
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVitrimerization of crosslinked elastomers: a mechanochemical approach for recycling thermoset polymers†
Alireza
Bandegi
 a,
Thomas G.
Gray
*b,
Sarah
Mitchell
c,
Amin
Jamei Oskouei
a,
Michelle K.
Sing
c,
Jayme
Kennedy
c,
Kimberly
Miller McLoughlin
c and
Ica
Manas-Zloczower
*a
a,
Thomas G.
Gray
*b,
Sarah
Mitchell
c,
Amin
Jamei Oskouei
a,
Michelle K.
Sing
c,
Jayme
Kennedy
c,
Kimberly
Miller McLoughlin
c and
Ica
Manas-Zloczower
*a
aDepartment of Macromolecular Science and Engineering, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH 44106, USA. E-mail: ixm@case.edu
bDepartment of Chemistry, Case Western Reserve University, 10900 Euclid Avenue, Cleveland, OH 44106, USA. E-mail: txg37@case.edu
cBraskem America, 550 Technology Drive, Pittsburgh, PA 15219, USA
First published on 2nd June 2023
Abstract
Crosslinked poly (ethylene-vinyl acetate) (EVA) is extensively used in a wide range of applications including solar cell encapsulates and athletic shoes. However, recycling large amounts of crosslinked EVA (or EVA thermoset) waste is still a global challenge. Converting a permanent crosslinked network into a dynamic exchangeable network through a vitrimerization process is a cost effective and environmentally friendly method for recycling thermoset waste already present in the market. In this work, crosslinked EVA is vitrimerized with a catalyst via a mechanochemical approach. Zinc acetate is added as a catalyst and poly(vinyl alcohol) as a feedstock of hydroxyl groups to promote a dynamic exchange reaction that occurs via a transesterification mechanism. The vitrimerized EVA was reprocessed via extrusion and compression molding three times at low temperatures without additional catalyst or hydroxyl groups. This study shows that converting crosslinked EVAs into vitrimers using a mechanochemical approach is effective for three crosslinked EVA samples with different topology and vinyl acetate content enabling EVA waste to be recycled and reprocessed into value-added products.
Introduction
Poly(ethylene-vinyl acetate) (EVA) is a copolymer of ethylene and vinyl acetate (VA) frequently used as a commodity plastic. The properties of EVA are mainly controlled by the VA content.1–3 In most applications, EVA is used as a crosslinked material that has been cured in the presence of a free radical initiator such as peroxide to form a three-dimensional network. Crosslinked EVA materials are used in a range of applications such as insulation materials, cables, photovoltaic modules, and shoe soles.4–6 However, crosslinked EVAs cannot be recycled or reused due to their high thermal and chemical stability.One way to address the challenge of EVA recyclability involves converting the permanently crosslinked network into a covalent adaptive network (CAN). In CANs, dynamic crosslinks are incorporated into a polymer network to promote an exchange reaction that leads to topology rearrangement of the polymer network. Consequently, CANs can be reprocessed similar to thermoplastic materials without loss in mechanical properties.7–9 In 2011, Leibler and co-workers proposed a new class of materials, vitrimers, by adopting CANs in a crosslinked polyester network using a classical transesterification reaction.10 They showed that the efficient exchange reactions in vitrimers allow for topological rearrangement at high temperatures and result in rapid stress relaxation while preserving network integrity.10 Subsequently, different types of vitrimers based on dynamic chemistries such as dioxaborolane metathesis,11 boronic ester,12 vinylogous urethanes,13 transesterification14 and disufides15 have been developed. Each of these chemistries relax their stress under imposed strain which indicates reprocessability. Most vitrimers can be reprocessed through conventional techniques for processing thermoplastic materials such as extrusion,13 melt blowing,16,17 injection molding,10 and compression molding.18
In previous work, a vitrimer-like crosslinked EVA was developed through cross-linking thermoplastic EVA with a dynamic cross-linker (i.e. triethyl borate).19 The EVA vitrimer showed enhanced thermal stability and mechanical properties with up to two times enhancement in Young's modulus and storage modulus compared with the thermoplastic EVA.19 However, this method is only applicable for thermoplastic EVAs. While the presence of dynamic crosslinks enhances the properties of thermoplastic EVA while preserving processability, the triethyl borate crosslinker cannot be applied to permanently crosslinked EVA using commercially relevant techniques. Recent work has shown that it is possible to recycle thermoset waste by incorporating dynamic crosslinks while ball milling to produce a vitrimer.20 Ball milling is a more viable approach as it is more economical, environmentally friendly, and commercially scalable.20–28 The incorporation of an appropriate catalyst while ball milling a permanently crosslinked network results in the formation of metal–ligand bonds. The vitrimerization happens through compression molding of the ball milled powder at elevated temperatures which results in the formation of dynamic crosslinked network.
Herein, a mechanochemical approach was used to recycle crosslinked EVAs into high value-added products. The presence of ester groups in the EVA networks allows for vitrimerimation to be accomplished with transesterification catalysts, specifically zinc acetate, a highly efficient and non-toxic catalyst.23,27 To enable a transesterification reaction, a feedstock of hydroxyl groups must be added in the ball milling stage.23,29–31 Vitrimerization of crosslinked EVAs was achieved by cryomilling crosslinked EVA powder, zinc acetate as the catalyst, and poly(vinyl alcohol) (PVOH) as the feedstock for OH groups, followed by compression molding. Fourier transform infrared spectroscopy (FTIR) confirms the formation of ester–zinc complexes during vitrimerization, which is also supported by DFT calculations. Furthermore, stress relaxation experiments demonstrate the existence of dynamic crosslinked networks in the vitrimerized samples. The vitrimerized EVA were also reprocessed at least three times without loss in mechanical properties to demonstrate the recyclability of the networks.
Experimental section
Materials
All three EVA grades were supplied by Braskem. PVOH, zinc acetate, and dicumyl peroxide (DCP) were purchased from Sigma-Aldrich and used as received.Compounding and crosslinking
A micro-compounder (Xplore) was used to mix DCP with EVA granules. Temperature zones were held constant at 100 °C (close to melting temperature of EVA), the screw speed was 30 rpm, and the die port was held open. Each extrudate strand was collected, chopped (3–5 mm), and fed to the micro-compounder for a second pass. All EVA/DCP blends were crosslinked using compression molding (Carver press) at 175 °C for 15 minutes under constant pressure of 1 MPa. The molds were 1 mm thick. Molar feed ratios are reported in Table 1.| Sample | Wt% VA (1H-NMR)32 | Nominal MFRa (190 °C) | M w (g mol−1)32 | [VA]/[DCP] (mol mol−1) | PVOH (wt%) | Zinc acetate (wt%) |
|---|---|---|---|---|---|---|
| a Melt flow rate from datasheet. b Molecular weight of initial thermoplastic EVAs. | ||||||
| EVA-V-1 | 19 | 8.0 | 83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 679 679 |
30 ± 0.03 | 12.3 | 2.5 |
| EVA-V-2 | 22 | 2.1 | 103591 |
35 ± 0.03 | 14.1 | 2.9 |
| EVA-V-3 | 9 | 2.0 | 100604 |
14 ± 0.07 | 6.5 | 1.4 |
Vitrimerization process
Cryomilled powders (<200 μm) were obtained by cryomilling fine EVA particles (<1 mm) with catalyst (zinc acetate) and PVOH in a cryomill tank (Retsch Cryomill), purged with N2 (g). Each cryomilling process was 47 minutes with 3 cycles of grinding for 15 minutes at a frequency of 30 Hz and 2 intermediate grindings for 1 minute at frequency of 5 Hz. The cryomilled powders did not contain any large particle and showed a narrow size distribution of fine powders (<200 μm) (Fig. S1, ESI†). Compression molding of the cryomilled powder mixtures (1.5–2 gram) was performed at 175 °C and 13.5 MPa with 10 minutes preheating and 15 minutes heating and applying pressure in the mold (area of 4.9 cm2 and thickness of 1 mm), to obtain the vitrimerized samples (Fig. 1).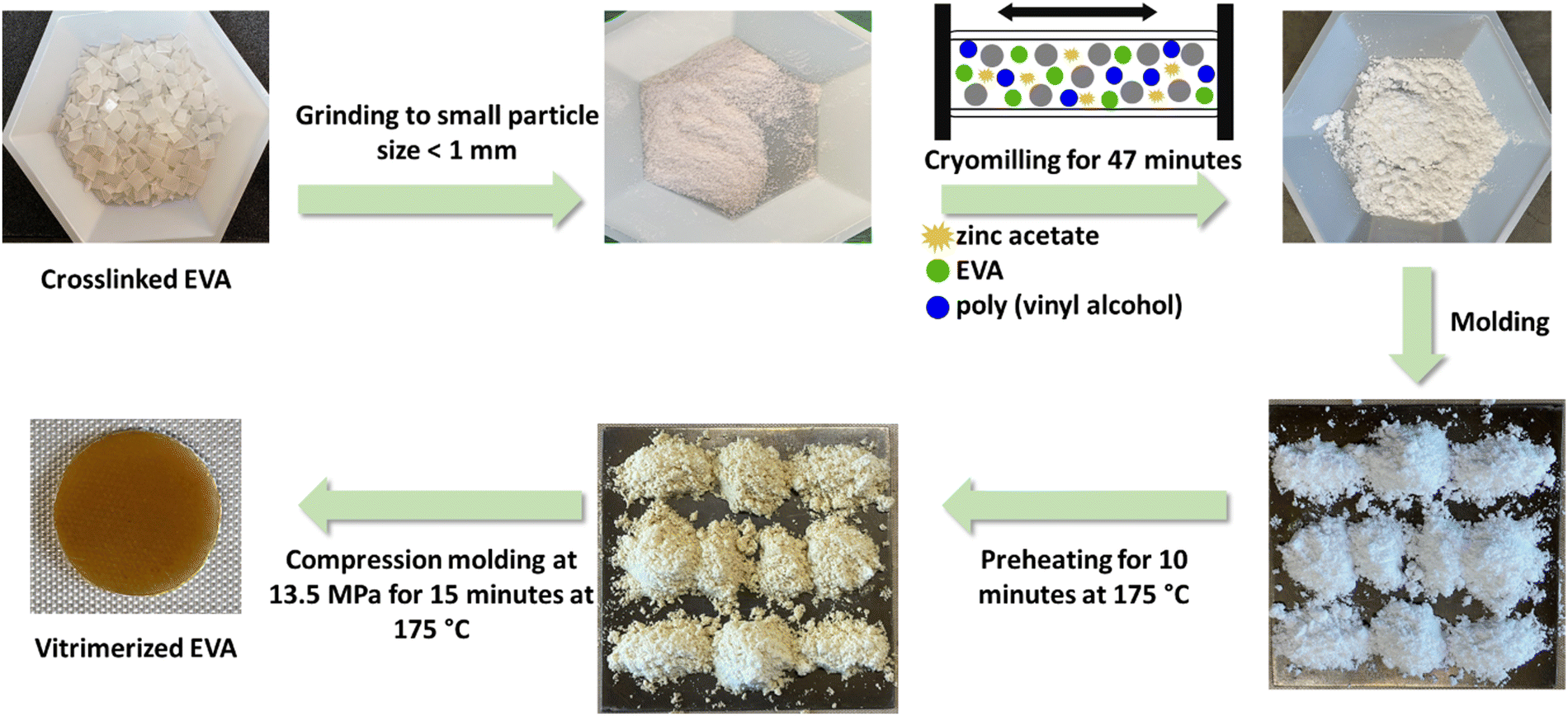 | ||
| Fig. 1 Vitrimerization process of crosslinked EVA. | ||
Reprocessing procedure
First, the compression molded vitrimerized samples were cut into small pieces (3–5 mm) and then extruded using a counter-rotating minilab twin screw extruder operating at 120 °C and screw speed of 10 rpm with a residence time of 3 minutes. Then, the extruded strands were cut into small pieces (3–5 mm, 0.5 gram) and compression molded (area of 4.9 cm2 and thickness of 1 mm) at 175 °C for 15 minutes at 13.5 MPa. These samples were cut again into small pieces and compression molded again under the same processing conditions.Characterization
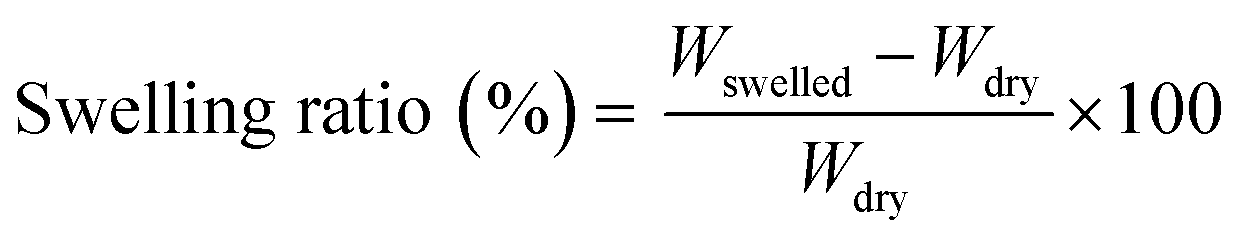
Calculations
Spin-restricted density-functional theory computations were performed within Gaussian16 rev. A.03.33 Calculations utilized the PBE0 exchange–correlation functional of Perdew, Burke, and Ernzerhof,34 and the def2tzvp basis set.35,36 Optimizations proceeded using tight convergence criteria and an ultrafine integration grid. Harmonic vibrational frequencies confirmed stationary points as minima or saddle points of the potential energy hypersurface. All calculations include Grimme's empirical dispersion correction with Becke–Johnson damping.37 Standard Mulliken and Mayer population analyses were performed with the AOMix-CDA program of Gorelsky.38,39Results and discussion
Formation of EVA vitrimer
The vitrimerization process was performed with three different crosslinked EVAs with varying VA content and molecular weight (Table 1), a transesterification catalyst, and feedstock hydroxy groups. To initiate vitrimerization, the dynamic crosslinks are installed into the crosslinked EVA network through cryomilling crosslinked EVAs with the transesterification catalyst and feedstock hydroxy groups. For each vitrimerization, the concentration of transesterification catalyst (zinc acetate) was kept constant, with 8 mol% zinc acetate respective to the VA content. The concentration of feedstock hydroxy groups (PVOH) was also kept constant with a 1.5 molar ratio of hydroxyl groups to VA groups ([OH]/[VA] = 1.5). During cryomilling, zinc atoms from the zinc acetate coordinate to VA groups throughout the crosslinked EVA networks to form ester–zinc complexes (Fig. 2(a)). These complexes serve as dynamic crosslink sites throughout the crosslinked network and will undergo a transesterification reaction at high temperatures with hydroxy groups from PVOH.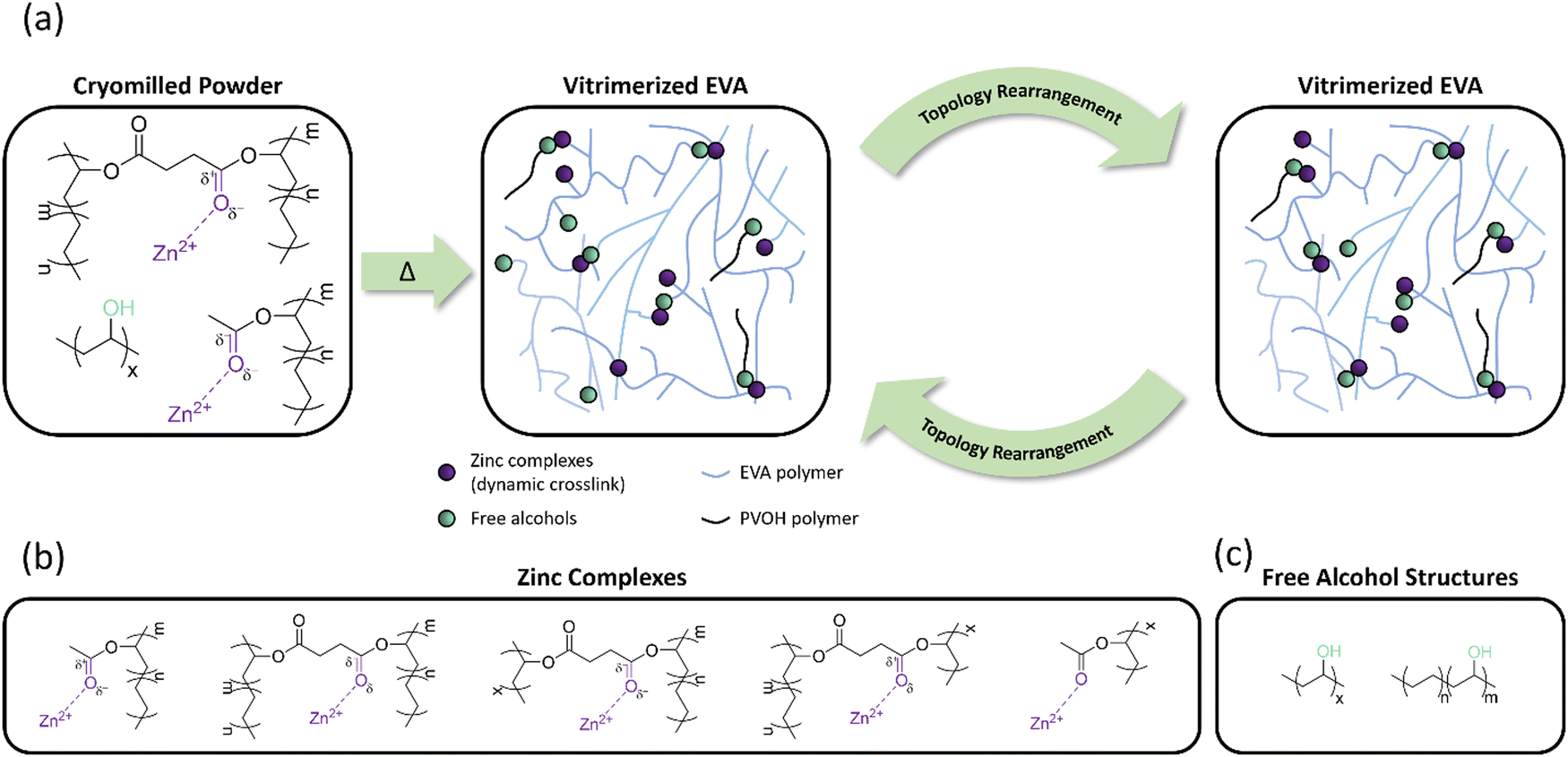 | ||
| Fig. 2 (a) Vitrimerization of cryomilled powder of crosslinked EVA and topology rearrangement within the vitrimerized network during reprocessing at elevated temperatures. (b) Potential zinc-complexes in the vitrimerized network. (c) Potential free hydroxyl groups in the vitrimerized network. | ||
FTIR was used to confirm the formation of ester–zinc complexes and the need for feedstock hydroxy groups (Fig. 3(a)). In the initially crosslinked EVA spectra there is no peak related to hydroxyl groups (3200 to 3600 cm−1), indicating the need to add a source of hydroxy groups via PVOH.20,21 The peak related to ester–zinc complex appears at 1600–1500 cm−1 with low and broad intensity relative to the ester functional groups (Fig. 3(b)).40 This peak is present in each of the three EVA vitrimer spectra, whereas as no peak is present for the initially crosslinked EVA samples. This indicates the formation of ester–zinc complexes throughout the network after cryomilling.
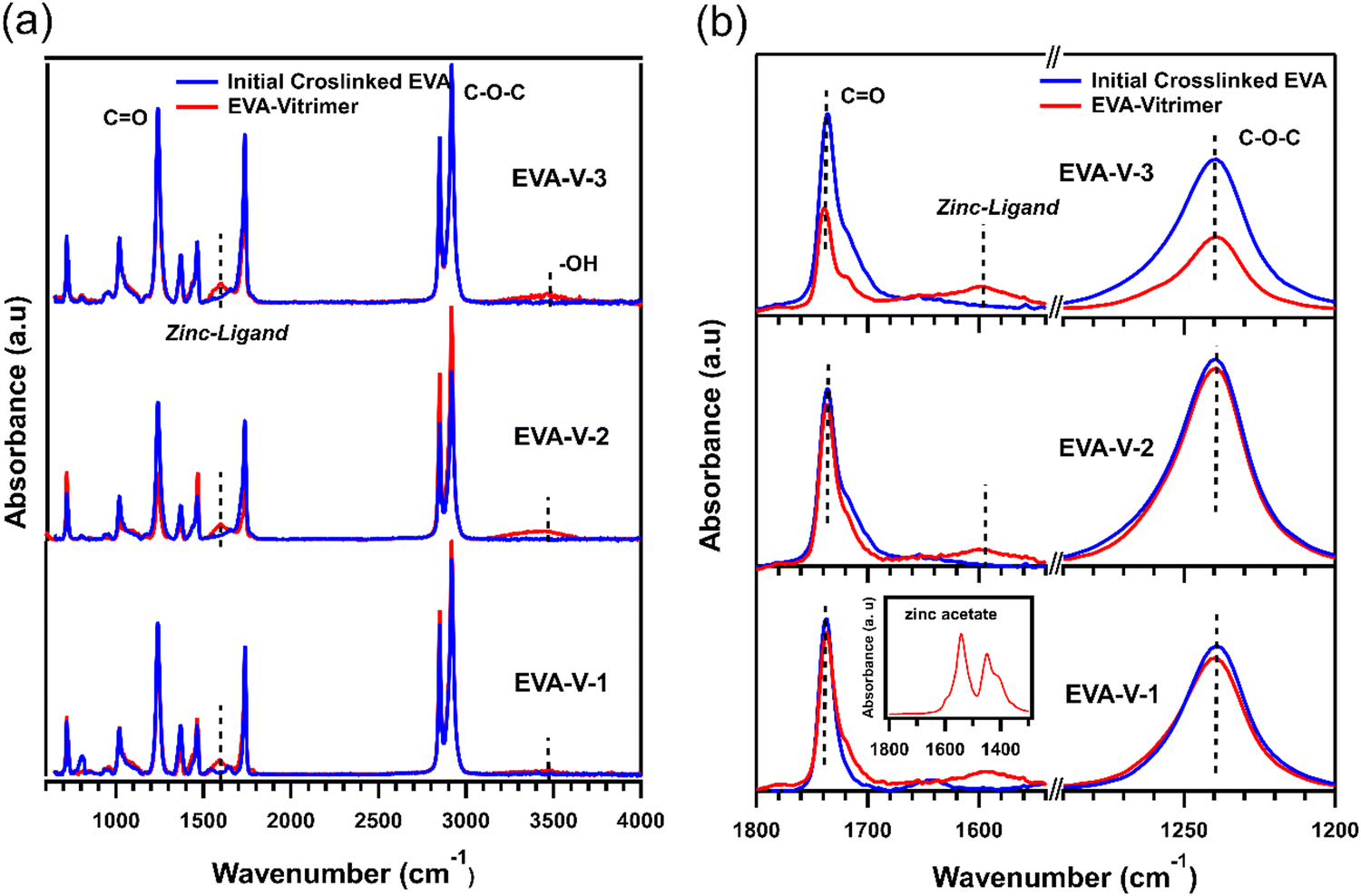 | ||
| Fig. 3 (a) Full FTIR spectra of initial crosslinked and vitrimerized EVAs, (b) the magnified spectrum range related to ester functional groups (1737 cm−1) and zinc–ligand complex (1600–1500 cm−1). The inset shows the magnified spectrum related to zinc acetate. | ||
Once cryomilling occurs and dynamic crosslinks are inserted throughout the crosslinked network, a transesterification reaction is initiated via compression molding the cryomilled powders. The transesterification reaction occurs between ester–zinc complexes and free hydroxy groups present in the system (Fig. 2). Since zinc acetate can complex to both free and crosslinked VA groups, both types of ester–zinc complexes can contribute to the transesterification reactions during the vitrimerization process resulting in a complex vitrimerized network. After the first processing cycle of the vitrimerized network the systems consist of a complex mixture of ester–zinc complexes and free hydroxyl groups (Fig. 2(b) and (c)). Therefore, topology rearrangement of vitrimerized EVA occurs through a combination of exchange reactions between any ester and hydroxyl group present in the system at elevated temperatures during compression molding.
To confirm the formation of a vitrimer network, stress relaxation for each of the vitrimerized EVAs was obtained (Fig. 4a). Characteristic of a vitrimer network, exchange reactions are activated at high temperatures leading to topology rearrangements and stress relaxation. Each vitrimerized EVA exhibited stress relaxation at different starting temperatures. For comparison, the stress relaxation of the initial crosslinked EVAs, EVAs cryomilled without catalyst, EVAs cryomilled without PVOH, and EVAs cryomilled without catalyst and PVOH were measured. Each of these control samples exhibited slower relaxation rates compared to the vitrimerized EVAs (Fig. S2, ESI†). The stress relaxation results confirm that the crosslinked EVA networks were successfully vitrimerized and bond exchange reactions occurred within the crosslinked networks.
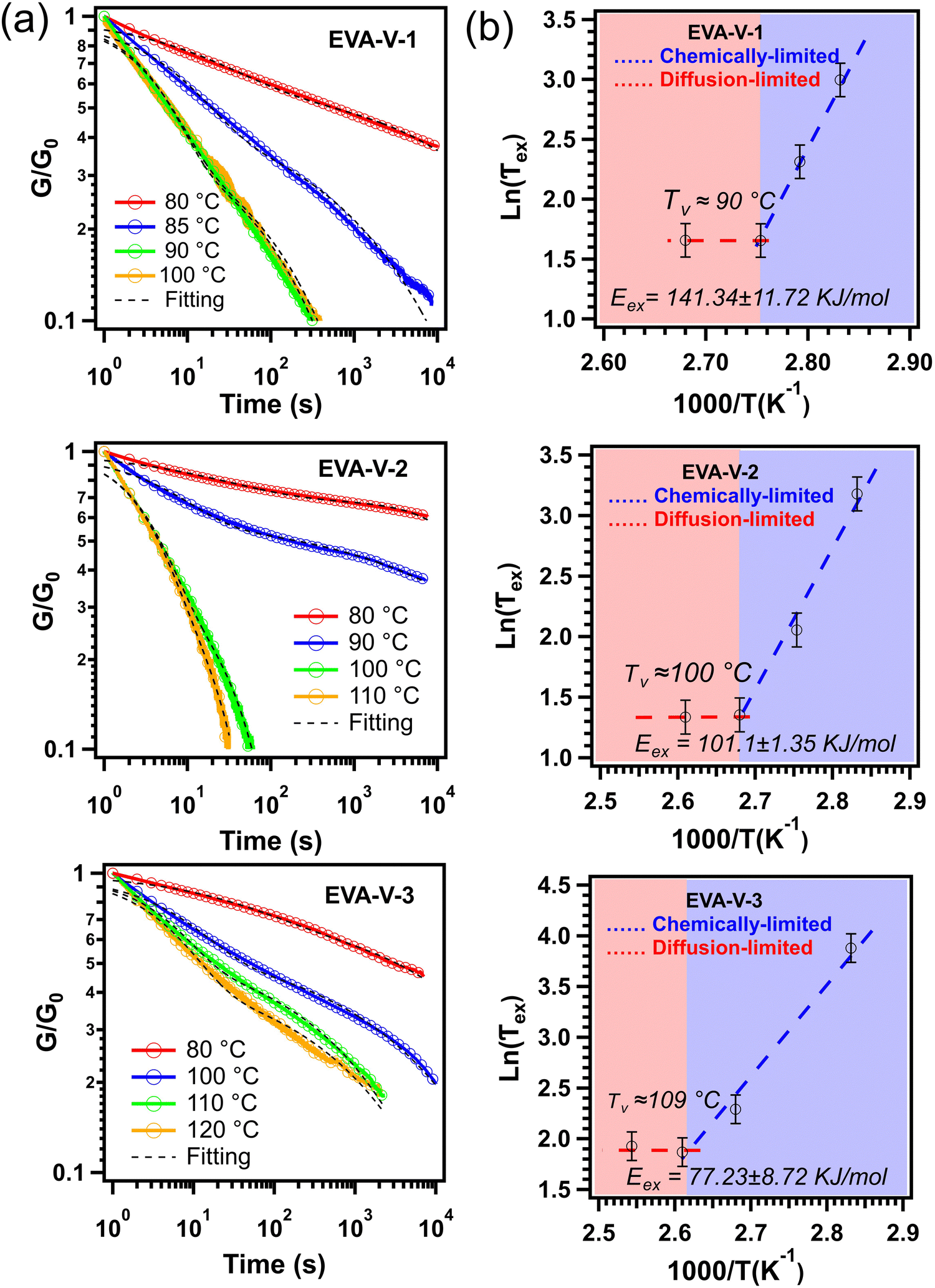 | ||
| Fig. 4 (a) Stress relaxation of vitrimerized EVAs at different temperatures and (b) Arrhenius plots of the measured relaxation times (black dashed lines are fitting to eqn (1)). The G0 is considered as initial stress at time equal to one second (t = 1 s). The error bars for the relaxation times graphed in the plots to calculate the activation energy reflect the sample variability, which was determined by performing stress relaxation on five separate samples of EVA-V-3 at 120 °C (Fig. S2d, ESI†). | ||
In most studies on vitrimer systems, a characteristic relaxation time, τ*(T), is obtained from a simple Maxwell model and is considered as the time required to relax the stress to 1/e of its initial value.41–45 In this work, due to the sharp transition at the initial stage of stress relaxation, a single exponential decay cannot describe quantitatively the stress relaxation of the vitrimerized networks. Previous work has demonstrated that for more complex vitrimer systems, stress relaxation can be deconvoluted into two characteristic times: a long time corresponding to the network relaxation (“net”) and a short time corresponding to the bond exchange reaction (“ex”).46,47 Given the commercial scale of the starting materials and their resultant dispersity, it also fits that the characteristic relaxation times would reflect a distribution of times and thus the relaxation behavior should follow a Kohlrausch–Williams–Watts (KWW) stretched exponential decay (1):
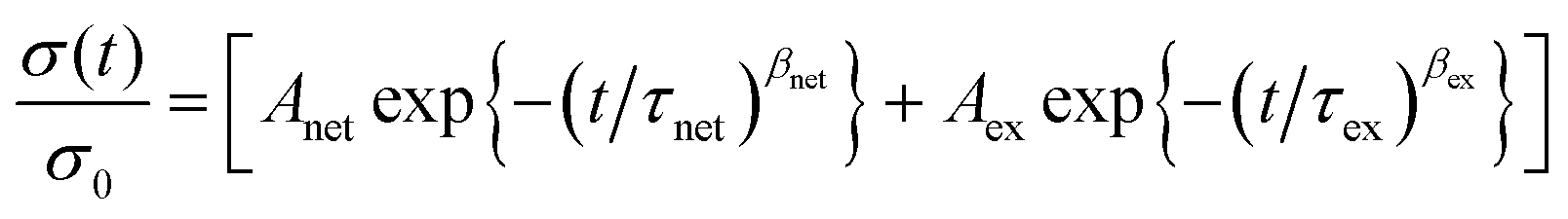 | (1) |
| τ*(T) = τ0*(T)exp(−Eex/RT) | (2) |
The activation energy values obtained from the slope in the chemically-limited regime (blue dashed line) for the vitrimerized EVA samples are consistent with values reported in literature (70 to 150 kJ mol−1) for the activation energy of transesterification bond exchange reactions.53 The variation between the three EVA materials could be attributed to various factors including differences in topology, molecular weight, and initial crosslinked density. This has been seen in literature for vinylogous urethane vitrimer networks, where changing the molecular weights and the nature of the matrix resulted in activation energies ranging from 68 to 149 kJ mol−1.54 However, more structure–reactivity studies are required to obtain a better understanding of the factors that influence the properties of the vitrimerized samples.
Reprocessing vitrimerized EVAs
With the confirmation of vitrimerized samples via FTIR and stress relaxation, the reprocessability of the vitrimerized EVAs was explored using extrusion and compression molding, both conventional techniques for processing thermoplastic materials (Fig. 5). Subsequent reprocessing steps could be performed without the addition of extra catalyst or PVOH at temperatures as low as 120 °C, which is in line with the vitrimerization temperatures determined via stress relaxation to be in the range of 90–110 °C. To further confirm the need for both the zinc acetate catalyst and PVOH feedstock to vitrimerize crosslinked EVA, samples were cryomilled with different combinations of feedstock and catalyst. Samples cryomilled with just feedstock PVOH (no catalyst) were not reprocessable via extrusion and did not retain a network structure (Fig. S3, ESI†). Samples with only zinc acetate (no PVOH) could also not be reprocessed and high temperature solvent swelling resulted in the material dissolving, indicating that a vitrimerized network was not formed. | ||
| Fig. 5 Reprocessing of vitrimerized EVA through extrusion and compression molding. | ||
DMA results confirm that the vitrimerized and reprocessed samples exhibit the elastic properties of crosslinked networks indicated by the presence of a high temperature rubbery plateau (Fig. 6). However, the vitrimerized and reprocessed samples show lower rubbery plateau moduli compared to initial crosslinked EVAs, indicative of a lower crosslinking density in the polymer network.19,55 This lower crosslinking density is confirmed via solvent swelling experiments at high temperature (100 °C), where the vitrimerized and reprocessed samples exhibit a higher swelling ratio relative to initial crosslinked EVAs (Table S1, ESI†). Encouragingly, at room temperature, the reprocessed samples maintain or even exceed the storage modulus of the initially vitrimerized EVAs (Fig. 6). Previous studies have shown that room temperature mechanical properties of EVA are driven by crystallinity and rigid amorphous contributions.32 Retention of the room temperature storage modulus indicates the retention of the integrity of the crystalline and amorphous regions after vitrimerization and reprocessing.
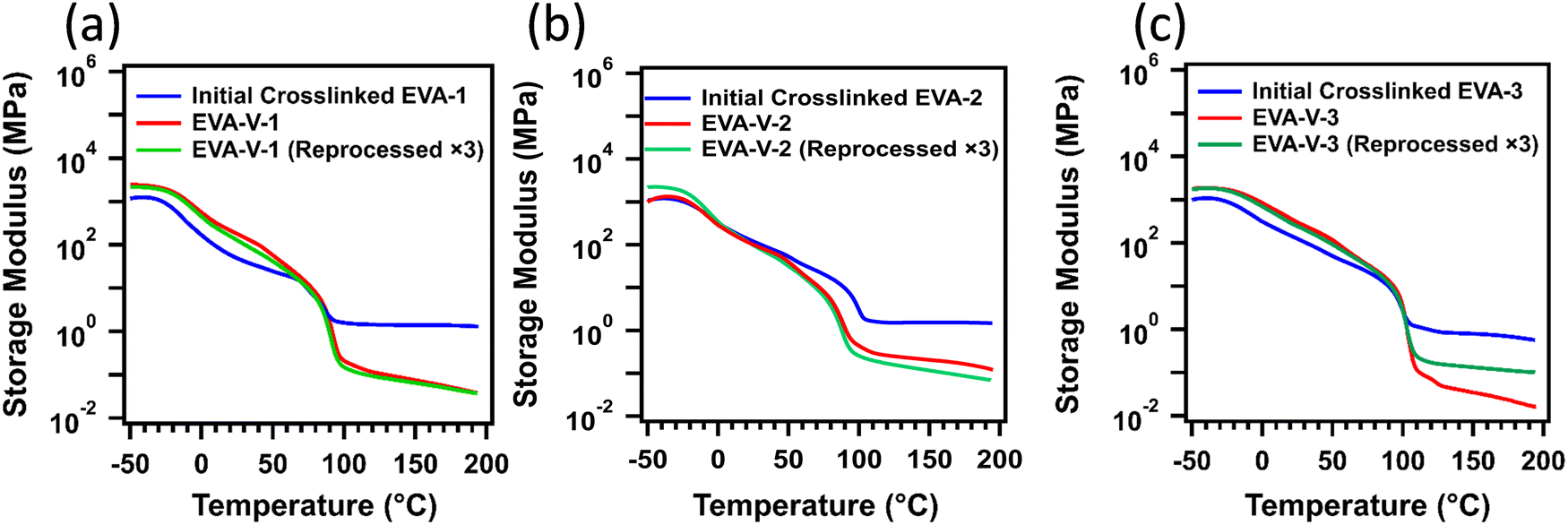 | ||
| Fig. 6 DMA results for the initial crosslinked and vitrimerized EVAs; (a) EVA 1; (b) EVA 2; (c) EVA 3. | ||
Computational results
To further develop and model the vitrimerization of crosslinked EVA via cryomilling, DFT calculations were performed using mononuclear zinc acetates as minimal models of Zn2+ embedded in EVA polymer networks. Zinc(II) is a closed-shell d10 metal ion with no attendant ligand-field stabilization energy56 and is a borderline hard–soft Lewis acid57 that polarizes bound carbonyl ligands. Due to zinc(II) being essentially redox-inert, it cannot inherently generate radicals (radicals that could form during cryomilling). For the simplicity of modeling purposes, primary alcohols were modeled as methanol (pKa in water is 15.5) and secondary alcohols as isopropyl alcohol (pKa in water is 16.5).58 The secondary alcohol model is a better comparison to the experimental vitrimerization process due to PVOH being a secondary alcohol. The corresponding esters are modeled as methyl acetate and isopropyl acetate, allowing for degenerate exchange, which occurs in vitrimers where the reaction coordinate is identical in the forward and reverse directions.In the vitrimerization process, cryomilling is used to initially install zinc acetate into the crosslinked EVA by zinc acetate binding with VA groups present in the polymer network. To computationally observe this structure zinc was modeled as five-coordinate zinc, where zinc binds the acetate group through the respective carbonyl oxygen in the optimized structure, Fig. 7(a) (isopropyl acetate) and Fig. S4a (ESI†) (methyl acetate). The coordination geometry of zinc is essentially square pyramidal, with both acetate counterions binding bidentately. Shown in Fig. S5 (ESI†) is a partial Kohn–Sham orbital energy level diagram of the five-coordinate methyl acetate complex, which indicates a high degree of electrostatic interaction between zinc and the ester ligand, with occupied orbitals originating on the zinc acetate fragment. The lowest unoccupied Kohn–Sham orbital (LUMO) resides almost entirely on the bound methyl acetate, with 57% of orbital density on the carbonyl carbon. This orbital composition is consistent with the expected electrophilicity at carbon of a bound ester. The natural atomic charge59 of the carbonyl carbon is +0.82 for the isopropyl acetate complex and +0.83 for the methyl acetate complex.
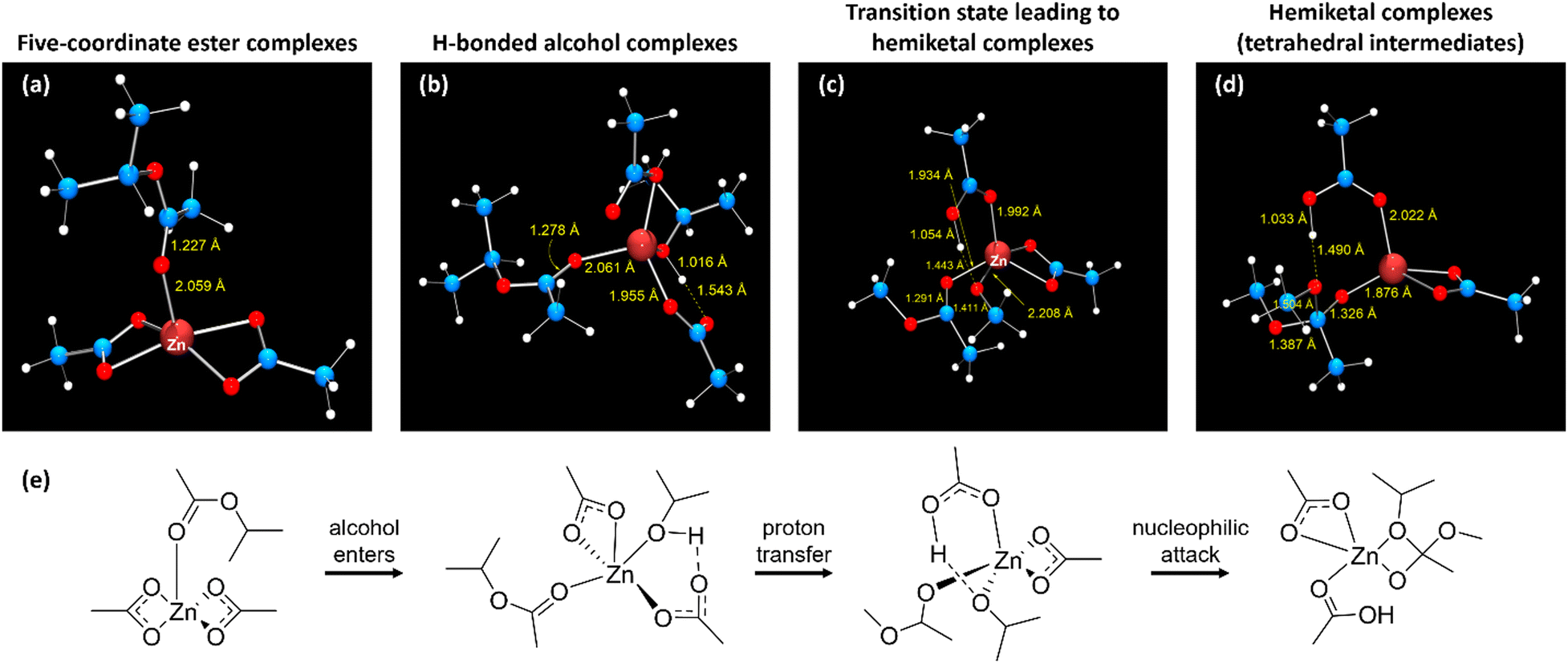 | ||
| Fig. 7 Optimized geometries for the isopropyl acetate complex; (a) five-coordinate ester complex; (b) hydrogen-bonded alcohol complex were alcohol complexes to zinc; (c) transition state leading indicating a proton transfer prior to forming tetrahedral carbon; (d) hemiketal complex (tetrahedral intermediate); (e) step-by-step formation of hemiketal complex. Legend: large red spheres (zinc), small red spheres (oxygen), blue spheres (carbon), and white spheres (hydrogen). | ||
The cryomilled powder also includes PVOH (feedstock of hydroxy groups), so attempts to locate a six-coordinate complex where the alcohol and ester simultaneously bind zinc were attempted. The optimized geometries led to the alcohol decomplexing along with one oxygen of one acetate ligand, resulting in a κ1-acetate accepting a hydrogen bond from the alcohol ligand, Fig. 7(b) (isopropyl acetate) and Fig. S4 (ESI†) (methyl acetate). So, the best model of the cryomilled powder consists of a zinc complex where zinc is five-coordinate, with three oxygen donors provided by acetates, one from the ester carbonyl, and one from the bound alcohol. The hydrogen-bonded proton of the isopropyl alcohol complex shows a Mayer bond order60,61 with the acetate oxygen (H-bond acceptor) of 0.21; the bond order to the alcohol oxygen is 0.84; further indicating that the Brønsted acidic alcohol is not dissociated. Corresponding bond orders in the methyl alcohol complex are 0.21 (H–acetate O) and 0.71 (H–alcohol O). Formation of the hydrogen bond cause little to no change in the electrophilicity of the ester carbonyl carbon. Natural atomic charges are 0.85 for the isopropyl alcohol complex and 0.82 for the methanol complex.
To model the second part of the vitrimerization process, where transesterification occurs via compression molding, transition states for nucleophilic attack of the bound alcohol to the bound ester were located by synchronous transit-guided quasi-Newton methods, as proposed by Schlegel and co-workers.62 Harmonic vibrational frequency calculations validated that the converged structures are saddle points of the potential energy hypersurface. Optimized transition state geometries for the isopropyl complex appear as Fig. 7(c) and Fig. S4c (ESI†) for the methyl complex. The structure show that formation of the hemiketal entails proton transfer from the alcohol oxygen to the dangling (κ1) carboxylate. The nucleophilic (alcohol) oxygen enters within bonding range of the ester carbon, which pyramidalizes. The transition state lies 78.7 kJ mol−1 above the separated reactants for the isopropyl ester and 97.9 kJ mol−1 methyl ester, in terms of free energy (activation energy).
Formation of the hemiketal (tetrahedral intermediate) simultaneously with proton transfer to the κ1 acetate.63 The tetrahedral intermediate binds as a chelating anion toward zinc and the coordination geometry is best described as distorted square pyramidal. Converged geometries of hemiketal complexes appear as Fig. 7(d) for the isopropyl complex and Fig. S4d (ESI†) for the methyl complex. Fig. S6 (ESI†) depicts a partial Kohn–Sham orbital energy level diagram of the hemiketal complex formed from methanol and methyl acetate. The diagram suggests strong electrostatic bonding, with frontier orbitals tightly localized on the hemiketal (HOMO) or κ2-acetate; there is little mixing between them. The HOMO now resides on the (anionic) hemiketal, as expected for a nucleophile; and the LUMO resides on the more electrophilic κ2-acetate, with minimal density on zinc. This result complements earlier conclusions about the need for supporting basic sites in catalyzed transesterifications.63 Additionally, the methodology developed here to confirm the mechanism for vitrimer formation in crosslinked EVAs using a zinc acetate catalyst and PVOH feedstock could be used as a framework to explore alternative catalyst systems that increase the industrial relevance of the resultant vitrimers.
Conclusion
A series of permanently crosslinked EVAs with different VA content and a range of molecular weights were vitrimerized via cryomilling crosslinked EVA with zinc acetate and PVOH. The vitrimerized EVAs relaxed stress rapidly at temperatures as low as 100 °C and were therefore reprocessable through conventional processing methods for thermoplastic polymers such as extrusion and compression molding at temperatures as low as 120 °C. The vitrimerized samples could be reprocessed at least three times without additional catalyst or feedstock hydroxy groups. It was found that the vitrimerization process requires both the addition of zinc acetate and PVOH to enable a transesterification reaction.DFT calculations support the need for the –OH feedstock and indicate approximate activation free energies of 78.7 kJ mol−1 for zinc acetate-mediated transesterification of secondary alcohols. Formation of the tetrahedral intermediate occurs via proton transfer concomitant with nucleophilic attack of the alcohol oxygen to the ester carbon. The presence of hydrogen bonds with the acetate groups indicate that the carboxylate ligands are not passive in the transesterification reaction. Additionally, DFT modelling developed in this study can provide a framework for future catalyst selection supporting method development at industrially relevant timescales.
Futures studies are underway to understand how factors such as initial crosslink density, VA content, molecular weight, and system topology may affect the transesterification exchange reaction activation energy and Tv. Additionally, to further elucidate the effect of VA content, molecular weight, and system topology on mechanical properties of the vitrimerized and reprocessed systems. This study opens a new avenue to design a platform for recycling thermoset EVA waste already present in the market and provide guidance for future work to tune the properties of recycled EVAs.
Disclaimer
This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This material is based upon work supported by the U.S. Department of Energy's Office of Energy Efficiency and Renewable Energy (EERE) under the Advanced Manufacturing Office Award Number DE-EE0007897 awarded to the REMADE Institute, a division of Sustainable Manufacturing Innovation Alliance Corp. This material is based upon work supported by the US Department of Energy's Office of Energy Efficiency and Renewable Energy (EER) under the Advanced Manufacturing Office Award Number 20-10-RR-4029.References
- M. Alexandre, G. Beyer, C. Henrist, R. Cloots, A. Rulmont, R. Jérôme and P. Dubois, Preparation and Properties of Layered Silicate Nanocomposites Based on Ethylene Vinyl Acetate Copolymers, Macromol. Rapid Commun., 2001, 22(8), 643–646, DOI:10.1002/1521-3927(20010501)22:8<643::AID-MARC643>3.0.CO;2-%23.
- M. Brogly, M. Nardin and J. Schultz, Effect of Vinylacetate Content on Crystallinity and Second-Order Transitions in Ethylene—Vinylacetate Copolymers, J. Appl. Polym. Sci., 1997, 64(10), 1903–1912, DOI:10.1002/(SICI)1097-4628(19970606)64:10<1903::AID-APP4>3.0.CO;2-M.
- S.-S. Choi and Y. Y. Chung, Considering Factors for Analysis of Crosslink Density of Poly(Ethylene-Co-Vinyl Acetate) Compounds, Polym. Test., 2018, 66, 312–318, DOI:10.1016/j.polymertesting.2018.01.038.
- K. Agroui, G. Collins and J. Farenc, Measurement of Glass Transition Temperature of Crosslinked EVA Encapsulant by Thermal Analysis for Photovoltaic Application, Renewable Energy, 2012, 43, 218–223, DOI:10.1016/j.renene.2011.11.015.
- J. Mosnácek, A. A. Basfar, T. M. Shukri and M. A. Bahattab, Poly(Ethylene Vinyl Acetate) (EVA)/Low Density Polyethylene (LDPE)/Ammonium Polyphosphate (APP) Composites Cross-Linked by Dicumyl Peroxide for Wire and Cable Applications, Polym. J., 2008, 40(5), 460–464, DOI:10.1295/polymj.PJ2007153.
- L. Wang, Y. Hong and J. X. Li, Durability of Running Shoes with Ethylene Vinyl Acetate or Polyurethane Midsoles, J. Sports Sci., 2012, 30(16), 1787–1792, DOI:10.1080/02640414.2012.723819.
- W. Zou, J. Dong, Y. Luo, Q. Zhao and T. Xie, Dynamic Covalent Polymer Networks: From Old Chemistry to Modern Day Innovations, Adv. Mater., 2017, 29(14), 1606100, DOI:10.1002/adma.201606100.
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Enabling Applications of Covalent Adaptable Networks, Annu. Rev. Chem. Biomol. Eng., 2019, 10(1), 175–198, DOI:10.1146/annurev-chembioeng-060718-030217.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Dynamic Covalent Chemistry, Angew. Chem., Int. Ed., 2002, 41(6), 898–952, DOI:10.1002/1521-3773(20020315)41:6<898::aid-anie898>3.0.co;2-e.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Silica-like Malleable Materials from Permanent Organic Networks, Science, 2011, 334(6058), 965–968, DOI:10.1126/science.1212648.
- M. Röttger, T. Domenech, R. van der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, High-Performance Vitrimers from Commodity Thermoplastics through Dioxaborolane Metathesis, Science, 2017, 356(6333), 62–65, DOI:10.1126/science.aah5281.
- A. P. Bapat, B. S. Sumerlin and A. Sutti, Bulk Network Polymers with Dynamic B–O Bonds: Healable and Reprocessable Materials, Mater. Horiz., 2020, 7(3), 694–714, 10.1039/C9MH01223K.
- C. Taplan, M. Guerre, J. M. Winne and F. E. Du Prez, Fast Processing of Highly Crosslinked, Low-Viscosity Vitrimers, Mater. Horiz., 2020, 7(1), 104–110, 10.1039/C9MH01062A.
- Q. Shi, K. Yu, X. Kuang, X. Mu, C. K. Dunn, M. L. Dunn, T. Wang and H. Jerry Qi, Recyclable 3D Printing of Vitrimer Epoxy, Mater. Horiz., 2017, 4(4), 598–607, 10.1039/C7MH00043J.
- A. Ruiz de Luzuriaga, R. Martin, N. Markaide, A. Rekondo, G. Cabañero, J. Rodríguez and I. Odriozola, Epoxy Resin with Exchangeable Disulfide Crosslinks to Obtain Reprocessable, Repairable and Recyclable Fiber-Reinforced Thermoset Composites, Mater. Horiz., 2016, 3(3), 241–247, 10.1039/C6MH00029K.
- K. Jin, S. Kim, J. Xu, F. S. Bates and C. J. Ellison, Melt-Blown Cross-Linked Fibers from Thermally Reversible Diels–Alder Polymer Networks, ACS Macro Lett., 2018, 7(11), 1339–1345, DOI:10.1021/acsmacrolett.8b00685.
- K. Jin, A. Banerji, D. Kitto, F. S. Bates and C. J. Ellison, Mechanically Robust and Recyclable Cross-Linked Fibers from Melt Blown Anthracene-Functionalized Commodity Polymers, ACS Appl. Mater. Interfaces, 2019, 11(13), 12863–12870, DOI:10.1021/acsami.9b00209.
- M. Delahaye, J. M. Winne and F. E. Du Prez, Internal Catalysis in Covalent Adaptable Networks: Phthalate Monoester Transesterification As a Versatile Dynamic Cross-Linking Chemistry, J. Am. Chem. Soc., 2019, 141(38), 15277–15287, DOI:10.1021/jacs.9b07269.
- H. Guo, L. Yue, G. Rui and I. Manas-Zloczower, Recycling Poly(Ethylene-Vinyl Acetate) with Improved Properties through Dynamic Cross-Linking, Macromolecules, 2020, 53(1), 458–464, DOI:10.1021/acs.macromol.9b02281.
- L. Yue, H. Guo, A. Kennedy, A. Patel, X. Gong, T. Ju, T. Gray and I. Manas-Zloczower, Vitrimerization: Converting Thermoset Polymers into Vitrimers, ACS Macro Lett., 2020, 9(6), 836–842, DOI:10.1021/acsmacrolett.0c00299.
- A. Bandegi, M. Amirkhosravi, H. Meng, M. K. R. Aghjeh and I. Manas-Zloczower, Vitrimerization of Crosslinked Unsaturated Polyester Resins: A Mechanochemical Approach to Recycle and Reprocess Thermosets, Glob. Challenges, 2022, 2200036, DOI:10.1002/gch2.202200036.
- L. Yue, V. Solouki Bonab, D. Yuan, A. Patel, V. Karimkhani and I. Manas-Zloczower, Vitrimerization: A Novel Concept to Reprocess and Recycle Thermoset Waste via Dynamic Chemistry, Glob. Challenges, 2019, 3, DOI:10.1002/gch2.201800076.
- L. Yue, M. Amirkhosravi, K. Ke, T. G. Gray and I. Manas-Zloczower, Cellulose Nanocrystals: Accelerator and Reinforcing Filler for Epoxy Vitrimerization, ACS Appl. Mater. Interfaces, 2021, 13(2), 3419–3425, DOI:10.1021/acsami.0c19350.
- A. Bandegi, M. Montemayor and I. Manas-Zloczower, Vitrimerization of Rigid Thermoset Polyurethane Foams: A Mechanochemical Method to Recycle and Reprocess Thermosets, Polym. Adv. Technol., 2022, 33(10), 3750–3758, DOI:10.1002/pat.5827.
- L. Yue, M. Amirkhosravi, X. Gong, T. G. Gray and I. Manas-Zloczower, Recycling Epoxy by Vitrimerization: Influence of an Initial Thermoset Chemical Structure, ACS Sustainable Chem. Eng., 2020, 8(33), 12706–12712, DOI:10.1021/acssuschemeng.0c04815.
- L. Yue, K. Ke, M. Amirkhosravi, T. G. Gray and I. Manas-Zloczower, Catalyst-Free Mechanochemical Recycling of Biobased Epoxy with Cellulose Nanocrystals, ACS Appl. Bio Mater., 2021, 4(5), 4176–4183, DOI:10.1021/acsabm.0c01670.
- L. Yue, V. S. Bonab, A. Patel, D. Yuan, V. Karimkhani and I. Manas-Zloczower, Dynamic Networks for Recycling Thermoset Polymers. US Pat., 10822467, 2020 Search PubMed.
- I. Manas-Zloczower and L. Yue, One-Step, Solvent-Free Method for Recycling and Reprocessing Thermoset Polymers with Tunable Properties, US Pat., 11339268, 2022 Search PubMed.
- M. Capelot, D. Montarnal, F. Tournilhac and L. Leibler, Metal-Catalyzed Transesterification for Healing and Assembling of Thermosets, J. Am. Chem. Soc., 2012, 134(18), 7664–7667, DOI:10.1021/ja302894k.
- A. Demongeot, S. J. Mougnier, S. Okada, C. Soulié-Ziakovic and F. Tournilhac, Coordination and Catalysis of Zn2+ in Epoxy-Based Vitrimers, Polym. Chem., 2016, 7(27), 4486–4493, 10.1039/C6PY00752J.
- J. Han, T. Liu, C. Hao, S. Zhang, B. Guo and J. Zhang, A Catalyst-Free Epoxy Vitrimer System Based on Multifunctional Hyperbranched Polymer, Macromolecules, 2018, 51(17), 6789–6799, DOI:10.1021/acs.macromol.8b01424.
- K. M. McLoughlin, A. J. Oskouei, M. K. Sing, A. Bandegi, S. Mitchell, J. Kennedy, T. G. Gray and I. Manas-Zloczower, Thermomechanical Properties of Cross-Linked EVA: A Holistic Approach, ACS Appl. Polym. Mater., 2023, 5(2), 1430–1439, DOI:10.1021/acsapm.2c01928.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, G16_C01. p Gaussian 16, Revision A.03, Gaussian, Inc., Wallin., 2016 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- F. Weigend and R. Ahlrichs, Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305, 10.1039/B508541A.
- F. Weigend, Accurate Coulomb-Fitting Basis Sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8(9), 1057–1065, 10.1039/B515623H.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32(7), 1456–1465, DOI:10.1002/jcc.21759.
- S. I. Gorelsky, AOMix: Program for Molecular Orbital Analysis, University of Ottawa, version 6.94, 2018, https://www.sg-chem.net Search PubMed.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187–196 CrossRef CAS.
- T. Hoang, N. T. Chinh, N. T. T. Trang, T. T. X. Hang, D. T. M. Thanh, D. V. Hung, C.-S. Ha and M. Aufray, Effects of Maleic Anhydride Grafted Ethylene/Vinyl Acetate Copolymer (EVA) on the Properties of EVA/Silica Nanocomposites, Macromol. Res., 2013, 21(11), 1210–1217, DOI:10.1007/s13233-013-1157-8.
- R. L. Snyder, D. J. Fortman, G. X. De Hoe, M. A. Hillmyer and W. R. Dichtel, Reprocessable Acid-Degradable Polycarbonate Vitrimers, Macromolecules, 2018, 51(2), 389–397, DOI:10.1021/acs.macromol.7b02299.
- J. S. A. Ishibashi and J. A. Kalow, Vitrimeric Silicone Elastomers Enabled by Dynamic Meldrum's Acid-Derived Cross-Links, ACS Macro Lett., 2018, 7(4), 482–486, DOI:10.1021/acsmacrolett.8b00166.
- W. Denissen, G. Rivero, R. Nicolaÿ, L. Leibler, J. M. Winne and F. E. Du Prez, Vinylogous Urethane Vitrimers, Adv. Funct. Mater., 2015, 25(16), 2451–2457, DOI:10.1002/adfm.201404553.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, Catalytic Control of the Vitrimer Glass Transition, ACS Macro Lett., 2012, 1(7), 789–792, DOI:10.1021/mz300239f.
- J. P. Brutman, P. A. Delgado and M. A. Hillmyer, Polylactide Vitrimers, ACS Macro Lett., 2014, 3(7), 607–610, DOI:10.1021/mz500269w.
- L. Imbernon, S. Norvez and L. Leibler, Stress Relaxation and Self-Adhesion of Rubbers with Exchangeable Links, Macromolecules, 2016, 49(6), 2172–2178, DOI:10.1021/acs.macromol.5b02751.
- M. Hayashi and L. Chen, Functionalization of Triblock Copolymer Elastomers by Cross-Linking the End Blocks via Trans-N-Alkylation-Based Exchangeable Bonds, Polym. Chem., 2020, 11(10), 1713–1719, 10.1039/C9PY01759C.
- K. S. Fancey, A Mechanical Model for Creep, Recovery and Stress Relaxation in Polymeric Materials, J. Mater. Sci., 2005, 40(18), 4827–4831, DOI:10.1007/s10853-005-2020-x.
- J. D. Ferry, Viscoelastic Properties of Polymers, John Wiley & Sons, 1980 Search PubMed.
- L. Li, X. Chen, K. Jin and J. M. Torkelson, Vitrimers Designed Both To Strongly Suppress Creep and To Recover Original Cross-Link Density after Reprocessing: Quantitative Theory and Experiments, Macromolecules, 2018, 51(15), 5537–5546, DOI:10.1021/acs.macromol.8b00922.
- A. Dhinojwala, J. C. Hooker and J. M. Torkelson, Retardation of Rotational Reorientation Dynamics in Polymers near the Glass Transition: A Novel Study over Eleven Decades in Time Using Second-Order Non-Linear Optics, J. Non. Cryst. Solids, 1994, 172–174, 286–296, DOI:10.1016/0022-3093(94)90447-2.
- A. M. Hubbard, Y. Ren, D. Konkolewicz, A. Sarvestani, C. R. Picu, G. S. Kedziora, A. Roy, V. Varshney and D. Nepal, Vitrimer Transition Temperature Identification: Coupling Various Thermomechanical Methodologies, ACS Appl. Polym. Mater., 2021, 3(4), 1756–1766, DOI:10.1021/acsapm.0c01290.
- T. Kimura and M. Hayashi, One-Shot Transformation of Ordinary Polyesters into Vitrimers: Decomposition-Triggered Cross-Linking and Assistance of Dynamic Covalent Bonds, J. Mater. Chem. A, 2022, 10(34), 17406–17414, 10.1039/D2TA04110C.
- Y. Spiesschaert, C. Taplan, L. Stricker, M. Guerre, J. M. Winne and F. E. Du Prez, Influence of the Polymer Matrix on the Viscoelastic Behaviour of Vitrimers, Polym. Chem., 2020, 11(33), 5377–5385, 10.1039/D0PY00114G.
- J. Liu (Daniel), H.-J. Sue, Z. J. Thompson, F. S. Bates, M. Dettloff, G. Jacob, N. Verghese and H. Pham, Effect of Crosslink Density on Fracture Behavior of Model Epoxies Containing Block Copolymer Nanoparticles, Polymer, 2009, 50(19), 4683–4689, DOI:10.1016/j.polymer.2009.05.006.
- G. Parkin, Synthetic Analogues Relevant to the Structure and Function of Zinc Enzymes, Chem. Rev., 2004, 104(2), 699–768, DOI:10.1021/cr0206263.
- R. G. Pearson, Hard and Soft Acids and Bases, J. Am. Chem. Soc., 1963, 85(22), 3533–3539, DOI:10.1021/ja00905a001.
- F. G. Bordwell, Equilibrium Acidities in Dimethyl Sulfoxide Solution, Acc. Chem. Res., 1988, 21(12), 456–463, DOI:10.1021/ar00156a004.
- A. E. Reed, R. B. Weinstock and F. Weinhold, Natural Population Analysis, J. Chem. Phys., 1985, 83(2), 735–746, DOI:10.1063/1.449486.
- I. Mayer, Charge, Bond Order and Valence in the Ab Initio SCF Theory, Chem. Phys. Lett., 1983, 97(3), 270–274, DOI:10.1016/0009-2614(83)80005-0.
- A. J. Bridgeman, G. Cavigliasso, L. R. Ireland and J. Rothery, The Mayer Bond Order as a Tool in Inorganic Chemistry, J. Chem. Soc., Dalton Trans., 2001, 14, 2095–2108, 10.1039/B102094N.
- C. Peng and H. Bernhard Schlegel, Combining Synchronous Transit and Quasi-Newton Methods to Find Transition States, Isr. J. Chem., 1993, 33(4), 449–454, DOI:10.1002/ijch.199300051.
- S. Bhusal, C. Oh, Y. Kang, V. Varshney, Y. Ren, D. Nepal, A. Roy and G. Kedziora, Transesterification in Vitrimer Polymers Using Bifunctional Catalysts: Modeled with Solution-Phase Experimental Rates and Theoretical Analysis of Efficiency and Mechanisms, J. Phys. Chem. B, 2021, 125(9), 2411–2424, DOI:10.1021/acs.jpcb.0c10403.
Footnote |
| † Electronic supplementary information (ESI) available: The particle size distribution of cryomilled powders, swelling results, stress relaxation of initial crosslinked EVAs, fitting parameters to eqn (1), and optimized cartesian coordinates (Å) in .xyz format. See DOI: https://doi.org/10.1039/d3ma00098b |
| This journal is © The Royal Society of Chemistry 2023 |