
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLiquid–liquid phase separation for microencapsulation of native cytokine to enhance immune activation†
Zhenhua
Hu‡
a,
Li
Cheng‡
b,
Qiuling
Chen
a,
Tianqing
Xin
b and
Xiaoyan
Wu
 *b
*b
aDepartment of Health and Nursing, Nanfang College of Sun Yat-sen University, Guangzhou, China
bDepartment of Pediatrics, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China. E-mail: xwu@hust.edu.cn
First published on 20th October 2023
Abstract
Therapeutic cytokines have achieved remarkable success in combination immunotherapy against malignant tumors. Nonetheless, their short half-life in the bloodstream leads to poor compliance and hinders their clinical effectiveness. While some pegylated cytokines have been developed to extend their circulation half-life, structural modifications often alter cytokines’ receptor affinities, reduce their activity, and even pose the risk of reversing their clinical effects. Therefore, there is an urgent need to develop new long-acting cytokines with stable blood concentration profiles and high activity. Inspired by the biomimetic partition process of liquid–liquid phase separation (LLPS) in living cells, we designed a method for microencapsulating cytokines into polymer microparticles driven by LLPS, enabling sustained delivery of native cytokines with high activity. Initially, we created a PEG/dextran aqueous biphasic system by mixing porous microparticles loaded with dextran-70 kDa and a PEG-20 kDa solution. Next, we introduced GM-CSF as a model cytokine into the biphasic system, allowing it to be distributed into the dextran-rich phase under the driving force of LLPS. We then sealed the porous microparticles to complete the microencapsulation of GM-CSF, resulting in GM-CSF/LLPS-MP. GM-CSF/LLPS-MP demonstrated a consistent release of native GM-CSF over a two-week period, promoting dendritic cell differentiation and function. Moreover, it enhanced the synergistic inhibitory effect of GM-CSF and PD-1 antibodies on melanoma tumors compared to GM-CSF solution. These findings offer proof of concept that liquid–liquid phase separation is an effective method for achieving the microencapsulation of native cytokines, thereby enhancing immune activation.
Introduction
Immune checkpoint inhibitor (ICI) therapy has achieved remarkable clinical effects in cancer immunotherapy over the past few years, but unfortunately, many patients have limited responses and are resistant to ICI therapy.1,2 The primary reasons for this are often attributed to the immunosuppressive tumor microenvironment (TEM), which may lack the necessary survival factors for T cells or induce T cell exhaustion.3,4 As potent immune-modulating molecules, cytokines can reverse the immunosuppressive TEM.5,6 Recently, the number of clinical trials combining cytokines synergistically with ICI has exponentially increased.7,8 However, due to their short half-life, cytokines require frequent injections, resulting in wide fluctuations in blood concentration, poor compliance, and other influences on their clinical effects.9,10 Up to this point, two primary strategies have been employed to extend the blood half-life of cytokines. One strategy involves structural modification, as exemplified by pegylated Interferon Alfa-2b, which has found use in clinical settings.11,12 However, modification of the structure usually changes the affinity of cytokines to their receptors and results in a loss of almost 40–70% of activities,13 which increases the injection dose and cost. Furthermore, PEGylation of cytokines carries the risk of reversing their activities and producing the opposite clinical results. For example, a new site-specific PEGylation of interleukin-2 (IL-2) recently demonstrated immunosuppressive effects contrary to the immunostimulant effects of Bempegaldesleukin (NKTR-214), which is the first PEGylated IL-2 and has shown the ability to activate and proliferate CD8+ T cells and NK cells in clinical trials.14,15 It adds more uncertainty, insecurity, and complexity to the strategy of PEGylation of cytokines. The other strategy is to microencapsulate cytokines into polymeric microparticles to sustainably release native cytokines. The release profile of polymeric microparticles can be close to zero-order and can reduce wide fluctuations in the blood concentration of cytokines.16,17 However, the microencapsulation of cytokines into polymeric microparticles for sustained delivery has yet to be successful in the clinic. The reason is that some of the conditions in the manufacturing process of microparticles, such as homogenization, sonication, extrusion, and exposure to organic solvents, can cause cytokine degradation and denaturation.18,19 Besides, cytokines are vulnerable to impairment by acids generated from the degradation of polymeric microparticles.20 Therefore, it is imperative to develop a new strategy to make the sustained delivery of cytokines through the advanced microparticles system available in the clinic.Recently, liquid–liquid phase separation (LLPS) has been discovered to drive the condensation of biomacromolecules into non-membrane compartments in the cytoplasm, which is involved in various cell functions.21–23 Inspired by this natural condensation process of biomacromolecules in live cells, we proposed to microencapsulate cytokines into pre-fabricated porous microparticles through the biomimetic process of LLPS to achieve a steady release of native cytokines. This way, we can separate the loading process of cytokines from the manufacturing process of microparticles, thus avoiding damage to the cytokines from adverse factors during the preparation process of microparticles. Moreover, the biomimetic LLPS can efficiently condense cytokines into the pores of microparticles, thus allowing for the microencapsulation of cytokines with high activities.
LLPS usually happens in an aqueous biphasic system composed of two kinds of hydrophilic polymers.24,25 The classic aqueous biphasic system is composed of (polyethylene glycol) (PEG) and dextran.26,27 The two phases have different affinities for the biomolecules. The PEG phase has repellency to biomolecules, and dextran has a high affinity with them. Biomolecules will be distributed in the dextran-rich phase under the driving of LLPS when they are added into the PEG/dextran aqueous biphasic system.27 In this approach, we pre-fabricate dextran-loaded porous microparticles and then mix them with a PEG-20 kDa solution to create the PEG/dextran aqueous biphasic system. When we introduce cytokines into the PEG-20 kDa solution, the cytokines are partitioned into the dextran-rich phase located within the microparticles due to the driving force of LLPS. Finally, we seal the pores of the microparticles to complete the microencapsulation of cytokines.
GM-CSF, an FDA-approved cytokine, is widely recognized as a critical factor for the generation of DCs and DC development from both bone marrow and spleen cell cultures,28 and has exhibited a synergistic effect with immune checkpoint inhibitors (ICIs) in the treatment of melanoma in Phase 2 clinical trials.29,30 However, GM-CSF must be injected daily in each 14 days treatment cycle due to its short half-life in vivo. In this work, we use GM-CSF as a model cytokine drug to test the concept that microencapsulation of cytokine driven by the biomimetic process of LLPS is a novel and practical approach to develop sustained delivery of native cytokine, which could potentially enhance its application in cancer immunotherapy.
Materials and methods
Materials
Dextran-70 kDa, poly(ethylene glycol) (PEG)-20 kDa, PEG-400Da, pluronic(R) F-127, and poly(vinyl alcohol) (PVA) (87–90% hydrolyzed, average molecular weight 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–70000) were obtained from Sigma Aldrich, China. Poly(D,L-lactide-co-glycolide) (PLGA5050 2.5 A, inherent viscosity 0.16–0.24 dL g−1) was obtained from Evonik Degussa, China. GM-CSF and PD-1 antibody were obtained from Sino Biological, China. Albumin–fluorescein isothiocyanate conjugate (FTIC-BSA), Dextran, Texas Red™, 70000 MW, and MicroBCA were obtained from Thermo Fisher, China.
000–70000) were obtained from Sigma Aldrich, China. Poly(D,L-lactide-co-glycolide) (PLGA5050 2.5 A, inherent viscosity 0.16–0.24 dL g−1) was obtained from Evonik Degussa, China. GM-CSF and PD-1 antibody were obtained from Sino Biological, China. Albumin–fluorescein isothiocyanate conjugate (FTIC-BSA), Dextran, Texas Red™, 70000 MW, and MicroBCA were obtained from Thermo Fisher, China.
Preparation of dextran-loaded porous microparticles
We weighed the appropriate amount of PLGA5050 2.5 A polymer and F-127 and completely dissolved them in 1 mL of dichloromethane. Subsequently, we weighed the appropriate amount of dextran and suspended it in the above 1 mL of dichloromethane solution under a vortex. Next, the suspension was added to 6 mL of 4% PVA solution and emulsified at a speed of 20000 rpm for 1.5 minutes using a homogenizer. The emulsion was then transferred to 100 mL of 0.5% sodium chloride and allowed to solidify with continuous stirring. Finally, the solidified microparticles were collected through centrifugation, washed three times with distilled water, and then subjected to freeze-drying for storage.
Microencapsulation of cargos into LLPS-MP
We weighed 100 mg of dried dextran-preloaded porous microparticles and placed them into a vial. We added 2 mL of PEG-20 kDa solution into the vial and mixed it thoroughly to achieve homogeneity. We then added BSA or cytokine solution at a concentration of 1 mg mL−1 to the mixture and gently shook it for 2 hours on an ice bath. We then collected the microparticles through centrifugation and immersed them in different concentration of PEG-400Da solutions to seal the pores. After that, we collected the LLPS-MP through centrifugation, rinsed it with distilled water, and then subjected it to freeze-drying for storage.Distribution of FITC labeled BSA in LLPS-MP
We loaded FITC-labeled BSA into Texas-red labeled dextran-preloaded porous microparticles using the LLPS-MP preparation process. We used confocal laser scanning microscopy (CLSM) images to observe the distribution of BSA and dextran inside the LLPS-MP. The samples for CLSM were prepared following the methods outlined in our previous work.31Investigation on the pore closure condition for LLPS-MP
We prepared different concentrations of PEG-400Da solution and weighed an appropriate number of dextran-loaded porous microparticles. We incubated the porous microparticles with the PEG-400Da solution at different temperatures, took out the microparticle samples at different time points, washed them with distilled water three times, and then took a small number of pieces for scanning electron microscopy as per the previous method.31Determination of loading content and encapsulation efficiency of LLPS-MP
We weighed 10 mg of lyophilized LLPS-MP and placed it into a 1.5 mL centrifuge tube. We added 1 mL of acetonitrile and vortexed to dissolve the microparticles. We then centrifuged at 8000 rpm for 10 minutes and removed the supernatant as much as possible. We evaporated the residual acetonitrile to dry, and dissolved the powder in 1 mL of deionized water. We measured the BSA amount using the MicroBCA method, and calculated the loading content of LLPS-MP. In this way, we also extrapolated the encapsulation efficiency of LLPS-MP. Loading content (LC) = actual drug content/mass of microparticles × 100%, Encapsulation efficiency (EE) = experimental LC/theoretical LC × 100%.In vitro release of LLPS-MP
We weighed 20.0 mg of lyophilized LLPS-MP into a vial and added 1 mL of PBS (pH = 7.4) containing 0.01% sodium azide as a release medium. We placed the vial in a 37 °C air bath shaker and shook it at 100 rpm. We took out 1 mL of release medium at predetermined times and added 1 mL of fresh medium. We centrifuged the released sample at 4 °C at 5000 rpm for 5 minutes and took out the supernatant for the test. We measured the BSA concentration by a micro BCA method. We tested the in vitro release of GM-CSF/LPPS-MP in the same condition and measured the released GM-CSF by the micro-BCA or ELISA method.Quantification of soluble acids from LLPS-MP
We weighed 50 mg of freeze-dried LLPS-MP recycled from in vitro release, dissolved it in 0.5 ml of chloroform, and added 2 ml of distilled water. We mixed the solution under a mild vortex and allowed the biphasic solution to stand for 10 minutes. After centrifuging at 4000 rpm for 5 minutes at 4 °C, we quickly removed the upper water layer and added freshwater. We repeated the extraction process three times and finally collected the water phases together. The total extracted soluble acids from LLPS-MP were determined by titration with 0.1 M NaOH solution. Additionally, pH in the release medium was measured using a pH electrode.CD spectrum analysis of the sample released from LLPS-MP
The intact BSA standard solution and BSA sample released from microspheres on days 1, 7, and 14 were prepared at the same concentration. The circular dichroism (CD) value was detected using a J-500A Jasco (Tokyo, Japan) instrument at a far-ultraviolet CD spectrum with a 200–280 nm band. The wave scanning speed was 50 nm min−1 at room temperature.Cell preparation and in vitro stimulation
Bone marrow (BM) cells from C57BL/6 mice were isolated by flushing femurs with 5 mL of 2% FBS in PBS. The BM cells were centrifuged once and then resuspended in 5 mL of Lyse RBC buffer (Invitrogen) at room temperature for 4–5 minutes. The reaction was stopped with 10–20 mL of PBS. The cells were strained through a 70 μm filter and resuspended in RPMI 1640 medium with 10 ng mL−1 GM-CSF. BM cells were seeded at 5 mL per well in 6-well tissue culture plates. Fresh medium with GM-CSF was added and replated every three days. Loosely adherent cells were present on day 7. In some experiments, DCs were cultured as above but in a medium containing GM-CSF/LLPS-MP. DC cells were harvested and stained with specific antibodies for the indicated cell surface markers. For activation stimulation, differentiated DCs were resuspended at 0.5 × 106 mL−1 in fresh medium with a TLR agonist, LPS (1 g mL−1) (Sigma-Aldrich), and plated at 1 mL per well in 48-well plates and cultured for 20 hours.32 The supernatants were collected, and the level of TNF-α, IL-12p70, and IL-10 were determined using ELISA kits (BD Biosciences).In vivo mouse study
All the animal experiments obtained ethical approval from the Experimental Animal Ethics of Huazhong University of Science and Technology. Healthy C57BL/6 mice were treated with GM-CSF solution and GM-CSF/LLPS-MP. GM-CSF was injected subcutaneously at a dose of 9 μg kg−1 for seven days continuously, and GM-CSF/LLPS-MP was injected subcutaneously at a dose of 150 μg kg−1 GM-CSF on the first day. Spleen tissues were isolated, and the total number of viable splenic leukocytes was determined using a hemocytometer count on day 7. Single cells from spleen suspensions were stained with anti-CD11c-FITC antibody, and the percentage of CD11c+ DC was analyzed by flow cytometry.33 The spleen tissues were also stained with H&E. DC-enriched splenocytes from treated mice were incubated with Lucifer yellow (1 mg mL−1) for 1 hour at 37 °C and stained with CD11c and CD11b antibodies and analyzed by flow cytometry to measure pinocytosis of Lucifer yellow, as indicated by mean fluorescence intensity (MFI).34 C57BL/6 mice were implanted subcutaneously with 1 × 106 B16 melanoma cells on the flank.35 At 3, 6, and 9 days after tumor cell inoculation, respectively, saline, blank MP, GM-CSF, anti-PD1, and anti-PD1 combined with GM-CSF solution or GM-CSF/LLPS-MP were administered subcutaneously into the flank of mice. GM-CSF and anti-PD1 were at 9 μg kg−1 and 10 mg kg−1, respectively. GM-CSF/LLPS-MP was at the equivalent dose of 150 μg kg−1 GM-CSF. The sizes were measured seven times every two days, and tumor volumes were calculated using the formula: 1/2 (length × width2). In addition, the indicated cytokine levels in serum were measured after five days of inoculation using enzyme-linked immunosorbent assay (ELISA). The survival curve could be surveyed using Kaplan–Meier analysis. For the histology study, tumors in different groups were resected and performed with H&E staining after the last time of treatment.Statistical analysis
Data analysis was performed using GraphPad Prism (Version 8.0). All data are expressed as means ± SEM. Statistical comparisons among different groups were performed using one-way analysis of variance (ANOVA) followed by Tukey's post hoc test. Statistically significance was considered at p < 0.05 as customary.Results and discussion
Microencapsulation of biomacromolecules driven by liquid–liquid phase separation into porous microparticle
Microencapsulation driven by liquid–liquid phase separation (LLPS) was performed following the schematic illustration in Fig. 1(A). As a model protein molecule, fluorescein isothiocyanate (FITC)-labeled bovine serum albumin (BSA) was employed to study the distribution of biomacromolecules in a PEG-20 kDa/dextran-70 kDa aqueous biphasic system. The ratio of PEG/dextran was located in the two-phase range, as shown in the phase diagram of Fig. S1 (ESI†). Firstly, solutions of FITC-BSA and dextran labeled with rhodamine B isothiocyanate (RITC-dextran) were separately added to the PEG-20 kDa solution. We observed that FITC-BSA accumulated at the upper phase of the PEG-20 kDa solution, while RITC-dextran accumulated at the lower phase of the PEG-20 kDa solution (Fig. 1(B)). When FITC-BSA and RITC-dextran were added to the PEG-20 kDa solution, FITC-BSA was primarily richer at the bottom and merged with the RITC-dextran phase, resulting in the formation of a yellow layer. This showed that the biomacromolecule was favored by the dextran-rich phase and was repelled by the PEG-rich phase in the PEG/dextran aqueous biphasic system. Next, we determined the percentages of BSA distributed in the dextran-rich phase in different PEG/dextran biphasic systems to find the suitable ratio of PEG-20 kDa and dextran-70 kDa for the LLPS loading process. The partition percentage of BSA had no significant difference in the same concentration of PEG solution when the dextran concentration increased from 0.25% to 10% (Fig. 1(C)). However, the partition percentage of BSA rose from 75% to 95% when the PEG-20 kDa concentration increased from 10% to 15%.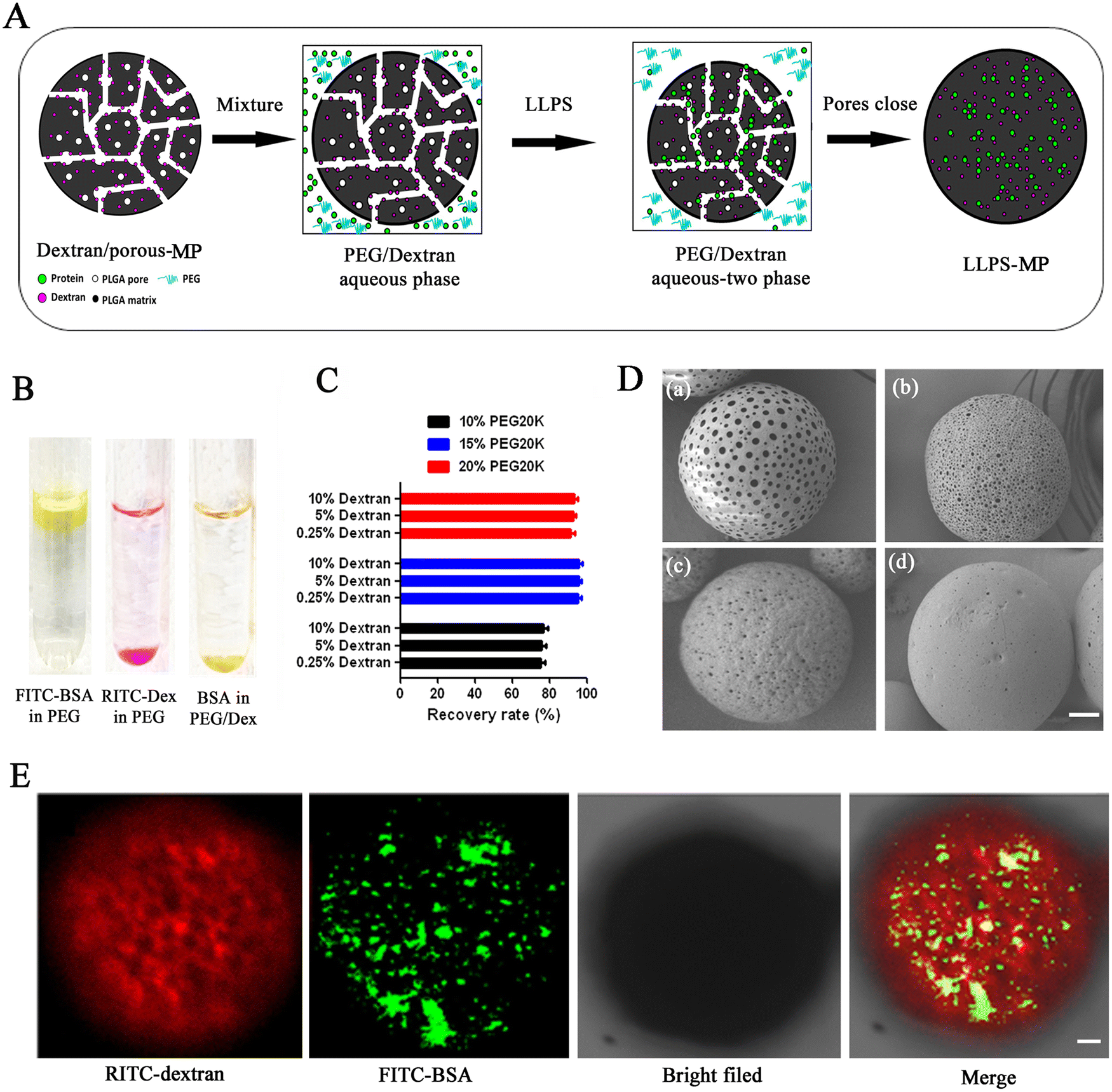 | ||
| Fig. 1 Microencapsulation of biomacromolecules driven by LLPS into porous microparticles. (A) Schematic illustration of the microencapsulation process of LLPS-MP. (B) The distribution of FITC-BSA and RITC-dextran-70 kDa in PEG-20 kDa solution. (C) BSA recovery rates from the dextran-rich phase in different PEG/dextran systems (n = 3). (D) SEM images of dextran-loaded porous microparticles with different size pores. (a)–(d) microparticles from formulations containing 10%, 25%, 40%, 55% F-127, respectively. The scale bar is 10 μm. (E) Confocal images of FITC-BSA microencapsulated in RITC-dextran pe-loaded microparticle under the driving of LLPS. The scale bar is 5 μm. | ||
Nevertheless, the partition percentage slightly decreased when the concentration of PEG-20 kDa further increased to 20%. Hence, we opted for a 15% PEG-20 kDa solution for the LLPS loading process. Next, we preloaded dextran-70 kDa into porous microparticles with different sizes of pores using the traditional emulsion-solvent evaporation method.36 The sizes of the pores on the surface of the porous microparticles ranged from 300 nm to 3000 nm, as measured by SEM (Fig. 1(D)). We identified the optimal pore size (approximately 500 nm), as illustrated in Fig. 1(D)-c, for the loading process. To observe the loading process intuitively under the driving of LLPS, we mixed RITC-dextran preloaded porous microparticles with a 15% PEG-20 kDa solution to form a PEG/dextran aqueous biphasic system. There was almost no red RITC-dextran diffusing out from the porous microparticles. When green FITC-BSA was added into the PEG-20 kDa solution, most of the FITC-BSA accessed into the pores of the microparticles under the driving of LLPS after two hours. The yellow overlay in the confocal images showed that the distribution of FITC-BSA was co-located with the RITC-dextran inside of the microparticles (Fig. 1(E)). These results indicated that LLPS is a practical approach for loading BSA into the dextran-rich phase inside of the microparticles.
Optimization of process parameters of LLPS-MP
To accomplish the last step of microencapsulation, we need to seal the pores of porous microparticles after loading. To close the pores of polymeric microparticles, the polymer chains need to be rearranged above the Tg temperature of the polymer. However, the typical glass transition temperature (Tg) of the polymer is often higher than 37 °C, which is not conducive to the stability of biomolecules. A plasticizer can reduce the Tg of the polymer to help the closure of pores at a lower, more moderate temperature. However, most plasticizers are organic solvents that can denature biomolecules. In this work, we were surprised to find that liquid PEG-400Da exhibited excellent compatibility with biomolecules, had the ability to dissolve the PLGA polymer, and served as an effective for pore closure at room temperature. After incubation in 95% PEG-400Da solution at 25 °C for 15 minutes (condition of closure-1), the pores on the surface of microparticles were closed, but the internal pores remained unchanged, as shown in Fig. S2 (ESI†). After incubation in 90% PEG-400Da solution at 42 °C for 2 hours (condition of closure-2), the surface pores were closed, and the sizes of internal pores became smaller, although they were not completely closed. Finally, after incubation in 80% PEG-400Da solution at 42 °C for 4 hours (condition of closure-3), both the surface and internal pores were successfully closed. Notably, the surface pores could be effectively sealed under the above three closure conditions.Next, we proceeded to evaluate the loading content (LC) and encapsulation efficiency (EE), as well as the in vitro release profiles of BSA/LLPS-MP after pore closure. The LC and EE of BSA/LLPS-MP decreased with increasing incubation time in the PEG-400 Da solution. Only BSA/LLPS-MP treated with the closure-1 condition (Closure-1-MP) exhibited an EE over 80% (Fig. 2(A)). The in vitro release results showed that Closure-1-MP achieved an accumulative release percentage of more than 90% after 14 days, while Closure-2-MP and Closure-3-MP displayed incomplete releases of less than 80% (Fig. 2(B)).
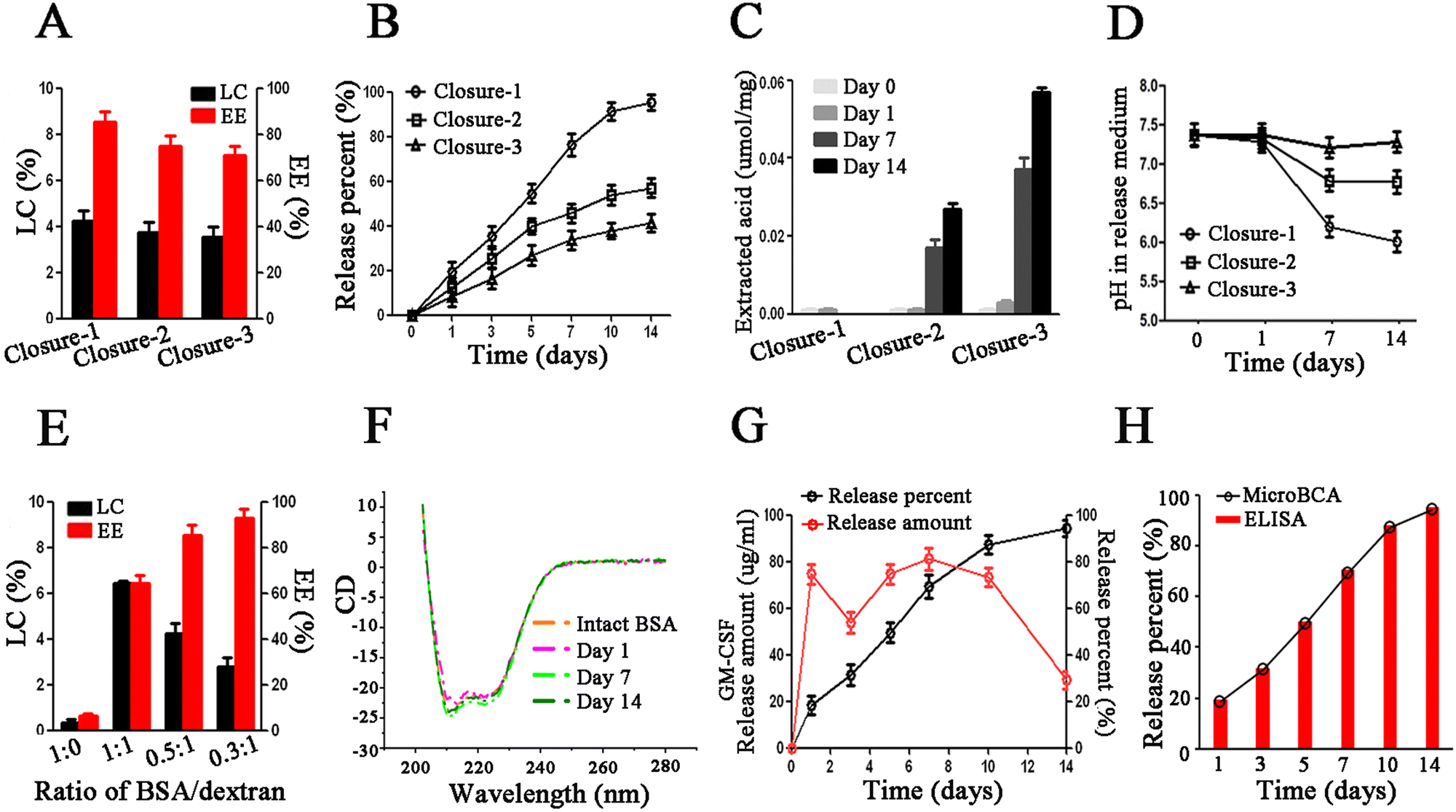 | ||
| Fig. 2 Optimization of process parameters of LLPS-MP. (A) Effects of pore-closure conditions on load content (LC) and encapsulation efficiency (EE) of BSA/LLPS-MP (n = 3). Closure-1:95% PEG-400 Da at 25 °C for 15 min; closure-2:90% PEG-400 Da at 42 °C for 2 hours; 80% PEG-400 Da at 42 °C for 4 hours. (B) In vitro release of BSA/LLPS-MP prepared under different pore-closure conditions (n = 3). (C) Extracted water-soluble acid from BSA/LLPS-MP during in vitro release (n = 3). (D) pH in in vitro release medium (n = 3). (E) LC and EE of BSA/LLPS-MP from the formulation with different ratios of BSA/dextran (n = 3). (F) CD spectrum of intact BSA as control and BSA released from BSA/LLPS-MP on days 1, 7, and 14. (G) In vitro release percent and amount of GM-CSF/LLPS-MP (n = 3). (H) Comparative analysis of in vitro release profiles of GM-CSF/LLPS-MP measured by MicroBCA and ELISA methods. | ||
The acid generated during polymer degradation is the major harmful factor that denatures biomacromolecules.20 Rapid excretion of internal acid from polymer microparticles could protect the loaded biomacromolecules from acid-induced damage.31 BSA/LLPS-MP was recycled at different release time points, and the internal soluble acid was extracted and measured. During the release period, there was almost negligible soluble acid inside Closure-1-MP (Fig. 2(C)). No soluble acid was observed in Closure-2-MP and Closure-3-MP on release days 0 and 1, and soluble acid significantly increased after seven days of the release in these two groups. These results suggested that the soluble acid during the release accumulated in Closure-2-MP and Closure-3-MP but could be rapidly excreted in Closure-1-MP. This point could be explained by the lower pH value in the release medium from Closure-1-MP after seven days of release compared with the other two groups (Fig. 2(D)). It can be explained that the internal pores of Closure-1-MP facilitated the immediate diffusion of acid out of the microparticles, preventing damage to cytokines by acid during the release period. In short, Closure-1-MP exhibited high EE, complete release, and rapid acid excretion, making the Closure-1 condition the preferred choice for sealing BSA/LLPS-MP.
Next, we investigated the effect of the ratio of BSA/dextran on the LC and EE of LLPS-MP. The LC of BSA/LLPS-MP loaded with 10% dextran increased with the increasing input amount of BSA, but the EE exhibited an opposite trend (Fig. 2(E)). Therefore, the ratio of BSA/dextran at 0.5:1 was chosen to balance the LC and EE. Especially, LC and EE of BSA/LLPS-MP without dextran were very low, even though the input amount of BSA was large enough. A similar low LC was observed when the PEG-20 kDa solution was replaced by a water solution in the loading process. These results showed that LLPS in a PEG/dextran aqueous biphasic system was indispensable for the microencapsulation of cytokines. The reason is that, as hydrophilic molecules, cytokines prefer to stay in a water solution and hardly access the hydrophobic pores of microparticles by themselves. It is noteworthy that a potent bridge built under the driving of LLPS allowed cytokines to rapidly partition into their favored dextran-rich phase inside microparticles. This microencapsulation driven by LLPS is a biomimetic partition process expected to protect the activity of biomolecules.
To evaluate the protection of biomolecules, we measured the CD spectrums of BSA recycled and released from BSA/LLPS-MP. The data showed that BSA's integral structures were kept well during the preparation and release process of BSA/LLPS-MP (Fig. 2(F)). This suggested that LLPS-MP could microencapsulate biomolecules while preserving their native activity. Next, we used GM-CSF as a model cytokine to test the optimized formulation and parameters of LLPS-MP and termed the microencapsulated GM-CSF as GM-CSF/LLPS-MP. The average LC and EE of GM-CSF/LLPS-MP were 3.85% and 85.4%, respectively. Finally, we determined the in vitro physical release using MicroBCA and the in vitro bioactive release using ELISA. GM-CSF/LLPS-MP exhibited a steady release profile close to zero order over 14 days (Fig. 2(G)). The coincident profiles of physical release and bioactive release of GM-CSF/LLPS-MP showed that released GM-CSF had an intact structure and native activity (Fig. 2(H)). Thus, we could confirm that GM-CSF/LLPS-MP could release native GM-CSF sustainably for two weeks.
GM-CSF/LLPS-MP enhanced dendritic cell differentiation and function
Next, we evaluated the sustained activation of the dendritic cells (DCs) by GM-CSF/LLPS-MP. First, BM-derived DCs were cultured with different GM-CSF formulations for seven days and harvested to analyze the expression of markers for mature DCs, including CD80 and CD86.37 Fresh GM-CSF solutions were replenished into the medium every three days, while GM-CSF/LLPS-MP was only added on the first day. After seven days of incubation, the expression of CD80 and CD86 in GM-CSF solution-treated cells was nearly three to four-fold more than that in PBS or blank-MP treated cells, but it was almost two to two-and-a-half-fold less than that in cells treated with GM-CSF/LLPS-MP (Fig. 3(A)). This meant that GM-CSF/LLPS-MP doubled the number of mature DCs, compared with GM-CSF/solution. Next, the function of the mature DCs was evaluated through further stimulation by lipopolysaccharide (LPS) for 20 hours.38 The representative Th1 and Th2 cytokines (IL-12p70, TNF-α, IL-10) in the supernatant were collected and measured. GM-CSF/LLPS-MP increased these secreted cytokines from DCs nearly three to four-fold compared with GM-CSF/solution (Fig. 3(B)–(D)). These results showed that GM-CSF/LLPS-MP exhibited significantly enhanced mature differentiation and function of DCs thanks to its sustained activation on DCs. | ||
| Fig. 3 GM-CSF/LLPS-MP enhanced dendritic cell differentiation and function. Bone marrow (BM) derived dendritic cells (DCs) were cultured with different GM-CSF formulations for seven days and were harvested and stained with antibodies for the indicated cell surface markers. (A) Mature DCs markers were determined using flow cytometry. Black peaks show the background staining. Red peaks show specific fluorescence for CD80 and CD86 markers. (B)–(D) Accumulation of representative Th1 and Th2 cytokines in the supernatant of harvested DCs was measured after 20 h of stimulation by LPS (1 μg mL−1) (n = 4). | ||
GM-CSF/LLPS-MP increased immune activation in the mouse spleen
To evaluate in vivo enhanced immune activation by GM-CSF/LLPS-MP, we isolated mouse spleen cells after seven days of treatment and performed hematoxylin and eosin (H&E) staining. H&E images from mice treated with PBS and blank MP demonstrated well-organized follicular structures with the normal boundary between red and white pulp. However, the boundary of the splenic follicles in mice treated with GM-CSF solution became unclear due to cellular infiltration (Fig. 4(A)). The splenic follicular structure in mice treated with GM-CSF/LLPS-MP had been disrupted by diffuse infiltration of mononuclear leukocytes. The cellular infiltration in splenic follicles suggested an increased number of splenic cells. Hemocytometer counting confirmed that GM-CSF/LLPS-MP increased the number of splenic cells by two-fold compared with the GM-CSF solution (Fig. 4(B)). Furthermore, the flow cytometer analysis showed that GM-CSF/LLPS-MP increased the percentage of splenic DCs two-fold more than that in the GM-CSF solution-treated group (Fig. 4(C)). Additionally, GM-CSF/LLPS-MP also enhanced the antigen capture by splenic DCs. GM-CSF/LLPS-MP increased the up-regulation level of pinocytosis of Lucifer yellow by splenic DCs almost two-fold more than that in the GM-CSF solution-treated group (Fig. 4(D)). These results further supported that GM-CSF/LLPS-MP could provide more robust immune activation in the mouse spleen than the GM-CSF solution.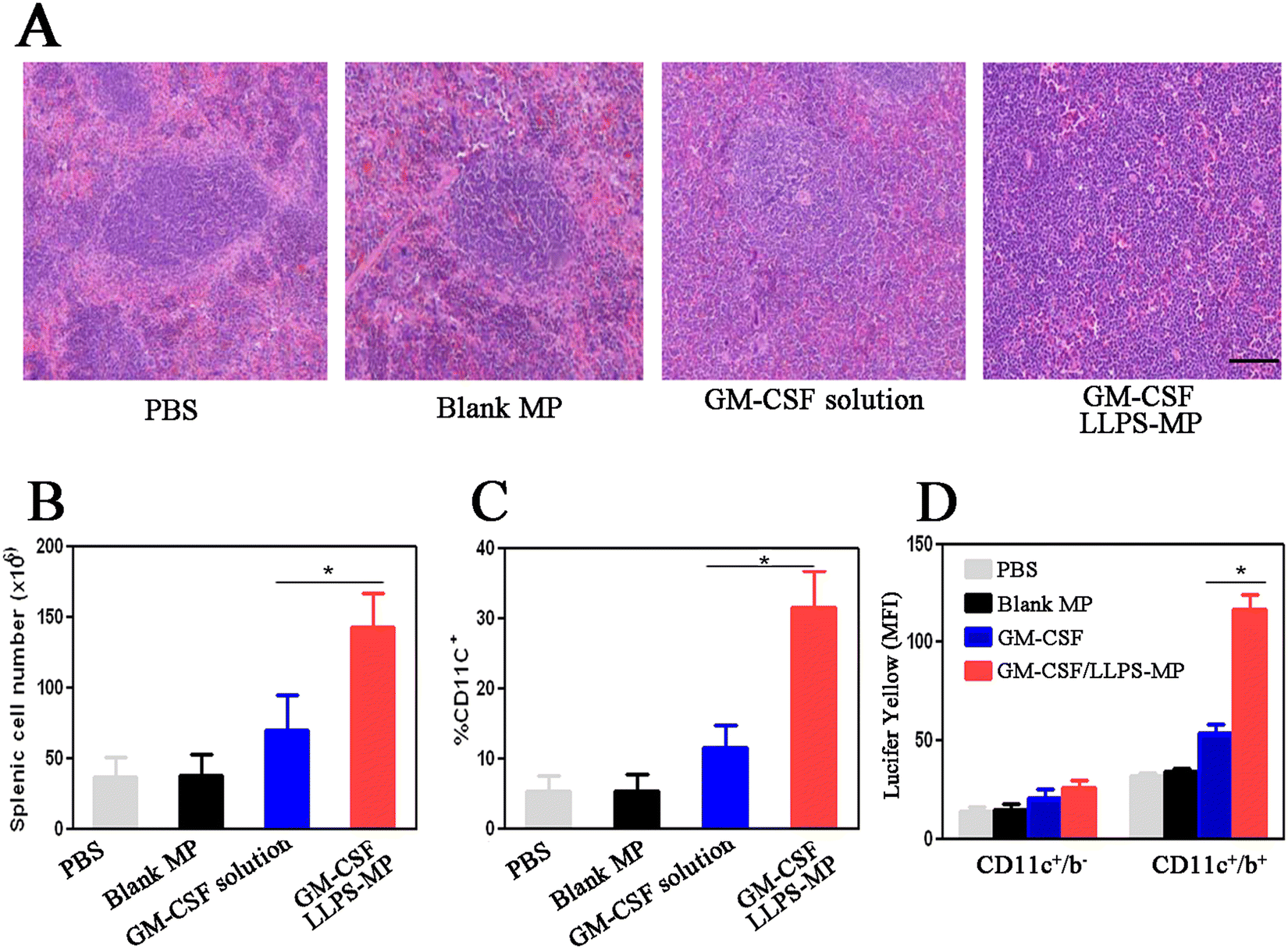 | ||
| Fig. 4 GM-CSF/LLPS-MP increased immune activation in the mouse spleen. C57BL/6 mice were treated with different GM-CSF formulations for seven days. (A) Spleens from treated mice were isolated and stained with hematoxylin and eosin (H&E). The scale bar is 100 μm. (B) The total cell number of splenic leukocytes from treated mice. (C) The percentage of CD11c+ DCs from the spleen in treated mice (n = 3). (D) The antigen capture by splenic DC was evaluated through pinocytosis of Lucifer yellow. Staining with CD11c and CD11b was performed to identify DC subsets, as determined by mean fluorescence intensity (MFI). (n = 3). | ||
GM-CSF/LLPS-MP enhanced the synergistic inhibition effect of GM-CSF and PD-1 antibody on melanoma tumors
Next, we examined the synergistic antitumor effects of GM-CSF/LLPS-MP combined with PD-1 antibody in a melanoma tumor mouse model. On day three after the inoculation of tumor cells, mice were treated with various therapeutic regimens, as described in the Methods section. After five days of treatment, serum samples from all groups were collected and the concentrations of the representative Th1 cytokines INF-γ, TNF-α, IL-2, and IL-12p70 were measured.39 The combination of GM-CSF solution and PD-1 antibody exhibited synergistic effects on the secretion of these cytokines compared to either alone (Fig. 5(A) and (D)). The levels of cytokines were further elevated in the combination of GM-CSF/LLPS-MP and PD-1 antibody. GM-CSF/LLPS-MP also enhanced the synergistic effects of GM-CSF solution combined with PD-1 antibody on tumor growth inhibition and the extended survival rate. We strictly immunized mice with the various GM-CSF formulations after inoculated tumor cells and monitored tumor volume and survival. The combination of GM-CSF solution and PD-1 antibody showed around 60% inhibited rate of tumor volume compared with the saline group (Fig. 5(E)). The inhibition rate was increased to 87% by the combination GM-CSF/LLPS-MP and PD-1 antibody. Furthermore, we monitored the survival of all the groups, and the results showed the GM-CSF/LLPS-MP combined group had the highest survival rates of 85% (Fig. 5(F)). The histology images of tumor sections after treatment showed that more lymphocytes accumulated in mice treated with GM-CSF/LLPS-MP combined group compared with other groups, as indicated by the black arrows in Fig. 5(G). This indicated more robust immune activation in the GM-CSF/LLPS-MP treated group and forecasted better regression after treatment.40,41 In short, GM-CSF/LLPS-MP significantly enhanced the synergistic effects of GM-CSF and PD-1 antibodies on tumor inhibition compared to the GM-CSF solution. | ||
| Fig. 5 GM-CSF/LLPS-MP enhanced the synergistic inhibition effect of GM-CSF and PD-1 antibody on melanoma tumors. (A)–(D) The serum concentration of the representative secreted immune cytokines, including INF-γ, TNF-α, IL-2, and IL-12p70 in all groups of treated mice (n = 8). (E) and (F) Tumor growth and overall survival curves of melanoma tumor burden mice after treatment with various GM-CSF formulations or combined with PD-1 antibody (n = 8). (G) Representative images of H&E-stained tumor tissues showing increased lymphocyte infiltration (as indicated by the black arrow) in mice from GM-CSF/LLPS-MP combined with the anti-PD1 treated group (n = 4). The scale bar is 100 μm. | ||
Conclusion
In conclusion, GM-CSF/LLPS-MP exhibited more potent stimulation of DCs differentiation compared to GM-CSF solution, thanks to its sustained immune activation. Furthermore, the synergistic immune effects of the combination of GM-CSF and PD-1 antibodies against melanoma tumors were further enhanced by GM-CSF/LLPS-MP. This study underscores the effectiveness of microencapsulating cytokines driven by LLPS as a viable approach for achieving sustained delivery of native cytokines. LLPS-MP presents itself as a promising translational platform for the development of long-acting cytokines to enhance their efficacy in cancer immunotherapy. Our forthcoming studies will delve deeper into the pharmacokinetics (PK) of GM-CSF/LLPS-MP and aim to establish correlations between PK and pharmacodynamics (PD) to further validate the biweekly injection frequency of GM-CSF/LLPS-MP.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Nanfang College of Sun Yat-sen University for providing funding (2021BQ012) and thank the foundation (funding number: 2019020701011504) from China's Wuhan Municipal Science and Technology Bureau and the foundation (funding number: 2023BCB026) from Hubei Provincial Department of Science and Technology Project.References
- A. J. Schoenfeld and M. D. Hellmann, Cancer Cell, 2020, 37, 443–455 CrossRef CAS PubMed.
- D. Romero, Nat. Rev. Clin. Oncol., 2021, 18, 194 CrossRef PubMed.
- T. Tang, X. Huang, G. Zhang, Z. Hong, X. Bai and T. Liang, Signal Transduction Targeted Ther., 2021, 6, 72 CrossRef PubMed.
- J. S. O’Donnell, M. W. L. Teng and M. J. Smyth, Nat. Rev. Clin. Oncol., 2019, 16, 151–167 CrossRef PubMed.
- D. Xue, E. Hsu, Y. X. Fu and H. Peng, Antibody Ther., 2021, 4, 123–133 CrossRef CAS PubMed.
- K. G. Nguyen, M. R. Vrabel, S. M. Mantooth, J. J. Hopkins, E. S. Wagner, T. A. Gabaldon and D. A. Zaharoff, Front. Immunol., 2020, 11, 575597 CrossRef CAS PubMed.
- D. J. Propper and F. R. Balkwill, Nat. Rev. Clin. Oncol., 2022, 19, 237–253 CrossRef CAS PubMed.
- T. A. Waldmann, Cold Spring Harbor Perspect. Biol., 2018, 10, a028472 CrossRef CAS PubMed.
- P. Berraondo, M. F. Sanmamed, M. C. Ochoa, I. Etxeberria, M. A. Aznar, J. L. Pérez-Gracia, M. E. Rodríguez-Ruiz, M. Ponz-Sarvise, E. Castañón and I. Melero, Br. J. Cancer, 2019, 120, 6–15 CrossRef CAS PubMed.
- L. Bonati and L. Tang, Curr. Opin. Chem. Biol., 2021, 62, 43–52 CrossRef CAS PubMed.
- F. Carrat, F. Bani-Sadr, S. Pol, E. Rosenthal, F. Lunel-Fabiani, A. Benzekri, P. Morand, C. Goujard, G. Pialoux, L. Piroth, D. Salmon-Céron, C. Degott, P. Cacoub and C. Perronne, JAMA, 2004, 292, 2839–2848 CrossRef CAS PubMed.
- P. Murer and D. Neri, New Biotechnol., 2019, 52, 42–53 CrossRef CAS PubMed.
- F. M. Veronese and A. Mero, BioDrugs, 2008, 22, 315–329 CrossRef CAS PubMed.
- B. Zhang, J. Sun, Y. Wang, D. Ji, Y. Yuan, S. Li, Y. Sun, Y. Hou, P. Li, L. Zhao, F. Yu, W. Ma, B. Cheng, L. Wu, J. Hu, M. Wang, W. Song, X. Li, H. Li, Y. Fei, H. Chen, L. Zhang, G. C. Tsokos, D. Zhou and X. Zhang, Nat. Biomed. Eng., 2021, 5, 1288–1305 CrossRef CAS PubMed.
- A. Diab, N. M. Tannir, S. E. Bentebibel, P. Hwu, V. Papadimitrakopoulou, C. Haymaker, H. M. Kluger, S. N. Gettinger, M. Sznol, S. S. Tykodi, B. D. Curti, M. A. Tagliaferri, J. Zalevsky, A. L. Hannah, U. Hoch, S. Aung, C. Fanton, A. Rizwan, E. Iacucci, Y. Liao, C. Bernatchez, M. E. Hurwitz and D. C. Cho, Cancer Discovery, 2020, 10, 1158–1173 CrossRef CAS PubMed.
- K. Inoue, H. Onishi, Y. Kato, T. Michiura, K. Nakai, M. Sato, K. Yamamichi, Y. Machida and Y. Nakane, Cancer Chemother. Pharmacol., 2004, 53, 415–422 CrossRef CAS PubMed.
- M. L. Laracuente, M. H. Yu and K. J. McHugh, J. Controlled Release, 2020, 327, 834–856 CrossRef CAS PubMed.
- F. Wu and T. Jin, AAPS PharmSciTech, 2008, 9, 1218–1229 CrossRef CAS PubMed.
- S. E. Reinhold, K. G. Desai, L. Zhang, K. F. Olsen and S. P. Schwendeman, Angew. Chem., Int. Ed., 2012, 51, 10800–10803 CrossRef CAS PubMed.
- H. Tamber, P. Johansen, H. P. Merkle and B. Gander, Adv. Drug Delivery Rev., 2005, 57, 357–376 CrossRef CAS PubMed.
- A. A. Hyman, C. A. Weber and F. Jülicher, Annu. Rev. Cell Dev. Biol., 2014, 30, 39–58 CrossRef CAS PubMed.
- S. Alberti, A. Gladfelter and T. Mittag, Cell, 2019, 176, 419–434 CrossRef CAS PubMed.
- P. H. Peng, K. W. Hsu and K. J. Wu, Am. J. Cancer Res., 2021, 11, 3766–3776 CAS.
- B. Y. Zaslavsky, L. A. Ferreira and V. N. Uversky, Biomolecules, 2019, 9, 473 CrossRef CAS PubMed.
- C. Yuan, A. Levin, W. Chen, R. Xing, Q. Zou, T. W. Herling, P. K. Challa, T. P. J. Knowles and X. Yan, Angew. Chem., Int. Ed., 2019, 58, 18116–18123 CrossRef CAS PubMed.
- C. D. Crowe and C. D. Keating, Interface Focus, 2018, 8, 20180032 CrossRef PubMed.
- C. D. Keating, Acc. Chem. Res., 2012, 45, 2114–2124 CrossRef CAS PubMed.
- L. Conti and S. Gessani, Immunobiology, 2008, 213, 859–870 CrossRef CAS PubMed.
- C. S. Fishburn, J. Pharm. Sci., 2008, 97, 4167–4183 CrossRef CAS PubMed.
- H. L. Kaufman, C. E. Ruby, T. Hughes and C. L. Slingluff, Jr., J. Immunother. Cancer., 2014, 2, 11 CrossRef PubMed.
- Y. Liu, A. H. Ghassemi, W. E. Hennink and S. P. Schwendeman, Biomaterials, 2012, 33, 7584–7593 CrossRef CAS PubMed.
- F. Zhou, B. Ciric, G. X. Zhang and A. Rostami, Clin. Exp. Immunol., 2014, 178, 447–458 CrossRef CAS PubMed.
- A. Basu, G. Chakrabarti, A. Saha and S. Bandyopadhyay, Immunology, 2000, 99, 305–313 CrossRef CAS PubMed.
- S. K. Basak, A. Harui, M. Stolina, S. Sharma, K. Mitani, S. M. Dubinett and M. D. Roth, Blood, 2002, 99, 2869–2879 CrossRef CAS PubMed.
- K. E. Ugen, M. A. Kutzler, B. Marrero, J. Westover, D. Coppola, D. B. Weiner and R. Heller, Cancer Gene Ther., 2006, 13, 969–974 CrossRef CAS PubMed.
- J. W. McGinity and P. B. O’Donnell, Adv. Drug Delivery Rev., 1997, 28, 25–42 CrossRef PubMed.
- Y. Xu, Y. Zhan, A. M. Lew, S. H. Naik and M. H. Kershaw, J. Immunol., 2007, 179, 7577–7584 CrossRef CAS PubMed.
- I. Bekeredjian-Ding, S. I. Roth, S. Gilles, T. Giese, A. Ablasser, V. Hornung, S. Endres and G. Hartmann, J. Immunol., 2006, 176, 7438–7446 CrossRef CAS PubMed.
- H. Tian, G. Shi, Q. Wang, Y. Li, Q. Yang, C. Li, G. Yang, M. Wu, Q. Xie, S. Zhang, Y. Yang, R. Xiang, D. Yu, Y. Wei and H. Deng, Signal Transduction Targeted Ther., 2016, 1, 16025 CrossRef PubMed.
- F. Maibach, H. Sadozai, S. M. Seyed Jafari, R. E. Hunger and M. Schenk, Front. Immunol., 2020, 11, 2105 CrossRef CAS PubMed.
- T. R. Fadel, F. A. Sharp, N. Vudattu, R. Ragheb, J. Garyu, D. Kim, E. Hong, N. Li, G. L. Haller, L. D. Pfefferle, S. Justesen, K. C. Herold and T. M. Fahmy, Nat. Nanotechnol., 2014, 9, 639–647 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00405h |
| ‡ Z. H. and L. C. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |