
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConducting poly(3,4-ethylenedioxythiophene) materials with sustainable carrageenan counter-ions and their thermoelectric properties†
Zhongnan
Duan
 ab,
Joseph
Phillips
a,
Letizia
Liirò-Peluso
a,
Simon
Woodward
a,
Oleg
Makarovsky
b,
Michael P.
Weir
b,
H. Jessica
Pereira‡
*a and
David B.
Amabilino
*c
ab,
Joseph
Phillips
a,
Letizia
Liirò-Peluso
a,
Simon
Woodward
a,
Oleg
Makarovsky
b,
Michael P.
Weir
b,
H. Jessica
Pereira‡
*a and
David B.
Amabilino
*c
aSchool of Chemistry, The GSK Carbon Neutral Laboratories for Sustainable Chemistry, University of Nottingham, Triumph Road, NG7 2TU, UK. E-mail: H.J.Pereira@soton.ac.uk
bSchool of Physics and Astronomy, University of Nottingham, University Park, NG7 2RD, UK
cInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Consejo Superior de Investigaciones Científicas, Campus Universitari de Bellaterra, 08193 Cerdanyola del Vallès, Spain. E-mail: amabilino@icmab.es
First published on 9th October 2023
Abstract
The preparation and properties of conducting polymers comprising poly(3,4-ethylenedioxythiophene) (PEDOT) and two types of carrageenan – each on their own or combined – as counter-ions are described. The aim of the work is to provide alternative, more sustainable materials that can complement the existing variety of conducting polymers based on the same doped poly(thiophene) derivative. The materials were prepared using chemical oxidation of the 3,4-ethylenedioxythiophene (EDOT) monomer in water. The naturally-occurring polymers kappa- and lambda-carrageenan (bearing one and three sulphate groups per disaccharide monomer unit, respectively) were present during the polymerisation, and are proved to be present in the final composite by infrared and X-ray photoelectron spectroscopies and matrix-assisted laser desorption-ionisation mass spectrometry. The materials produced in this work show good conductivity in thin film form by casting from suspensions (between 1 and 14 S cm−1) and in addition show thermoelectric properties that make them attractive for a range of functionalities.
Introduction
The electrically conducting poly(3,4-ethylenedioxythiophene) poly(styrenesulfonate) (PEDOT:PSS, Fig. 1) is an exemplary functional organic material.1,2 The parent PEDOT, discovered3 in 1992, has found widespread applications as an anti-static coating,4 as a component of electroluminescent5 and electrochromic6 devices, capacitors,7 and printed electronics in general.8 While the conducting9 and thermoelectric10 properties of PEDOT:PSS are excellent for a purely organic system, we felt that improvements might be made in the sustainability and even performance by replacing the petrochemically-derived PSS with a readily available natural product.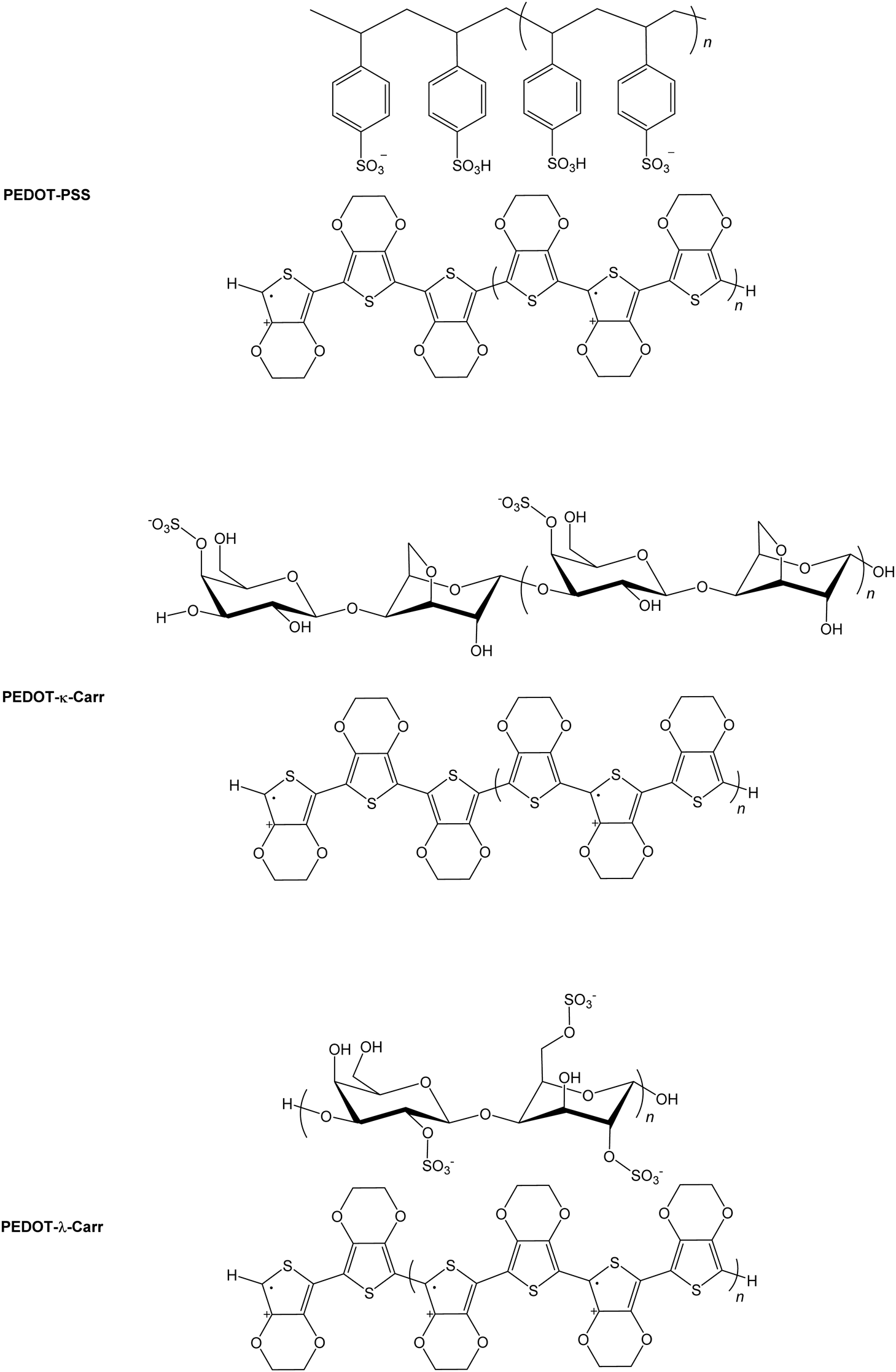 | ||
| Fig. 1 Chemical structures of conducting polymers. The simplified chemical structures of the widely used conducting polymer PEDOT:PSS and the materials prepared in this work, where Carrs are used in place of the synthetic poly(styrene) derivative. | ||
Carrageenans (Carrs) are a family of commercially available and widely used multiply sulphated linear poly(saccharide)s that are extracted from seaweeds.11,12 The variety of derivatives of the linear polymers arise from the number and position(s) of sulphate ester group(s) attachment, and the nature of the 4-linked galactopyranose unit. The materials are generally soluble in warm water. They are used mainly in the food industry,13 but also have applications in medical treatments14 and various films and coatings.15 For our purpose, their existence as sulphate appended polymers made them attractive as replacements for PSS in conducting materials because of the like charge in the sidechains of the macromolecules.
Here, we demonstrate that the use of Carrs, in place of the synthetic PSS, readily affords materials with excellent properties. We focussed on the λ and κ-Carrs. Other materials incorporating Carr and PEDOT have been prepared before,16 in some cases where PEDOT:PSS was immobilized using the poly(saccharide) as a framework,17 or by using the Carr as a template for PEDOT growth.18 One study on the formation of PEDOT-Carr materials focussed on the stabilisation of the dispersion.16 Our aim is to establish materials that can approach the universality of PEDOT:PSS using natural poly(saccharide)s as a sustainable replacement, where the material can be processed through a dispersion. Our results will show that this aim is feasible.
