
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cross-linkable and resorbable PEDOT-based ink using a hyaluronic acid derivative as dopant for flexible bioelectronic devices†
Maxime
Leprince
ab,
Simon
Regal
c,
Pascal
Mailley
a,
Fabien
Sauter-Starace
c,
Isabelle
Texier
 *a and
Rachel
Auzély-Velty
b
*a and
Rachel
Auzély-Velty
b
aUniv. Grenoble Alpes, CEA, LETI, DTBS, F-38000 Grenoble, France. E-mail: isabelle.texier-nogues@cea.fr
bUniv. Grenoble Alpes, CNRS, CERMAV, F-38000 Grenoble, France
cUniv. Grenoble Alpes, CEA, LETI, CLINATEC, F-38000 Grenoble, France
First published on 28th July 2023
Abstract
Transient soft electronics that eliminate secondary surgery for device removal and enable effective integration into the body show great promise for diagnostic and therapeutic applications. Here, a novel cross-linkable and resorbable poly(3,4-ethylenedioxy)thiophene (PEDOT)-based ink using a hyaluronic acid (HA) derivative as dopant was designed and used to build a flexible and resorbable all-organic bioelectronic device. The HA derivative possesses structural features that enable it to degrade in few weeks, to serve as a dopant of PEDOT and to prepare water-resistant conductive patterns by photo-induced thiol-ene cross-linking. This cross-linking method, using poly(ethylene glycol) as crosslinker, afforded films with an initial conductivity of 0.6 S cm−1, which could be enhanced up to 4.6 S cm−1 after successive wetting/drying cycles. The resulting cross-linked films did not present cytotoxic effect for fibroblasts. Furthermore, the ink could be formulated to be inkjet-printed to create conductive tracks with sinusoidal wave patterns and good uniformity on a degradable poly(L-lactide-co-glycolide) (PLGA) film. PEDOT-based micropatterns embedded in PLGA could withstand flexion and torsion without breaking the conductivity, and could maintain conductivity properties until degradation of the device (at ∼45 days).
1. Introduction
Soft flexible bioelectronics, interfacing with the biological world, have attracted considerable interest in healthcare applications ranging from the monitoring to diagnosis and therapy.1–4 From materials point of view, soft bioelectronic devices are composed of inorganic materials, metals or conducting polymers, as well as soft substrates/encapsulants. In addition to flexibility, required for improving contact and interaction with biological tissues, these devices should provide stable electronic communication with the surrounding biological systems, at least within the prescribed time frames if there are intended to degrade. Such devices, that can be safely absorbed by the body after they have completed their therapeutic or diagnostic functions (transient bioelectronics), become appealing for medical applications as they eliminate the risks related to surgical retrieval and reduce the chronic foreign body reaction.2,5–7Among materials used to establish electronic communication, conducting polymers (CPs) have become a popular choice due to their mixed ionic and electronic conductivity.3,8,9 This indeed gives them the ability to convert ionic signals, typical of the biological world, to electronic signals, unlike inorganic materials, which are not permeable to ions. The predominant CP used in the fabrication of bioelectronic devices consists in poly(3,4-ethylenedioxythiophene) doped with poly(styrene sulfonate) (PEDOT:PSS).10 PEDOT:PSS is commercially available as waterborne dispersion (“ink”) with tunable conductivity, chemical stability, and easy processability.11 As a flexible versatile material, PEDOT:PSS can be processed using various printing techniques such as inkjet, extrusion, screen printing and its combination with other polymers, solvents and/or conductive materials enables to tune its functionality and conductive properties for a wide range of (bio)-electronic applications.12–15 Moreover, previous reports have demonstrated in vitro biocompatibility of PEDOT:PSS.16 However, its long-term in vivo toxicity and biodegradation, with potential release of acidic PSS degradation products, are still unclear in spite of the use of PEDOT:PSS as a functional coating of metal electrodes to record human electrophysiological signals.17–19 In order to circumvent potential problems associated with PSS, we recently designed a hyaluronic acid (HA)-based dopant to produce a conductive and resorbable PEDOT-based ink.20 HA was selected as it is a natural polysaccharide ubiquitous in the body and widely used for biomedical applications.21 Its modification with both sulfates and phenylboronic acid (PBA) moieties, mimicking the chemical functions of PSS, resulted in a HAS-PBA derivative that could efficiently dope the PEDOT chains and HAS could degrade in few weeks in physiological conditions. In addition, the new PEDOT:HAS-PBA ink was found to be biocompatible and could be easily processed by inkjet printing.
However, in bioelectronics, conductive materials are exposed to wet media. Therefore, for PEDOT:HAS-PBA to be suitable for bioelectronics, it should be stable in such conditions, and keep its conductive properties unchanged when wet. In the case of PEDOT:PSS, cross-linking is necessary to prevent delamination and re-dispersion of conducting films placed in aqueous physiological conditions for long-term applications.22,23 An usual cross-linking strategy of PEDOT-PSS films relies on the use of methoxysilane-based molecules such as (3-glycidyloxypropyl)trimethoxysilane (GOPS).22,24,25 However, GOPS cross-linking requires high curing temperatures (140 °C for 1 h), which are not compatible with fragile biomolecules like HA. Moreover, it has negative impact on the conductivity values, due to the natural insulating properties of the siloxane network.22,25
Based on this, we hypothesized that introducing photo-reactive cross-linking groups to HAS-PBA backbone would circumvent these limitations for preparing degradable cross-linked conductive PEDOT:HAS-PBA structures. Therefore, we synthesized an alkene functional HAS-PBA derivative (HAS-PBA-PEGene) by introducing alkene groups on HAS-PBA through an oligo(ethylene glycol) spacer arm for further photo-induced thiol-ene cross-linking.
We herein describe the chemical design of this cross-linkable and resorbable conductive ink and its use in the fabrication of a flexible and resorbable all-organic micro-patterned bioelectronic device by combining inkjet-printing and photo-induced thiol-ene cross-linking on a degradable poly(L-lactide-co-glycolide) (PLGA) film (Fig. 1). PLGA was selected as soft substrate and encapsulation layer as it is an important polymer matrix used for devices aimed to applications with finite implant life-time.26 We demonstrate the functionality and degradability of the bioelectronic device by performing conductivity measurements over time.
 | ||
| Fig. 1 Design of degradable cross-linked conductive patterns based on PEDOT:HAS-PBA-PEGene on a flexible resorbable PLGA sheet by combining inkjet-printing and photo-induced thiol-ene cross-linking. | ||
2. Experimental
2.1. Materials
Sodium hyaluronate (Mw = 219 kg mol−1, Đ = 1.4) was obtained from Lifecore Biomedical (Chaska, Minesotta, USA). 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMTMM), 3-aminophenylboronic acid hemisulfate salt (3APBA), pentenoic anhydride, N,N-diisopropylethylamine, 3,4-ethylenedioxythiophene (EDOT), dodecylbenzene sulfonic acid (DBSA), ammonium persulfate (APS), phosphate buffered saline (PBS), 3-mercaptopropyltrimethoxysilane (MPTMS), Resomer® RG 504 poly(D,L-lactide-co-glycolide) (PLGA), lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP), 1,2-dithiothreitol (DTT) and O-(2-aminoethyl)-O′-[2-(Boc-amino)ethyl]deca(ethylene glycol), were obtained from Merck (Saint-Quentin Fallavier, France). Hydrochloric acid and glycerol were purchased from Fischer Scientific and iron(II) sulfate heptahydrate (FeSO4), from Alfa Aesar. Poly(ethylene glycol) bis(thiol) (Mn = 3500 g mol−1) was purchased from Interchim. All the organic solvents were provided from VWR International (Fontenay-sous-Bois, France). All reagents were used as received without further purification. The water used in all the experiments was purified by an Elga Purelab purification system. The sulfated HA sample (HAS) with a DS of 4 (i.e. 4 sulfate groups per repeating disaccharide unit) was synthesized as previously described.20 The heterobifunctional oligo(ethylene glycol) spacer arm with an alkene group at one end, and an amine group at the other end (referred to as “PEGene”) was synthesized as described in the ESI† (Fig. S1).2.2. Synthesis of polymers and surface modifications
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm using an Ultra Turrax T-10 basic disperser with a S 10 N-8G dispersing tool (Roth, Karlsruhe, Germany). After mixing, the solution was stirred at room temperature under nitrogen until the pH was stable and below 1.4–1.5 (∼18 h). The medium was dialyzed against deionized water with a 6–8 kDa dialysis membrane (Spectrum Laboratories, CA, USA). Then, the pH was adjusted to 7.4 by addition of 1 M NaOH. The medium was then filtered successively through 3 μm, 1.2 μm and 0.8 μm cellulose acetate membrane filters, to remove large aggregates. The PEDOT:HAS-PBA was then recovered by freeze-drying as a deep blue powder, with a yield of ∼94%.
000 rpm using an Ultra Turrax T-10 basic disperser with a S 10 N-8G dispersing tool (Roth, Karlsruhe, Germany). After mixing, the solution was stirred at room temperature under nitrogen until the pH was stable and below 1.4–1.5 (∼18 h). The medium was dialyzed against deionized water with a 6–8 kDa dialysis membrane (Spectrum Laboratories, CA, USA). Then, the pH was adjusted to 7.4 by addition of 1 M NaOH. The medium was then filtered successively through 3 μm, 1.2 μm and 0.8 μm cellulose acetate membrane filters, to remove large aggregates. The PEDOT:HAS-PBA was then recovered by freeze-drying as a deep blue powder, with a yield of ∼94%.
2.3. Ink film deposition on glass slides.
Slides were washed with acetone and ethanol. Lyophilized PEDOT-based ink was dispersed in water at 13 g L−1 and the suspension was vigorously stirred for 10 min and sonicated in an ultrasound bath for 30 s. 60 μL of suspension were deposited on a glass slide inside a silicone ring of 1 cm inner diameter. The ink was dried at room temperature, then the silicone ring was removed from the glass substrate.2.4. Ink formulation for inkjet printing
A water solution “S” containing DBSA (2 g L−1) and glycerol (2.3 vol%) was prepared. PEDOT:HAS-PBA-PEGene (10 mg) was dissolved in S (932.4 μL). The resulting mixture was then mixed with 17.6 μL of a solution of PEG-(SH)2 in water (100 g L−1) ([thiol]/[alkene] = 0.9), and 50 μL of a solution of LAP in water (20 g L−1). The surface tension of the ink formulation thus obtained was measured with the pending drop technique as 32 ± 0.4 mN m−1. Its shear viscosity was found to be 0.02 Pa.s at 10 s−1 and 25 °C. The ink was vortexed and sonicated for 30 s in an ultrasound bath before printing.2.5. Preparation of PLGA thin film
The fabrication of the PLGA thin film was adapted from the protocol described by Nakamura et al.28 Briefly, 295 mg of PLGA was dissolved in acetone solution at 5 wt%. The solution was stirred for 30 min to ensure complete dissolution of the PLGA, and poured in a 100 mm diameter aluminum-weighing dish. The solution was cured at 60 °C for 1 h to achieve homogenous 20 μm thick PLGA film. After curing, the film was peeled off and cut for the printing.2.6. Characterization of materials
2.7. Characterization of the formulated ink
A TA Instruments Rheometer AR 2000ex with a cone-plate geometry (cone 4 cm–4°) was used to measure the shear viscosity of the formulated PEDOT:HAS-PBA-PEGene/PEG-(SH)2 ink. Shear viscosity experiments were performed in steady-state flow mode.The surface tension was measured with a Krüss Drop Shape Analyzer DSA100 using the pendant drop method and a Young–Laplace model.
2.8. Inkjet printing
The conductive tracks were printed using a Dimatix Material Printer DMP-2831 from Fujifilm with 10 pL droplet size cartridges. After vortexing the ink and 30 sec of sonication, the cartridge was filled with 1 mL of PEDOT:HAS-PBA-PEGene/PEG-(SH)2 formulation. Conductive lines, with a total length of 1.55 cm and a width of 0.3 mm, were printed on PLGA film (Fig. 2(A)). Just before the printing, the PLGA film was activated by O2 plasma at 250 W for 5 min (MVD100Applied MST, San Jose, USA). The PEDOT:HAS-PBA-PEGene/PEG-based ink was printed with a 25 μm drop spacing, corresponding to a printer resolution of 1016 dots per inch (dpi). This value was optimized for the superposition of individual droplets to form a continuous layer, which is desirable to obtain low resistance tracks. After printing 50 layers, the tracks were exposed to UV light (λ = 405 nm, power = 75 mW cm−2) for 1 min 30 (UVA-Hand, Honle and dried overnight (Fig. 2(B))).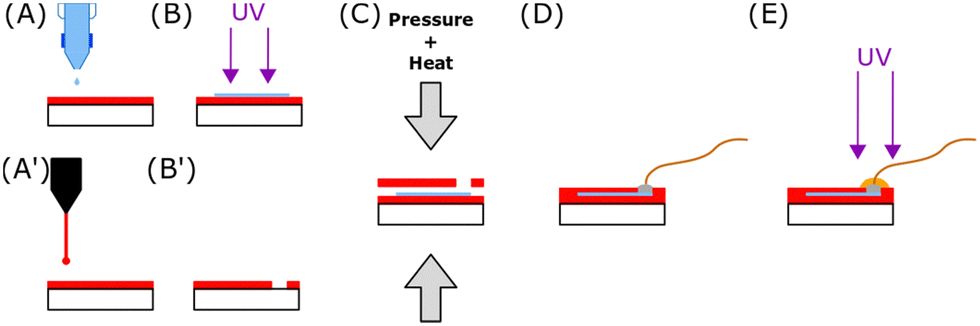 | ||
| Fig. 2 Device fabrication process. (A) Inkjet printing of PEDOT:HAS-PBA-PEGene ink on PLGA film. (B) UV cross-linking of the deposited film and drying overnight. (A′) Laser patterning of a second PLGA layer to create contact opening. (B′) PLGA layer with opening for electrical contact. (C) Thermal and pressure sealing process of the device. (D) Connection of a stainless steel wire with conductive epoxy. (E) Insulation of the electrical contact with an epoxy glue. | ||
2.9. Device fabrication
Opening was performed with laser cutting (Speedy 400 Trotec, Austria) on a second PLGA layer (Fig. 2(A′) and (B′)). Both PLGA layers were manually aligned. Homemade press was used to seal the two films together in an oven at 100 °C for 5 min (Fig. 2(C)). The applied torque was 10 Nm. A conductive epoxy (ELECOLIT 3025, Panacol-Elosol GmbH, Germany) was applied between the printing pads and a 100 μm diameter stainless steel wire (Fig. 2(D)). The epoxy was cured for 24 h at room temperature. A UV insulating epoxy (EpoTek OG116-31, Epoxy Technology, Inc., United states) was applied onto the conductive epoxy. The epoxy was exposed to UV light for 1 min 30 (λ = 405 nm, power = 75 mW cm−2) (Fig. 2(E)). The other extremity of the stainless steel wire was welded to a connector after immersion for 3 s in Indalloy flux #2 (Indium corporation, United State), using Indalloy 121 tin (Indium corporation, United State).2.10. Cell viability
The experiments were performed in compliance with the 10993-5 standard. The samples were put in contact with Dulbecco's modified Eagle's medium (DMEM) without phenol red in a glass container under agitation. The ratio of sample to extraction medium was 6 cm2 mL−1, and three samples/condition (n = 3) were used. First, medium extraction was performed after 24 h of contact with the sample at 37 °C and 5% CO2, followed by sampling and medium replacement every 3 days for 30 days.The metabolic activity, and hence the viability, of dermal fibroblasts in the presence of the extracted media was assessed by a colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. 96-Well plates were seeded with 5000 fibroblasts per well (n = 10 wells per condition). After 24 h of culture, 100 μL of the extracted media were added per well at 37 °C for 24 hours. After incubation, the medium was removed and replaced by a 0.5 mg mL−1 tetrazolium salt (MTT) solution. The plates were incubated at 37 °C and 5% CO2. After 4 h, the MTT solution was withdrawn and the formazan crystals were dissolved in dimethyl sulfoxide (DMSO). Finally, the optical densities at 550 nm were measured using a microplate reader (Multiskan™ FC, ThermoScientific™). The absorbance at 550 nm was directly proportional to the number of living fibroblasts, and the results (mean ± SEM) expressed in percentage of viability compared to controls.
