
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA multivariate metal–organic framework based pH-responsive dual-drug delivery system for chemotherapy and chemodynamic therapy†
Muhammad Usman
Akbar
 ab,
Arslan
Akbar
b,
Umair Ali Khan
Saddozai
c,
Malik Ihsan Ullah
Khan
d,
Muhammad
Zaheer
*b and
Muhammad
Badar
*a
ab,
Arslan
Akbar
b,
Umair Ali Khan
Saddozai
c,
Malik Ihsan Ullah
Khan
d,
Muhammad
Zaheer
*b and
Muhammad
Badar
*a
aGomal Center of Biochemistry and Biotechnology, Gomal University, Dera Ismail Khan, 29050, Pakistan. E-mail: mbadar@gu.edu.pk
bDepartment of Chemistry and Chemical Engineering, Syed Babar Ali School of Science and Engineering, Lahore University of Management Sciences (LUMS), Lahore, 54792, Pakistan. E-mail: muhammad.zaheer@lums.edu.pk
cDepartment of Preventive Medicine, Institute of Biomedical Informatics, Bioinformatics Center, Henan Provincial Engineering Center for Tumor Molecular Medicine, School of Basic Medical Sciences, Henan University, Kaifeng 475004, China
dInstitute of Molecular Biology and Biotechnology, The University of Lahore, Lahore, 54000, Pakistan
First published on 12th October 2023
Abstract
Combination therapy has emerged as a promising strategy due to its synergistic therapeutic pathways that enhance anticancer efficacy and limit the emergence of drug resistance. In this work, MIL-88B type multivariate (MTV-1) nanocarriers based on a mixed linker (1,4-benzenedicarboxylic acid and biphenyl-4,4′-dicarboxylic acid) and metals (iron and cobalt) were synthesized. The presence of the distinct linkers modified the pore makeup of MTV-1 and facilitated the co-encapsulation of two anticancer drugs of varying molecular sizes: 5-fluorouracil (5-FU) and curcumin (CUR). The drug loading measurements on MTV-1@5-FU + CUR represented a loading capacity of 15.9 wt% for 5-FU and 9.3 wt% for CUR, respectively. They further exhibited a pH-responsive drug release pattern with higher concentrations of 5-FU and CUR released at pH 5.5 (simulating cancer microenvironment) compared to pH 7.4 (physiological environment). Moreover, we also demonstrated that MTV-1 MOFs, due to the presence of mixed valence metal ions, could exhibit peroxidase-like activity and catalyze H2O2 decomposition to produce ˙OH radicals for chemodynamic therapy. Cell cytotoxicity assays exhibited significant inhibitory effects of MTV-1@5-Fu + CUR against HepG2 cells with an IC50 of 78.7 μg mL−1. With dual-drug loading, pH-responsive release, and chemodynamic therapy, MTV-1 shows excellent potential for multifunctional anticancer treatment.
Introduction
Cancer has remained one of the leading causes of death worldwide.1 The main hurdles to overcoming this issue lie in the widely used traditional chemotherapies based on monotherapeutics.2 The poor delivery mechanisms and insufficient distribution of drugs in an intracellular environment have aggravated the problem.3 The emergence of drug resistance, target site mutagenesis, and interconnected resistance pathways further mitigates the efficacy of drugs designed to act on singular molecular pathways.4 Recently, using multiple drugs against a single target, also known as combination therapy, has shown promising results in alleviating these problems.5–8 Combination therapy offers multiple therapeutics in a single treatment and benefits from their distinct modes of action for enhanced anticancer activity.9 Various drug delivery carriers, such as liposomes,10 supramolecular nanoparticles,11 polymers,12 layered double hydroxides (LDHs),13 and mesoporous silica,14 have been employed for multi-drug delivery. Among all the present nanocarriers, metal–organic frameworks (MOFs), have attracted much attention for drug delivery applications.15–20 MOFs are constructed through the coordination between metal ions and organic linkers, also known as secondary building units (SBUs).21 When used as drug delivery systems (DDSs), MOFs can prolong the drug release period,22 enhance drug solubility,23 improve the antitumor effect,24 and bring a pharmacokinetic change in the drug delivery pattern.25,26Moreover, synthetic or post-synthetic modifications can turn the MOFs into stimuli-responsive nanocarriers to release their payload against external (temperature, irradiation, and pressure) or internal (redox reaction, pH, ATP, and H2S) stimuli.27–31 Additionally, chemical constituents of MOFs can be manipulated to utilize higher levels of reactive oxygen species (ROS) produced in cancer cells for chemodynamic therapy.32–35 Compared to normal cells, cancer cells generate more ROS (especially H2O2) stress due to mitochondrial malfunction and metabolic activity.36–38 ROS is usually classified into singlet oxygen (1O2), hydroxyl radicals (˙OH), hydrogen peroxide (H2O2) and superoxide anion radical (O2˙−).39,40 Moreover, with coordination complexes containing divalent or trivalent metal ions such as Iron (Fe), Cobalt (Co), Manganese (Mn), Cerium (Ce) and Copper (Cu), H2O2 can be decomposed into highly potent ˙OH radicals through Fenton and Fenton-like reactions.41,42 These ˙OH radicals are highly reactive and can cause lipid peroxidation, DNA damage and oxidation of proteins.43–45 Compared to monometallic molecules, the presence of two or more different metals in coordination compounds, in many cases, has demonstrated enhanced catalytic performance due to the synergistic effect between the metals.46,47 For instance, the presence of a Fe–Cu complex (heme proteins) and Mo–Fe proteins in the human body plays an essential role in cytochrome and nitrogenase oxidation metabolic pathways.48,49 The coexistence of different metals and their synergism in some cases becomes indispensable for efficient catalytic properties of the enzymes.50 Similar to naturally found bimetallic enzymes, mixed metal MOFs have also demonstrated improved catalytic performance due to the synergistic effect between different metals.51,52 The Fenton-like performance of Fe-based MOFs largely depends upon the concentration of divalent Fe2+ in the system and cycling between Fe2+ and Fe3+ and vice versa.53 However, achieving an appropriate ratio of Fe2+ and Fe3+ metals in MOF synthesis is troublesome and leads to the formation of an amorphous product or requires thermal reduction resulting in defective MOFs.54 Therefore, synthesizing mixed metal MOFs with controlled architecture using pre-synthesized mixed metal clusters containing trivalent Fe3+ and divalent second metal (MII) is considered a viable option. The pre-synthesized mixed-metal cluster exhibits a highly defined stoichiometric ratio of both metals, plus the excellent electron transfer between different metals further improves the catalytic performance of the designed framework.55
Although MOFs have made some progress in chemodynamic therapy,56 their use for combination therapy is limited.57 One of the main reasons behind the low utility of MOFs for combination therapy is the use of analogous SBUs (organic/inorganic) in MOF synthesis, leaving less room for pore engineering and modifications.58,59 To overcome this, multi-component/multivariate (MTV) MOFs have recently emerged as an alternative solution to the problem.60,61 In MTV MOFs, more than one type of SBU (linker and metal) are intertwined chemically in a singular framework; maintaining the structural traits of the parent MOF while altering the physico-chemical environment.62,63 Using multiple SBUs helps tailor the pore environment to co-encapsulate different drugs and release them through engineered chemical functionalities.64,65 Minor alterations in the SBUs can bring macroscopic changes and result in a variety of MTV-MOFs synthesized with targeted properties.66 Due to these characteristics, MTV-MOFs have been proving their mettle in the fields of gas storage,67 luminescence sensing,68 and heterogeneous catalysis.69 However, their potential for multi-drug delivery and chemodynamic therapy hasn’t been fully explored.
5-Fluorouracil (5-FU) is a hydrophilic (pyrimidine analog) anticancer drug used to treat a variety of tumors through DNA/RNA disruption.70 However, 5-FU is non-specific to targets, and its rapid degradation rate (5–10 min) leads to the inadequate presence of the drug in cancer cells,71 which has reduced its efficacy and led to the emergence of multi-drug resistance (MDR).72 To overcome this issue, 5-FU has been combined with other drugs, such as paclitaxel, cisplatin, and curcumin (CUR), to improve cytotoxicity and reduce dosage requirements.73–75 CUR, a natural polyphenol chemotherapeutic agent, has shown great potential for antioxidant, antimicrobial, and antitumor purposes.76–79 CUR induces anticancer effects by initiating various signalling (e.g., PI3K/Akt, STAT3, and NF-kB) pathways inducing apoptosis.80,81 However, free CUR cannot be administered directly due to its poor bioavailability and high hydrophobicity, requiring carriers for the delivery to the target site.82
To co-encapsulate 5-FU and CUR by pore engineering, we synthesized MTV-1 MOFs with variable pore sizes by introducing a sizeable ratio of biphenyl 4-4, dicarboxylic acid (BPDC) into mono linker FeCo-MIL-88B MOFs through an isoreticular expansion strategy. Fe and Co-based mixed-metal clusters were used in the synthesis process due to their accessible Lewis acid sites for drug adsorption and peroxidase-like activity of both metals. The drug loading capacity (DLC) and drug loading efficiency (DLE) of MTV-1@5-FU + CUR were measured to be 15.9 and 18.1 wt% for 5-FU and 9.3 and 10.5 wt% for CUR. Mathematical kinetic release models were applied to fit the nanocarriers’ drug release pattern at different pHs (pH = 7.4 for normal tissues and pH = 5.5 for the tumor cell environment). Moreover, the in vitro cytotoxicity profile of MTV-1 based on chemotherapy and chemodynamic therapy was evaluated against HEK-293 and HepG2 cell lines.
Materials and methods
Materials
Iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O), cobalt(II) nitrate hexahydrate (Co(NO3)3·6H2O), 1,4-benzenedicarboxylic acid (BDC), biphenyl-4,4′-dicarboxylic acid (BPDC), sodium acetate trihydrate (CH3COONa⋯3H2O), 5-fluorouracil (5-FU), curcumin (CUR), 3,3′,5,5′-tetramethylbenzidine (TMB) and N,N-dimethylformamide (DMF), glutaraldehyde, crystal violet, glacial acetic acid, Tween-80 and phosphate buffer saline (PBS) tablets were purchased from Sigma-Aldrich. Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin–streptomycin (pen-strep), trypsin-EDTA, L-glutamine and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were obtained from Gibco, Invitrogen. All of the chemicals were used as received.Characterization
The morphology of the samples was characterized using a scanning electron microscope (SEM) FEI NOVA Nano SEM 450 equipped with energy dispersive X-ray spectroscopy (EDX). Samples were coated with gold prior to imaging. The metal composition and leaching analysis was performed through inductively coupled plasma-optical emission spectroscopy (ICP-OES) using an Agilent Technologies 5110-svdv ICP-OES system. Zeta potential (ZP) was determined in water medium at room temperature using Malvern zetasizer (Nano ZS, Malvern). The crystal structure was analysed through an X-ray diffraction (XRD) pattern over a 2θ range from 5–50° obtained on BRUKER D2 Phaser using Ni-filtered Cu-Kα irradiation (λ = 1.5406 Å). The Brunauer–Emmett–Teller (BET) method was used to calculate the surface area at 77 K using a Quantachrome Nova 2200e. Bruker Alpha Platinum ATR was used to perform infrared spectroscopic studies in the 400–4000 cm−1 range. The thermal behaviour of the samples was assessed through Thermogravimetric analysis (TGA) using a TA instrument in the range of 10 to 600 °C (10° min−1 ramp) under an N2 atmosphere. UV-Vis spectroscopic studies were performed to verify drug loading, release, and TMB oxidation using a spectrophotometer (Shimadzu UV-1800). The fluorometric measurements in the TA probe were carried out on a PerkinElmer Enspire 2300 multimode reader at an emission wavelength of 435 nm (λex 315 nm). Cellular uptake and intracellular ROS-based studies through fluorescence imaging were performed using a confocal laser scanning microscope (CLSM) LSM-880 ZEISS, Jena, Germany.Synthesis of FeCo-clusters
Bi-metallic (FeCo) clusters were synthesized using a previously reported method with slight modifications.83 A solution of CH3COONa·3H2O (6 g, 0.044 mol) in 10 mL of deionized water was prepared (solution-1). Additionally, Fe(NO3)3·9H2O (1.142 g, 0.0028 mol) and Co(NO3)2·6H2O (4.154 g, 0.014 mol) were mixed separately in 10 mL of deionized water (solution-2). Later, solution-1 was added dropwise into a filtered and stirred solution-2. The mixture was allowed to react for 24 h with mild stirring at room temperature. After 24 h, the brown precipitates were filtered and washed with a small amount of ethanol. Later, the filtrate was allowed to air dry at room temperature.Synthesis of nanocarriers
To synthesize the FeCo-MIL-88B MOF, equal masses of the FeCo-cluster (200 mg) and BDC (200 mg) were dissolved separately in 11 mL of DMF each. Samples were sonicated until the cluster and linker were dissolved. Later, the linker solution was added dropwise into the stirred solution of the cluster containing an additional 1 mL of glacial acetic acid. The mixture solution was kept under mild stirring until a homogenous solution was obtained that was transferred to an autoclave with a 50 mL capacity and incubated for 24 h at 120 °C. After 24 h, precipitates were collected through centrifugation and washed thrice with DMF and ethanol. The same methodology synthesized four additional MOFs except for the addition of different concentrations of BPDC in the linker mixture. The BPDC to BDC percentage in additional experiments was kept at 25, 50, 75, and 100%, and the synthesized MOFs were subsequently called MTV-1, MTV-2, MTV-3, and FeCo-MIL-88D.Drug loading and release
All nanocarriers were activated under vacuum at 100 °C for 24 h before drug loading experiments. Initially, three different types of synthesized MOFs were selected for drug-loading experiments, namely FeCo-MIL-88B, MTV-1, and FeCo-MIL-88D. Briefly, 60 mg of activated MOFs were dispersed in 30 mL of 5-FU (2 mg mL−1) and CUR (2 mg mL−1) solution and left for 48 h under mild stirring. After 48 h, drug-loaded MOFs were isolated through centrifugation. The absorbance of the supernatant was determined using a UV-Vis spectrophotometer with the corresponding characteristic peaks of 5-FU (265 nm) and CUR (435 nm). The calibration curve of 5-FU and CUR in ethanol was applied to determine the drug loading capacity (DLC) and drug loading efficiency (DLE) according to the following equations:84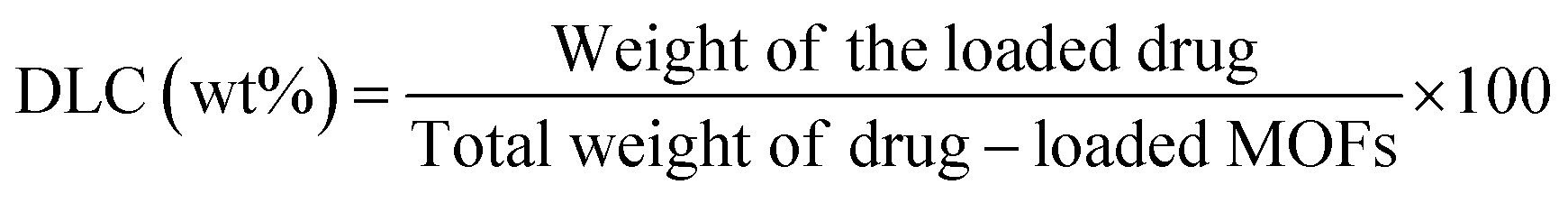 | (1) |
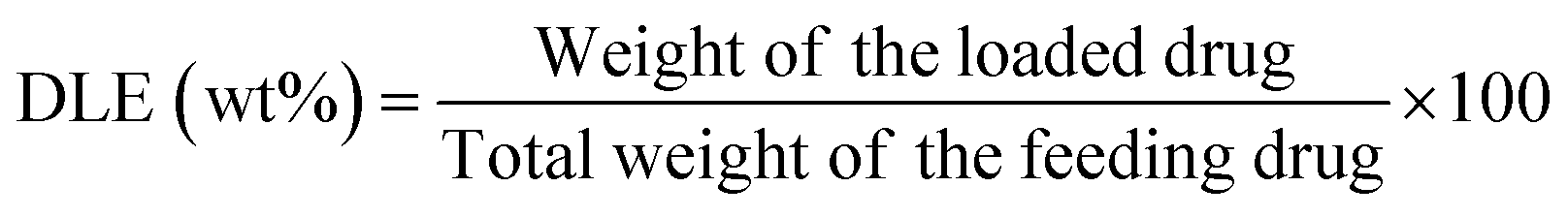 | (2) |
Based on the drug loading experiments (Table S2, ESI†) only MTV-1 MOFs loaded with 5-FU and CUR were further used for drug release studies. Drug release experiments were conducted under different pH conditions (5.5 and 7.4) in PBS with 1% (v/v) Tween 80. Briefly, a concentrated solution of MTV-1@5-FU + CUR (15 mg) was inserted into a dialysis bag (3.5 kDa MWCO) and dialyzed against 30 mL of PBS + 1% (v/v) Tween 80 solutions of pH 5.5 and 7.4. At predetermined time intervals, 1 mL of the solution containing the released drug was withdrawn and replaced with an equal amount of preheated PBS + 1% (v/v) Tween 80 solution to keep the volume constant. The isolated solutions were analyzed using UV-Vis spectroscopy to measure the amount of 5-FU and CUR according to the calibration curve of each drug drawn in the same buffer. The experiments were performed in duplicate, and the results are presented as an average. The cumulative drug release percentage was expressed as
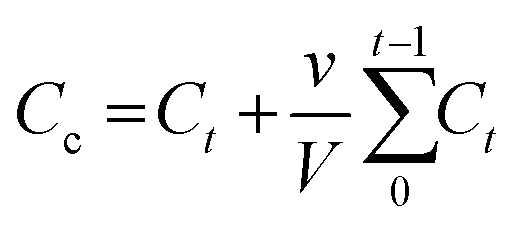 | (3) |
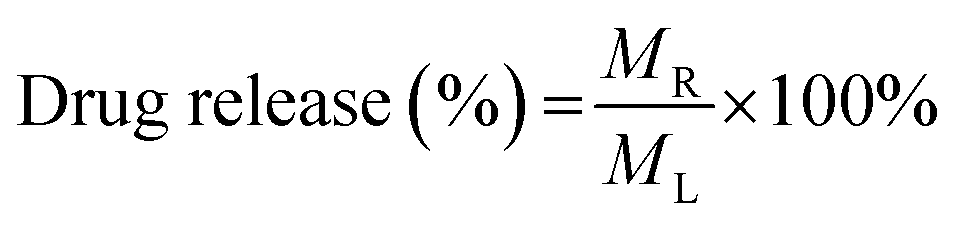 | (4) |
In eqn (3), Cc represents an adjusted concentration of CUR at time t, Ct represents the measured concentration of 5-FU/CUR at time t, v is the volume of the withdrawn sample, and V is the volume of the release solution. In eqn (4), MR represents the released drug, and ML represents the loaded drug concentrations.
Cell culture
Human embryonic kidney cells (HEK-293 cells) and Human Hepatoma cells (HepG2 cells) were supplied by The University of Lahore (UOL) Cell Culture Collection (UCCC). The cells were cultured in DMEM supplemented with 10% Hi-FBS, 1% Pen-strep (100 IU mL−1 penicillin and 100 μg mL−1 streptomycin), 2 mM L-glutamine, and 1% non-essential amino acids. Cells were grown in cell culture flasks in a humidified atmosphere at 37 °C supplemented with 5% CO2. Cells were sub-cultured in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 split ratio with 0.25% trypsin-EDTA (1–2 mL) treatment every 3–5 days.
:3 split ratio with 0.25% trypsin-EDTA (1–2 mL) treatment every 3–5 days.
Cell viability test
The in vitro cytotoxicity of the samples against HEK-293 and HepG2 cell lines was evaluated through MTT assay.85 Briefly, HepG2 and HEK-293 cells (100 μL) with a density of 1 × 104 were seeded (triplicates) in a 96-well plate. The cells were incubated for 24 h at 37 °C in a CO2 incubator. After that, the cell culture medium was removed, and different concentrations (3.9 to 500 μg mL−1) of 5-FU, CUR, MTV-1, and MTV-1@5-FU + CUR dissolved in the culture medium were added to the cells. In comparison, the cells in the control groups were not treated with any of the compounds. After 48 h of incubation, 10 μL of MTT (12 mM) reagent was added to each well, and the cells were allowed to incubate for another 4 h. After that, the medium was removed, and formazan was dissolved by adding 100 μL of DMSO. The absorbance was recorded at 570 nm using a PerkinElmer Enspire 2300 multimode reader (Germany). The treatment groups’ IC50 (half maximal inhibitory concentration) values were calculated through non-linear regression using a dose–response curve. To evaluate cell viability in the presence of H2O2 for peroxidase-like activity, the cells were co-cultured with different concentrations of MTV-1, H2O2 (200 μM), and MTV-1 + H2O2 (200 μM) according to the same procedure.Colony formation assay
The long-term cytotoxic profile of the synthesized system was evaluated through a colony formation assay. Briefly, HepG2 cells with a density of 1000 cells per well were seeded into 6-well plates in 2 mL of medium containing 10% FBS. The cells were allowed to initiate colonies for two days. Later, these cells were treated with the desired concentrations of 5-FU, CUR, MTV-1, and MTV-1@5-FU + CUR for eight days under a humidified atmosphere at 37 °C in 5% CO2. The cells were rinsed with PBS. A mixture of 0.5% crystal violet and 6% glutaraldehyde was added, and cells were left for 30 min. Later, the added mixture was removed carefully and plates containing colonies were left to dry at room temperature. After that, photographs were taken and colonies were counted.Cellular uptake studies
Due to the intrinsic green fluorescence of CUR, CLSM imaging was used to investigate the cellular uptake of MTV-1@5-FU + CUR. For this, the HepG2 cells with a density of 1 × 104 were grown in 24 well plates for 24 h. After that, the previous medium was replaced with fresh medium containing MTV-1@5-FU + CUR solution (40 μg mL−1). The cells were allowed to incubate for 48 h. Then, the culture medium was sucked out, and cells were washed with PBS twice and fixed with 4% formalin. Later, cells were stained with DAPI solution for 25 min in the dark at 37 °C and visualized under CLSM.Peroxidase-like activity assay
The peroxidase-like activity was determined by the amount of TMB oxidation that occurred in the process.86 Briefly, 5 ml of weak acidic PBS (pH 5.5) was prepared containing different amounts of MTV-1 MOFs (0, 20, 40, 60, 80, and 100 μg mL−1), TMB (0.25 mM), and H2O2 (1 mM). The mixture was incubated for 10 min at 37 °C. After that, UV-vis spectroscopic spectra of the samples at a 652 nm wavelength corresponding to the oxidized TMB in the solution were obtained. Furthermore, the peroxidase-like activity of the MTV-1 MOFs was further checked by varying the pH (4–8) and temperature (30–60 °C) of the solution, keeping the concentration of the MOFs (60 μg mL−1) constant.Intracellular ROS generation
HepG2 cells with a density of 3 × 104 cells were seeded into a 12-well confocal dish and incubated for 24 h at 37 °C (5% CO2) for intracellular ROS detection. After 24, the previous medium was replaced with fresh and different treatments of PBS, MTV-1 (50 μg mL−1) + H2O2 (100 μM) at pH 7.4 and 6.5 were applied to the cells for 4 h. Then, cells were washed with fresh medium, followed by the addition of DCFH-DA solution (25 μM), and incubated for another 1 h under dark conditions. Later, the cells were washed with PBS, and nuclei were stained with DAPI. The intracellular ˙OH generation was examined under a CLSM by measuring the fluorescence of DCFH (λex = 488 nm, λem = 525 nm).Metal leaching experiment
MTV-1 (0.50 mg mL−1) was immersed in phosphate buffers (pH 7.4 and 5.5) at 37 °C for 1, 2, 4, 6, 12 and 24 h.87 After each predetermined interval of time, samples were centrifuged for 20 min at 5000 rpm, and the supernatant collected was filtered through a Dismic (0.2 μm) syringe filter to remove any solids present. The concentration of the leached metals was determined through ICP-OES at room temperature.Terephthalic acid probing technique
To detect ˙OH generation by the decomposition of H2O2, three different treatment groups containing TA + H2O2, TA + MTV-1, and TA + MTV-1 + H2O2 were incubated in PBS (5.5) at 37 °C. The concentration of MTV-1 (50 μg mL−1), TA (2 mM), and H2O2 (1 mM) was kept constant in each treatment group. Each treatment group was allowed to react for 2 h, and photoluminescence spectra under excitation of a wavelength of 315 nm and emission wavelength of 435 nm were recorded using a fluorescence spectrometer.88Statistical analysis
All of the statistical analyses were performed using GraphPad Prism version 8.0. Data from the MTT assay was presented as mean ± standard deviation. The Kruskal–Wallis test, followed by Dunn's multiple comparison analysis, was performed to evaluate the statistically significant treatments from the control groups. A p-value equal to 0.05 or less was considered significant. The degree of significance from the control group is demonstrated as ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05. The IC50 values were calculated by fitting the cell viability data into non-linear regression analysis and evaluating it through a dose–response curve.Kinetic studies
Mathematical kinetic release models are applied to better understand the underlying mechanism of drug release from the nanocarriers.89 Some of the widely used kinetic release models, such as zero-order, first-order, Higuchi, Korsmeyer–Peppas, and Hixson–Crowell, were applied in our study.90,91 The formulae of these models are available in Table S4 (ESI†).Results and discussion
Synthesis and characterization
Fabrication of MTV-MOFs consisted of a two-step approach. At first, a FeCo(μ3-O) cluster was synthesized following a previously published route.83 During the synthesis procedure, metal ions in the cluster are arranged triangularly and bridged with a central oxygen (μ3-O) atom. The octahedrally coordinated metal ions are connected to the solvent molecules (−OH, H2O, and Cl−) at their terminal positions. Acetate ions act as a bridge between each pair of metal ions allowing the cluster to adopt a specified tri-nuclear geometry.92In the second step, pure phase FeCo-MIL-88B, FeCo-MIL-88D, and MTV-MOFs were synthesized by reacting the FeCo(μ3-O) cluster with individual and mixed linkers (BDC and BPDC) of varying stoichiometry. During the reaction, acetate ligands of the cluster were replaced by the carboxylates of reacting linkers in a dissociative manner, resulting in the formation of the desired MOF.93 A total of five MOFs were synthesized based on the percentage mixing of the additional BPDC linker (0, 25, 50, 75 and 100%) and termed FeCo-MIL-88B, MTV-1, MTV-2, MTV-3, and FeCo-MIL-88D respectively. Various analytical techniques were utilized to characterize the synthesized nanocarriers. The samples’ morphological appearance and metal composition were assessed through SEM, EDX, and ICP-OES. The SEM micrographs of the FeCo(μ3-O) cluster show irregular morphology and a polycrystalline nature (Fig. S1a and b, ESI†), while the EDX, elemental maps (Fig. S1c and d, ESI†), and ICP-OES analysis of the metal composition revealed the homogenous distribution of Fe and Co in a 2:1 ratio (Table S1, ESI†). Similarly, SEM micrographs of the nanocarriers synthesized from the same cluster revealed hexagonal rod-shaped morphology similar to MIL-88B topology (Fig. 1).94 However, a gradual increase in the size of crystals from FeCo-MIL-88B (190 ± 18 nm) to MTV-1 (364 ± 26 nm), MTV-2 (680 ± 32 nm), MTV-3 (1015 ± 58 nm) and FeCo-MIL-88D (1630 ± 62 nm) was observed by increasing the concentration of BPDC in the reaction mixture. The elemental composition of the MTV-1 MOFs in EDX and elemental mapping (Fig. 2a and b) also confirmed the homogenous distribution of both metals with a 2:1 ratio of Fe and Co.
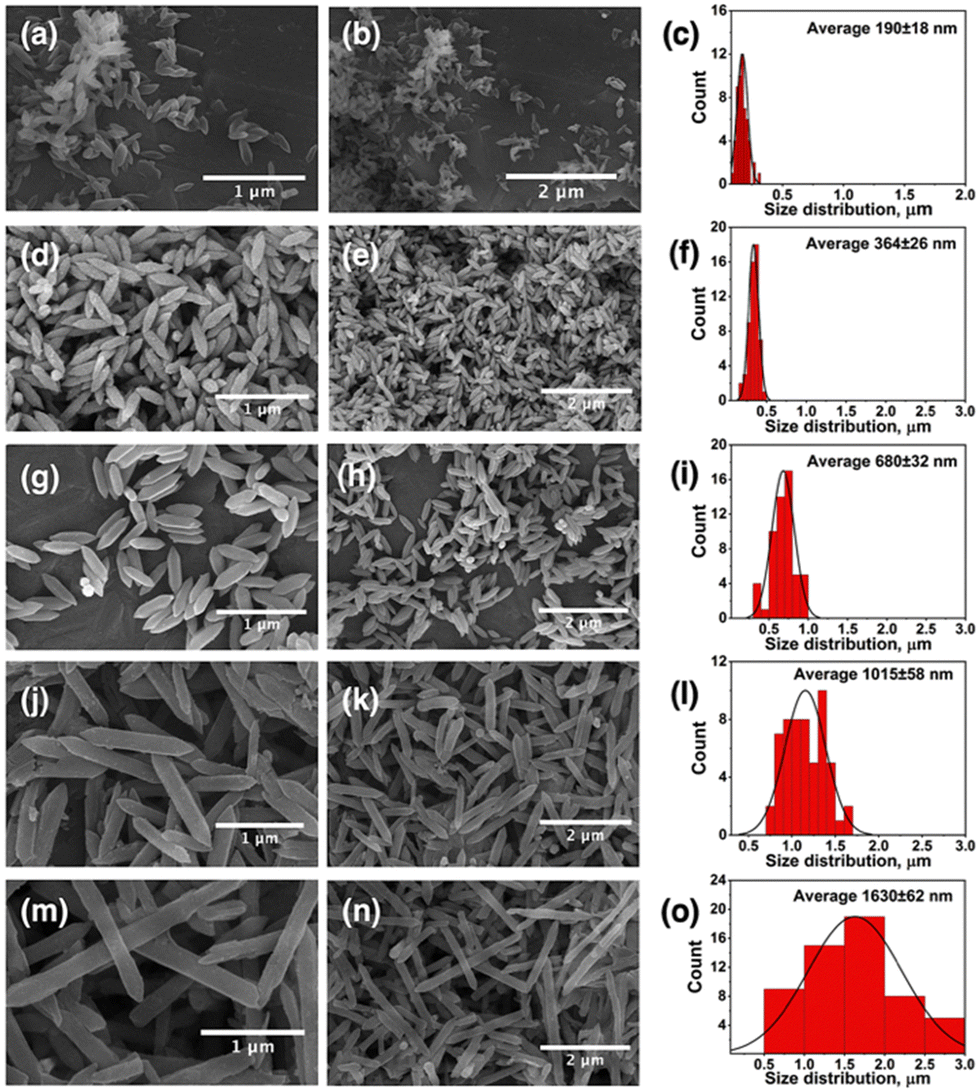 | ||
| Fig. 1 SEM images of synthesized MOFs with their corresponding size distribution charts. (a)–(c) FeCo-MIL-88B, (d)–(f) MTV-1, (g)–(i) MTV-2, (j)–(l) MTV-3, and (m)–(o) FeCo-MIL-88D. | ||
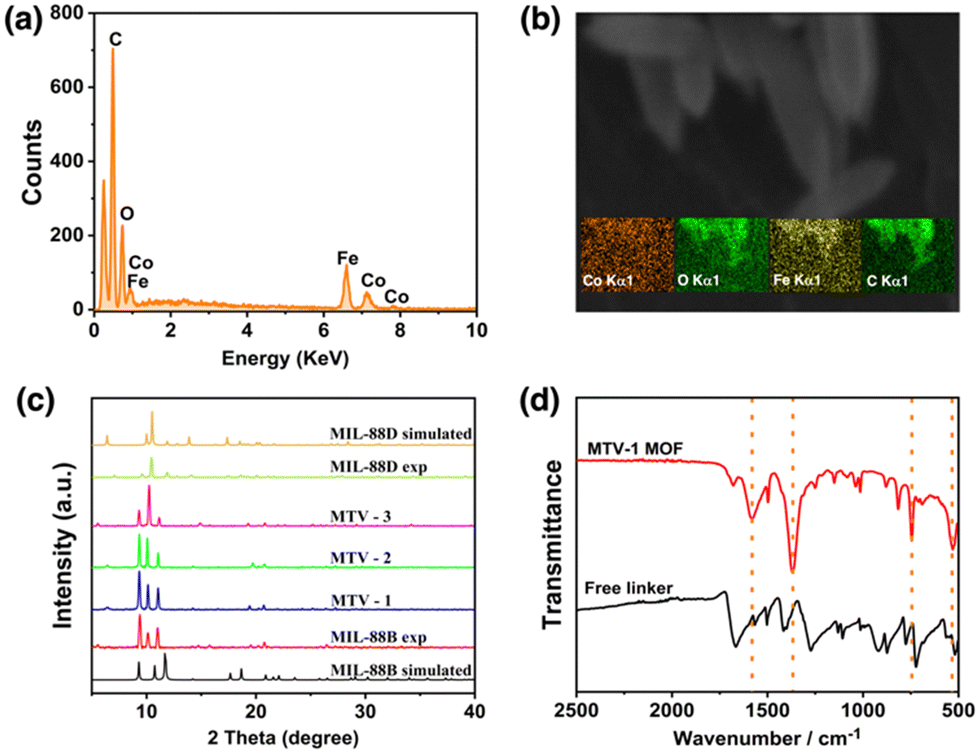 | ||
| Fig. 2 The EDX (a) and elemental maps (b) of MTV-1. The PXRD pattern (c) of simulated and experimental MOFs, and FT-IR spectra (d) of MTV-1. | ||
The PXRD patterns of the simulated and synthesized FeCo-cluster are shown in Fig. S2a (ESI†). The synthesized PXRD pattern of the cluster matched well with the reported FeCo-cluster in the literature.83 The PXRD pattern of the synthesized mono-linker MOFs exhibited high crystallinity evident from sharp peaks that matched well with the simulated MIL-88B and MIL-88D MOFs (Fig. 2c).95,96 The synthesized MOFs revealed three characteristic peaks at 9.4 and 10.14° (2theta), corresponding to 002 and 100 planes. The third peak at 11.1° was attributed to the 101 plane of the open structure MIL-88B MOFs due to entrapped solvent molecules.97
The FT-IR spectra of the FeCo-cluster are shown in Fig. S2b (ESI†). In the FT-IR spectrum, the stretching at 726 cm−1 and 524 cm−1 represent the FeCo–O bonds in the cluster. The absence of a band at ∼600 cm−1 corresponding to the monometallic Fe3O cluster represents the broken D3h symmetry to C2v upon incorporating one Co atom in the structure, while the bands at 1577 cm−1 and 1409 cm−1 are related to the O–C–O asymmetric and symmetric stretching of the acetate ligands.98,99 The reduction in the band gap from 320 to <240 cm−1 of the asymmetric and symmetric (1373 and 1606 cm−1) vibrations of MTV-1 MOFs shows the coordination of the linker with metal clusters and the formation of MOFs.100 The IR stretching around 1680 cm−1 in MTV-1 corresponds to the presence of coordinated DMF molecules in the structure (Fig. 2d). Based on the crystal size and cellular uptake kinetics,101 MTV-1 MOFs (25% of BPDC) were only used for subsequent experiments in the study.
Fabrication of MTV-1@5-FU + CUR
The FT-IR spectra of 5-FU, CUR, MTV-1@5-FU + CUR, and activated MTV-1 are shown in Fig. 3a. In the FT-IR spectra of activated MTV-1, no peak around 1680 cm−1 related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of DMF was observed indicating the activation of MTV-1 due to the evacuation of solvent molecules. In the FT-IR spectra of 5-FU, peaks found at 1731, 1240, and 800 cm−1 correspond to the C–N, C–O, and C–F stretching modes also found in MTV-1@5-FU + CUR.102 The characteristic peaks related to CUR can also be seen in the FT-IR spectra of MTV-1@5-FU + CUR, indicating its incorporation.103 According to the literature, CUR can coordinate with the central metal in a diketo form.104 Since MTV-1 is composed of an oxo-centered trinuclear metal cluster (SBU) coordinated to removable H2O molecules, some of these H2O molecules are eliminated upon activation, leaving open metal sites/coordinatively unsaturated sites (CUSs).105 The CUR molecules are capable of coordinating with these CUSs. As shown in the FT-IR spectra of free CUR (Fig. 3a), the vibrational stretching at 1496 and 1424 cm−1 indicates the existence of a free carbonyl group of enol and diketone form of CUR. In the FT-IR spectra of MTV-1@5-FU + CUR, the enolic form peak at 1424 cm−1 is absent, hinting at CUR's presence in diketo form. Some of the characteristic peaks of 5-FU and CUR present in MTV-1@5-FU + CUR are listed in Table S3 (ESI†).
O group of DMF was observed indicating the activation of MTV-1 due to the evacuation of solvent molecules. In the FT-IR spectra of 5-FU, peaks found at 1731, 1240, and 800 cm−1 correspond to the C–N, C–O, and C–F stretching modes also found in MTV-1@5-FU + CUR.102 The characteristic peaks related to CUR can also be seen in the FT-IR spectra of MTV-1@5-FU + CUR, indicating its incorporation.103 According to the literature, CUR can coordinate with the central metal in a diketo form.104 Since MTV-1 is composed of an oxo-centered trinuclear metal cluster (SBU) coordinated to removable H2O molecules, some of these H2O molecules are eliminated upon activation, leaving open metal sites/coordinatively unsaturated sites (CUSs).105 The CUR molecules are capable of coordinating with these CUSs. As shown in the FT-IR spectra of free CUR (Fig. 3a), the vibrational stretching at 1496 and 1424 cm−1 indicates the existence of a free carbonyl group of enol and diketone form of CUR. In the FT-IR spectra of MTV-1@5-FU + CUR, the enolic form peak at 1424 cm−1 is absent, hinting at CUR's presence in diketo form. Some of the characteristic peaks of 5-FU and CUR present in MTV-1@5-FU + CUR are listed in Table S3 (ESI†).
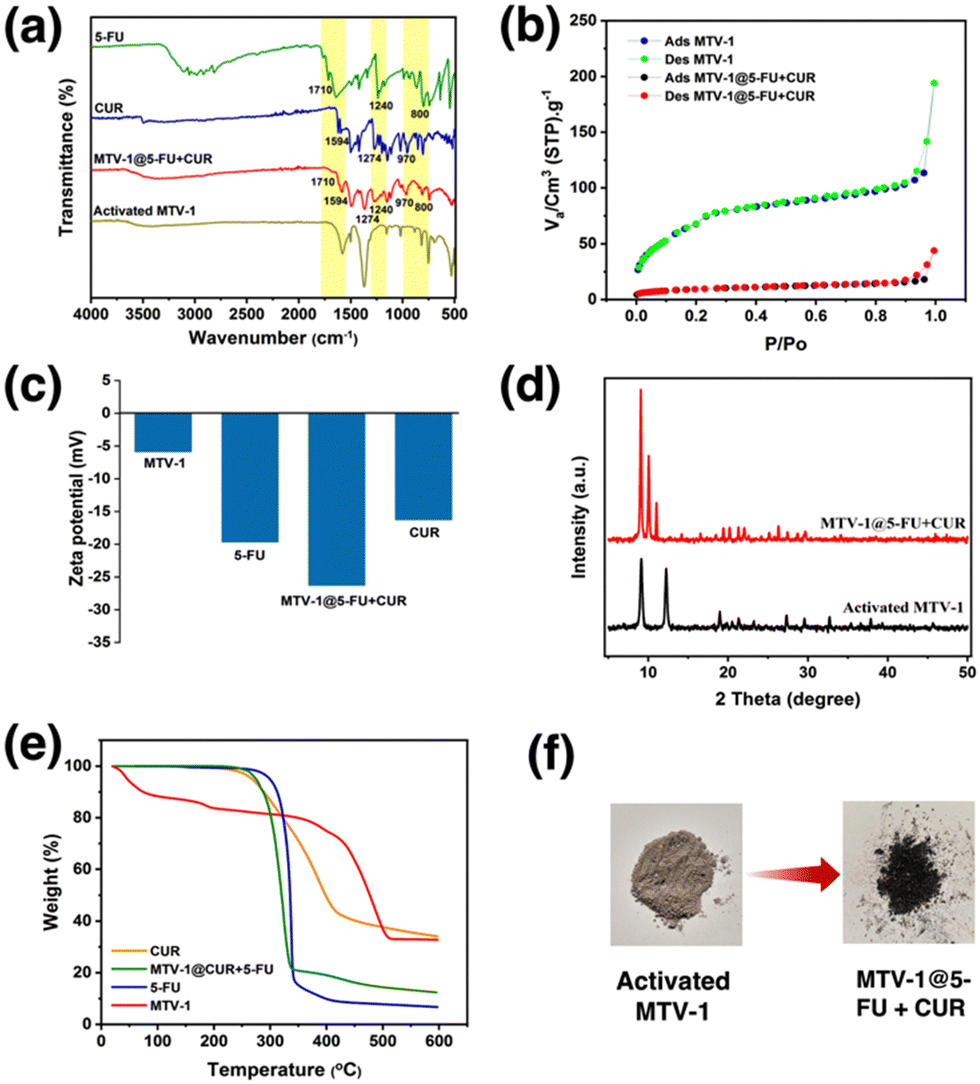 | ||
| Fig. 3 FT-IR spectra (a), N2 adsorption–desorption isotherm (b), zeta potential results (c), PXRD pattern (d), and TGA curves (e) of MTV-1 before and after drug loading. Digital photographs (f) of activated and dual-drug loaded MTV-1 MOFs. | ||
The nitrogen sorption measurements exhibited differences in the BET surface due to the presence of distinct pores with FeCo-MIL-88B (110 m2 g−1), MTV-1 (192 m2 g−1) and FeCo-MIL-88D (382 m2 g−1) (Fig. S3, ESI†). The increase in the surface area and pore heterogeneity can be observed after adding BPDC into the FeCo-MIL-88B MOFs. However, the surface area of the MTV-1 MOFs decreased from 192 to 29 m2 g−1 after drug loading, highlighting the occupation of pores by drug molecules (Fig. 3b). Drug loading was also confirmed by electro-kinetic potential/zeta potential results (Fig. 3c). The reduced potential of MTV-1 after 5-FU and CUR loading suggests drug loading not just into the pores (confirmed by a reduction in BET surface area) but also on the surface of MTV-1.106,107
The PXRD pattern of activated MTV-1 and MTV-1@5-FU + CUR is shown in Fig. 3d. A shift from 10.8 to 12° (2theta) was observed in thermally activated samples due to cage shrinkage and elimination of trapped solvent molecules based on Bragg's law.108 No significant change in the PXRD pattern of the activated MTV-1 and MTV-1@5-FU + CUR was observed. However, a shift in the peak of the 101 plane from 12° to 11.05° and the emergence of a peak at 10.4° of the 100 plane correspond to the open form of the structure due to reverse breathing and drug encapsulation.109 Moreover, no extra peaks related to the crystalline form of drug molecules were seen in the PXRD pattern of MTV-1@5-FU + CUR, suggesting the presence of drug molecules in an amorphous form.
The TGA analysis of the samples is shown in Fig. 3e. The mass loss below the 300 °C temperature range was attributed to the evacuation of coordinated solvent/water molecules inside the structure. In contrast, the second mass loss from ∼340 to 510 °C was due to the decomposition of the linkers and structural collapse.110,111 The drug-loaded MTV-1@5-FU + CUR did not exhibit an initial mass loss pattern like MTV-1 and presented significant mass loss in the range of 270–340 °C related to the decomposition of drug molecules.112,113 The TGA analysis further concludes the successful encapsulation of the drug molecules into the structure of MTV-1.
The amount of drugs loaded on the nanocarriers was checked through UV-vis spectroscopy based on the calibration curves of 5-FU (λ = 265 nm) and CUR (λ = 425 nm) in ethanol (Fig. S4 and S5, ESI†). The drug loading capacity (DLC) of the nanocarriers for 5-FU and CUR was found to be 15.9 and 9.3 wt%, while the drug loading efficiency (DLE) for 5-FU and CUR were 18.1 and 10.5 wt%, respectively.
Drug release
The pH-responsive drug release from MTV-1@5-FU + CUR was investigated in PBS buffer solutions under physiological (pH 7.4) and tumor microenvironment (pH 5.5) conditions (Fig. 4). It is well known that the cancer cell environment is acidic; thus, developing pH-responsive nanocarriers could be an effective strategy to enhance the selectivity and efficacy of therapeutics.114,115 The dual-drug release profile of MTV-1@5-FU + CUR was monitored for 48 h based on the calibration curves of 5-FU and CUR in PBS +1%v/v Tween-80 (Fig. S6 and S7, ESI†). The release profile indicated a burst release of both drugs during the initial eight hours, releasing more than 21 and 37% of the loaded 5-FU and CUR at pH 5.5. The burst release of the drug during the first few hours can be attributed to the adsorbed drug molecules on the surface of the framework.19 After the initial burst release, a gradual increase in the release was observed, leading to 50% of the loaded 5-FU being released after 32 h and CUR after 18 h at pH 5.5. While at pH 7.4, the percentage of 5-FU and CUR release stood at only 18% and 21% at the same time intervals.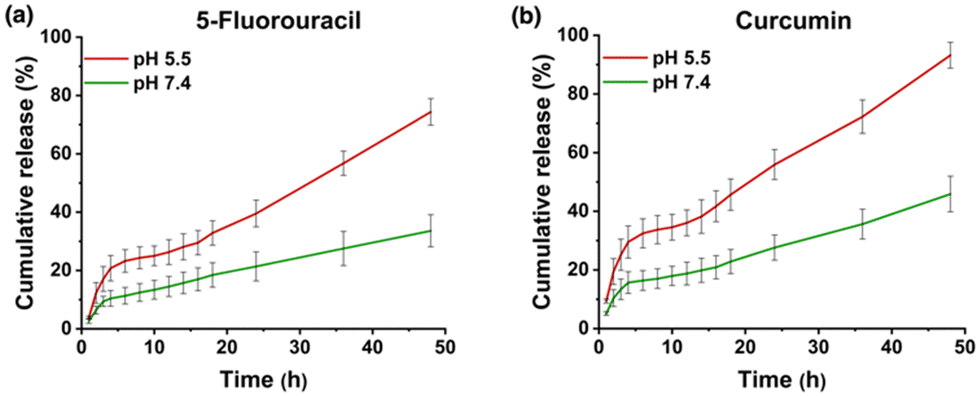 | ||
| Fig. 4 Drug release profile of MTV-1@5-FU + CUR for 5-FU (a) and CUR (b) at different pH values (5.5 and 7.4). | ||
The rapid drug release by the pH-responsive nanocarriers at lower pH is helpful in the accumulation of therapeutics in tumorigenic tissues, enhances efficacy, and reduces cytotoxicity to healthy cells.116 The faster release of CUR from the carriers can be correlated with the protonation of CUR at lower pH.117 The diketo oxygen atoms in the CUR act as proton sponges in an acidic environment with proton abundance resulting in a higher release of the drug from the carriers.118 While 5-FU weakly binds to MOFs, which causes its quicker release in the simulated environment.119 Another reason behind the rapid release of drug molecules under acidic pH (5.5) could be the MOF's linker protonation and structural decomposition.120 As seen in the PXRD pattern of the MTV-1 samples soaked in PBS (5.5 and 7.4) for 48 h (Fig. 5a), the samples soaked in PBS (7.4) mostly retained their crystalline character. In comparison, the PXRD pattern of the nanocarriers immersed in PBS (5.5) resulted in the loss of prominent peaks related to the parent MOF. Additionally, extra peaks emerged around 17, 26, 31, and 45° (2theta), indicating the formation of Fe/Co phosphates.121,122 The strong affinity of phosphate ions in PBS towards the accessible Fe/Co sites results in the release of carboxylate ligands and degradation of the structure.123
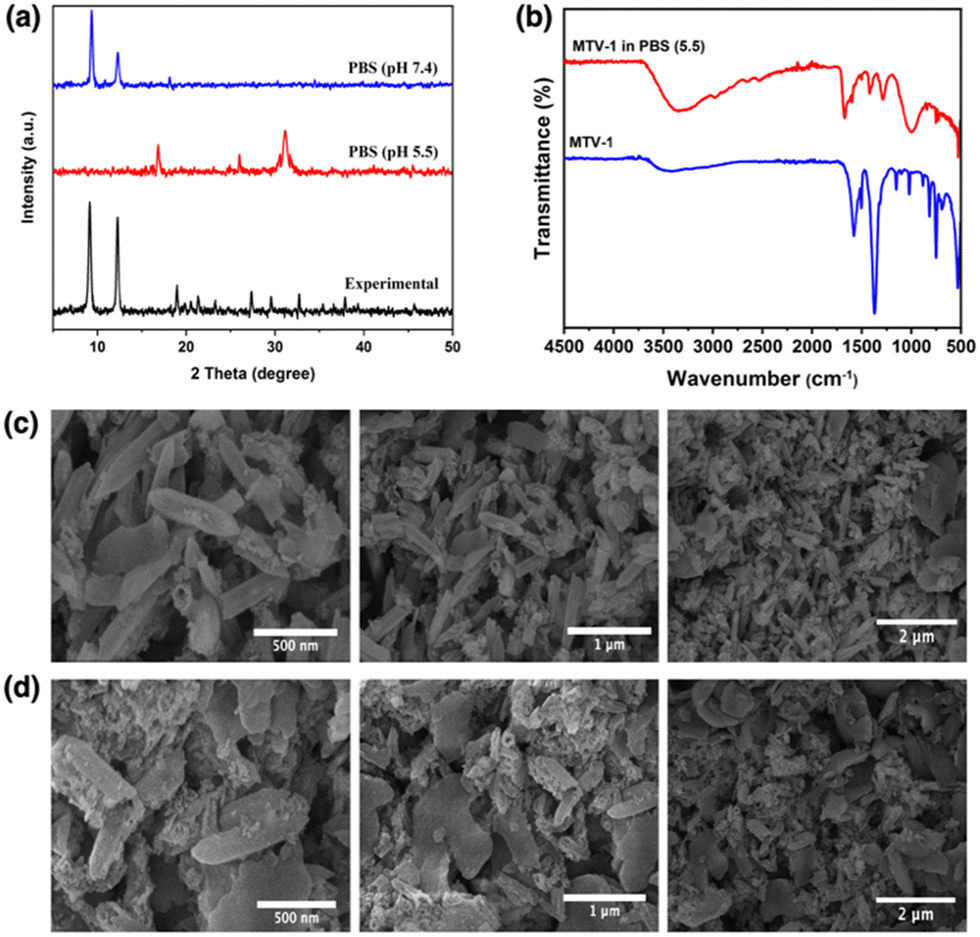 | ||
| Fig. 5 PXRD pattern (a) of MTV-1 immersed in PBS (5.5 and 7.4), FT-IR spectra (b) of MTV-1 immersed in PBS (5.5), and SEM images of MTV-1 immersed in buffers of pH = 7.4 (c) and pH = 5.5 (d). | ||
FT-IR further evaluated this to understand the variation in the atomic connectivity of the MTV-1 structure under PBS. As shown in Fig. 5b, significant changes in the vibrational stretching can be observed in the samples immersed in PBS (5.5). A visible decrease in the intensity of the metal-oxygen bonds around 725 cm−1 and 524 cm−1 is found in the spectra. The vibrational bands of the free linker around 1680 cm−1 (antisymmetric) and 1288 cm−1 (symmetric) can also be seen in the spectra of PBS-immersed samples.124 Moreover, the broad band found around 1100 cm−1 to 920 cm−1 is related to the antisymmetric stretching of PO43− groups.125 Considering the strong attraction of phosphate groups towards polyvalent cations and the presence of the uncoordinated ligands’ footprints, the data supports the analogy of drug release through structural decomposition.
SEM analysis also investigated the structural anomaly of the MTV-1 after immersion into different pHs. As shown in Fig. 5c, slight distortions in the morphology were found in the samples immersed in PBS (7.4), but the overall structure was retained. However, amorphous structures without appropriate morphology can be seen in samples immersed in PBS (5.5), which further supports the theory of rapid drug release due to structural decomposition (Fig. 5d).
Kinetic study
The drug release from a carrier depends on various factors, such as drug movement, carrier degradation, swelling, and interaction with guest molecules.126 To better understand the drug release process, data from drug release studies were fitted into different mathematical kinetic release models and their formulas are presented in Table S4 (ESI†). These models are described below.Zero-order
In zero-order release models, the release of encapsulated drug is time-dependent and follows a constant rate irrespective of the concentration of the drug.127 The obtained data from the release studies are fitted by plotting cumulative drug release (%) against time. M0 is the drug's initial concentration, Mt is the drug released over time t, and K0 is the zero-order constant.First-order
First-order release models describe that the relationship between time and concentration is always concentration-dependent. According to this model, the release amount of the drug always depends on the remaining drug concentration inside the carriers and tends to decrease over time.128M0 is the initial concentration of the drug dissolved, Mt is the concentration of the drug released at time t, and K1 is the first-order constant. The data is presented by plotting the log of cumulative (%) drug remaining against time.Higuchi model
The Higuchi release model is based on Fick's law of diffusion and states that the drug release rate depends on diffusion. It is applied when the drug's initial concentration exceeds its solubility in the matrix.129 In this model, Mt is the drug concentration released at time t, and KH is the release constant.Korsmeyer–Peppas model
The Korsmeyer–Peppas model of drug release is applied when the release mechanism is unknown, the drug release is from a polymeric system, or two or more phenomena govern the drug release process.130 The data are represented by plotting the log of cumulative drug release (%) against the log of time. Where Mt/M∞ denotes fractional drug release at time t, kKP is the Korsmeyer–Peppas release constant, and n is the release exponent.Hixson–Crowell model
The volume and area of the carriers govern the Hixson–Crowell drug release model. It is applied when the drug release from the carriers is limited to the dissolution rate rather than diffusion.131 The data is represented by plotting the cube root of the remaining drug (%) against time. Mt/M∞ represents fractional drug release, and KHC is the Hixson–Crowell release constant.The results obtained by fitting the different kinetic models (Fig. S8 and S9, ESI†) to the in vitro drug release profiles are presented in Table 1. It was found that drug release under normal pH favours zero order, first order, and the Hixson–Crowell model. In contrast, the correlation coefficient (R2) was found to be higher in zero order for drug release in acidic pH. The R2 value for acidic pH release was also higher in the Hixson–Crowell and Higuchi model. It can be concluded from the application of mathematical release models and their representative R2 values that the main driving forces behind drug release at both pHs are diffusion and structural disintegration of MTV-1 MOFs.
| Drug | pH | Parameters | Zero-order | First-order | Higuchi model | Korsmeyer–Peppas | Hixson–Crowell |
|---|---|---|---|---|---|---|---|
| 5-FU | 5.5 | R 2 | 0.9688 | 0.8821 | 0.9447 | 0.9343 | 0.9399 |
| 7.4 | 0.9644 | 0.9709 | 0.9432 | 0.9285 | 0.9707 | ||
| CUR | 5.5 | 0.9648 | 0.9419 | 0.9303 | 0.8973 | 0.9585 | |
| 7.4 | 0.9602 | 0.977 | 0.9792 | 0.9417 | 0.9723 |
Cytotoxicity assay and cellular uptake
The cytotoxicity effect of free 5-FU, CUR, MTV-1, and MTV-1@5-FU + CUR was carried out against HEK-293 and HepG2 cell lines using an MTT assay.132 The cell viability results indicate dose-dependent growth inhibition of both cell lines. As shown in Fig. 6, with the increase in the concentration of treating agents, the viability of the cells decreased after 48 h. First, the cytotoxic profile of all treating agents was evaluated against HEK-293. The biocompatibility of any carrier against normal cells is always considered an essential step in developing efficient DDS.133 The nanocarriers, which are target-specific, biodegradable, and present lower cytotoxic effects against normal cells, are mostly considered suitable for drug delivery applications.13,101,134 Interestingly, MTV-1 did not show severe effects on the growth of HEK-293 cells and presented higher biocompatibility with an IC50 of 295.4 μg mL−1. However, a lower percentage of viable cells was observed when HEK-293 cells were treated with the MTV-1@5-FU + CUR compared to free 5-FU and CUR.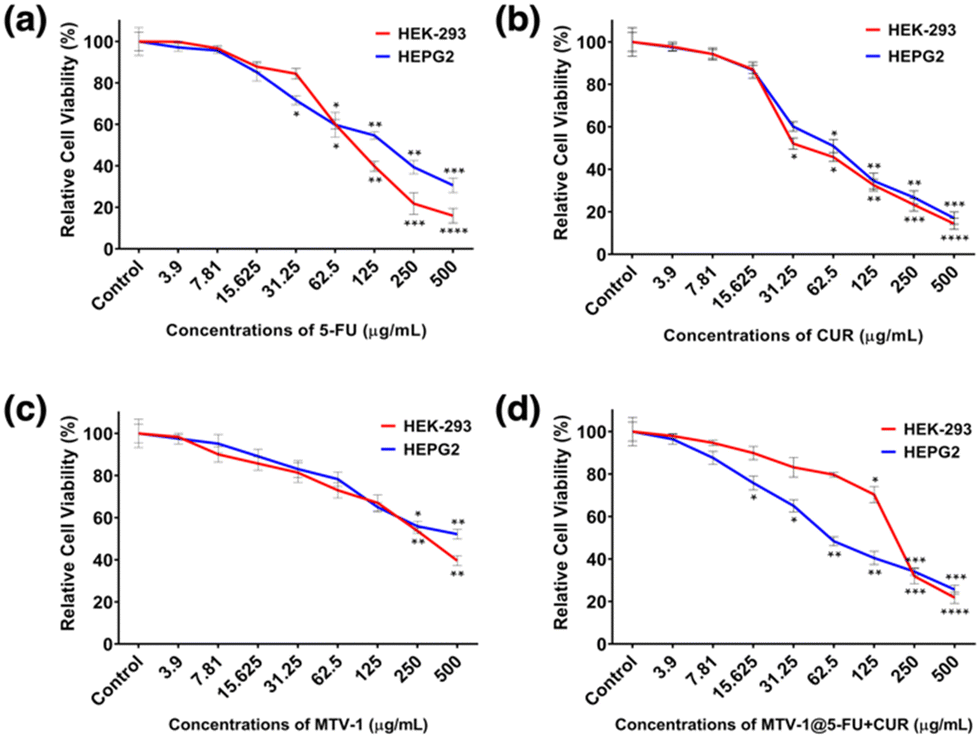 | ||
| Fig. 6 Results related to the percentage cell viability of HEK-293 and HepG2 cell lines against different concentrations of 5-FU (a), CUR (b), MTV-1 (c), and MTV-1@5-FU + CUR (d) after 48 h. The degree of significance related to treatment groups against control groups in each cell line is shown as ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05. | ||
The estimated IC50 values of the treating agents are shown in Table 2. In the case of HepG2 cells, a similar cell viability trend was observed for 5-FU, CUR, and MTV-1 with slight variation in the IC50 values. The MTV-1@5-FU + CUR exhibited enhanced cytotoxic effects against HepG2 cells compared to free 5-FU and CUR. Although the difference in the IC50 values of the applied MTV-1@5-FU + CUR and free drugs is not huge, it is essential to note the bioavailability of a free drug vs. an encapsulated one. Free drugs are readily available in the system, and the incubation time of a cytotoxicity assay might be enough to check their cytotoxicity. In contrast, an extended period of time is required to assess the cytotoxicity of a DDS as the drug release is time-constrained and not fully available to the system immediately.135
| Cell lines | Treatment groups (μg mL−1) | MTV-1@5-FU + CUR | ||
|---|---|---|---|---|
| 5-FU | CUR | MTV-1 | ||
| HEK-293 | 138.6 | 57.6 | 295.4 | 175.3 |
| HepG2 | 93.9 | 68.5 | 423.9 | 78.7 |
The statistically significant effects against the control groups for each treatment were represented as ****p ≤ 0.0001, ***p ≤ 0.001, **p ≤ 0.01, and *p ≤ 0.05. The IC50 values for MTV-1@5-FU + CUR against HepG2 cells (78.7 μg mL−1) were found to be lower than HEK-293 (175.3 μg mL−1). The cytotoxicity profile against the normal cells could be improved using surface chemistry to modify the nanocarriers with targeted biopolymer coatings reported in the literature.136–139
To evaluate the long-term anticancer efficacy of free 5-FU, CUR, MTV-1, and MTV-1@5-FU + CUR, colony-forming assay was performed in HepG2 cancer cell lines. The cells were treated with equal doses of 125 μg mL−1 of the samples and allowed to incubate for eight days. As shown in Fig. S10 (ESI†) a significant decrease in the number of colonies (>10-fold) in the cells treated with MTV-1@5-FU + CUR was observed compared with the control, free 5-FU, CUR, and MTV-1. These results were found in agreement with previous studies, which observed that nanocarriers encapsulated with chemotherapeutics showed a higher reduction in the colony formation ability of cancer cells compared with the free drug molecules.140 Another reason behind the higher anti-colony formation activity of the MTV-1@5-FU + CUR is the synergistic effect of 5-FU and CUR against cancer cells. According to previous studies, dihydropyrimidine dehydrogenase (DPYD) expression is considered one of the major determinants in limiting 5-FU catabolism and efficacy.141 While CUR is known to induce the P53 pathway which plays a key role in downregulating DPYD expression. Therefore, CUR synergistically enhances the cytotoxicity of 5-FU by indirect downregulation of DPYD expression.142
The potential of MTV-1 to be used as DDS was also evaluated through cellular uptake studies based on the intrinsic green fluorescence of CUR.143 The fluorescence imaging results for the cellular uptake of MTV-1@5-Fu + CUR into HepG2 cells are presented in Fig. 7; the blue fluorescence represents the cell's nucleus stained by DAPI, and the detected green fluorescence is of CUR. The merged overlay image reveals the successful internalization of the MTV-1@5-FU + CUR by HepG2 cells, as green fluorescence of the CUR can be observed in the cytoplasmic area. Generally, the therapeutic ability of any anticancer drug depends on its effective delivery into the cytoplasm, accumulation, and retention for a sustained period.144 Based on the results, the significant cytotoxicity of MTV-1@5-FU + CUR observed against HepG2 cell lines compared to free drugs could also be attributed to the successful internalization, accumulation and sustained release of both drugs encapsulated into the nanocarriers. Together, these results further suggest the potential of MTV-1@5-FU + CUR to be used as an effective DDS against HepG2 cancer cells.
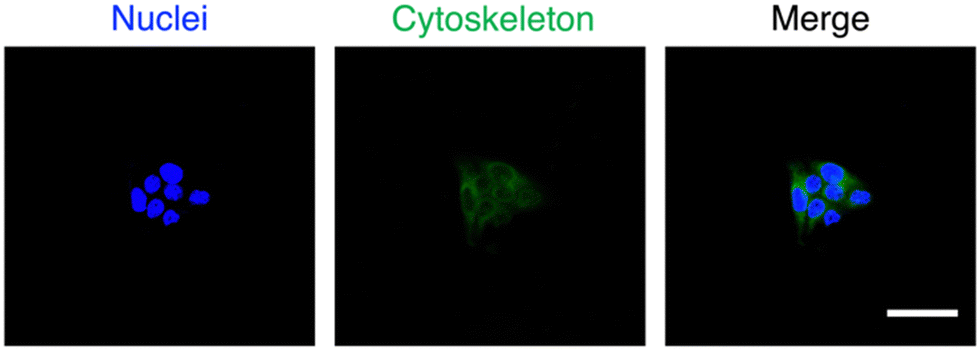 | ||
| Fig. 7 The fluorescence images of cell nucleus (a) with blue fluorescence, stained by DAPI, MTV-1@5-FU + CUR (green fluorescence) (b), and overlay image (c) with scale bars = 20 μm. | ||
Peroxidase like activity
Due to the frequent utilization of Fe and Co-based compounds as catalysts for redox reactions,145,146 MTV-1 was investigated for their peroxidase-like characteristics using the 3,3′,5,5′-tetramethylbenzidine (TMB) oxidation method (Scheme 1).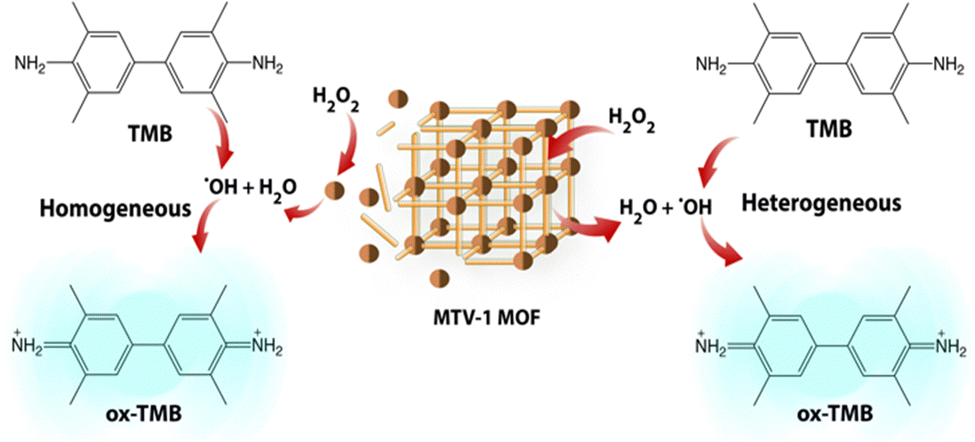 | ||
| Scheme 1 Schematic representation of two-way channelled peroxidase-like activity of MVT-1 MOFs. | ||
TMB is a classic colourless chromogenic agent that tends to transform into a green-coloured oxidized form (ox-TMB) in the presence of H2O2 and a catalyst.147 Mechanistic studies have revealed that H2O2 is first decomposed into highly reactive ˙OH radicals by the catalyst, and these ˙OH radicals in the second step oxidize the TMB present in the system.148 As shown in Fig. 8a, no ox-TMB absorbance was observed at 652 nm when the system only contained H2O2 + TMB. In contrast, adding MOFs into the buffer system comprising H2O2 + TMB resulted in the change of colour and appearance of the absorption peak at 652 nm. This suggested that the peroxidase-like activity of our MOFs is similar to that of horseradish peroxidase (HRP). The desirable catalytic performance of the MTV-1 was also investigated by varying different factors, such as the system's catalyst concentration, temperature, and pH. In general, the catalytic reaction is accelerated by increasing the concentration of the catalyst, which was also observed in our case. The increase in catalyst concentration also increased the conversion of TMB into ox-TMB with increased colour intensity (Fig. 8a). Also, the optimal pH and temperature for the reaction were around 4 and 55 °C (Fig. 8b and c). The lower absorbance intensity of ox-TMB near physiological pH (7.4) verified the lesser ˙OH production under normal conditions, indicating the safety for normal tissues. On the contrary, the ox-TMB absorbance intensity enhanced with an increase in the system's acidity, which marked the higher catalytic activity of MTV-1 under acidic (tumor) conditions.
 | ||
| Fig. 8 Kinetic analyses of TMB oxidation by keeping the concentrations of TMB and H2O2 constant but varying the concentrations of MTV-1 MOFs (a), keeping all of the reactants constant but varying the pH (b), and temperature (c). Digital photographs of TMB colour change at different concentrations of MTV-1 (d), Fluorescence spectra of TA present in various reaction systems (e), cell viability of HepG2 cell line relative to the presence of MTV-1 with or without H2O2 (f) and confocal images of DCFH-DA and DAPI stained cells for intracellular ˙OH detection (g) scale bar = 20 μm. | ||
Another reason behind the higher catalytic activity at lower pH is the leaching of free metal ions into the system. As confirmed by the ICP-OES studies, higher metal leaching activity was observed in the PBS (5.5) soaked MTV-1 samples as compared to PBS (7.4) (Fig. S11, ESI†). Various studies have observed that the reaction constant of free metal ions is many folds higher compared to the one coordinated to the structures.149
In this way, our MOFs not only produced ˙OH radicals at the surface (heterogeneous catalysis) but also through the leached Fe and Co metal ions in an acidic buffer (homogenous catalysis). These results further indicated the acid-sensitive intelligent Fenton-like catalytic performance of MTV-1. The higher release of Co ions compared to Fe ions is due to the weak CoII–O coordination bonds prone to disintegration in the presence of competing water molecules in the medium.150–152 Based on the hard and soft acid–base (HSAB) principle, under similar coordination environments, high-valent metals (Fe3+, Cr3+, Zr4+, etc.) containing high charge density known as hard acids tend to make strong coordination bonds with oxygen donor ligands (carboxylate ligands) known as hard bases. Although carboxylate linkers have a lower pKa, high valent metal ions are linker-hungry for the maintenance of charge balance.153 This leads to higher connectivity numbers of metal clusters with more linkers to further improve the resultant MOF stability.154 However, low-valent metal ions, including Co2+, Zn2+, Ni2+, etc., are known as soft acids. Their coordination is usually more stable with N-containing linkers (soft bases) than oxygen donating linkers. In our case, the presence of one Co2+ per cluster resulted in loose coordination with BDC and BPDC, as both are O-donating linkers.155 Therefore, in the presence of competing phosphate groups and water molecules, Co2+ ions were released faster as compared to high valent Fe3+ ions that have a strong coordination with the linker.123,150
To investigate the production of ˙OH radicals during the catalytic oxidation of TMB, a terephthalic acid (TA) probing technique was utilized.156 The TA can quickly react with ˙OH and oxidize into a highly fluorescent 2-hydroxy terephthalic acid with characteristic fluorescence emission at 430 nm.157 In our case, no fluorescence emissions of HTA were observed when H2O2 + TA or MTV-1 + TA were used. However, when MTV-1 was added to the H2O2 + TA solution, sharp fluorescence emission was monitored at 430 nm, suggesting the production of ˙OH during the oxidation process (Fig. 8e).
We further verified the Fenton-like catalytic performance of MTV-1 through a cell viability assay. As shown in Fig. 8f, less cell viability is observed in the MTV-1 treatment groups with the H2O2 compared to MTV-1 and H2O2 alone. This indicates the more potent anticancer activity of MTV-1 MOFs in the presence of H2O2 available in higher concentrations inside cancer cells. The intracellular ROS probe (DCFH-DA) further proved the ˙OH production in the HepG2 cellular environment. The MTV-1 gave a weak fluorescence signal at pH 7.4 compared to no signals from the control group. However, significant green emissions were recorded at pH 6.5, indicating the production of intracellular ˙OH in large amounts (Fig. 8g). The intracellular ROS studies signify the safety profile of MTV-1 against normal cells as very little signals of ROS production were detected at pH = 7.4 mimicking the physiological environment. Some of the recently reported MOFs for multifunctional co-delivery of drugs are presented in Table S5 (ESI†). Based on the results obtained, MTV-1, with pH-responsive dual-drug delivery and smart peroxidase-like activity (Scheme 2), could be used as a potential candidate for synergistic anticancer therapy.
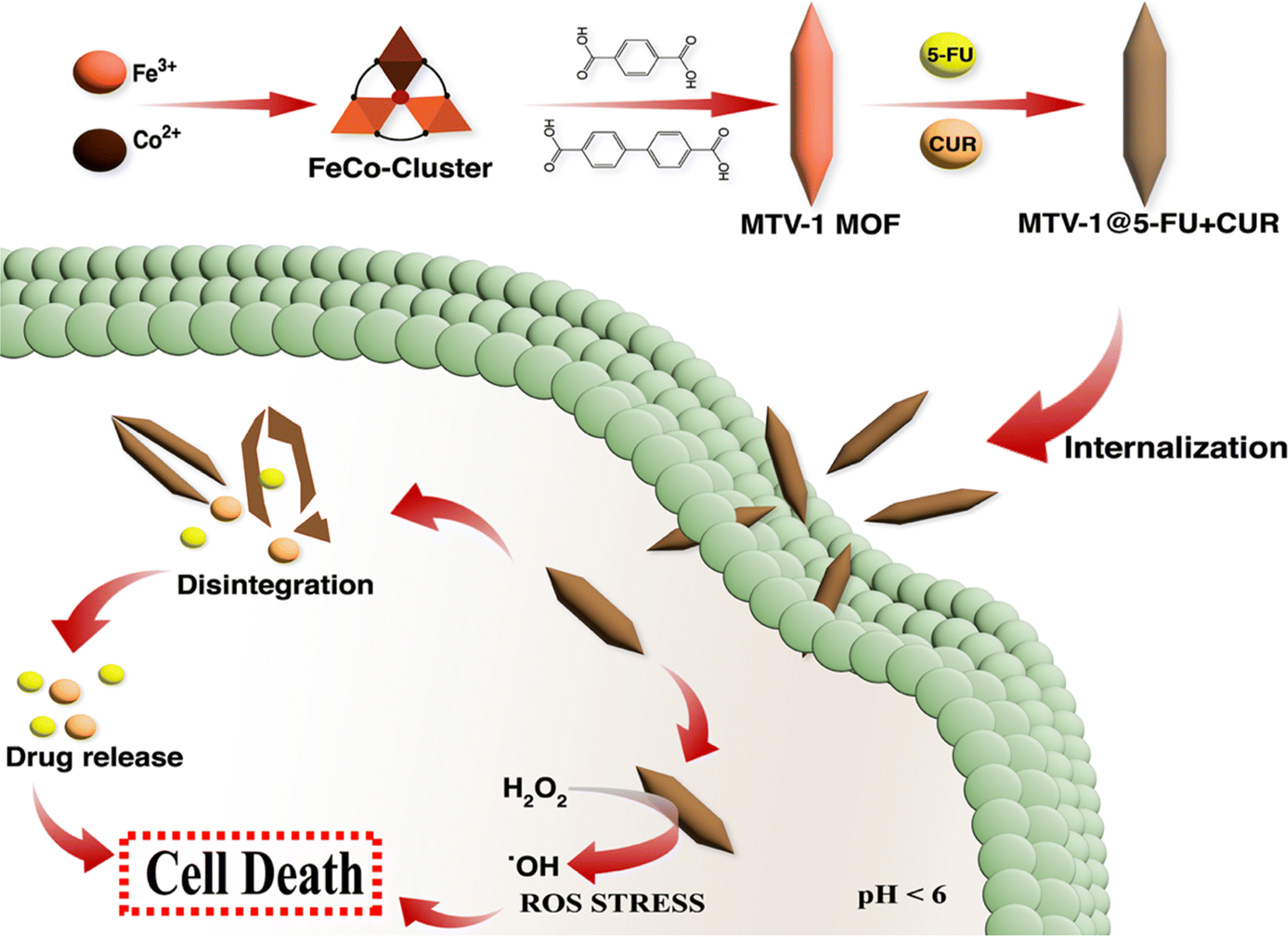 | ||
| Scheme 2 Schematic illustration of MTV-1 MOF synthesis, the mechanism of pH-responsive drug delivery and ROS mediated multidimensional anticancer activity. | ||
Conclusion
We have established multivariate MOF carriers based on a mixed-ligand and metals approach. Using mixed ligands of varying lengths generated pores of varying sizes and facilitated the co-encapsulation of two anticancer drugs of different sizes. Drug loading and release experiments demonstrated that MTV-1 MOFs could carry a load of 15.9 and 9.3 wt% of 5-FU and CUR, respectively. These nanocarriers further exhibited pH-responsive drug release with higher concentrations of drugs released under cancer microenvironment conditions (pH 5.5) compared to conditions imitating a typical physiological environment (pH 7.4). The rapid release under a cancer cell environment is due to the collapse of the MTV-1's structure, confirmed by FT-IR, SEM, and XRD measurements. It shows the potential of MTV-1's to target cancer cells effectively. Moreover, the peroxidase-like catalytic activity of MTV-1 MOF ensured enhanced anticancer activity against HepG2 cells through a combination of chemotherapy and chemodynamic therapy. This work expands the dimensions of designing MOFs for co-encapsulation and delivery of multiple drugs through pore engineering. The results suggest the potential of multivariate MOFs to be used as potential candidates of DDS for multifunctional anticancer therapy.Author contributions
M.U. Akbar: methodology, analysis and manuscript writing. A. Akbar: methodology. UAK. Saddozai: methodology and analysis. MIU. Khan: methodology. M. Zaheer: project supervision, funds management, manuscript editing and review. M. Badar: supervision, manuscript editing and review.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This work was supported by the Faculty Initiative Fund (grant no. FIF-739) by the Lahore University of Management Sciences (LUMS). M.U.A acknowledges LUMS and Gomal University for providing technical support and a curriculum base for the investigations in the project. The authors also extend their gratitude to The University of Lahore and Henan University for providing technical support in the project.References
- K. B. Tran, J. J. Lang, K. Compton, R. Xu, A. R. Acheson, H. J. Henrikson, J. M. Kocarnik, L. Penberthy, A. Aali and Q. Abbas, Lancet, 2022, 400, 563–591 CrossRef PubMed.
- D. Plana, A. C. Palmer and P. K. Sorger, Cancer Discovery, 2022, 12, 606–624 CrossRef CAS PubMed.
- W. Liu, A. Dong, B. Wang and H. Zhang, Adv. Sci., 2021, 8, 2003033 CrossRef CAS PubMed.
- N. Vasan, J. Baselga and D. M. Hyman, Nature, 2019, 575, 299–309 CrossRef CAS PubMed.
- H. Meng, W. X. Mai, H. Zhang, M. Xue, T. Xia, S. Lin, X. Wang, Y. Zhao, Z. Ji and J. I. Zink, ACS Nano, 2013, 7, 994–1005 CrossRef CAS PubMed.
- S. Karimifard, N. Rezaei, E. Jamshidifar, S. Moradi Falah Langeroodi, M. Abdihaji, A. Mansouri, M. Hosseini, N. Ahmadkhani, Z. Rahmati and M. Heydari, ACS Appl. Nano Mater., 2022, 5, 8811–8825 CrossRef CAS.
- Q. Pan, J. Tian, H. Zhu, L. Hong, Z. Mao, J. M. Oliveira, R. L. Reis and X. Li, ACS Biomater. Sci. Eng., 2020, 6, 2175–2185 CrossRef CAS PubMed.
- F. Yaghoubi, N. S. H. Motlagh, S. M. Naghib, F. Haghiralsadat, H. Z. Jaliani and A. Moradi, Sci. Rep., 2022, 12(1), 1959 CrossRef CAS PubMed.
- Y. Wu, S. Lv, Y. Li, H. He, Y. Ji, M. Zheng, Y. Liu and L. Yin, Biomater. Sci., 2020, 8, 949–959 RSC.
- L. Zhang, Z. Lin, Y. Chen, D. Gao, P. Wang, Y. Lin, Y. Wang, F. Wang, Y. Han and H. Yuan, Eur. J. Pharm. Sci., 2022, 174, 106199 CrossRef CAS PubMed.
- X. Liu, C. Wang, H. Ma, F. Yu, F. Hu and H. Yuan, Adv. Healthcare Mater., 2019, 8, 1801486 CrossRef PubMed.
- X. Wan, J. J. Beaudoin, N. Vinod, Y. Min, N. Makita, H. Bludau, R. Jordan, A. Wang, M. Sokolsky and A. V. Kabanov, Biomaterials, 2019, 192, 1–14 CrossRef CAS PubMed.
- F. Duda, M. Kieke, F. Waltz, M. E. Schweinefuß, M. Badar, P. P. Müller, K.-H. Esser, T. Lenarz, P. Behrens and N. K. Prenzler, J. Mater. Sci.: Mater. Med., 2015, 26, 1–8 CrossRef CAS PubMed.
- J. Yan, X. Xu, J. Zhou, C. Liu, L. Zhang, D. Wang, F. Yang and H. Zhang, ACS Appl. Bio Mater., 2020, 3, 1216–1225 CrossRef CAS PubMed.
- R. Cui, P. Zhao, Y. Yan, G. Bao, A. Damirin and Z. Liu, Inorg. Chem., 2021, 60, 1664–1671 CrossRef CAS PubMed.
- M. Cai, W. Liang, K. Wang, D. Yin, T. Fu, R. Zhu, C. Qu, X. Dong, J. Ni and X. Yin, ACS Appl. Mater. Interfaces, 2022, 14, 36366–36378 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., 2006, 118, 6120–6124 CrossRef.
- S. Begum, Z. Hassan, S. Bräse, C. Wöll and M. Tsotsalas, Acc. Chem. Res., 2019, 52, 1598–1610 CrossRef CAS PubMed.
- S. Tarasi, A. Ramazani, A. Morsali, M.-L. Hu, S. Ghafghazi, R. Tarasi and Y. Ahmadi, Inorg. Chem., 2022, 61, 13125–13132 CrossRef CAS PubMed.
- X. Zhao, S. He, B. Li, B. Liu, Y. Shi, W. Cong, F. Gao, J. Li, F. Wang, K. Liu, C. Sheng, J. Su and H. Hu, Nano Lett., 2023, 23(3), 863–871 CrossRef CAS PubMed.
- A. Dutta, Y. Pan, J.-Q. Liu and A. Kumar, Coord. Chem. Rev., 2021, 445, 214074 CrossRef CAS.
- R. C. Huxford, J. Della Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef CAS PubMed.
- R. K. Alavijeh and K. Akhbari, Colloids Surf., B, 2022, 212, 112340 CrossRef PubMed.
- Q. Jiang, M. Zhang, Q. Sun, D. Yin, Z. Xuan and Y. Yang, Mol. Pharmaceutics, 2021, 18, 3026–3036 CrossRef CAS PubMed.
- S. Lawson, K. Newport, N. Pederniera, A. A. Rownaghi and F. Rezaei, ACS Appl. Bio. Mater., 2021, 4, 3423–3432 CrossRef CAS PubMed.
- C.-Y. Sun, C. Qin, X.-L. Wang and Z.-M. Su, Expert Opin. Drug Delivery, 2013, 10, 89–101 CrossRef CAS PubMed.
- W. Cai, J. Wang, C. Chu, W. Chen, C. Wu and G. Liu, Adv. Sci., 2019, 6, 1801526 CrossRef PubMed.
- Y. Zhang, F. Wang, E. Ju, Z. Liu, Z. Chen, J. Ren and X. Qu, Adv. Funct. Mater., 2016, 26, 6454–6461 CrossRef CAS.
- F. Oroojalian, S. Karimzadeh, S. Javanbakht, M. Hejazi, B. Baradaran, T. J. Webster, A. Mokhtarzadeh, R. S. Varma, P. Kesharwani and A. Sahebkar, Mater. Today, 2022, 57, 192–224 CrossRef CAS.
- J. Dong, V. Wee and D. Zhao, Nat. Mater., 2022, 21, 1334–1340 CrossRef CAS PubMed.
- M. U. Akbar, M. Badar and M. Zaheer, ACS Omega, 2022, 7, 32588–32598 CrossRef CAS PubMed.
- K. Li, J. Yang and J. Gu, Chem. Mater., 2021, 33, 2198–2205 CrossRef CAS.
- Y. Zhong, X. Li, J. Chen, X. Wang, L. Wei, L. Fang, A. Kumar, S. Zhuang and J. Liu, Dalton Trans., 2020, 49, 11045–11058 RSC.
- J. Yang, S. Ma, R. Xu, Y. Wei, J. Zhang, T. Zuo, Z. Wang, H. Deng, N. Yang and Q. Shen, J. Controlled Release, 2021, 334, 21–33 CrossRef CAS PubMed.
- B. Yang, L. Ding, H. Yao, Y. Chen and J. Shi, Adv. Mater., 2020, 32, 1907152 CrossRef CAS PubMed.
- G.-Y. Liou and P. Storz, Free Radical Res., 2010, 44, 479–496 CrossRef CAS PubMed.
- J. Wang and J. Yi, Cancer Biol. Ther., 2008, 7, 1875–1884 CrossRef CAS PubMed.
- H. Nakamura and K. Takada, Cancer Sci., 2021, 112, 3945–3952 CrossRef CAS PubMed.
- E. Novo and M. Parola, Fibrog. Tissue Repair, 2008, 1, 1–58 CrossRef PubMed.
- H. Ranji-Burachaloo, P. A. Gurr, D. E. Dunstan and G. G. Qiao, ACS Nano, 2018, 12, 11819–11837 CrossRef CAS PubMed.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2014, 275, 121–135 CrossRef CAS PubMed.
- M. Cheng, C. Lai, Y. Liu, G. Zeng, D. Huang, C. Zhang, L. Qin, L. Hu, C. Zhou and W. Xiong, Coord. Chem. Rev., 2018, 368, 80–92 CrossRef CAS.
- X. Fu, Y. Zhang, G. Zhang, X. Li, S. Ni and J. Cui, Appl. Mater. Today, 2022, 26, 101353 CrossRef.
- S. Koo, O. K. Park, J. Kim, S. I. Han, T. Y. Yoo, N. Lee, Y. G. Kim, H. Kim, C. Lim and J.-S. Bae, ACS Nano, 2022, 16, 2535–2545 CrossRef CAS PubMed.
- N. Xu, Q. Huang, L. Shi, J. Wang, X. Li, W. Guo, D. Yan, T. Ni, Z. Yang and Y. Yan, Dalton Trans., 2023, 52, 1687–1701 RSC.
- Q. Shao, P. Wang and X. Huang, Adv. Funct. Mater., 2019, 29, 1806419 CrossRef.
- S.-M. Hwang, S. J. Han, H.-G. Park, H. Lee, K. An, K.-W. Jun and S. K. Kim, ACS Catal., 2021, 11, 2267–2278 CrossRef CAS.
- Y. S. Lee, M. Yuan, R. Cai, K. Lim and S. D. Minteer, ACS Catal., 2020, 10, 6854–6861 CrossRef CAS.
- S. Chatterjee, K. Sengupta, S. Hematian, K. D. Karlin and A. Dey, J. Am. Chem. Soc., 2015, 137, 12897–12905 CrossRef CAS PubMed.
- W. N. Lanzilotta and L. C. Seefeldt, Biochemistry, 1997, 36, 12976–12983 CrossRef CAS PubMed.
- M. Chen, Z. Long, R. Dong, L. Wang, J. Zhang, S. Li, X. Zhao, X. Hou, H. Shao and X. Jiang, Small, 2020, 16, 1906240 CrossRef CAS PubMed.
- Q. Wu, M. S. Siddique, Y. Guo, M. Wu, Y. Yang and H. Yang, Appl. Catal., B, 2021, 286, 119950 CrossRef CAS.
- X. Zhao, N. Zhang, T. Yang, D. Liu, X. Jing, D. Wang, Z. Yang, Y. Xie and L. Meng, ACS Appl. Mater. Interfaces, 2021, 13, 36106–36116 CrossRef CAS PubMed.
- J. D. Evans, C. J. Sumby and C. J. Doonan, Chem. Soc. Rev., 2014, 43, 5933–5951 RSC.
- S. Wongsakulphasatch, F. Nouar, J. Rodriguez, L. Scott, C. Le Guillouzer, T. Devic, P. Horcajada, J.-M. Grenèche, P. Llewellyn and A. Vimont, Chem. Commun., 2015, 51, 10194–10197 RSC.
- C. Cao, X. Wang, N. Yang, X. Song and X. Dong, Chem. Sci., 2022, 13, 863–889 RSC.
- R. Singh, A. Prasad, B. Kumar, S. Kumari, R. K. Sahu and S. T. Hedau, ChemistrySelect, 2022, 7, e202201288 CrossRef CAS.
- M. X. Wu and Y. W. Yang, Adv. Mater., 2017, 29, 1606134 CrossRef PubMed.
- H. D. Lawson, S. P. Walton and C. Chan, ACS Appl. Mater. Interfaces, 2021, 13, 7004–7020 CrossRef CAS PubMed.
- H. Furukawa, U. Müller and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 3417–3430 CrossRef CAS PubMed.
- M. Chen, J. Zhang, J. Qi, R. Dong, H. Liu, D. Wu, H. Shao and X. Jiang, ACS Nano, 2022, 16, 7732–7744 CrossRef CAS PubMed.
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC.
- S. J. Lee, C. Doussot, A. Baux, L. Liu, G. B. Jameson, C. Richardson, J. J. Pak, F. Trousselet, F.-X. Coudert and S. G. Telfer, Chem. Mater., 2016, 28, 368–375 CrossRef CAS.
- T. M. Osborn Popp, A. Z. Plantz, O. M. Yaghi and J. A. Reimer, Chem. Phys. Chem., 2020, 21, 32–35 CrossRef CAS PubMed.
- Y. Fu, Z. Kang, W. Cao, J. Yin, Y. Tu, J. Li, H. Guan, Y. Wang, Q. Wang and X. Kong, Angew. Chem., 2021, 133, 7798–7806 CrossRef.
- L. Zhu, F. Hu, B. Sun, S. Gu, T. Gao and G. Zhou, Adv. Sustainable Syst., 2023, 7, 2200394 CrossRef CAS.
- Y.-B. Zhang, H. Furukawa, N. Ko, W. Nie, H. J. Park, S. Okajima, K. E. Cordova, H. Deng, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2015, 137, 2641–2650 CrossRef CAS PubMed.
- Y.-N. Zeng, H.-Q. Zheng, J.-F. Gu, G.-J. Cao, W.-E. Zhuang, J.-D. Lin, R. Cao and Z.-J. Lin, Inorg. Chem., 2019, 58, 13360–13369 CrossRef CAS PubMed.
- C.-C. Cao, C.-X. Chen, Z.-W. Wei, Q.-F. Qiu, N.-X. Zhu, Y.-Y. Xiong, J.-J. Jiang, D. Wang and C.-Y. Su, J. Am. Chem. Soc., 2019, 141, 2589–2593 CrossRef CAS PubMed.
- M. Carlsson, M. Gustavsson, G.-Z. Hu, E. Murén and H. Ronne, PLoS One, 2013, 8, e52094 CrossRef CAS PubMed.
- N. Leelakanok, S. Geary and A. Salem, J. Pharm. Sci., 2018, 107, 513–528 CrossRef CAS PubMed.
- D. B. Longley, D. P. Harkin and P. G. Johnston, Nat. Rev. Cancer, 2003, 3, 330–338 CrossRef CAS PubMed.
- M. H. Jouybari, S. Hosseini, K. Mahboobnia, L. A. Boloursaz, M. Moradi and M. Irani, Colloids Surf., B, 2019, 179, 495–504 CrossRef PubMed.
- Q. Fu, J. Wang and H. Liu, Drug Delivery, 2020, 27, 1535–1543 CrossRef CAS PubMed.
- S. Pakian, F. Radmanesh, H. Sadeghi-Abandansari and M.-R. Nabid, ACS Appl. Polym. Mater., 2022, 4, 8238–8252 CrossRef CAS.
- S. Dhanavel, E. Nivethaa, V. Narayanan and A. Stephen, Mater. Sci. Eng., C, 2017, 75, 1399–1410 CrossRef CAS PubMed.
- P. RS, A. Mal, S. K. Valvi, R. Srivastava, A. De and R. Bandyopadhyaya, ACS Appl. Bio. Mater., 2020, 3, 4643–4654 CrossRef CAS PubMed.
- P. Ratrey, S. V. Dalvi and A. Mishra, ACS Omega, 2020, 5, 19004–19013 CrossRef CAS PubMed.
- M. Takahashi, S. Uechi, K. Takara, Y. Asikin and K. Wada, J. Agric. Food Chem., 2009, 57, 9141–9146 CrossRef CAS PubMed.
- B. H. Choi, C. G. Kim, Y. Lim, S. Y. Shin and Y. H. Lee, Cancer Lett., 2008, 259, 111–118 CrossRef CAS PubMed.
- G. A. Borges, S. T. Elias, B. Amorim, C. L. de Lima, R. D. Coletta, R. M. Castilho, C. H. Squarize and E. N. S. Guerra, Phytother. Res., 2020, 34, 3311–3324 CrossRef CAS PubMed.
- C. Mohanty, M. Das and S. K. Sahoo, Expert Opin. Drug Delivery, 2012, 9, 1347–1364 CrossRef CAS PubMed.
- K. R. Sanchez-Lievanos, M. Tariq, W. W. Brennessel and K. E. Knowles, Dalton Trans., 2020, 49, 16348–16358 RSC.
- M. Parsaei and K. Akhbari, Inorg. Chem., 2022, 61, 14528–14543 CrossRef CAS PubMed.
- A. Gerbino, G. Schena, S. Milano, L. Milella, A. F. Barbosa, F. Armentano, G. Procino, M. Svelto and M. Carmosino, PLoS One, 2016, 11, e0156021 CrossRef PubMed.
- W. Ni, L. Zhang, H. Zhang, C. Zhang, K. Jiang and X. Cao, Inorg. Chem., 2022, 61, 3281–3287 CrossRef CAS PubMed.
- Z. Wang, A. Von Dem Bussche, P. K. Kabadi, A. B. Kane and R. H. Hurt, ACS Nano, 2013, 7, 8715–8727 CrossRef CAS PubMed.
- H. Ranji-Burachaloo, F. Karimi, K. Xie, Q. Fu, P. A. Gurr, D. E. Dunstan and G. G. Qiao, ACS Appl. Mater. Interfaces, 2017, 9, 33599–33608 CrossRef CAS PubMed.
- M. L. Bruschi, Strategies to modify the drug release from pharmaceutical systems, Woodhead Publishing, 2015 Search PubMed.
- J. Dredán, I. Antal and I. Rácz, Int. J. Pharm., 1996, 145, 61–64 CrossRef.
- G.-H. Son, B.-J. Lee and C.-W. Cho, J. Pharm. Invest., 2017, 47, 287–296 CrossRef CAS.
- L. Peng, M. Asgari, P. Mieville, P. Schouwink, S. Bulut, D. T. Sun, Z. Zhou, P. Pattison, W. Van Beek and W. L. Queen, ACS Appl. Mater. Interfaces, 2017, 9, 23957–23966 CrossRef CAS PubMed.
- Q. Liu, H. Cong and H. Deng, J. Am. Chem. Soc., 2016, 138, 13822–13825 CrossRef CAS PubMed.
- L. Yang, T. Zhao, I. Boldog, C. Janiak, X.-Y. Yang, Q. Li, Y.-J. Zhou, Y. Xia, D.-W. Lai and Y.-J. Liu, Dalton Trans., 2019, 48, 989–996 RSC.
- G.-T. Vuong, M.-H. Pham and T.-O. Do, Dalton Trans., 2013, 42, 550–557 RSC.
- C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828–1831 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Férey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS PubMed.
- M.-H. Pham, G.-T. Vuong, A.-T. Vu and T.-O. Do, Langmuir, 2011, 27, 15261–15267 CrossRef CAS PubMed.
- T.-z Zhang, Y. Lu, Y.-g Li, Z. Zhang, W.-l Chen, H. Fu and E.-b Wang, Inorg. Chim. Acta, 2012, 384, 219–224 CrossRef CAS.
- B. Iqbal, A. Laybourn and M. Zaheer, Ceram. Int., 2021, 47, 12433–12441 CrossRef CAS.
- N. Singh, S. Qutub and N. M. Khashab, J. Mater. Chem. B, 2021, 9, 5925–5934 RSC.
- A. R. Chowdhuri, D. Laha, S. Pal, P. Karmakar and S. K. Sahu, Dalton Trans., 2016, 45, 18120–18132 RSC.
- A. Tiwari, A. Singh, N. Garg and J. K. Randhawa, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed.
- P. George, R. K. Das and P. Chowdhury, Microporous Mesoporous Mater., 2019, 281, 161–171 CrossRef CAS.
- J. N. Hall and P. Bollini, React. Chem. Eng., 2019, 4, 207–222 RSC.
- S. Bhattacharjee, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- B. Lei, M. Wang, Z. Jiang, W. Qi, R. Su and Z. He, ACS Appl. Mater. Interfaces, 2018, 10, 16698–16706 CrossRef CAS PubMed.
- W. L. Bragg, Scientia, 1929, 23 Search PubMed.
- P. Horcajada, F. Salles, S. Wuttke, T. Devic, D. Heurtaux, G. Maurin, A. Vimont, M. Daturi, O. David and E. Magnier, J. Am. Chem. Soc., 2011, 133, 17839–17847 CrossRef CAS PubMed.
- R. I. Walton, A. S. Munn, N. Guillou and F. Millange, Chem. – Eur. J., 2011, 17, 7069–7079 CrossRef CAS PubMed.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem. – Eur. J., 2011, 17, 6643–6651 CrossRef CAS PubMed.
- J. Gandara-Loe, B. E. Souza, A. Missyul, G. Giraldo, J.-C. Tan and J. Silvestre-Albero, ACS Appl. Mater. Interfaces, 2020, 12, 30189–30197 CrossRef CAS PubMed.
- S. Rojas, I. Colinet, D. Cunha, T. Hidalgo, F. Salles, C. Serre, N. Guillou and P. Horcajada, ACS Omega, 2018, 3, 2994–3003 CrossRef CAS PubMed.
- X. Zhang, Y. Dong, X. Zeng, X. Liang, X. Li, W. Tao, H. Chen, Y. Jiang, L. Mei and S.-S. Feng, Biomaterials, 2014, 35, 1932–1943 CrossRef CAS PubMed.
- Y. Yang, W. Zhu, L. Cheng, R. Cai, X. Yi, J. He, X. Pan, L. Yang, K. Yang and Z. Liu, Biomaterials, 2020, 246, 119971 CrossRef CAS PubMed.
- I. A. Lázaro and R. S. Forgan, Coord. Chem. Rev., 2019, 380, 230–259 CrossRef.
- H. Kalita, B. P. Kumar, S. Konar, S. Tantubay, M. K. Mahto, M. Mandal and A. Pathak, Mater. Sci. Eng., C, 2016, 60, 84–91 CrossRef CAS PubMed.
- K. Akulov, R. Simkovitch, Y. Erez, R. Gepshtein, T. Schwartz and D. Huppert, J. Phys. Chem. A, 2014, 118, 2470–2479 CrossRef CAS PubMed.
- X. G. Wang, L. Xu, M. J. Li and X. Z. Zhang, Angew. Chem., Int. Ed., 2020, 59, 18078–18086 CrossRef CAS PubMed.
- C. Adhikari, A. Mishra, D. Nayak and A. Chakraborty, J. Drug Delivery Sci. Technol., 2018, 47, 1–11 CrossRef CAS.
- C.-Z. Yuan, Y.-F. Jiang, Z. Wang, X. Xie, Z.-K. Yang, A. B. Yousaf and A.-W. Xu, J. Mater. Chem. A, 2016, 4, 8155–8160 RSC.
- A. M. Beale and G. Sankar, J. Mater. Chem., 2002, 12, 3064–3072 RSC.
- X. Li, L. Lachmanski, S. Safi, S. Sene, C. Serre, J.-M. Grenèche, J. Zhang and R. Gref, Sci. Rep., 2017, 7, 13142 CrossRef CAS PubMed.
- Y.-S. Lin and K.-S. Lin, Microporous Mesoporous Mater., 2021, 328, 111456 CrossRef CAS.
- M. d J. Velásquez-Hernández, R. Ricco, F. Carraro, F. T. Limpoco, M. Linares-Moreau, E. Leitner, H. Wiltsche, J. Rattenberger, H. Schröttner and P. Frühwirt, CrystEngComm, 2019, 21, 4538–4544 RSC.
- X. Cai, M. Jin, L. Yao, B. He, S. Ahmed, W. Safdar, I. Ahmad, D.-B. Cheng, Z. Lei and T. Sun, J. Mater. Chem. B, 2022, 716–733 Search PubMed.
- M.-L. Laracuente, H. Y. Marina and K. J. McHugh, J. Controlled Release, 2020, 327, 834–856 CrossRef CAS PubMed.
- P. Mazurek, M. A. Brook and A. L. Skov, Langmuir, 2018, 34, 11559–11566 CrossRef CAS PubMed.
- F. R. Wibowo, O. A. Saputra, W. W. Lestari, M. Koketsu, R. R. Mukti and R. Martien, ACS Omega, 2020, 5, 4261–4269 CrossRef CAS PubMed.
- A. Saravanan, M. Maruthapandi, P. Das, S. Ganguly, S. Margel, J. H. Luong and A. Gedanken, ACS Appl. Bio Mater., 2020, 3, 8023–8031 CrossRef CAS PubMed.
- C.-Y. Loo, R. Rohanizadeh, P. M. Young, D. Traini, R. Cavaliere, C. B. Whitchurch and W.-H. Lee, J. Agric. Food Chem., 2016, 64, 2513–2522 CrossRef CAS PubMed.
- Y.-L. Luo, Y.-S. Shiao and Y.-F. Huang, ACS Nano, 2011, 5, 7796–7804 CrossRef CAS PubMed.
- C. Y. Ang, S. Y. Tan and Y. Zhao, Org. Biomol. Chem., 2014, 12, 4776–4806 RSC.
- M. U. Akbar, S. Khattak, M. I. Khan, U. A. K. Saddozai, N. Ali, A. F. AlAsmari, M. Zaheer and M. Badar, Front. Pharmacol., 2023, 14 DOI:10.3389/fphar.2023.1265440.
- P. F. Gu, H. Xu, B. W. Sui, J. X. Gou, L. K. Meng, F. Sun, X. J. Wang, N. Qi, Y. Zhang and H. B. He, Int. J. Nanomed., 2012, 109–122 CrossRef CAS PubMed.
- M. Parsaei and K. Akhbari, Inorg. Chem., 2022, 61, 19354–19368 CrossRef CAS PubMed.
- S. Karimi and H. Namazi, Int. J. Pharm., 2023, 634, 122675 CrossRef CAS PubMed.
- J. Wu, S. Jiang, W. Xie, Y. Xue, M. Qiao, X. Yang, X. Zhang, Q. Wan, J. Wang and J. Chen, J. Mater. Chem. B, 2022, 10, 8535–8548 RSC.
- S. Mallakpour, F. Sirous and C. M. Hussain, New J. Chem., 2021, 45, 8409–8426 RSC.
- B. Mansoori, A. Mohammadi, F. Abedi-Gaballu, S. Abbaspour, M. Ghasabi, R. Yekta, S. Shirjang, G. Dehghan, M. R. Hamblin and B. Baradaran, J. Cell. Physiol., 2020, 235, 6817–6830 CrossRef CAS PubMed.
- P. Gokare, N. K. Finnberg, P. H. Abbosh, J. Dai, M. E. Murphy and W. S. El-Deiry, Sci. Rep., 2017, 7, 9711 CrossRef PubMed.
- W. Ni, Z. Li, Z. Liu, Y. Ji, L. Wu, S. Sun, X. Jian and X. Gao, J. Pharm. Sci., 2019, 108, 1284–1295 CrossRef CAS PubMed.
- A. Sahu, N. Kasoju and U. Bora, Biomacromolecules, 2008, 9, 2905–2912 CrossRef CAS PubMed.
- M. M. Yallapu, M. Jaggi and S. C. Chauhan, Colloids Surf., B, 2010, 79, 113–125 CrossRef CAS PubMed.
- T. Yang, D. Yu, D. Wang, T. Yang, Z. Li, M. Wu, M. Petru and J. Crittenden, Appl. Catal., B, 2021, 286, 119859 CrossRef CAS.
- X. Liang, D. Wang, Z. Zhao, T. Li, Y. Gao and C. Hu, Adv. Funct. Mater., 2022, 32, 2203001 CrossRef CAS.
- W. Shi, Q. Wang, Y. Long, Z. Cheng, S. Chen, H. Zheng and Y. Huang, Chem. Commun., 2011, 47, 6695–6697 RSC.
- H. Jiang, Z. Chen, H. Cao and Y. Huang, Analyst, 2012, 137, 5560–5564 RSC.
- M. Hermanek, R. Zboril, I. Medrik, J. Pechousek and C. Gregor, J. Am. Chem. Soc., 2007, 129, 10929–10936 CrossRef CAS PubMed.
- N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS PubMed.
- J. A. Greathouse and M. D. Allendorf, J. Am. Chem. Soc., 2006, 128, 10678–10679 CrossRef CAS PubMed.
- N. C. Burtch, I. M. Walton, J. T. Hungerford, C. R. Morelock, Y. Jiao, J. Heinen, Y.-S. Chen, A. A. Yakovenko, W. Xu and D. Dubbeldam, Nat. Chem., 2020, 12, 186–192 CrossRef CAS PubMed.
- M. Ding, X. Cai and H.-L. Jiang, Chem. Sci., 2019, 10, 10209–10230 RSC.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- X. C. Huang, Y. Y. Lin, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS PubMed.
- S. K. Maji, A. K. Dutta, D. N. Srivastava, P. Paul, A. Mondal and B. Adhikary, J. Mol. Catal. A: Chem., 2012, 358, 1–9 CrossRef CAS.
- K.-I. Ishibashi, A. Fujishima, T. Watanabe and K. Hashimoto, J. Photochem. Photobiol., A, 2000, 134, 139–142 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00389b |
| This journal is © The Royal Society of Chemistry 2023 |