
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unsubstituted thiophene–diketopyrrolopyrrole conjugated polymer thin films via oxidative chemical vapor deposition – electronic behavior†
Marek K.
Charyton
 a,
Tobias
Reiker
bc,
Kamil
Kotwica
de,
Monika
Góra
f,
Helmut
Zacharias
b and
Nicolas D.
Boscher
*a
a,
Tobias
Reiker
bc,
Kamil
Kotwica
de,
Monika
Góra
f,
Helmut
Zacharias
b and
Nicolas D.
Boscher
*a
aMaterials Research and Technology Department, Luxembourg Institute of Science and Technology, L-4362 Esch-sur-Alzette, Luxembourg. E-mail: nicolas.boscher@list.lu
bCenter for Soft Nanoscience, University of Münster, Münster 48149, Germany
cPhysics Institute, University of Münster, Münster 48149, Germany
dFaculty of Chemistry, Warsaw University of Technology, 00-664 Warszawa, Poland
eInstitute of Physical Chemistry, Polish Academy of Sciences, 01-224 Warszawa, Poland
fFaculty of Chemistry, University of Warsaw, 02-093 Warszawa, Poland
First published on 23rd May 2023
Abstract
Conjugated polymers (CPs) based on diketopyrrolopyrrole (DPP) constitue an important class of high-performance organic semiconductors. While N-alkylation of DPP is required for the solution-based synthesis of DPP-based CPs, oxidative chemical vapour deposition (oCVD) is shown to provide a straightforward solventless and scalable approach for the facile polymerisation of N-unsubstituted DPP. The oCVD reaction of 3,6-di(2-thienyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (TDPP) yields polymer thin films combining H-bonding and conjugated covalent bonds, resulting in a superior conductivity and a systematic increase of the lifetime of electronically excited states compared to N-alkylated counterparts.
Introduction
Serendipitously discovered in 1974,1 diketopyrrolopyrrole (DPP) rapidly reached commercialization as high-performance pigments for paints, inks and plastics (e.g. Ferrari red pigment). Beyond their intense coloration and wide range of colour shade,2,3 the industrial and commercial success of DPP derivatives was mainly dictated by their low fabrication costs, low dissolvability and excellent thermal2 and photo-stability (8th-grade light fastness).4–6 For example, DPP pigments based on a DPP core flanked by two aromatic moieties can readily be produced from the reaction of inexpensive aromatic nitriles with succinic acid ester.4 Along the past decade, DPPs have also attracted a growing interest for organic electronic,7 photovoltaic7,8 and photoelectrocatalytic applications9 due to their intrinsic physical and chemical properties. DPPs’ properties are strongly influenced by the substituents attached to the core (e.g. phenyl, pyridyl, thienyl, furanyl, seleneyl)10 and N-positions (mainly alkyl groups). DPPs can either be used as single molecules, e.g. photosensitizers on metal oxide surfaces,9 or (co)polymerised, e.g. p-type, n-type or ambipolar polymeric semiconductors for organic field effect transistors (OFETs).11,12 Several studies have highlighted the cooperative effect promoted by H-bonds13 or conjugated covalent bonds11,12,14–17 between DPP derivatives on their functional properties. Particularly, DPP-based conjugated polymers (CPs) are benchmark CPs for high-performance organic semiconductors.7 However, N-alkylation, which can significantly increase the production costs, is required to prevent π–π stacking and ensure the solubility and processability of DPP-based CPs.11,12,14–17 In addition, the synthesis of DPP-based CPs implies multistep processes catalysed by expensive and toxic derivatives (e.g. tin and palladium-based compounds).11,12,15–17 Furthermore, purification of DPP-based CPs remain limited to column chromatography,11,12,16 which further increases costs and constitutes an additional hurdle to scaling-up.Oxidative chemical vapor deposition (oCVD), which relies on the vapor phase transport of aromatic compounds and their subsequent reaction with an oxidizing agent to readily form conjugated polymers directly in thin film form,18 constitutes an attractive alternative to solution-based methods. oCVD possesses several assets, such as conformality, substrate independence and excellent control of the film parameters (thickness, porosity).18,19 In recent years, oCVD has been successfully used in a broad range of research fields and applications, including photovoltaic,20 catalysis,21 energy storage,22 and gas sensing.23 Noteworthy, Gharahcheshmeh et al. used water-assisted oCVD to engineering the π–π stacking in poly(3,4-ethylenedioxythiophene) (PEDOT) thin films and reached a record-high electrical conductivity of 7520 S cm−1.24 Indeed, macromolecular conformation is well known to play a significant role on the properties of polymeric materials.25–28 Particularly, reduced intermolecular distances can yield superior charge carrier density and mobility, improved delocalization and electrical conductivities.29–31 Being a solvent-free process, oCVD suppresses the need for solubilizing substituents that both hinder π–π stacking and prevent the integration of more advanced groups. Recently, Bengasi et al. implemented the oCVD strategy for the simultaneous synthesis and deposition of porphyrin-based CPs.32 The decoupling of the porphyrin substituents from the synthesis requirement allowed the synthesis of porphyrin-based CPs bearing unexplored groups.21,30 Particularly, the use of small substituents, i.e. phenyl, enabled to reduce intermolecular distances and improve π–π stacking,23,30 yielding superior conductivities for porphyrin-based CPs.30
Taking advantage of the potential of oCVD, we investigate in this work the oCVD reaction of DPP derivatives with the aim to enable both the formation of conjugated covalent bonds and H-bonds in DPP-based CPs thin films. To this end, we selected both N-unsubstituted and N-alkylated DPP derivatives flanked with electron-rich thienyl moieties that provide active sites for oxidative polymerization. The occurrence of the oxidative coupling reaction was evidenced by ultraviolet-visible-near infrared (UV-Vis-NIR) spectroscopy and laser desorption ionization high-resolution mass spectrometry (LDI-HRMS). The influence of the N-substituent on the electronic properties of the DPP-based CPs thin films was evaluated using two-photon photoelectron spectroscopy (2PPE). 2PPE uses a first photon to excite an electron into an intermediate state and a second photon for probing. By varying the time delays between pump and probe pulse, 2PPE allows the exploration of excited states dynamics. In contrast to purely optical spectroscopy methods, optically dark states can be investigated directly. Further, the high surface sensitivity, inherent to the method, makes 2PPE suitable for thin film characterisation.33 The data obtained give direct access to the fundamental processes of charge generation in organic semiconducting devices, e.g. exciton,34–38 triplet33,39–44 and polaron generation,45–47 which are controlled by the macromolecular conformation. Indeed, vibronic and electronic coupling is influenced by the molecular structure, and π–π stacking can induce a photocurrent enhancement.34,35,48–53
Results and discussion
Gas phase synthesis of thiophene–diketopyrrolopyrrole conjugated polymer thin films
Our investigations towards the chemical vapor deposition of thienyl-bridged diketopyrrolopyrrole polymers began with the oCVD reaction of 3,6-di(2-thienyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (TDPP) (Table S1, ESI†). Electron-rich thienyl groups are selected as core-substituents to provide active sites for the intermolecular oxidative polymerization. Among the commercially available thienyl core substituted DPP derivatives, TDPP constitutes the simplest motif, bearing no substituent at its axial positions (N-positions). Alkyl chains, which are commonly used as axial substituents to grant solubility to TDPP-based polymers (blocking H-bonding and reducing π–π stacking),11,12,14–17 are not required in oCVD since the synthesis, deposition and integration are performed simultaneously.18,21,23,30,32 Iron(III) chloride (FeCl3) was selected due to its ability to promote the dehydrogenative coupling of both thiophene compounds54 and heterocyclic macrocycles.21,23,30,32 Prior to its sublimation under oxidative conditions, the thermal stability of TDPP was controlled by thermogravimetry (Fig. S1, ESI†), which reveals a stability up to at least 300 °C. The oCVD reaction of TDPP and FeCl3 (Scheme 1) was undertaken under reduced pressure (10−3 mbar) in a custom-built reactor equipped with two crucibles used to simultaneously sublime TDPP (255 °C) and FeCl3 (170 °C) towards a heated stage (200 °C) on which were placed the substrates (Scheme S1, ESI†).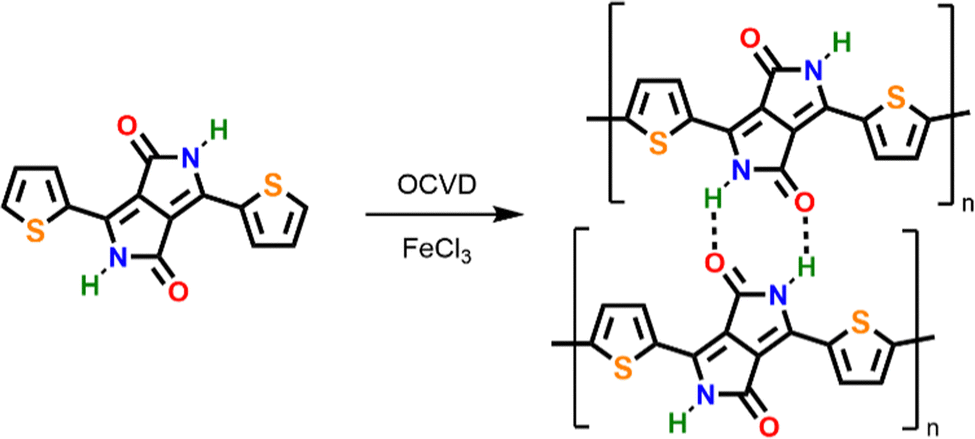 | ||
| Scheme 1 Molecular structures of TDPP (left) and the conjugated polymer (right) formed from the oxidative intermolecular coupling reaction of TDPP through the α position of the thienyl substituent in the presence of FeCl3. Due to the unsubstituted axial positions of TDPP, hydrogen bonds between TDPP monomers, oligomers and polymer chains are also possible and can yield strong intermolecular interactions. One should note here that intermolecular coupling may also occur through the normally less-reactive β position of thienyl substituent. | ||
The oCVD reaction of TDPP with FeCl3 leads to the formation of deep blue coatings (denoted pTDPP) that contrasts with the vivid pink color of the coatings obtained from the sublimation of TDPP (denoted sTDPP) (Fig. 1). Such a color change, from pink to blue, suggests the expansion of the chromophore and the formation of TDPP oligomers. Indeed, Casutt et al. previously described that TDPP dimers exhibit dark violet color,3 whereas the parental Boc-substituted TDPP monomer exhibits a yellow-orange coloration. UV/Vis/NIR spectroscopy, performed on coated borosilicate glass substrates, confirms significant differences in the absorption spectrum of the deep blue pTDPP coating with respect to the vivid pink sTDPP coating. Particularly, upon reaction with FeCl3, the absorption maximum undergoes a bathochromic shift from 564 nm in sTDPP to 650 nm in pTDPP. The bathochromic shift of the absorption maximum, assigned to the π–π* transition, suggests the enlargement of the conjugation length through an oxidative polymerization process. This band is observed at a slightly lower wavelength than previously reported ones for DPP copolymers that are often copolymerized with multiple donor units to enhance donor–acceptor interactions and further reduce band gap.55–57 One can note here the bathochromic shift of the main absorption peaks (514 and 564 nm) of the sTDPP coating by comparison to the positions of the main absorption peaks of TDPP in solution (488 and 525 nm).58 Importantly, pTDPP exhibits a broad absorption in the NIR region, up to 2400 nm (Fig. 1). Upon doping with FeCl3, positive polarons or positive bipolarons form and induce the generation of localized electronic states, above and below the valence and conduction bands, respectively. Therefore the transitions related to these new localized electronic states yield absorption bands at higher wavelengths. Particularly, the NIR absorption band observed at high doping levels can be assigned to the transition related to bipolarons. According to the observations previously reported for hydrogen bonded TDPP dimers,3 the pTDPP coating is insoluble in common organic solvents, including dimethyl sulfoxide (DMSO) (Fig. S2, ESI†). This contrasts with the sTDPP coating which is fully soluble in DMSO (Fig. S2 and S3, ESI†).
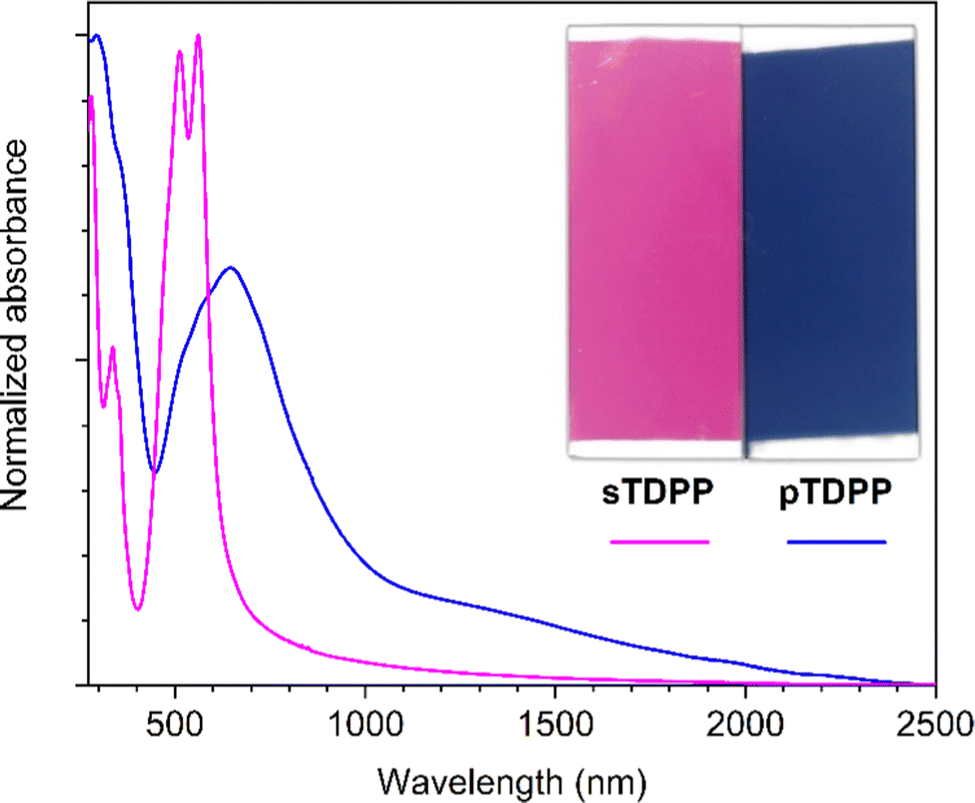 | ||
| Fig. 1 Optical images (inset) and UV/Vis/NIR absorption spectra of the sublimed sTDPP coating (pink) and oCVD pTDPP coating (blue) prepared from TDPP on glass substrates. | ||
LDI-HRMS analysis directly performed on the pTDPP coating reveals the formation of TDPP oligomers confirming the successful intermolecular coupling of TDPP under oCVD conditions (Fig. 2). Up to tetrameric oligomers are observed in the mass spectrum of pTDPP, while only monomeric species are observed for sTDPP. One should note here that LDI-HRMS analysis, with an instrumental limit of 4000 m/z, does not provide an exhaustive view into the mass distribution. Therefore, formation of longer oligomers, with masses outside the instrumental limit, is conceivable. Moreover, the intensities related to the detected species are not directly related to their abundance. Unfortunately, the insolubility of the pTDPP coating (Fig. S2, ESI†) prohibits GPC analysis and a thorough characterization of the oCVD coating. In addition to the oxidative coupling of TDPP, LDI-HRMS evidences the integration of one to several chlorines (Fig. 2b), which could also contribute to the very weak solubility of pTDPP. Analysis of the Cl 1s XPS spectrum confirms the presence of an organic chloride environment related to the chlorination of the TDPP oligomeric and polymeric species (Fig. S4, ESI†). Chlorination is a well-known side reaction in oCVD processes that involve metal chlorides as oxidants.59
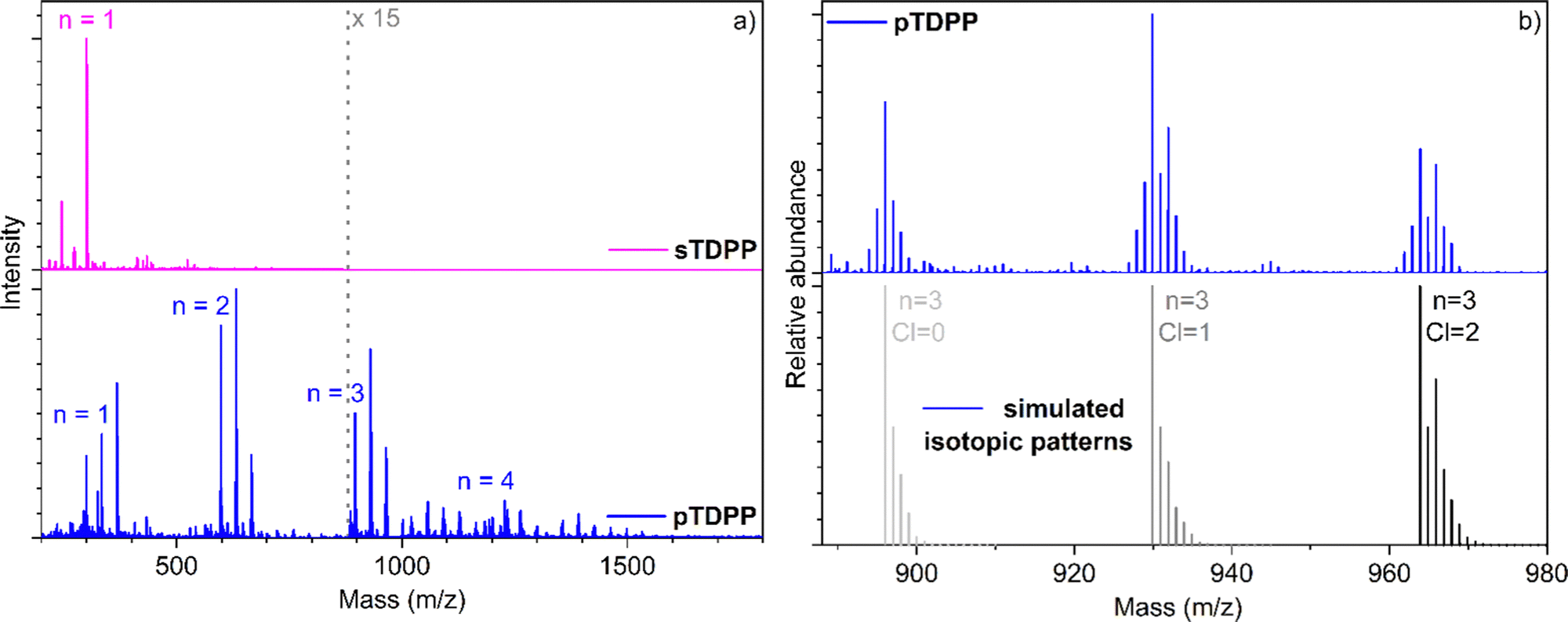 | ||
| Fig. 2 (a) LDI-HRMS spectra of the sublimed sTDPP coating (pink) and oCVD pTDPP coating (blue) prepared from TDPP. (b) LDI-HRMS spectra of the trimeric region tha shows integration of chlorine atoms, a well-known side reaction in oCVD when using chlorinated oxidants. Experimental LDI-HRMS spectrum of pTDPP is displayed (blue) alongside simulated isotopic patterns of trimeric species bearing 0 ([(TDPP)3–2H]+, light grey), 1 ([(TDPP)3–3H + 1Cl]+, dark grey) and 2 chlorine atoms ([(TDPP)3–4H + 2Cl]+, black). | ||
With the aim to perform a GPC analysis and undoubtedly demonstrate the oxidative polymerization of TDPP in oCVD, we investigated the oCVD reaction of 2,5-bis(2-ethylhexyl)-3,6-di(2-thienyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (BEHTDPP). In accordance with previous solution-based studies, the grafting of long alkyl chains to the axial positions of the TDPP motif is foreseen to confer solubility to the formed polymeric chains by reducing π–π intermolecular interactions between the aromatics systems, and by preventing hydrogen bonding between amide functional groups.11,12,14–17 In addition to the 2-ethylhexyl group, we also investigated the oCVD reaction of TDPP substituted with one or two tert-butyloxycarbonyl (boc) protecting groups. However, the low-pressure (10−3 mbar) and temperature (200 °C) operating conditions required to supply the TDPP-based monomers from the vapor phase in the proposed CVD approach yield to the removal of the Boc group during sublimation step (Fig. S1, ESI†).
Similarly to what is observed for TDPP, the oCVD reaction of BEHTDPP with FeCl3 (Table S1 and Scheme S2, ESI†) yields the formation of coatings (denoted pBEHTDPP) whose color (brownish to greyish) contrasts with the color (pinkish) of the coatings prepared from the sole sublimation of BEHTDPP (denoted sBEHTDPP) (Fig. S5, ESI†). UV/Vis/NIR spectroscopy analysis confirmed significant differences in the absorption spectrum of sBEHTDPP and pBEHTDPP coating (Fig. S5, ESI†). Such as for TDPP, upon reaction with FeCl3, the absorption maximum undergoes a bathochromic shift from 506 nm in sBEHTDPP to 658 nm in pBEHTDPP. Although less pronounced than for pTDPP (Fig. 1), pBEHTDPP also exhibits absorption in the NIR region, up to 2200 nm (Fig. S5, ESI†). LDI-HRMS analysis directly performed on the pBEHTDPP coating reveals the formation of BEHTDPP oligomers, confirming the successful intermolecular coupling of BEHTDPP (Fig. 3a). Up to trimeric species are observed in the mass spectrum of pBEHTDPP, while only monomeric species are observed for sBEHTDPP. The GPC analysis of pBEHTDPP (Fig. 3b) confirms the formation of polymeric chains under oCVD condition. Satisfactorily, monomer and short oligomers are not detected by GPC which only displays a broad peak pointing to masses up to 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. Given the similarities in the UV/Vis/NIR (Fig. 1 & Fig. S5, ESI†) and LDI-HRMS spectra (Fig. 2 and 3a) of pTDPP and pBEHTDPP, one may assume that the pTDPP samples are also composed of polymer chains with molecular weights in the range of tens of thousands g mol−1.
000 g mol−1. Given the similarities in the UV/Vis/NIR (Fig. 1 & Fig. S5, ESI†) and LDI-HRMS spectra (Fig. 2 and 3a) of pTDPP and pBEHTDPP, one may assume that the pTDPP samples are also composed of polymer chains with molecular weights in the range of tens of thousands g mol−1.
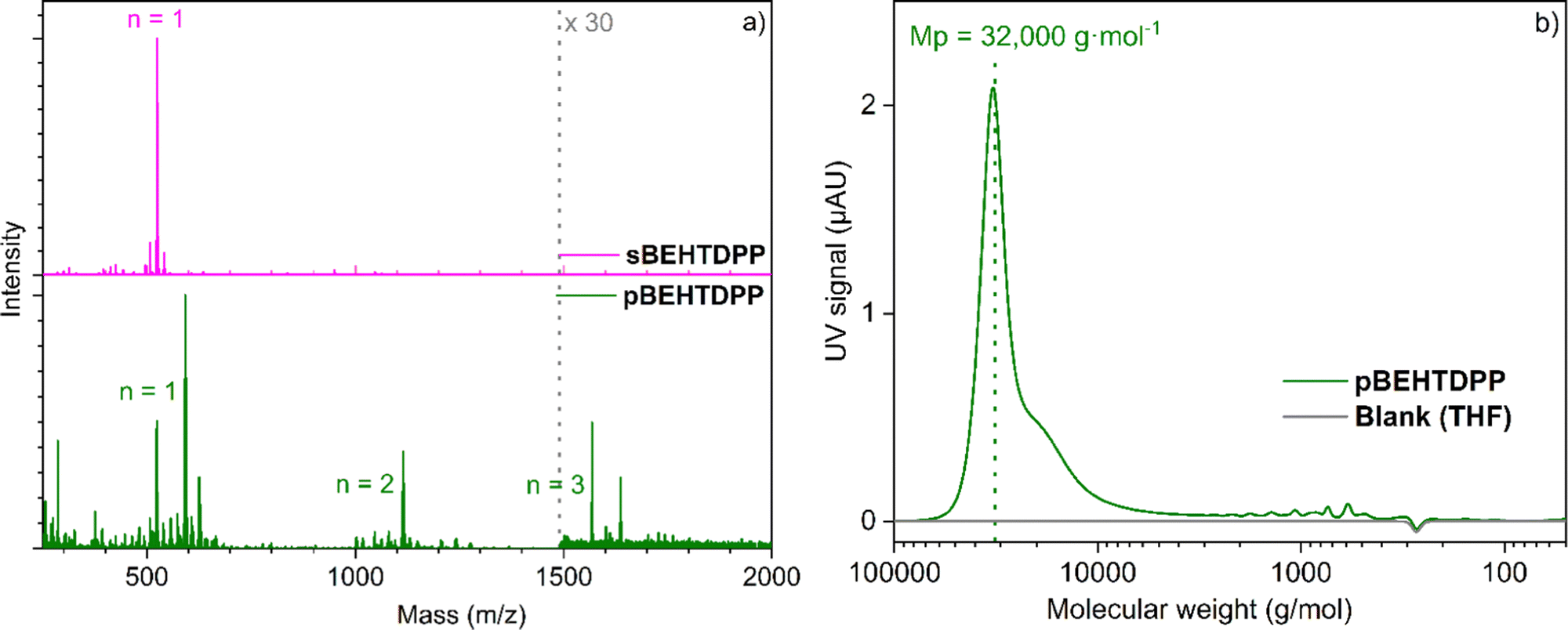 | ||
| Fig. 3 (a) LDI-HRMS spectra of the sublimed sBEHTDPP coating (pink) and (b) oCVD pBEHTDPP coating (grey) prepared from TDPP and (b) GPC spectrum of pBEDTDPP. | ||
Scanning electron microscopy (SEM) and atomic force microscopy (AFM) of sTDPP and pTDPP reveal the formation of rather dense coatings with a rugged surface (Fig. 4). Upon polymerization, the size of the main features observed at the surface increase from 50 to 100 nm, respectively. Contrarily, sBEHTDPP and pBEHTDPP exhibit far less features at their surface (Fig. S6 and S7, ESI†), which strongly contrast with the morphology of the coatings grown from TDPP. Indeed, the long alkyl chains attached to the axial positions of the TDPP motif in BEHTDPP reduce the π–π interactions. Beyond enabling a higher solubility, reduction of π–π stacking is also responsible for the lower sticking coefficient of the BEHTDPP monomeric units, yielding a lower sublimation temperature despite of a higher molecular weight (155 °C for BEHTDPP against 255 °C for TDPP) and a higher mobility at the surface of the substrate. Thus, in addition of influencing the packing arrangement of the DPP-based compounds,60 alkyl axial substituents can affect their mobility and supramolecular aggregation. Interestingly in the perspective of optoelectronic applications, pTDPP exhibit a low surface roughness with an arithmetic mean height (Sa), i.e. mean difference in height from the mean plane, close to 2 nm.
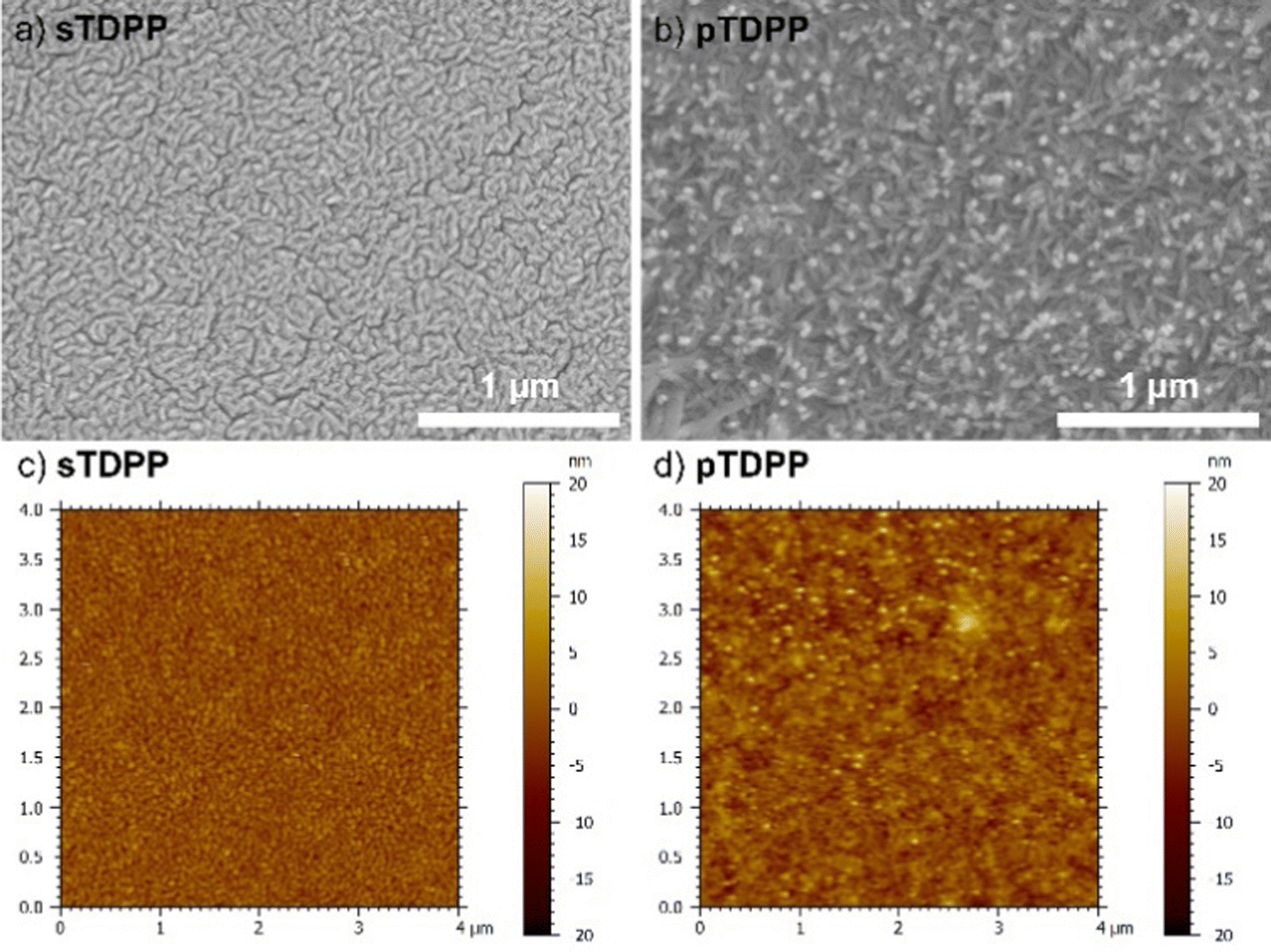 | ||
| Fig. 4 (a and b) High magnification SEM images and (c and d) AFM topography images of the sublimed sTDPP coating (a and c) and oCVD pTDPP coating (b and d) prepared from TDPP. | ||
Electronic properties of the thiophene–diketopyrrolopyrrole conjugated polymer thin films
Fostered by the straightforward synthetic and deposition approach reported above, the following section is exploring the electronic and electrochemical behavior of thin films of unsubstituted thiophene–diketopyrrolopyrrole conjugated polymer (pTDPP). The current–voltage (I(V)) characteristics of sTDPP and pTDPP deposited onto chips patterned with interdigitated electrodes revealed an ohmic behaviour (Fig. 5a). As expected, the dehydrogenative coupling of the TDPP units, which yield the formation of a conjugated backbone, induces a strong increase of the electrical conductivity from 7.8 × 10−10 S cm−1 for sTDPP to 1.7 × 10−3 S cm−1 in pTDPP. Noteworthy, the oCVD pBEHTDPP coating exhibits a reduced conductivity (9.7 × 10−5 S cm−1) compared to pTDPP (Fig. 5b and Fig. S8, ESI†). The discrepancy between the conductivities of pTDPP and pBEHTDPP coatings likely arises from a reduced intermolecular π–π stacking in the presence of long axial alkyl substituents. Limited intermolecular π–π stacking due to bulky substituents is a strong limitation into the photovoltaic or electronic performances of conjugated polymers.61 Thus, the ability to readily polymerize and deposit diketopyrrolopyrrole-based polymer irrespective of their substituents should rapidly improve the properties of these materials.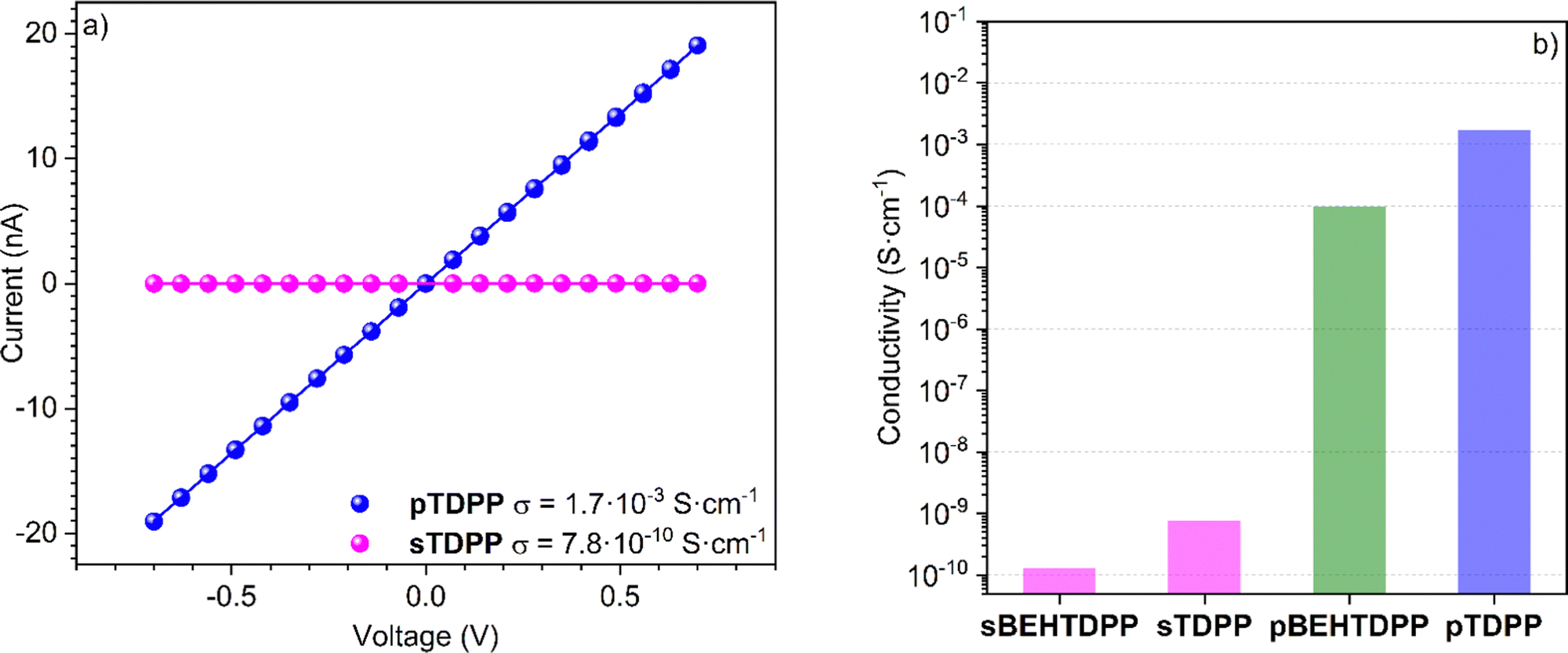 | ||
| Fig. 5 (a) Lateral electrical conductivity measurement of sublimed sTDPP coating (pink dots) and oCVD pTDPP coating (blue dots) on interdigitated chips. Pink and blue solid lines show the linear fits used to determine the conductivities. (b) Histogram of electrical conductivity values for the sublimed and oCVD coatings. | ||
Such as for other conjugated polymers, the repetition of σ and π bonds along the oligomeric and polymeric chains constituting pTDPP is expected to give rise to a delocalization of π orbitals and of an energy level splitting of the π bonds. Analysis of the valence band region of the XPS spectra allows to estimate the valence band (VB) or highest occupied molecular orbital (HOMO) position of the sTDPP and pTDPP. In particular, the valence band maximum (VBM), determined from the extrapolation of the linear fit of the leading edges of the spectra to the baseline, is shifted from 1.53 eV (sTDPP) to 0.99 eV (pTDPP) upon oxidative polymerisation (Fig. S9, ESI†). This observation is consistent with the expansion of π-electronic system known to raise the energy of the HOMO.
From the photoelectron spectra of the maximum 2PPE signals shown in Fig. 6, one can observe a clean broad spectrum with photoelectrons up to Ekin = 1.5 eV for pTDPP, and the typical increasing intensity towards lower kinetic energy, usually caused by secondary electrons. Comparing the spectrum of pTDPP with highest 2PPE intensity, which occurs at a pump – probe delay of Δτ = 300 fs, with the background spectrum (pump pulse first), a clear 2PPE feature is identified, centred around Ekin = 0.4–0.8 eV. The spectrum for pBEHTDPP shows a very similar pattern, but somewhat higher in intensity and with a more pronounced peak from secondary electrons at lower kinetic energies.