2.11. Characterization of electrode degradation
Printed devices were immersed, excepted the electrical contact, in water at 37 °C for 48 days. The water level was kept constant during the entire measurement period. The electrical resistance of the tracks was measured every 4 h with 34972A LXI Data Acquisition (Keysight, United State).3. Results and discussion
3.1. Synthesis of HAS-PBA-PEGene
For the synthesis of HAS-PBA-PEGene, we used an heterobifunctional oligo(ethylene glycol) spacer arm with a pentenamide group at one end, and an amine group at the other end (Fig. 3(a)). The presence of this hydrophilic spacer appeared to be necessary to overcome the steric hindrance of the sulfate groups of the HAS derivative possessing a degree of sulfation (DSs) of 4. Indeed, our first attempt to cross-link HAS-PBA esterified with pentenoate29 by a photoinduced thiol-ene reaction using a poly(ethylene glycol)-bis(thiol) derivative (PEG-(SH)2) as a cross-linker was unsuccessful. The PEGene derivative was synthesized in two steps from commercial O-(2-aminoethyl)-O′-[2-(Boc-amino)ethyl]deca(ethylene glycol) (H2N-(EG)11-NH-Boc) as described in the ESI† (Fig. S1). PEGene was grafted on HAS simultaneously with 3APBA by an amide coupling reaction using DMTMM as a coupling agent. The coupling yields of PEGene and 3APBA (40% and ∼100%, respectively) were the same as those obtained when these amine derivatives were grafted separately. The resulting HAS-PBA-PEGene conjugate was isolated in a 90% yield after purification by a diafiltration process and freeze-drying. Successful grafting of PEGene and 3APBA on HA was confirmed by 1H NMR spectroscopy (Fig. S2, ESI†), which also allowed the determination of the degrees of substitution in PEGene and 3APBA. DS values of 0.3 and 0.17 were found for PBA and PEGene, respectively. | ||
| Fig. 3 Synthesis of the PEDOT:HAS-PBA-PEGene ink. (a) Functionalization of HAS-PBA with PEGene by an amine–acid coupling reaction. (b) Oxidative polymerization of EDOT in the presence of HAS-PBA-PEGene allowing to obtain the PEDOT:HAS-PBA-PEGene ink. (c) Photograph of the PEDOT:HAS-PBA-PEGene ink suspension in water. (d) Relationship between the (EDOT + HAS-PBA-PEGene) concentration during the PEDOT polymerization and the average diameter of PEDOT:HAS-PBA-PEGene particles. | ||
3.2. Oxidative polymerization of EDOT in the presence of HAS-PBA-PEGene and cross-linking
In PEDOT:PSS, ethylenedioxythiophene (EDOT) is oxidatively polymerized in the presence of PSS whose function is to stabilize the p-doped cationic PEDOT and allow the dispersion of PEDOT in water by formation of a polyelectrolyte complex. Herein, a similar procedure was applied to synthesize the PEDOT/HAS-PBA-PEGene ink. The polymerization conditions were derived from those optimized for the synthesis of the PEDOT/HAS-PBA ink.20 HAS-PBA-PEGene and EDOT were first dissolved in water. The molar ratio EDOT/HAS-PBA-PEGene was set to 4, which corresponds to two EDOT per saccharide unit. The ammonium persulfate (APS) oxidant and the iron II sulfate catalyst were then added (Fig. 3(b)). During polymerization, the reaction medium became deep blue as a result of PEDOT formation (Fig. 3(c)). The polymerization was considered as finished when the pH became constant, usually ∼1.5, within 18 h. After pH neutralization, purification by dialysis, and freeze-drying, the ink was recovered as a solid deep blue powder with a yield comprised between 80 and 95%. DLS (Dynamic Light Scattering) analysis of the PEDOT:HAS-PBA-PEGene ink at a concentration of 0.08 g L−1 in water revealed that the EDOT + HAS-PBA-PEGene concentration during ink polymerization had a significant impact on the final size of ink dispersion particles (Fig. 3(d)). The results obtained from DLS analysis led us to conclude that the ink prepared using an (EDOT + HAS-PBA-PEGene) concentration of 20 g L−1 was suitable for inkjet printing as it exhibited particle size of 370 nm.To investigate the cross-linking ability of HAS-PBA-PEGene, this compound was mixed in aqueous solution with poly(ethylene glycol)bis-thiol (PEG-(SH)2) as a cross-linker and a water-soluble photoinitiator (LAP), and exposed to UV-light (λ = 405 nm) after deposition onto a glass slide without surface modification. After drying overnight and immersion in water, a free-standing film was obtained (Fig. 4(a)), which confirmed successful cross-linking of HAS-PBA-PEGene.
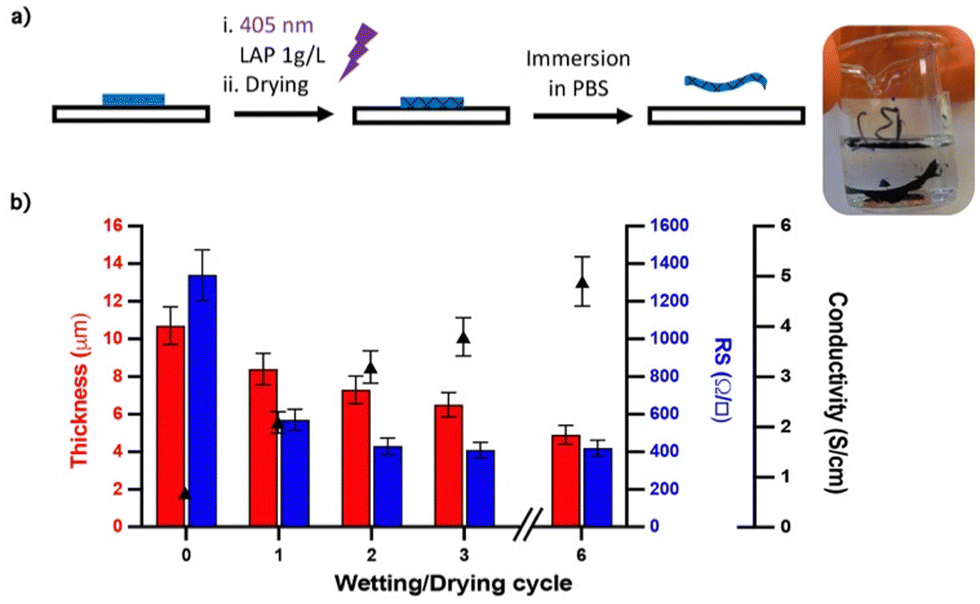 | ||
| Fig. 4 Synthesis and conductivity properties of cross-linked PEDOT:HAS-PBA-PEGene films. (a) UV cross-linking of PEDOT:HAS-PBA-PEGene films. (b) Effect of wetting and drying cycles on the conductivity and thickness of cross-linked PEDOT:HAS-PBA-PEGene/PEG films. | ||
3.3. Conductivity of PEDOT:HAS-PBA-PEGene before and after cross-linking
