DOI:
10.1039/D3MA00076A
(Paper)
Mater. Adv., 2023,
4, 2340-2353
In situ fabrication and design of a novel electrochemical sensor based on the Ag3.84Sn3S8@rGO nanocomposite for competitive ultra-detection of metronidazole in human urine†
Received
16th February 2023
, Accepted 20th April 2023
First published on 24th April 2023
Abstract
It is indispensable to develop an efficient technology for reducing contaminants in wastewater. Thus, we have developed a novel nanocomposite catalyst via the in situ formation of non-stoichiometric Ag3.84Sn3S8 on reduced graphene oxide (rGO) sheets. Significantly, we varied the concentration of rGO between 0.05 to 0.5% to get the optimal concentration (0.25%). To examine the application of the nanocomposite as a potential catalyst, we performed visible-light-induced mineralization of the antibiotic drug metronidazole (MNZ) and found the optimized efficiency. Based on this prospect, we have designed an electrochemical sensor for the ultra-detection of MNZ. The proposed sensor shows an excellent linear range (0.01–1500 nM), outstanding sensitivity (48 μA nM−1 cm−2), a significant limit of detection (LOD) (4 pM), excellent anti-interference ability (also in real samples) and good reproducibility.
1. Introduction
Nanostructured metal sulfides have garnered significant attention as promising electro- and photocatalysts due to their eco-friendliness and exceptional physicochemical properties.1 In this context, group 11 sulfides (Cu, Ag, and Au) are unique due to their ability to form many polymorphs owing to the very common +1 state. These sulfides are proven by crystallographic methods to be electrochemically stable in nature.2 One of the interesting examples is Ag2S, which can exist as three polymorphs (α, β and γ-Ag2S) within a very close temperature interval.3 Among them, the cubic γ−Ag2S is metal-rich and non-stoichiometric, and this type of material has several advantages: (i) it shows significantly large electronic conductivity of nearly 1.3 × 103 Ω−1 cm−1; (ii) it can prevent phase transformation during the intercalation of foreign metal ions owing to the crystallographic structural stability imparted by the presence of only M–S bonds and no S–S bond;4 (iii) it helps the formation of multi-metallic sulfides, which can provide more redox reaction sites, and offers notable stability in acids, as well as alkaline media;5 (iv) due to its non-stoichiometric nature, S vacancies are created, which might ease the adsorption of molecules on its catalytic surface and accelerate the desired catalysis.6
In this regard, recently, Hu et al. have discovered that S vacancies can be easily created by a non-stoichiometric synthetic approach and play a crucial role in the adsorption of N2 onto the catalytic active sites of Zn0.1Sn0.1Cd0.8S for the photofixation process.6 The study involved the calcination of the Zn0.1Sn0.1Cd0.8S nanocrystal in a saturated O2 atmosphere to obtain Zn0.1Sn0.1Cd0.8SO.6 This kind of approach has not been considered for Ag-based non-stoichiometric ternary metal sulfides yet.
Further, this type of nanocrystal can be grown on any material with an sp2 carbon backbone like graphene derivatives, such as graphene oxide (GO) and reduced graphene oxide (rGO), due to their various advantages: (i) the backbone gives thermal and chemical stability and supports the nanocrystal to grow on its surface; (ii) they can improve the catalytically active surface area, resulting in enhanced adsorption capacity, and (iii) accelerate electron mobility, easing the catalytic process. In this context, with less oxygenated functional groups, rGO shows physicochemical properties closer to the pristine graphene and hence shows superior catalytic advantages.7–11
A solvothermal process is crucial to this kind of nanocomposites because it (i) favors successful interaction between the precursors, (ii) facilitates in situ fabrication (iii) gives a spectrum of geometries via the modulation of size, shape and overall morphology.12–14
In recent years, the research community has shown interest in developing newly engineered semiconductor multi-component catalysts that can detect multiple organic pollutants. Among the chemicals found in pharmaceutical waste, metronidazole (MNZ) has gained enormous importance as it is extensively used in the medical field.15 Long exposure to this drug causes very dangerous mutagenic diseases in living things, resulting in cancer.16 It is important to note that, being a non-biodegradable entity, MNZ is hardly adsorbed on the soil molecules, and this is the reason its specific detection and removal are challenging concerns.17–21
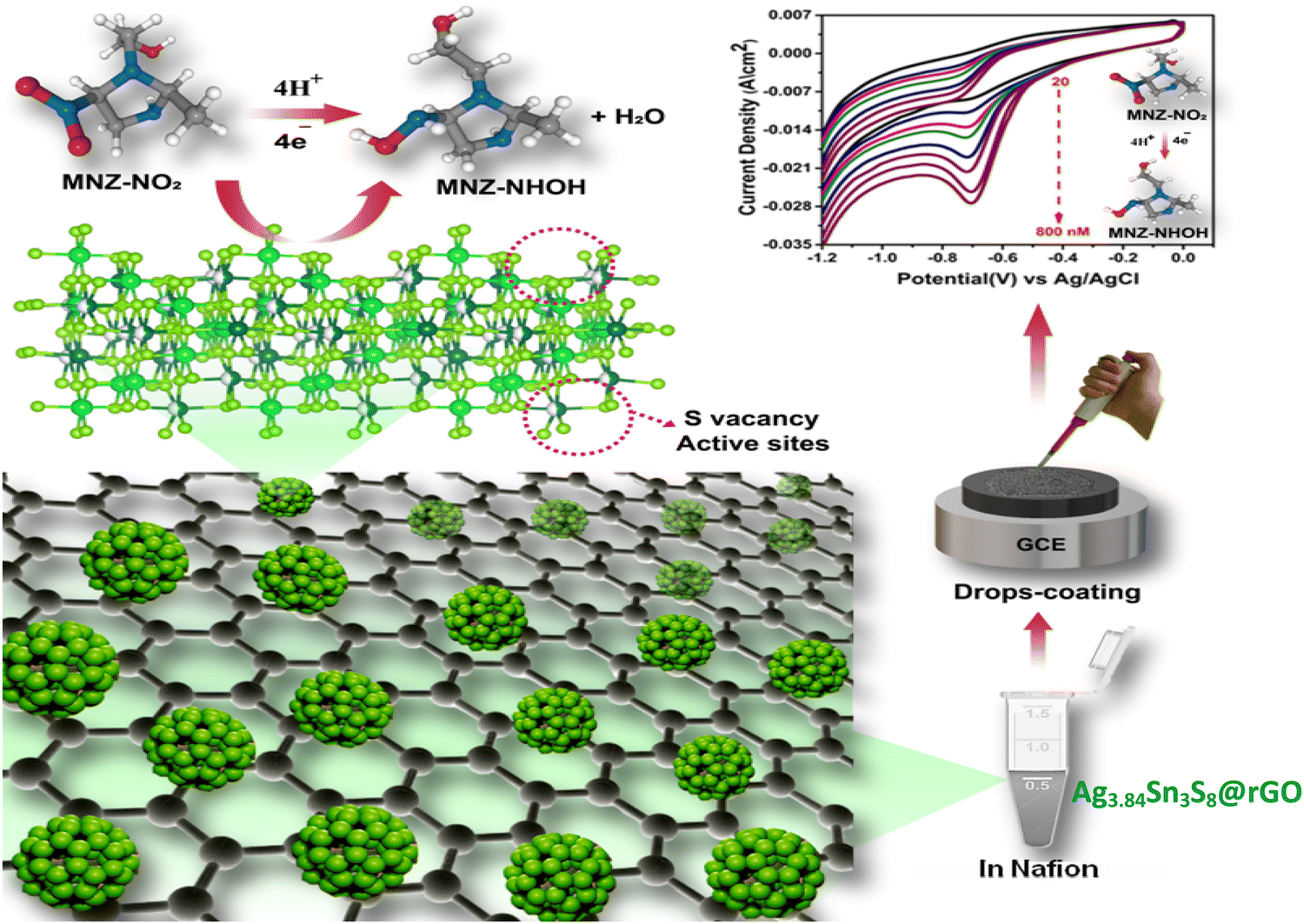
Here, we have aimed to: (1) achieve the targeted synthesis of non-stoichiometric Ag3.84Sn3S8 onto reduced graphene oxide (rGO) to prepare a nanocomposite (NC) via a facile solvothermal route, wherein the reduction of GO and the growth of the nanocrystal would be taken place in situ, (2) explore the importance of the desired S vacancies in catalysis, (3) test and optimize rGO by extensive photophysical, photochemical and electrochemical characterizations for enhanced catalytic activity. Finally, we have explored the efficiency of the NC as an excellent electrocatalytic sensor for the ultra-detection of the antibiotic drug MNZ. To the best of our knowledge, to date, no study has been performed on the activation and mineralization of a nitrogenous antibiotic drug on a non-stoichiometric nanocatalyst.
2. Experimental section
2.1. The general technique to prepare a non-stoichiometric metal sulfide nanocrystal
In this context, the characteristics of metals and the initial metallic molar ratio have a significant role. For example, to get the composition of M1x+δ Mx2 S, M1 must be comparatively harder point than M2 and the molar ratio (M1/M2) must be taken >1.22
2.2. Preparation of the nanocatalyst
The chemicals and materials used are mentioned in the ESI.† A 100 mL suspension of GO (prepared by the Hummers’ method)23 in ethanol was taken in a 250 mL beaker at a concentration of 0.5 mg mL−1. To this suspension, AgNO3, SnCl4·2H2O and CH4N2S were added in a molar ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:10 and stirred for 1 hour at room temperature (rt) for homogenization and then transferred into a 250 mL autoclave for solvothermal decomposition at 200 °C for 10 h. Thereafter, the mixture was cooled down to rt, and 5 mmol L-ascorbic acid was added. For uniformity, the mixture was vigorously stirred for 1 h. Then, again the autoclave was sealed and kept at 200 °C for an additional 5 h. The greyish-black precipitate was separated by the addition of EtOH and washed with EtOH/DI H2O (1:1) 5 times by centrifugation. Then, the material was dried under a vacuum in a furnace at 10 °C for 20 h. The as-synthesized material is assigned as Ag3.84Sn3S8@rGO0.5. A similar process was employed to prepare three more nanocomposites (rGO0.25, rGO0.1, and rGO0.05) by varying the GO concentration between 0.5–0.05 mg mL−1. Bare Ag3.84Sn3S8 was obtained under similar conditions. To probe the presence of S vacancies, the nanocrystal was subjected to calcination at 300 °C for 3 h in a saturated O2 atmosphere, as discussed in ‘Electronic spectral analysis (3.2.6)’. The details of the physical experiments, photocatalytic measurements and electrode fabrication are elaborated in ESI.†
:5:10 and stirred for 1 hour at room temperature (rt) for homogenization and then transferred into a 250 mL autoclave for solvothermal decomposition at 200 °C for 10 h. Thereafter, the mixture was cooled down to rt, and 5 mmol L-ascorbic acid was added. For uniformity, the mixture was vigorously stirred for 1 h. Then, again the autoclave was sealed and kept at 200 °C for an additional 5 h. The greyish-black precipitate was separated by the addition of EtOH and washed with EtOH/DI H2O (1:1) 5 times by centrifugation. Then, the material was dried under a vacuum in a furnace at 10 °C for 20 h. The as-synthesized material is assigned as Ag3.84Sn3S8@rGO0.5. A similar process was employed to prepare three more nanocomposites (rGO0.25, rGO0.1, and rGO0.05) by varying the GO concentration between 0.5–0.05 mg mL−1. Bare Ag3.84Sn3S8 was obtained under similar conditions. To probe the presence of S vacancies, the nanocrystal was subjected to calcination at 300 °C for 3 h in a saturated O2 atmosphere, as discussed in ‘Electronic spectral analysis (3.2.6)’. The details of the physical experiments, photocatalytic measurements and electrode fabrication are elaborated in ESI.†
3. Results and discussion
3.1. The non-stoichiometric synthetic approach
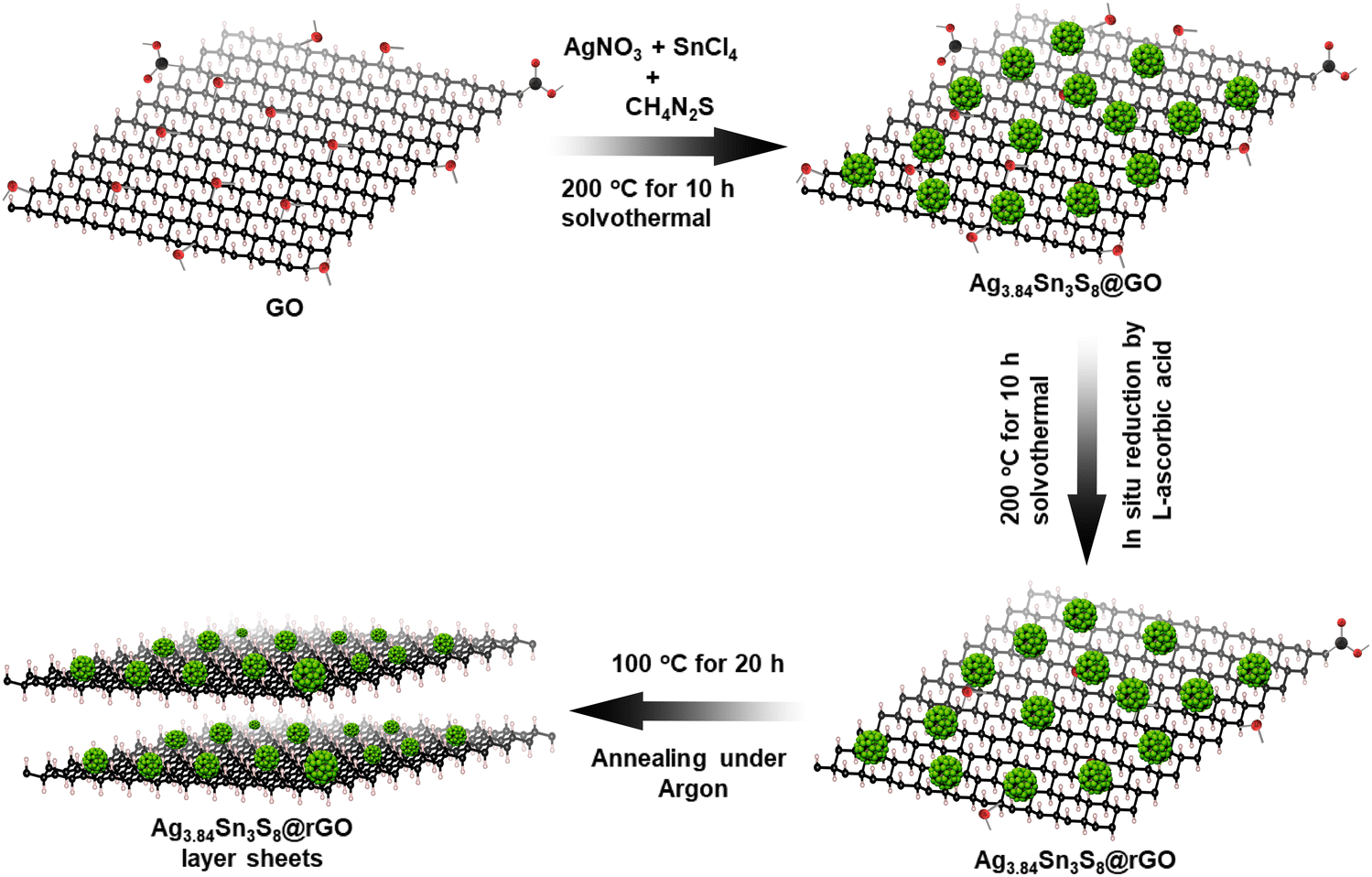
Here, we used an Ag/Sn molar ratio of 1.4 to prepare Ag3.84Sn3S8.24,25 Additionally, to fabricate with rGO, a similar in situ synthesis approach was employed, and here, we used CH4N2S to allow the slow dissociation of S for uniformity (Scheme 1).26
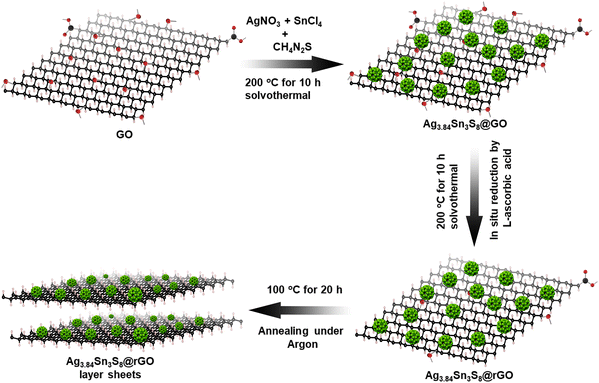 |
| | Scheme 1 The in situ synthetic route to obtain the NC. | |
3.2. Characterization
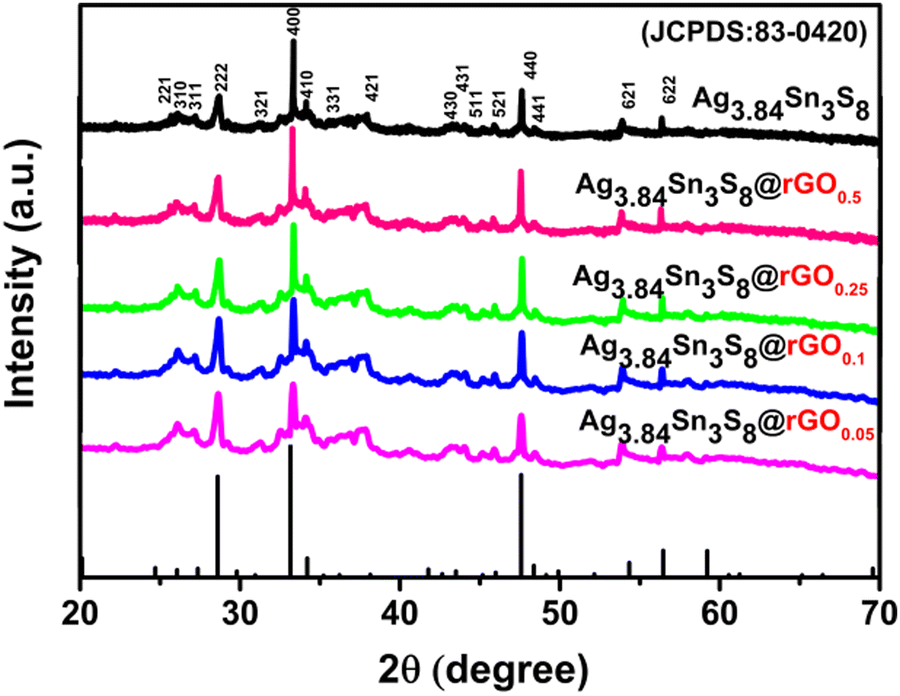
3.2.1. Powder X-ray diffraction (PXRD) analysis.
The PXRD patterns [Fig. 1] were completely in accordance with JCPDS No: 83-0420 and ICSD No: 079510. The NCs belonged to a cubic system having the space group P4132 (a:10.80, b, c and α = β = γ). The literature suggests that the percentage of rGO can significantly affect the surface energy of the nanocrystal grown on its surface, indicating interfacial interaction between Ag3.84Sn3S8 and rGO.27 Generally, the surface energies of the crystalline planes of a cubic crystal follow the order {110} > {100} > {111}.23 Here, we compared the relative intensity of (440) with respect to the highest intense peak (400), which are the crystallographic equivalent planes of (110) and (100), respectively. The intensity ratio between (440) and (400) (I440/I400) of the materials were as follows: Ag3.84Sn3S8@rGO0.25 > Ag3.84Sn3S8@rGO0.5 > Ag3.84Sn3S8@rGO0.1 > Ag3.84Sn3S8@rGO0.05. This result indicates that Ag3.84Sn3S8@rGO0.25 might have the highest surface energy.
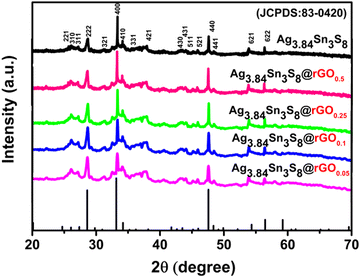 |
| | Fig. 1 The PXRD pattern analysis of Ag3.84Sn3S8 and the Ag3.84Sn3S8@rGO(0.05–0.5) NCs. | |
3.2.2. FTIR spectral analysis.
The in situ reduction of GO and the formation of NCs were supported by the characteristic peaks observed in the FTIR spectra (Fig. S1, ESI†). Fig. S1 (ESI†) shows that, in the case of GO, the characteristic peak at 3408 cm−1 was due to the O–H stretching vibration, and the peak at 1735 cm−1 was that of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of COOH, which resides at the edge of the GO sheets. A peak appeared at 1620 cm−1 due to the O–H bending and ring vibrations of the epoxide groups. Further, the peak at 1282 cm−1 supported the existence of C–O–C. The characteristic peak at 1055 cm−1 was due to C–O (epoxy/alkoxy).1 The intensities of the characteristic peaks were substantially reduced due to the reduction of GO to rGO, and the oxygenated functional groups readily vanished, supporting the formation of the Ag3.84Sn3S8@rGO NC. This result indicates interfacial binding between Ag3.84Sn3S8 and rGO.27
O group of COOH, which resides at the edge of the GO sheets. A peak appeared at 1620 cm−1 due to the O–H bending and ring vibrations of the epoxide groups. Further, the peak at 1282 cm−1 supported the existence of C–O–C. The characteristic peak at 1055 cm−1 was due to C–O (epoxy/alkoxy).1 The intensities of the characteristic peaks were substantially reduced due to the reduction of GO to rGO, and the oxygenated functional groups readily vanished, supporting the formation of the Ag3.84Sn3S8@rGO NC. This result indicates interfacial binding between Ag3.84Sn3S8 and rGO.27
3.2.3. EDX analysis.
The EDX [Fig. S2 and S3 correspond to the pure sample and NCs, respectively, ESI†] study confirmed the presence of the desired composition [Table S1, ESI†].
3.2.4. TEM image analysis.
The size, shape and morphology of the Ag3.84Sn3S8@rGO0.5 NC are depicted in Fig. 2. Fig. 2(a) represents rGO, and Fig. 2(b) and (c) show that the NCs were uniformly distributed on the rGO nanosheet. The particle size distribution [Fig. 2(b), inset] confirmed that the average size of the particles was between 45–50 nm, and the shape obtained was quasi-spherical [Fig. 2(d)]. The HRTEM image showed [Fig. 2(e)] distinct lattice fringes, confirming the perfect crystallinity of the NC. The d-spacing of 0.325 nm was assigned to the crystallographic lattice spacing between the neighboring 311 planes, which is completely consistent with the PXRD results. The SAED pattern [Fig. 2(f)] analysis also supported the observation from the PXRD analysis. The in situ reduction of GO and the excellent dispersion of nanoparticles on the surface of rGO might be a result of the reciprocity between Ag3.84Sn3S8 and the oxygenated functional groups of GO. It can be explained as follows: while the reacting metal ions engage with the functional groups of GO during synthesis and the sp2 carbon backbone supports the nucleation and growth of the nanomaterial, simultaneously the growing nanoparticles restrict the aggregation of the rGO sheets.28,29 Both these phenomena are supported by the following observations: the uniform average size of all materials [Fig. S4 (a), (b), (c) and (d), ESI†] and the absence of the rGO peak in the PXRD patterns [Fig. 1].
 |
| | Fig. 2 The TEM images of (a) rGO and (b) Ag3.84Sn3S8@rGO0.5 in low magnification and (c and d) high magnification, respectively; the particle size distribution of the NC is given in the inset. (e) The HRTEM image and (f) the corresponding SAED pattern. | |
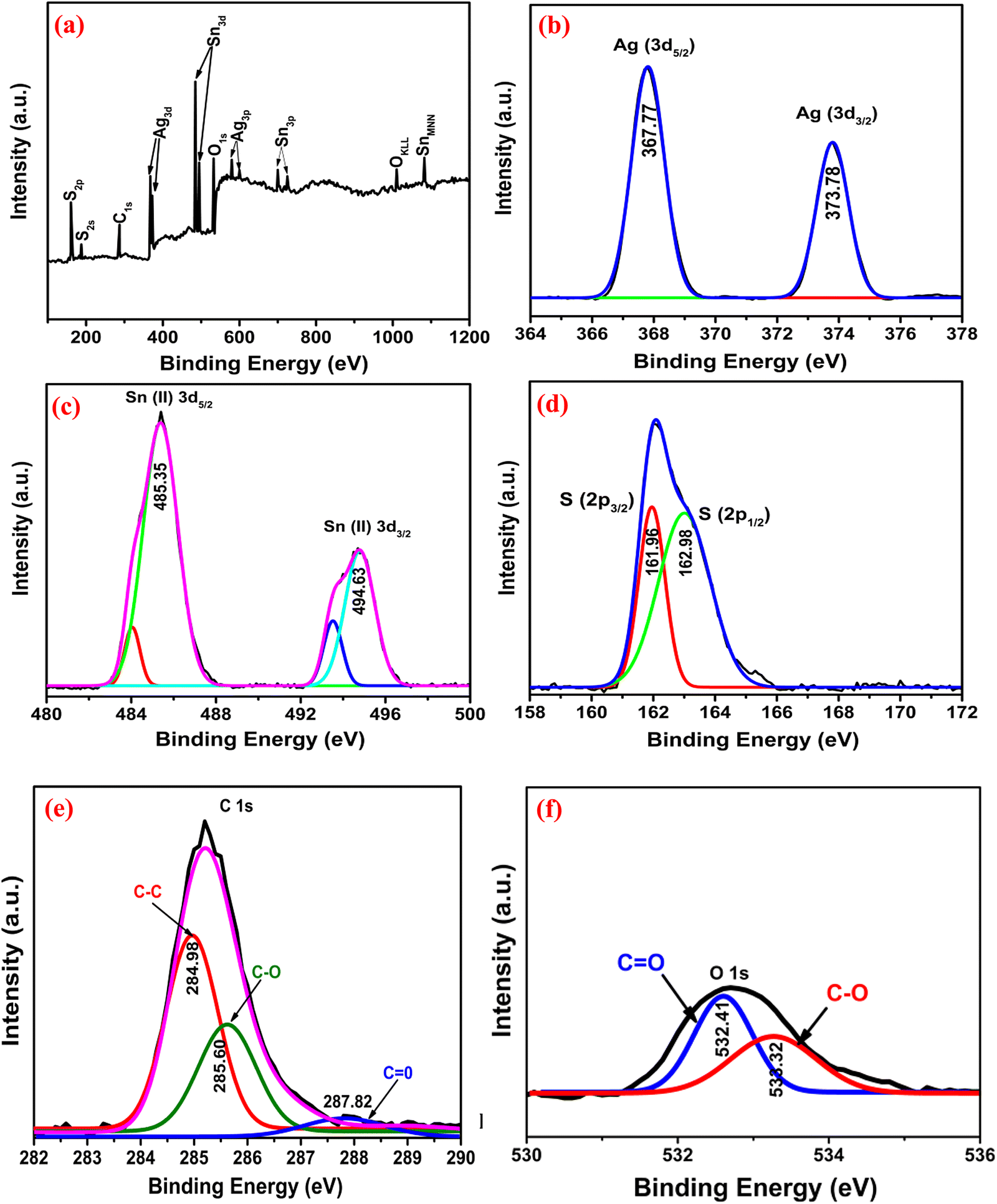
3.2.5. XPS analysis.
To further explicate the chemical composition and valence state, XPS analyses were performed [Fig. 3]. Fig. 3(a) shows the full survey scan, confirming the presence of all elements. Fig. 3(b) shows the high-resolution spectrum of Ag 3d presenting two characteristic doublets at 367.77 and 373.78 eV, which indicate Ag(I) 3d5/2 and 3d3/2, respectively.30 The Sn 3d spectrum [Fig. 3(c)] showed doublet spin–orbit peaks at 485.35 and 494.63 eV corresponding to Sn(II) 3d5/2 and 3d3/2. In the S 2p [Fig. 3(d)] region, the peaks at 161.96 and 162.98 eV correspond to S 2p3/2 and 2p1/2, indicating the bonding of S−2 with the metal ions.31 The C 1s spectrum [Fig. 3(e)] could be split into characteristic peaks assigned to C–C (284.98 eV), C–O (285.60 eV) and CO (287.82 eV) of rGO. The reduction of GO was further confirmed by the higher peak intensity of C–C than those of C–O and CO.18 In Fig. 3(f), the peaks at 532.41 and 533.32 eV are present due to O−2 and O−1, respectively.32
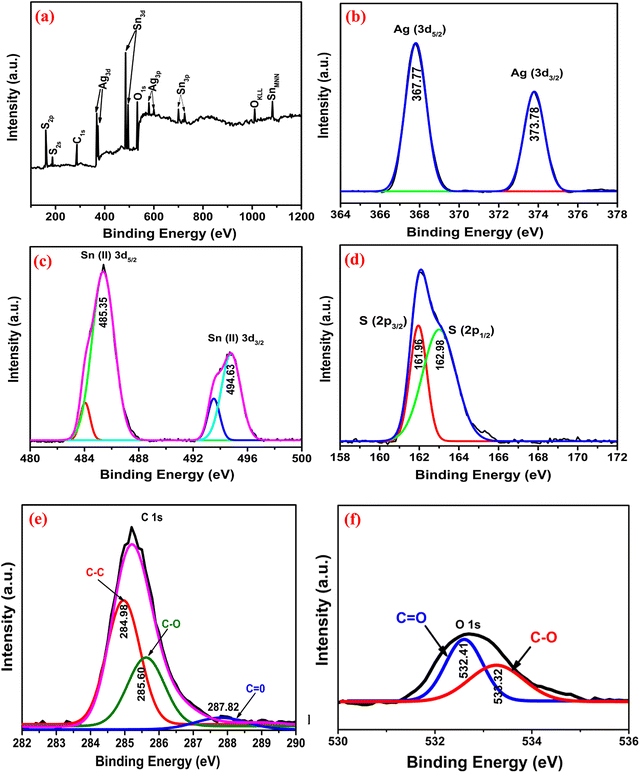 |
| | Fig. 3 The XPS spectra of Ag3.84Sn3S8@rGO0.5: (a) Full scan, (b) Ag, (c) Sn, (d) S, (e) C, and (f) O. | |
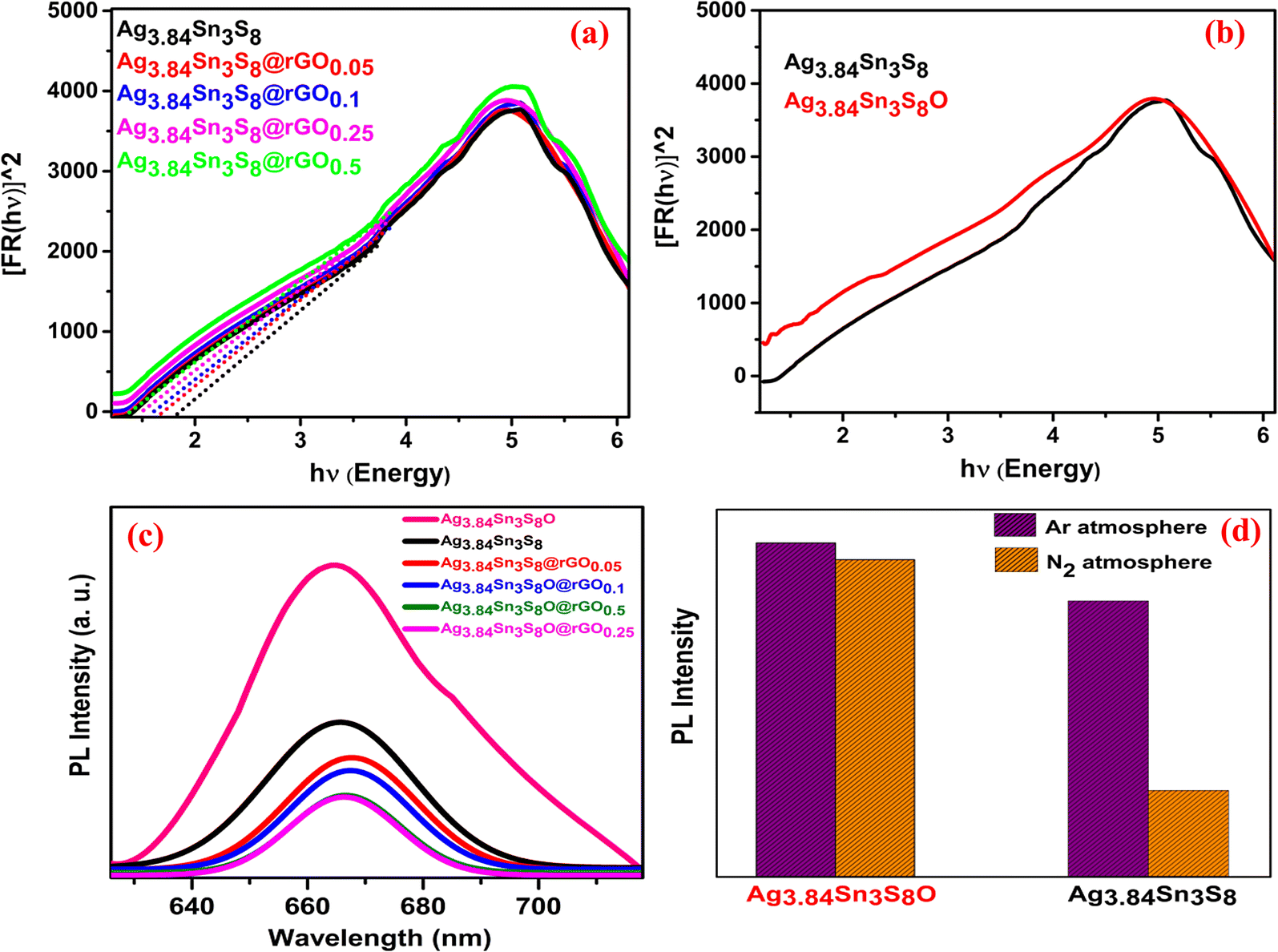
3.2.6. Optical studies to analyze the presence and importance of S vacancies.
In the DRS study, [Fig. 4(a)] for Ag3.84Sn3S8, absorption took place at 670 nm, and as the rGO concentration increased, absorption was slightly red-shifted, which reflected on the value of bandgaps (Table 1).33,34 The lowering of the bandgap can be attributed to an electronic interaction between rGO and Ag3.84Sn3S8 during in situ synthesis. On calcination under O2 saturation, the position of the absorption band edge shifted from 1.850 (Ag3.84Sn3S8) to 0.72 eV (Ag3.84Sn3S8O) attributed to O doping in the non-stoichiometric crystal lattice site.6 This O doping in the lattice ultimately led to changes in the electronic properties [Fig. 4(b)]; consequently, the characteristic absorption tail feature of Ag3.84Sn3S8 vanished. This result convincingly suggests the presence of S vacancies in the crystal lattice.6 It is well-known that the lowering of PL intensity of a particular nanocrystal upon modification is a result of higher e−–h+ pair charge separation; therefore, we analyzed the samples further by PL spectroscopy. In our study, the peaks of the wide PL bands were located between 665–670 nm [Fig. 4(c)], which is likely due to the band edge emission because the corresponding energy associated with this gap is nearly equal to the bandgap obtained from the DRS analysis. The highest PL intensity obtained for Ag3.84Sn3S8O indicates the total occupation of S vacancies by O, and thus, the least charge separation happens, which strongly supports that the S vacancies play a crucial role as electron trappers in the crystal. Further, to confirm our findings, we measured PL under saturated Ar and N2 atmospheres for Ag3.84Sn3S8 and Ag3.84Sn3S8O only [Fig. 4(d)]. In the case of Ag3.84Sn3S8O, there was no such change in the PL intensity in the two different atmospheres, signifying that the S vacancies were already occupied by O atoms and no further elements could get attached to the crystal. For Ag3.84Sn3S8, there was a significant change in PL intensity in the two different atmospheres, and the overall intensities were also quite low compared with the intensities of Ag3.84Sn3S8O. This result suggests that there were enough S vacancies to adsorb the Ar and N2 molecule in Ag3.84Sn3S8, and the trapped electrons (generated through S vacancies) had a chance to travel from the catalyst to the adsorbed foreign element to facilitate catalysis. It is important to note that if the foreign element is a nitrogenous molecule, then all bonding orbitals would be occupied, and naturally, the electrons from the catalyst will go to the anti-bonding orbitals, triggering electronic level changes in the molecule.6
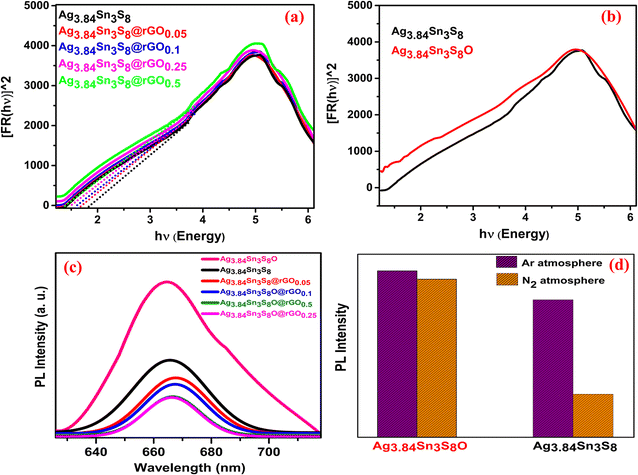 |
| | Fig. 4 The UV-Vis DRS spectra of the (a) synthesized catalysts and (b) Ag3.84Sn3S8 and Ag3.84Sn3S8O (c) The PL spectra of the catalysts; (d) The PL intensity comparison of Ag3.84Sn3S8 and Ag3.84Sn3S8O in the presence of Ar and N2. | |
Table 1 The band gaps (Eg) obtained from the DRS study
| Materials |
E
g (eV) |
| Ag3.84Sn3S8 |
1.850 |
| Ag3.84Sn3S8@rGO0.05 |
1.681 |
| Ag3.84Sn3S8@rGO0.1 |
1.565 |
| Ag3.84Sn3S8@rGO0.25 |
1.460 |
| Ag3.84Sn3S8@rGO0.5 |
1.334 |
| Ag3.84Sn3S8O |
0.72 |
3.2.7. Electrochemical Mott–Schottky (M–S) analysis.
To investigate the probable energy profile diagram, M–S analysis [Fig. S5, ESI†] was performed at 1000 Hz. There was a slight positive shift (Table S2, ESI†) in the flat band (fb) potential, indicating band bending; the small change supports the similar bulk structure of the NC,35 and the positive slope confirms the n-type semiconducting property.36,37 It was noticeable that with increasing concentration of rGO, the carrier density (e−) increased and reach the optimum as Ag3.84Sn3S8@rGO0.25 had the highest value. Considering that the optical bandgap narrowed from (1.85 eV), as well as fb (−0.8 V), the work function of rGO (−0.09 V) and redox potential of MNZ (−0.05 V), it can be assumed that the possibility of e−–h+ pair recombination is significantly minimized in the presence of rGO, and charge carriers (e−) might get dislocated from the CB to rGO, profoundly facilitating the reduction probability of MNZ [Fig. S6, ESI†].
3.2.8. BET isotherm analysis.
To analyze the active reaction sites, BET analysis was performed using the N2 adsorption–desorption isotherms (77 K) [Fig. S7(a–f), ESI†]. The obtained isotherms matched well with Type IV hysteresis and revealed a mesoporous structure. The specific surface areas and pore sizes are tabulated in Table S3 and discussed in ESI† (line no: 131–136). The high surface areas of the base materials indicate the presence of S vacancies in the non-stoichiometric crystal lattice. The increase in the surface area upon incorporating rGO supports interfacial binding between Ag3.84Sn3S8 and rGO.29
3.2.9. Electrochemical impedance spectroscopy (EIS).
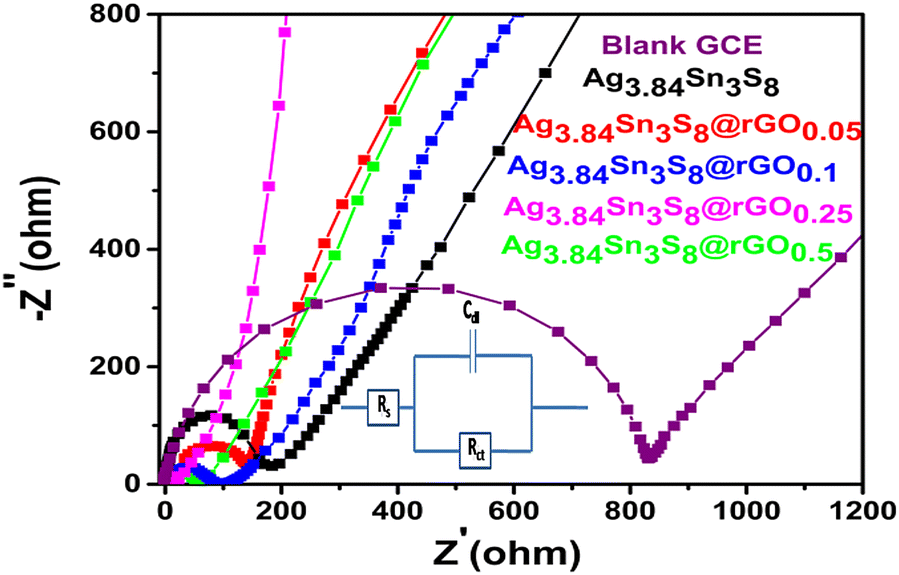
The Nyquist plots [Fig. 5] gave a clear idea about the ease of charge transfer between the electrode–electrolyte interfaces.38–41 The arc radii decreased from blank GCE to pure Ag3.84Sn3S8 followed by the composites fabricated, which further confirmed that rGO incorporation efficiently separated the e−–h+ pairs and exceptionally reduced the Rct. This study also supported the optimization result from M–S analysis, as the smallest arc radius was observed for Ag3.84Sn3S8@rGO0.25 with the least Rct [Table 2]. The best-fitted Randles equivalent circuit model is shown in the inset of Fig. 5.
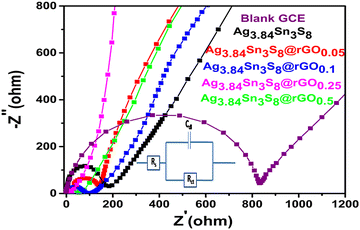 |
| | Fig. 5 The Nyquist plots of the blank and GCEs fabricated using the as-synthesized materials. | |
Table 2 The resistance parameters obtained from the Nyquist plots
| Materials |
R
s (Ohm) |
R
ct (Ohm) |
C
dl (μF cm−2) |
| Bare GCE |
118.28 |
752.25 |
217.95 |
| Ag3.84Sn3S8 |
115.01 |
165.60 |
51.60 |
| Ag3.84Sn3S8@rGO0.05 |
110.04 |
132.30 |
36.21 |
| Ag3.84Sn3S8@rGO0.1 |
109.25 |
91.68 |
21.58 |
| Ag3.84Sn3S8@rGO0.25 |
106.96 |
18.20 |
5.43 |
| Ag3.84Sn3S8@rGO0.5 |
107.54 |
56.71 |
14.12 |
3.3. The electrocatalytic detection of MNZ
3.3.1. Testing of the catalytic performance.
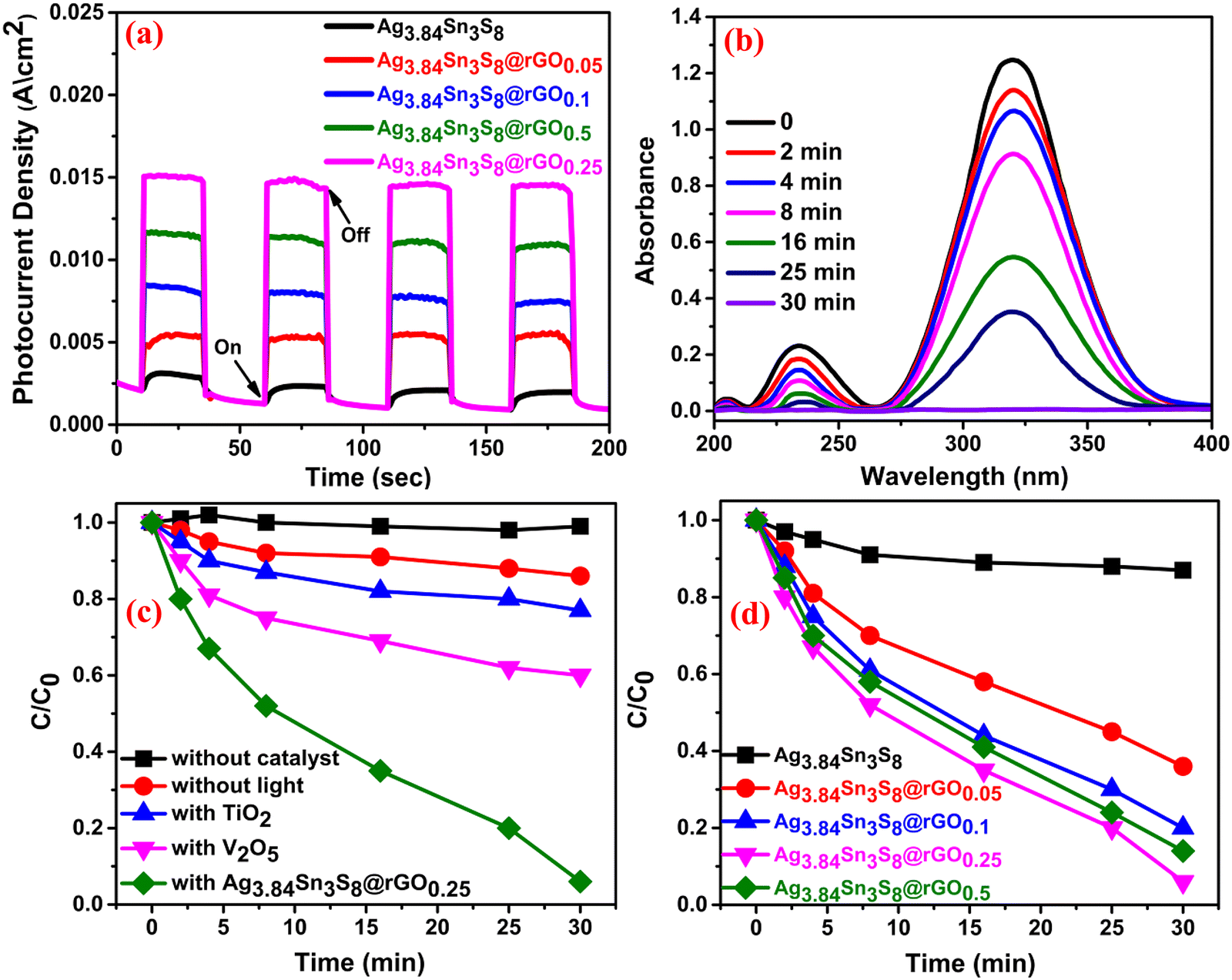
Before delving into the electrosensing property, we investigated the catalytic applicability of the NCs. Accordingly, the transient photocurrent characteristics of the ITO-coated glass modified using the catalysts were measured by chronoamperometry under chopped light illumination [Fig. 6(a)] (elaborated in ESI†). The study was performed by dipping the fabricated slides into N2 saturated 0.05 (M) PBS. The result suggested that with the introduction of rGO, the e−–h+ pairs were efficiently separated and the charge carrier (e−) possessed a longer lifetime [Fig. 6(a)], which is also supported by the optical analysis. The high photocurrent densities further confirmed the excellent conductivity of the composites, and the highest value was observed for Ag3.84Sn3S8@rGO0.25, which is consistent with the M–S and EIS studies. This result thus motivated us to examine the photocatalytic performance of Ag3.84Sn3S8@rGO0.25 in MNZ degradation. Fig. 6(b) shows the performance under visible-light irradiation, and the characteristic absorption peak of MNZ (∼318 nm) vanishes by ∼98% due to degradation within 30 minutes, indicating complete degradation. Interestingly, no other noticeable peak was generated during the course of this photocatalysis, suggesting that no intermediate with absorption energy corresponding to 318 nm was formed. For comparison with commercial V2O5 and TiO2, photocatalysis [Fig. 6(c)] was performed in exactly the same way, and only 33% and 22% degradation were observed, respectively. Additionally, the reactions were also performed without the catalyst and irradiation for a complete evaluation. We found that without the catalyst, no degradation happened, indicating the profound stability of MNZ under visible light, and in a similar manner, only 11% degradation of MNZ was observed without light. The comparative study revealed [Fig. 6(d)] the following order of performance: Ag3.84Sn3S8@rGO0.25 (98%) > Ag3.84Sn3S8@rGO0.50 (90%) > Ag3.84Sn3S8@rGO0.1 (79%) > Ag3.84Sn3S8@rGO0.05 (65%) > Ag3.84Sn3S8 (12%). The probable mechanism is explained through the band energy diagram [Fig. S6, ESI†] of n-type Ag3.84Sn3S8, which possesses photogenerated negatively charged carriers (e−) in the CB and h+ in the VB. Interestingly, the incorporation of rGO significantly reduces the possibility of recombination as the e− are transferred to rGO due to its energetically favorable alignment with the CB of the catalyst owing to extended π conjugation.29 However, beyond a certain percentage of black rGO, (i) it starts shielding the incident photon, thus preventing charge pair generation; (ii) it may reduce the pore volume of the nanoparticles, resulting in low adsorption of the analyte in the solution; (iii) it may mask the catalytically active sites, retarding the overall catalytic process.42 The plausible reaction pathway is depicted in ESI† (eqn (S1)–(S7)).43–45
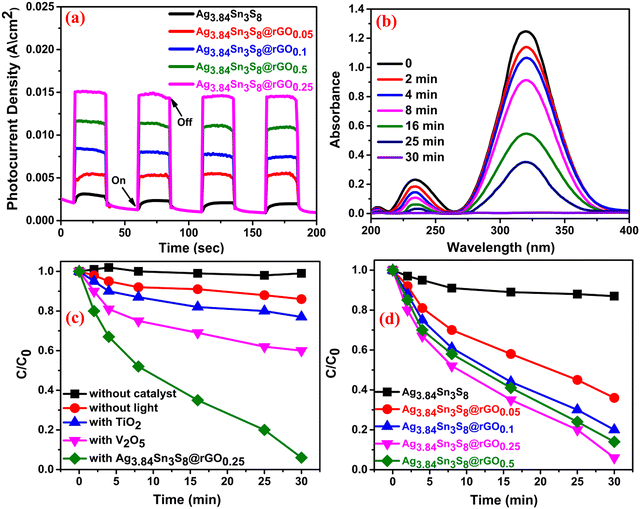 |
| | Fig. 6 (a) Photocurrent study of the as-synthesized catalysts. (b) The time-dependent photocatalytic performance curves of Ag3.84Sn3S8@rGO0.25. A comparison of Ag3.84Sn3S8@rGO0.25 with (c) different commercial catalysts and (d) the as-synthesized catalysts. | |
3.3.2. Photocatalytic reaction rate analysis.
The rate constants for individual catalysts [Table S4, ESI†] were obtained [Fig. S8(a), ESI†] by employing the pseudo-first-order kinetics eqn (1):The highest rate constant 0.0825 min−1 was obtained for Ag3.84Sn3S8@rGO0.25, as expected.
The effect of catalyst dosage on the performance was studied by varying the concentration from 5–80 mg mL−1 [Fig. S8(b), ESI†]. It was found that beyond the concentration of 20 mg mL−1, the activity was readily reduced. This might be due to several reasons: (1) a decrease in interfacial area between the catalyst and the solution, (2) the light may be scattered by the aggregated catalyst beyond this concentration and (3) the number of active sites might sufficiently reduce beyond the optimized rGO concentration.
3.3.3. Reactive oxygen species determination to support the plausible mechanism.
To identify the actual reactive species involved in the reaction, scavengers of hydroxyl radicals (˙OH), superoxide radicals (O2˙−) and holes (h+) were added to the reaction mixture during photocatalysis. We used n-butanol (n-BuOH), benzoquinone (BZQ) and triethylamine (TEA) for ˙OH, O2˙− and h+ respectively46–48 [Fig. S8(c), ESI†]. Interestingly, the efficiency was greatly suppressed in the presence of BZQ and n-BuOH but not in the case of TEA, confirming the predominant involvement of O2˙− and ˙OH only. The efficiencies obtained were as follows: 70% (TEA), 13% (BZQ) and 31% (n-BuOH).
3.3.4. Photocatalytic stability measurement.
To establish the stability and reproducibility of the catalysts for practical application, we investigated their performance through five catalytic cycles [Fig. S8(d), ESI†], and the efficiencies obtained were in the range of 98–88%, which indicates the higher accumulation of MNZ on the catalytic surface, inhibiting the further generation of reactive species. Overall, the results suggest acceptable stability, and a comparative analysis is presented [Table S5, ESI†]. From the table, we can see that though some low-cost catalysts are available, in terms of catalyst dosage, the total time for complete degradation and the value of the rate constant, our catalyst shows overall significant activity.
3.3.5. Electrochemical response of the proposed sensor and electrochemical active surface area (ECSA) determination.
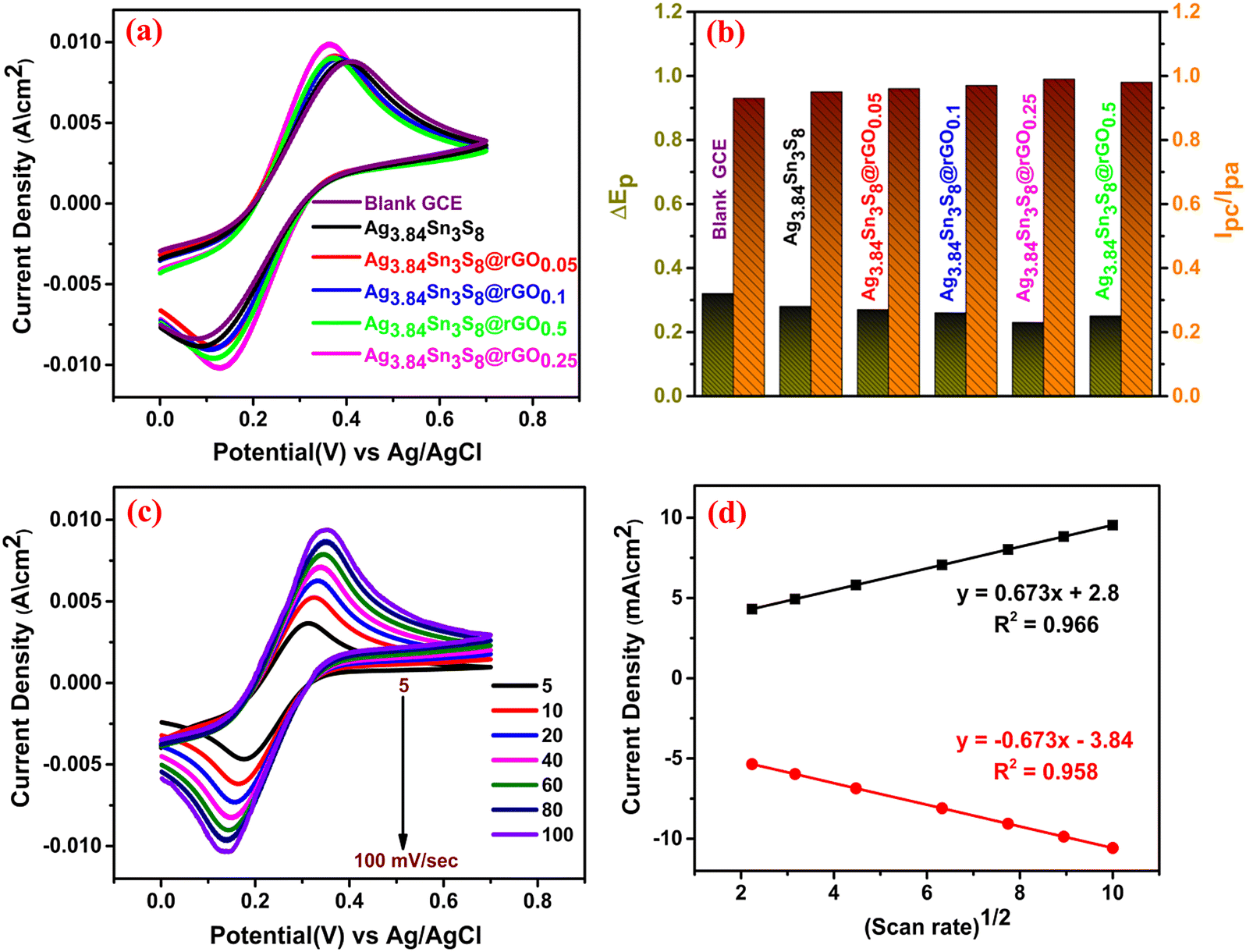
To examine the electrochemical response of the proposed sensors, we recorded the CV curves of six different fabricated electrodes, including the bare GCE [Fig. 7(a)]. These measurements were carried out in 0.1(M) KCl electrolyte containing 10 (mM) [Fe(CN)6]3−/4− at a scan rate of 100 mV sec−1. The electrocatalytic reaction on the electrode surface can be outlined as follows;| | | [Fe(CN)6]−4 → [Fe(CN)6]−3 + e (oxidation) | (2) |
| | | [Fe(CN)6]−3 + e → [Fe(CN)6]−4 (reduction) | (3) |
Eqn (2) and (3) represent the anodic/cathodic peaks obtained in the CV curves. The symmetrical nature and peak current densities confirmed the improved capacitive nature with an increase in rGO. Here, ΔEp and Ipc/Ipa were also optimized [optimum ΔEp ≈ 0.24 and (Ic/Ia) ≈ 1] for Ag3.84Sn3S8@rGO0.25 [Fig. 7(b)]. This is probably due to several determining factors, such as the significant electron/mass transfer capability, a large number of surface active reaction sites owing to the S vacancies, the lowest resistance and excellent conductivity, which are already confirmed by the BET analysis (large surface area) [Fig. S7, ESI†] and EIS [Fig. 5].49,50 Hence, the optimum electrode i.e. Ag3.84Sn3S8@rGO0.25 was chosen for the fabrication of the working electrode. To calculate the ECSA, CV was performed by varying the scan rates from 5–100 mV sec−1 under similar reaction conditions [Fig. 7(c)], and the peak current kept increasing with the increase in scan rate. The asymptotic line observed in the curve of peak current density vs. (scan rate)1/2 [Fig. 7(d)] conveniently satisfied the Randles–Sevcik equation51 (eqn (4)), from which the ECSAs of all the samples were calculated [Table 3]. The result clearly suggests that the highest ECSA and surface architecture of Ag3.84Sn3S8@rGO0.25 had an extraordinary effect on its electrocatalytic performance.| | | Ip = 2.69 × 105n3/2AD1/2Cγ½ | (4) |
where Ip is the peak current density, n is the number of migrating electrons, A is the ECSA of the working electrode, D is the diffusion coefficient of the electrolyte, C is the concentration of the ([Fe(CN)6]3−/4− ion and γ is the operating scan rate.
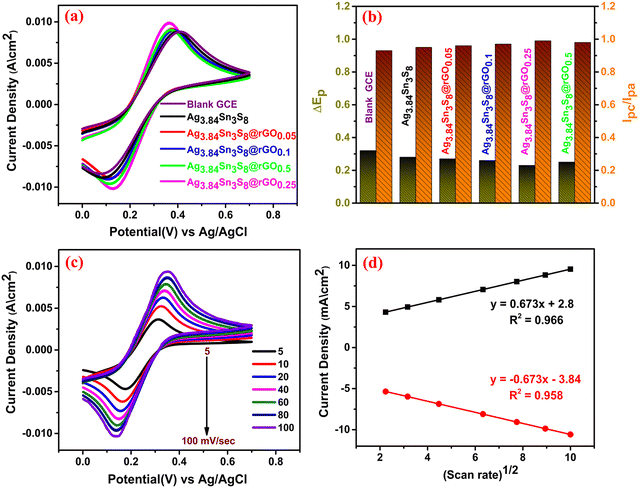 |
| | Fig. 7 (a) The CV curves, (b) ΔEp and (Ipc/Ipa) of the blank and fabricated GCEs; (c) CV of Ag3.84Sn3S8@rGO0.25 with scan rate variation; (d) the current density vs. (scan rate)1/2 plot. | |
Table 3 The ECSA values of pure Ag3.84Sn3S8 and the respective NCs
| Materials |
ECSA (cm2) |
| Ag3.84Sn3S8 |
0.0200 |
| Ag3.84Sn3S8@rGO0.05 |
0.0218 |
| Ag3.84Sn3S8@rGO0.1 |
0.224 |
| Ag3.84Sn3S8@rGO0.25 |
0.0241 |
| Ag3.84Sn3S8@rGO0.5 |
0.0229 |
3.3.6. Electrochemical ultra-sensing of MNZ.
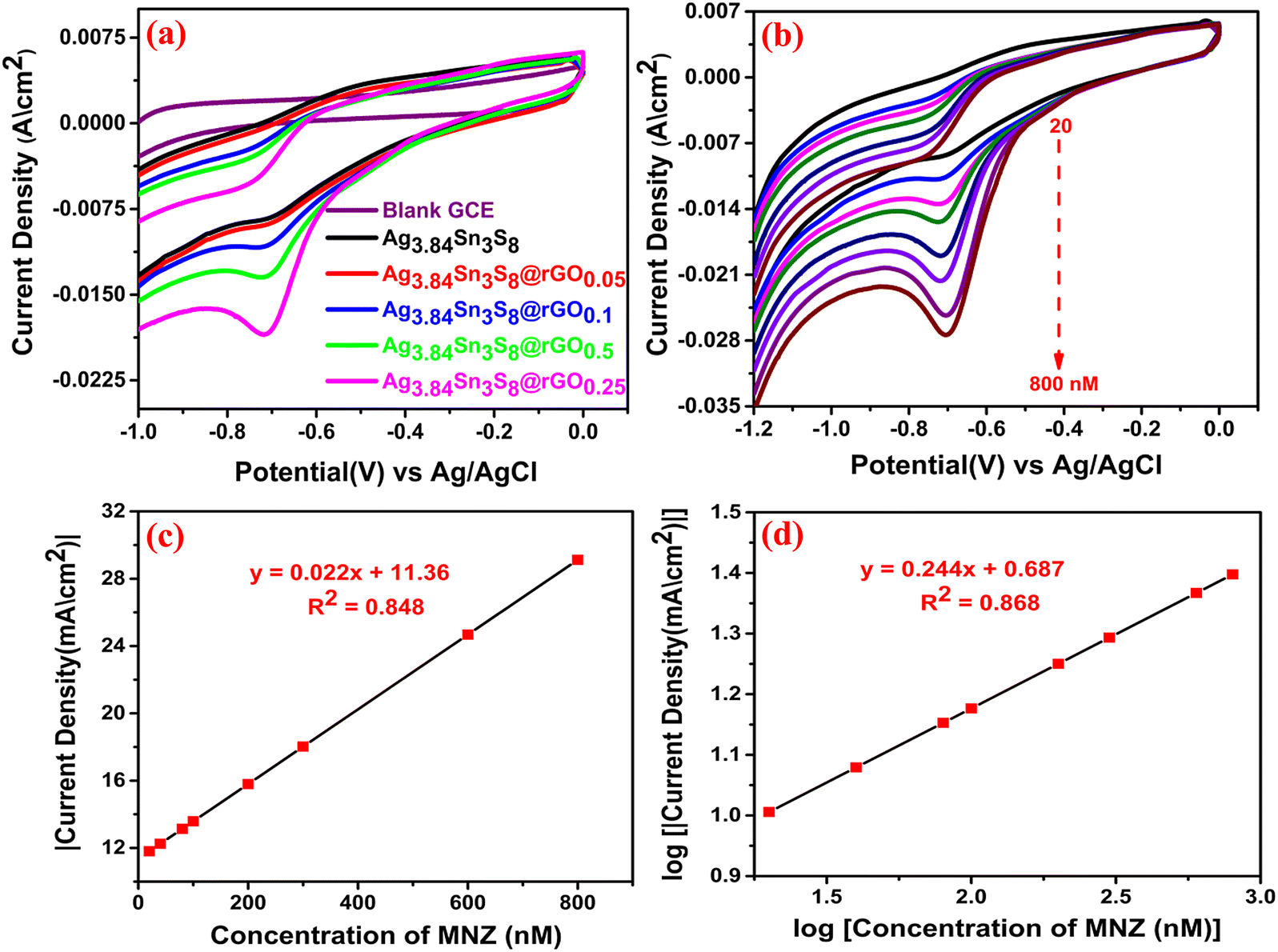
To fulfill our main objective, we investigated the electrochemical MNZ ultra-sensing capacity of the GCEs fabricated using the as-synthesized catalysts. The CV responses [Fig. 8(a)] of the bare and modified GCEs were recorded in 0.05 (M) PBS (pH = 7) containing 500 nM MNZ at a scan rate of 50 mV sec−1. It was clearly seen that in the case of bare GCE, no peak appeared at the characteristic reduction potential (−0.7 V) of MNZ. Again, the peak current density was observed (20 mA cm−2) for Ag3.84Sn3S8@rGO0.25 at −0.7 V, reflecting our previous observations (S vacancies, the highest surface area, feasible band alignment, the highest charge carrier density, and the lowest electrochemical resistance among the tested catalysts).
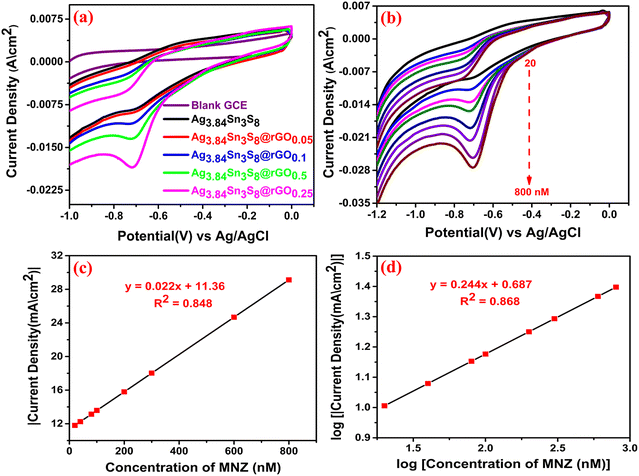 |
| | Fig. 8 (a) CV of the GCEs at 500 nM MNZ; (b) The CV curves of Ag3.84Sn3S8@rGO0.25 at varying MNZ concentrations. The calibration plots: (c) |current density| vs MNZ concentration and (d) log of current density vs. log of MNZ concentration. | |
The effect of the concentration of MNZ on the proposed sensor was further deeply analyzed in the concentration range of 20 to 800 nM in an N2-saturated electrolyte (to ensure an inert atmosphere within the cell) while keeping the reaction conditions the same [Fig. 8(b)]. The results suggested that the increasing quantities of MNZ were readily recognized by the proposed sensor. It is important to note that there was no oxidation peak in the reverse scan of CV, which confirmed the irreversible reduction of MNZ. The calibration plot of peak |current density| vs. concentration could be expressed by the linear regression equation: I (mA) = 0.022 (nM) + 11.36 with R2 = 0.848 [Fig. 8(c)]. Further, the logarithmic calibration plot (log of reduction peak current density vs. log of concentration) [Fig. 8(d)] also demonstrated a linear regression equation: I (log mA) = 0.244 (nM) + 0.687, R2 = 0.868. These results conveniently suggest that the sensing mechanism followed first-order kinetics.
3.3.7. The mechanistic investigation of the reaction pathway.
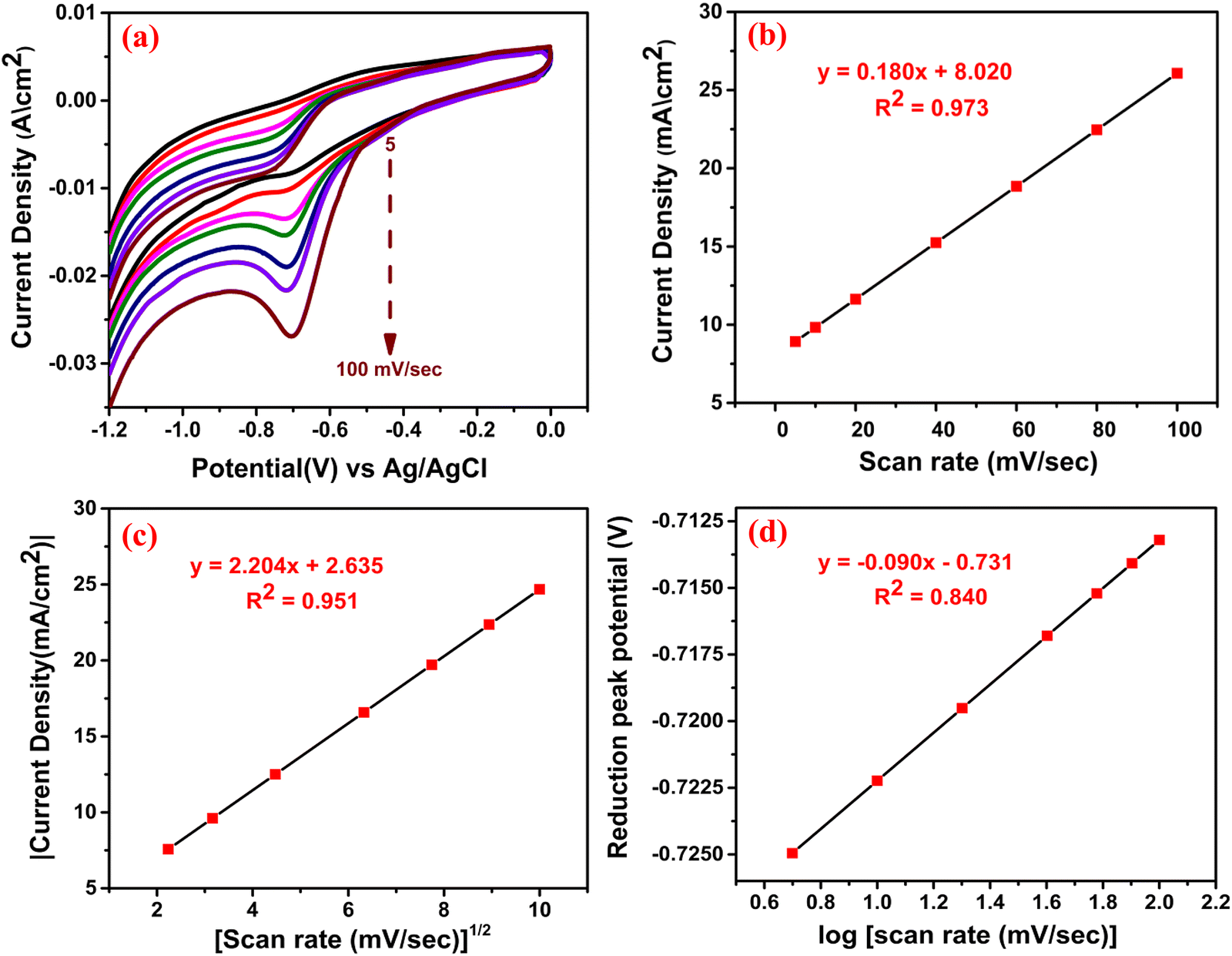
To explore the mechanistic aspect of the sensing reaction, we have emphasized the scan rate variation with current density and peak potential. The CV responses were studied in the presence of MNZ by varying the scan rates between 5–100 mV sec−1 [Fig. 9(a)] under similar reaction conditions. We could see that the peak current density increased with the increase in scan rate, and the calibration plot [Fig. 9(b)] of peak current density vs scan rate showed the linear regression equation: I (mA) = 0.180 (mV sec−1) + 8.020, R2 = 0.973. This result indicates that the electrochemical reduction was an adsorption-controlled phenomenon i.e. electrocatalytic surface-detained kinetics. On the other hand, the linear relation between peak current density and (scan rate)1/2 suggested [Fig. 9(c)] that the reaction might also proceed through diffusion-controlled kinetics according to the linear regression equation: I (mA) = 2.204 (mV sec−1)1/2 + 2.635, R2 = 0.951. Among these kinetic pathways, the most probable was determined based on the higher value of R.52 In our case, the higher value of R (0.986) is obtained in Fig. 9(b), which indicates that the reaction predominantly proceeds through surface adsorption rather than diffusion-controlled kinetics. This crucial observation again convincingly supports the adsorption of MNZ onto the active sites arising from S vacancies. The unit cell structure of the nanocrystal clearly let us visualize the presence of S vacancies, in which the foreign molecules can get adsorbed, and under electrochemical bias, the generation of charge carriers is facilitated by the rGO backbone for the molecule to undergo catalysis (discussed in 3.2.6) (Scheme 2). The charge transfer coefficient (α) was calculated from the slope of the Tafel plot of the cathodic peak potential vs. log of scan rate [Fig. 9(d)] according to eqn (5).53 The slope was obtained from the linear regression equation: E (V) = −0.090 (log scan rate (mV sec−1)) – 0.731, R2 = 0.840.| | | Slope = n(1 − α)F/2.3RT | (5) |
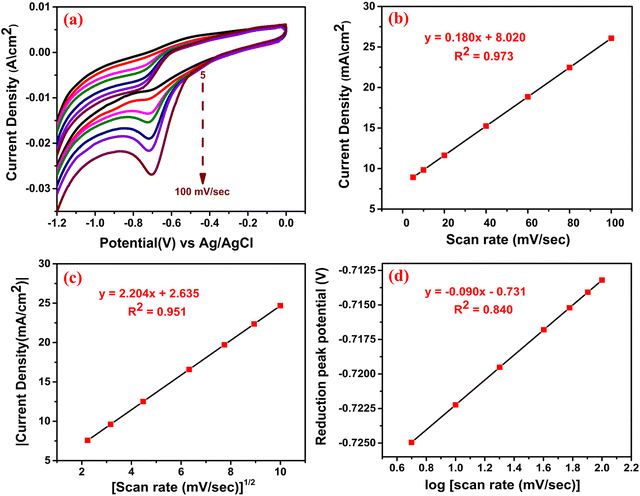 |
| | Fig. 9 (a) CV at different scan rates; the calibration plots for (b) peak current density vs scan rate and (c) peak current density vs (scan rate)1/2 and (d) peak potential vs log of scan rate. | |
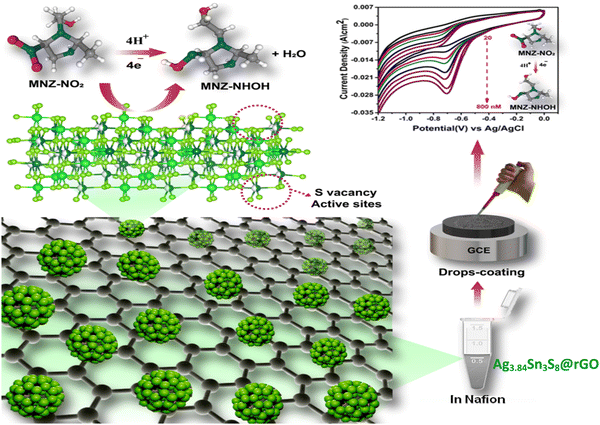 |
| | Scheme 2 The mechanistic pathway of MNZ reduction on vacant active sites (S vacancies) on the catalyst surface. | |
The corresponding terms in eqn (5) indicate standard parameters. ‘α’ was determined to be 0.99 by taking n = 4. The number of electrons (n) was calculated to be 3.89 based on eqn (6):52
| | | I = (2.99 × 105)n[(1−α)n]1/2ACoDo1/2ν1/2 | (6) |
The corresponding terms in eqn (6) carry their usual meaning.
Further, the adsorption quantity of MNZ was estimated using eqn (7):
where
Γ is the amount of MNZ adsorbed on the sensor, which was found to be 2.6 × 10
−6 mol cm
−2.
3.3.8. Determination of catalyst sensitivity and the limit of detection (LOD).
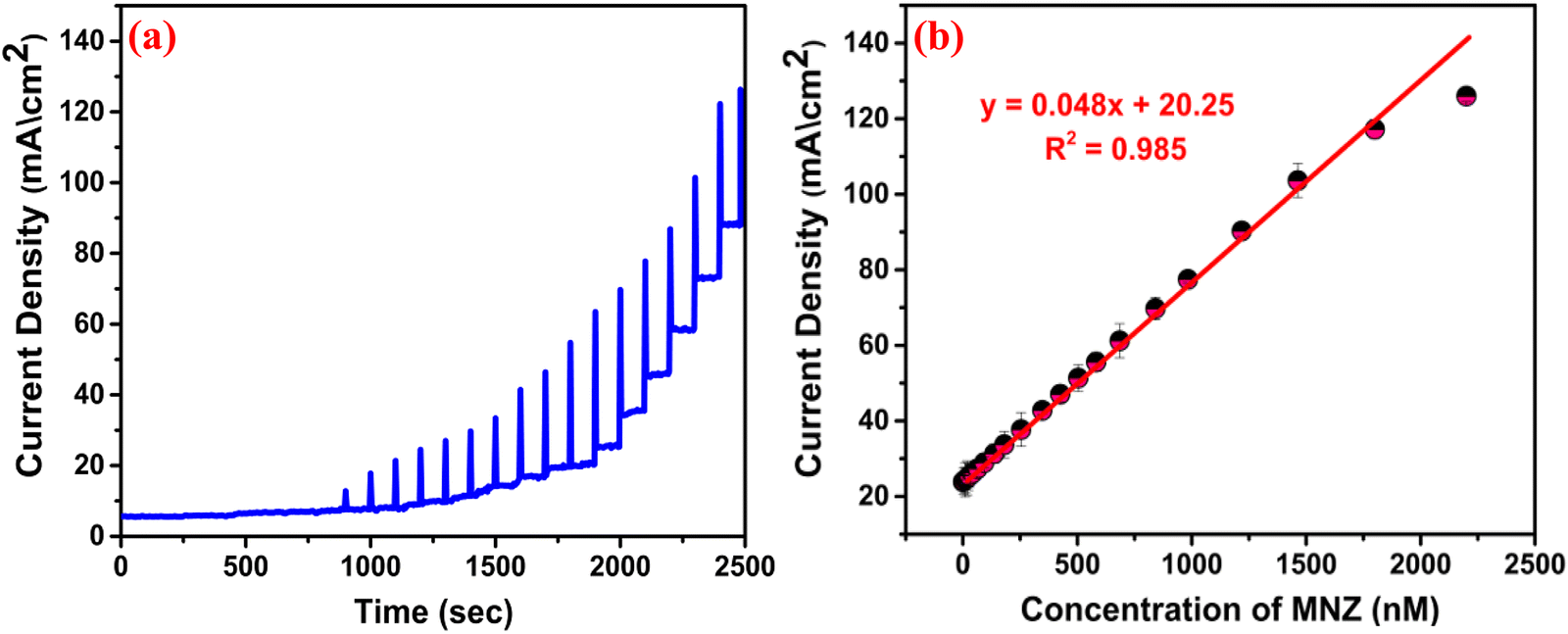
To determine the sensitivity and LOD, we performed chronoamperometric studies by maintaining the optimum electrochemical parameters [Fig. 10(a)], and a calibration plot of current density vs MNZ concentration was obtained [Fig. 10(b)]. The reduction potential was fixed at −0.7 V vs. Ag/AgCl, and the reductive current density increased with the amount of MNZ at concentrations ranging between 0.01–2500 nM, confirming the accumulation of the analyte on the sensor, that is, the adsorption of MNZ on to the crystal. Additionally, the calibration plot revealed a linear range from 0.01 to 1500 nM with the linear regression equation: I (mA) = 0.048 (nM) + 20.25, R2 = 0.985. Interestingly, at higher concentrations, the reduction current density decreased, which confirmed that beyond a certain extent of adsorption, the S vacancies get fully occupied by the MNZ molecules, which subsequently produce internal resistance and obstruct further adsorption, resulting in less diffusion of the molecules into the electrode. By following the IUPAC recommended method,52 from the slope, the LOD and sensitivity were calculated to be 4 pM and 48 μA.nM−1.cm−2, respectively.
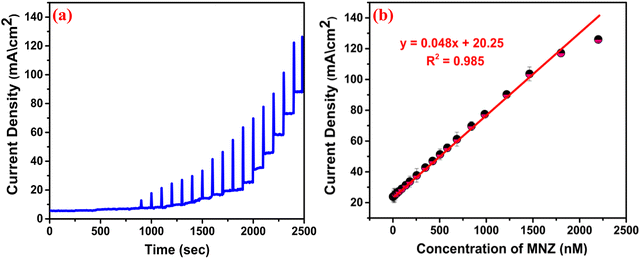 |
| | Fig. 10 (a) The chronoamperometry test; (b) The calibration plot for current density vs. MNZ concentration. | |
3.3.9. The factors affecting the electro-sensing performance.
Several factors were examined, including (i) pH, (ii) interfering agents, (iii) stability test by varying catalytic cycles, electrodes and duration of the experiment.
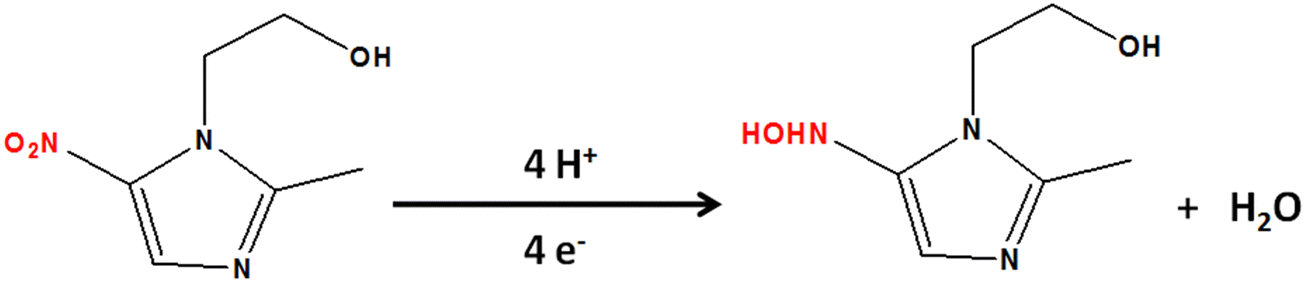
The influence of pH on the sensing performance was examined in the 3.0 to 11.0 range [Fig. S9(a), ESI†]. This study revealed that the electrochemical reduction was linearly dependent on pH. The highest cathodic peak current density was obtained at pH 7, and the calibration plot [Fig. S9(b), ESI†] for reduction peak potential vs pH revealed that, with increasing pH, the potential shifted toward negative values, indicating the protonation–deprotonation characteristics of MNZ. The corresponding linear regression equation could be expressed as E = −0.047pH – 0.700, R2 = 0.959. It is interesting to note that the slope value of −0.047 was quite close to the theoretical approximation (0.059) obtained using the Nernst equation, further confirming the involvement of an equal number of e− and H+ in the reduction of MNZ.50,54 The probable reaction pathway involves the reduction of the nitro group to nitroso, which is subsequently reduced to hydroxyl amine (Scheme 3).55
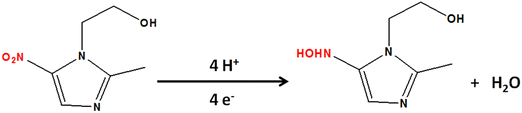 |
| | Scheme 3 The protonation–deprotonation step in the probable reduction reaction pathway of MNZ. | |
Anti-interference studies were performed to analyze the selectivity of the sensor toward MNZ base12d on chronoamperometric measurements in the presence of other co-interfering compounds individually in N2 saturated 0.05 (M) PBS (pH = 7.0). Here, we used the following co-interfering compounds: ornidazole (ODZ), ofloxacin (OXN), ciprofloxacin (CFX), tinidazole (TDE), imidazole (IZE), 5-nitroimidazole (5-NIZ), 4-nitrophenol (4-NP), 4-nitrobenzene (4-NB), Hg and uric acid (UA). The concentration of these compounds was kept 10 times higher than that of MNZ [Fig. 11(a)] to obtain a clear picture of selectivity. The obtained result conveniently confirms that the as-proposed sensor is perfectly suitable for the detection of MNZ in real samples.
 |
| | Fig. 11 (a) The anti-interference test: (1) MNZ (2) ODZ, (3) OXN, (4) CFX, (5) TDE, (6) IZE, (7) 5-NIZ, (8) 4-NP, (9) 4-NB, (10) Hg and (11) UA; (b) Repeatability test; (c) reproducibility test; (d) durability study. | |
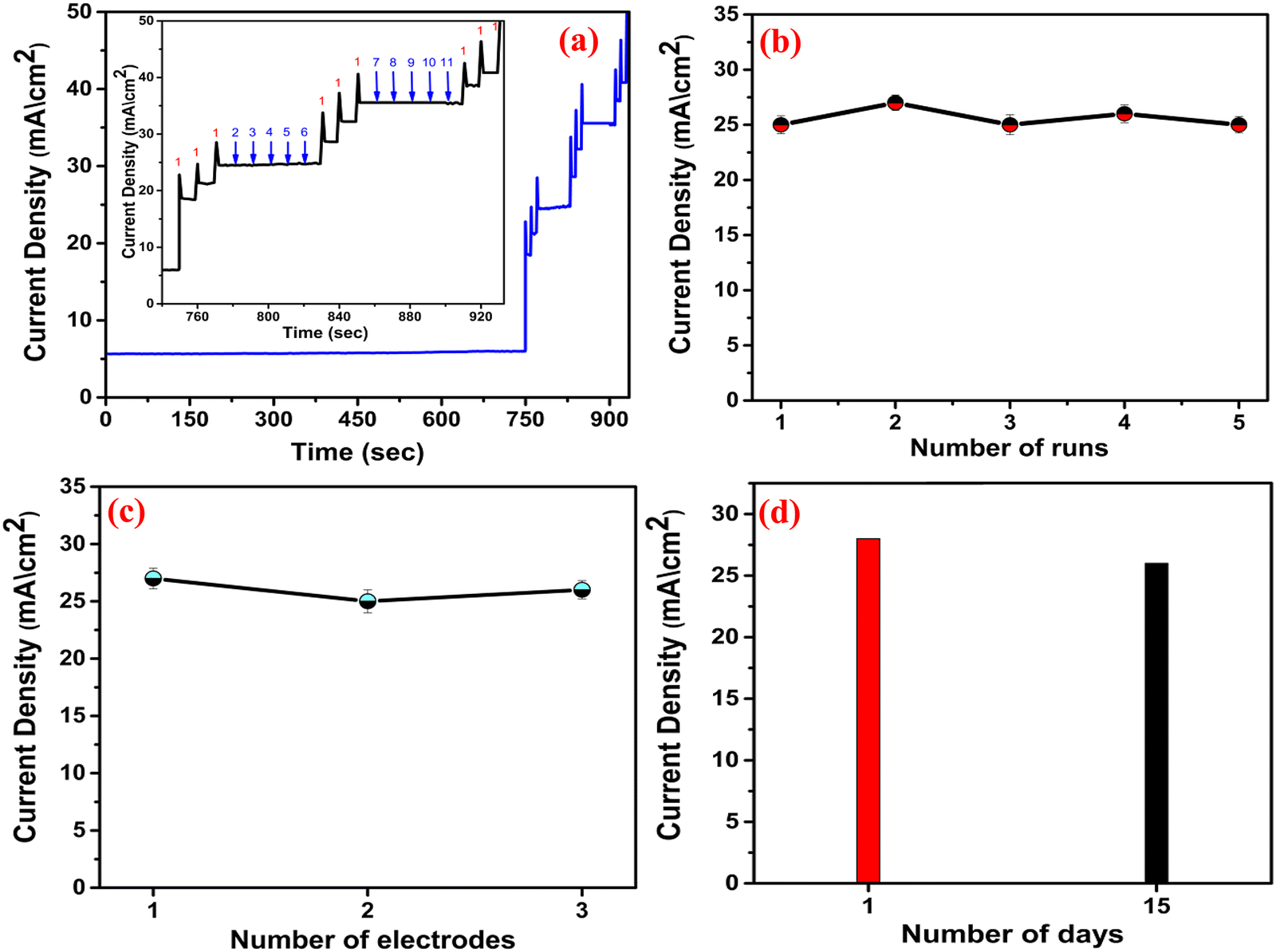
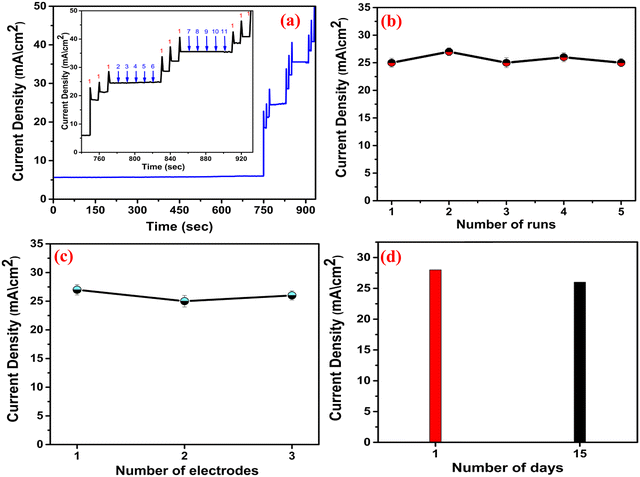
To examine the repeatability, CV was performed in five different catalytic runs [Fig. 11(b)] keeping the same experimental parameters (the concentration of MNZ was 500 nM). The relative standard deviation (RSD) was estimated to be 2.1%, which is quite excellent in terms of repeatability.32 The reproducibility was further evaluated by CV [Fig. 11(c)] using three different electrodes, and the RSD was obtained as 1.6%, suggesting significant accuracy.55 The durability was examined by carrying out two CV runs [Fig. 11(d)], and 96% of the initial current density was retained even after two weeks, indicating excellent long-term stability. Overall, the stability experiments indicate excellent anti-interfering behavior, outstanding repeatability, acceptable reproducibility and very good durability of the proposed sensor. The comparative analysis of the sensing performance of different catalysts is presented in Table 4, and in terms of the linear electrochemical sensing range, LOD and sensitivity, the proposed sensor is excellent.
Table 4 Comparative analysis of the electro-sensing of MNZ by different fabricated electrodes
| Fabricated electrodes |
Techniques |
Linear range (μM) |
LOD (μM) |
Sensitivity (μA μM−1 cm−2) |
Year |
Ref. |
| DMIP |
DPV |
0.04–200 |
0.0091 |
— |
2016 |
56
|
| MWCNTs/CTS-Ni |
DPV |
0.1–150 |
0.025 |
0.695 |
2017 |
57
|
| Cu(II)-CNP |
SWV |
0.02–1.6 |
0.004 |
|
2017 |
58
|
| GR-IL |
DPV |
0.1–25 |
0.047 |
|
2017 |
59
|
| Pt NS/PFFF |
DPV |
2.5–500 |
0.05 |
|
2017 |
60
|
| SDS-GR |
DPSV |
0.08–200 |
0.0085 |
|
2018 |
61
|
| Cu-poly(Cys) |
LSV |
0.5–400 |
0.37 |
|
2018 |
62
|
| ASPCE |
DPV |
0.05–563, 753–2873 |
0.01 |
|
2018 |
63
|
| SrMoSe2 |
DPV |
0.05–914.12 |
0.001 |
1.13 |
2018 |
64
|
| PrV/SCN |
Current-time |
0.001–2444.0 |
0.0008 |
1.386 |
2019 |
65
|
| CNF@Au NPs |
DPV |
0.1–2000 |
0.024 |
|
2022 |
66
|
| Ag3.84Sn3S8@rGO0.25 |
Current-time |
0.01–1500 nM |
4 pM |
48 μA nM−1 cm−2 |
2023 |
This work |
3.3.10. Real sample analysis.
The detailed electrochemical sensing experiments confirmed that the as-proposed sensor has the capability to ascertain the concentration of MNZ in real samples also. In this context, human urine and water were assigned as the backgrounds in the real sample analysis. The urine sample was obtained from a middle-aged (35 years old) healthy person, and water was collected from different lakes in our campus (IIEST, Shibpur) and diluted (1:1), followed by centrifugation to filter out the undesired impurities. Then, to get the spiked sample solutions, a definite amount of MNZ was mixed with the urine, as well as water samples (in an N2-saturated cell). The standard addition method was employed to analyze the whole recovery process. In the electrochemical sensing experiments, the recoveries were observed in the range between 98.7 and 99.9%, indicating excellent selectivity and sensitivity for MNZ in real samples [Table S6, ESI†].
4. Conclusion
In summary, a very simple in situ solvothermal method has been developed to prepare a novel catalyst: Ag3.84Sn3S8@rGO, varying the concentration of rGO (0.05–0.5). The detailed analyses confirm the formation of the NC along with its potential as an efficient electrocatalyst and further, ensure the optimization of its catalytic efficiency (Ag3.84Sn3S8@rGO0.25). We found an outstanding response (sensitivity: 48 μA nM−1 cm−2 & LOD: 4 pM) of the optimal catalyst toward electrochemical MNZ sensing. The extended electrochemical studies reveal the enhanced electrocatalytic efficiency, excellent selectivity, significant stability and real sample applicability. These notable features of the proposed sensor might be due to the S vacancies in its crystal lattice sites, large surface area, excellent charge transfer capacity due to the presence of rGO, large carrier density and convenient bandgap energy. This study opens up a new in situ synthetic strategy to prepare an NC, which can serve as an excellent nanocatalyst, toward environmental sustainability.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
Authors are thankful to Dr Divesh N. Srivastava, Principal Scientist, Analytical Division, Central Salt and Marine Chemicals Research Institute, Bhavnagar, India, for providing analytical facility. The author is indebted to UGC-RGNF (ID: RGNF-2015-17-SC-WES-690) for research funding. The authors are also grateful to the MHRD (India), the Department of Chemistry and Dr M. N. Dastur School of Materials Science and Engineering, IIEST, Shibpur for providing instrumental facilities.
References
- R. He, X. Huang and L. Feng, Energy Fuels, 2022, 36(13), 6675–6694 CrossRef CAS.
- H. Park, J. Kwon, J. Kim, K. Park, T. Song and U. Paik, Cryst. Growth Des., 2020, 20(5), 3325–3333 CrossRef CAS.
- S. I. Sadovnikov and A. I. Gusev, J. Mater. Chem. A, 2017, 5, 17676 RSC.
- Y. Xie, G. Bertoni, A. Riedinger, A. Sathya, M. Prato, S. Marras, R. Y. Tu, T. Pellegrino and L. Manna, Chem. Mater., 2015, 27, 7531–7537 CrossRef CAS PubMed.
- H. Park, J. Kwon, H. Choi, D. Shin, T. Song and X. W. D. Lou, ACS Nano, 2018, 12, 2827–2837 CrossRef CAS PubMed.
- S. Hu, X. Chen, Q. Li, Y. Zhao and W. Mao, Catal. Sci. Technol., 2016, 6, 5884–5890 RSC.
- X. Li, Y. Wei, C. Ma, H. Jiang, M. Gao, S. Zhang, W. Liu, P. Huo, H. Wang and L. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 11755–11764 CrossRef CAS PubMed.
- T. Iftikhar, Y. Xu, A. Aziz, G. Ashraf, G. Li, M. Asif, F. Xiao and H. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 31462–31473 CrossRef CAS PubMed.
- S. Gupta and V. (Ravi) Subramanian, ACS Appl. Mater. Interfaces, 2014, 6(21), 18597–18608 CrossRef CAS PubMed.
- G. Xie, K. Zhang, B. Guo, Q. Liu, L. Fang and J. R. Gong, Adv. Mater., 2013, 25, 3820–3839 CrossRef CAS PubMed.
- M. N. Hossain, J. Wen and A. Chen, Sci. Rep., 2017, 7, 3184 CrossRef PubMed.
- Y. Gu, S. Sun, Y. Liu, M. Dong and Q. Yang, ACS Omega, 2019, 4(18), 17672–17683 CrossRef CAS PubMed.
- X. Bai, L. Li, H. Liu, L. Tan, T. Liu and X. Meng, ACS Appl. Mater. Interfaces, 2015, 7(2), 1308–1317 CrossRef CAS PubMed.
- D. A. Brewster, Y. Bian and K. E. Knowles, Chem. Mater., 2020, 32(5), 2004–2013 CrossRef CAS.
- P. Ganguly, S. Mathew, L. Clarizia, S. Kumar R, A. Akande, S. J. Hinder, A. Breen and S. C. Pillai, ACS Omega, 2020, 5, 406–421 CrossRef CAS PubMed.
- M. Baek, E. J. Kim, S. W. Hong, W. Kim and K. Yong, Photochem. Photobiol. Sci., 2017, 16, 1792–1800 CrossRef CAS PubMed.
- P. Ganguly, S. Mathew, L. Clarizia, S. Kumar R, A. Akande, S. Hinder, A. Breen and S. C. Pillai, Appl. Catal., B, 2019, 253, 401–418 CrossRef CAS.
- A. N. Anju, N. Sultana, S. M. A. Nayem, A. Awal, S. C. Roy, M. A. Aziz and A. J. S. Ahammad, J. Electron. Mater., 2022, 51, 2877–2888 CrossRef CAS.
- T. Yu, L. Glennon, O. Fenelon and C. B. Breslin, Talanta, 2023, 251, 123758 CrossRef CAS PubMed.
- R. Karim, P. Saha, R. Akter, R. Falguni, R. A. Shital, A. Awal, M. A. Mamun and Dr. A. J. S. Ahammad, ChemistrySelect, 2023, 8(9), e202204174 CrossRef CAS.
- M. Darbandi, M. F. Mohajer, M. Eynollahi and K. Asadpour-Zeynali, Chem. Phys., 2022, 560, 111590 CrossRef CAS.
- C. Balischewski, H.-S. Choi, K. Behrens, A. Beqiraj, T. Körzdörfer, A. Geßner, A. Wedel and A. Taubert, ChemistryOpen, 2021, 10, 272–295 CrossRef PubMed.
- B. Panigrahy and S. Srivastava, New J. Chem., 2016, 40, 3370–3384 RSC.
-
E. Panha, H. Araki, Y. Akaki, M. Nakashima, T. Yamaguchi, S. Seto and S. Nakamura, 2 Thin-Film Compound Semiconductor PV., 2016.
- S. Nakamura, P. Eang, T. Yamaguchi, S. Seto, Y. Akaki, H. Katagiri and H. Araki, Phys. Status Solidi A, 2019, 216, 1800872 CrossRef.
- B. Yang, X. Zuo, P. Chen, L. Zhou, X. Yang, H. Zhang, G. Li, M. Wu, S. Jin and X. S. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 137–143 CrossRef CAS PubMed.
- Z. Chen, S. Liu, M.-Q. Yang and Y.-J. Xu, ACS Appl. Mater. Interfaces, 2013, 5(10), 4309–4319 CrossRef CAS PubMed.
- B. Yang, X. Zuo, P. Chen, L. Zhou, X. Yang, H. Zhang, G. Li, M. Wu, S. Jin and X. S. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 137–143 CrossRef CAS PubMed.
- A. Mondal, A. Prabhakaran, S. Gupta and V. R. Subramanian, ACS Omega, 2021, 6, 8734–8743 CrossRef CAS PubMed.
- J. Satra, U. K. Ghorui, P. Mondal, G. R. Bhadu and B. Adhikary, Dalton Trans., 2020, 49, 9464–9479 RSC.
- A. Rauf, M. S. Arif Sher Shah, J. Y. Lee, C. H. Chung, J. W. Bae and P. J. Yoo, RSC Adv., 2017, 7, 30533–30541 RSC.
- W. Li, J. Guo, L. Cai, W. Qi, Y. Sun, J. L. Xu, M. Sun, H. Zhu, L. Xiang, D. Xie and T. Ren, Sensors Actuators, B Chem., 2019, 290, 443–452 CrossRef CAS.
- H. Huang, X. Li, J. Wang, F. Dong, P. K. Chu, T. Zhang and Y. Zhang, ACS Catal., 2015, 5, 4094–4103 CrossRef CAS.
- U. K. Ghorui, J. Satra, P. Mondal, S. Mardanya, A. Sarkar, D. N. Srivastava, B. Adhikary and A. Mondal, Mater. Adv., 2021, 2(12), 4041–4057 RSC.
- D. Chaudhary, S. Singh, V. D. Vankar and N. Khare, J. Photochem. Photobiol., A, 2018, 351, 154–161 CrossRef CAS.
- P. Mondal, J. Satra, U. K. Ghorui, N. Saha, D. N. Srivastava and B. Adhikary, ACS Appl. Nano Mater., 2018, 1(11), 6015–6026 CrossRef CAS.
- P. Mondal, J. Satra, D. N. Srivastava, G. R. Bhadu and B. Adhikary, ACS Catal., 2021, 11, 3687–3703 CrossRef CAS.
- M. Sakthivel, R. Sukanya, S.-M. Chen and B. Dinesh, J. Phys. Chem. C, 2018, 122, 12474–12484 CrossRef CAS.
- J. Satra, P. Mondal, U. K. Ghorui and B. Adhikary, Sol. Energy Mater. Sol. Cells, 2019, 195, 24–33 CrossRef CAS.
-
J. Satra and B. Adhikary, Advances in Energy Research, Springer Nature, 2020, vol. 1, pp. 81–88 DOI:10.1007/978-981-15-2666-4_9.
- U. K. Ghorui, P. Mondal, J. Satra, B. Adhikary and A. Mondal, Mater. Sci. Semicond. Process., 2022, 143, 106559 CrossRef CAS.
- J. Zhang, J. Yu, M. Jaroniec and J. R. Gong, Nano Lett., 2012, 12, 4584–4589 CrossRef CAS PubMed.
- H. B. Ammar, M. B. Brahim, R. Abdelhédi and Y. Samet, J. Mol. Catal. A: Chem., 2016, 420, 222–227 CrossRef CAS.
- T. Pérez, S. Garcia-Segura, A. El-Ghenymy, J. L. Nava and E. Brillas, Electrochim. Acta, 2015, 165, 173–181 CrossRef.
- C. Ding, K. Fu, Y. Pan, J. Liu, H. Deng and J. Shi, Catalysts, 2020, 10, 1097 CrossRef CAS.
- Q. Xiao, Z. Si, J. Zhang, C. Xiao and X. Tan, J. Hazard. Mater., 2008, 150, 62–67 CrossRef CAS PubMed.
- P. Wang, J. Xian, J. Chen, Y. He, J. Wang, W. Li, Y. Shao and D. Li, Appl. Catal., B, 2014, 144, 644–653 CrossRef CAS.
- J. Wang, P. Wang, Y. Cao, J. Chen, W. Li, Y. Shao, Y. Zheng and D. Li, Appl. Catal., B, 2013, 136–137, 94–102 CrossRef CAS.
- J. Johny, S. Sepulveda-Guzman, B. Krishnan, D. A. Avellaneda, J. A. Aguilar Martinez and S. Shaji, Chem. Phys. Chem., 2016, 18, 1061–1068 CrossRef PubMed.
- X. Liu, S. P. C. Hsu, W.-C. Liu, Y.-M. Wang, X. Liu, C.-S. Lo, Y.-C. Lin, S. C. Nabilla, Z. Li, Y. Hong, C. Lin, Y. Li, G. Zhao and R.-J. Chung, Nanoscale Res. Lett., 2019, 14(189), 1–9 CAS.
- P. Mondal, U. K. Ghorui, J. Satra, S. Mardanya, D. N. Srivastava, G. R. Bhadu and B. Adhikary, ACS Appl. Nano Mater., 2020, 3(4), 3876–3891 CrossRef CAS.
-
C. M. A. Brett and A. M. O. Brett, Electrochemistry Principles, Methods, and Applications, Oxford University Press, Oxford, UK, 1st edn, 1993 Search PubMed.
- S. Ramaraj, S. M. Chen, B. Dinesh and K. H. Chen, J. Taiwan Inst. Chem. Eng., 2018, 82, 64–74 CrossRef.
- N. Karikalan, R. Karthik, S. M. Chen and H. A. Chen, Sci. Rep., 2017, 7, 1–10 CrossRef CAS PubMed.
- T. Kokulnathan and S.-M. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 7893–7905 CrossRef CAS PubMed.
- N. Xiao, J. Deng, J. Cheng, S. Ju, H. Zhao, J. Xie, D. Qian and J. He, Biosens. Bioelectron., 2016, 81, 54–60 CrossRef CAS PubMed.
- A. Mao, H. Li, L. Yu and X. Hu, J. Electroanal. Chem., 2017, 799, 257–262 CrossRef CAS.
- E. Shahnazari-Shahrezaie and A. Nezamzadeh-Ejhieh, RSC Adv., 2017, 7, 14247–14253 RSC.
- J. Peng, C. Hou and X. Hu, Sens. Actuators, B, 2017, 169, 81–87 CrossRef.
- J. Huang, X. Shen, R. Wang, Q. Zeng and L. Wang, RSC Adv., 2017, 7, 535–542 RSC.
- M. Zhu, H. Ye, M. Lai, J. Ye, J. Kuang, Y. Chen, J. Wang and Q. Mei, Int. J. Electrochem. Sci., 2018, 13, 4100–4114 CrossRef CAS.
- Y. Gu, X. Yan, W. Liu, C. Li, R. Chen, L. Tang, Z. Zhang and M. Yang, Electrochim. Acta, 2018, 152, 108–116 CrossRef.
- P. Sundaresan, T. W. Chen, S. M. Chen, T. W. Tseng and X. H. Liu, Int. J. Electrochem. Sci., 2018, 13, 1441–1451 CrossRef CAS PubMed.
- M. Sakthivel, R. Sukanya, S. M. Chen and B. Dinesh, J. Phys. Chem. C, 2018, 122, 12474–12484 CrossRef CAS.
- T. Kokulnathan and S. M. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 7893–7905 CrossRef CAS PubMed.
- L. Zhang, M. Yin, J. Qiu, T. Qiu, Y. Chen, S. Qi, X. Wei, X. Tian and D. Xu, Biosens. Bioelectron.: X, 2022, 10, 100102 CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Papri
Mondal
a,
Papri
Mondal