Results and discussion
Preparation of the conducting polymers
The conducting polymers were prepared using iron(III) as the oxidant,19,20 by fully dissolving the chloride salt (FeCl3) in deionised water, followed by dropwise addition to a degassed mixture of 3,4-ethylenedioxythiophene (EDOT) with the respective Carr in deionised water (Scheme 1). After an hour, the rapidly stirred dark blue solutions were allowed to cool and the material was then fractionated. For reasons that will become apparent, the reactions were carried out at ratio of EDOT to Carrs and iron of 1:1:1, 4:1:1, 5:1:1, 7:1:1, 8:1:1 and 16:1:1 (in this last ratio, there are sixteen EDOT monomers for every iron ion and Carr disaccharide monomer unit) and at concentrations of 9.5 and 19 mM of iron salt and Carr in deionized water. The ratio of oxidant (iron salt) to EDOT was varied so as to produce polymers with different numbers of charge carriers, because of varying levels of oxidation of the thiophene residues, and the proportion of the Carr to iron salt was kept constant (we assume that all the iron salt leads to a cation radical that is compensated for by the anionic polymer). The Carr concentration is calculated taking the disaccharide monomer as the molecular weight. At higher concentrations than this value, the products precipitate and agglomerate very quickly, giving an intractable material.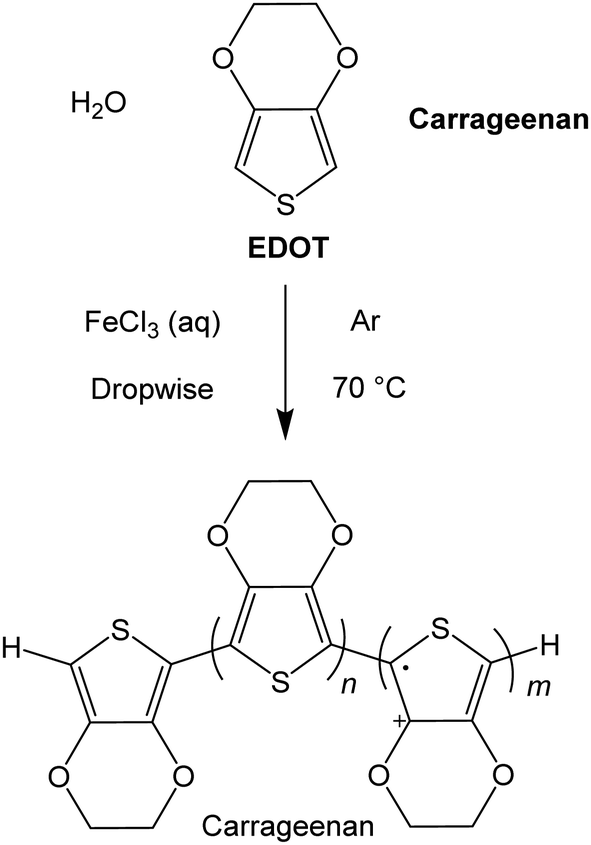 | ||
| Scheme 1 A general synthetic route for the materials prepared here, where Carrageenan refers to either the λ or κ poly(saccharide). | ||
Before the fractionation, a sample of the crude reaction mixture was kept (and noted as fraction A), and the remainder of the material was separated into various fractions by centrifugation (Scheme 2). After removal of the clear blue supernatant liquid (∼80% of the top layer), the part containing the particles at the bottom of the centrifuge tube was diluted and stored as fraction B in the first case. This process was replicated by dilution and repeated centrifugation, providing seven other fractions (labelled here C-I).
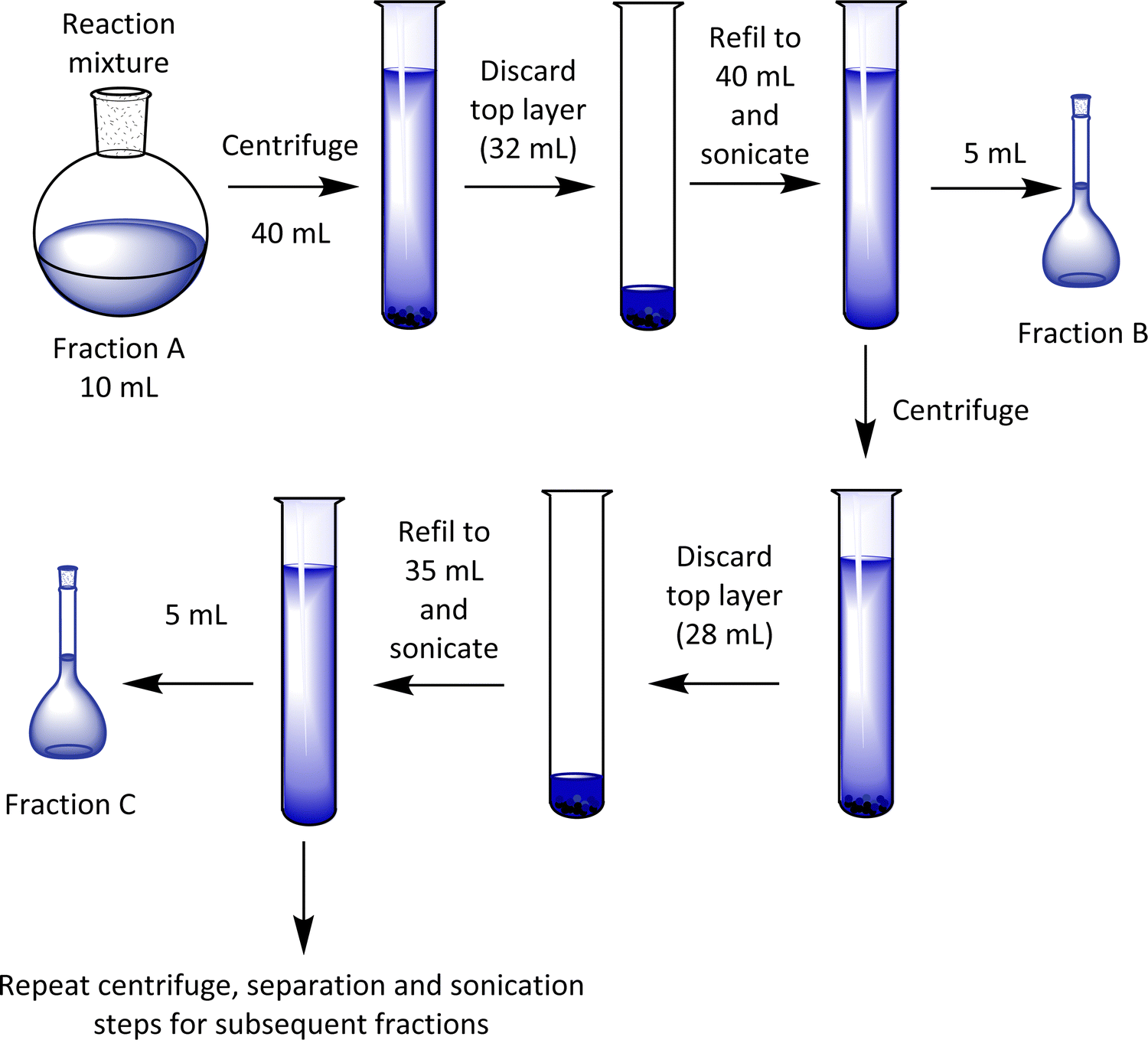 | ||
| Scheme 2 Cartoons showing the separation of fractions of the PEDOT-Carr materials. | ||
The materials can be characterised initially by spectroscopic techniques, most obviously and quickly by the absorption spectra of the suspensions (vide infra), although in our research we sought to focus on the materials that showed the most interesting film-forming ability and electrical properties so as to identify the most promising fractions for giving high conductivity materials that formed homogeneous films once cast onto a substrate. Therefore, we present the film morphology and conductivity first, followed by the characterisation of the spectroscopic properties of the materials in the following sections.
Film morphology and electrical characterization
The suspensions of all the products were cast onto cleaned borosilicate glass slides, where 60 μL of each fraction was drop-cast onto glass with the liquid contained to the desired shape by Kapton tape and an adhesive putty, and the samples were dried in a sealed environment with high humidity (we will refer to it as the “greenhouse method”) and then dried in air. The greenhouse method comprised evaporation in an atmosphere previously saturated with water vapour. For some of the samples, a cracked morphology was observed in the thin films (vide infra), but this drop-casting proved a more reliable and consistent way to obtain a relatively even distribution of material compared with open drying in air or spin-coating. When those latter two methods were used, cracked films were obtained almost without exception, or the films were extremely thin (sub-100 nm).Initial morphology assessment was performed using reflection optical microscopy images (see ESI,† Fig. S2–S4). To judge the wider film homogeneity and detailed morphological features scanning electron microscopy (SEM) of iridium-coated samples were employed (see ESI,† Fig. S5–S9). At the microscopic level, the morphology of the cast films using the sealed high humidity environment (that are between 1 and 3 microns thick) depends on the nature of the Carr used as the counter-ion (see Fig. 2 for fraction E). In general, the most interesting materials were those between fraction C and F, as these are those isolated in the greatest quantity and with the most reliable and facile film-forming characteristics. The fraction E of the materials prepared from both the pure λ- and κ-Carr shows an apparently rough texture with a great deal of fine detail comprising of apparent colloidal strings. These strings form a dense mesh on the substrate when viewed at the scale of a few microns by SEM. There are superficial cracks, but these do not propagate to the interface with the glass support. On the other hand, fraction E of the material prepared using the mixture of Carrs forms an apparently smoother film with somewhat larger colloidal particles (Fig. 2). The morphology of the materials at both the scale of a few and hundreds of microns does depend greatly on the fraction of the material taken (see ESI† Fig. S5–S9).
 | ||
Fig. 2 Morphology of conducting polymers by SEM. The images show scanning electron micrographs of iridium-coated films prepared using PEDOT-κ-, λ-, and Mix-Carrs from the reactions with 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry and always fraction E. We have chosen to present areas with superficial cracks; there are also smooth areas (see the ESI,† Fig. S5–S9). :1 stoichiometry and always fraction E. We have chosen to present areas with superficial cracks; there are also smooth areas (see the ESI,† Fig. S5–S9). | ||
Atomic force microscopy (AFM) was also used to study the materials at a smaller scale and higher resolution than SEM, and to observe the effect of sonication on the dispersions of the polymer particles in water. Generally speaking, the topographic morphology of the films prepared using the sealed high humidity environment and deposited onto clean glass is similar to that observed via SEM, where a complicated pattern of contiguous spherical objects forms interconnected strings over the surface of the material for the pure carrageenan-based materials (Fig. 3–5, and ESI,† Fig. S10–S13). Although slight differences in morphology are observed at a smaller scale (500 × 500 nm range), the roughness of the films is low (<65 nm for an area of 4 × 4 μm). Both PEDOT-κ-Carr and PEDOT-λ-Carr films exhibit apparently similar morphology on the large scale, and both films show depressions, but overall they are homogeneous as evidenced by the phase signal (Fig. 3 and 4, top right inset). However, these AFM images indicate that fraction E of PEDOT-κ-Carr (Fig. 3) forms a smoother film compared to other fractions (Fig. S11, ESI†) as well as other types: PEDOT-λ-Carr and PEDOT-Mix-Carr (ESI,† Fig. S12 and S13). Fractions E (Fig. 4) and D (ESI† Fig. S12) of PEDOT-λ-Carr show negligible differences with fraction E being marginally smoother than that of D. The PEDOT-Mix-Carr derived films are rougher than the corresponding neat materials with films prepared from fraction D (ESI,† Fig. S13) being smoother compared to fraction E (Fig. 5). The corresponding phase images exhibit a continuous and uniform distribution of the material investigated even in areas where the topography images show deep depressions. Notably, the phase images of the PEDOT-Mix-Carr film shows a distinct change in colour (dark or white lines) at the edges of the grains/particulates and this can be attributed to the asperities at the air interface.
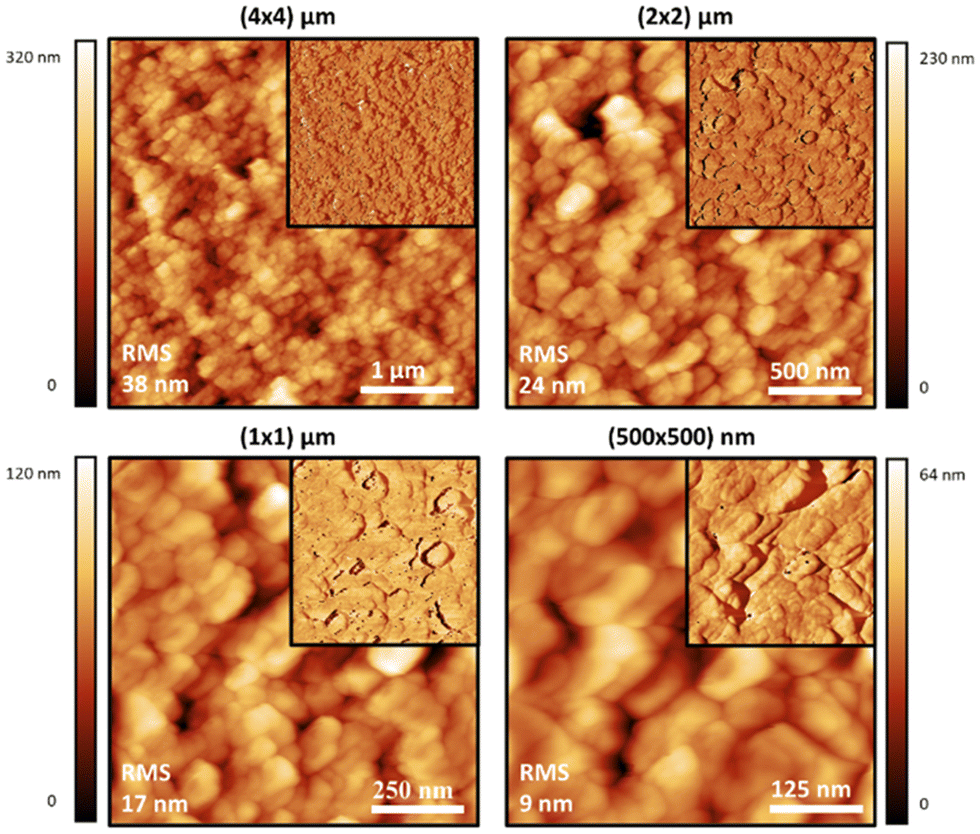 | ||
| Fig. 3 Intermittent contact-mode AFM images of films prepared on glass substrates using an aqueous solution of the PEDOT-κ-Carr (E) formed in the 5:1 stoichiometry reaction which has been sonicated for 10 minutes. Topographic AFM images at different scan sizes and corresponding phase images as top right insets. Roughness is indicated in each image. | ||
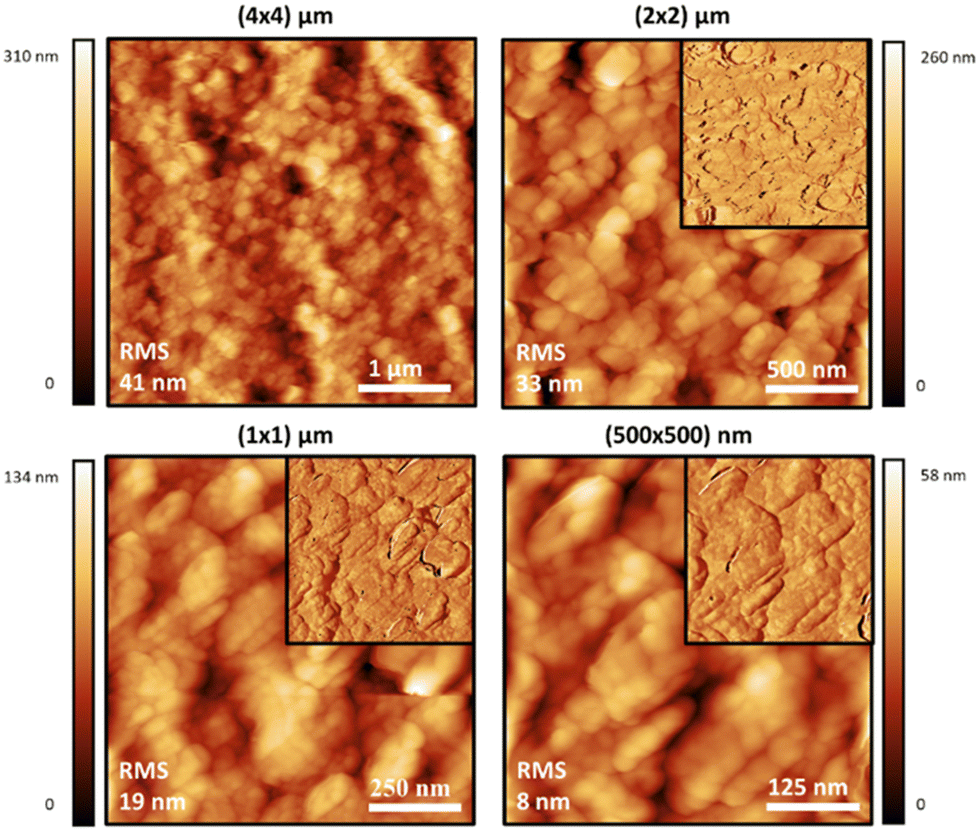 | ||
| Fig. 4 Intermittent contact-mode AFM images of films prepared on glass substrates using an aqueous solution of the PEDOT-λ-Carr (E) formed in the 5:1 stoichiometry reaction which has been sonicated for 10 minutes. Topographic AFM images at different scan sizes and corresponding phase images as top right insets. Roughness is indicated in each image. | ||
 | ||
| Fig. 5 Intermittent contact-mode AFM images of films prepared on glass substrates using an aqueous solution of the PEDOT-Mix-Carr (E) formed in the 5:1 stoichiometry reaction which has been sonicated for 10 minutes. Topographic AFM images at different scan sizes and corresponding phase images as top right insets. Roughness is indicated in each image. | ||
For the electrical characterisation, films deposited on glass were transferred to a non-magnetic PCB (printed circuit board) material holder and connected to the gold-plated contact pins using silver paint and silver wire at four positions perpendicular to the long axis of the strip of the conducting polymer (Fig. 6, top row). The conductivity was measured using the 4-terminal technique which is a typical method to measure the resistance and conductivity of thin films and enables elimination of contact resistance.21 There are four independent electrodes which cross the whole width of the film providing uniform current density across the sample, with current applied to the outer two terminals and the measurement of the resulting potential between the inner two terminals. The conductivity measurements were carried out at room temperature (approximately 20 °C) in ambient air. The values of conductivity for the various fractions will be discussed in the coming paragraphs, and in this context the macroscopic film features should be considered. Apart from the microscopic features of the films discussed above, that vary slightly in their morphology, some of the films show cracks of varying severity, that we have been unable to avoid to date. The phenomenon of cracking in the films is quite common in organic polymer casting.22 The appearance of cracks could be caused by unfavourable solvent evaporation rate, thickness inhomogeneities across the sample and variation in transverse stresses of the drop-cast film.22 For instance, for film thickness below a critical value, final films can be homogeneous while thicker ones have cracks. For the PEDOT-Carr samples reported here, the suspensions with 19 mM concentration cast onto UV/O3 treated glass substrates to give narrow strips using the greenhouse method (to make the evaporation rate slower than in open air) are relatively homogeneous but some minor cracks are still present.
 | ||
| Fig. 6 The non-magnetic PCB holder with a sample connected through four wires bound to a typical film of the conducting polymer composite (top). Reflection optical micrographs of PEDOT-κ-Carr fractions showing the variety of bulk morphologies, with and without cracks (bottom). | ||
Three factors could influence the behaviour of the materials and have an effect on the film characteristics and the resulting conductivity of the PEDOT-Carr materials: (i) the fraction of the material obtained from the reactions, (ii) the ratio of reagents used in their preparation and (iii) the overall concentration. So, the materials with the following ratios of EDOT to Carr of 1:1, 4:1, 5:1, 7:1, 8:1, and 16:1 have very different conductivities, and the films prepared using concentrations of 9.5 and 19 mM of iron salt and Carr in deionized water are also distinct.
The conductivity of the films of the various fractions of PEDOT-κ-Carr prepared using equimolar amounts of oxidant and EDOT and at concentrations of either 9.5 or 19 mM show greatest values in fractions F or E, respectively (Table 1). All of the fractions of the material prepared at 19 mM show an order of magnitude higher conductivity than those prepared at 9.5 mM. The data also show a trend that is, in general, the fractions D, E and F have the higher conductivities across all the samples.
| Fraction of the PEDOT-κ-Carr material | Morphology of films from 9.5 mM reaction | Conductivity of films from 9.5 mM reaction (× 10−2)/S cm−1 | Morphology of films from 19 mM reaction | Conductivity of films from 19 mM reaction (× 10−2)/S cm−1 |
|---|---|---|---|---|
| C | US | 5.1 | MIC | 29 |
| D | US | 4.5 | MIC | 40 |
| E | US | 5.5 | US | 58 |
| F | US | 8.4 | MIC | 31 |
| G | US | 3.5 | MAC | 25 |
The ratio of reagents influences the conductivity of the materials greatly. At a higher concentration of reagents, the most conducting material produced using the κ-Carr poly(saccharide) was achieved with a 5:1 ratio of EDOT monomer to polyanion (Table 2). Across this group of materials, fraction E was slightly more conducting, although the morphology of the materials appeared to be the same. Because the most conducting material produced from κ-Carr poly(saccharide) was achieved with a 5:1 ratio of EDOT monomer to polyanion, we explored the λ-Carr and a mixture of the two sugars.
| Ratio of EDOT:κ-Carr in reaction mixture | Conductivity of fraction D films/S cm−1 | Morphology of fraction D films | Conductivity of fraction E films/S cm−1 | Morphology of fraction E films |
|---|---|---|---|---|
| 1:1 | 0.40 | US | 0.58 | US |
| 4:1 | 2.55 | MIC | 1.63 | MIC |
| 5:1 | 5.60 | MIC | 6.88 | US |
| 7:1 | 1.93 | MIC | 2.43 | MIC |
| 8:1 | 1.86 | MAC | 2.34 | MAC |
| 16:1 | 0.50 | MAC | 2.26 | US |
The effect of the ratio of reagents is seen most dramatically for the materials incorporating the mixture of sugars, which present the highest conductivity of the materials we have explored to date. The materials derived from pure κ-Carr (Table 2) while having conductivities of the same order of magnitude, are approximately half the value of the conducting polymer made with the mixture of Carrs (Table 3), and one third to the pure λ-Carr (Table 4).
| Fraction of the PEDOT-Mix-Carr material | Morphology | Conductivity/S cm−1 |
|---|---|---|
| A | US | 2.91 |
| B | MIC | 6.18 |
| C | MIC | 9.55 |
| D | MIC | 11.79 |
| E | MIC | 8.03 |
| F | MAC | 6.82 |
| G | MIC | 5.41 |
| H | MIC | 5.47 |
| I | MAC | 2.08 |
| Fraction of the PEDOT-λ-Carr material | Morphology | Conductivity/S cm−1 |
|---|---|---|
| C | MIC | 8.27 |
| D | MIC | 9.10 |
| E | MIC | 11.35 |
| F | MAC | 13.88 |
With all these data on morphology and conductivity pointing to certain materials as leads for high conductivity, we now present the spectroscopic characterization of the samples to show the similarities and differences that affect their behaviour.
Spectroscopic characterization
The suspensions of all the products, at representative stages of separation, were characterised using various techniques to ensure the composition and nature of the materials. All the composites are very dark blue, and UV-visible-near infrared (UV-Vis-NIR) absorption spectroscopy gave a qualitative idea of the purity of the samples regarding the content of unreacted monomers. EDOT has absorption bands in the UV region, at around 230 nm and 255 nm.23 This feature is seen most intensely in the UV-Vis-NIR absorption spectra of fraction A from all the reactions (ESI,† Fig. S16–S19), and is also apparent in higher fractions, depending mainly on the stoichiometry of the reagents. The EDOT is evidently removed by fractionation, as the absorption band decreases with greater separation (from fraction A to I, the peak of EDOT at around 280 nm becomes less significant). The pure Carrs show a broad rise in absorbance below 300 nm and deeper into the UV and no absorption peaks in the visible light region (ESI,† Fig. S20). The polymer composite has the characteristic charge transfer absorption band in the NIR that corresponds to the doped conducting PEDOT component.24 The intensity of this band varies according to the oxidant:EDOT ratio used in the polymerisation reaction (ESI,† Fig. S21), indicating different charge carrier densities. A very broad band centred at approximately 800 nm is seen followed by a continuous increase in intensity into the NIR until the limit of the spectrometer used (1200 nm). For the polymer based on PEDOT-κ-Carr, the absorption from EDOT is around 230 nm and 255 nm. But the absorption from the PEDOT radicals is very limited. For the polymer PEDOT-λ-Carr, apart from the EDOT peaks, the absorption from the PEDOT radicals is very obvious in the NIR region.Electron paramagnetic resonance (EPR) spectroscopy proved particularly useful for assessing the paramagnetic nature of the materials as well as the presence of iron ions as an impurity in the polymer. Representative spectra of samples produced from the polymerisation with iron(III) as an oxidant are shown in Fig. 7. The resonance at g = 4.21225 in the spectrum of PEDOT-κ-Carr fraction A is characteristic of Fe(III) (aq) in an octahedral environment (as a hexa-aqua complex, for example)26 and the other much sharper signal, at g = 2.002, is assigned to the radical present in the chains of doped-PEDOT. The EPR spectrum of PEDOT-κ-Carr (H) contains no detectable Fe(III) (aq) and a strong sharp resonance at g = 2.002, which corresponds to the doped-PEDOT. The g-factor for doped-PEDOT in both suspensions are very close to one another and to the literature values27,28 for the material (near the g-factor of the free electron, 2.002).
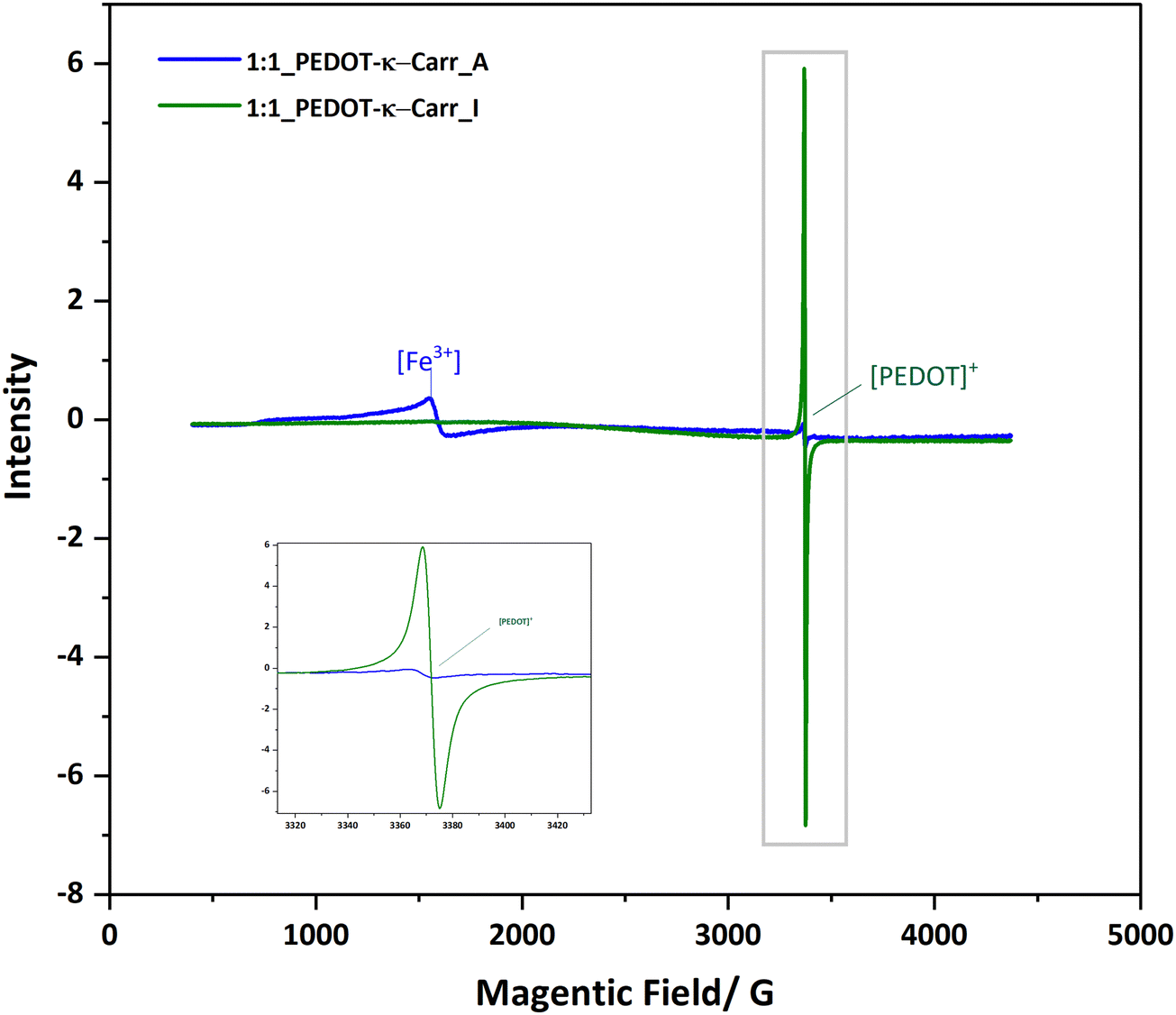 | ||
| Fig. 7 EPR spectra of two representative samples from the iron(III) catalysed reaction recorded at 77 K. | ||
In addition to removing inorganic salts, the centrifugation–dilution procedure (Scheme 2) is shown to give a material with a stronger EPR signal corresponding to the doped-PEDOT radicals. The intensity is shown via the area of the peak in the ESI† (Fig. S22–S25). With the same concentration and measurement conditions, a significant difference between the area of the [PEDOT]+ peaks are probably a result of the reduction of the iron salt after the centrifugations as well as differences in the doping level of the materials.
Matrix-assisted laster desorption-ionization time of flight spectrometry (MALDI-TOF MS) was used to explore the nature of the Carr in the samples. The spectra (ESI,† Fig. S26–S33) show characteristic repeat units displayed, 140 Daltons for the EDOT oligomer series in the positive mode, and corresponding carbohydrate signals in the negative mode. The MALDI-TOF MS spectra of the 5:1 reaction PEDOT-κ-Carr fraction E in a linear negative mode (ESI† Fig. S29) shows the repeat unit of 388 Daltons from the whole κ-Carr monomeric unit. Also, the 145 Dalton fragment separations are from the units from the 3,6-anhydro-D-galactose of the κ-Carr, which is a feature of this poly(saccharide). A difference of 128 Daltons corresponds to a loss of one oxygen atom from the 3,6-anhydro-D-galactose unit (ESI,† Fig. S26). The MALDI-TOF MS spectra of the 5:1 reaction PEDOT-λ-Carr fraction E in the linear negative mode (ESI,† Fig. S31) shows peaks corresponding to the repeating unit of 560 Daltons from the whole λ-Carr monomeric unit. Also, the 257 Daltons fragment is from the 3-linked-D-galactopyranose 2-sulfate (G2S) of the λ-Carr, and the 241 and 225 unit fragments are all from G2S which lose one and two oxygen atoms respectively. For the Mix-Carr (κ-Carr and λ-Carr) composite material, the linear negative mode shows (ESI,† Fig. S32) the presence of fragments from both κ-Carr and λ-Carr components.
To explore the chemical purity of the samples and the stoichiometry of the composite materials, X-ray photoelectron spectroscopy (XPS) was employed (Fig. 8). The pure Carr spectra show the peaks expected for the poly(saccharide) structures with appended sulphate groups, which have potassium counter-ions in part as seen in the C 1s region corresponding to pure κ-Carr. The κ-Carr is shown as a representative example (Fig. 8), where those features can be seen (the other spectra are given in the ESI,† Fig. S34–S36).
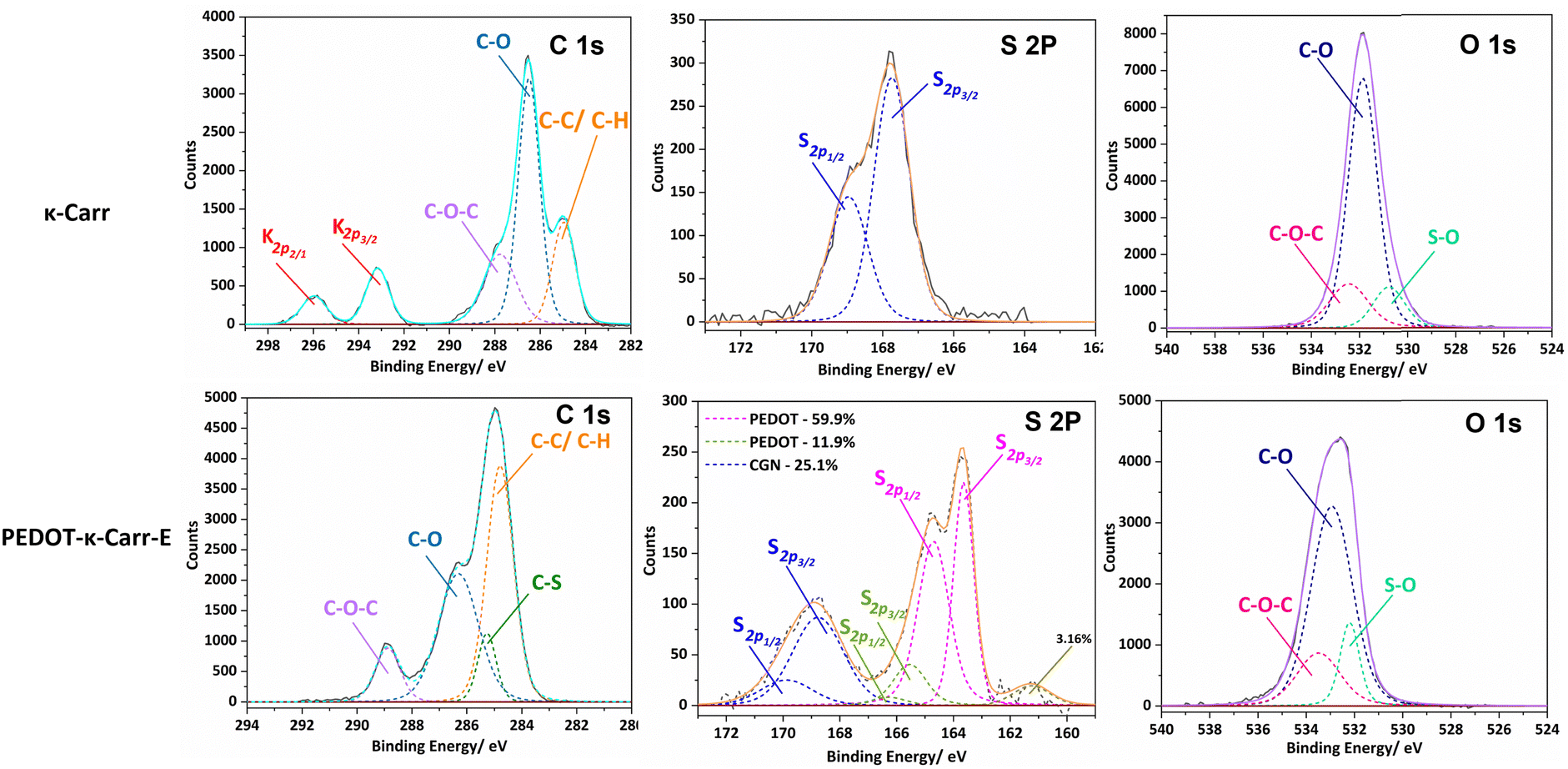 | ||
| Fig. 8 Relevant regions of the XPS spectra of κ-Carr (top) and PEDOT-κ-Carr-E (bottom). In the C1s region, the as-used κ-Carr shows peaks from potassium that is present as a counter-ion. | ||
The spectra of the PEDOT-Carr composites show peaks from the two components, that are particularly evident in the oxygen 1s and sulphur 2p regions. The C 1s peak of PEDOT-κ-Carr composites indicate the presence of C–C/C–H, C–O and C–O–C environments as evidenced by peaks appearing at 285 eV, 286.6 eV and 289 eV which are also present in pure κ-Carr. Also, all the PEDOT-Carr composites show an additional peak corresponding to C–S (285.9 eV) which corroborates the presence of PEDOT in the material. The presence of PEDOT also increases the relative intensities of peaks corresponding to C–C/C–H and C–O compared to the C1s spectra of pure Carrs.
The known PEDOT:PSS XPS spectrum for the sulphur (S2p) envelope29 has overlapping spin orbit doublet peaks (S 2p3/2 and 2p1/2) around 163.5 eV and 165 eV corresponding to the sulphur in the thiophene moiety in PEDOT, and these are also observed in the PEDOT-Carr materials. The overlap of the sulphur XPS spectrum of κ-Carr30 with PEDOT-κ-Carr is evident, with S 2p3/2 and 2p1/2 appearing at 169 eV and 170 eV respectively. Additionally, a third doublet is also observed for PEDOT-Carr materials (Fig. 8 and 9) which could correspond to sulphur atoms in undoped PEDOT moieties.31 The comparison with PEDOT-κ-Carr-E (Fig. 8) and PEDOT-κ-Carr-I (Fig. 9) show that the content of Carr reduced following the centrifugation separation steps (25.1% vs. 22.4%).
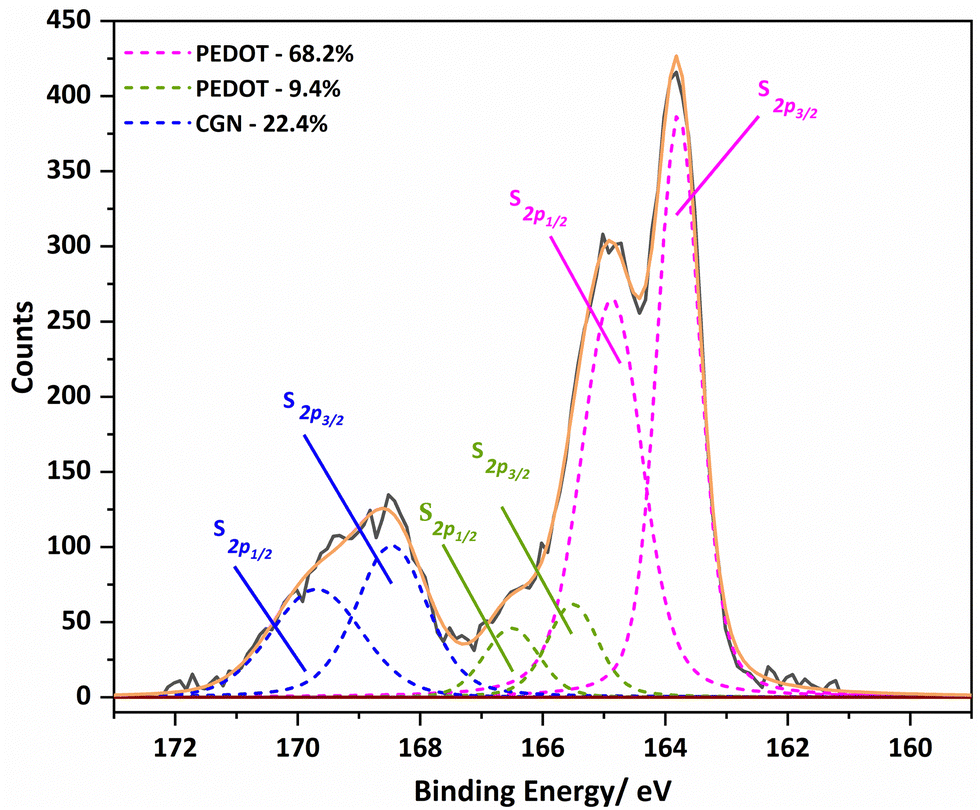 | ||
| Fig. 9 The S 2p region of the XPS spectra of PEDOT-κ-Carr-I with the fitted curve. | ||
The O 1s spectra for PEDOT-Carr materials are similar to the O 1s spectra of the respective Carrs (ESI,† Fig. S34–S36) used as counter-ions except for changes in relative proportions of peaks corresponding to C–O regions caused by the contribution from PEDOT moieties.
The Raman spectra of all the PEDOT materials isolated in this study show the characteristic resonance enhanced bands of the doped conducting polymer.32,33 The most intense signal is the symmetrical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching at approximately 1435 cm−1 that is flanked at a higher wavenumber by the antisymmetric CC stretching (at around 1505 and 1560 cm−1) and at lower wavenumber by the thiophene C–C stretching (1366 cm−1). The other bands seen in the spectra (see Fig. 10 for the fractions E of λ-, κ- and Mix-Carr materials) are all characteristic of doped PEDOT, while the enhanced nature of these bands obscures any signal from the sugar counter-ion.34 The different fractions of the polymers (ESI,† Fig. S37–S39) show quite similar bands, with fraction A having the largest blue shift and therefore the more benzoid structure.35 Fractions E and I have practically identical signals more in line with the quinoid structure of the conducting polymer backbone.36,37
C stretching at approximately 1435 cm−1 that is flanked at a higher wavenumber by the antisymmetric CC stretching (at around 1505 and 1560 cm−1) and at lower wavenumber by the thiophene C–C stretching (1366 cm−1). The other bands seen in the spectra (see Fig. 10 for the fractions E of λ-, κ- and Mix-Carr materials) are all characteristic of doped PEDOT, while the enhanced nature of these bands obscures any signal from the sugar counter-ion.34 The different fractions of the polymers (ESI,† Fig. S37–S39) show quite similar bands, with fraction A having the largest blue shift and therefore the more benzoid structure.35 Fractions E and I have practically identical signals more in line with the quinoid structure of the conducting polymer backbone.36,37
 | ||
| Fig. 10 Raman spectra of films of fractions E of the PEDOT-Carr materials. | ||
Fourier-transform infrared (FT-IR) spectroscopy showed the characteristic carbohydrate absorbance bands in the region 1270–600 cm−1, where the position and intensity of the bands are specific for each poly(saccharide) and were assigned based on previous literature.38,39 The spectra of the composites prepared here are difficult to acquire because of the intense IR absorption arising from the absorption of the PEDOT component,29 which has main peaks for the thiophene C–C bonds at approximately 1516, 1400 and 1350 cm−1 and the C–S bond at about 850, 700 and 580 cm−1 respectively.24
All the FT-IR spectra of the composites (ESI,† Fig. S40–S43) indicate the presence of the PEDOT and of the Carrs, that show an absorption band at around 1250 cm−1 corresponding to the ester sulphate groups. The bands of κ-Carr at 1150 cm−1 and 1050–1010 cm−1 are assigned to C–O and C–C stretching vibrations of the pyranose ring, and the band at around 930 cm−1 to the C–O bond in 3,6-anhydro-D-galactose, which is a feature that defines this poly(saccharide). The band at 845 cm−1 is related to D-galactose-4-sulphate of κ-Carr, and bands at 770, 741 and 694 cm−1 are assigned to the skeleton bending of pyranose (ESI,† Fig. S40). For the PEDOT-κ-Carr materials from the 5:1 reaction, the band around 930 cm−1 can only be seen in κ-Carr and PEDOT-κ-Carr fraction A, and it is less evident in the subsequent fractions (B–I). This observation implies a ring-opening of κ-Carr during the formation of the composites (ESI† Fig. S41).38 For the PEDOT-λ-Carr materials (Fig. S41, ESI†), the band at around 840 cm−1 is related to D-galactose-2-sulfate of the poly(saccharide), and those at about 770, 741 and 694 cm−1 are from the pyranose units. The peak appearing at 920 cm−1 in the λ-Carr spectra (ESI† Fig. S40) is much weaker than that of the κ-Carr since the 5-membered ring of 3,6-anhydro-D-galactopyranose is cleaved and sulfated.38
While there are poly(thiophene) derivatives that display induced circular dichroism (CD) in their structures,40 in the present materials this kind of effect was negligible. This observation is consistent with a model of nanophase separation of the π-functional part and the carbohydrate counter-ion, where the chiral Carr apparently has a very minor influence on the organisation of the doped PEDOT that stacks together in an essentially achiral form in the colloid.
Thermoelectric properties
The thermoelectric properties of the cast strips of the conducting materials were determined by drop casting the PEDOT-Carr suspension on Kapton films and drying under the greenhouse environment. Once the films were dry, they were cut into strips which were 10 × 5 mm, then two silver electrodes were painted across the whole width with a 2 mm gap between them (ESI† Fig. S44). A representative plot of the induced potential as the temperature gradient is increased over time is shown in Fig. 11. A typical thermoelectric response of a PEDOT type material is seen in this graph,41 whereby an increment in the temperature difference across the sample results in an increase in the induced potential.42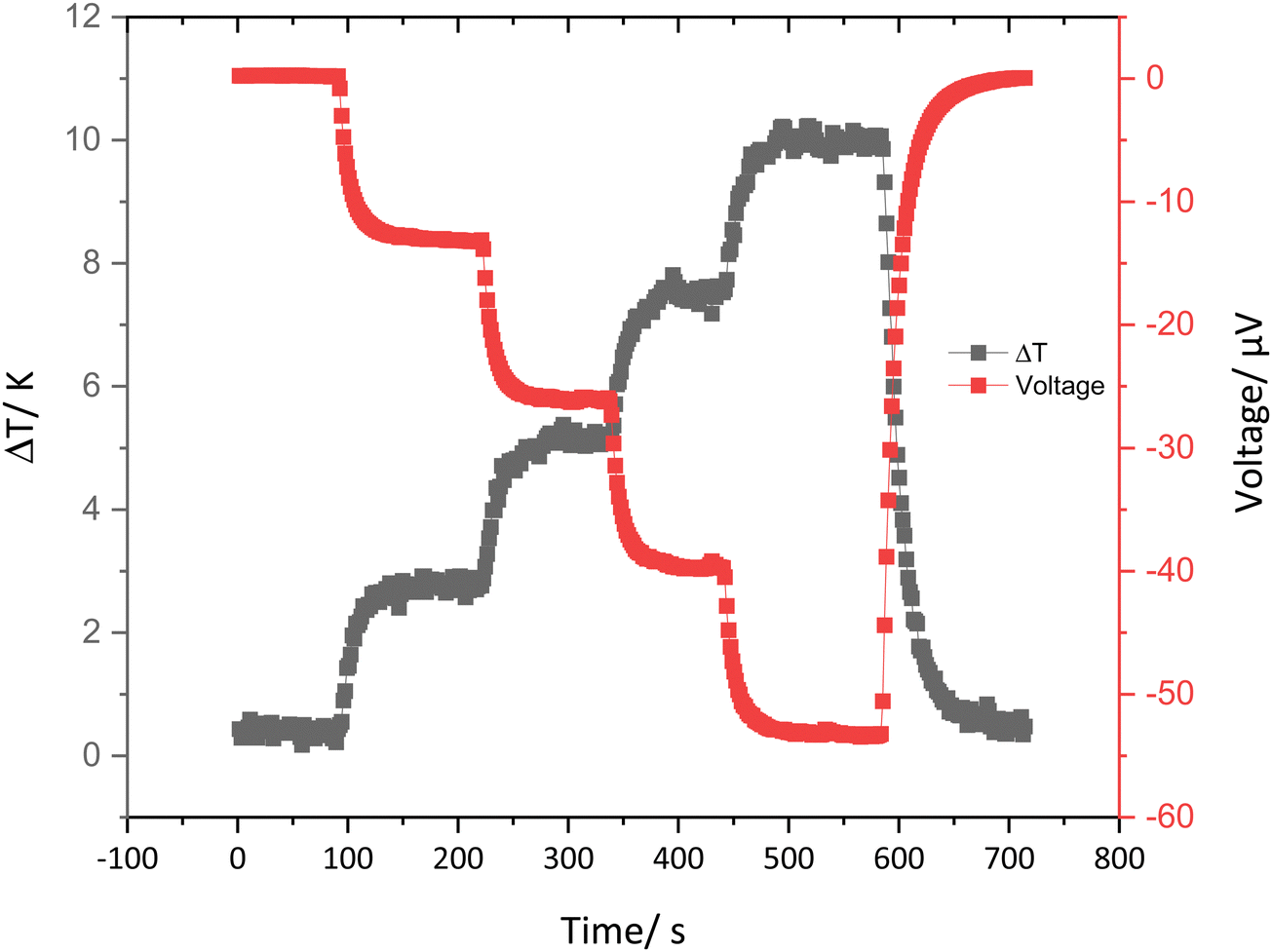 | ||
| Fig. 11 A representative graph showing the ΔT and induced voltage against time for 5:1 PEDOT-κ-Carr (D). | ||
From the data collected on this kind of plot, the Seebeck coefficient can be calculated for the range of materials prepared here. The Seebeck coefficient is a factor that can measure the induced thermoelectric voltage as a function of a temperature difference across the material.43 The data given in Table 5 show that, in general, the values are of the same order – if somewhat lower in magnitude – to the Seebeck coefficient and electrical conductivity of pristine PEDOT:PSS (15–18 μV K−1 and 0.2–1 S cm−1).44,45 Nonetheless, these results do show that materials with thermoelectric properties similar to the standard material can be achieved using a more sustainable counter-ion to the conducting polymer component.
| Seebeck coefficient S (μV K−1) | Fraction C | Fraction D | Fraction E | Fraction F |
|---|---|---|---|---|
| Polymer composite | ||||
| PEDOT-κ-Carr | 5.6 | 7.1 | 1.3 | 11.5 |
| PEDOT-λ Carr | 5.5 | 5.2 | 5.6 | 5.6 |
| PEDOT-Mix-Carr | 7.8 | 7.5 | 8.2 | 6.5 |
Conclusions
It is demonstrated here that naturally occurring carrageenans are viable alternatives for PSS in PEDOT materials. The new systems are electrically conducting and behave as thermoelectric materials,46 showing that the combination of a functional synthetic polymer and natural poly(saccharide) is a promising combination for applications of this kind of conducting material. The morphology of the materials as films on glass depends on the composition of the composites, the smooth films comprise of fused particles. A range of doping levels can be achieved by varying the stoichiometry in the reactions leading to the polymers. This work concentrates on the anion component, and surely others are desirable for the source of the conducting polymer component and the catalyst used to make it. Apart from the step we have made to a more sustainable route, we believe that these new materials may have other characteristics that make them an interesting alternative for other applications of this family of functional polymer materials.Author contributions
The manuscript was written through contributions of all authors. ZD principally and JP preliminarily, purified and characterized the materials. LLP performed the AFM experiments. MPW and OM helped in device fabrication, supervising ZD. SW collaborated and helped in coordination and supervision. HJP supervised ZD and helped in the collection of the XPS data. DBA conceptualized, supervised and coordinated the research and attained funding. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
ZD thanks the China Scholarship Council for a PhD fellowship and the University of Nottingham for a Vice-Chancellor's award. This work was supported by the Propulsion Futures Beacon of Excellence in the University of Nottingham. MPW acknowledges funding from a Nottingham Research Fellowship from the University of Nottingham. DBA thanks the “Severo Ochoa” program for Centres of Excellence CEX2019-000917-S. The authors thank the Nanoscale and Microscale Research Centre (nmRC), Prof. Peter H. Beton for granting access to the Asylum Cypher S AFM in the School of Physics and Astronomy, Dr E. Stephen Davies for recording the EPR spectra, Dr Graham Rance for measuring the Raman spectra and Ben Pointer-Gleadhill in the mass spectrometry service in the School of Chemistry at the University of Nottingham.References
- C. K. Chiang, C. R. Fincher, Y. W. Park, A. J. Heeger, H. Shirakawa, E. J. Louis, S. C. Gau and A. G. MacDiarmid, Phys. Rev. Lett., 1977, 39, 1098–1101 CrossRef CAS.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc. Chem. Commun., 1977, 578–580 RSC.
- G. Heywang and F. Jonas, Adv. Mater., 1992, 4, 116–118 CrossRef CAS.
- F. Jonas and J. T. Morrison, Synth. Met., 1997, 85, 1397–1398 CrossRef CAS.
- Y. Liu, Y. Zhao, S. Xu and S. Cao, Polymer, 2015, 77, 42–47 CrossRef CAS.
- M. Zhao, Q. Zhao, B. Li, H. Xue, H. Pang and C. Chen, Nanoscale, 2017, 9, 15206–15225 RSC.
- R. R. Søndergaard, M. Hösel and F. C. Krebs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 16–34 CrossRef.
- J. C. Gustafsson, B. Liedberg and O. Inganäs, Solid State Ionics, 1994, 69, 145–152 CrossRef CAS.
- Y. H. Kim, J. Lee, S. Hofmann, M. C. Gather, L. Müller-Meskamp and K. Leo, Adv. Funct. Mater., 2013, 23, 3763–3769 CrossRef CAS.
- N. Satoh, M. Otsuka, T. Ohki, A. Ohi, Y. Sakurai, Y. Yamashita and T. Mori, Sci. Technol. Adv. Mater., 2018, 19, 517–525 CrossRef CAS PubMed.
- R. J. Tye, Carbohydr. Polym., 1989, 10, 259–280 CrossRef CAS.
- J. L. Jiang, W. Z. Zhang, W. X. Ni and J. W. Shao, Carbohydr. Polym., 2021, 257, 117642 CrossRef CAS PubMed.
- M. B. Aga, A. H. Dar, G. A. Nayik, P. S. Panesar, F. Allai, S. A. Khan, R. Shams, J. F. Kennedy and A. Altaf, Int. J. Biol. Macromol., 2021, 192, 197–209 CrossRef CAS PubMed.
- B. B. Sedayu, M. J. Cran and S. W. Bigger, Carbohydr. Polym., 2019, 216, 287–302 CrossRef CAS PubMed.
- G. A. Paula, N. M. B. Benevides, A. P. Cunha, A. V. de Oliveira, A. M. B. Pinto, J. P. S. Morais and H. M. C. Azeredo, Food Hydrocoll., 2015, 47, 140–145 CrossRef CAS.
- B. G. Kim, J. H. Lim, J. Y. Kim, C. Frances Glover, T. Watson, D. Bryant, A. M. Obeidat, A. C. Rastogi, A. W. M. Diah, S. Saehana and C. I. Holdsworth, J. Phys. Conf. Ser., 2019, 1242, 012007 CrossRef.
- H. Rastin, B. Zhang, J. Bi, K. Hassan, T. T. Tung and D. Losic, J. Mater. Chem. B, 2020, 8, 5862–5876 RSC.
- R. Zamora-Sequeira, I. Ardao, R. Starbird and C. A. García-González, Carbohydr. Polym., 2018, 189, 304–312 CrossRef CAS PubMed.
- R. Corradi and S. P. Armes, Synth. Met., 1997, 84, 453–454 CrossRef CAS.
- M. Lefebvre, Z. Qi, D. Rana and P. G. Pickup, Chem. Mater., 1999, 11, 262–268 CrossRef CAS.
- E. J. Zimney, G. H. B. Dommett, R. S. Ruoff and D. A. Dikin, Meas. Sci. Technol., 2007, 18, 2067–2073 CrossRef CAS.
- B. S. Tomar, A. Shahin and M. S. Tirumkudulu, Soft Matter, 2020, 16, 3476–3484 RSC.
- Z. Cui, C. Coletta, T. Bahry, J. L. Marignier, J. M. Guigner, M. Gervais, S. Baiz, F. Goubard and S. Remita, Mater. Chem. Front., 2017, 1, 879–892 RSC.
- R. Ben Ishay, Y. Harel, R. Lavi and J. P. Lellouche, RSC Adv., 2016, 6, 89585–89598 RSC.
- R. Berger, J. Kliava, E. M. Yahiaoui, J. C. Bissey, P. K. Zinsou and P. Béziade, J. Non. Cryst. Solids, 1995, 180, 151–163 CrossRef CAS.
- I. Persson, J. Solution Chem., 2018, 47, 797–805 CrossRef CAS PubMed.
- A. Zykwinska, W. Domagala, B. Pilawa and M. Lapkowski, Electrochim. Acta, 2005, 50, 1625–1633 CrossRef CAS.
- W. Domagala, B. Pilawa and M. Lapkowski, Electrochim. Acta, 2008, 53, 4580–4590 CrossRef CAS.
- A. Elschner, S. Kirchmeyer, W. Lovenich, U. Merker and K. Reuter, PEDOT: principles and applications of an intrinsically conductive polymer, 2010 Search PubMed.
- S. B. R. Berton, G. A. M. de Jesus, R. M. Sabino, J. P. Monteiro, S. A. S. Venter, M. L. Bruschi, K. C. Popat, M. Matsushita, A. F. Martins and E. G. Bonafé, Carbohydr. Res., 2020, 487, 107883 CrossRef CAS PubMed.
- M. Fabretto, K. Zuber, C. Hall, P. Murphy and H. J. Griesser, J. Mater. Chem., 2009, 19, 7871–7878 RSC.
- S. Garreau, G. Louarn, J. P. Buisson, G. Froyer and S. Lefrant, Macromolecules, 1999, 32, 6807–6812 CrossRef CAS.
- J. Ouyang, Q. Xu, C. W. Chu, Y. Yang, G. Li and J. Shinar, Polymer, 2004, 45, 8443–8450 CrossRef CAS.
- P. D. A. Pudney, T. M. Hancewicz and D. G. Cunningham, Spectroscopy, 2002, 16, 217–225 CrossRef CAS.
- A. Matsuda, K. G. Nakamura and K. Kondo, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 1–4 Search PubMed.
- W. W. Chiu, J. Travaš-Sejdić, R. P. Cooney and G. A. Bowmaker, Synth. Met., 2005, 155, 80–88 CrossRef CAS.
- I. Cruz-Cruz, M. Reyes-Reyes and R. López-Sandoval, Thin Solid Films, 2013, 531, 385–390 CrossRef CAS.
- M. Şen and E. N. Erboz, Food Res. Int., 2010, 43, 1361–1364 CrossRef.
- M. Černá, A. S. Barros, A. Nunes, S. M. Rocha, I. Delgadillo, J. Čopíková and M. A. Coimbra, Carbohydr. Polym., 2003, 51, 383–389 CrossRef.
- F. G. Bäcklund, A. Elfwing, C. Musumeci, F. Ajjan, V. Babenko, W. Dzwolak, N. Solina and O. Inganäs, Soft Matter, 2017, 13, 4412–4417 RSC.
- C. Liu, J. Xu, B. Lu, R. Yue and F. Kong, J. Electron. Mater., 2012, 41, 639–645 CrossRef CAS.
- X. Wu, N. Gao, H. Jia and Y. Wang, Chem. – Asian J., 2021, 16, 129–141 CrossRef CAS PubMed.
- F. X. Jiang, J. K. Xu, B. Y. Lu, Y. Xie, R. J. Huang and L. F. Li, Chin. Phys. Lett., 2008, 25, 2202–2205 CrossRef CAS.
- Z. Fan and J. Y. Ouyang, Adv. Electronic Mater., 2019, 5, 1800769 CrossRef CAS.
- M. Z. Ali, K. M. K. Ishak, M. A. M. Zawawi, M. Jaafar and Z. Ahmad, Synth. Met., 2022, 286, 117037 CrossRef CAS.
- M. Campoy-Quiles, Phil. Trans. R. Soc. A, 2019, 377, 20180352 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00547j |
| ‡ Current Address: School of Electronics and Computer Science, University of Southampton, Southampton SO17 1BJ, UK. |
| This journal is © The Royal Society of Chemistry 2023 |