We first measured the conductivity of films based on PEDOT:HAS-PBA-PEGene alone and cross-linked with PEG-(SH)2 using the 4-point probe method.30 Interestingly, the film based on PEDOT:HAS-PBA-PEGene alone displayed a conductivity σ of 3.8 S cm−1, better than that of PEDOT:HAS-PBA (σ = 1.5 S cm−1).20 Previous reports on the addition of PEG to PEDOT:PSS have shown a similar beneficial effect with conductivity enhancement.31 After cross-linking, the conductivity was decreased to 0.6 S cm−1. This result could be attributed to two main reasons. First, it may be related to a morphology change caused by the cross-linking of the dopant chains leading to a reduced charge carrier mobility, as reported by previous studies on PEDOT:PSS cross-linking.22,25 Second, the addition of PEG cross-linker inside the sample could reduce the electrical conductivity of the film by acting as an insulating fraction.3.4. Effect of wetting and drying cycles on the conductivity of cross-linked PEDOT:HAS-PBA-PEGene/PEG films
To investigate the effect of wetting/drying cycles on film conductivity, the PEDOT:HAS-PBA-PEGene film was simultaneously cross-linked and covalently immobilized on a glass slide functionalized with thiol functions via thiol-ene coupling. After drying overnight, the grafted film was immersed in water for 1 h, then dried, several times. The film thickness and conductivity were measured after each wetting/drying cycle. As shown in Fig. 4(b), with increasing number of wetting/drying cycles, the film conductivity significantly increased. This increase of film conductivity appeared to be due not only to a reduction of its thickness, from 10.7 μm to 4.9 μm, but also to the rapid decrease of sheet resistance after the first cycle (Fig. 4(b)). These combined results of improved conductivity, reduced thickness and sheet resistance, indicate partial removal of excess HAS-PBA-PEGene and/or PEG cross-linker together with re-ordering and conformational changes of PEDOT chains. To further clarify these mechanisms, atomic force microscopy (AFM), X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy experiments were conducted.Fig. 5(a) and (b) show AFM topology images of cross-linked PEDOT:HAS-PBA-PEGene/PEG films before and after immersion in water and drying. While the surface of the pristine film was relatively flat and covered with grains of different sizes, it appeared much more “wavy”, and without grains, after the wetting/drying step. These images thus reveal clear morphological changes of the film, which could be attributed to partial removal of HAS-PBA-PEGene associated with a rearrangement of the polymer chains and enhanced clustering into PEDOT domains.
 | ||
| Fig. 5 Characterization of cross-linked PEDOT:HAS-PBA-PEGene/PEG films before and after immersion in water and drying. (a) AFM images of films before (left) and after immersion in water and drying (right). (b) S(2p) XPS spectra and (c) Raman spectra of films before and after immersion in water and drying. | ||
The assumption of HAS-PBA-PEGene removal by immersion in water was supported by XPS experiments. Fig. 5(c) displays the XPS spectra of cross-linked PEDOT:HAS-PBA-PEGene/PEG films with no treatment and after one wetting/drying cycle. S(2p) peaks were observed in both films at the binding energy of 166.2 and 162.2 eV. The peak at 166.2 eV corresponds to the sulfur signal from the sulfate of HAS-PBA-PEGene while that at 162.2 eV corresponds to the sulfur signals from the thioether of the PEG cross-linker and of PEDOT thiophene.32 The calculated surface ratio of HAS-PBA-PEGene to PEDOT/PEG cross-linker for the film with and without wetting/drying cycle was 1.80 and 0.98, respectively, showing that HAS-PBA-PEGene content was markedly reduced by 46% as a result of the wetting/drying cycle.
Finally, Raman spectroscopy demonstrated that the polymeric structure was also affected by wetting and drying treatments. Fig. 5(d) shows the Raman spectra of the cross-linked PEDOT:HAS-PBA-PEGene/PEG films before and after a wetting/drying cycle. The band between 1400 and 1450 cm−1 is due to Cα![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cβ symmetric stretching vibration.33 The 1425.2 cm−1 peak for the pristine film was red-shifted to 1423.4 cm−1 after treatment. The same phenomenon was observed with ethylene glycol-treated PEDOT:PSS films.34 The red shifting of the Raman spectra indicates that the resonant structure of the PEDOT chain was changed from a benzoid to a quinoid structure. The benzoid structure may be the favorite structure for a coil conformation and the quinoid structure may be the favorite structure for a linear or expanded-coil structure.35 At the molecular level, the conductivity enhancement observed after wetting/drying may be due to the conformational change of PEDOT chains in the PEDOT:HAS-PBA-PEGene/PEG film, from the benzenoid to the quinoid structure.
Cβ symmetric stretching vibration.33 The 1425.2 cm−1 peak for the pristine film was red-shifted to 1423.4 cm−1 after treatment. The same phenomenon was observed with ethylene glycol-treated PEDOT:PSS films.34 The red shifting of the Raman spectra indicates that the resonant structure of the PEDOT chain was changed from a benzoid to a quinoid structure. The benzoid structure may be the favorite structure for a coil conformation and the quinoid structure may be the favorite structure for a linear or expanded-coil structure.35 At the molecular level, the conductivity enhancement observed after wetting/drying may be due to the conformational change of PEDOT chains in the PEDOT:HAS-PBA-PEGene/PEG film, from the benzenoid to the quinoid structure.
3.5. Inkjet printing on glass versus biopolymer film
The fabrication of the PEDOT:HAS-PBA-PEGene micropatterns on PLGA substrate exploited inkjet printing, as it is a cost-effective and mask-less process, and enables the printing of water-based conductive inks without damaging degradable polyester substrates.36 In this process, the physical properties of the ink, such as the surface tension and the rheological properties (i.e. shear viscosity), have a significant impact on the printability behavior. Therefore, the PEDOT:HAS-PBA-PEGene/PEG-(SH)2 formulation was optimized to be printed with our Dimatix DMP-2800 inkjet printer using conditions similar to those used for the PEDOT:HAS-PBA ink.20 In addition to adding DBSA as a surfactant, glycerol was incorporated in the formulation to prevent premature drying of the ink in the printhead nozzles.All printed devices were composed of 50 layers and were exposed to UV light (λ = 405 nm) and dried overnight. First, 7 × 7 mm2 squares were printed on glass slides and PLGA films to measure the conductivity of the ink. After printing, the conductivity was decreased to 0.05 S cm−1. This result can be attributed to two main reasons. First, the addition of glycerol in the solution, which is not evaporated during device drying at room temperature, could act as an insulator within the conductive matrix. Second, the printed technique created micro cracks in the film during drying (see ESI,† Fig. S3).
Fig. 6(a) and (b) show conductive tracks with straight and serpentine shapes, respectively, printed onto PLGA. The linewidth of the serpentine and straight tracks on the PLGA film was measured as about 291 and 360 μm, respectively, with good uniformity. The thickness of the 50 layer-polymer tracks was 1.13 μm (i.e., average height per layer of 22.6 nm). Fig. 6(c) displays the complete device after the packaging process (see Experimental section). The thickness of the device was about 40 μm. Optical digital microscopy images of a 500 μm conductive tracks at higher magnification showed that they remained uniform after encapsulation (Fig. S4, ESI†). Fig. 6(d) shows the advantage of using an all-polymeric device to withstand flexion (curvature radius in the order of 10 mm) without breaking the conductivity.
 | ||
| Fig. 6 Conductive track printed on PLGA substrates. (a) Serpentine line (the insert is a zoom on the track); (b) straight line; (c) completed device after fabrication; (d) photographic images of device under bending. | ||
3.6. Biocompatibility, stability, and biodegradability of cross-linked PEDOT:HAS-PBA-PEGene/PEG tracks onto PLGA films
L929 mouse fibroblasts showed good cell viability (>80%) on cross-linked PEDOT:HAS-PBA-PEGene/PEG films coated onto PLGA over 4 weeks, with no significant difference with the control (PLGA) (Fig. S5, ESI†). Next, in order to assess the stability and biodegradability of the device in aqueous environment, measurements of the electrical resistance were performed for a period of 48 days. Fig. 7(a) shows the variation of the electrical resistance over time for two devices. The good stability of the conductive track was observed up to 45 days after the beginning of the immersion. This step was followed by a sudden increase of the electrical resistance, correlated to a fast degradation of the device, caused by the bulk erosion of PLGA. Indeed, PLGA undergoes homogeneous erosion via hydrolysis of the ester linkages in the polymer backbone for a prolonged period, followed by a sudden mass loss.37–39 This degradation was visually confirmed by Fig. 7(b), which shows photographs of a device collected at various times after immersion in water at 37 °C. We notice that the device was totally degraded at 48 days in physiological conditions. The observed degradation time scale of the device was consistent with previous reports for films based on PLGA with a lactide/glycolide ratio of 50/50.38 The hydrolysis of ester bonds in PLGA backbone could also be accompanied by auto-hydrolysis of HAS-PBA-PEGene that cleaved the polysaccharide chain into smaller oligomers as reported previously,20 but with a time lag. Although the degradation rate of HAS-PBA-PEGene in aqueous solution could not be determined exactly, it may be similar to its HAS precursor (40 ± 10% in two weeks),20 as it is related to the presence of sulfate groups on HA. However, its degradation rate could be modulated by the PLGA encapsulation and electrostatic complexation with positively charged PEDOT.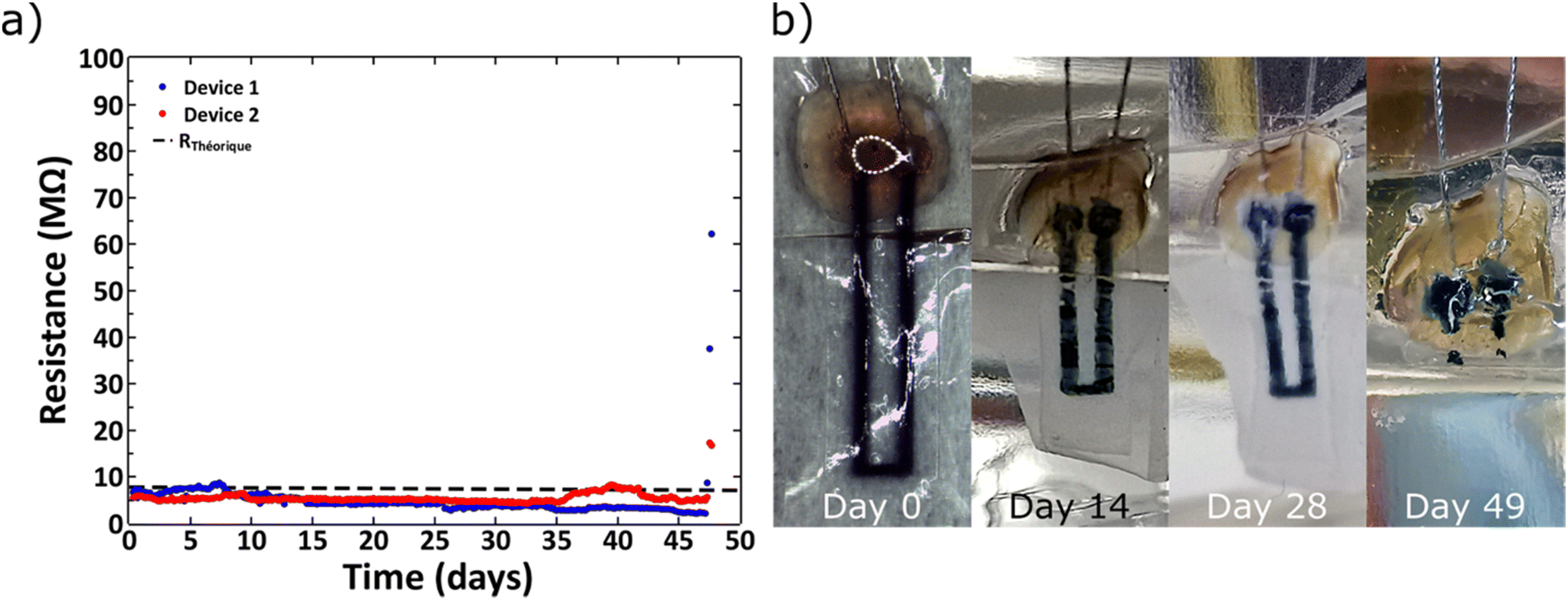 | ||
| Fig. 7 Electrical characterization of the device. (a) Evolution of the resistance during the device immersion in water at 37 °C. The dotted line indicates the theoretical resistance calculated from the device geometric parameters and the conductive properties of the ink; (b) photographic images of the evolution of the device degradation over time. | ||
The measured resistance before hydration was 7.30 and 5.03 MΩ for the devices 1 and 2, respectively. These values were consistent with the theoretical resistance (RTh) calculated with the following formula:
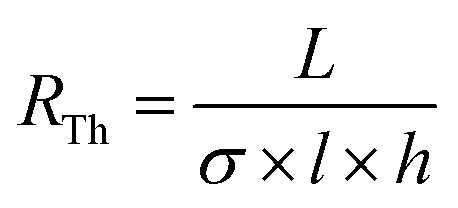
A rapid increase of the electrical resistance was observed at ∼45 days of degradation time, at which point the structural integrity of devices was lost.
4. Conclusions
We successfully developed a cross-linkable, printable, and resorbable PEDOT ink, using a suitably modified HA derivative. The resulting UV cross-linked films using PEG as cross-linker did not solubilize in aqueous solution and did not present cytotoxic effects on fibroblasts. Conductivity measurements showed a beneficial effect of PEG used as cross-linker, and a conductivity enhancement after wetting/drying. In addition, well-defined micro-patterns of PEDOT:HAS-PBA-PEGene could be created on PLGA films by inkjet-printing, allowing the fabrication of flexible and resorbable micro-patterned bioelectronic devices. Indeed, conducting tracks embedded in PLGA could withstand at least flexion and torsion related to handling without breaking the conductivity, and could maintain conductivity properties until degradation of the device. All together, these results show that this new ink may have promising applications as interface material for biological signal measurements by transient devices. In the future, the ink could also be used for conductive tracks after an increase of its intrinsic conductivity with an adapted post-treatment.Author contributions
Maxime Leprince: investigation, writing – original draft. Simon Regal: investigation, writing – original draft. Pascal Mailley: conceptualization, writing – review & editing. Fabien Sauter-Starace: conceptualization, validation, writing – review & editing, supervision. Isabelle Texier: conceptualization, validation, writing – review & editing, supervision. Rachel Auzély-Velty: conceptualization, validation, writing – original draft, writing – review & editing, supervision. All authors have read and given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the French National Research Agency in the framework of the STRETCH project (ANR-8-CE19-0018-01). LETI-DTBS and CERMAV are supported by the French National Research Agency in the framework of the LabEx Arcane (grant ANR-17-EURE-0003) and Glyco@Alps (ANR-15-IDEX-02) programs. We acknowledge the help of Denis Mariolle, Lukasz Borowik, and Denis Rouchon, respectively for AFM, XPS, and Raman spectroscopy analysis performed at the Platform for Nano Characterization (PFNC) at CEA-LETI. We also thank I. Jeacomine (CERMAV-CNRS) for her support at the NMR platform of ICMG (FR2607).References
- G. Balakrishnan, J. Song, C. Mou and C. J. Bettinger, Adv. Mater., 2022, 34, 2106787 CrossRef CAS PubMed.
- O. Bettucci, G. M. Matrone and F. Santoro, Adv. Mater. Technol., 2022, 7, 2100293 CrossRef CAS.
- T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379–385 CrossRef CAS PubMed.
- S. Wang, J. Y. Oh, J. Xu, H. Tran and Z. Bao, Acc. Chem. Res., 2018, 51, 1033–1045 CrossRef CAS PubMed.
- A. Fanelli and D. Ghezzi, Curr. Opin. Biotechnol, 2021, 72, 22–28 CrossRef CAS PubMed.
- V. R. Feig, H. Tran and Z. Bao, ACS Cent. Sci., 2018, 4, 337–348 CrossRef CAS PubMed.
- S. Lee, S. M. Silva, L. M. Caballero Aguilar, T. Eom, S. E. Moulton and B. S. Shim, J. Mater. Chem. B, 2022, 10, 8575–8595 RSC.
- S. Inal, J. Rivnay, A.-O. Suiu, G. G. Malliaras and I. McCulloch, Acc. Chem. Res., 2018, 51, 1368–1376 CrossRef CAS PubMed.
- D. T. Simon, E. O. Gabrielsson, K. Tybrandt and M. Berggren, Chem. Rev., 2016, 116, 13009–13041 CrossRef CAS PubMed.
- L. V. Kayser and D. J. Lipomi, Adv. Mater., 2019, 31, 1806133 CrossRef PubMed.
- L. B. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- T. Cheng, X.-L. Yang, S. Yang, L. Li, Z.-T. Liu, J. Qu, C.-F. Meng, X.-C. Li, Y.-Z. Zhang and W.-Y. Lai, Adv. Funct. Mater., 2023, 33, 2210997 CrossRef CAS.