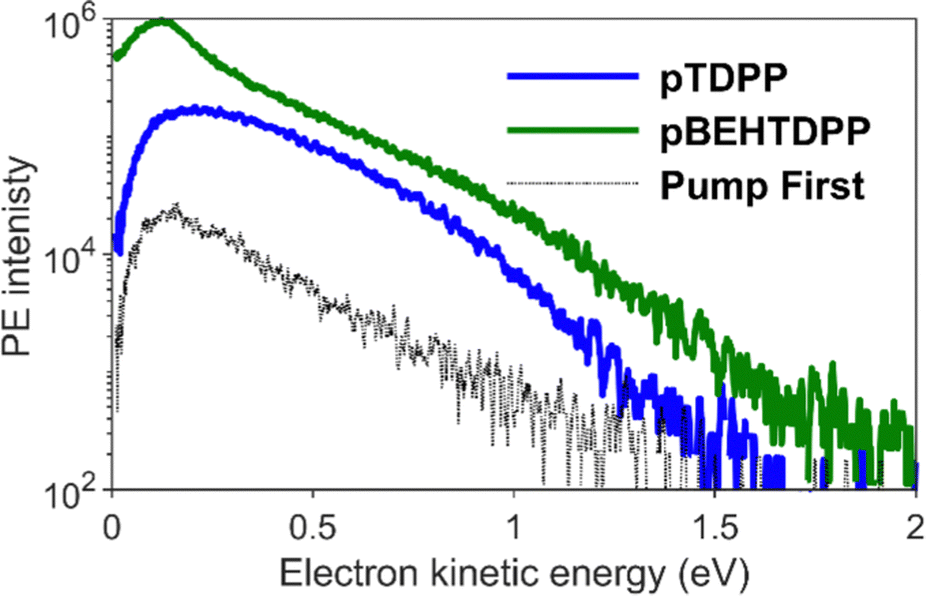 | ||
| Fig. 6 Photoelectron spectra of pTDPP (blue) and pBEHTDPP (green) measured with an excitation of hvpump = 1.88 eV and hvprobe = 4.8 eV at a delay of Δτ = 300 fs, where the 2PPE signal strength reaches its maximum. For comparison, a spectrum of pTDPP is shown in black where the sample pulse arrives at the sample before the probe pulse. | ||
The time-resolved dynamics were analysed as a multi-exponential decay and can be described adequately by four distinguishable individual lifetimes. It is worth noting that a fourfold exponential decay model, in its simplicity, represents the most straightforward approach. However, we want to mention commonly employed alternative methods which consider that a rapidly decaying state feeds a longer living state. In this cascaded model, a longer living state therefore has initially zero intensity and rises with a time constant which reflects the lifetime of the rapid decay.62 From a physical point of view, the overall excited state dynamics in conjugated polymers can accurately be depicted by a set of rate equations featuring interconnected states.63–66 Following extensive testing, a cascaded model and approaches with fitted rate equations resulted not in substantially improved or different lifetimes and their relative intensities. Further, there is no clear indication of rising components in the tr-2PPE data, see also the two-dimensional presentation in Fig. S10 (ESI†). Thus, we conclude that our more phenomenological description is entirely adequate.
For both pTDPP and pBEHTDPP, an ultrafast lifetime in the femtosecond scale (τ1 ∼ 100 fs), two intermediates with a few picoseconds τ2 ∼ 1 ps, τ3 ∼ 5 ps, and a very long lifetime τ4 ≫ 50 ps, beyond our measurement range, are obtained. These lifetimes τk and also their amplitudes Ak are of similar orders of magnitude for both samples, but depend on the electron kinetic energy. Fig. 7a shows the fitted decay times τ1, τ2, τ3 for pTDPP (blue data) and pBEHTDPP (green data) in the range of low kinetic energies from Ekin = 0.2 eV up to higher kinetic energies of Ekin = 1.0 eV, each integrated over a width of ΔEkin = 100 meV. The general trend shows a slight increase for all three independent lifetimes towards lower kinetic energies. The lifetimes for pTDPP are consistently longer than those of pBEHDPP. Moreover, the difference is larger for lower kinetic energies, which is particularly evident in the ultrafast lifetime τ1. For pBEHDPP the short lifetime, τ1 ∼ 60 fs, is on the order of the pump and probe pulse cross-correlation and probably limited by this. For pTDPP, the short lifetime of τ1 ∼ 120 fs is nearly twice as long. This difference in the accumulation of the charge carrier generation can also be seen clearly in the inset of Fig. 7c.
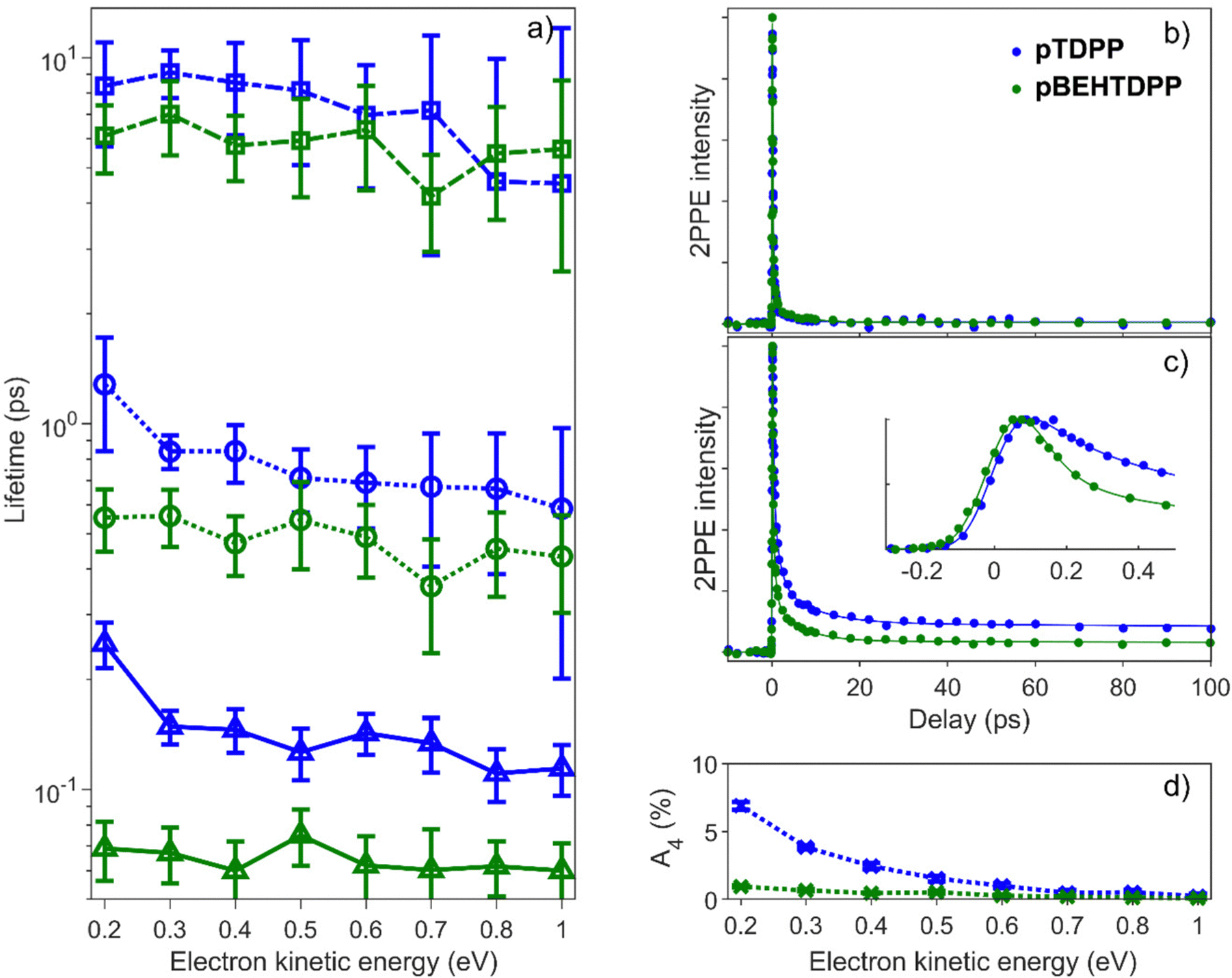 | ||
| Fig. 7 Time resolved evolution of the 2PPE signal measured on pTDPP (blue) and pBEHTDPP (green) with an excitation of hvpump = 1.88 eV and hvprobe = 4.8 eV, (a) show the trend of the three fitted lifetimes as a function of the kinetic energy in the 2PPE spectrum, (b) decay for Ekin = 1 eV, (c) decay for Ekin = 0.2 eV. Solid lines correspond to the respective multi-exponential fit, the inset shows a zoom around the temporal overlap. Panel (d) depicts the contribution of the longest lifetime to the signal as a function of kinetic electron energy. | ||
The temporal evolution of the 2PPE signal for high (Fig. 7b) and for low kinetic energies (Fig. 7c) was recorded and fitted. The decay curves for high kinetic energies are almost identical for the polymer with (pBEHTDPP) and without (pTDPP) N-substituents (Fig. 7b). It can be described almost completely by a double exponential decay (A4 ∼ 0% and A3 ∼ 0%, Fig. S11, ESI†). In contrast, at low energies (Fig. 7c) a long living component manifests itself in an offset in the data for pTDPP compared to the polymer with N-alkyl substituents (pBEHTDPP) at lifetimes Δτ > 10 ps. The decay of this long living component cannot accurately be measured by our delay stage and is therefore set as constant to infinity. However, the difference in the offset is clearly visible in the development of its amplitude A4 in Fig. 7d.
This long living component, accounted by A4, typically reflects a variety of charge generating quasiparticles. Evidence is mounting that free charge carriers can be generated quasi instantaneously in conjugated polymers upon excitation. Moreover, the correct arrangement of TDPP molecules can enable singlet fission, which, in particular, is a promising mechanism for increasing the efficiency of organic solar cell components.63,64,67–69 For TDPP and derivates, up to 200% triplet exciton quantum efficiency have been measured,64 interestingly, in connection with a higher yield for entities with shorter side chains. Singlet fission occurs within tens of picoseconds and can be stabilised by expanded π-skeletons of the molecules.70 Polarons and triplet excitons typically have lifetimes in the nano- to microsecond range.39,43,67,71 Even though it is not possible to distinguish both directly, the offset at long lifetimes is indicative for an increased charge carrier generation. Another indication is the course of the relative amplitudes with varying electron energy. Whereas the high-energy range can be described biexponentially, it is evident that for lower kinetic energies the relative contributions of the fast lifetimes decrease and the contributions of the slower ones increase, in particular for pTDPP. The lower kinetic energies represent states between the VBM and the LUMO, typically charge transfer excitons, polarons and triplets. Under the assumption of a binding energy of EBLUMO = −3.9 eV and a photon energy of the probe pulse of hvprobe = 4.8 eV, the LUMO is represented in the 2PPE spectra at a kinetic energy of Ekin = 0.9 eV.
The temporal evolution at energies around and above the LUMO shows for both samples a much faster decay than at lower energies. In particular, the ultrafast decays τ1 and τ2 are present with A1 ∼ 90% and A2 ∼ 10% (Fig. S11, ESI†). The decay in the range of τ1 ∼ 100 fs is predominantly associated with the excitation of higher vibronic states.41,72 Exciton self-trapping, polaron (pair) formation,45,47,65,73,74 and local relaxations are also expected on this time scale, although the former is probably even below our temporal resolution (<30 fs).36,40,45 The decay times of the LUMO then results in τ2 = (0.6 ± 0.4) ps and τ2 = (0.5 ± 0.1) ps for pTDPP and pBEHTDPP, respectively, and are therefore not significantly different. Fig. 7a shows, however, that for both τ1 and τ2, the lifetimes for pTDPP are systematically longer over the entire kinetic energy range than those of pBEHTDPP. We ascribe this phenomenon to the absence and presence, respectively, of the side chains. The additional alkyl groups in pBEHTDPP probably cause a broader spatial distribution of the excited state over the backbone, and therefore opens additional pathways to undergo a fast relaxation. On one hand, this leads to an increased lifetime in pTDPP of the initial excitation, while on the other hand, its relative contribution A1 is decreased (Fig. S11, ESI†).
For the comparison of the medium-long lifetime τ3, the same systematic behaviour is observed. For pTDPP and at lower kinetic energies, this lifetime is slightly longer, than that of pBEHTDPP. The relative proportion of this decay component also increases towards lower kinetic energies. Whereas this relative amount remains rather small for pBEHTDPP (increases from A3 = 1% to a maximum of 4%) it grows to 12% for pTDPP. Lifetimes of approx. 5–12 ps are typical for hot intramolecular charge transfer processes,36,45 and facilitate free charge generation.45,74,75 In addition, this lifetime is of the order of that for generation of the 1(T1T1)- state measured for several thin films of TDPP monomers. This generation time was measured to be a few tens of picoseconds for different kinds of side chains.64,67 Roy and Dasgupta measured the formation of a triplet pair from the first excited state within about 700 fs in TDPP nanoparticles, remarkedly close to the lifetime τ2 for both polymers in the present measurement.36 The energy of the monomer TDPP triplet is about 1.1 eV above the VBM and appears around Ekin = 0.2 eV in the 2PPE spectra, the energy region where the contributions of A3 and A4 are also increasing.43,49,64 Shivhare et al. measured a negligible offset in the picosecond time scale for thiophene/DPP polymers, but an enhanced offset in combination with PCBM, and attributed this to the charge concentration.66 The longevity of charge signatures facilitated through enhanced hydrogen bonds is in accord with the results by Ávila-Rovelo et al. in thiophene-capped DPP molecules.52
The ability to synthesize N-unsubstituted thiophene–diketopyrrolopyrrole conjugated polymers and readily deposit them in the form smooth, dense and thickness-controlled thin films constitutes a breakthrough in the field of DPP-based conjugated polymers. Particularly, the simplicity of the proposed CVD method contrasts with recently developed solution-based approach towards the synthesis of DPP-based blue-colored pigments, which involves (i) tetrahydrofuran, chloroform, and toluene as solvents, (ii) di-tert-butyl decarbonate, N-bromosuccinimide and tetrakis(triphenylphosphine) palladium for grafting boc groups and enabling the Stille coupling, and (iii) a thermal treatment (190 °C) in the presence trifluoromethylsulfonic acid to ensure a complete removal of the boc group.3 Thus, the oCVD reaction of DPP derivatives should allow to go far beyond the state-of-the-art of DPP-based materials by eliminating the restrictions dictated by solution-based approaches and allowing the synthesis, engineering and integration of DPP-based conjugated polymers for optoelectronic and catalytic applications. Further improvements may also involve the use of liquid oxidants, such as antimony pentachloride (SbCl5) and vanadium oxytrichloride (VOCl3),76–78 in replacement of solid oxidants like FeCl3 or CuCl2 whose by-products remain in the oCVD coatings.59 Moreover, the delivery rate of liquid oxidants can be more precisely regulated, resulting in better control of the oxidant to monomer ratio.
Conclusions
In summary, the proposed CVD method provides a straightforward solventless and scalable approach for the facile synthesis, engineering and integration of DPP-based conjugated polymers for thin films applications. In this first demonstration, and in contrast to solution-based strategies, the oxidative polymerization and H-bonding of an N-unsubstituted DPP derivative was successfully achieved. Noteworthy, the new conjugated TDPP polymer was readily deposited as uniform and low-roughness thin films on various substrates. Interestingly, the DPP-based conjugated polymer (pTDPP) without N-alkyl substituent exhibits a higher conductivity and a slight but systematic increase of all lifetimes, notably an increase in the fraction of the very long-lived excited species associated with the free charge carriers or their precursors.Experimental
Oxidative chemical vapor deposition
The oCVD experiments were performed in a custom-built oCVD reactor equipped with two evaporators (Scheme S1, ESI†). Evaporators were loaded with 3,6-di(2-thienyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (TDPP) or 2,5-bis(2-ethylhexyl)-3,6-di(2-thienyl)-2,5-dihydropyrrolo[3,4-c]pyrrole-1,4-dione (BEHTDPP) and iron (III) chloride (FeCl3) and heated to 255 °C, 155 °C and 170 °C, respectively (Table S1, ESI†). FeCl3 was obtained from Sigma-Aldrich (97%) and TDPP79 and BEHTDPP80 were synthesized according to literature. The chamber was fed with argon and maintained to a pressure of 10−3 mbar. Deposition time was set to 30 min for all experiments. Microscope borosilicate glass slides (Menzel-Gläser), silicon wafers (Siltronix), FTO-coated glass and chips patterned with interdigitated gold electrodes (Fraunhofer IPMS) were employed.Thin film characterization
The thickness of the films was measured by using an Alpha Step D-500 profilometer from KLAvTencor. Optical absorbance was measured in the range 250–2500 nm using an UV/Vis/NIR spectrophotometer with a 150 mm diameter integrating sphere (PerkinElmer, Lambda 950). Atmospheric-pressure laser desorption/ionization high-resolution mass spectrometry (AP-LDI-HRMS) was performed directly on the as-deposited films without any matrix deposition using a LTQ/Orbitrap Elite Hybrid Linear Ion Trap-Orbitrap mass spectrometer from Thermo Scientific coupled AP-MALDI (ng) UHR source from MassTech Inc. equipped with a 355 nm Nd:YAG laser. An in-source decay (ISD) of 70 V was applied to the films to prevent the detection of non-covalent TDPP or BEHTDPP clusters that could interfere with the detection of the desorbed oligomers. A maximum injection time of 800 ms and a resolving power of 240000 at m/z 400 within the normal mass range (m/z 300–1000) and the high mass range (m/z 1000–4000) were employed for the HRMS analyses.
The electrical conductivity was evaluated from the films deposited on chips patterned with interdigitated gold electrodes and stored in the laboratory under ambient conditions. The current–voltage scans were recorded using a two-point probe and conductivity was evaluated using the Ohm's law. Cyclic voltammograms of the films deposited on FTO-coated glass were registered using an Autolab potentiostat, which was also used for the spectroelectrochemical studies. In order to determine the background current, the CV curves were registered for bare FTO-coated glass. The currents recorded for the polymer films were at least two orders of magnitude larger than that observed for uncovered FTO glass substrates.
The UV/Vis/NIR spectra were recorded using Cary 5000 spectrophotometer. After cleaning and rinsing with dichloromethane and drying, the films deposited on FTO-coated glass were placed under an argon atmosphere in a measuring cell equipped with Ag/Ag+ reference electrode and filled with 0.1 M Bu4NPF6/acetonitrile electrolyte. The spectroelectrochemical measurements were performed in quasi-static mode during which the working electrode was polarized in small steps. After each potential change, a wait time was applied until the measured current dropped to zero or to negligible value.
UPS spectra were measured with a hemispherical analyser (Specs Phoibos 150) and a cold cathode capillary discharge Helium lamp (He-I, hv = 21.22 eV). The valence band maximum is given against the Fermi energy of the system. The sample was placed in vacuum with a base pressure of 2 × 10−10 mbar.
A fibre laser (Active Fiber Systems, λ = 1030 nm, repetition rate frep = 500 kHz, τFWHM < 40 fs pulse duration) is split into two separate output beams. One beam is converted to the 4th harmonic (s-polarized, λ = 257 nm, hv = 4.8 eV, τFWHM = 65 ± 5 fs) by two optically nonlinear BBO crystals and utilized as the probe pulse. The other beam seeds a non-collinear optical parametric amplifier (NOPA) which generates pulses of about τ = 30 fs duration in the near infrared (Light Conversion, Orpheus N, tuneable λ = 650–950 nm, hν = 1.9–1.3 eV, p-polarized). The frequency-converted pulses pass through two prism compressors to compensate for the dispersion of the optical paths. Before the pulses are spatially and temporally superimposed using a dielectric mirror, the infrared beam passes through a high-precision delay stage with a resolution of Δτ < 1 fs (Physik Instrumente, PI M-405.DG). The sample is placed in an ultra-high vacuum chamber, and the kinetic energy of the emitted photoelectrons is analysed using a time-of-flight tube (eTOF) equipped with a delay line detector (Surface Concept, DLD 6565-4Q). A fourfold exponential function (1) was convoluted with a Gaussian corresponding to the width of the correlation of pump and probe pulse, τcx = 70 ± 5 fs, and was fitted to the time-resolved 2PPE signal. Here, τ4 was fixed to infinity, as the lifetime Δτ » 50 ps exceeds the range of the delay stage.
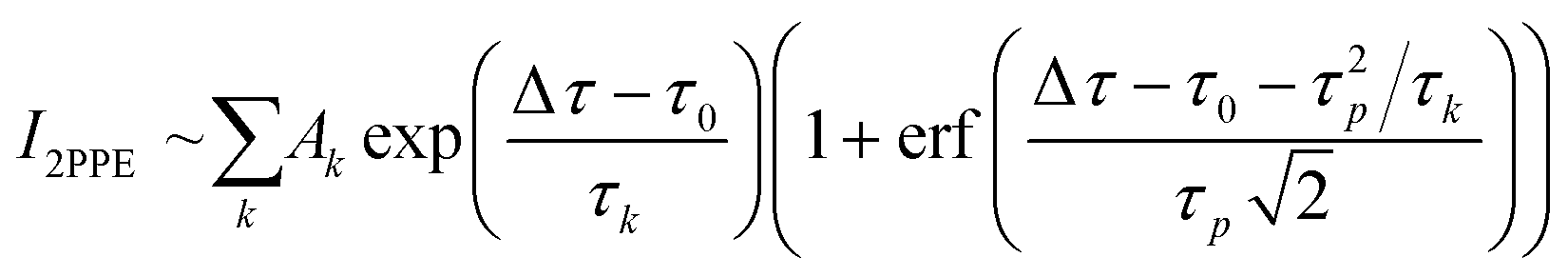 | (1) |
Author contributions
M. K. C.: conceptualization, synthesis, investigation, formal analysis, writing – original draft; T. K.: investigation, formal analysis, writing – original draft; K. K.: investigation, formal analysis, writing – review and editing; M. G.: synthesis, writing – review and editing; H. Z.: writing – review and editing; N. D. B.: conceptualization, methodology, investigation, formal analysis, funding acquisition, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 101031568 (TODAM project) and the National Science Centre of Poland, grant No 2019/32/C/ST5/00179. Dr G. Frache, P. Grysan, B. Marcolini, R. Vaudemont, Dr J. Guillot and C. Vergne from LIST are acknowledged for data collection and insightful discussions.References
- D. G. Farnum, G. Mehta, G. G. I. Moore and F. P. Siegal, Tetrahedron Lett., 1974, 15, 2549–2552 CrossRef.
- O. Zeman, V. Pelikan and J. Pachman, ACS Sustainable Chem. Eng., 2022, 10, 4788–4791 CrossRef CAS.
- M. Casutt, B. Dittmar, H. Makowska, T. Marszalek, S. Kushida, U. H. F. Bunz, J. Freudenberg, D. Jänsch and K. Müllen, Chem. – Eur. J., 2019, 25, 2723–2728 CrossRef CAS PubMed.
- A. Iqbal, M. Jost, R. Kirchmayr, J. Pfenninger, A. Rochat and O. Wallquist, Bull. Soc. Chim. Belg., 1988, 97, 615–644 CrossRef CAS.
- S. A. L. Shaikh, S. S. Birajdar, S. D. Ambore, A. L. Puyad, P. Vijayanand, S. V. Bhosale and S. V. Bhosale, Results Chem., 2022, 4, 100473 CrossRef CAS.
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS.
- Q. Liu, S. E. Bottle and P. Sonar, Adv. Mater., 2020, 32, 1903882 CrossRef CAS PubMed.
- X. Song, N. Gasparini, M. M. Nahid, S. H. K. Paleti, C. Li, W. Li, H. Ade and D. Baran, Adv. Funct. Mater., 2019, 29, 1902441 CrossRef.
- K. P. Sokol, W. E. Robinson, J. Warnan, N. Kornienko, M. M. Nowaczyk, A. Ruff, J. Z. Zhang and E. Reisner, Nat. Energy, 2018, 3, 944–951 CrossRef CAS.
- L. Shen, Z. Tang, X. Wang, H. Liu, Y. Chen and X. Li, Phys. Chem. Chem. Phys., 2018, 20, 22997–23006 RSC.
- B. Lim, H. Sun, J. Lee and Y.-Y. Noh, Sci. Rep., 2017, 7, 164 CrossRef PubMed.
- K. Guo, J. Bai, Y. Jiang, Z. Wang, Y. Sui, Y. Deng, Y. Han, H. Tian and Y. Geng, Adv. Funct. Mater., 2018, 28, 1801097 CrossRef.
- E. D. Głowacki, H. Coskun, M. A. Blood-Forsythe, U. Monkowius, L. Leonat, M. Grzybowski, D. Gryko, M. S. White, A. Aspuru-Guzik and N. S. Sariciftci, Org. Electron., 2014, 15, 3521–3528 CrossRef PubMed.
- T. Reiker, Z. Liu, C. Winter, N. F. Kleimeier, D. Zhang and H. Zacharias, J. Phys. Chem. C, 2021, 125, 5572–5580 CrossRef CAS.
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Acc. Chem. Res., 2016, 49, 78–85 CrossRef CAS PubMed.
- R. Diao, H. Ye, Z. Yang, S. Zhang, K. Kong and J. Hua, Polym. Chem., 2019, 10, 6473–6480 RSC.
- M. Li, J. Li, D. Di Carlo Rasi, F. J. M. Colberts, J. Wang, G. H. L. Heintges, B. Lin, W. Li, W. Ma, M. M. Wienk and R. A. J. Janssen, Adv. Energy Mater., 2018, 8, 1800550 CrossRef.
- M. Heydari Gharahcheshmeh and K. K. Gleason, Adv. Mater. Interfaces, 2019, 6, 1801564 CrossRef.
- K. Baba, G. Bengasi, F. Loyer, J. P. C. Fernandes, D. El Assad, O. De Castro and N. D. Boscher, ACS Appl. Mater. Interfaces, 2020, 12, 37732–37740 CrossRef CAS PubMed.
- R. M. Howden, E. J. Flores, V. Bulović and K. K. Gleason, Org. Electron., 2013, 14, 2257–2268 CrossRef CAS.
- D. Bansal, D. Cardenas-Morcoso and N. D. Boscher, J. Mater. Chem. A, 2023, 11, 5188–5198 RSC.
- P. Moni, J. Lau, A. C. Mohr, T. C. Lin, S. H. Tolbert, B. Dunn and K. K. Gleason, ACS Appl. Energy Mater., 2018, 1, 7093–7105 CrossRef CAS.
- G. Bengasi, R. Meunier-Prest, K. Baba, A. Kumar, A. L. Pellegrino, N. D. Boscher and M. Bouvet, Adv. Electron. Mater., 2020, 6, 2000812 CrossRef CAS.
- M. Heydari Gharahcheshmeh, M. T. Robinson, E. F. Gleason and K. K. Gleason, Adv. Funct. Mater., 2021, 31, 2008712 CrossRef CAS.
- B. J. Schwartz, Annu. Rev. Phys. Chem., 2003, 54, 141–172 CrossRef CAS PubMed.
- S. Chaudhry, Y. Wu, Z. Cao, S. Li, J. L. Canada, X. Gu, C. Risko and J. Mei, Macromolecules, 2021, 54, 8207–8219 CrossRef CAS.
- K. Gu and Y.-L. Loo, J. Polym. Sci., Part B: Polym. Phys., 2019, 57, 1559–1571 CrossRef CAS.
- H. Li, M. E. DeCoster, C. Ming, M. Wang, Y. Chen, P. E. Hopkins, L. Chen and H. E. Katz, Macromolecules, 2019, 52, 9804–9812 CrossRef CAS.
- X. Wang, X. Zhang, L. Sun, D. Lee, S. Lee, M. Wang, J. Zhao, Y. Shao-Horn, M. Dincǎ, T. Palacios and K. K. Gleason, Sci. Adv., 2018, 4, eaat5780 CrossRef CAS PubMed.
- G. Bengasi, J. S. Desport, K. Baba, J. P. Cosas Fernandes, O. De Castro, K. Heinze and N. D. Boscher, RSC Adv., 2020, 10, 7048–7057 RSC.
- J. F. J. Ponder, S. A. Gregory, A. Atassi, A. K. Menon, A. W. Lang, L. R. Savagian, J. R. Reynolds and S. K. Yee, J. Am. Chem. Soc., 2022, 144, 1351–1360 CrossRef CAS PubMed.
- G. Bengasi, K. Baba, G. Frache, J. Desport, P. Gratia, K. Heinze and N. D. Boscher, Angew. Chem., Int. Ed., 2019, 58, 2103–2108 CrossRef CAS PubMed.
- G. D. Hale, S. J. Oldenburg and N. J. Halas, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 55, R16069 CrossRef CAS.
- X.-H. Jin, M. B. Price, J. R. Finnegan, C. E. Boott, J. M. Richter, A. Rao, S. M. Menke, R. H. Friend, G. R. Whittell and I. Manners, Science, 2018, 360, 897–900 CrossRef CAS PubMed.
- A. De Sio, X. T. Nguyen and C. Lienau, Z. Naturforsch., A: Phys. Sci., 2019, 74, 721–737 CrossRef CAS.
- P. Roy and J. Dasgupta, Pure Appl. Chem., 2020, 92, 707–716 CrossRef CAS.
- E. Varene and P. Tegeder, Appl. Phys. A: Mater. Sci. Process., 2012, 107, 13–16 CrossRef CAS.
- L. Bogner, Z. Yang, S. Baum, M. Corso, R. Fitzner, P. Bäuerle, K. J. Franke, J. I. Pascual and P. Tegeder, J. Phys. Chem. C, 2016, 120, 27268–27275 CrossRef CAS.
- J. R. Ochsmann, D. Chandran, D. W. Gehrig, H. Anwar, P. K. Madathil, K.-S. Lee and F. Laquai, Macromol. Rapid Commun., 2015, 36, 1122–1128 CrossRef CAS PubMed.
- A. E. Jailaubekov, A. P. Willard, J. R. Tritsch, W.-L. Chan, N. Sai, R. Gearba, L. G. Kaake, K. J. Williams, K. Leung, P. J. Rossky and X.-Y. Zhu, Nat. Mater., 2013, 12, 66–73 CrossRef CAS PubMed.
- A. A. Paraecattil and N. Banerji, J. Am. Chem. Soc., 2014, 136, 1472–1482 CrossRef CAS PubMed.
- T. A. Ford, I. Avilov, D. Beljonne and N. C. Greenham, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 125212 CrossRef.
- B. P. Karsten, R. K. M. Bouwer, J. C. Hummelen, R. M. Williams and R. A. J. Janssen, Photochem. Photobiol. Sci., 2010, 9, 1055–1065 CrossRef CAS PubMed.
- S. Vempati, L. Bogner, C. Richter, J.-C. Deinert, L. Foglia, L. Gierster and J. Stähler, J. Chem. Phys., 2020, 152, 74715 CrossRef CAS PubMed.
- P. Roy, A. Jha, V. B. Yasarapudi, T. Ram, B. Puttaraju, S. Patil and J. Dasgupta, Nat. Commun., 2017, 8, 1716 CrossRef PubMed.
- E. L. Frankevich, A. A. Lymarev, I. Sokolik, F. E. Karasz, S. Blumstengel, R. H. Baughman and H. H. Hörhold, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 9320–9324 CrossRef CAS PubMed.
- P. B. Miranda, D. Moses and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 81201 CrossRef.
- I. Meager, R. S. Ashraf, S. Mollinger, B. C. Schroeder, H. Bronstein, D. Beatrup, M. S. Vezie, T. Kirchartz, A. Salleo, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2013, 135, 11537–11540 CrossRef CAS PubMed.
- M. Kirkus, L. Wang, S. Mothy, D. Beljonne, J. Cornil, R. A. J. Janssen and S. C. J. Meskers, J. Phys. Chem. A, 2012, 116, 7927–7936 CrossRef CAS PubMed.
- O. G. Reid, J. A. N. Malik, G. Latini, S. Dayal, N. Kopidakis, C. Silva, N. Stingelin and G. Rumbles, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 27–37 CrossRef CAS.
- R. Meng and R. Zhu, Sci. Rep., 2022, 12, 10087 CrossRef CAS PubMed.
- N. R. Ávila-Rovelo, G. Martinez, W. Matsuda, S. Sinn, P. Lévêque, D. Schwaller, P. Mésini, S. Seki and A. Ruiz-Carretero, J. Phys. Chem. C, 2022, 126, 10932–10939 CrossRef.
- R. H. Friend, D. D. C. Bradley and P. D. Townsend, J. Phys. D: Appl. Phys., 1987, 20, 1367 CrossRef CAS.
- H. Goktas, X. Wang, N. D. Boscher, S. Torosian and K. K. Gleason, J. Mater. Chem. C, 2016, 4, 3403–3414 RSC.
- M. M. Wienk, M. Turbiez, J. Gilot and R. A. J. Janssen, Adv. Mater., 2008, 20, 2556–2560 CrossRef CAS.
- M. Gora, W. Krzywiec, J. Mieczkowski, E. C. Rodrigues Maia, G. Louarn, M. Zagorska and A. Pron, Electrochim. Acta, 2014, 144, 211–220 CrossRef CAS.
- C. H. Woo, P. M. Beaujuge, T. W. Holcombe, O. P. Lee and J. M. J. Fréchet, J. Am. Chem. Soc., 2010, 132, 15547–15549 CrossRef CAS PubMed.
- R. Feng, N. Sato, T. Yasuda, H. Furuta and S. Shimizu, Chem. Commun., 2020, 56, 2975–2978 RSC.
- K. Baba, G. Bengasi, D. El Assad, P. Grysan, E. Lentzen, K. Heinze, G. Frache and N. D. Boscher, Eur. J. Org. Chem., 2019, 2019, 2368–2375 CrossRef CAS.
- M. Stolte, S.-L. Suraru, P. Diemer, T. He, C. Burschka, U. Zschieschang, H. Klauk and F. Würthner, Adv. Funct. Mater., 2016, 26, 7415–7422 CrossRef CAS.
- J. Kesters, P. Verstappen, M. Kelchtermans, L. Lutsen, D. Vanderzande and W. Maes, Adv. Energy Mater., 2015, 5, 1500218 CrossRef.
- B. Stadtmüller, S. Emmerich, D. Jungkenn, N. Haag, M. Rollinger, S. Eich, M. Maniraj, M. Aeschlimann, M. Cinchetti and S. Mathias, Nat. Commun., 2019, 10, 1470 CrossRef PubMed.
- C. M. Mauck, P. E. Hartnett, Y.-L. Wu, C. E. Miller, T. J. Marks and M. R. Wasielewski, Chem. Mater., 2017, 29, 6810–6817 CrossRef CAS.
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749–11761 CrossRef CAS PubMed.
- D. W. Polak, M. T. do Casal, J. M. Toldo, X. Hu, G. Amoruso, O. Pomeranc, M. Heeney, M. Barbatti, M. N. R. Ashfold and T. A. A. Oliver, Phys. Chem. Chem. Phys., 2022, 24, 20138–20151 RSC.
- R. Shivhare, G. J. Moore, A. Hofacker, S. Hutsch, Y. Zhong, M. Hambsch, T. Erdmann, A. Kiriy, S. C. B. Mannsfeld, F. Ortmann and N. Banerji, Adv. Mater., 2022, 34, 2101784 CrossRef CAS PubMed.
- A. M. Levine, G. He, G. Bu, P. Ramos, F. Wu, A. Soliman, J. Serrano, D. Pietraru, C. Chan, J. D. Batteas, M. Kowalczyk, S. J. Jang, B. L. Nannenga, M. Y. Sfeir, E. H. R. Tsai and A. B. Braunschweig, J. Phys. Chem. C, 2021, 125, 12207–12213 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- P. E. Hartnett, E. A. Margulies, C. M. Mauck, S. A. Miller, Y. Wu, Y.-L. Wu, T. J. Marks and M. R. Wasielewski, J. Phys. Chem. B, 2016, 120, 1357–1366 CrossRef CAS PubMed.
- L. Wang, W. Jiang, S. Guo, S. Wang, M. Zhang, Z. Liu, G. Wang, Y. Miao, L. Yan, J.-Y. Shao, Y.-W. Zhong, Z. Liu, D. Zhang, H. Fu and J. Yao, Chem. Sci., 2022, 13, 13907–13913 RSC.
- A. C. Rosenfeldt, B. Göhler and H. Zacharias, J. Chem. Phys., 2010, 133, 234704 CrossRef PubMed.
- T. Wang and W.-L. Chan, J. Phys. Chem. Lett., 2014, 5, 1812–1818 CrossRef CAS PubMed.
- J. Li, H. Cao, Z. Zhang, S. Liu and Y. Xia, Photonics, 2022, 9, 689 CrossRef CAS.
- R. Tautz, E. Da Como, T. Limmer, J. Feldmann, H.-J. Egelhaaf, E. von Hauff, V. Lemaur, D. Beljonne, S. Yilmaz, I. Dumsch, S. Allard and U. Scherf, Nat. Commun., 2012, 3, 970 CrossRef PubMed.
- B. S. Rolczynski, J. M. Szarko, H. J. Son, Y. Liang, L. Yu and L. X. Chen, J. Am. Chem. Soc., 2012, 134, 4142–4152 CrossRef CAS PubMed.
- M. Mirabedin, H. Vergnes, N. Caussé, C. Vahlas and B. Caussat, Appl. Surf. Sci., 2021, 554, 149501 CrossRef CAS.
- F. Muralter, A. M. Coclite and K. K. S. Lau, Adv. Electron. Mater., 2021, 7, 2000871 CrossRef CAS.
- S. Kaviani, M. Mohammadi Ghaleni, E. Tavakoli and S. Nejati, ACS Appl. Polym. Mater., 2019, 1, 552–560 CrossRef CAS.
- S. Stas, S. Sergeyev and Y. Geerts, Tetrahedron, 2010, 66, 1837–1845 CrossRef CAS.
- A. B. Tamayo, M. Tantiwiwat, B. Walker and T.-Q. Nguyen, J. Phys. Chem. C, 2008, 112, 15543–15552 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00197k |
| This journal is © The Royal Society of Chemistry 2023 |