
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A simple, sustainable route to flexible microporous carbon cloth for energy storage applications†
Thria
Alkhaldi
 ab,
L. Scott
Blankenship
a and
Robert
Mokaya
*a
ab,
L. Scott
Blankenship
a and
Robert
Mokaya
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: r.mokaya@nottingham.ac.uk
bDepartment of Chemistry, Jeddah University, Jeddah 23442, Saudi Arabia
First published on 21st July 2023
Abstract
Activated carbon cloth (ACC) has the potential to be extremely useful in gas capture and storage applications as it combines high porosity, robustness, and flexibility with ease of handling. While it has been produced by a few researchers, the synthesis methods used to date either do not yield a product with high porosity, or if appropriate textural properties are achieved the synthesis is complex and arduous. Following a systematic study, we show that an almost exclusively microporous flexible ACC can be achieved with surface area >1900 m2 g−1via stabilisation with NH4Cl only, followed by activation with benign activating agent potassium oxalate (PO). After extensive optimisation and simplification of the process, it was found that the stabilisation step can be omitted in a synthesis route requiring only a simple carbonisation step to produce a flexible microporous carbon with surface area >2200 m2 g−1, thus further reducing the need for additional solvents and reagents. The CO2 and CH4 uptake of the ACCs developed in this work is comparable to that previously reported for flexible porous carbons prepared via more complicated routes and the porosity of the ACCs can be tuned to specific gas uptake applications according to the synthesis conditions.
1 Introduction
Activated carbons are widely employed as effective adsorbent materials for the storage of adsorbates such as H2, CH4, and CO2 due to their large specific surface areas (SSA), good chemical stability and low cost. When nanoporous activated carbon is used for these applications, it must still be treated to generate a functional material (e.g., membranes, thin films) from carbon powder. This results in a lowering of the specific surface area and specific pore volume, due to the effects of aggregation and pore blockage.1 Since these textural properties play a key role in energy storage applications, it would be highly desirable to develop a cost-effective and simple processing procedure for a lightweight, mechanically and chemically stable, flexible, non-fragile material without losing porosity. This may exist in the form of activated carbon cloth (ACC),2,3 whose conformability and flexibility make it much more versatile and robust as a potential liner in gas storage tanks. Recently, ACCs have been suggested as such a flexible form of activated carbon that may function as an efficient gas adsorbent. In the case of use as absorbents for energy related gases (e.g., CO2, CH4 and H2), such flexible activated carbon cloth would be much easier to handle than powder and granular forms. They would offer the processability and mechanical properties required for the construction of more efficient storage tank designs incorporating the carbons as coatings or liners inside tanks. In addition a continuous material like carbon cloth does not have interparticle voids which are present in powders. Adsorption of small molecules is associated mainly with micropores and narrow mesopores, thus the porosity attributed to these larger interparticle voids is not particularly useful in this type of application.4–10 Thus ACCs may provide a method to maximising ‘useful’ porosity.With respect to possible precursors for ACCs, previous work has inspired by reported syntheses of the related material, carbon fibre (CF).11,12 While both polyacrylonitrile and cellulose-based materials have been used,13–16 the latter are today favoured for their lower production cost and the fact that they do not derive from fossil fuels.11,17 In particular, synthetic textiles are more suitable than natural cellulosic fibres due to due to their structural regularity as well as lack of impurities (especially lignin and hemicellulose) which can result in low yield and poor porosity in derived materials.11,18,19 As such the regenerated cellulosic (structure shown in Fig. 1) fibre, viscose rayon (VR) has become the precursor of choice in ACC synthesis.11,12,20,21
 | ||
| Fig. 1 The structure of cellulose, of which VR fibres are composed.22 | ||
Preparation of ACC from VR has been attempted by a number of methods.2,3,20,21,23–25 The principal hurdle is to facilitate development of porosity and removal of heteroatoms (principally O) via carbonisation from the original textile, without impinging on the structural integrity of the material at the macro scale. As such, some researchers have favoured pyrolysis of VR following impregnation with reagents such as chloride and phosphate salts in order to promote porogenesis via dehydration reactions4,26,27 over the depolymerisation that occurs on thermal treatment of textiles.2,3,23,28 When successful, these reagents can be simultaneously considered stabilisers and porogens. Huidoboro et al. found that pore size could be tuned in these processes by selection of the appropriate dehydrating agent; reagents such as ZnCl2 and H3PO4 promote micropore formation, whereas ACCs derived via impregnation with Na2HPO4 resulted in principally mesoporous products.3 Due to the low porosity of these ACCs (maximum reported surface area, ABET is 643 m2 g−1)23 Kostoglou et al. used physical activation with CO2 to produce a microporous ACC with surface area of over 1200 m2 g−1 from VR impregnated with ZnCl2 and NH4Cl.21 As is typical for activation with CO24,29 this unfortunately came at the cost of a very high pyrolysis temperature of 930 °C. On the other hand, activation of biomass with KOH has been found to reliably produce activated carbon powder with ultrahigh porosity,1,4,30,31 thus Attia and co-workers used this technique to produce ACCs from a stabilised textile with surface area and pore volume over 1900 m2 g−1 and 0.80 mL g−1 respectively. However, prior to stabilisation with ZnCl2 and NH4Cl, the surface of the VR was decorated with polypyrrole nanoparticles.20 It is therefore unclear whether the improvements in the porosity of the resulting ACC are a result of activation of the polypyrrole or the VR itself, as the former has been successfully used to produce highly porous activated carbons.32
As described above, the current state-of-the-art methods for preparing cellulose-based ACCs involve a number of steps that consume significant energy, chemicals and time. Sustainable, efficient syntheses are achievable by (among other factors) the minimisation of the number of reagents and the number of steps used. Thus far, moderate to highly porous ACC has only been produced from VR in two or more steps, i.e. (i) impregnation with ZnCl2 and NH4Cl and (ii) activation with either CO2 or KOH.20,21 Therefore, this work details attempts at simplifying and improving the sustainability of the synthesis of ACC, via three broad methods; (i) omitting the stabilisation step, i.e. negating the use of ZnCl2 and NH4Cl which has to date been considered to be a crucial step in the preparation of ACCs that preserve the flexibility of activated carbon fabric, (ii) not using an additional activating agent, and (iii) use of the less caustic activating agent potassium oxalate (PO), which has previously been shown to give comparable porosity in activated carbons to the more commonly used KOH.33–35 Due to porosity of the resulting ACCs and the sparsity of the literature on the gas uptake capacity of ACCs in general, we explored the CO2 and CH4 uptake of the materials prepared in this work.
2 Experimental
Dyed viscose rayon (VR) with a 2/2 twill weave was obtained from a local fabric shop.2.1 Synthesis
Synthesis of ACC was performed via five methods, each starting with a piece of VR (7 × 7 cm). The titles of subsections below reflect the pre-treatment of VR prior to thermal treatment (i.e. activation). In all cases, activation was attempted by heating the sample in a tube furnace under N2 (60 mL min−1) at some temperature for 1 h. Ramp rate to the target temperature was 5 °C min−1. In order to remove any contaminants left by the porogen(s), all samples were subsequently washed by stirring in aqueous HCl solution (400 mL, 10 vol%) for 2 h, then rinsed with sufficient deionised water to give neutral washings. Washed material was then dried overnight at 100 °C.Cognisant that ZnCl2 has been extensively exploited as an activating agent (porogen),23,26,27,36,37 the above procedure was modified such that the stabilised VR was activated without the use of KOH. The concentration of the NH4Cl/ZnCl2 solution was varied by changing the volume of water used (100, 50 or 10 mL) and activation was carried out at 630 or 700 °C following a ramp rate of 5 °C min−1. Following washing and drying as above, samples derived via this method were designated VRnzZ-X-T, where the (capital) Z indicates that ZnCl2 also takes the role of an activating agent and X and T indicate the volume of stabilising solution and activation temperature respectively. Thus, a sample prepared using 50 mL of NH4Cl/ZnCl2 solution at 700 °C is designated as VRnzZ-50-700.
In a further modification of the synthesis route, potassium oxalate (PO) was investigated as an alternative activating agent. Stabilisation of VR was performed by immersion in 10 mL of an aqueous solution of 0.28 M NH4Cl for 1 h at room temperature, and then drying at 60 °C. The NH4Cl-stabilised VR was soaked in 1.2 M PO solution and then dried at 60 °C for 30 min. Activation of the NH4Cl-stabilised VR/PO mixture was then performed at 750, 800 or 900 °C for 1 h following a ramp rate of 5 °C min−1. Following washing and drying as described above, the resulting samples were designated VRnP-T, where n signifies stabilisation with NH4Cl, P indicates activation with potassium oxalate and T is the activation temperature. Thus a sample activated at 800 °C is designated as VRnP-800.
2.2 Material characterisation
CHN elemental analysis was performed on a CE440 Elemental Analyser (Exeter Analytical). For assessment of sample graphititic nature and identification of any crystalline materials, XRD measurements were taken on a PANalytical X'Pet Pro diffractometer using CuKα X-rays (40k, 40 m) with a wavelength of 1.5418 Å, at 2θ between 2 to 80°. Angular and time step size between scans were 0.02° and 50 s respectively. SEM images were collected on a JEOL JSM-7100F with an accelerating voltage of 5 kV.To assess porosity, N2 (Air products, technical grade) sorption isotherms were measured at −196 °C on a Micrometrics 3flex sorptometer. Isotherm measurement was in the relative pressure range 1 × 10−8 to 1, with Po determined by the pressure of liquid N2 in the reference tube. Up to relative pressure of 4 × 10−4 the equilibration interval was 45 s; full details of the analysis procedure are available in the ESI,† Table S1. Following isotherm measurement, freespace (void volume) was measured using He (Air products, technical grade) at both ambient and anlysis temperature. Prior to analysis, the carbon samples were degassed on a Micromeritics Smart VacPrep under vacuum (at least 1 × 10−4 mbar). After vaccuum was achieved, samples were then heated at 10 °C min−1 to 90 °C for 1 h, then ramped at 10 °C min−1 to 200 °C and held for 12 h, before being allowed to cool under vaccuum. Sample tubes were then backfilled with N2 prior to transferring to the sorptometer. On the analysis port, samples were again degassed for 1 h at a vacuum of at least 1 × 10−5 at ambient temperature, and 1 × 10−8 at cryogenic temperature.
From the isotherms, surface area (ABET) was determined using the Rouquerol criteria38via the BETSI method so as to improve reproducibility.39 Further details of the method as well as the results can be found in the ESI† Section S6, including graphical outputs showing the relative pressure range used for each isotherm and filter parameters used. Total pore volume was determined using the single point method on the isothermal plateau (relative pressure ∼0.9), and micropore volume and micropore surface area were calculated with the t-plot method using the carbon black STSA thickness curve. Pore size distributions (PSDs) were calculated using the SAIEUS software and the 2D-NLDFT heterogeneous surface kernel with a fitting parameter, λ of 3.5 for all isotherms.40,41 This kernel was used as it accounts for the chemical heterogeneity present in ACs and ACCs. Further details can be found in the ESI,† Section S7 alongside a graphical display of the fits. Values of overall porosity, i.e. ANLDFT and VNLDFT can then be extracted from cumulative PSDs as well as porosity within some range of pore widths; i.e. micro-, ultramicroporosity etc.
2.3 Gas uptake measurements
Gravimetric gas uptake isotherms were measured at 25.00 ± 0.01 °C controlled by a constant temperature, flowing water bath on a XEMIS gravimetric sorption analyser (Hiden). Sample masses were at least 50 mg, and the mass measurement precision of XEMIS is 0.1 μg with a mass stability of 1 μg. CO2 (Air products, Industrial grade) and CH4 (BOC, purity 99.9995%) isotherms were measured up to 40 and 100 bar respectively. The pressure precision for any isotherm point is 0.02% at any point, and the base vaccuum pessure is < 1 × 10−6 mbar. Equilibrium uptake was determined by identifying the asymptote of rate of change in uptake after a relaxation of 99% (tolerance 2%). Data was sampled at an interval of 1 s. Prior to isotherm measurement, samples were degassed in-situ at 240 °C (ramp rate 10 °C min−1) for at least 4 h until sample mass was stable.Measurements on the XEMIS give excess uptakes, i.e. the uptake of gas above that which would have been taken up in a volume equivalent to that of the total pore volume at the same pressure and temperature but without the attractive interactions between gas molecules and pore walls of the adsorbate. This requires skeletal density, which was mesured using He pycnometry on a Micromertics Accupyc II. For CH4 storage studies, total (absolute) quantity adsorbed, Qt is a more practical measure. The total mass adsorbed (mt) may be estimated from the excess mass of CH4 adsorbed by 1 g of adsorbent, mexc (g) from eqn (1);42,43
| mt = mexc + dCH4Vt | (1) |
3 Results and discussion
Elemental analysis of VR indicated C and H content of 40 and 6 wt%. O content can thus be assumed to be 54 wt%. As shown in Fig. 2(a), VR is made of woven fibres that are relatively continuous (Fig. 2(c)). This general structural continuity exhibited in the textile must be maintained in the activated products in order to retain flexibility. | ||
| Fig. 2 SEM images of VR taken at magnification of (a) × 40, (b) × 100, and (c) × 4500. | ||
3.1 KOH-activation
The direct activation of VR with KOH resulted in the product, VRd being entirely powder activated carbon. The C-content of VRd was 72 wt% (cf. 41 wt% for VR) while the O- and H- content were reduced from 53 to 26 wt% and 6 to 1 wt% respectively. Regarding porosity, VRd exhibited a fully reversible nitrogen sorption isotherm with type I characteristics (see Fig. S28, ESI†) indicating that the carbon was mainly microporous. The pore size distribution curve of VRd indicates the presence of micropores (Fig. S28, ESI†). VRd had a surface area of 944 m2 g−1 with 80% (753 m2 g−1) arising from micropores. This process highlighted the fact that KOH-activation was overly harsh, and therefore not a suitable choice as an activating agent for the direct preparation of flexible ACC.In the next synthesis attempt, VR was first stabilised with NH4Cl and ZnCl2 prior to KOH activation. The stabilisation gave gave more encouraging results; flexibility of the precursor was retained after stabilisation, and the resultant product (VRnzK-700) retained flexibility in some areas of the cloth following activation with KOH, however significant portions of the cloth hardened (Fig. S2, ESI†). This indicates that the flexibility of the similar material created by Attia et al. using a similar stabilisation procedure but with the inclusion of polypyrrole nanoparticles can be at least partially attributed to the polypyrrole.20 It should be noted that, as shown in Table 1, the flexible portion of VRnzK-700 has significantly higher C, (69 vs. 56 wt%) as well as N (5.8 wt% vs. nil) content compared to the hard (powdered) portion. This indicates adsorption of NH4Cl into at least some regions of the precursor, resulting in the evident partial stabilisation. The powder and flexible portions of VRnzK-700 have nitrogen sorption isotherms of similar shape (see Fig. S28, ESI†), but the former has higher surface area; 1263 vs. 847 m2 g−1, and pore volume 0.8 vs. 0.6 cm3 g−1. Although both parts have comparable pore size distributions (Fig. S28, ESI†), the powder form has higher levels of microporosity at 85 vs. 75% for surface area (i.e., 1076 cf. 647 m2 g−1) and 63 vs. 50% for pore volume (i.e. 0.5 cf. 0.3 cm3 g−1). The relatively low content of C in both portions of VRnzK-700 relative to VRd indicates that some part of the stabilisation procedure may prevent the degradation of volatile oxygen rich moieties during activation/pyrolysis. Further data for VRd and VRnzK-700 can be found in the ESI† (Fig. S5, S6, S28 and Table S2), but the remainder of this work discusses the flexible products. It is clear that KOH is not the best candidate for activation of VR to flexible ACC, even with the presence of stabilising reagents. The corrosive nature of KOH clearly limits the stabilising effects of NH4Cl and ZnCl2, thus a gentler activation strategy is required.
| Sample | C | H | N | O |
|---|---|---|---|---|
| VR | 41 | 6.0 | 0.0 | 54 |
| VRd | 72 | 1.0 | 0.0 | 26 |
| VRnzK-700 (hard) | 56 | 0.5 | 0.0 | 44 |
| VRnzK-700 (flexible) | 69 | 1.6 | 5.8 | 24 |
3.2 Flexible ACCs
The three remaining routes, i.e. stabilisation of VR with NH4Cl and ZnCl2 followed by thermal treatment to facilitate activation with the latter (ZnCl2) rather than using KOH (Fig. S3, ESI†); stabilisation of VR with NH4Cl only followed by activation with PO (Fig. S4, ESI†); and carbonisation followed by PO activation (series VRcP-T) all resulted in fully flexible ACC regardless of changes in activation temperature, concentration of stabilising solution, and/or amount of porogen (where applicable).| Sample | C | H | N | O |
|---|---|---|---|---|
| VRnzZ-10-630 | 58 | 1.5 | 6.8 | 34 |
| VRnzZ-50-630 | 60 | 1.6 | 6.1 | 32 |
| VRnzZ-100-630 | 81 | 1.6 | 4.3 | 14 |
| VRnzZ-10-700 | 69 | 1.5 | 5.8 | 24 |
| VRnzZ-50-700 | 74 | 0.5 | 3.8 | 21 |
| Sample | C | H | N | O |
|---|---|---|---|---|
| VRnP-750 | 76 | 0.1 | 0.0 | 24 |
| VRnP-800 | 82 | 0.9 | 0.3 | 17 |
| VRnP-900 | 84 | 1.0 | 0.5 | 15 |
| VRc | 85 | 0.5 | 0.2 | 15 |
| VRcP-650 | 82 | 0.3 | 0.4 | 17 |
| VRcP-700 | 83 | 0.3 | 0.3 | 17 |
| VRcP-750 | 90 | 0.0 | 0.1 | 10 |
| VRcP-800 | 87 | 0.3 | 0.3 | 12 |
| VRcP-900 | 82 | 0.2 | 0.3 | 18 |
On the other hand, use of PO as activating agent appears to almost completely eliminate any N from ACCs derived from NH4Cl-stabilised VR (series VRnP-T, see Table 3). The N content for some VRnP-T samples is above nil, but this is also the case for the samples derived without NH4Cl (VRc and VRcP-T) so this very low N content can be ascribed to small amounts of contamination. The loss of N must then be attributed to reactions between NH4Cl and PO, possibly forming salts of cyanide and/or cyanate as has been observed previously for pyrolytic reactions involving metal salts and N-enrich precursor mixtures.48,49 As should be expected, the C/O ratio can be increased for these samples simply by increasing the activation temperature. The C content and consequently the C/O ratio can be increased for flexible ACCs simply by increasing the activation temperature.
Direct pyrolysis at 800 °C of VR to yield VRc more than doubles the C content to 85 wt% (Table 3), which is also slightly higher than that for all of the NH4Cl-stabilised, PO-activated ACCs (series VRnP-T). This is because the degradative depolymerisation reactions taking place on heating naked VR are not competing with reactions arising from to the presence of stabiliser or, indeed an activating agent. However on activation of VRc with PO, the relationship between C/O ratio and activation temperature is somewhat more complex. Firstly, activation at 650 and 700 °C results in a lower C content than that of VRc. A similar trend has previously been reported for the PO-activation at 600 °C of a highly carbon rich precursor.50 On the other hand, for activation at 750 and 800 °C, the C content increases up to 90 wt% which is more than that of both VRc and the equivalent NH4Cl-stabilised ACCs which are activated at the same temperatures. VRcP-900 however has a C content of only 82 wt% which is lower than that of both VRc and VRnP-900. As such, it can be inferred that PO does not solely have an oxidative effect, i.e. the production of K2CO3 on reactions with C which is subsequently removed during washing. Indeed at both low and high temperatures, activation of VRc with PO appears to result in the incorporation of O-rich moieties into the structure of the resultant ACC. While it is common for C content to plateau at high activation temperatures using K-salt porogens including PO,35,50,51 to our knowledge this decrease in C at high temperatures has thus far only been reported for some carbons derived from cellulose acetate.30
XRD analysis of the flexible ACCs (Fig. S5, ESI†) confirmed the absence of any retained inorganic matter as there were no sharp peaks indicative of crystalline material. This confirms that the washing step is effective in ensuring that the ACCs are purely carbonaceous. This is confirmed by TGA - see Fig. S6 (ESI†). Regarding the nature of the carbon cloths, the XRD patterns of VRnzZ-X-T samples (Fig. S5, ESI†) show broad peaks at 2ϑ of 23 and 43° which correspond to the (002) and (100) reflections of turbostratic carbon. The broad nature of the peaks indicates that the carbon cloths are essentially amorphous rather than graphitic, which is consistent with what is expected for activated carbons. The XRD patterns of the PO-activated VRnP-T and VRcP-T series of flexible ACCs (Fig. S7b and c, ESI†) indicate that they are also essentially amorphous with very low levels of graphitisation. The ACCs prepared at a low activation temperature (750 °C) appear to have a comparatively higher of graphitisation, which is consistent with higher temperatures disrupting any graphitic domains that may be present.
3.3 Morphology
Exemplar SEM images of the flexible ACCs can be found in Fig. 3 (and Fig. S7–S10, ESI†). In general, the fibrous structure of the original VR (Fig. 2) can still be seen, which explains the fact that the products remain flexible. However the morphology and integrity of the fibers is disrupted. The extent of the disruption may relate to both the harshness of activation conditions and/or which porogens/stabilisers were used in the synthesis. Some breakage of fibers is evident for ACCs that were derived using stabilising reagents observed in Fig. 3 for sample VRnzZ-10-700 and VRnP-900. This is also evident for other ACCs in series VRnzZ-X-T (Fig. S7, ESI†) and VRnP-T (Fig. S8, ESI†) though the disruption appears to be more extensive for the former. Apart from this, there is clear evidence of the formation of grooves and holes in the fibres in the case of VRnzZ-X-T samples as shown in Fig. 3 and Fig. S9 (ESI†) which is not seen for series VRnP-T (Fig. 3 (middle row) and Fig. S7, ESI†). This indicates that the use of ZnCl2 as a porogen results in some etching of the fibres, however etching (at least that which is visible with SEM) does not seem to occur for activation with PO. The extent of the etching and/or breakage does appear to be related to the dilution, X, of the NH4Cl/ZnCl2 solution used in the synthesis of VRnzZ-X-T. This can be discerned by comparing the morphology of samples synthesised with a total stabilising solution volume of 50 and 10 mL (see Fig. 3 and Fig. S7–S10, ESI†) where the higher concentration in the latter caused greater etching and breakage of the fibres. Finally, for all synthesis routes activation temperature alone does not appear to have a great effect on the structure of the derived samples. | ||
| Fig. 3 SEM images of the three series of flexible ACCs selected to show effects of the synthesis regime and harshness of activation on the fibrous structure; VRnzZ-X-T (top row), VRnP-T (middle row), and VRcP-T (bottom row). | ||
Samples derived via the sequential carbonisation and PO activation of VR appear to suffer only limited breakage of the fibres, or only small amounts of etching of the surface as shown in Fig. 3 for VRcP-750 and VRcP-900, and which is indeed the case for all VRcP-T samples (Fig. S10, ESI†). There is, however, some mild deformation relative to the pristine VR fibres (cf.Fig. 2 and Fig. 3 (bottom row) and Fig. S10, ESI†). This indicates that the two heating stages were mild and did not cause extensive damage to the VR fibres unlike what is observed with use of the so-called ‘stabilising’ reagents, especially ZnCl2. This further confirms the relative gentleness/mildness of activation with PO with respect to the retention of the fibrous structure of VR. The temperature of activation with PO with or without a stabilisation step does not seem to significantly affect the morphology of the derived ACCs as shown in Fig. 3 and Fig. S7–S10 (ESI†). While there are some minor differences in morphology between series VRnP-T (Fig. 3 middle row and Fig. S9, ESI†) and VRcP-T (Fig. 3 bottom row and Fig. S10, ESI†), with the latter showing slightly higher retention of structural intgerety, there is not sufficient evidence to indicate major differences with respect to effects on morphology of PO-activated ACCs derived by using NH4Cl stabilisation of VR versus carbonisation at 800 °C.
 approaches 1, most evident for samples in series VRnzZ-X-T (Fig. 4(a)), indicating adsorption on the external surface.
approaches 1, most evident for samples in series VRnzZ-X-T (Fig. 4(a)), indicating adsorption on the external surface.
| Sample | A BET/m2 g−1 | V sp/cm3 g−1 | SAD/m2 cm−3 | A NLDFT/m2 g−1 | V NLDFT/cm3 g−1 | ||||
|---|---|---|---|---|---|---|---|---|---|
| VRnzZ-10-630 | 947 | (909, 96%) | 0.39 | (0.35, 91%) | 2460 | 819 | (737, 90%) | 0.36 | (0.30, 82%) |
| VRnzZ-50-630 | 1222 | (1208, 99%) | 0.47 | (0.47, 99%) | 2600 | 1051 | (968, 98%) | 0.44 | (0.40, 90%) |
| VRnzZ-100-630 | 926 | (883, 95%) | 0.40 | (0.36, 90%) | 2344 | 1157 | (1152, 100%) | 0.38 | (0.36, 95%) |
| VRnzZ-10-700 | 598 | (581, 97%) | 0.24 | (0.23, 95%) | 2471 | 854 | (852, 100%) | 0.25 | (0.24, 97%) |
| VRnzZ-50-700 | 965 | (893, 93%) | 0.41 | (0.35, 84%) | 2354 | 1232 | (1174, 95%) | 0.42 | (0.34, 82%) |
| VRnP-750 | 1933 | (1841, 95%) | 0.80 | (0.72, 90%) | 2416 | 1735 | (1601, 92%) | 0.77 | (0.62, 81%) |
| VRnP-800 | 993 | (963, 97%) | 0.40 | (0.37, 93%) | 2489 | 1148 | (1117, 97%) | 0.38 | (0.35, 92%) |
| VRnP-900 | 1095 | (1058, 97%) | 0.45 | (0.41, 93%) | 2455 | 1125 | (1037, 92%) | 0.44 | (0.35, 81%) |
| VRcP-650 | 942 | (876, 93%) | 0.42 | (0.36, 86%) | 2243 | 1159 | (1044, 90%) | 0.43 | (0.32, 74%) |
| VRcP-700 | 1013 | (966, 95%) | 0.44 | (0.40, 91%) | 2302 | 1233 | (1098, 89%) | 0.45 | (0.33, 74%) |
| VRcP-750 | 2226 | (1803, 81%) | 1.26 | (0.80, 63%) | 1767 | 2105 | (1452, 69%) | 1.17 | (0.50, 43%) |
| VRcP-800 | 713 | (657, 92%) | 0.30 | (0.26, 87%) | 2377 | 830 | (767, 92%) | 0.31 | (0.24, 77%) |
| VRcP-900 | 701 | (663, 95%) | 0.29 | (0.26, 90%) | 2417 | 816 | (789, 97%) | 0.29 | (0.25, 87%) |
 | ||
| Fig. 4 N2 isotherms for flexible ACCs (a), (c) and (e) and PSDs derived via fitting to the 2D-NLDFT heterogeneous surface kernel (b), (d) and (f) for sample series VRnzZ-X-T (top row), VRnP-T (middle row) and VRcP-T (bottom row). | ||
For VRnzZ-X-T samples, the surface area is in the range of 598–1222 m2 g−1 and is highest for sample VRnzZ-50-630. The pore volume is between 0.24 and 0.47 cm3 g−1, which gives very high surface area density (SAD, ratio of surface area to pore volume) of between 2315 and 2600 m2 cm−3. The high SAD is consistent with the high microporosity of the carbons with the proportion of microporosity ranging from 93 to 99% for surface area and 84 to 99% for pore volume. In comparison, the surface area of VRnP-T ACCs ranges from 993 to 1933 m2 g−1 with pore volume of 0.40 to 0.80 cm3 g−1. This gives SAD of between 2416 to 2482 m2 cm−3, and equally high levels of microporosity (Table 4). The textural data in Table 4 suggests that a temperature of ∼750 °C is most suitable for activation via this route as sample VRnP-750 has surface area and pore volume that is almost double that achieved at 800 and 900 °C but without loss of microporosity. A similar temperature effect, with optimal performance at 750 °C, is observed for VRcP-T samples. All VRcP-T samples have SAD above 2240 m2 cm−3 except for VRcP-750 (1767 m2 cm−3), a trend that is also observed for the level of microporosity.
In terms of overall porosity (Table 4), samples activated using PO at 750 °C (VRnP-750 and VRcP-750) outperform samples activated with ZnCl2, or those activated at other temperatures. Indeed, the ABET and Vsp of these samples are approximately twice that of any other sample in this work. A similar effect is seen for total values of ANLDFT and VNLDFT, though less pronounced.§ The greater maximal porosity of PO-activated materials relative to those activated with ZnCl2 is a result of the difference in their activation mechanisms, in that ZnCl2 is limited in its ability to create pores by the availability of moieties present in the precursor which are susceptible to dehydration reactions. Indeed the relatively high heteroatom content of VRnzZ-X-T (see Table 2) samples is indicative of the fact that a relatively low proportion of moieties susceptible to dehydration reactions were in fact eliminated via dehydration. In the case of PO-activated samples, the maximisation of porosity at 750 °C can be attributed to an optimisation in the balance of mobility of K+ ions with the effects of overactivation at high temperatures.
Most of the flexible ACC samples are almost entirely microporous - regardless of activating/stabilising reagents and conditions - according to t-plot calculations (at least 92 and 85% in terms of ABET and Vsp respectively), the key outlier being VRcP-750 having approximately a third of its pore volume made up by pores of widths larger than 20 Å. Corresponding NLDFT-derived values of porosity were used to estimate porosity below 10 Å as this has been shown to be important for low pressure uptakes of both CO2 and CH4, and a similar trend is exhibited as in the classical measures of porosity; all samples except VRcP-750 have the majority of their porosity arising from pores of size <10 Å. This is also evident when comparing PSDs of the samples, as shown in Fig. 4(c), with VRcP-750 clearly exhibiting a broad, hierarchical PSD unlike the other samples - as can be expected due to the shape of the isotherm. The origin of the broad porosity in VRnP-750 relative to the narrow PSDs of the other VRnP-T ACCs is perhaps a result of activation at 750 °C facilitating oxidative porogenesis to occur, thus forming supermicropores and mesopores. Regarding the VRcP-T series, it appears that 750 °C whilst facilitating creation of supermicropores and mesopores, is however, still not severe enough to lead to overactivation of VRc, which prevents the destruction of the aforementioned larger pores. Raising the activation temperature to ≥800 °C, however does seem to overactivate VRc.
Activation of NH4Cl-stabilised VR at 750 °C (VRnP-750) appears to yield pores centred at 8 Å while activation at 800 and 900 °C generates smaller pores (Fig. 4(d)). This likely contributes to the overall high porosity for VRnP-750. Typically, pore sizes shift to larger values with increasing activation temperature which is the opposite of the trend seen for VRnP-T. To further explore this unusual trend, it is useful to look to variations in the elemental composition of the VRnP-T samples (see Table 3). VRnP-750 has a significantly higher O content (24 wt%) than the other two samples (both ∼16 wt%), indicating that reactions of porogen-derived compounds to remove oxygen-rich moieties are more extensive at higher temperatures during PO-activation of NH4Cl-stabilised VR. On the other hand, for the samples derived via carbonisation followed by activation with PO (VRcP-T series), while the broadening in PSD follows the same trend as VRnP-T series, the oxygen content is at its minimum for the sample activated at 750 °C. This indicates that the stabilisation effects for which NH4Cl has been employed in previous steps include limiting reactions of porogen with the aforementioned O-rich groups during pyrolysis, which in turn limits the porosity development achievable at higher temperatures, which is consistent with the data for VRnzZ-X-T series in Table 2. While referring to the variation O-content, it is important to note that while the 2D-NLDFT heterogeneous surface kernel does account for chemical heteoregeniety of the pore,41 it may not account for the differences in O-content across different samples. Thus, the exact position of pore widths has a certain degree of uncertainty.
For samples derived by stabilisation with NH4Cl and activation with ZnCl2, there are only small variations in total porosity and indeed microporosity (Table 4). However, there appears to be a relationship between synthesis conditions and the pore size; samples VRnzZ-50-630 and VRnzZ-10-630 have PSD centred at 8 to 9 Å while all other ACCs in the series have have the the majority of pores with size ≤6 Å. It has previously been shown that increasing amount of ZnCl2 during activation of biomass can lead to higher overall porosity, and shifting of micropore widths to higher values.52,53 This can explain the larger pores for VRnzZ-50-630 and VRnzZ-10-630, but less so the apparent temperature-dependence of the PSD. As the size and distribution of pores formed by ZnCl2 activation of the NH4Cl-stabilised VR is known principally to be driven by dehydration around ZnCl2 and its hydrates, it is possible that at 700 °C any water associated with the ZnCl2 crystal structure is driven off before porogenesis occurs, thus yielding smaller pores.
As stated above, the aim of the present work was to simplify the preparation of ACCs. This has been successfully achieved as we have been able to prepare fully flexible, cloth-like ACCs via a much simpler route that negates the need for stabilisation and only requires carbonisation of VR followed by conventional activation with PO. The use of PO, as a milder activating agent rather than KOH, which is harsher, is key in achieving flexible ACCs. Our route simplifies the synthesis route (fewer steps, fewer reagents as only PO is needed), and lower energy use. In additon to the advantage of a simpler process, our routes yield ACCs with enhanced porosity with surface area and pore volume of up to 2226 m2 g−1 and 1.26 cm3 g−1, respectively. These textural properties are higher than those previously achieved for current state-of-the-art ACCs prepared via more complicated synthesis routes.20,23,25 This porosity also exceeds that of the commercially available VR-derived ACC, Calgon Zorflex (ABET of up to 1300 m2 g−1 and Vsp of up to 0.44 cm3 g−1),54 as well as other commercial ACCs derived from other precursors (ABET in range 600 to 1700 m2 g−1 and Vsp in range 0.27 to 0.64 cm3 g−1).13,14,15,16 In the next section we explore the gas (CH4 and CO2) uptake of the present ACCs and compare them to previous benchmark ACC materials.
3.3.2.1 CO2 capture. Excess isotherms for CO2 uptake on the selected ACCs are shown in Fig. 5 with tabulated uptake values in Table 5. It is clear that the high overall porosity (Table 4 and Fig. 4) of VRcP-750 means it outcompetes the other two in terms of CO2 capacity at high pressures, reaching 16.3 mmol g−1 at 40 bar, in line with previous reports.5,57 It is apparent that the porosity and/or compositions of the other two samples show some benefit for low pressure CO2 uptake as shown in Fig. 5(b). We have recently shown that at pressures below 10 bar, volume in pores of width less than 10 Å are highly associated with CO2 uptake at 25 °C.57 This serves to explain why VRnP-750 outperforms VRcP-750 at sufficiently low pressures, having pore volume of 0.62 cm3 g−1 as compared with 0.50 cm3 g−1 in this width range (see Table 4). At very low pressures (∼0.10 bar), N-rich (6.1 wt%) VRnzZ-50-630 becomes competitive compared to the other two samples despite having approximately half the overall porosity. While the effects of N-doping versus pore size on CO2 uptake remain a subject of debate,8,58 recent work by Burrow et al. suggests that the N-content becomes more important at very low pressures56 - a phenomenon which appears to be confirmed by the results shown here.
 | ||
| Fig. 5 Excess gravimetric CO2 uptake isotherms measured at 25 °C up to 40 bar, with a linear (a) and logarithmic (b) pressure scale. | ||
| Sample | Q/mmol g−1 | ||||
|---|---|---|---|---|---|
| 0.15 | 1.0 | 20 | 40 | W | |
| VRcP-750 | 0.63 | 2.9 | 14.0 | 16.3 | 13.4 |
| VRnP-750 | 1.1 | 4.1 | 12.3 | 13.3 | 9.2 |
| VRnzZ-50-630 | 1.2 | 3.5 | 8.9 | 9.5 | 6.0 |
Until now, the best ambient temperature CO2 uptakes in ACCs have been reported by Attia and co-workers via the activation of a ZnCl2/NH4Cl-stabilised VR-polypyrrole composite with KOH, which achieved CO2 uptake capacities of 4.2 and 14.3 mmol g−1 at 1.0 and 20 bar respectively.20 Our methods yield flexible ACCs with similar uptakes but eliminate the need for the formation of the polypyrrole composite, as well as the necessity of using ZnCl2 and/or NH4Cl. Additionally, our simpler synthesis routes use the milder PO as activating agent. Furthermore, a flexible ACC just as suited for high pressure (20 bar) uptake can be achieved by eliminating the stabilisation step in favour of a carbonisation step. This further reduces the number of reagents needed in the synthesis of ACC from VR. Attia et al. attributed their material's high CO2 capacity at 20 bar to a combination of high surface area and N-doping (up to 10 wt%).20 However, considering sample VRcP-750 contains no N and has similar porosity to the best performing sample in the aformentioned work, while achieving the same excess CO2 uptake, it can be assumed that porosity is likely the determining factor for CO2 capture at 20 bar.
3.3.2.2 CH4 storage. CH4 uptake isotherms for the three samples are shown in Fig. 6 and the uptake at various pressures is given in Table 6. It should be noted that for VRcP-750 and VRnP-750 there are apparent uptake maxima in the isotherms at approximately 61 and 90 bar respectively (see Fig. 6(a)). This is a phenomenon observed commonly in high pressure adsorption of supercritical fluids, and indicates that the density of bulk or free CH4 is increasing more quickly with pressure than the density of adsorbed CH4 (pore density).43,59–61 The fact that this maximum occurs at a higher pressure for VRnP-750 than for VRcP-750, and is not visible for VRnzZ-50-630 indicates the the rate of pressure-dependent change in pore density of methane (ρads) at a given pressure varies in the order VRcP-750 < VRnP-750 < VRnzZ-50-630. For VRnzZ-50-630 the maximum likely exists ca. 100 bar where it has been frequently observed on shale samples.43,62,63 The differences in ρads between these three samples is probably best ascribed to differences in pore structure, as supercritical adsorption only takes place via micropore adsorption followed by monolayer formation in mesopores.43ρads is thus higher in samples that are proportionally more microporous, whereas adsorption in mesopores hardly contributes to the adsorbed gas density.
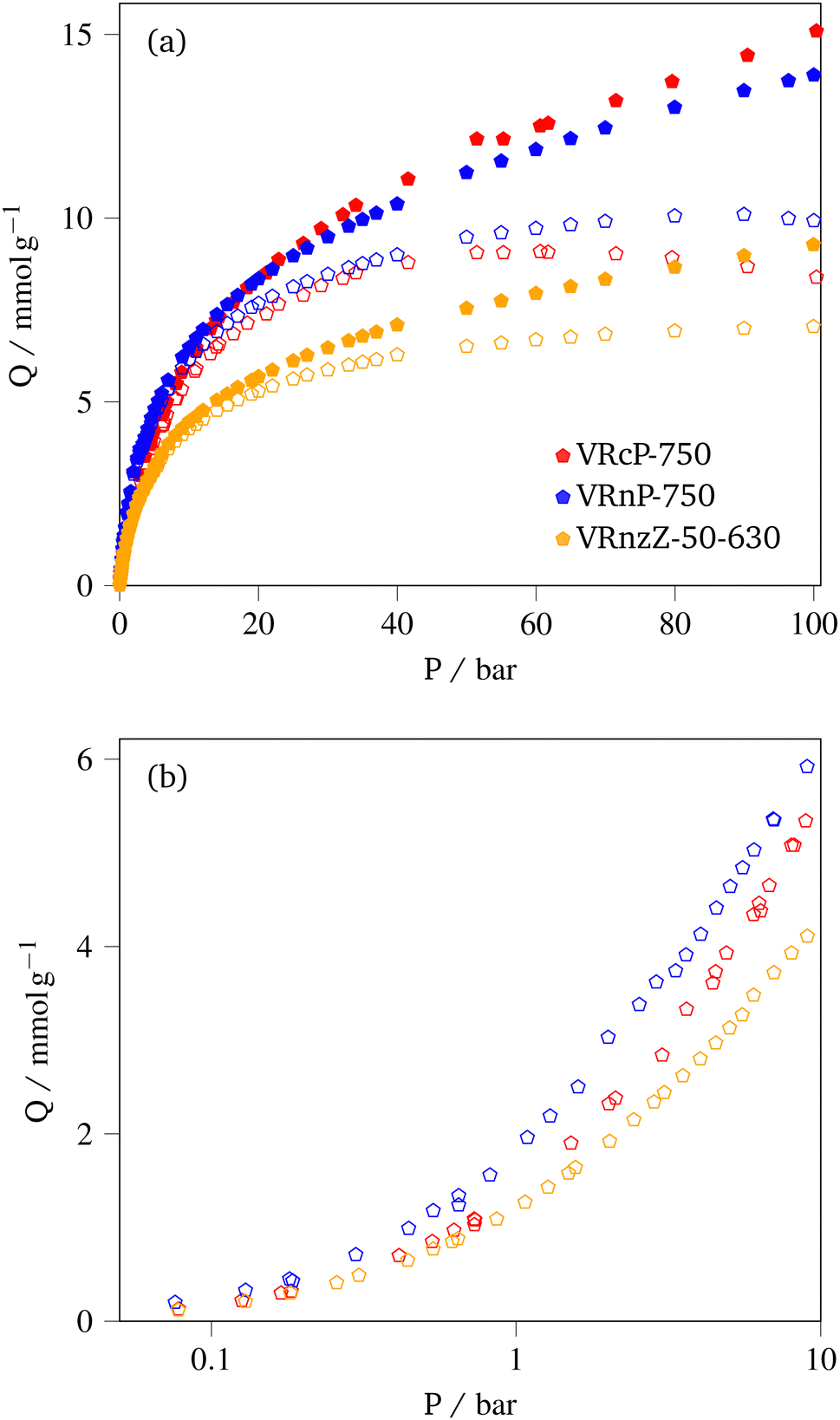 | ||
| Fig. 6 Gravimetric CH4 uptake isotherms measured at 25 °C up to 100 bar, with a linear pressure scale (a) and up to 10 bar with a logarithmic pressure scale (b). Filled symbols indicate total uptake and empty symbols are excess. Total uptake is not included in (b) as total uptakes are only relevant at high pressures. | ||
| Sample | Q/mmol g−1 | ||||||
|---|---|---|---|---|---|---|---|
| 1.0 | 20 | 35 | Max | ||||
| VRcP-750 | 1.3 | 7.3 | (8.3) | 8.2 | (10.4) | 8.9 | (14.8) |
| VRnP-750 | 1.9 | 7.7 | (8.3) | 8.8 | (10.0) | 10.2 | (13.8) |
| VRnzZ-50-630 | 1.2 | 5.3 | (5.6) | 6.1 | (6.8) | 7.1 | (9.2) |
Total uptake, Qt at high pressure is the more relevant parameter for CH4 storage applications. While VRnP-750 noticeably outperforms VRcP-750 in terms of excess CH4 storage at pressures under 20 bar, the latter exceeds the former in terms of high pressure total CH4 uptaake. This is because of the high overall pore volume of VRcP-750, which outweighs its relatively low porosity in the width range 8.0 to 15 Å. Similarly, the significantly lower capacity of VRnzZ-50-630 can be attributed to a much lower pore volume, only becoming competitive with the PO-activated samples at very low pressures. Unlike for CO2 capture, the composition of the samples plays little role in improving CH4 uptake thus the relatively high low-pressure uptake must be solely attributed to porosity. In a similar manner to CO2 uptake, the PO-activated samples achieve similar high and low pressure CH4 uptakes to those reported by Attia and co-workers,20 despite our much simpler synthetic methods. Indeed, stabilisation of VR can be completely foregone to achieve greater total capacity than found previously, at 20 bar.
4 Conclusions
The development of flexible nanoporous and high surface-area carbon materials may show promise for applications involving the adsorption of small molecules. However, thus far the typical synthesis from viscose rayon (VR) is complex and unsustainable, requiring so-called stabilising reagents (ZnCl2 and NH4Cl) as well as corrosive activating agents such as KOH, and in some cases the formation of composites with VR prior to activation. In this study, a series of mechanically stable and flexible activated carbon cloths (ACCs) were prepared with the view to iteratively eliminate many of the steps and reagents used in previous studies. We found that the use of toxic ZnCl2 in the stabilisation step hinders the development of the porosity important for CO2 capture and CH4 storage. Furthermore, KOH can be replaced as activating agent by potassium oxalate (PO) to yield highly microporous carbons, and has the advantage of not risking compromising flexibility of the ACC product. Upon optimisation of reaction conditions, a flexible ACC was achieved with impressive surface area and pore volume of 1933 m2 g−1 and 0.80 cm3 g−1 respectively of which 95% is in micropores. This resulted in excellent CH4 uptake at 25 °C of 1.9 and 9.3 mmol g−1 at 1.0 and 35 bar respectively. Furthermore, the material may be interesting for ambient CO2 capture as an uptake of 4.2 mmol g−1 at 25 °C and 1 bar was achieved. Synthesis protocols can be further simplified by elimination of the stabilisation step altogether in favour of a short pyrolysis step that, surprisingly, further increased the porosity. The resulting PO-activated flexible ACC has surface area of 2226 m2 g−1 and pore volume of 1.26 cm3 g−1. However, approximately a third of this porosity is in mesopores, resulting in a material suited for high pressure CO2 capture applications with uptake of 16.3 mmol g−1 at ambient temperature and 40 bar. The porosities and gravimetric gas uptake capacities found here are comparable with those reported previously for ACCs which are derived through far more complex procedures.Author contributions
Thria Alkhaldi: conceptualization, methodology, formal analysis, investigation, writing – review & editing. L. Scott Blankenship: writing – original draft, formal analysis, visualization, writing – review & editing, validation. Robert Mokaya: conceptualisation, writing – review & editing, supervision, funding acquisition, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Jeddah University, Kingdom of Saudi Arabia, for funding a PhD studentship for Thria Alkhaldi. RM thanks the Royal Society for a Research Grant, and for a Royal Society Wolfson Research Merit Award. Additionally we thank Theo Ching for helium pycnometry measurements.References
- D. Lozano-Castello, J. A. Monge, M. Lillo, D. Amoros and A. Solano, Fuel, 2002, 81, 1777–1803 CrossRef CAS.
- B. M. Babic, S. K. Milonjic, M. J. Polovina and B. V. K. Kaludierovič, Carbon, 1999, 37, 477–481 CrossRef CAS.
- A. Huidobro, A. Pastor and F. Rodrıguez-Reinoso, Carbon, 2001, 39, 389–398 CrossRef CAS.
- L. S. Blankenship and R. Mokaya, Mater. Adv., 2022, 3, 1905–1930 RSC.
- V. Presser, J. McDonough, S. H. Yeon and Y. Gogotsi, Energy Environ. Sci., 2011, 4, 3059–3066 RSC.
- M. Cox and R. Mokaya, Sustainable Energy Fuels, 2017, 1, 1414–1424 RSC.
- K. R. Matranga, A. Stella, A. L. Myers and E. D. Glandt, Sep. Sci. Technol., 1992, 27, 1825–1836 CrossRef CAS.
- B. Adeniran and R. Mokaya, Chem. Mater., 2016, 28, 994–1001 CrossRef CAS.
- S. Reljic, E. Jardim, C. Cuadrado-Collados, M. Bayona, M. Martinez-Escandell, J. Silvestre-Albero and F. Rodríguez-Reinoso, Porous Materials: Theory and Its Application for Environmental Remediation, 2021, vol. 139 Search PubMed.
- S. Biloe, V. Goetz and A. Guillot, Carbon, 2002, 40, 1295–1308 CrossRef CAS.
- E. Frank, L. M. Steudle, D. Ingildeev, J. M. Spörl and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2014, 53, 5262–5298 CrossRef CAS PubMed.
- A. G. Dumanlı and A. H. Windle, J. Mater. Sci., 2012, 47, 4236–4250 CrossRef.
- N. Kostoglou, C. Koczwara, C. Prehal, V. Terziyska, B. Babic, B. Matovic, G. Constantinides, C. Tampaxis, G. Charalambopoulou and T. Steriotis, et al. , Nano Energy, 2017, 40, 49–64 CrossRef CAS.
- J. V. Nabais, T. Canário, P. Carrott and M. R. Carrott, J. Porous Mater., 2007, 14, 181–190 CrossRef CAS.
- C. Kim, P. Srimuk, J. Lee, S. Fleischmann, M. Aslan and V. Presser, Carbon, 2017, 122, 329–335 CrossRef CAS.
- M. E. Ramos, P. R. Bonelli, A. L. Cukierman, M. R. Carrott and P. Carrott, J. Hazard. Mater., 2010, 177, 175–182 CrossRef CAS PubMed.
- P. Morgan, Carbon Fibers and Their Composites, CRC press, 2005 Search PubMed.
- J.-B. Donnet and R. C. Bansal, Carbon Fibers, CRC Press, 1998 Search PubMed.
- J. Hearle, J. Polym. Sci., 1958, 28, 432–435 CrossRef CAS.
- N. F. Attia, M. Jung, J. Park, H. Jang, K. Lee and H. Oh, Chem. Eng. J., 2020, 379, 122367 CrossRef CAS.
- N. Kostoglou, C. Koczwara, C. Prehal, V. Terziyska, B. Babic, B. Matovic, G. Constantinides, C. Tampaxis, G. Charalambopoulou, T. Steriotis, S. Hinder, M. Baker, K. Polychronopoulou, C. Doumanidis, O. Paris, C. Mitterer and C. Rebholz, Nano Energy, 2017, 40, 49–64 CrossRef CAS.
- NEUROtiker, Structure of cellulose (chain conformation), 2007, https://upload.wikimedia.org/wikipedia/commons/0/07/Cellulose_Sessel.svg.
- G. Rodríguez-Blanco, L. Giraldo and J. C. Moreno-Piraján, Appl. Surf. Sci., 2010, 256, 5221–5225 CrossRef.
- X. Huang, Materials, 2009, 2, 2369–2403 CrossRef CAS.
- M. Jung, J. Park, S. Y. Cho, S. E. Elashery, N. F. Attia and H. Oh, Surf. Interfaces, 2021, 23, 100960 CrossRef CAS.
- P. E. Hock and M. A. A. Zaini, Acta Chim. Slovaca, 2018, 11, 99–106 CrossRef CAS.
- M. A. A. Zaini and M. S. M. Shaid, Hung. J. Ind. Chem., 2017, 44, 129–133 Search PubMed.
- F. Suárez-García, A. Martínez-Alonso and J. M. D. Tascón, Carbon, 2004, 42, 1419–1426 CrossRef.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 RSC.
- L. S. Blankenship, N. Balahmar and R. Mokaya, Nat. Commun., 2017, 8, 1–12 CrossRef PubMed.
- N. Balahmar and R. Mokaya, J. Mater. Chem. A, 2019, 7, 17466–17479 RSC.
- M. Sevilla, R. Mokaya and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 2930–2936 RSC.
- M. Sevilla, A. S. M. Al-Jumialy, A. B. Fuertes and R. Mokaya, ACS Appl. Mater. Interfaces, 2018, 10, 1623–1633 CrossRef CAS PubMed.
- A. Altwala and R. Mokaya, RSC Adv., 2022, 12, 20080–20087 RSC.
- M. Sevilla, G. A. Ferrero and A. B. Fuertes, Carbon, 2017, 114, 50–58 CrossRef CAS.
- P. T. Williams and A. R. Reed, J. Anal. Appl. Pyrolysis, 2004, 71, 971–986 CrossRef CAS.
- M. A. A. Zaini, T. W. Meng, M. J. Kamaruddin, S. H. M. Setapar and M. A. C. Yunus, Sains Malays., 2014, 43, 1421–1428 Search PubMed.
- J. Rouquerol, P. Llewellyn and F. Rouquerol, Stud. Surf. Sci. Catal., 2007, 49–56 CrossRef CAS.
- J. W. Osterrieth, J. Rampersad, D. Madden, N. Rampal, L. Skoric, B. Connolly, M. D. Allendorf, V. Stavila, J. L. Snider and R. Ameloot, et al. , Adv. Mater., 2022, 2201502 CrossRef CAS PubMed.
- J. Jagiello, Langmuir, 1994, 10, 2778–2785 CrossRef CAS.
- J. Jagiello and J. P. Olivier, Carbon, 2013, 55, 70–80 CrossRef CAS.
- D. Do and H. Do, Carbon, 2003, 41, 1777–1791 CrossRef CAS.
- T. F. Rexer, M. J. Benham, A. C. Aplin and K. M. Thomas, Energy Fuels, 2013, 27, 3099–3109 CrossRef CAS.
- I. H. Bell, J. Wronski, S. Quoilin and V. Lemort, Ind. Eng. Chem. Res., 2014, 53, 2498–2508 CrossRef CAS PubMed.
- Z. Yang, Y. Xia, X. Sun and R. Mokaya, J. Phys. Chem. B, 2006, 110, 18424–18431 CrossRef CAS PubMed.
- J. Singh, S. Basu and H. Bhunia, J. Taiwan Inst. Chem. Eng., 2019, 102, 438–447 CrossRef CAS.
- Y. Li, S. Wang, B. Wang, Y. Wang and J. Wei, Nanomaterials, 2020, 10, 174 CrossRef CAS PubMed.
- E. Fuente, R. Gil, R. Giron, M. Lillo-Ródenas, M. Montes-Moran, M. Martin and A. Linares-Solano, Carbon, 2010, 48, 1032–1037 CrossRef CAS.
- N. Tsubouchi, M. Nishio and Y. Mochizuki, Appl. Surf. Sci., 2016, 371, 301–306 CrossRef CAS.
- A. Altwala and R. Mokaya, Energy Environ. Sci., 2020, 13, 2967–2978 RSC.
- A. M. Aljumialy and R. Mokaya, Mater. Adv., 2020, 1, 3267–3280 RSC.
- Q. Qian, M. Machida, M. Aikawa and H. Tatsumoto, J. Mater. Cycles Waste Manage., 2008, 10, 53–61 CrossRef CAS.
- F. Caturla, M. Molina-Sabio and F. Rodriguez-Reinoso, Carbon, 1991, 29, 999–1007 CrossRef CAS.
- R. Kaiser, A. Kulczyk, D. Rich, R. J. Willey, J. Minicucci and B. MacIver, Ind. Eng. Chem. Res., 2007, 46, 6126–6132 CrossRef CAS.
- E. A. Hirst, A. Taylor and R. Mokaya, J. Mater. Chem. A, 2018, 6, 12393–12403 RSC.
- J. N. Burrow, J. E. Eichler, Y. Wang, D. C. Calabro and C. B. Mullins, J. Mater. Chem. A, 2022, 10, 24649–24661 RSC.
- L. S. Blankenship, N. Albeladi, T. Alkhaldi, A. Madkhali and R. Mokaya, Energy Adv., 2022, 1, 1009–1020 RSC.
- M. Sevilla, J. B. Parra and A. B. Fuertes, ACS Appl. Mater. Interfaces, 2013, 5, 6360–6368 CrossRef CAS PubMed.
- A. L. Myers and P. A. Monson, Adsorption, 2014, 20, 591–622 CrossRef CAS.
- D. Do, H. Do, C. Fan and D. Nicholson, Langmuir, 2010, 26, 4796–4806 CrossRef CAS PubMed.
- S. Zhou, H. Xue, Y. Ning, W. Guo and Q. Zhang, Fuel, 2018, 211, 140–148 CrossRef CAS.
- X. Tang, N. Ripepi, N. P. Stadie, L. Yu and M. R. Hall, Fuel, 2016, 185, 10–17 CrossRef CAS.
- H. Tian, T. Li, T. Zhang and X. Xiao, Int. J. Coal Geol., 2016, 156, 36–49 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: One PDF file with all referenced supporting information, as well as isotherms in.aif format, and a pyhton module for the calculation of total adsorption from excess available at https://github.com/sblanky/total_adsorption. See DOI: https://doi.org/10.1039/d3ma00157a |
| ‡ Vt - derived using porosimetry - is used as an estimate for Vads, the adsorbed volume of CH4, in the absence of a preferable method. |
| § This is perhaps due to the fact that these calculations only take into account porosity in pores smaller than 500 Å, i.e. excluding the external surface. Furthermore the NLDFT kernel used in this work accounts for surface heterogeneity, and the BET method is a poor method of determining surface area in microporous materialsi as it only accounts for adsorption of N2 onto a single surface.38–41 This may account for the fact that the range of values for ANLDFT is smaller than that for ABET. On the other hand, values for VNLDFT and Vsp are similar, as the assumptions used in determination of Vsp do not exclude micropores. |
| This journal is © The Royal Society of Chemistry 2023 |