- M. Criado-Gonzalez, A. Dominguez-Alfaro, N. Lopez-Larrea, N. Alegret and D. Mecerreyes, ACS Appl. Polym. Mater., 2021, 3, 2865–2883 CrossRef CAS PubMed.
- D. Li, W.-Y. Lai, Y.-Z. Zhang and W. Huang, Adv. Mater., 2018, 30, 1704738 CrossRef PubMed.
- L. Zhou, M. Yu, X. Chen, S. Nie, W.-Y. Lai, W. Su, Z. Cui and W. Huang, Adv. Funct. Mater., 2018, 28, 1705955 CrossRef.
- S. Stritesky, A. Markova, J. Vitecek, E. Safarikova, M. Hrabal, L. Kubac, L. Kubala, M. Weiter and M. Vala, J. Biomed. Mater. Res., Part A, 2018, 106, 1121–1128 CrossRef CAS PubMed.
- M. Ganji, E. Kaestner, J. Hermiz, N. Rogers, A. Tanaka, D. Cleary, S. H. Lee, J. Snider, M. Halgren, G. R. Cosgrove, B. S. Carter, D. Barba, I. Uguz, G. G. Malliaras, S. S. Cash, V. Gilja, E. Halgren and S. A. Dayeh, Adv. Funct. Mater., 2018, 28, 1700232 CrossRef.
- D. Khodagholy, J. N. Gelinas, T. Thesen, W. Doyle, O. Devinsky, G. G. Malliaras and G. Buzsáki, Nat. Neurosci., 2015, 18, 310–315 CrossRef CAS PubMed.
- D. A.-O. Khodagholy, J. N. Gelinas, Z. Zhao, M. Yeh, M. Long, J. D. Greenlee, W. A.-O. Doyle, O. Devinsky and G. Buzsáki, Sci. Adv., 2016, 2, e1601027 CrossRef PubMed.
- M. Leprince, P. Mailley, L. Choisnard, R. Auzely-Velty and I. Texier, Carbohydr. Polym., 2023, 301, 120345 CrossRef CAS PubMed.
- A. Fallacara, E. Baldini, S. Manfredini and S. Vertuani, Polymers, 2018, 10, 701 CrossRef PubMed.
- E. Stavrinidou, P. Leleux, H. Rajaona, D. Khodagholy, J. Rivnay, M. Lindau, S. Sanaur and G. G. Malliaras, Adv. Mater., 2013, 25, 4488–4493 CrossRef CAS PubMed.
- S. Zhang, E. Hubis, C. Girard, P. Kumar, J. DeFranco and F. Cicoira, J. Mater. Chem. C, 2016, 4, 1382–1385 RSC.
- O. Berezhetska, B. Liberelle, G. De Crescenzo and F. Cicoira, J. Mater. Chem. B, 2015, 3, 5087–5094 RSC.
- A. Hakansson, S. Han, S. Wang, J. Lu, S. Braun, M. Fahlman, M. Berggren, X. Crispin and S. Fabiano, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 814–820 CrossRef CAS.
- G. D. Cha, D. Kang, J. Lee and D.-H. Kim, Adv. Healthcare Mater., 2019, 8, 1801660 CrossRef PubMed.
- T. Figueiredo, J. Jing, I. Jeacomine, J. Olsson, T. Gerfaud, J.-G. Boiteau, C. Rome, C. Harris and R. Auzely-Velty, Biomacromolecules, 2020, 21, 230–239 CrossRef CAS PubMed.
- Y. Nakamura, S. Horiuchi and Y. Nishioka, Jpn. J. Appl. Phys., 2018, 57, 02CD02 CrossRef.
- J. Mergy, A. Fournier, E. Hachet and R. Auzély-Velty, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4019–4028 CrossRef CAS.
- M. Fabretto, K. Zuber, C. Jariego-Moncunill and P. Murphy, Macromol. Chem. Phys., 2011, 212, 2173–2180 CrossRef CAS.
- D. Alemu Mengistie, P.-C. Wang and C.-W. Chu, J. Mater. Chem. A, 2013, 1, 9907–9915 RSC.
- X. Crispin, F. L. E. Jakobsson, A. Crispin, P. C. M. Grim, P. Andersson, A. Volodin, C. Van Haesendonck, M. Van der Auweraer, W. R. Salaneck and M. Berggren, Chem. Mater., 2006, 18, 4354–4360 CrossRef CAS.
- J. Ouyang, C.-W. Chu, F.-C. Chen, Q. Xu and Y. Yang, Adv. Funct. Mater., 2005, 15, 203–208 CrossRef CAS.
- J. Ouyang, Q. Xu, C.-W. Chu, Y. Yang, G. Li and J. Shinar, Polymer, 2004, 45, 8443–8450 CrossRef CAS.
- I. Lee, S. Park, Y. S. Lee, Y. Kim, M. H. Kang and C. Yun, Langmuir, 2023, 39, 1600–1610 CrossRef CAS PubMed.
- L. Ferlauto, P. Vagni, A. Fanelli, E. G. Zollinger, K. Monsorno, R. C. Paolicelli and D. Ghezzi, Biomaterials, 2021, 274, 120889 CrossRef CAS PubMed.
- J. Li, P. Nemes and J. Guo, J. Biomed. Mater. Res., Part B, 2018, 106, 1129–1137 CrossRef CAS PubMed.
- L. Lu, C. A. Garcia and A. G. Mikos, J. Biomed. Mater. Res., 1999, 46, 236–244 CrossRef CAS PubMed.
- E. Vey, C. Rodger, J. Booth, M. Claybourn, A. F. Miller and A. Saiani, Polym. Degrad. Stab., 2011, 96, 1882–1889 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of the PEGene derivative; 1HNMR spectrum of PEGene; optical microscopy image of ink-jet printed PEDOT:HAS-PBA-PEGene; cell viability. See DOI: https://doi.org/10.1039/d3ma00170a |
| This journal is © The Royal Society of Chemistry 2023 |