
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral amplification induced by self-assembly of different aggregation states in liquid crystal block copolymer films with chiral response†
Jianan
Yuan‡
b,
Xuemin
Lu‡
*a,
Xiaojie
He‡
b,
Feng
Zheng‡
b and
Qinghua
Lu‡
 *a
*a
aSchool of Chemistry and Chemical Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China. E-mail: xueminlu@sjtu.edu.cn; qhlu@sjtu.edu.cn
bSchool of Chemical Science and Engineering, Tongji University, Shanghai, 200092, China
First published on 14th April 2023
Abstract
Chiral films with a high optical asymmetry show substantial potential in organic optoelectronics and other chiroptical fields. However, the fabrication of chiral films with high circular dichroism and chiral response remains challenging. Herein, hierarchical nanostructures with controllable optical asymmetry and chiral response were formed in a liquid crystalline block copolymer film (LC-BCP) induced by the enantiomers dibenzoyl tartaric acid (DiBTA) or dibenzyl ester tartaric acid (DiBETA). A high chiral amplification was achieved with an asymmetry factor (gabs) of up to 0.079 when DiBTA was used, which was 10-fold larger than when DiBETA was used. The difference in gabs was verified by the different stacking modes between the exogenous chiral molecules and LC-BCP. Specifically, DiBTA caused a J-aggregation self-assembly, while DiBETA resulted in an H-aggregation self-assembly. Additionally, the obtained chiral films presented an excellent chiral response to photo and thermal stimuli, allowing the chiral switch rapidly and reversibly. This study provides a pathway to the design of chiral film materials with high optical activity and stimuli response for potential applications in diverse fields such as chiral recognition, storage, metamaterials, and so on.
Introduction
Inspired by the versatility of helical structures in nature,1 scientists have used sophisticated synthetic techniques to prepare helical polymers for various applications, such as asymmetric catalysis,2,3 circularly polarized luminescence (CPL),4–8 and metamaterials.9 The absorption dissymmetry factor (gabs), defined as, gabs = 2(AL − AR)/(AL + AR), is a parameter to evaluate the properties of chiral materials, AL and AR are the absorbance for left and right polarized light, respectively.10 Generally, for intrinsic chiral polymers, especially chiral π-conjugated polymers, the gabs value is in the range of 10−2 to 10−4 due to the exciton coupling of the helical π-conjugated backbones,11,12 and the gabs values can be further amplificated by endowing the chiral π-conjugated polymers with liquid crystalline property.13 However, the most popular approach to the preparation of high gabs films is by introducing a chiral liquid crystal into the chiral self-assembly system, where the ordered self-assembly property of the liquid crystals can lead to the formation of higher chiral optical activity (gabs < 10−1).14,15 In addition, the helical assembly nanostructure can also effectively amplify the gabs of chiral films, namely, chiral amplification caused by chiral structures.16 Notably, the phase separation of chiral block copolymer films is an elegant way to amplify the chirality of films by constructing helical nanophases.17Recently, helical nanostructure in achiral block copolymer (BCP) system have been constructed by the addition of exogenous chiral molecules and is considered a promising approach to fabricate chiral films due to the variety of chiral nanostructures and convenient process control.18–25 Watkins et al. first reported the formation of a helical nanostructure in an achiral PEO-b-PtBA using enantiopure tartaric acid (TA).26 In our previous reports, chiral helical structures could be an effective template to control the chiral arrangement of fluorescent molecules, thereby leading to circularly polarized luminescence.27 Ho et al. also reported the formation of helical nanoribbons with long pitches in PS-b-P2VP using enantiomeric mandelic acid as the chiral molecules.28 Although recent progress has been made in this chiral structure area, chiral films present a relatively low asymmetry factor compared to the intrinsic chiral polymer assembly system. The gabs ranges from 10−2 to 10−5, which may be due to the weak chiral assembly resulting in the poor regularity of helical stacking. Therefore, it is worth exploring the effectiveness of enhancing the phase-separation self-assembly and helical stacking of BCP to achieve chiral amplification. However, the relationship between chiral amplification and helically self-assembled structures has rarely been reported for achiral polymer systems.
To realize the stimulus response of chiral information is also an important basic technology to expand the application of chiral films, for example, chiral storage and memory materials, but few studies have been conducted in this area. Recently, several research groups attempted to introduce azobenzene groups into the BCP helical self-assembly to modulate chirality,29–31 however, the chiral supramolecular assembly in azobenzene BCPs exhibited low gabs.
In this study, a chiral response film with an amplified chirality was prepared by the co-assembly of the enantiomers dibenzoyl tartaric acid (DiBTA) and enantiomers dibenzyl ester tartaric acid (DiBETA), with a liquid crystalline block copolymer (LC-BCP), poly (ethyleneoxide)-b-poly (methyl acrylate) bearing azobenzene side chain (PEO-b-PMA (Azo)). The chirality transfer was first achieved through the interactions between small chiral molecules and segments of LC-BCP via hydrogen bonding and π–π stacking. The J-type stacking of chiral molecules and LC-BCP segments induced a prominent chiral amplification effect with a gabs of up to 0.079 (Scheme 1). Moreover, the chiral film was characterized by repeated chiral erasure and reconstruction based on azobenzene photoisomerization and liquid crystal rearrangement. This elegant strategy of building large gabs chiral films with chiral switching features may provide a promising avenue to produce advanced chiral materials.
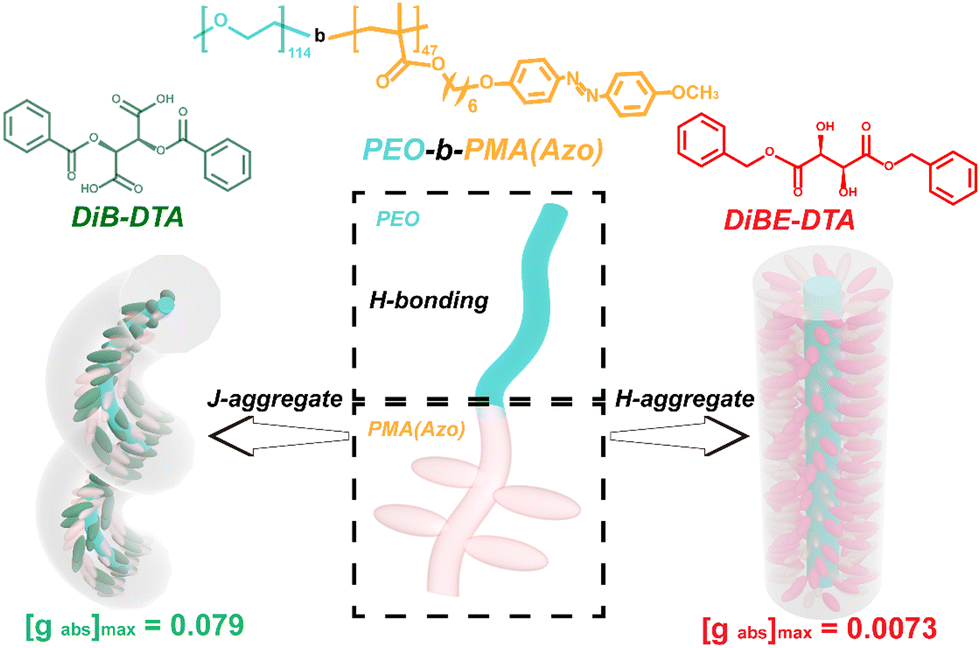 | ||
| Scheme 1 Schematic diagram of the stacking manners in chiral DiB-DTA- or chiral DiBE-DTA-doped LC-BCP films, and their corresponding gabs values. | ||
Results and discussion
Preparation and characterization of chiral composite films
The LC-BCP, PEO114-PMA (Azo)47 (Mw = 24.2 kg mol−1, PDI = 1.198), was prepared according to our previous work.32 Then, the LC-BCP, DiBTA, and DiBETA were dissolved in THF. After the solids were completely dissolved, the DiBTA and DiBETA solutions were added to the LC-BCP solutions at a molar ratio (DiBTA: LC-BCP = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; DiBETA: LC-BCP = 1:1) and stirred at room temperature overnight. The THF solution of DiBTA-or DiBETA-doped LC-BCP was then filtered through a 0.45 μm polytetrafluoroethylene microporous membrane. An appropriate amount of the solution was drop-coated or spin-coated on a quartz plate. After the solvent was evaporated at room temperature, the film was annealed on a hot plate at 125 °C.
:1; DiBETA: LC-BCP = 1:1) and stirred at room temperature overnight. The THF solution of DiBTA-or DiBETA-doped LC-BCP was then filtered through a 0.45 μm polytetrafluoroethylene microporous membrane. An appropriate amount of the solution was drop-coated or spin-coated on a quartz plate. After the solvent was evaporated at room temperature, the film was annealed on a hot plate at 125 °C.
Circular dichroism (CD) is used to detect chiral information in composite films, and during the testing process, the sample was rotated by 45-, 90-, and 180 degrees to eliminate the effect of linear dichroism. Fig. 1 presents the CD spectra of the composite films, which were different from the CD spectra of the neat DiBTA (Fig. S1a, ESI†). The first peak at 213 nm in Fig. 1a could be attributed to the exciton coupling between DiBTA and LC-BCP. Similarly, the cotton effect at 220 nm in Fig. 1b corresponds to the interaction between DiBETA and LC-BCP. We found that the absorption peak of the azobenzene at 275–425 nm shows an intense cotton effect, which indicated that the chiral was successfully transferred to the PMA (Azo) block.
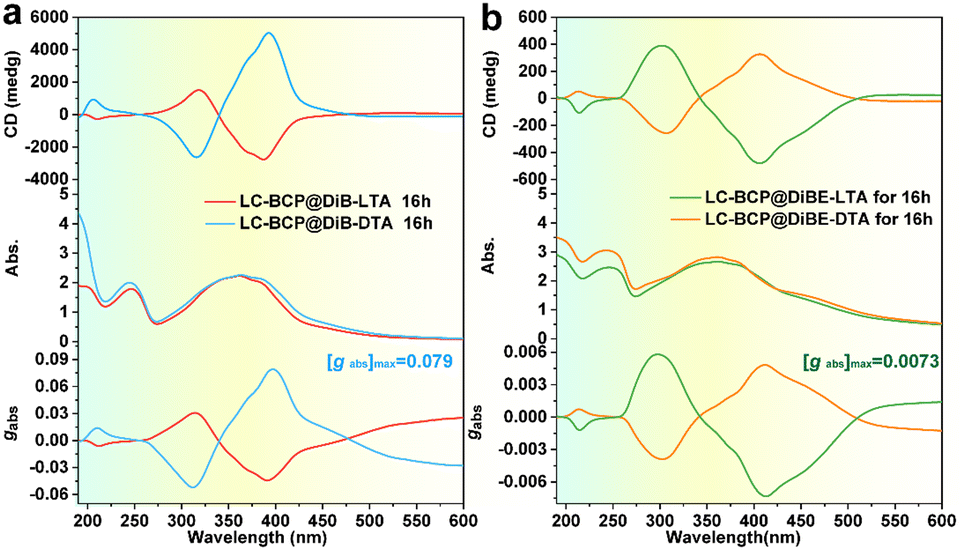 | ||
| Fig. 1 CD and UV-vis spectra of LC-BCP@DiB-D/LTA (a) and LC-BCP@DiBE-D/LTA (b) after thermal annealing at 125 °C for 16 h, and the corresponding gabs. | ||
Remarkably, the gabs of the azobenzene chiral peak of the LC-BCP@DiBTA film (up to 0.079) was one order of magnitude larger than that of the LC-BCP@DiBETA film, although both films were prepared under the same conditions. We also discussed the influence of different chiral molecules induced polymer film thickness on chiral information. The thickness of the film prepared by the drop casting method is ca. 2 μm. The results showed that there was no significant change in the gabs of micrometer-level drop-coated films, and the gabs of LC-BCP@DiBTA films were still higher than LC-BCP@DiBETA films (Fig. S2, ESI†). To the best of our knowledge, this is the largest gabs for block copolymer-based chiral films induced by an exogenous chiral environment.32,33 It is conceivable that the high gabs is closely related to the stacking manner of the helical supramolecular assembly in the LC-BCP composite system.34 Therefore, the interaction between DiBTA and LC-BCP during the chiral transfer process may be a key factor for chiral differences.
Driving force of chirality transfer and mechanism of differential chirality amplification
To understand the chiral transfer and the chiral differences in LC-BCP doped with DiBTA or DiBETA, the interactions between DiBTA and DiBETA with PEO block and PMA (Azo) block were further explored. First, the interaction between the PEO homopolymer and DiBTA was investigated using infrared spectroscopy (IR) and differential scanning calorimetry (DSC). By comparing the IR spectra of PEO, PEO@DiBTA, and DiBTA, we observed that the characteristic peaks of the carboxyl hydroxyl groups of DiBTA shifted from 3545 cm−1 to 3509 cm−1 and 3194 cm−1 to 3173 cm−1, respectively, which was due to the hydrogen bonding between the carboxyl hydroxyls of DiBTA and PEO (Fig. 2a). In general, the hydrogen bonding interactions between chiral molecules and PEO can inhibit the crystallization of the PEO phase.35 The DSC results indicated the formation of hydrogen bonds between the DiBTA and PEO in LC-BCP due to the disappearance of PEO melting and crystalline peak (Fig. 2b and Fig. S3, ESI†).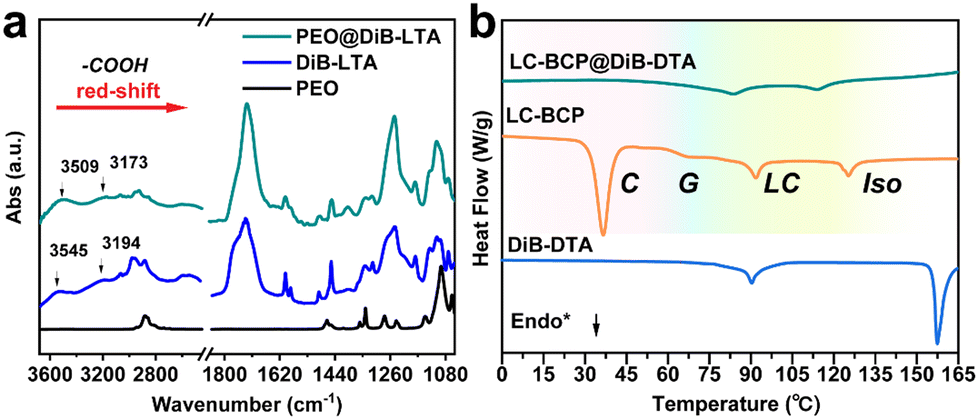 | ||
| Fig. 2 (a) IR spectrum of PEO, DiB-LTA, and PEO@DiB-LTA, respectively. (b) DSC heating curve of LC-BCP, DiB-DTA, and LC-BCP@DiB-DTA; C: crystalline phase; G: glass transition; LC: liquid crystal phase; Iso: isotropic phase. | ||
Similarly, for PEO@DiBE-DTA, the peaks for the hydroxyls in the IR spectra shifted from 3488 cm−1 to 3273 cm−1 and 3432 cm−1 to 3175 cm−1 relative to the neat DiBE-DTA. Those peaks were also broadened, suggesting that the hydroxyl groups of DiBE-DTA formed hydrogen bonds with the ether group of PEO (Fig. S4a, ESI†). The decrease in the crystalline peak of PEO in the DSC spectra of the LC-BCP@DiBE-DTA films also supports this hypothesis (Fig. S4b, ESI†). Therefore, hydrogen bonding between chiral molecules and LC-BCP is likely an important driving force for intermolecular chiral transfer. According to the previous report,31 the chiral expression of the PMA (Azo) block in BCP could be achieved due to the hydrogen bonding between the hydroxyl groups of chiral molecules with the carbonyl groups of the side chains of azobenzene.
Herein, the interaction between DiBTA and the side chain carbonyl group of PMA (Azo) was detected using the monomer Me6 as the model compound. In general, the formation of hydrogen bonding between molecules containing carboxyl and hydroxyl groups leads to a blue shift of the carbonyl and hydroxyl peaks.36 However, the peaks attributed to azobenzene and the carbonyl peaks of molecule were not shifted in the IR spectra for both Me6 samples as before and after doping with DiB-LTA or DiBE-LTA (Fig. S5, ESI†). Given the high crystallinity and the strong π–π interactions between chiral molecules, a possible mechanism for the chiral expression of the PMA (Azo) block is the π–π stacking between the DiBTA or DiBETA and the PMA (Azo) block.37–39 To verify this mechanism, UV spectroscopy and nuclear magnetic resonance (NMR) were used to investigate the supramolecular stacking of PMA (Azo) blocks with DiBTA.
In contrast to the azobenzene absorption peak of the neat LC-BCP at 357 nm, the characteristic UV peak of LC-BCP@DiB-DTA was red-shifted to 378 nm, and the characteristic peak of the benzene ring of DiB-DTA was also red-shifted from 232 nm to 253 nm (Fig. 3a). These results indicated the J-type stacking manner of DiBTA with the PMA (Azo) block. This finding was further confirmed by the NMR data.40 Significant high-field shifts were observed for the hydrogen protons in the chiral carbon and benzene ring of the chiral molecule, and the hydrogen protons of azobenzene ring in LC-BCP due to misaligned parallel stacking between DiBTA and azobenzene (Fig. 3b). In contrast, the formation of H-aggregation by face-to-face stacking of DiBETA with azobenzene resulted in a blue shift in the UV spectrum of LC-BCP@DiBE-DTA (Fig. S6a, ESI†). Meanwhile, the hydrogen protons of the chiral carbon of DiBETA shifted to the down-field, as are the hydrogen protons of the azobenzene ring of LC-BCP and benzene ring of the chiral molecule. (Fig. S6b, ESI†). Thus, we concluded that the chiral transfer in the composite systems arose from π–π stacking between chiral molecules and the azobenzene group of BCP. However, the different types of stacking between DiBTA or DiBETA and LC-BCP resulted in different gabs.
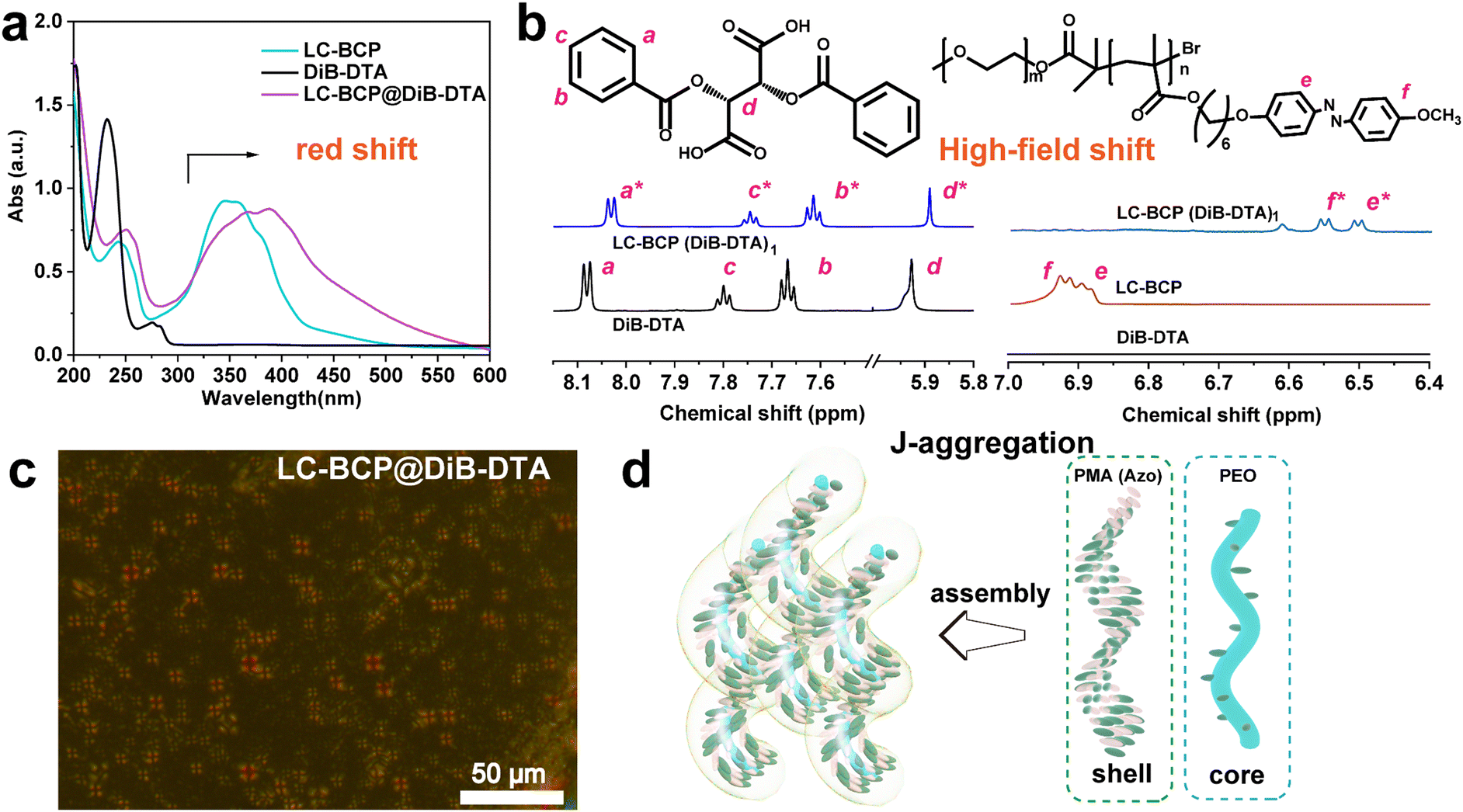 | ||
| Fig. 3 (a) UV spectra and (b) Locally amplified 1H NMR spectra of LC-BCP, DiB-DTA, and LC-BCP@DiB-DTA with a molar ratio of 1. (c) POM picture of LC-BCP@ DiB-DTA at 90 °C. (d) Schematic diagram of the formation of the hierarchical helical structure. | ||
Vibrational circular dichroism (VCD) is an essential tool for determining the chirality of chain conformations. LC-BCP@DiB-LTA and LC-BCP@DiB-DTA films showed mirror VCD signals, benzene skeleton vibrations, carbon–carbon single bonds and carbon–oxygen single bonds (Fig. S7a, ESI†), in contrast to the composite films of LC-BCP doped with the enantiomeric DiBETA, which showed no significant chiral signal in Fig. S7b (ESI†). This result indicates the LC-BCP@DiB-DTA or DiB-LTA films exhibit a high level of chain conformational chirality. As shown in Fig. 3c, polarized optical microscopy (POM) of LC-BCP@DiB-DTA revealed a typical fan-shaped texture of a helical liquid crystal,41 whereas, LC-BCP@DiBE-DTA exhibited a typical columnar nematic schlieren texture (Fig. S8, ESI†). Based on these results, we speculated that the synergistic effect, including the interaction between DiBTA and PEO block by hydrogen bonding (helical core) and the interaction between DiBTA and azobenzene blocks by π–π chiral stacking (shell), caused hierarchical chiral assembly (Fig. 3d).
Different aggregation induced hierarchical chiral morphology
Transmission electron microscopy (TEM) was used to investigate the morphology of the assembled LC-BCP@DiBTA film. To enhance the contrast of the PEO phase during TEM, the PEO phase was stained using RuO4. The bright areas observed in the TEM dark field images are the PEO phase, while the dark areas are the PMA (Azo) phase. It can be seen that the PEO block was a discontinuous phase that was dispersed throughout the main phase of PMMA. The PEO exhibited left helical structures in the LC-BCP@DiB-DTA film (Fig. 4a) and right helical structures in the LC-BCP@DiB-LTA film (Fig. 4c). The small angle X-ray scattering profile also showed typical columnar scattering peaks for LC-BCP@DiB-DTA, and the d-spacing of PEO columns was ∼20 nm (Fig. S9, ESI†). TEM bright field images showed that the diameters of the helices ranged from 22 to 25 nm (Fig. S10, ESI†). As previously mentioned, the hydrogen bonding between DiBTA and PEO blocks caused PEO to form a helical structure in the annealing process of film preparation. Then, molecular dynamics was used to simulate the formation of the PEO@DiBTA helix. First, we built the initial structure, with 15 PEO repeat units and 15 chiral molecules. To eliminate the unreasonable structure, the geometric optimization of the model was performed using smart algorithms. Then, NVT and NPT dynamic simulations were performed to obtain an energy-optimized structure, and a helical structure locally appeared in the polymer chains. After quenching the dynamics simulation, a regular helix with lower energy was formed in which the chiral molecules were arranged around the PEO chain by H-bonding (Fig. S11 and Table S1, ESI†). | ||
| Fig. 4 Dark field TEM image in scanning mode, the inset represents the PEO phase structure (a and c) and AFM image (b and d), the sketch map is the aggregate structure of PMA (Azo) and DiBTA around the PEO helix phase in LC-BCP@DiB-DTA film and LC-BCP@DiB-LTA film, respectively. The sample slice was prepared using a microtome with a thickness of ∼100 nm. | ||
The three-dimensional helical structure was observed by AFM (Fig. 4b and d). The diameter of the helix (∼30 nm) is larger than that of the helical structure of PEO from the TEM image (Fig. S12, ESI†), thus, suggesting that the PMA(Azo) block (bright portion) was further assembled around the PEO helix. As the PMA (Azo) block was covalently bonded to the PEO block, the azobenzene could form J-aggregations through the introduction of DiBTA around the PEO helix. In contrast, we found that the supramolecular assembly of LC-BCP@DiBETA exhibited a columnar structure rather than helices (Fig. S13, ESI†).
To further assess the different stacking arrangements in DiBTA- and DiBETA-doped systems, the stacking energies of azobenzene and DiBTA or DiBETA were calculated by molecular dynamic (Table 1). The result showed that Azo-DiBETA exhibited no helical arrangement, and the energy required for the assembly was larger than that of the AZO-DiBTA helical system (Fig. 5). Therefore, the helix consisting of the J-type stacking of Azo-DiBTA exhibited optimal energy (Table 1). The large gabs in LC-BCP@DiBTA likely originated from the PMA (Azo) block gathering around the PEO@DiBTA in J-aggregations mode.
| Model | E 0 | E 1 | E 2 | E 3 |
|---|---|---|---|---|
| Units: kcal mol−1 | ||||
| AZO-DiBTA | 50.46 | −49.38 | −36.04 | −44.28 |
| AZO-DiBETA | 210.67 | 86.72 | 127.32 | 105.21 |
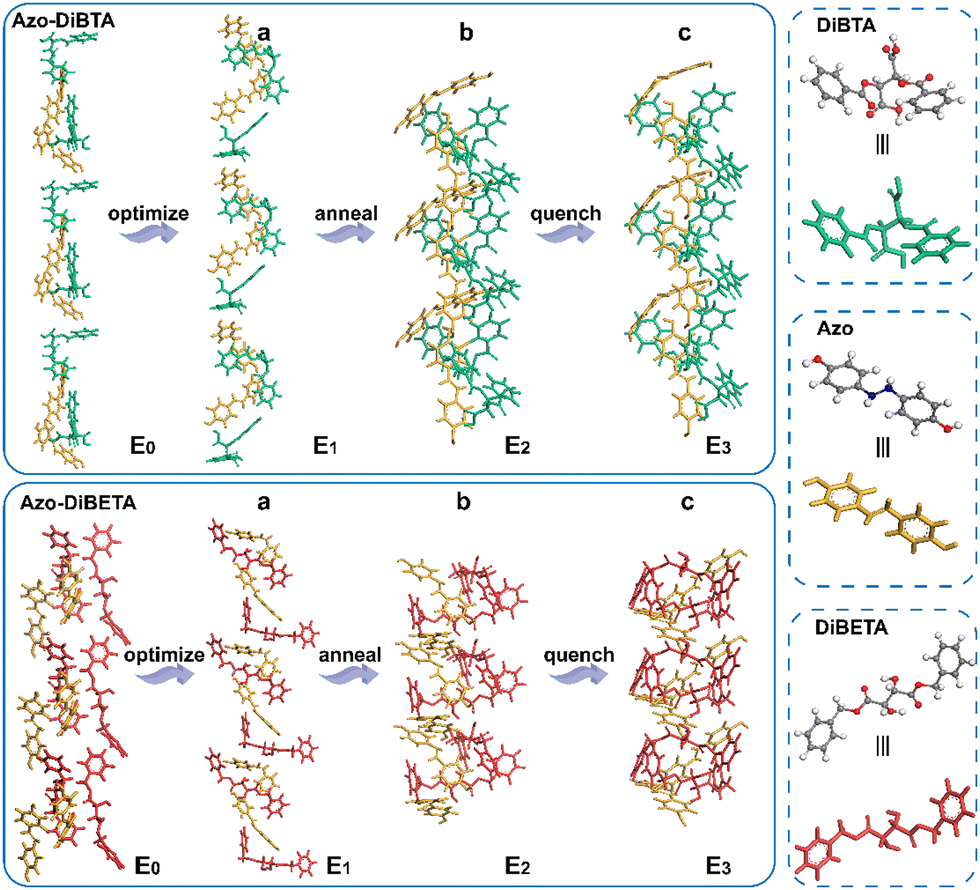 | ||
| Fig. 5 Energy-optimized structure (a), NVT, and NPT annealing dynamic simulation (b), and quenching dynamic simulation (c). | ||
Chiral reversible response
According to the previous report, the chiral polymer can be endowed with controllable properties through photo and thermal cis-trans isomerization of azobenzene.29,42 When the annealed LC-BCP@DiBTA film was exposed to UV light at 365 nm for 10 s, the CD signal of azobenzene disappeared from 275 to 425 nm due to the disruption of the organization of the azobenzene liquid crystal, only PEO columns generated by microphase separation were retained (Fig. 6b). Meanwhile, the original in plane-oriented columnar structure was changed to an out of plane-oriented structure. Such a phenomenon has also been revealed in other reports.43,44 Moreover, the helical structure of the azobenzene groups could be recovered by thermal annealing at the liquid crystal phase temperature (90–120 °C). The CD signal of azobenzene was also recovered following 10 s of thermal annealing, and the gabs gradually increased over time (Fig. S14, ESI†). This finding could be due to the conversion of cis-azobenzene to trans-azobenzene upon heating, followed by the recovery of helical π–π stacking. After 2 h of thermal annealing, the hierarchical helical assembly morphology was also reconstructed (Fig. 6c). Thus, the chiral response film exhibits rapid conversion under photo-thermal conditions and may be a potential material for chiral optical storage materials.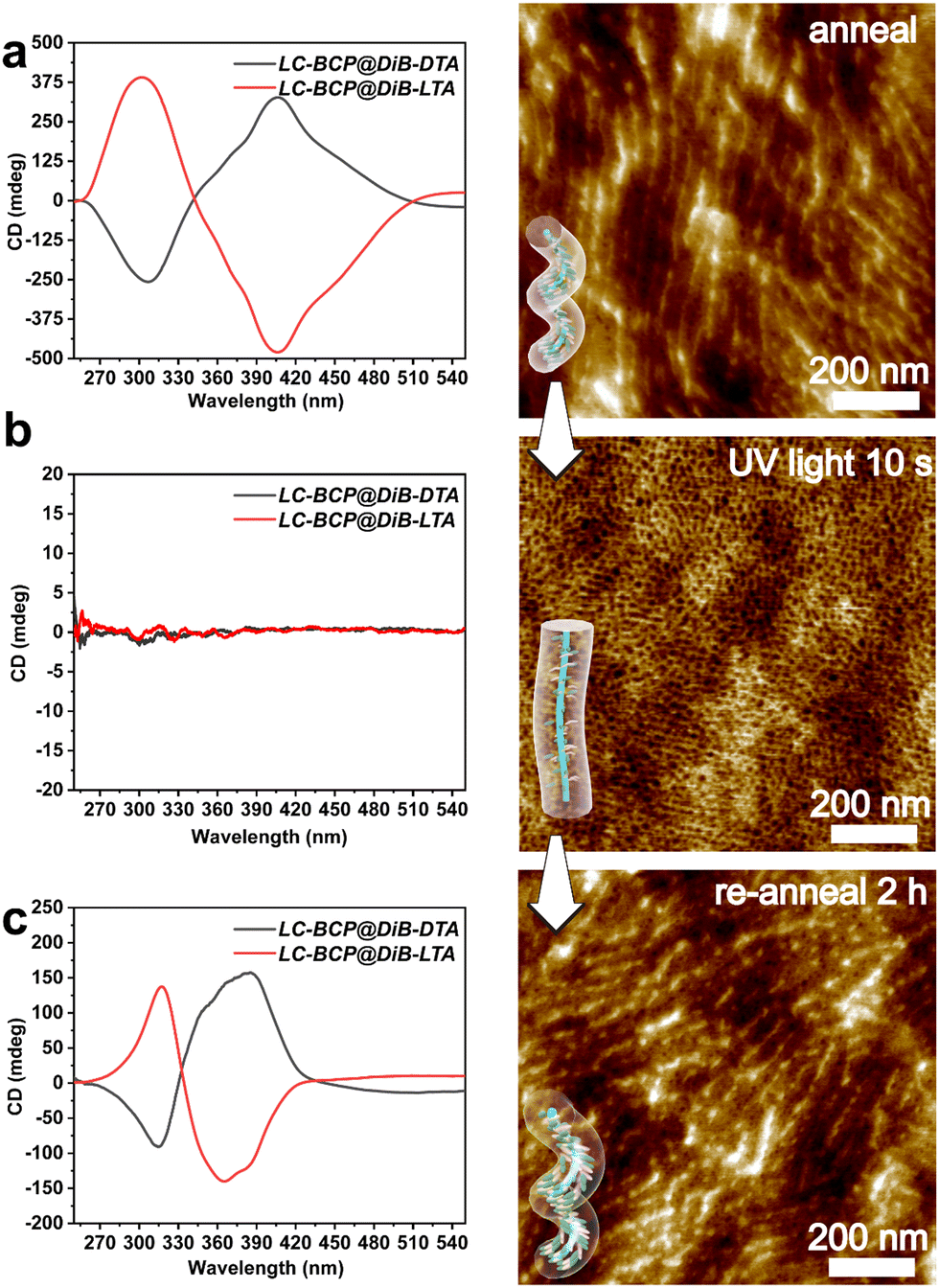 | ||
| Fig. 6 CD spectra and AFM image of LC-BCP@DiB-LTA film before (a) and after (b) UV irradiation (10 s, 365 nm,150 mW cm−2), and the recovered helical structure after re-annealing for 2 h (c). | ||
Conclusions
In summary, we constructed supramolecular helical structures with different self-assemblies in LC-BCP induced by exogenous chiral molecules. Hydrogen bonding between the chiral molecule DiBTA/DiBETA and PEO, and π–π stacking between azobenzene and DiBTA/DiBETA, induced the chiral transfer and chiral amplification. Specifically, the supramolecular J-aggregation of DiBTA with LC-BCP induced the formation of supramolecular helical structures, resulting in chiral films with high gabs. In addition, such films exhibited chiral switching properties, and the chiral information could be optically erased and thermally recovered. This strategy provided a pathway for the fabrication of chiral optical materials and memory storage materials with large asymmetries.Experiment and methods
Characterization
Nuclear magnetic resonance (NMR) spectroscopy was carried out on a 600 MHz AVANCE III HD NMR spectrometer. Deuterated chloroform was used as the NMR solvent.Polarizing microscopy (POM, Leica DM LP) was used to observe the texture of liquid-crystal block copolymers.
Differential scanning calorimetry (DSC) was performed on a Discovery DSC 250 instrument. The thermodynamic properties of the pure polymer films and polymer films doped with chiral molecules were characterized under a nitrogen atmosphere at a heating and cooling rate of 10 °C min−1.
Circular dichroism (CD) spectra were acquired on a JASCO J-1500 and UV-vis spectra were recorded on a Shimadzu UV-2600 UV-vis spectrometer. The bulk films of PEO-PMA (Azo) blended with DiB-D/LTA or DiBE-D/LTA were prepared by drop-casting a tetrahydrofuran solution on quartz slides. The films were dried at room temperature, followed by thermal annealing below the clear point of the liquid-crystal block copolymers. During the testing process, the sample was rotated by 45-, 90-, and 180 degrees to eliminate the effect of linear dichroism.
The infrared spectra (IR) were recorded on a Thermofisher Fourier IS-5 transform (FT) IR spectrometer. The samples of PEO-PMA (Azo), DiBTA, PEO-PMA (Azo) blended with DiB-DTA, and PEO-PMA (Azo) blended with DiB-LTA were prepared in THF at a concentration of ∼10 mg mL−1. The THF solution was applied dropwise to the BaF2 substrate and the annealing conditions were the same as those used to prepare the CD film.
The vibrational circular dichroism (VCD) spectra were acquired on a vibrational circular dichroism spectrometer (Chiral IR-2X).
Small angle X-ray scattering (SAXS) measurements were performed at the Shanghai Synchrotron Radiation Facility (SSRF).
Transmission electron microscopy (TEM) measurements were obtained on a JEOL JEM 2100 TEM instrument and Talos F200S G2 TEM instrument with an accelerating voltage of 200 kV. The annealed films were embedded in epoxy resin and cured overnight at 60 °C. Thin sections of ∼100 nm was prepared using a Leica ultra-cut UCT microtome. The inner morphology was assessed using atomic force microscopy (AFM) on a Dimension FastScan Bio microscope.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (grant no. 21574081, 21975156 and 51733007).Notes and references
- F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS PubMed.
- K. Akagi, T. Yamashita, K. Horie, M. Goh and M. Yamamoto, Adv. Mater., 2020, 32, 1906665 CrossRef CAS PubMed.
- M. Goh, M. Kyotani and K. Akagi, J. Am. Chem. Soc., 2007, 129, 8519–8527 CrossRef CAS PubMed.
- N. Lu, X. Gao, M. Pan, B. Zhao and J. Deng, Macromolecules, 2020, 53, 8041–8049 CrossRef CAS.
- J. Hong, S. Kim, G. Park, Y. Lee, H. Kim, S. Kim, T.-W. Lee, C. Kim and Y. You, Chem. Sci., 2021, 12, 8668–8681 RSC.
- M. Pan, R. Zhao, B. Zhao and J. Deng, Macromolecules, 2021, 54, 5043–5052 CrossRef CAS.
- H. Yamamoto, T. Inagaki, J. Park, S. Yoshida, K. Kaneko, T. Hanasaki and K. Akagi, Macromolecules, 2021, 54, 8977–8986 CrossRef CAS.
- B. Zhao, X. Gao, K. Pan and J. Deng, ACS Nano, 2021, 15, 7463–7471 CrossRef CAS PubMed.
- S. Vignolini, N. A. Yufa, P. S. Cunha, S. Guldin, I. Rushkin, M. Stefik, K. Hur, U. Wiesner, J. J. Baumberg and U. Steiner, Adv. Mater., 2012, 24, 23–27 CrossRef PubMed.
- G. Albano, G. Pescitelli and L. Di Bari, Chem. Rev., 2020, 120, 10145–10243 CrossRef CAS PubMed.
- B. Nowacki, H. Oh, C. Zanlorenzi, H. Jee, A. Baev, P. N. Prasad and L. Akcelrud, Macromolecules, 2013, 46, 7158–7165 CrossRef CAS.
- Y. Wang, Y. Li, S. Liu, F. Li, C. Zhu, S. Li and Y. Cheng, Macromolecules, 2016, 49, 5444–5451 CrossRef CAS.
- K. Watanabe, I. Osaka, S. Yorozuya and K. Akagi, Chem. Mater., 2012, 24, 1011–1024 CrossRef CAS.
- B. A. San Jose, S. Matsushita and K. Akagi, J. Am. Chem. Soc., 2012, 134, 19795–19807 CrossRef CAS PubMed.
- K. Liu, Y. Shen, X. Li, Y. Zhang, Y. Quan and Y. Cheng, Chem. Commun., 2020, 56, 12829–12832 RSC.
- H.-F. Wang, K.-C. Yang, W.-C. Hsu, J.-Y. Lee, J.-T. Hsu, G. M. Grason, E. L. Thomas, J.-C. Tsai and R.-M. Ho, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4080–4089 CrossRef CAS PubMed.
- R.-M. Ho, M. C. Li, S.-C. Lin, H.-F. Wang, Y.-D. Lee, H. Hasegawa and E. L. Thomas, J. Am. Chem. Soc., 2012, 134, 10974–10986 CrossRef CAS PubMed.
- W. Zhang, K. Yoshida, M. Fujiki and X. Zhu, Macromolecules, 2011, 44, 5105–5111 CrossRef CAS.
- Y. Zhao, N. A. Abdul Rahim, Y. Xia, M. Fujiki, B. Song, Z. Zhang, W. Zhang and X. Zhu, Macromolecules, 2016, 49, 3214–3221 CrossRef CAS.
- L. Yang, F. Wang, D.-I. Y. Auphedeous and C. Feng, Nanoscale, 2019, 11, 14210–14215 RSC.
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1900110 CrossRef CAS PubMed.
- Y. Wang, X. Li, F. Li, W.-Y. Sun, C. Zhu and Y. Cheng, Chem. Commun., 2017, 53, 7505–7508 RSC.
- T. Wen, H.-F. Wang, M.-C. Li and R.-M. Ho, Acc. Chem. Res., 2017, 50, 1011–1021 CrossRef CAS PubMed.
- S. Huang, Y. Chen, S. Ma and H. Yu, Angew. Chem., Int. Ed., 2018, 57, 12524–12528 CrossRef CAS PubMed.
- R. M. Ho, Y. W. Chiang, C. K. Chen, H. W. Wang, H. Hasegawa, S. Akasaka, E. L. Thomas, C. Burger and B. S. Hsiao, J. Am. Chem. Soc., 2009, 131, 18533–18542 CrossRef CAS PubMed.
- L. Yao, X. Lu, S. Chen and J. J. Watkins, Macromolecules, 2014, 47, 6547–6553 CrossRef CAS.
- Q. Li, J. Yuan, H. Liang, F. Zheng, X. Lu, C. Yu and Q. Lu, ACS Nano, 2020, 14, 8939–8948 CrossRef CAS PubMed.
- C.-C. Yang, P. Puneet, I. M. Lin, Y.-W. Chiang and R.-M. Ho, Giant, 2021, 7, 100059 CrossRef CAS.
- T. Miao, X. Cheng, H. Ma, Z. He, Z. Zhang, N. Zhou, W. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2021, 60, 18566–18571 CrossRef CAS PubMed.
- X. Cheng, T. Miao, L. Yin, Y. Ji, Y. Li, Z. Zhang, W. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2020, 59, 9669–9677 CrossRef CAS PubMed.
- J. Yuan, X. Lu, Q. Li, Z. Lü and Q. Lu, Angew. Chem., Int. Ed., 2021, 60, 12308–12312 CrossRef CAS PubMed.
- J. Yuan, X. Lu, S. Zhang, F. Zheng, Q. Deng, L. Han and Q. Lu, Macromolecules, 2022, 55, 1566–1575 CrossRef CAS.
- R. M. Tejedor, L. Oriol, J. L. Serrano, F. PartalUreña and J. J. LópezGonzález, Adv. Funct. Mater., 2007, 17, 3486–3492 CrossRef CAS.
- J. Lu, Y. Xue, K. Bernardino, N.-N. Zhang, W. R. Gomes, N. S. Ramesar, S. Liu, Z. Hu, T. Sun, A. F. D. Moura, N. A. Kotov and K. Liu, Science, 2021, 371, 1368–1374 CrossRef CAS PubMed.
- L. Yao and J. J. Watkins, ACS Nano, 2013, 7, 1513–1523 CrossRef CAS PubMed.
- B. Su, S. Wang, Y. Wu, X. Chen, Y. Song and L. Jiang, Adv. Mater., 2012, 24, 2780–2785 CrossRef CAS PubMed.
- B. Roy, T. Noguchi, Y. Tsuchiya, D. Yoshihara, T. Yamamoto and S. Shinkai, J. Mater. Chem. C, 2015, 3, 2310–2318 RSC.
- S. J. George, R. de Bruijn, Ž. Tomović, B. Van Averbeke, D. Beljonne, R. Lazzaroni, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2012, 134, 17789–17796 CrossRef CAS PubMed.
- S. Huang, H. Yu and Q. Li, Adv. Sci., 2021, 8, 2002132 CrossRef PubMed.
- R. Wang, X. Li, J. Bai, J. Zhang, A. Liu and X. Wan, Macromolecules, 2014, 47, 1553–1562 CrossRef CAS.
- K. Yano, Y. Itoh, F. Araoka, G. Watanabe, T. Hikima and T. Aida, Science, 2019, 363, 161–165 CrossRef CAS PubMed.
- Z. Liu, Y. Yao, X. Tao, J. Wei and S. Lin, ACS Macro Lett., 2021, 10, 1174–1179 CrossRef CAS PubMed.
- S. Nagano, Y. Koizuka, T. Murase, M. Sano, Y. Shinohara, Y. Amemiya and T. Seki, Angew. Chem., Int. Ed., 2012, 51, 5884–5888 CrossRef CAS PubMed.
- T. Seki, J. Mater. Chem. C, 2016, 4, 7895–7910 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00103b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |