
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoron nitride embedded in chitosan hydrogel as a hydrophobic, promising metal-free, sustainable antibacterial material†
Nisrine
Hammi
ab,
Marta
Kędzierska
c,
Natalia
Wrońska
d,
Nadia
Katir
a,
Jeremy
Dhainaut
 b,
Sebastien
Royer
b,
Katarzyna
Lisowska
d,
Maria
Bryszewska
c,
Katarzyna
Miłowska
c and
Abdelkrim
El Kadib
*a
b,
Sebastien
Royer
b,
Katarzyna
Lisowska
d,
Maria
Bryszewska
c,
Katarzyna
Miłowska
c and
Abdelkrim
El Kadib
*a
aEuromed Research Center, Engineering Division, Euro-Med University of Fes (UEMF), Route de Meknes, Rond-point de Bensouda, 30070, Fès, Morocco. E-mail: a.elkadib@ueuromed.org
bUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181-UCCS-Unité de Catalyse et Chimie du Solide, F-59000 Lille, France
cDepartment of General Biophysics, Faculty of Biology and Environmental Protection, University of Lodz, 141/143 Pomorska Street, 90-236 Lodz, Poland
dDepartment of Industrial Microbiology and Biotechnology, Faculty of Biology and Environmental Protection, University of Lodz, 12/16 Banacha Street, 90-236 Lodz, Poland
First published on 20th September 2023
Abstract
Evaporation-induced co-assembly of boron nitride-exfoliated chitosan hydrogel provides micrometer-thick boron nitride-filled chitosan nanocomposite films. Owing to the favorable interfacial interaction of NH2 belonging to chitosan and boron center in boron nitride (BN) sheets, a loading of up to 60 wt% of boron nitride could be entrapped inside, without compromising the film quality or its flexibility. Notably, increasing boron nitride loading alters the wettability of the resulting films and improves its mechanical and thermal properties. Enhanced biological response (e.g., antibacterial properties) also correlates with the entrapped amount of boron nitride, which highlights the potential use of the latter as metal-free, antibacterial surface coating materials.
1. Introduction
The urgent need for more efficient antibacterial and antiviral materials is driven by the widespread of bacteria and virus and the emergence of antibiotic-resistant organisms.1 Most antibacterial and antiviral materials are based on metal and metal oxide-containing nanomaterials.1b,2 Popular examples are silver nanoparticles, zinc oxide, copper oxide, and titanium dioxide.3 While they are proven to be effective, the implementation of these metal-based nanomaterials is fraught with challenges, such as high toxicity, that hinders daily use in places hosting vulnerable persons (e.g., hospitals, nursing home, and kindergarten)4 and their possible accumulation in the environment raises concerns.5 Additional drawback lies on the poor understanding of the exact nature of the antibacterial site (nanoparticles versus clusters and/or ionic atoms), rendering it difficult to establishment a clear correlation between the structure and biological activity.6 Alternatively, synthetic polymers and reactive organic building-blocks,7 particularly those featuring ammonium in their side chains, display promising biological response with additional assets of being structurally well-defined, fully degradable, and less toxic.8 Unfortunately, tailorable functional polymeric architectures required for such purpose are not often easily accessible, with their development being impeded by the dearth of efficient synthetic methods. In most optimistic scenarios, the cost associated with the synthesis of potential derivatives overwhelms to a major extent their benefits.9An alternative solution to these man-made ammonium-based polymers could be the direct use of naturally occurring biomass and more preferably those discarded as bio-waste to avoid additional synthetic chemistry.10 Chitosan meets these requirements owing to the abundance of its source (chitin) for which trivial deacetylation affords an auspicious polymeric backbone of linked 2-amino-2-deoxy-D-glucan and 2-acetamidodeoxy-D-glucan.11 Chitosan is the sole accessible cationic natural polymer, making its interaction with negatively charged species highly favorable. Chitosan also coordinates Lewis acid metals and clusters,12 allowing its use for metal sequestration and heterogeneous catalysis.13 Besides, chitosan can be handled on demand and shaped as transparent film with controllable thickness.14 This flexibility in configuring chitosan substructure14,15 avoids the tyranny of shaping the final material into a suitable end use device, a step that is recognized as very challenging to derive laboratory-successful materials for the market.
Though suffering from low chemical, thermal, and mechanical stability, some of these properties can be improved by associating chitosan with a variety of nano-sized objects, including hydroxyapatite, carbon derivatives (graphene and carbon nanotubes), clay, metal and metal oxide nanoparticles, and cellulose-based micro- and nanocrystals.16 These materials have been used for antibacterial purposes, with a remarkable synergistic contribution of the filler to their biological response in few cases. Unfortunately, some of these fillers lead simultaneously to the hemolysis of human red blood cells, low biocompatibility, and high cytotoxicity, making the exploration of new and innovative filler candidates necessary.
Hitherto marginally explored for the above-mentioned purposes, boron nitride (BN) displays many exciting features that could open more possibilities as metal-free antibacterial materials. Boron nitride is structurally similar to graphene and carbon nanotubes, with additional advantages of being highly polarized and sensitive to radicals. Besides, early studies have claimed that BN nanotubes (NTs) and hexagonal BN are more cytocompatible than their carbon counterparts in vitro and in vivo.17
While graphene and carbon nanotubes have been the focus of extensive biological studies, boron nitride seems to be forgotten, in spite of the above cited advantages.18 We recently initiated a research program revolving around the use of chitosan marine waste for the preparation of sustainable bio-based packaging and antibacterial materials.19 We have explored the holistic association of chitosan with a variety of sophisticated objects including single and mixed metal oxides,20 lamellar and tubular clay nanoparticles,21 surface-modified graphene oxide,22 metal–organic framework,23 and phosphorylated cellulose nanocrystals.24 Prompted by the inherent properties of boron nitride, we herein embarked to investigate the possible embedding of boron nitride in chitosan films through evaporation-induced assembly of their colloidal solution to conceive radical-sensitive packaging films. We have specifically assessed the effect of BN loading on the mechanical, thermal, and chemical properties of the resulting boron nitride-filled chitosan films (CS-BNx-f). We have also evaluated some of their biological properties, including their antibacterial activity against Escherichia coli and Staphylococcus aureus, hemolysis, and cytotoxicity.
2. Experimental
2.1 Materials and reagents
Commercially available reagents and solvents were purchased from Across and Sigma-Aldrich (St. Louis, MO, USA). Chitosan of medium molecular weight (viscosity: 200–800 cps and a deacetylation degree of 75–85%) was purchased from Sigma-Aldrich (Hamburg, Germany). h-BN (CAS number 10043-11-5) was purchased from Sigma-Aldrich.2.2 Preparation of boron nitride-filled chitosan nanocomposite films
A total of 0.1 g chitosan was dissolved in 8 mL aqueous acetic acid (1% v/v). The boron nitride suspension was prepared by dispersing a proper amount of boron nitride in hot water with sonication for 1 h and then gradually added to the chitosan solution, followed by stirring at ambient temperature for 24 h. Boron nitride-filled chitosan solutions were subsequently poured into a plastic Petri dish and dried under room temperature to form films. For comparison, pure chitosan films were also prepared in the same way but without the addition of boron nitride. The obtained films were denoted as CS-BNx-f (x refers to the BN content: x = 1, 3, 5, 10, 30, 40, and 60 wt%).2.3 Characterization
Fourier-transform infrared spectra (ATR FT-IR) were obtained with a PerkinElmer Spectrum 100FT-IR spectrometer on neat samples. X-Ray diffraction (XRD) was performed using a Bruker X-ray AXS D8 Advance diffractometer in Bragg–Brentano configuration and equipped with a LynxEye Super Speed detector. XRD patterns were recorded with Cu Kα radiation (λ = 0.154 nm, 40 kV, 30 mA) in the 10–80° 2θ range with a 0.05° 2θ step. Scanning transmission electron microscopy–energy dispersive X-ray spectroscopy (STEM-EDX) was conducted using a FEG TEM/STEM system (Titan Themis FEI) operated at 300 kV. The microscope was equipped with a monochromator, a super-X windowless four-quadrant silicon drift detector (SDD) for the STEM-EDX mapping, and a probe Cs corrector allowing spatial resolution of about 65 pm. Thermogravimetric analyses (TGA) under air were done with a thermal analyzer instrument Q500 in the range of 25–900 °C with the ramp of 5 °C min−1. Contact angle measurements were recorded using a dynamic contact angle meter (KRUSS GmbH Easy Drop, Kruss GmbH, Hamburg, Germany) equipped with a charge-coupled device camera and using an image capture program employing scat software (VCA Optima, AST Products, Billerica, MA, USA). The cut film (3 cm × 3 cm) was fixed on the top of a dynamic support. A droplet (3 μL) was placed on the film surface, and the change in the contact angles was treated by the software (VCA Optima, AST Products, Billerica, MA, USA) of the apparatus. Each measurement was repeated four times, and their average was considered. Tensile tests of the long films were performed on an Instron testing machine, Model 4466. The samples were drawn at a crosshead speed of 10 mm min−1 using a 100 N load cell at room temperature. A single bundle of films was partially locked at its ends in a stiff cardboard paper, giving a gauge length of 25 mm. Each sample diameter was calculated from an average of three values using a Mitutoyo electronic micrometer.2.4 Biological activity of boron nitride-filled chitosan nanocomposite films
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2000 (with modifications). Bacteria were cultured on Luria–Bertani (LB) medium at 37 °C on a rotary shaker. After the incubation, the test inoculum of S. aureus and E. coli, containing 1 × 105 colony-forming units (CFU per mL) in 500-fold diluted LB medium, was prepared. Next, the bacterial suspension was applied to the tested films of 1 cm × 1 cm. Native, unmodified chitosan films were analyzed as a control sample. After dripping the suspension of selected bacteria on the tested materials (chitosan films), each sample was covered with a sterile film (1 cm × 1 cm). Next, the samples were incubated in the moist chamber for 24 h at 37 °C. After incubation, the samples were put in the sterile tube containing phosphate buffer and vortexed. After that, the films were removed from the tubes, and with the remaining solution, a serial dilution was performed in phosphate buffer. Out of each dilution, 100 μL bacterial suspension was seeded on agar plates and incubated for 24 h at 37 °C. Next, the viable cells of S. aureus or E. coli were counted. Each type of film was tested in triplicate and analyzed individually in three independent experiments.
:2000 (with modifications). Bacteria were cultured on Luria–Bertani (LB) medium at 37 °C on a rotary shaker. After the incubation, the test inoculum of S. aureus and E. coli, containing 1 × 105 colony-forming units (CFU per mL) in 500-fold diluted LB medium, was prepared. Next, the bacterial suspension was applied to the tested films of 1 cm × 1 cm. Native, unmodified chitosan films were analyzed as a control sample. After dripping the suspension of selected bacteria on the tested materials (chitosan films), each sample was covered with a sterile film (1 cm × 1 cm). Next, the samples were incubated in the moist chamber for 24 h at 37 °C. After incubation, the samples were put in the sterile tube containing phosphate buffer and vortexed. After that, the films were removed from the tubes, and with the remaining solution, a serial dilution was performed in phosphate buffer. Out of each dilution, 100 μL bacterial suspension was seeded on agar plates and incubated for 24 h at 37 °C. Next, the viable cells of S. aureus or E. coli were counted. Each type of film was tested in triplicate and analyzed individually in three independent experiments.
| % Hemolysis = As/Ac × 100% |
| Adsorption of Hb = 100% − (As/Ac × 100%) |
| % inhibition = (A0 − A)/A0 * 100% |
3. Results and discussion
3.1 Preparation and characterization of boron nitride-filled chitosan films (CS-BNx-f)
As depicted in Scheme 1, nanocomposite films were prepared starting from water-soluble chitosan solution mixed with a solution of dispersed boron nitride (h-BN) of known weight concentration. The resulting solution was cast onto a clean Petri dish for 24 h until the total evaporation of the solvent. Whatever the amount of h-BN engaged in the initial solution (from 1 wt% to 60 wt%, final dry weight), homogenous, stable, and flexible CS-BNx-f films were obtained, as illustrated in Scheme 1c. The shift from transparent to opaque white films reflects the increased amounts of the filler in the dry composites. The successful entrapment of such a large loading of boron nitride, up to 60 wt%, without compromising the film-forming properties of chitosan or its flexibility could be rooted in the strong interfacial interaction occurring between the filler and the polymer backbone. Considering the highest affinity of NH2 to boron through base-to-acid coordination (NH2 → B), it is likely that a similar scenario occurs between the pendant amino groups of chitosan and boron atoms located on the flat sheets, which provide the driving force to disperse the inorganic filler even at high loading. In support of these assumptions, fast sedimentation occurs for native h-BN dispersed in aqueous medium [5 mg in 12 mL of H2O] after 24 h while the CS-BN mixture stays stable for an extended time of at least seven days (Scheme 1d).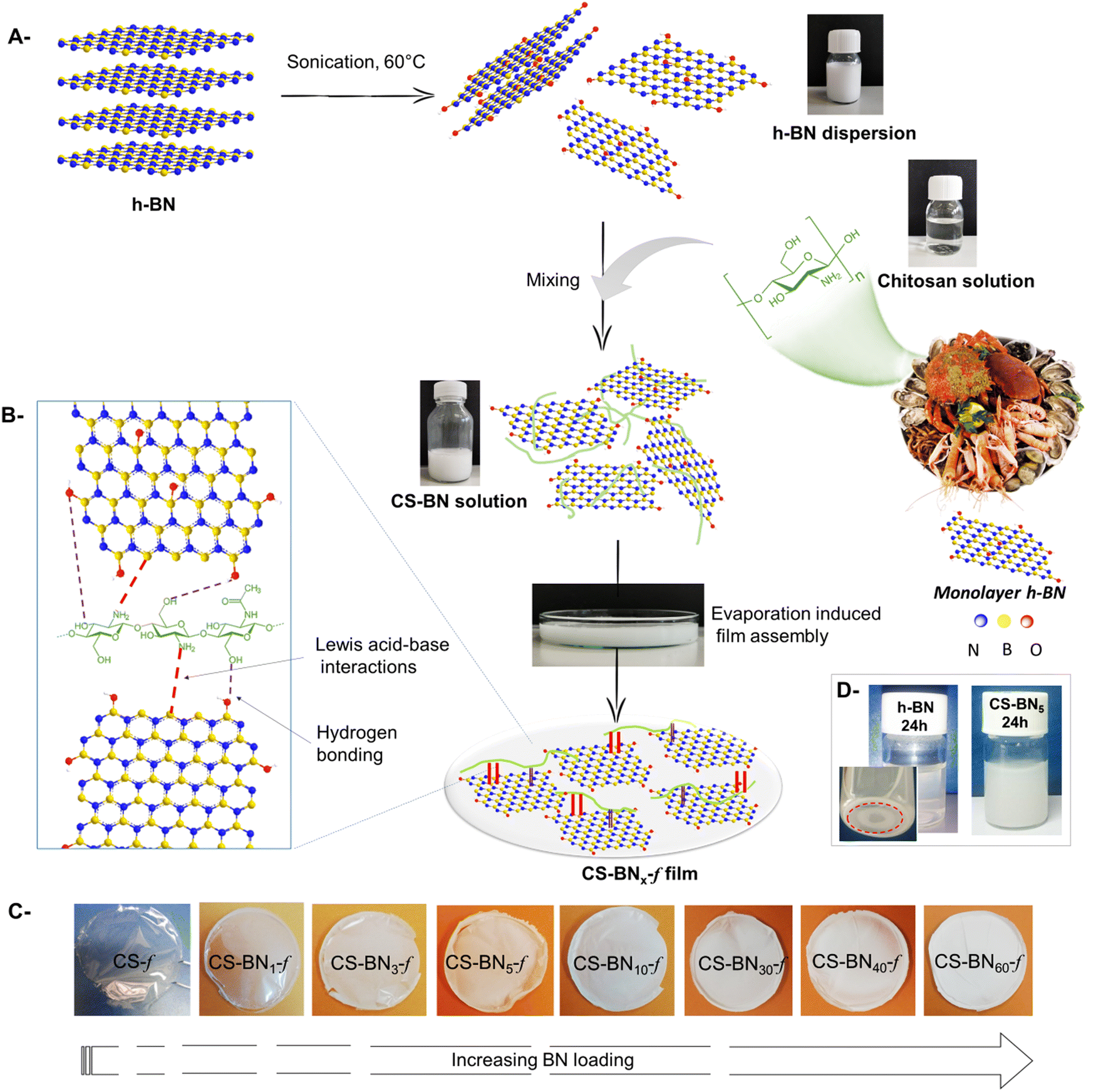 | ||
| Scheme 1 (a) Illustration of the multistep preparation procedure for boron nitride-embedded chitosan hydrogels. (b) Chemical structure and interfacial interaction of chitosan and boron nitride. (c) The as-prepared hybrid films (denoted as CS-BNx-f) with increasing BN amount. Left to right: 0, 1, 3, 5, 10, 30, 40, and 60 wt%. (d) Digital photos of h-BN and CS-BN5 acidic-aqueous solution after 24 h. | ||
The FTIR spectra of CS-BNx-f shows the fingerprint of both chitosan and h-BN (Fig. S1, ESI†). All the composite films show typical absorption peaks of chitosan, namely, the amine band (NH2) at 1568 cm−1 and the carbonyl band (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–NHMe) at 1643 cm−1.25 The strong signature of h-BN at 762 cm−1, attributed to B–N stretching, was present in all the CS-BNx-f composite films, thus bringing a firm evidence for the incorporation of boron nitride inside the film-forming chitosan network.26 A significant increase in the intensity of the two stretching bonds of BN at 1309 cm−1 and 762 cm−1 occurs with increasing amount of BN engaged in the initial mixture. We have also noticed a minor shift of the two characteristic vibrations of chitosan (CO–NH and NH2) with respect to those in CS-BNx-f, as illustrated in Fig. S1 (ESI†). These variations suggest the occurrence of strong interfacial interactions between the two components, which seems in agreement with the long-term stability of exfoliated boron nitride/chitosan colloidal solution.21a,23
O–NHMe) at 1643 cm−1.25 The strong signature of h-BN at 762 cm−1, attributed to B–N stretching, was present in all the CS-BNx-f composite films, thus bringing a firm evidence for the incorporation of boron nitride inside the film-forming chitosan network.26 A significant increase in the intensity of the two stretching bonds of BN at 1309 cm−1 and 762 cm−1 occurs with increasing amount of BN engaged in the initial mixture. We have also noticed a minor shift of the two characteristic vibrations of chitosan (CO–NH and NH2) with respect to those in CS-BNx-f, as illustrated in Fig. S1 (ESI†). These variations suggest the occurrence of strong interfacial interactions between the two components, which seems in agreement with the long-term stability of exfoliated boron nitride/chitosan colloidal solution.21a,23
The XRD diffraction peaks of CS-BNx-f films show sharp and well-resolved crystalline peaks that match the pristine structure of hexagonal boron nitride (Fig. S2, ESI†). This is in contrast to native chitosan structure, which exhibits only a broad amorphous peak at 20.4°.25,27 The two sharp peaks observed at 2θ values of 26° and 55° are respectively attributed to the [002] and [004] crystallographic planes of h-BN, and the d-spacing value of the nanoparticles calculated for the peak at 2θ value of 26.7° was found to be 0.33 nm. Additionally, the intensity of the peak at 26.7° increases with increasing amount of BN loaded inside the CS-BNx-f films, suggesting that boron nitride particulates preserve their inherent crystallinity inside of the film-forming network even at high concentrations of BN nanoparticles.28
The morphology of native chitosan films and its analogues CS-BNx-f was next investigated using scanning electron microscopy (SEM), as shown in Fig. 1 and Fig. S4 (ESI†). The surface of the native chitosan film was smooth and regular without appreciable cracks (Fig. S4a, ESI†), while that of CS-BN1-f, CS-BN5-f, CS-BN10-f, and CS-BN30-f showed a rough surface, with the highest fraction of h-BN being dispersed in the film and a very small amount of h-BN aggregates (Fig. S4b, ESI†). The roughness of the surface can be moreover confirmed by comparing the thickness of the films, with the native chitosan being the least thick (6 μm) while that of the most loaded CS-BN60-f was 23 μm thicker (Table S5, ESI†).
 | ||
| Fig. 1 Microstructural investigation of boron nitride-filled chitosan hydrogels. (a) SEM images of: CS-BN10-f, CS-BN30-f, and CS-BN60-f and onset (the corresponding digital photos for each material); (b) Mapping the location of nitrogen, boron, carbon, and oxygen through SEM/EDX analysis; (c) Energy dispersive X-ray diffraction spectra showing the abundance of boron, carbon, nitrogen, and oxygen. | ||
Elemental mapping analysis undertaken for CS-BN30-f revealed the uniform presence of boron, nitrogen, carbon, and oxygen within the network (Fig. 1b and c). The homogeneous distribution of BN inside the films was reached through successful dispersion and exfoliation of the filler inside, as previously commented. When the loading of BN nanolayers was further increased as in the case of CS-BN60-f, the microstructure of aligned BN nanoflakes becomes visible, indicating that the structure-directing assembly of BN particulates caused by their mutual interaction with the functionalities of the chitosan framework.
Improved thermal stability was noticed for the CS-BNx-f films with increasing filler amount from 1 to 60 wt% compared to the native chitosan films (Fig. 2a and Fig. S3, ESI†). The half weight decomposition temperature provides valuable information about the thermal stability of the newly prepared films. Native chitosan exhibits a T50 of 338 °C, which is the lowest temperature within the series, while CS-BN60-f displays a T50 of 495 °C, with the most delayed half weight degradation among the films. On the other hand, a complete decomposition was experienced for chitosan at 700 °C; a significant char residue was observed in case of the CS-BNx-f films, which correlates well with the amount of BN engaged in the initial solution (Table S5, ESI†). No weight loss could be observed for native boron nitride due to its high thermal stability. Obviously, the entrapment of BN inside the films delays the polymer degradation and improves its thermal resistance, which open a new channel of possibilities as sustainable flame-retardant polymeric materials.29
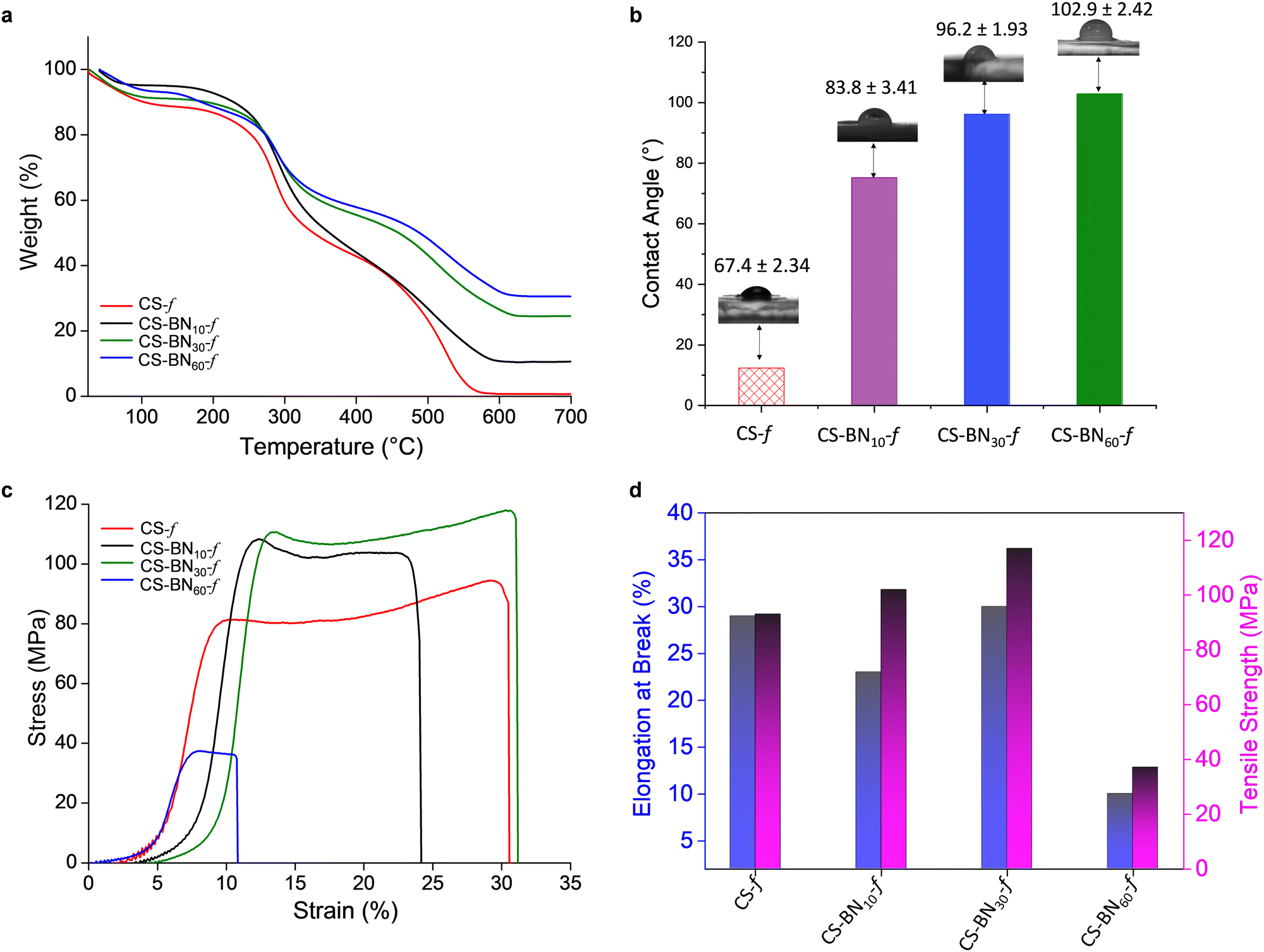 | ||
| Fig. 2 Elucidating the impact of boron nitride loading on the key properties of BN-embedded chitosan hydrogels: (a) Thermogravimetric analysis; (b) contact angle measurement; (c) tensile stress–strain curves; and (d) tensile strength and elongation at break of CS-f, CS-BN10-f, CS-BN30-f, and CS-BN60-f films. Values are averaged based on four repeated tests. | ||
Contact angle measurements show a dramatic change in the surface wettability of CS-BNx-f nanocomposites compared to the pristine chitosan film (Fig. 2b). As the amount of the filler increases, the water contact angle follows the same trend, reaching 102.9° for CS-BN60-f compared to 67.4° for native chitosan film. This corresponds to a significant shift of the Δθ value equal to 35°. The increased surface roughness in CS-BNx-f associated with the rigidity of the sheets that hinder the hydrogen-bonding network of the polysaccharides from being exposed to the surface reduces the inherent hydrophilicity of the initial film and switches the surface properties to water repellent and hydrophobic,22a which render CS-BNx-f nanocomposites more adaptable to moisture-resistant applications.
Fig. 2c shows the tensile stress vs. strain curves for native chitosan CS and its CS-BNx-f congeners. During the deformation of the films, three phases were observed on the stress–strain curves, namely, the elastic region, the stable stage (plateau), and the final stage where the stress decreased rapidly.30 At the beginning of the loading process, the stress–strain curve is nonlinear, which is a typical feature of soft polymer materials.31Fig. 2d displays the tensile strength and elongation at break. The composite film containing 30 wt% BN layers (CS-BN30-f) exhibited the highest tensile strength of about 119 MPa and a maximum elongation at break of 30%. These two respective values present an improvement of 26% and 7% to those recorded for neat chitosan film. This record outperforms the ones reached using graphene oxide CS-GO-f (60 MPa)32 and phosphorylated cellulose nanocrystals CS@PN-CNC-f (53 MPa),24 respectively. Besides, in most cases, an improvement in tensile strength has been often counterbalanced by a significant decrease in the elongation at break properties, which is not the case with CS-BN30-f. This unprecedented result indeed opens more possibilities for reinforcing chitosan-based nanocomoposites.24 Impressively, CS-BN30-f displayed the highest tensile strength among all boron nitride-filled polymer nanocomposites reported in the literature so far (Table S6, ESI†). CS-BN60-f displayed the lowest tensile strength of 10 MPa and a brittle-like behavior because of the significant aggregation of BN layers through π–π stacking, as evidenced by SEM analysis. The presence of aggregated microparticles and tactoides within the polymer network is known to worsen the mechanical properties of reinforced polymer-based nanocomposites.
3.2 Biological activity of CS-BNx-f nanocomposites
For the biological activity, we intentionally focused on hydrogels containing a lower amount of boron nitride because of their suitability for packaging in terms of flexibility and transparency as well as the cost-effectiveness of the final material considering that chitosan bio-waste is cheaper compared to boron nitride. Besides, when the aqueous formulation was used as an antiseptic spray, optimally discrete and uniform coating could be obtained with a lower amount of boron nitride. We have consequently investigated the antibacterial activities of native chitosan CS-f and marginally-loaded CS-BNx-f nanocomposites (i.e., CS-BN1-f, CS-BN3-f, and CS-BN5-f) against Escherichia coli and Staphylococcus aureus (Fig. 3a). Native CS-f used as a control did not inhibit the growth of E. coli or S. aureus. The low antibacterial activity of chitosan once shaped as films is already documented and mainly attributed to the inaccessibility of NH2 groups on the flattened surface.20,33 In turn, CS-BN1-f, CS-BN3-f, and CS-BN5-f displayed good antibacterial activities against E. coli and even nearly excellent activity against S. aureus. The antibacterial activity increased with increasing amounts of BN nanolayers in the film: CS-BN5-f > CS-BN3-f > CS-BN1-f. A similar trend was observed for both Gram-positive and Gram-negative bacteria. These results are very interesting given that in the literature, appreciable antibacterial activity was noticed only after filler conjugation when cellulose and graphene oxide are entrapped in chitosan hydrogel films.22b,24 Herein, commercially available boron nitride was used as such without the additional step of functionalization that could add cost and interfere with the biocompatibility.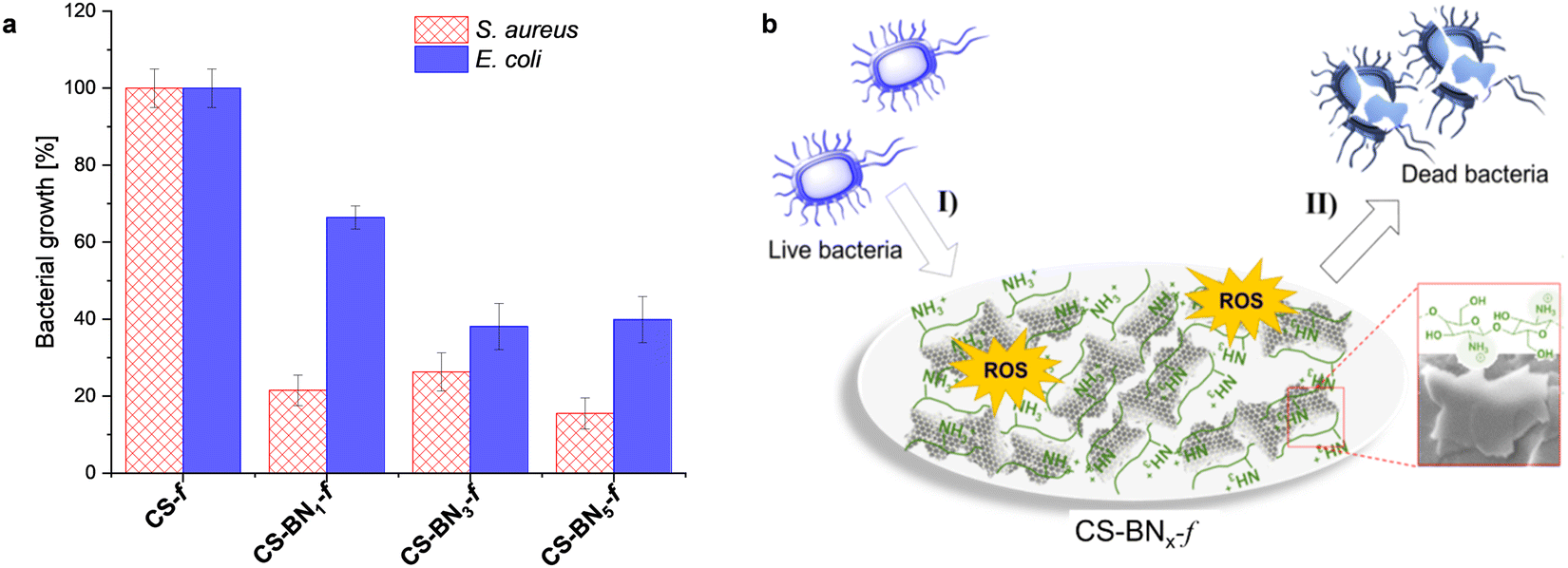 | ||
| Fig. 3 (a) Antibacterial activity of the resulting nanocomposite films: Inhibition (%) of Escherichia coli and Staphylococcus aureus after 24 h incubation. (b) Plausible mechanism illustrating the interplay of the CS-BNx-f nanocomposite with bacteria. | ||
Common claims in the literature focused primarily on the cationic groups of chitosan (either NH2 or ammonium) as responsible sites that interact with negatively charged components present on the bacterial surface, leading to membrane disturbance.34 However, the also literature highlighted the role of the surface wettability, with hydrophobic materials being more efficient to eradicate bacteria.35 This parameter appears in concordance with the wettability enhancement of chitosan films noticed with the increased loading of boron nitride. A close look at the literature shows the importance of the partnered polymer, with no significant decrease in bacterial density being observed for Pluronic P123, Pluronic F127, and ammonium oleate-coated boron nitride, while polyethylenimine-coated boron nitride resulted in significant antibacterial activity.36
We indeed tentatively propose that the bacteria are damaged by the flattened BN surface, which leads to the death of cells.37 A suggested mechanism of the CS-BN1-f films interacting with the bacterial cells is shown in Fig. 3b. The antimicrobial process concerns mainly two steps: first, the bacteria are attracted onto the surface of the CS-BNx-f nanocomposite by the strong electrostatic interaction between the positive charge belonging to chitosan and the negative charge from the bacterial cell. Thereafter, on the one hand, BN nanoplates with numerous sharp edges on the surface penetrate and disrupt the cell membranes of the bacteria and cause its inactivation.17b,37 On the other hand, the possible existence of unsaturated B atoms (B radicals) at the BN nanoplates edges could trigger the generation of ROS, leading to cell death.38 However, as will be commented later, these materials also display interesting antioxidant activity, which could attenuate the damaging effects of ROS and delay many events that contribute to cellular aging.39
The hemolysis of human erythrocytes was evaluated by measuring the hemoglobin content after incubating the cells with CS-BNx-f films for 3 and 24 h. The hemolysis results for all CS-BNx-f films are presented in Fig. 4a. While all the CS-BNx-f films induced hemolysis upon initial inspection, the hemolysis percentage did not exceed 7% after incubation for 3 h. After 24 h of incubation, the percentage of hemolysis increased up to 8.5%. The hemolysis rate also increased with increasing concentrations of BN nanolayers (from 1 wt% to 3 wt%) in the CS-BNx-f composites. However, a further increase in the BN nanolayer contents up to 5 wt% led to a slight decrease in the hemolysis rate, which could be attributed to the higher hydrophobicity and reduced affinity of bacterial cells.40 Notably, all the CS-BNx-f films demonstrated lower hemolytic activity compared to the already reported chitosan films filled with graphene oxide, CS-GO (7–7.5%), and those filled with nanocellulose (10–16%).22b,24,32 As hemolysis was not dependent on the incubation time, we investigated the possible hemoglobin adsorption on the surface of the films (Fig. 4b). It is widely accepted that BN nanosheets adhere to the cell membrane, disrupting the membrane integrity and causing membrane leakage.41 However, contact angle measurements revealed that the BN nanolayers increased the hydrophobicity of the chitosan film, which may reduce the cell affinity to CS-BNx-f, thus decreasing the possibility of cell-killing. Therefore, the percentage of hemolysis after 24 h may not reflect real hemolytic activity but rather the accumulation of hemoglobin on the surface of boron nitride composites.
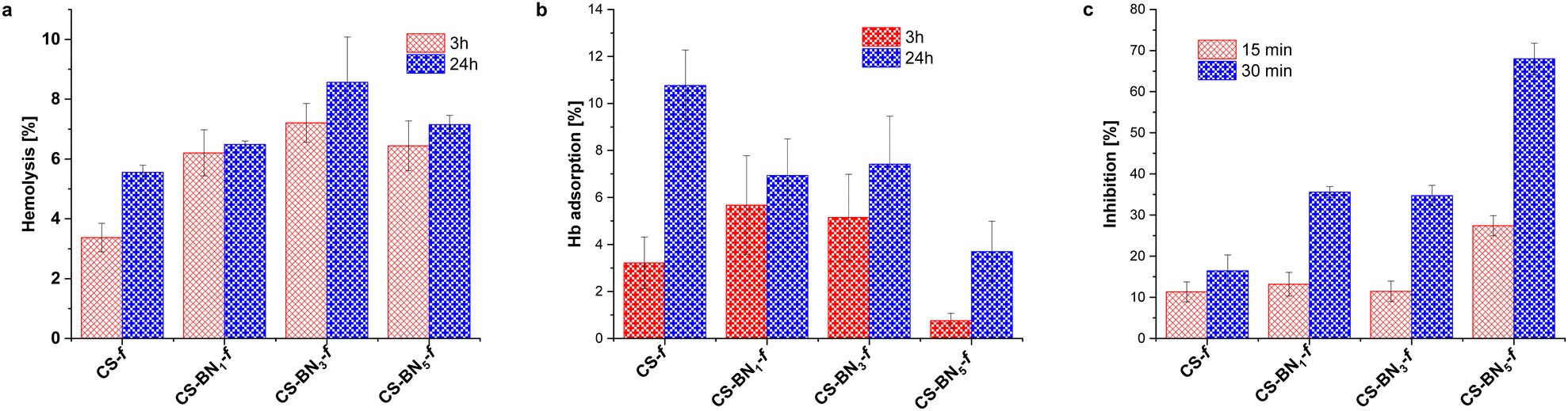 | ||
| Fig. 4 (a) Hemolysis of erythrocytes after incubation with CS-BNx-f; (b) hemoglobin adsorption on the surface of boron nitride-embedded chitosan hydrogels; and (c) ABTS radical scavenging activity of CS-BNx-f. | ||
Next, the antioxidant activity of the CS-BNx-f films was assessed using the ABTS method, and the results showed a noteworthy increase in the antioxidant capacity of the films as the BN loading (1 wt%, 3 wt%, 5 wt%) increased, compared to the pure chitosan film. These findings are in line with the previously reported antioxidant activity of both BN and chitosan, as documented in the literature.28a,42
4. Conclusions
Herein, we describe a novel approach for the assembly of boron nitride-chitosan hydrogels through evaporation-induced co-assembly. Regardless of the amount of boron nitride, the resulting combination provides homogeneous, flexible, stable, and crack-free nanocomposite films. The strong interfacial interaction between the filler and the polymer backbone provides the driving force for the successful entrapment of up to 60 wt% of boron nitride without compromising the film-forming properties of the network or its flexibility. Increasing the amount of boron nitride significantly improves the thermal and mechanical properties along with significant alteration of the surface wettability. While native chitosan film was devoid of appreciable antibacterial activity, introducing a tiny amount of boron nitride (1 wt% to 5 wt%) imparted CS-BNx-f nanocomposites with good antibacterial activities against Gram positive Staphylococcus aureus and Gram negative Escherichia coli. A slight variation in hemolysis was noticed with the addition of BN nanolayers while a significant increase in the antioxidant capacity of CS-BNx-f nanocomposites was recorded with increasing BN loading. The results suggest the potential application of boron nitride-filled chitosan nanocomposites for the development of metal-free, antibacterial surface-coating materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AEK thanks l’Académie Hassan II des Sciences et Technique for funding “Nano-Bio-Mat” project. NH thanks UEMF for the Scholarship. The Chevreul Institute is thanked for its help in the development of this work through the ARCHI-CM project supported by the “Ministere de l’Enseignement Superieur de la Recherche et de l’Innovation”, the region “Hauts-de-France”, the ERDF program of the European Union and the “Metropole Europeenne de Lille. Adeline MARIN is warmly acknowledged for the mechanical testing.References
- (a) J. Fu, T. Liu, S. S. Binte Touhid, F. Fu and X. Liu, ACS Nano, 2023, 17, 1739–1763 CrossRef CAS PubMed; (b) S. M. Imani, L. Ladouceur, T. Marshall, R. Maclachlan, L. Soleymani and T. F. Didar, ACS Nano, 2020, 14, 12341–12369 CrossRef CAS PubMed.
- (a) J. Hodek, V. Zajícová, I. Lovětinská-Šlamborová, I. Stibor, J. Müllerová and J. J. B. M. Weber, BMC Microbiol., 2016, 16, 1–12 CrossRef PubMed; (b) Y. C. Ong, S. Roy, P. C. Andrews and G. Gasser, Chem. Rev., 2019, 119, 730–796 CrossRef CAS PubMed.
- (a) F. Paladini, M. Pollini, A. Sannino and L. Ambrosio, Biomacromolecules, 2015, 16, 1873–1885 CrossRef CAS PubMed; (b) G. Reina, S. Peng, L. Jacquemin, A. F. Andrade and A. Bianco, ACS Nano, 2020, 14, 9364–9388 CrossRef CAS PubMed; (c) A. Rana, S. Pathak, D.-K. Lim, S.-K. Kim, R. Srivastava, S. N. Sharma and R. Verma, ACS Appl. Nano Mater., 2023, 6, 8106–8134 CrossRef CAS; (d) M. L. Ermini and V. Voliani, ACS Nano, 2021, 15, 6008–6029 CrossRef CAS PubMed.
- N. Karim, S. Afroj, K. Lloyd, L. C. Oaten, D. V. Andreeva, C. Carr, A. D. Farmery, I.-D. Kim and K. S. Novoselov, ACS Nano, 2020, 14, 12313–12340 CrossRef CAS PubMed.
- V. Bhandari, S. Jose, P. Badanayak, A. Sankaran and V. Anandan, Ind. Eng. Chem. Res., 2022, 61, 86–101 CrossRef CAS.
- A. I. Ribeiro, A. M. Dias and A. Zille, ACS Appl. Nano Mater., 2022, 5, 3030–3064 CrossRef CAS PubMed.
- (a) S. Ladhari, N.-N. Vu, C. Boisvert, A. Saidi and P. Nguyen-Tri, ACS Appl. Bio Mater., 2023, 6, 1398–1430 CrossRef CAS PubMed; (b) F. Khan, N. Tabassum, N. I. Bamunuarachchi and Y.-M. Kim, J. Agric. Food Chem., 2022, 70, 4817–4838 CrossRef CAS PubMed; (c) F. Hui and C. Debiemme-Chouvy, Biomacromolecules, 2013, 14, 585–601 CrossRef CAS PubMed.
- (a) M. Haktaniyan and M. Bradley, Chem. Soc. Rev., 2022, 51, 8584–8611 RSC; (b) Y. Lu, X. Xu and J. Li, J. Mater. Chem. B, 2023, 11, 3338–3355 RSC; (c) A. Dong, Y.-J. Wang, Y. Gao, T. Gao and G. Gao, Chem. Rev., 2017, 117, 4806–4862 CrossRef CAS PubMed; (d) E.-R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed; (e) A. Jain, L. S. Duvvuri, S. Farah, N. Beyth, A. J. Domb and W. Khan, Adv. Healthcare Mater., 2014, 3, 1969–1985 CrossRef CAS PubMed.
- (a) B. H. Gan, J. Gaynord, S. M. Rowe, T. Deingruber and D. R. Spring, Chem. Soc. Rev., 2021, 50, 7820–7880 RSC; (b) A. S. Carlini, L. Adamiak and N. C. Gianneschi, Macromolecules, 2016, 49, 4379–4394 CrossRef CAS PubMed.
- B. Ates, S. Koytepe, A. Ulu, C. Gurses and V. K. Thakur, Chem. Rev., 2020, 120, 9304–9362 CrossRef CAS PubMed.
- (a) N. Yan and X. Chen, Nature, 2015, 524, 155–157 CrossRef CAS PubMed; (b) X. Chen, H. Yang and N. Yan, Chem. – Eur. J., 2016, 22, 13402–13421 CrossRef CAS PubMed.
- (a) A. E. Kadib, K. Molvinger, M. Bousmina and D. Brunel, J. Catal., 2010, 273, 147–155 CrossRef; (b) A. El Kadib and M. Bousmina, Chem. – Eur. J., 2012, 18, 8264–8277 CrossRef PubMed; (c) A. El Kadib, A. Primo, K. Molvinger, M. Bousmina and D. Brunel, Chem. – Eur. J., 2011, 17, 7940–7946 CrossRef CAS PubMed.
- (a) B. Boumhidi, N. Katir, J. El Haskouri, K. Draoui and A. El Kadib, New J. Chem., 2020, 44, 14136–14144 RSC; (b) Á. Molnár, Coord. Chem. Rev., 2019, 388, 126–171 CrossRef; (c) A. El Kadib, ChemSusChem, 2015, 8, 217–244 CrossRef CAS PubMed.
- S. Ladet, L. David and A. Domard, Nature, 2008, 452, 76–79 CrossRef CAS PubMed.
- (a) A. El Kadib, Chem. Rec., 2020, 20, 753–772 CrossRef CAS PubMed; (b) S. Takeshita, S. Zhao, W. J. Malfait and M. M. Koebel, Angew. Chem., Int. Ed., 2021, 60, 9828–9851 CrossRef CAS PubMed.
- (a) J. Li, X. Tian, T. Hua, J. Fu, M. Koo, W. Chan and T. Poon, ACS Appl. Bio Mater., 2021, 4, 4014–4038 CrossRef CAS PubMed; (b) H. Wang, J. Qian and F. Ding, J. Agric. Food Chem., 2018, 66, 395–413 CrossRef CAS PubMed; (c) L. Bonnaire, S. Sandra, T. Helgason, E. A. Decker, J. Weiss and D. J. McClements, J. Agric. Food Chem., 2008, 56, 3791–3797 CrossRef CAS PubMed; (d) S. Kumar, A. Mukherjee and J. Dutta, Trends Food Sci. Technol., 2020, 97, 196–209 CrossRef CAS; (e) J. Yu, D. Wang, N. Geetha, K. M. Khawar, S. Jogaiah and M. Mujtaba, Carbohydr. Polym., 2021, 261, 117904 CrossRef CAS PubMed.
- (a) S.-W. Xiong, P.-G. Fu, Q. Zou, L.-Y. Chen, M.-Y. Jiang, P. Zhang, Z.-G. Wang, L.-S. Cui, H. Guo and J.-G. Gai, ACS Appl. Mater. Interfaces, 2021, 13, 196–206 CrossRef CAS PubMed; (b) S. Pandit, K. Gaska, V. R. S. S. Mokkapati, S. Forsberg, M. Svensson, R. Kádár and I. Mijakovic, RSC Adv., 2019, 9, 33454–33459 RSC; (c) X. Chen, P. Wu, M. Rousseas, D. Okawa, Z. Gartner, A. Zettl and C. R. Bertozzi, J. Am. Chem. Soc., 2009, 131, 890–891 CrossRef CAS PubMed; (d) G. Ciofani, S. Danti, G. G. Genchi, B. Mazzolai and V. Mattoli, Small, 2013, 9, 1672–1685 CrossRef CAS PubMed.
- Q. Weng, X. Wang, X. Wang, Y. Bando and D. Golberg, Chem. Soc. Rev., 2016, 45, 3989–4012 RSC.
- A. El Kadib, N. Wrońska, K. Lisowska, A. Anouar, N. Katir, K. Miłowska, B. Bielska and M. Bryszewska, Functional Materials in Biomedical Applications, Jenny Stanford Publishing, 2023, pp. 1–50 Search PubMed.
- N. Hammi, N. Wrońska, N. Katir, K. Lisowska, N. Marcotte, T. Cacciaguerra, M. Bryszewska and A. El Kadib, ACS Appl. Bio Mater., 2019, 2, 61–69 CrossRef CAS PubMed.
- (a) S. Frindy, A. Primo, A. E. K. Qaiss, R. Bouhfid, M. Lahcini, H. Garcia, M. Bousmina and A. El Kadib, Carbohydr. Polym., 2016, 146, 353–361 CrossRef CAS PubMed; (b) J. Chabbi, O. Jennah, N. Katir, M. Lahcini, M. Bousmina and A. El Kadib, Carbohydr. Polym., 2018, 183, 287–293 CrossRef CAS PubMed.
- (a) S. Frindy, A. Primo, H. Ennajih, A. El Kacem Qaiss, R. Bouhfid, M. Lahcini, E. M. Essassi, H. Garcia and A. El Kadib, Carbohydr. Polym., 2017, 167, 297–305 CrossRef CAS PubMed; (b) J. Chabbi, A. Aqil, N. Katir, B. Vertruyen, C. Jerôme, M. Lahcini and A. El Kadib, Carbohydr. Polym., 2020, 230, 115634 CrossRef CAS PubMed.
- N. Hammi, S. El Hankari, N. Katir, N. Marcotte, K. Draoui, S. Royer and A. El Kadib, Microporous Mesoporous Mater., 2020, 306, 110429 CrossRef CAS.
- S. Blilid, M. Kędzierska, K. Miłowska, N. Wrońska, M. El Achaby, N. Katir, E. Belamie, B. Alonso, K. Lisowska, M. Lahcini, M. Bryszewska and A. El Kadib, ACS Sustainable Chem. Eng., 2020, 8, 18354–18365 CrossRef CAS.
- N. Hammi, S. Chen, A. Primo, S. Royer, H. Garcia and A. El Kadib, Green Chem., 2022, 24, 4533–4543 RSC.
- A. Mukheem, S. Shahabuddin, N. Akbar, A. Miskon, N. Muhamad Sarih, K. Sudesh, N. Ahmed Khan, R. Saidur and N. Sridewi, Nanomaterials, 2019, 9, 645 CrossRef CAS PubMed.
- L. Durai, P. Yadav, H. Pant, V. V. S. S. Srikanth and S. Badhulika, New J. Chem., 2020, 44, 15919–15927 RSC.
- (a) K. Behera, M. Kumari, Y.-H. Chang and F.-C. Chiu, Int. J. Biol. Macromol., 2021, 186, 135–144 CrossRef CAS PubMed; (b) J. Chen, J. Shang, F. Xue, Q. Wei, N. Xu and E. Ding, J. Polym. Res., 2019, 26, 264 CrossRef CAS.
- S. Blilid, M. Boundor, N. Katir, M. El Achaby, M. Lahcini, J. P. Majoral, M. Bousmina and A. El Kadib, Macromolecules, 2023, 56, 1223–1235 CrossRef CAS.
- L. L. Hu, L. W. Xiong and T. X. Yu, Int. J. Mech. Sci., 2019, 159, 116–125 CrossRef.
- J. Chen, X. Liu, X. L. Zeng, H. Y. Ye and G. Q. Zhang, Compos. Commun., 2022, 29, 101038 CrossRef.
- N. Wrońska, A. Anouar, M. El Achaby, K. Zawadzka, M. Kędzierska, K. Miłowska, N. Katir, K. Draoui, S. Różalska and I. Piwoński, Materials, 2020, 13, 998 CrossRef PubMed.
- N. Wrońska, N. Katir, K. Miłowska, N. Hammi, M. Nowak, M. Kędzierska, A. Anouar, K. Zawadzka, M. Bryszewska, A. El Kadib and K. Lisowska, Int. J. Mol. Sci., 2021, 22, 5839 CrossRef PubMed.
- D. Raafat and H.-G. Sahl, Microb. Biotechnol., 2009, 2, 186–201 CrossRef CAS PubMed.
- X. Liu, L. Chang, L. Peng, R. Bai, Y. Wei, C. Ma and H. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 48358–48364 CrossRef CAS PubMed.
- (a) A. Merlo, V. R. S. S. Mokkapati, S. Pandit and I. Mijakovic, Biomater. Sci., 2018, 6, 2298–2311 RSC; (b) C. Parra, F. Montero-Silva, R. Henríquez, M. Flores, C. Garín, C. Ramírez, M. Moreno, J. Correa, M. Seeger and P. Häberle, ACS Appl. Mater. Interfaces, 2015, 7, 6430–6437 CrossRef CAS PubMed.
- A. Raval, N. S. Yadav, S. Narwani, K. Somkuwar, V. Verma, H. Almubarak, S. M. Alqahtani, R. Tasleem, A. M. Luke, S. T. Kuriadom and M. I. Karobari, J. Funct. Biomater., 2023, 14, 201 CrossRef CAS PubMed.
- S. Mateti, C. S. Wong, Z. Liu, W. Yang, Y. Li, L. H. Li and Y. Chen, Nano Res., 2018, 11, 334–342 CrossRef CAS.
- B. Poljsak, D. Šuput and I. Milisav, Oxid. Med. Cell Longev., 2013, 2013, 956792 Search PubMed.
- K. Kuroda, G. A. Caputo and W. F. DeGrado, Eur. J. Chem., 2009, 15, 1123–1133 CrossRef CAS PubMed.
- X. Xie, Z. Hou, G. Duan, S. Zhang, H. Zhou, Z. Yang and R. Zhou, Colloids Surf., B, 2021, 203, 111765 CrossRef CAS PubMed.
- (a) A. Wan, Q. Xu, Y. Sun and H. Li, J. Agric. Food Chem., 2013, 61, 6921–6928 CrossRef CAS PubMed; (b) T. Sun, D. Zhou, J. Xie and F. Mao, Eur. Food Res. Technol., 2007, 225, 451–456 CrossRef CAS; (c) P. Ahmad, A. Khalid, M. U. Khandaker, F. Rehman, M. I. Khan, H. Ali, N. Muhammad, M. S. Kiyani, A. Sulieman, M. A. Rauf Khan, Z. Razzaq, A. Khan, S. Haq, Y. Saeed and M. I. Irshad, Mater. Sci. Semicond. Process., 2022, 141, 106419 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00445g |
| This journal is © The Royal Society of Chemistry 2023 |