
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoliposomes protecting antimicrobial peptides via membrane-fused incorporation to fight wound infection†
Hao
Xue‡
a,
Jiaying
Li‡
b,
Liwei
Zhang
a,
Xiaolu
Song
*a,
Hui
Shi
c,
Yonghai
Feng
a,
Shuai
Hou
a,
Zengkai
Wang
a,
Taofeng
Zhu
*b and
Lei
Liu
 *a
*a
aInstitute for Advanced Materials, Jiangsu University, China. E-mail: liul@ujs.edu.cn
bDepartment of General Practice, Yixing Hospital affiliated to Jiangsu University, Yixing 214200, China
cSchool of Medicine, Jiangsu University, China
First published on 3rd July 2023
Abstract
Due to the abuse of antibiotics, drug resistance has become an urgent problem in public health and clinical practice. Recently, antimicrobial peptides (AMPs) have been considered as excellent antibacterial agents that are not inclined to induce the drug resistance of bacteria, but they are limited by their easy hydrolysis by enzymes and high cytotoxicity. In this work, we designed a strategy using amphipathic α-helical peptides coupled with antimicrobial peptides (AMPs) to fuse with liposomes to form functional antimicrobial liposomes (FALs) as typical bio-colloids. FALs can overcome the drug resistance of bacteria, improve antibacterial performance and biosafety, and prevent enzymolysis of the AMPs to enhance their stability. In detail, the pancreatic-enzyme-treated AMPs exhibited no antibacterial activity against E. coli and S. aureus due to the hydrolysis of peptide. In contrast, the FALs still maintained excellent antibacterial performance against both E. coli and S. aureus after being treated with pancreatic enzyme, and could be further applied in the treatment of skin wounds infected with S. aureus on mice. Hematoxylin (HE) staining and proliferating cell nuclear antigen (PCNA) were adopted for histological analysis, proving that the FALs constructed by such a strategy had broad application prospects in both wound disinfection and the promotion of wound healing.
Introduction
In recent years, a large number of common and rare diseases are caused by bacteria, and antibiotics are widely used to kill bacteria. However, with the abuse of antibiotics, the level of bacterial drug resistance is gradually increasing,1 resulting in highly drug-resistant bacteria, multi-drug-resistant strains, and even the emergence of superbacteria. Although the use of antibiotics has revolutionized the fight against bacterial infections, the rapid adaptation of bacterial species and the abuse of antibiotics have led to more deaths. New antibacterial regimens different from existing antibiotics need to be developed urgently. Non-antibiotic drugs,2 antibiotic combinations,3 the development of adjuvant therapy4 and other methods have commonly been used to kill bacteria. However, these also bring a series of problems, such as clinical limitations and the possibility of increasing bacterial mutation. Therefore, new treatments need to be developed to combat bacterial resistance.There are several methods to overcome bacterial resistance to kill bacteria, including metal nanomaterials,5 quaternary ammonium salt polymers,6 reactive oxygen species (ROS),7 AMPs,8etc. Among them, AMPs are good choice for fighting bacteria with drug resistance. In addition to their significant bactericidal effect, AMPs can also inhibit biofilm formation9 and induce the dissolution of existing biofilms.10 AMPs have a wide range of antibacterial and immunomodulatory activities against infectious bacteria (Gram-positive bacteria and Gram-negative bacteria), viruses, and fungi. AMPs damage cell membranes through electrostatic interactions,11 making it difficult for bacteria to develop resistance compared with conventional antibiotics.12 Although the advantages of AMPs are significant,13,14 there are still some problems that need to be solved, such as poor biocompatibility,15 degradation by enzymes,16 the high production cost of long sequences,17 potential toxicity to eukaryotic cells18 and other problems. Therefore, a series of strategies are needed to improve the stability, reduce the toxicity, and improve the effectiveness of AMPs for further subsequent applications. For instance, modification of AMPs can improve their metabolism and structural stability.19 However, it is accompanied by a decrease in the antimicrobial activity of AMPs. Drug carriers, i.e., metal oxides, silicon-based nanoparticles and carbon-based nanomaterials, have been used for the delivery of AMPs,20 leading to low cytotoxicity and improved bioavailability of AMP, but the biocompatibility and metabolic pathways of the carriers still need to be further explored. Furthermore, the production of nano-antibiotics by AMP self-assembling has also been proposed recently, which requires complicated and precise design to both ensure the antimicrobial activity of the AMP and the self-assembling of coupled peptide moieties in the whole peptide.21 Therefore, a simple and efficient strategy to protect AMPs against hydrolysis and improve the biocompatibility while maintaining activity for disinfection is highly anticipated, but still challenging.
Liposomes are essentially a kind of phospholipid vesicle that spontaneously form closed lipid bilayer vesicles in water.22,23 Liposomes were discovered by scientists in the 1960s, and their potential as an effective drug delivery system was immediately recognized.24 Since then, scientists have been conducting various studies on liposomes, including the control of their active site, the duration of their circulation in the body, and the release time and location of the delivered drugs.25 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) has a variety of advantages, including biocompatibility, self-assembly ability, drug loading ability and a wide range of physicochemical and biophysical properties.26,27 At present, clinical studies have shown that lipid-based drug carriers have various advantages such as immunogenicity, low toxicity and side effects, and easy adsorption.28
Herein, we designed a new strategy using a fusion peptide to combine AMPs with liposomes to form FALs. It has many advantages, such as remarkable bactericidal performance, low cytotoxicity, good biocompatibility, and resistance to hydrolysis by enzymes. Liposomes are essentially a kind of phospholipid vesicle that spontaneously form closed lipid bilayer vesicles in water.29,30 Liposomes have a variety of advantages, including biocompatibility, self-assembly ability, drug loading ability and a wide range of physicochemical and biophysical properties.31,32 For the functional AMPs, we designed the peptide FWR33 (FAEKFKEAVKDYFAKFWDGSGRWWRWWRRWWRR) (Fig. 1a), which was composed of a 12-amino-acid AMP sequence (RWWRWWRRWWRR) (WR12)33 with broad-spectrum antibacterial activity and an amphipathic α-helical peptide (FAEKFKEAVKDYFAKFWD) sequence that works as a fusion peptide that can interact with the liposomes to form stable, spherical nanoparticles through self-assembly.34,35 WR12 has a broad-spectrum antibacterial activity and excellent antibacterial performance. It is also a short-chain AMP that is much cheaper than other AMPs and can therefore facilitate clinical applications. The as-prepared FALs can improve antibacterial performance and biosafety, as well as prevent enzymolysis of the AMPs to enhance their stability. FALs could be further applied in the treatment of the skin wounds of a mouse with S. aureus infection. Hematoxylin (HE) staining and proliferating cell nuclear antigen (PCNA) were adopted for histological analysis, and the results verified that the FALs constructed by this strategy had broad application prospects both in wound disinfection and the promotion of wound healing.
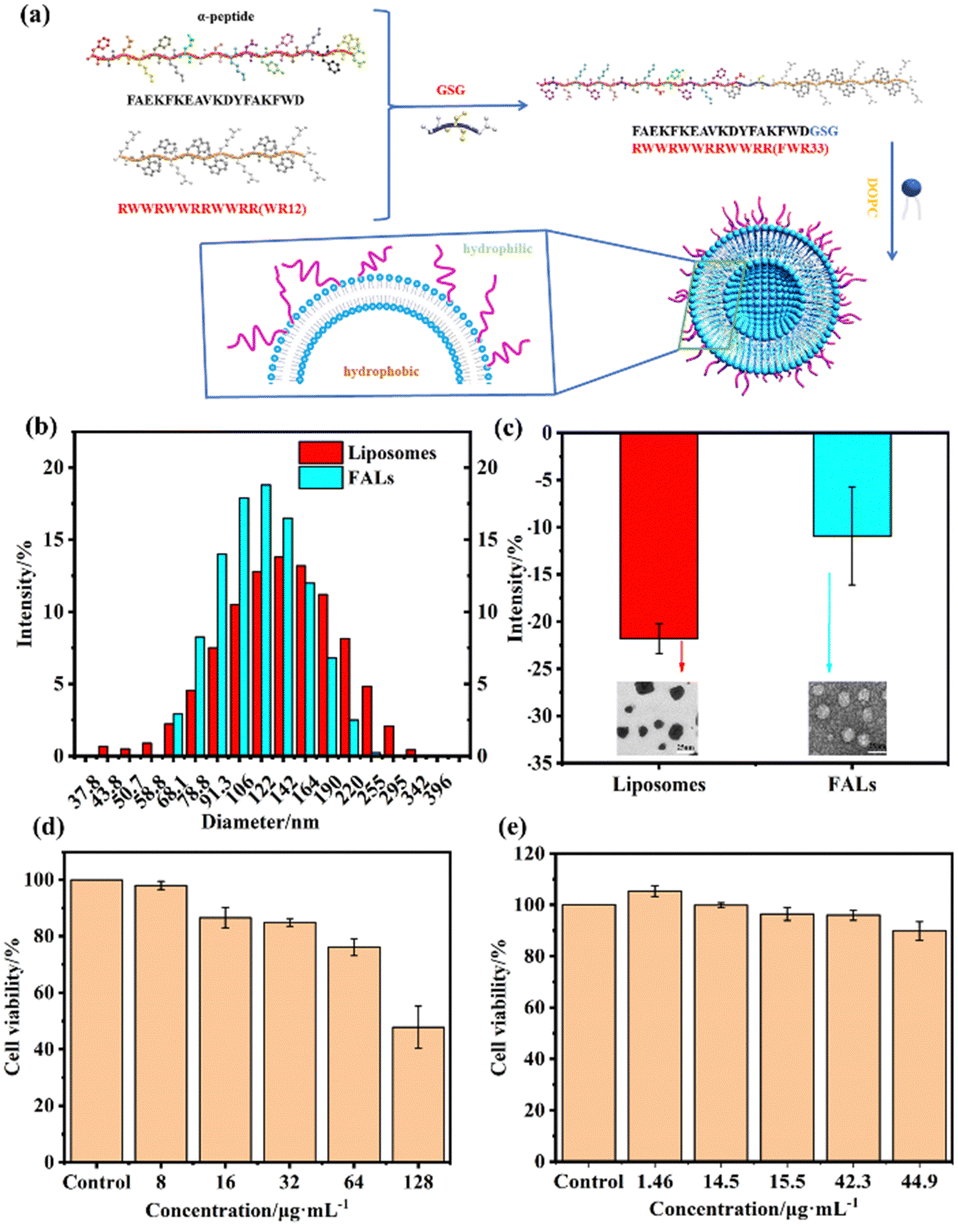 | ||
| Fig. 1 (a) Design and synthetic procedure of FALs. (b) Particle size of liposomes and FALs. (c) Zeta potential analysis of liposomes and FALs. (d) Cytotoxicity of FWR33. (e) Cytotoxicity of FALs after the ultrafiltration. | ||
Materials and methods
Materials
The phospholipid 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) was purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, USA). FWR33 (FAEKFKEAVKDYFAKFWDGSGRWWRWWRRWWRR) and WR12 (RWWRWWRRWWRR) were purchased from Qiangyao Biotechnology Co., Ltd. Hexafluoroisopropanol (HFIP) was purchased from Tokyo Huacheng Co., Ltd. Chloroform and phosphate buffer were purchased from Sinopharm Group Chemical Reagent Co., Ltd. Phosphotungstic acid negative staining solution (5% phosphotungstic acid negative staining solution) was purchased from Nanjing Senbeijia Biotechnology Co., Ltd. LB solid medium/LB agar (dry powder) was purchased from Beinong Yuhe Technology Development Co., Ltd. PC membranes 0.1 μm (100 pk−1) were purchased from Avanti/Whatman.Preparation of liposome solution
DOPC (100 mg) was fully dissolved in chloroform (10 mL) to form a liposome solution (10 mg mL−1). 50 μL of the above solution was transferred to a 1.5 mL centrifuge tube. The solvent was removed by vacuum. The residue was dissolved in 1 mL PBS (10 mM) to form the liposome solution (500 μg mL−1).Preparation of antimicrobial peptide solution
10 mg of WR12 was fully dissolved in 10 mL hexafluoroisopropanol (HFIP) to form a peptide solution (1 mg mL−1) after shaking on an oscillator for 12 h. The peptide solution (256 μL) was transferred to a 1.5 mL centrifuge tube. The solvent was removed by vacuum. The residue was dissolved in PBS (1 mL, 10 mM) to form the peptide solution (256 μg mL−1).Preparation of FWR33 solution
10 mg of FWR33 was fully dissolved in HFIP (10 mL) to form a peptide solution (256 μg mL−1) after shaking on an oscillator for 12 h. The peptide solution (256 μL) was transferred to a 1.5 mL centrifuge tube. The solvent was removed by vacuum. The residue was dissolved in PBS (1 mL, 10 mM) to form the FWR33 solution (256 μg mL−1).Synthesis of FALs
Liposome solution (0.25 mL, 500 μg mL−1), FWR33 solution (0.25 mL, 256 μg mL−1) and PBS (0.5 mL, 10 mM) were added to a 1.5 mL centrifuge tube, and the mixture was kept at −4 °C for 12 h. After extrusion, the FAL solution with good dispersion was prepared.Transmission electron microscopy (TEM)
The morphology of the FALs was characterized using a Tecnai12 transmission electron microscope (Philips, Netherlands) at an accelerating voltage of 120 kV. The sample preparation method was as follows: the sample was dropped onto a 230 mesh copper screen, and left for 15 min. After that, the sample was loaded onto the copper screen, and the liquid was removed using filter paper. The samples were stained with 10 μL of 1% uranyl acetate solution for 4 min and rinsed with 10 μL of deionized water. The liquid was removed using filter paper, and the samples were dried at room temperature.Circular dichroism (CD) spectra
The secondary structure changes of the liposomes, FWR33 and FALs were characterized via CD measurements, which were performed using a PTC-348W1 CD analyser. All measurements were conducted at room temperature, and the spectra were collected within the region of 190–250 nm. The slit-width was 2 nm, and the scan speed was 50 nm min−1. 500 μL of the FALs was added to an ultrafiltration tube (molecular weight 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000). The sample was centrifuged at a high speed of 12000 rpm, the lower clear solution was removed and the same amount of PBS was added to the residue. The operation was repeated three times. After the third ultrafiltration, the supernatant and subnatant were collected for further testing. For all samples, the signal of PBS was subtracted as the baseline. Each sample was added to a 1 mm colorimetric cell with a volume of 200 μL. All experiments were repeated three times. The FAL solution was ultrafiltrated, and the supernatant and subnatant were characterized by CD after the third ultrafiltration (Fig. 2b). The results showed that all the unreacted FWR33 had been removed and the FALs remained in the supernatant. The appearance of an additional α-helix secondary structure demonstrated that FWR33 was attached to the liposomes to form FALs.
000). The sample was centrifuged at a high speed of 12000 rpm, the lower clear solution was removed and the same amount of PBS was added to the residue. The operation was repeated three times. After the third ultrafiltration, the supernatant and subnatant were collected for further testing. For all samples, the signal of PBS was subtracted as the baseline. Each sample was added to a 1 mm colorimetric cell with a volume of 200 μL. All experiments were repeated three times. The FAL solution was ultrafiltrated, and the supernatant and subnatant were characterized by CD after the third ultrafiltration (Fig. 2b). The results showed that all the unreacted FWR33 had been removed and the FALs remained in the supernatant. The appearance of an additional α-helix secondary structure demonstrated that FWR33 was attached to the liposomes to form FALs.
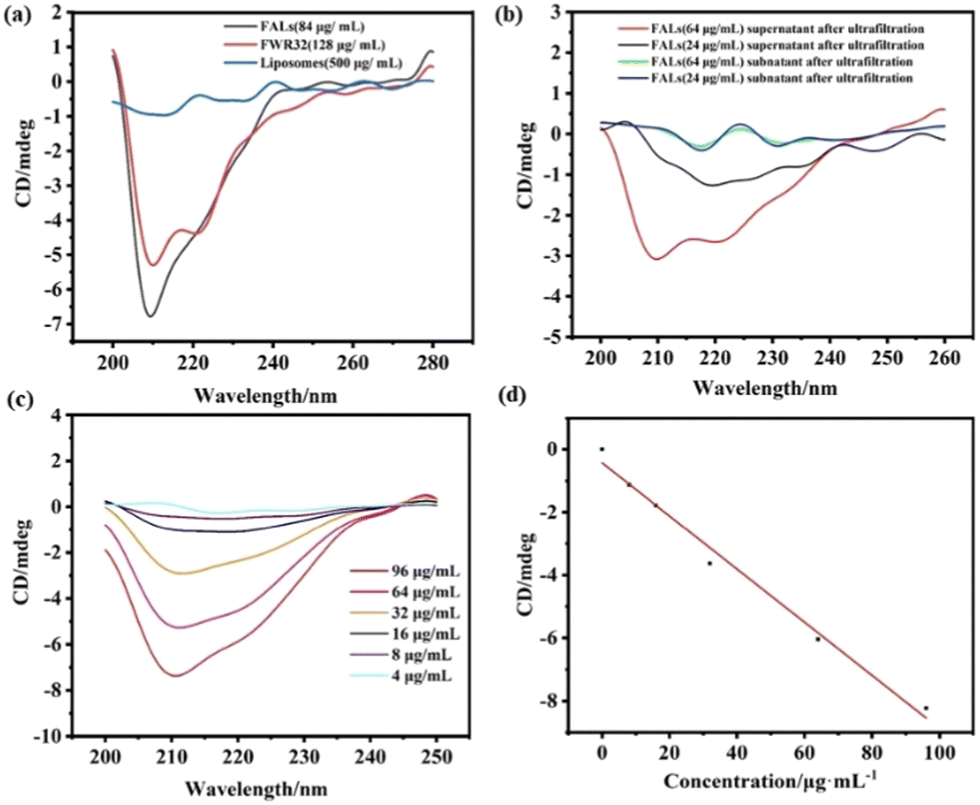 | ||
| Fig. 2 (a) CD of the three different materials. (b) CD of the supernatant and subnatant of the FALs after ultrafiltration (24 μg mL−1, 64 μg mL−1). (c) CD of FWR32 at 6 concentrations. (d) Curve fitting of FWR32 peaks. | ||
Calculation of loading efficiency of peptides on liposomes
The loading efficiency of FWR33 in liposomes was calculated using the standard curve method. Six different concentrations of FWR33 (4, 8, 16, 32, 64, 96 μg mL−1) were evaluated using CD (Fig. 2c). The value at 210 nm was recorded, and the standard curve of CD vs. concentration of FWR33 (Fig. 2d) was plotted for calculation of the loading efficiency.Dynamic light scattering (DLS) experiment
The particle size changes of the liposomes and FALs were measured using DLS, which was conducted using a ZetasizerNanoZS90. All samples were diluted with PBS to 1 mL and were added to a cuvette for testing. Each sample was tested three times at room temperature.Zeta potential measurement
The distribution of the surface charge of the FALs and liposomes in phosphate buffer solution was analysed using a ZetasizerNanoZS90. All samples were diluted with PBS to 1 mL and were added to a cuvette for testing. Each sample was tested three times at room temperature.Evaluation of antibacterial properties in vitro
Staphylococcus aureus (S. aureus, A TCC25923) and Escherichia coli (E. coli, A TCC25922) supplied by the Institute of Life Sciences of Jiangsu University were used as the model bacteria. The bacterial suspension (200 μL, 107 CFU mL−1) was added to the FALs (200 μL, 42.35 μg mL−1) in a 1.5 mL centrifuge tube. The tube was placed in a thermostatic oscillator (37 °C) for a certain time period (0 h, 1 h, 2 h, 3 h, 4 h). After that, the bacterial suspension was diluted approximately 103 times with PBS. 100 μL of the diluted bacterial suspension was rolled out onto the LB solid medium (Beinong Yuhe Technology Development Co.) and placed in a bacterial incubator (37 °C) for 18 h. Plate counting was used to measure the number of bacteria. The method for the determination of the antibacterial rate of WR12 is the same as the above method.Determinations of the antibacterial rate of trypsin-treated WR12 and trypsin-treated FALs: WR12 (200 μL, 128 μg mL−1) or FALs (200 μL, 42.35 μg mL−1) were mixed with trypsin (200 μL, 1 mg mL−1) for 2 h. 200 μL of the above solution was added to a bacterial suspension (200 μL, 107 CFU mL−1) in a 1.5 mL centrifuge tube. The tube was placed in a thermostatic oscillator (37 °C) for a certain time period (0 h, 1 h, 2 h, 3 h, 4 h). The bacterial suspension was diluted approximately 103 times with PBS. 100 μL of diluted bacterial suspension was rolled out onto the LB solid medium (Beinong Yuhe Technology Development Co.) and placed in the bacterial incubator (37 °C) for 18 h. Plate counting was used to measure the number of bacteria. The statistical differences were calculated using GraphPad Prism 9.
Microscopy characterization of bacteria
The treated E. coli and S. aureus were collected by centrifugation and washed with PBS. The bacterial suspension was then fixed in 2.5% glutaraldehyde and refrigerated at 4 °C for 6 h. After centrifugation, each sample was washed three times with PBS (10 mM) and dehydrated with gradient ethanol solution (50%, 70%, 80%, 90%, 95% and 100%) for 5 min. Finally, the bacterial solution was added dropwise onto a monocrystalline silicon chip and observed by SEM.Fluorescence staining of live/dead bacteria
SYTO 9 was used for green fluorescence staining of live bacteria, and propidium iodide (PI) was used for red fluorescence staining of dead bacteria. After the corresponding treatments, the centrifugally collected bacterial suspensions were stained with SYTO 9 and PI for 15 min at room temperature in the dark. The bacteria were observed with a confocal laser microscope (Dmi8, Germany).Cytotoxicity assay of FALs
100 μL of L929 mouse fibroblasts (50000 cells mL−1) was added to a 96-well plate and cultured for 24 h. Five concentrations (1.46 μg mL−1, 14 μg mL−1, 15.5 μg mL−1, 42.3 μg mL−1, and 44.6 μg mL−1) of FALs were exposed to UV light for 2 h and then added to the 96-well plate (10 microliters per well), respectively. After 24 h of incubation, 10 μL of CCK8 solution was added to the 96-well plate (10 microliters per well). After 2 h of incubation, the optical density (OD) was measured at 450 nm using a microplate reader (SYNERGY H4, USA).
In vivo wound healing assay
All animal experimental procedures were approved by the Jiangsu University committee and were carried out according to the guidelines of Jiangsu University Laboratory. All procedures conform to Chinese laws and the National Institutes of Health (NIH) Animal Ethics Committee. The ICR mice were provided by the Animal Laboratory Center of Jiangsu University. Circular skin lesions (7 mm in diameter) were made on the back skin of ICR male mice (30–40 g). The mice were divided into four groups randomly after a S. aureus suspension (20 μL, 108 CFU mL−1) was daubed onto the wound. After 24 h, the wounds of the mice were treated with four different materials: PBS (control), liposomes, WR12 and FALs. The wounds were observed and photographed daily. After 1 day of treatment, the mice were examined, and the wound tissues were collected. Plate counting was used to measure the number of bacteria. After 14 days of treatment, the back skin was harvested and the wounds were separated from the surrounding skin and immobilized in a 4% neutral formalin solution. The skin samples were embedded in paraffin, and sections (3–5 μm) were stained with hematoxylin and eosin (HE) for histological and immunohistochemical analysis.Results and discussion
Characterization of FALs and cell viability of FALs
To explore the successful synthesis of the FALs and the interaction effect between the designed peptides and liposomes, DLS was used to monitor the changes in particle size and static charge before and after the fusion between FWR33 and the liposomes (Fig. 1). Fig. 1b shows that the average particle size of the liposomes was 132.9 nm, and the PDI (dispersion coefficient) was 0.345. The average particle size of the FALs was 98.6 nm, and the PDI was 0.248. The TEM images of the liposomes and FALs are shown in Fig. S1 (ESI†). Compared with the average particle size of the liposomes, the particle size of the FALs became smaller due to the membrane fusion between the liposomes and FWR33. Meanwhile, the PDI was reduced from 0.345 to 0.248, and the dispersion coefficient was reduced to make the sample more stable.In addition to particle size analysis, Fig. 1c shows the Zeta potential analysis of the nanomaterials. In the FALs, the liposomes were negatively charged, the WR12 sequences were positively charged, and the other sequences were neutral. The average Zeta potential of the FALs was −10.92 mV, which was more positive compared to that of the liposomes (−21.8 mV), indicating that FWR33 was partially exposed on the outside of the liposomes.
In order to prove the self-assembly of FWR33 and liposomes in PBS (10 mM), the circular dichroism (CD) spectra of the liposomes, FWR33, and FALs were obtained (Fig. 2a). It was found that the secondary structure of the peptides in the FALs is different from that of the FWR33 peptides, showing significantly different peaks near 210 nm. Liposomes have no typical signal in CD spectra. The loading efficiency of FWR33 in the liposomes was calculated using the standard curve method (Fig. 2c and d), which was determined to be around 60% optimally for the four selected concentrations (Table S1, ESI†). For example, after ultrafiltration, when the sample with 64 μg mL−1 FWR33 was loaded, the actual concentration of FWR33 was 42.35 μg mL−1 and the loading efficiency was 64.60%. The details of the loading efficiency calculation are presented in the experimental section of the ESI,† (CD spectra and calculation of loading efficiency of peptides on liposomes).
In order to prevent cell and tissue function decline, biocompatibility is also an important indicator to take into consideration. Fig. 1d shows the cytotoxicity of the FALs at five concentrations. When the concentration of FALs was low, there was almost no cytotoxicity, and the cell survival rate reached more than 90% even at high concentration (44.9 μg mL−1). In contrast, the cell survival rate of WR12 was less than 85% even at a concentration of 32 μg mL−1. The results indicated that our FALs had better biocompatibility compared to WR12.
Evaluation of antibacterial performance of FALs against Gram-negative bacteria E. coli and Gram-positive bacteria S. aureus
To explore the antibacterial performance of the FALs, we treated the Gram-negative bacteria E. coli and Gram-positive bacteria S. aureus with WR12 and FALs to test the survival ratios of the bacteria. The antibacterial performance of WR12 against E. coli and S. aureus was very significant. After 2 h of interaction between WR12 and the bacteria, 92.48% of E. coli and 99.99% of S. aureus were killed. The numbers of E. coli and S. aureus both decreased by 99.99% after 4 h of treatment. However, pancreatic-enzyme-treated WR12 had no antibacterial performance against E. coli (Fig. 3a) and S. aureus (Fig. 4a) due to the hydrolysis of WR12 peptide. In contrast, the FALs still maintained a degree of antibacterial performance against E. coli (Fig. 3b) after being treated with pancreatic enzyme, and the bacterial viability decreased by about 86.13% after the treatment with the FALs for 4 h. Similarly, the numbers of S. aureus decreased by about 96.40% when the FALs were mixed with S. aureus for 4 h. The FALs prevent the enzymatic hydrolysis of AMPs, mainly due to the steric hindrance of the AMP brush on the liposome against the enzyme. Therefore, the FALs exhibited great antibacterial performance against E. coli and S. aureus even after being treated with pancreatic enzymes, which improved the stability of AMPs in utilization. Scanning electron microscopy (SEM) was used to observe the microscopic morphology of E. coli (Fig. 3c and d) and S. aureus (Fig. 4c and d) before and after treatment with the FALs. Normal E. coli were rod-shaped (Fig. 3c), and S. aureus appeared to be spherical without the wrinkle (Fig. 4c). After the treatment with the FALs, the morphology of the E. coli and S. aureus changed significantly due to damage to the cell membranes (Fig. 3d and 4d). It can be seen that the surface of E. coli and S. aureus was wrinkled and damaged, indicating the death of the bacteria. These results were further confirmed using a live/dead backlight bacterial viability kit. A red fluorescence image was obtained because the red dye PI fully entered the inside of the bacteria due to the membrane rupture. On the contrary, the images of live E. coli and S. aureus were both found to be green (Fig. 3e and 4e). | ||
| Fig. 3 (a and b) Assessment of E. coli growth by the spread plate method at 0 h, 1 h, 2 h, 3 h and 4 h for four samples (WR12, pancreatic-enzyme-treated WR12, FALs, pancreatic-enzyme-treated FALs). (*p < 0.05, ****p < 0.001) (c) SEM image of the control group of E. coli. (d) Colony image of the pancreatic-enzyme-treated FALs mixed with E. coli for 4 h. (e) Fluorescence image of the control group of E. coli obtained using a live/dead backlight bacterial viability kit. (f) Fluorescence image of the pancreatic-enzyme-treated FALs mixed with E. coli for 4 h using a live/dead backlight bacterial viability kit. | ||
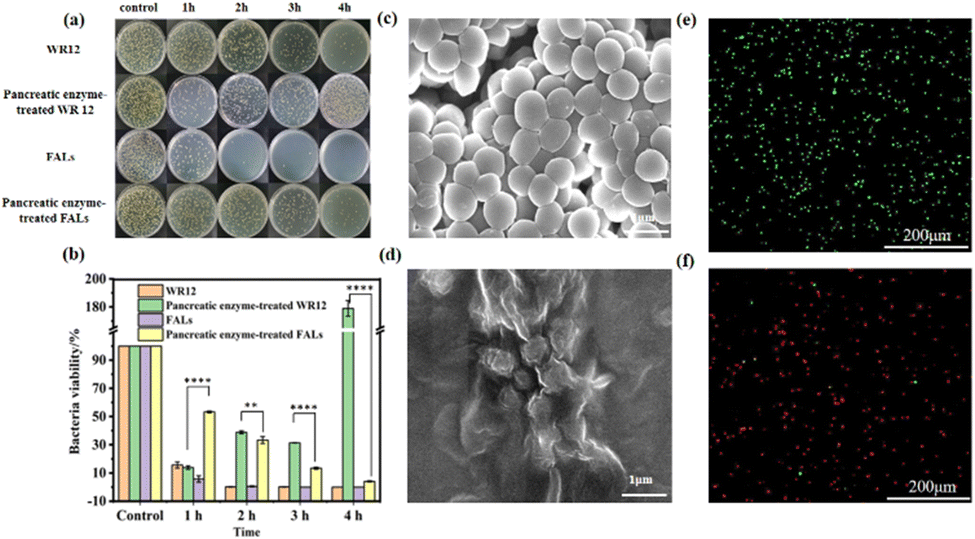 | ||
| Fig. 4 (a and b) Assessment of S. aureus growth by the spread plate method at 0 h, 1 h, 2 h, 3 h and 4 h for four samples (WR12, pancreatic-enzyme-treated WR12, FALs, pancreatic-enzyme-treated FALs). (**p < 0.01, ****p < 0.001) (c) SEM image of the control group of S. aureus. (d) Colony image of the pancreatic-enzyme-treated FALs mixed with S. aureus for 4 h. (e) Fluorescence image of the control group of S. aureus obtained using a live/dead backlight bacterial viability kit. (f) Fluorescence image of the pancreatic-enzyme-treated FALs mixed with S. aureus for 4 h using a live/dead backlight bacterial viability kit. | ||
Performance of FALs in the disinfection of an infected wound and the promotion of wound healing
In order to verify the performance of FALs in the disinfection and healing of infected wounds, we utilized a mouse skin wound model with S. aureus infection to conduct the experiment. The wounds were treated with PBS (control), liposomes, WR12 or FALs and were monitored and analysed at 3, 7, and 14 days. Generally, the wound size for the FAL group at the different time points was the smallest compared to those of the control group, liposome group and WR12 group (Fig. 5a). For example, on day 3, the wound size of the FAL group was 21.57%, which was smaller than that of the control group (52.80%), the liposome group (43.48%) and the WR12 group (31.40%) (Fig. 5b). Therefore, the wound healing of infected mice treated with FALs was significantly higher than those of the other treatment groups (Fig. 5c). Fig. 5a also shows the number of bacteria on the wound surface after 3 days of treatment. The number of bacteria in the FAL group was 1.15 × 106 CFU mL−1, which was much lower than that of the control group (7.80 × 106 CFU mL−1) and WR12 group (4.44 × 106 CFU mL−1). The amount of bacteria decreased by 85.26% in the FAL-treated group; in comparison, it decreased by only 43.59% after treatment with WR12. The results indicated that in this system, WR12 had a certain antibacterial activity; however, it was easily degraded without the protection of liposomes, resulting in weak antimicrobial activity and slow wound healing compared to the FAL group.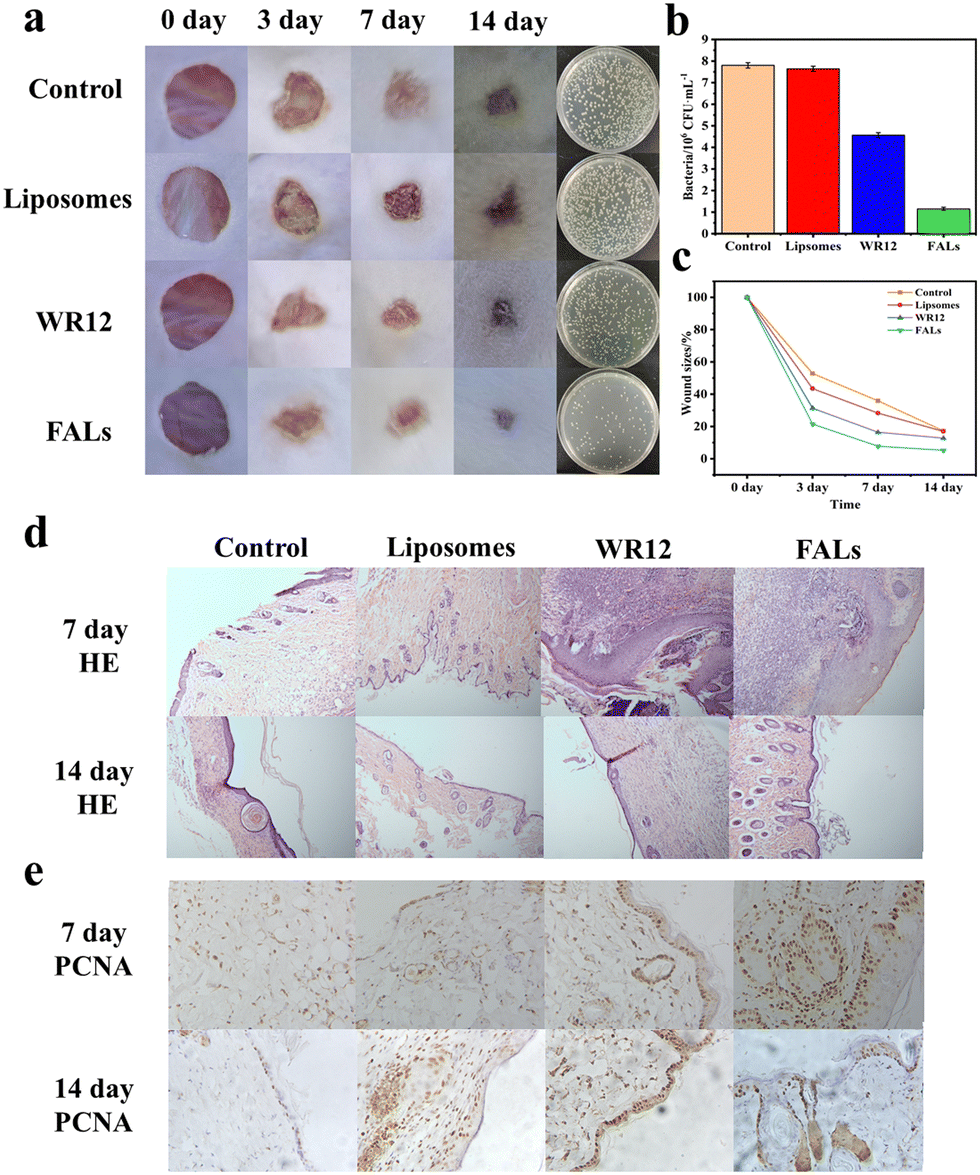 | ||
| Fig. 5 (a) Photographs of infected wounds on the backs of the mice given different treatments during the whole therapeutic process from day 0 to day 14, and of the bacteria separated from the different wound tissues that were cultured on agar plates for the different treatments. (b) Quantitative measurement of the wound size during the whole therapeutic process. (c) Number of bacteria surviving in the wound tissue of each sample on day 3. (d and e) HE and immunohistochemical staining performed on the wound tissues on day 7 and 14 of the treatment. | ||
Finally, to further study the therapeutic effect of the FALs on wounds, hematoxylin (HE) staining and proliferating cell nuclear antigen (PCNA) were adopted for histological analysis (Fig. 5d and e). The epithelization of the experimental group had basically been completed. Compared with those of the control group, the collagen fibers of the skin tissue in the FAL group were stably arranged, and some hair follicles had grown, which was no different from the normal tissue. The results of HE staining were consistent with the evaluation of wound healing in vivo. The results of immunohistochemistry staining for PCNA expression of proliferating nuclear antigen protein showed that, except for the FAL group, the expression level of PCNA in the other groups was lower (Fig. 5e). The number of PCNA-positive cells in the FAL group increased, and its cell proliferation activity was enhanced compared with that of the other groups, with particular promotion of the proliferation and metabolism of keratinocytes in the basal cells. In conclusion, the wound healing of mice treated with FALs exhibited a much better effect than the wound healing of the other treatment groups, indicating that the FALs can be better candidates and can be applied in the disinfection and wound healing.
Conclusions
We reported a simple strategy using a fusion peptide coupled with AMPs to fuse with liposomes to form FALs. Compared with the traditional AMP, it has more efficient bactericidal properties and almost no cytotoxicity. Additionally, it is not easily degraded by enzymes. A mouse wound model infected with S. aureus was used to prove that FALs can kill the bacteria on the infected wound and promote wound healing efficiently. These findings indicated that FALs constructed by this strategy have broad application prospects both in wound disinfection and promoting chronic wound healing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for financial support from the National Natural Science Foundation of China (22072060, 81802102) and Science Foundation of Wuxi Health Committee (No. BJ202220).Notes and references
- L. Imamovic and M. O. A. Sommer, Sci. Transl. Med., 2013, 5, 204ra132 CrossRef PubMed.
- B. S. F. Bazzaz, M. Fakori, B. Khameneh and H. Hosseinzadeh, J. Pharmacopuncture, 2019, 22(1), 49–54 CrossRef PubMed.
- M. Hagihara, J. L. Crandon and D. P. Nicolau, Expert Opin. Drug Saf., 2012, 11(2), 221–233 CrossRef CAS PubMed.
- L. S. Biswaro, M. G. da Costa Sousa, T. M. B. Rezende, S. C. Dias and O. L. Franco, Front. Microbiol., 2018, 9, 855–868 CrossRef PubMed.
- Y. Gao and Q. Ma, SmartMed., 2022, 1, e20220012 Search PubMed.
- C. Y. Lv, S. Q. Chen, Y. Xie, Z. W. Wei, L. Chen, J. X. Bao, C. He, W. F. Zhao, S. D. Sun and C. S. Zhao, J. Colloid Interface Sci., 2019, 556, 492–502 CrossRef CAS PubMed.
- X. Q. Qian, Y. Y. Zheng and Y. Chen, Adv. Mater., 2016, 28, 8097–8129 CrossRef CAS PubMed.
- C. Wang, T. T. Hong, P. F. Cui, J. H. Wang and J. Xia, Adv. Drug Delivery Rev., 2021, 175, 113818 CrossRef CAS PubMed.
- A. S. Birteksoz-Tan, Z. Zeybek, M. Hacioglu, P. B. Savage and C. Bozkurt-Guzel, J. Antibiot., 2019, 72(5), 291–297 CrossRef CAS PubMed.
- M. H. Semreen, M. I. El-Gamal, S. Abdin, H. Alkhazraji, L. Kamal, S. Hammad, F. El-Awady, D. Waleed and L. Kourbaj, Saudi Pharm. J., 2018, 26(3), 396–409 CrossRef PubMed.
- A. Hollmann, M. Martinez, P. Maturana, L. C. Semorile and P. C. Maffia, Front. Chem., 2018, 6, 13 CrossRef PubMed.
- M. L. Mangoni, A. M. McDermott and M. Zasloff, Exp. Dermatol., 2016, 25(3), 167–173 CrossRef CAS PubMed.
- T. M. A. Shafee, F. T. Lay, T. K. Phan, M. A. Anderson and M. D. Hulett, Cell. Mol. Life Sci., 2017, 74, 663–682 CrossRef CAS PubMed.
- R. E. Hancock and A. Patrzykat, Curr. Drug Targets: Infect. disord., 2002, 2, 79–83 CAS.
- D. Sun, J. Forsman and C. E. Woodward, J. Phys. Chem. B, 2017, 121, 10209–10214 CrossRef CAS PubMed.
- H. Ulvatne, Am. J. Clin. Dermatol., 2003, 4, 591–595 CrossRef PubMed.
- B. Bommarius, H. Jenssen, M. Elliott, J. Kindrachuk, M. Pasupuleti, H. Gieren, K. E. Jaeger, R. E. Hancock and D. Kalman, Peptides, 2010, 31, 1957–1965 CrossRef CAS PubMed.
- Y. Liscano, C. H. Salamanca, L. Vargas, S. Cantor, V. Laverde-Rojas and J. Oñate-Garzón, Antibiotics, 2019, 8, 238–253 CrossRef CAS PubMed.
- V. Teixeira, M. J. Feio and M. Bastos, Prog. Lipid Res., 2012, 51, 149–177 CrossRef CAS PubMed.
- M. Malmsten, Curr. Opin. Colloid Interface Sci., 2013, 18, 468–480 CrossRef CAS.
- L. Chen and J. F. Liang, Biomacromolecules, 2013, 14, 2326–2331 CrossRef CAS PubMed.
- F. Szoka, Jr. and D. Papahadjopoulos, Annu. Rev. Biophys. Bioeng., 1980, 9, 467–508 CrossRef PubMed.
- Y. H. Chan and S. G. Boxer, Curr. Opin. Chem. Biol., 2007, 11, 581–587 CrossRef CAS PubMed.
- M. J. Hope, M. B. Bally, L. D. Mayer, A. S. Janoff and P. R. Cullis, Chem. Phys. Lipids., 1986, 40(2), 89–107 CrossRef.
- Y. Jia, X. Wang, D. Hu, P. Wang, Q. Liu, X. Zhang, J. Jiang, X. Liu, Z. Sheng, B. Liu and H. Zheng, ACS Nano, 2019, 13(1), 386 CrossRef CAS PubMed.
- Y. Teramura, Biomaterials, 2015, 48, 119–128 CrossRef CAS PubMed.
- G. A. Koning, A. M. Eggermont, L. H. Lindner and T. L. ten Hagen, Pharm. Rep., 2010, 27, 1750–1754 CrossRef CAS PubMed.
- M. Zhao, X. F. Ding, J. Y. Shen, X. P. Zhang, X. W. Ding and B. Xu, J. Zhejiang Univ., Sci., B, 2017, 18(1), 15–26 CrossRef CAS PubMed.
- F. Szoka, Jr. and D. Papahadjopoulos, Annu. Rev. Biophys. Bioeng., 1980, 9, 467–508 CrossRef PubMed.
- Y. H. Chan and S. G. Boxer, Cur. Opin. Chem. Bio., 2007, 11(6), 581–587 CrossRef CAS PubMed.
- Y. Teramura, Biomaterials, 2015, 48, 119–128 CrossRef CAS PubMed.
- G. A. Koning, A. M. Eggermont, L. H. Lindner and T. L. ten Hagen, Pharm. Res., 2010, 27(8), 1750–1754 CrossRef CAS PubMed.
- M. F. Mohamed, A. Abdelkhalek and M. N. Seleem, Sci. Rep., 2016, 6, 29707–29720 CrossRef PubMed.
- L. M. Sun, A. P. Li, Y. Z. Hu, Y. Li, L. Shang and L. B. Zhang, Part. Part. Syst. Charact., 2019, 36, 1800420–1800425 CrossRef.
- X. R. Zhou, Y. M. Cao, Q. Zhang, X. B. Tian, H. Dong, L. Chen and S. Z. Luo, Int. J. Pharm., 2017, 528, 723–731 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00261f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |