. Huang, G. Fang and L. Zheng, Angew. Chem., 2022, 134, e202206420 CrossRef.
- J. Jiang, S. Song, J. Guo, J. Zhou and J. Li, Tetrahedron Lett., 2022, 98, 153820 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |